

Abstract
The difference in [3 + 2] cycloaddition reactivity between fac-[MO3(tacn)]+ (M = Re, 99Tc; tacn = 1,4,7-triazacyclononane) complexes has been reexamined with a selection of unsaturated substrates including sodium 4-vinylbenzenesulfonate, norbornene, 2-butyne, and 2-methyl-3-butyn-2-ol (2MByOH). None of the substrates was found to react with the Re cation in water at room temperature, whereas the 99Tc reagent cleanly yielded the [3 + 2] cycloadducts. Interestingly, a bis-adduct was obtained as the sole product for 2MByOH, reflecting the high reactivity of a 99TcO-enediolato monoadduct. On the basis of scalar relativistic and nonrelativistic density functional theory calculations of the reaction pathways, the dramatic difference in reactivity between the two metals has now been substantially attributed to differences in relativistic effects, which are much larger for the 5d metal. Furthermore, scalar-relativistic ΔG values were found to decrease along the series propene > norbornene > 2-butyne > dimethylketene, indicating major variations in the thermodynamic driving force as a function of the unsaturated substrate. The suggestion is made that scalar-relativistic effects, consisting of greater destabilization of the valence electrons of the 5d elements compared with those of the 4d elements, be viewed as a new design principle for novel 99mTc/Re radiopharmaceuticals, as well as more generally in heavy-element coordination chemistry.
Short abstract
Room temperature cycloaddition reactivity of fac-[99TcO3(tacn)]+ (tacn = 1,4,7-triazacyclononane) with a variety of unsaturated substrates and the lack of such reactivity for fac-[ReO3(tacn)]+ appears largely attributable to much stronger relativistic effects for Re relative to Tc, based on relativistic density functional theory calculations.
Introduction
Technetium-99m, a metastable nuclear isomer of technetium-99, is the most commonly used radioisotope in medicine, and the demand for 99mTc radiopharmaceuticals with novel biodistribution properties is considerable.1−4 A common early step toward the development of these products involves model chemistries with 99Tc and Re. Although the two elements are chemically very similar, they exhibit quantitative differences in reactivity, reflecting the somewhat greater stability (and lower reduction potentials) of the higher oxidation states of Re. In a seminal finding, Pearlstein and Davison in the 1980s showed that fac-[99TcVIIO3]+ complexes undergo [3 + 2] cycloadditions with olefins to yield 99TcVO diolate derivatives.5 The analogous ReVO-diolate species, in contrast, were found to be unstable, undergoing the opposite reaction when thermalized. We built on this finding to develop fac-[99mTcVIIO3]+ complexes as aqueous-phase labeling agents for olefins.6−8 The factors underlying the difference in reactivity between the two group 7 elements, however, have remained obscure. Physicochemical measurements at the Tromsø laboratory on analogous pairs of 4d and 5d metallocorroles,9−11 including those involving Mo12/W,1399TcVO14/ReVO,15 RuVIN16/OsVIN,17 and Ag/Au,18,19 suggested that relativistic effects might partly explain the difference in cycloaddition reactivity between 99m/99Tc and Re.20
Unfortunately, little is known about the importance of relativistic effects for transition-metal reactivity.21−23 For most of the 20th century, relativistic effects were not considered important for chemistry. Indeed, in 1929, Paul Dirac asserted that the only imperfections remaining in quantum mechanics “give rise to difficulties only when high-speed particles are involved, and are therefore of no importance in the consideration of atomic and molecular structure and ordinary chemical reactions in which it is, indeed, usually sufficiently accurate if one neglects relativity variation of mass and velocity and assumes only Coulomb forces between the various electrons and atomic nuclei.”24 This view started changing only in the 1970s.25,26 Today the importance of relativistic effects is well recognized for the static properties of sixth- and seventh-period elements.27 Relativity thus accounts for such well-known effects as the liquid state of Hg28 and the yellow color of elemental Au29 and Cs as well as a host of less well-known effects in heavy-element chemistry.30−33
Results and Discussion
Synthetic and Reactivity Studies
With the above as the backdrop, we chose to perform a comparative study of fac-[MO3(tacn)]+ (M = Re, 99Tc; tacn = 1,4,7-triazacyclononane) complexes with respect to their [3 + 2] cycloaddition reactivity with a selection of unsaturated substrates including sodium 4-vinylbenzenesulfonate, norbornene, 2-butyne, and 2-methyl-3-butyn-2-ol (2MByOH; Scheme 1). Because we already knew from our recent work that fac-[99TcO3(tacn)]+ reacts with a broad range of olefins to yield 99TcO-diolate products, we focused here particularly on complexes of the type fac-[ReO3(tacn)]X (X = Cl, BPh4).34 We verified that the Re complexes do not react with olefins and alkynes, as indeed was expected from Pearlstein and Davison’s original observations.6
Scheme 1. Cycloaddition of [99TcO3(tacn)]+ with Alkenes.

Because alkynes had not been examined as substrates until now, we chose to examine the interaction of the water-stable complex fac-[99TcO3(tacn)]Cl8 with the water-soluble propargylic alcohol 2MByOH. After the addition of 2 equiv of the propargylic alcohol to an aqueous solution of fac-[99TcO3(tacn)]Cl, a quick color change was observed from yellow to green. After stirring for 2 h at room temperature, the dinuclear complex [{99Tc(O)O2(tacn)}2(2MByOH)]Cl2 was isolated as the sole product following removal of all volatiles under high vacuum. No mononuclear intermediate was detected by either high-performance liquid chromatography (HPLC) or NMR. This finding suggests that the expected 99TcO-enediolate intermediate acts as a highly reactive substrate for a second equiv of fac-[99TcO3(tacn)]+ to yield the observed bis-adduct (Scheme 2).
Scheme 2. Double [3 + 2] Cycloaddition of Two fac-[99TcO3(tacn)]+ Cations with 2MByOH (Showing One of the Two Diastereomers Formed).

The Fourier transform infrared spectrum of [{99TcV(O)O2(tacn)}2(2MByOH)]Cl2 was found to exhibit a νTc=O band at 967 cm–1, considerably upshifted relative to that in [99TcO(tacn)(eg)]+ (949 cm–1; eg = ethane-1,2-diolato).35 Given that two symmetry-nonequivalent addition modes are conceivable for the second equiv of fac-[99TcO3(tacn)]+, 1H and 13C NMR spectroscopy of [{99TcV(O)O2(tacn)}2(2MByOH)]Cl2 understandably indicated the formation of two diastereomers in a 2:1 ratio (Scheme 3).36 Slow evaporation of an aqueous solution of the product in the presence of excess KBr led to crystallization of the major diastereomer of [{99TcV(O)O2(tacn)}2(2MByOH)]Br2 (isomer 1 in Scheme 3). Single-crystal X-ray diffraction analysis (Table 1 and Figure 1) revealed an intramolecular N4–H···O7 hydrogen bond, which, along with less overall steric crowding, appears to be responsible for the formation of isomer 1 as the major product. In contrast to the [3 + 2] cycloadducts of fac-[99TcO3(tacn)]+ with alkenes, slow decomposition of isomer 1 of the bisadduct (formation of ([TcO4]−) was observed over days.
Scheme 3. Observed Isomers of [{99TcV(O)O2(tacn)}2(2MByOH)]Cl2.

Table 1. Crystal Data and Structure Refinement for [{99TcV(O)O2(tacn)}2(2MByOH)]Br2·2.2H2O.
| empirical formula | C17H42Br2N6O9.20Tc2 |
| diffractometer | Xcalibur, Ruby diffractometer |
| wavelength (Å) | 0.71073 |
| fw | 833.58 |
| cryst syst | monoclinic |
| space group | P21/c |
| a (Å) | 16.5494(7) |
| b (Å) | 13.3352(5) |
| c (Å) | 14.509(2) |
| α (deg) | 90 |
| β (deg) | 114.955(11) |
| γ (deg) | 90 |
| volume (Å3) | 2903.1(6) |
| Z | 4 |
| density (calcd) (g cm–3) | 1.907 |
| temperature (K) | 183.1 |
| abs coeff (mm–1) | 3.758 |
| F(000) | 1662 |
| cryst size (mm3) | 0.234 × 0.145 × 0.075 |
| cryst description | green block |
| θ range for data collection (deg) | 2.715–30.508 |
| index ranges | –23 ≤ h ≤ 23, −19 ≤ k ≤ 19, −19 ≤ l ≤ 20 |
| reflns collected | 41201 |
| indep reflns | 8809 [R(int) = 0.0396) |
| reflns obsd | 7686 |
| criterion for observation | I > 2(I) |
| completeness to θ = 25.242° (%) | 99.0 |
| abs corrn | semiempirical from equivalents |
| max and min transmn | 1.000 and 0.789 |
| data/restraints/param | 8809/6/362 |
| GOF on F2 | 1.054 |
| final R indices [I > 2σ(I)] | R1 = 0.0469, wR2 = 0.1202 |
| R indices (all data) | R1 = 0.0550, wR2 = 0.1250 |
| largest diff peak and hole (e Å–3) | 2.533 and −2.461 |
| CCDC | 2071332 |
Figure 1.
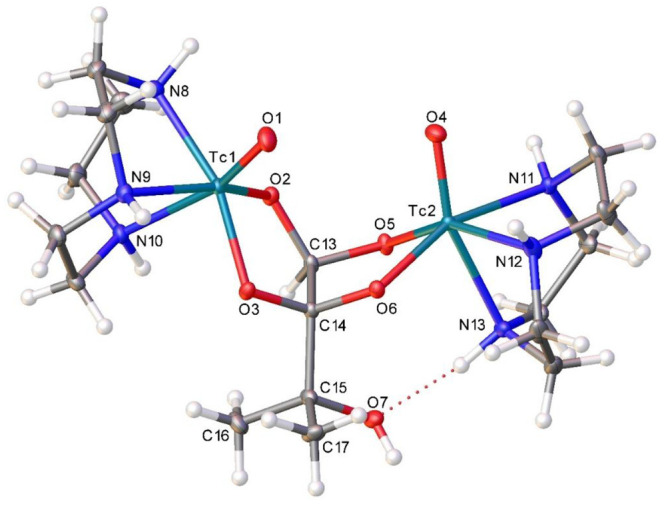
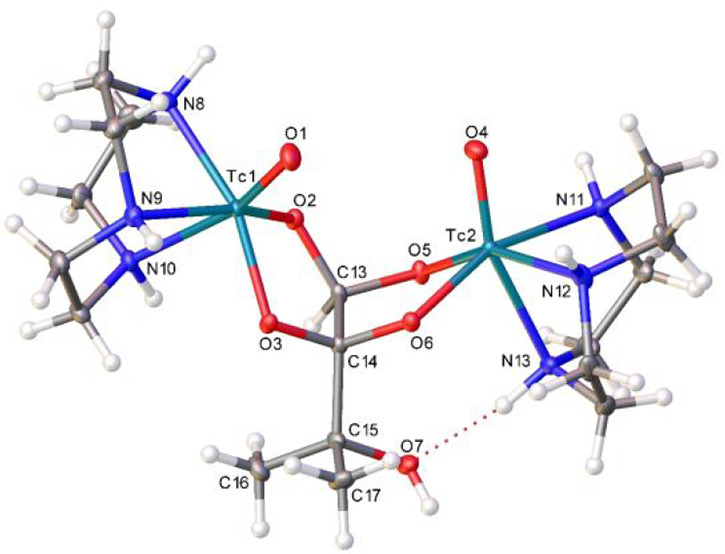
Thermal ellipsoid (50% probability) plot for [{99TcV(O)O2(tacn)}2(2MByOH))Br2. Bromide ions and water molecules have been omitted for clarity. Selected bond distances (Å) and angles (deg): Tc1–O1 1.661(3), Tc1–O2 1.926(3), Tc2–O4 1.665(3), Tc2–O5 1.946(3), Tc1–N8 2.163(4), Tc1–N9 2.175(4), Tc1–N10 2.295(4), Tc2–N11 2.185(3), Tc2–N12 2.147(4), Tc2–N13 2.250(4); O1–Tc1–O2 112.86(16), O2–Tc1–O3 81.73(12), O4–Tc2–O5 108.23(16), O5–Tc2–O6 81.42(12), O2–C13–O5 107.6(3), O3–C14–O6 108.8(3).
Theoretical Modeling
Relativistic and nonrelativistic density functional theory (DFT) calculations (typically with large all-electron STO-TZ2P basis sets; see Experimental Section for details) were used to investigate the [3 + 2] cycloaddition of the cationic complexes [MO3(tacn)]+ (M = Tc, Re) with four different olefins, namely, propene, dimethylketene, 2-butyne, and norbornene, in acetonitrile (MeCN) as a solvent (Table 2). Relativity was taken into account either via effective core potentials (ECPs) or with a scalar-relativistic treatment with the zeroth-order regular approximation (ZORA). Two-component spin–orbit relativistic calculations were undertaken in a few cases as random checks on the quality of the ECP and scalar-relativistic results; the latter results were indeed found to be adequate, with minimal differences relative to the spin–orbit calculations. The data in Table 1 led to the following conclusions.
Table 2. Scalar-Relativistic and Nonrelativistic DFT Energetics (eV) for Different Substrates in CH3CN.
| |
B3LYPscalara |
B3LYPnrela |
PBE0scalara |
PBE0nrela |
OPBE0scalara |
PBE-D2ECPb |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| substrate | metal | ΔG | ΔG⧧ | ΔG | ΔG⧧ | ΔG | ΔG⧧ | ΔG | ΔG⧧ | ΔG | ΔG⧧ | ΔG | ΔG⧧ |
| propene | Tc | –0.38 | 0.83 | –0.77 | 1.09 | –0.91 | 1.13 | –1.31 | 0.98 | –0.71 | 1.46 | –0.48 | 0.59 |
| Re | 1.28 | 1.86 | –0.81 | 1.10 | 0.33 | 1.69 | –1.38 | 0.98 | 0.54 | 2.01 | 0.55 | 1.09 | |
| dimethylketene | Tc | –1.44 | 1.28 | –1.83 | 1.17 | –2.02 | 1.20 | –2.42 | 1.10 | –1.77 | 1.62 | –1.47 | 0.50 |
| Re | –0.21 | 1.91 | –1.74 | 1.05 | –0.76 | 1.77 | –2.41 | 0.92 | –0.50 | 2.32 | –0.41 | 0.93 | |
| 2-butyne | Tc | –1.20 | 1.35 | –1.62 | 1.26 | –1.76 | 1.25 | –2.18 | 1.14 | –1.58 | 1.61 | –1.35 | 0.70 |
| Re | –0.21 | 1.91 | –1.50 | 1.25 | –0.48 | 1.74 | –2.10 | 1.09 | –0.30 | 2.07 | –0.21 | 1.11 | |
| norbornene | Tc | –0.73 | 1.10 | –1.14 | 0.92 | –1.25 | 0.98 | –1.67 | 0.83 | –1.00 | 1.35 | –0.96 | 0.32 |
| Re | 0.48 | 1.67 | –1.20 | 0.80 | –0.02 | 1.49 | –1.77 | 0.66 | 0.24 | 1.87 | 0.07 | 0.73 | |
Obtained with ADF.
Obtained with Gaussian.
Relativistic calculations indicate dramatically lower (in an algebraic sense) reaction free energies (ΔG) and free energies of activation (ΔG⧧) for Tc than for Re, consistent with the experimentally observed difference in reactivity between the two metals. These translate to substantially “earlier” transition states for Tc than for Re; in other bonds, key bonds affected by the reaction are rather similar in length to the starting materials for the Tc reactions compared with the Re reactions (Figure 2). In sharp contrast, nonrelativistic calculations (B3LYPnrel and PBE0nrel in Table 2) indicate similar ΔG and ΔG⧧ values for the two metals. The fact that these generalizations hold regardless of the exchange-correlation functional and the organic substrate indicates that the difference in reactivity between the two metals is largely a relativistic effect.
Figure 2.

Ball-and-stick diagrams, with key distances (Å), for the optimized PBE-D2ECP stationary points for the [3 + 2] cycloaddition of [MO3(tacn)]+ and norbornene. M = Tc (left), Re (right).
The above interpretation is supported by computations of the adiabatic electron affinities (EAs) for the M(VII) d0 complexes MeTcVIIO3 and MeReVIIO3 (Me = methyl). At the scalar relativistic level, the B3LYP values are 3.44 and 2.79 eV, respectively, i.e., the EA of the Tc(VII) complex is 650 meV higher than that of the Re(VII) complex. The scalar-relativistic PBE0 values are similar, 3.31 and 2.65 eV, as are the PBE-D2ECP values, 3.67 and 3.02 eV. At the nonrelativistic level, the B3LYP EAs are 3.64 and 3.32 eV, while the PBE0 EAs are 3.51 and 3.20 eV, respectively, which translates to a difference of just over 300 meV between the two metals. These results prove that the difference in the EAs or reduction potentials between the Tc(VII) and Re(VII) species is substantially ascribable to the relativistic destabilization of the Re 5d orbitals relative to the Tc 4d orbitals. Much the same considerations should apply to the cycloaddition reaction of interest in this study because it also involves a reduction, albeit a two-electron one, of the M(VII) centers.
Another key observation from Table 2 is that the ΔG values, which decrease along the series propene > norbornene > 2-butyne > dimethylketene, reflect dramatic variations in the thermodynamic driving force as a function of the olefinic substrate. In fact, for propene, all of the relativistic methods yield positive ΔG values, consistent with the experimental observation that simple, unstrained olefins do not react with cationic [ReVIIO3]+ reagents at room temperature.37 Interestingly, much smaller variations are observed among the ΔG⧧ values for the four substrates. Again, for Re, the calculations generally indicate the highest ΔG⧧ value for propene and lower values for dimethylketene and norbornene.
The above calculations are far from perfect. While the ΔG values are moderately consistent across different functionals (for the relativistic calculations), the ΔG⧧ values exhibit much wider variations. Of the different functionals examined, PBE-D2ECP appears to yield the lowest, and probably most realistic, ΔG⧧ values, which has also been observed in a DFT study of Ir-catalyzed reactions.38 Overall, our results underscore the need for substantial additional benchmarking of different functionals vis-à-vis transition-metal-mediated redox reactions, especially for 4d and 5d elements.
Conclusion
In earlier studies of metalloporphyrin-type compounds,9–11 we concluded that the difference in redox potential between analogous 4d and 5d complexes is largely attributable to scalar relativistic effects, much as calculated for ΔG and ΔG⧧ values in the present study. The greater relativistic destabilization of the valence electrons of the 5d elements compared with those of the 4d elements thus may be viewed as a reliable design principle for novel 99mTc radiopharmaceuticals, as well as more generally in heavy-element coordination chemistry. In other words, higher-valent technetium species such as pertechnetate or fac-[99/99mTcO3]+ derivatives should be much more easily reduced (i.e., accept electrons in their 4d orbitals) than isoelectronic Re species (where electrons would be added to 5d orbitals). This prediction—in this case, a postdiction—is nicely illustrated by the facile synthesis of 99(m)Tc(I) organometallic39 compounds via the reduction of pertechnetate, the analogous synthesis of Re(I) organometallics being far less facile. We look forward to seeing additional applications of relativity as a design principle in the synthesis of new classes of heavy/element coordination compounds.
Experimental Section
Instrumental Methods
IR spectra were measured as KBr pellets on a PerkinElmer BXII spectrometer. 1H and 13C NMR were recorded on a Bruker AV2-500 500-MHz spectrometer. Reactivity studies with Re compounds were performed on a Waters Acquity UPLC System coupled to a Bruker Daltonics HCTTM electrospray ionization mass spectrometer, using an Acquity UPLC BEH C18 1.7 μm (2.1 × 50 mm) column. Ultraperformance liquid chromatography (UPLC) solvents were formic acid (0.1% in Millipore water) (solvent A) and UPLC-grade MeCN (solvent B). Applied UPLC gradient: 0–0.5 min, 95% A and 5% B; 0.5–4.0 min, linear gradient from 95% A and 5% B to 0% A and 100% B; 4.0–5.0 min, 0% A and 100% B. The flow rate was 0.6 mL min–1. Detection was performed at 250 and 480 nm (DAD). Reactivity studies with Tc compounds were performed on a Merck Hitachi LaChrom L7100 pump coupled to a Merck Hitachi LaChrom L7200 tunable UV detector. The detection of radioactive 99Tc complexes was performed with an equipped Berthold LB508 radiodetector. Separations were achieved on a Macherey-Nagel C18 reversed-phase column (EC-250/3 Nucleosil 100-5 C18), using a gradient of triethylamine phosphate (TEAP)/MeCN as the eluent, with a flow rate of 0.5 mL min–1. TEAP method: t = 0–3 min, 100% TEAP; 3–3.1 min, 100–75% TEAP; 3.1–9 min, 75% TEAP; 9–9.1 min, 75–66% TEAP, 9.1–12 min, 66% TEAP; 12–12.1 min, 66–0% TEAP, 15–15.1 min, 0–100% TEAP; 15.1–18 min, 100% TEAP.
Synthetic and Reactivity Studies
Caution!99Tc is a weak β emitter. All experiments were performed in laboratories approved for working with low-level radioactive materials.
[99TcO3(tacn)]Cl was prepared as previously reported.40 Double-distilled water (dd-water) was used throughout. All chemicals were of reagent-grade quality or higher and were obtained from commercial suppliers.
Synthesis of [ReO3(tacn)][ReO4].41
Dirhenium heptoxide (520 mg, 1.1 mmol) was dissolved in dry tetrahydrofuran (THF; 5.0 mL). A solution of 1,4,7-triazacyclononane (125 mg, 0.96 mmol) in dry THF (1.0 mL) was added, and the resulting mixture was stirred for 30 min at room temperature. The colorless precipitate was filtered off and dried under vacuum. Yield: 98% (589 mg, 0.96 mmol).
Synthesis of [ReO3(tacn)](BPh4)
The aforementioned complex [ReO3(tacn)][ReO4] (188 mg, 0.31 mmol) was dissolved in distilled water (10 mL). A solution of sodium tetraphenylborate (210 mg, 0.61 mmol) dissolved in water (5 mL) was added, and the resulting mixture was stirred for 30 min at room temperature. The product precipitated as a pale-gray solid and was filtered off. Yield: 59% (121 mg, 0.18 mmol). Analytical data are in agreement with the literature.
Synthesis of [ReO3(tacn)]Cl
DOWEX-1 anion-exchange resin in chloride form (1000 mg) was washed with dd-water until the washings showed a pH of 7.0. The resin was then added to a solution of [ReO3(tacn)][ReO4] (183 mg, 0.3 mmol) in water (5.0 mL), and the suspension was stirred for 30 min at room temperature. The resin was filtered off, and [ReO3(tacn)]Cl was isolated by evaporation of the solvent under high vacuum. The successful exchange of [ReO4]− by Cl– was proven by IR and electrospray ionization mass spectrometry (negative mode). Yield: 66% (79 mg, 0.20 mmol). Analytical data are in agreement with the literature.42
Reactions of [ReO3(tacn)](BPh4) in MeCN with Alkenes and Alkynes
To a solution of [ReO3(tacn)](BPh4) (36 mg, 0.05 mmol) in MeCN (3.0 mL) was added the olefin or alkyne of interest (0.5 mmol), and the reaction mixture was stirred for 2 h at room temperature, followed by UPLC–MS analysis. If no reaction was observed, the temperature was raised to 85 °C for 2 h, and the reaction mixture was again analyzed by UPLC–MS. We found no evidence for the formation of a [3 + 2] cycloadduct for either norbornene or 2-butyne.
Reactions of [ReO3(tacn)]Cl in Water with Alkenes and Alkynes
To a solution of [ReO3(tacn)]Cl (18 mg, 0.05 mmol) dissolved in dd-water (2.0 mL) was added a water-soluble olefin or alkyne (0.5 mmol), and the reaction mixture was stirred for 2 h at room temperature, followed by UPLC–MS analysis. If no reaction was observed, the temperature was raised to 85 °C for 2 h, and the reaction mixture was again analyzed by UPLC–MS. We found no evidence for the formation of a [3 + 2] cycloadduct for either 2MByOH or sodium 4-vinylbenzenesulfonate.
Synthesis of [{99Tc(O)O2(tacn)}2(2MByOH)]Cl2
To a yellow solution of [99TcO3(tacn)]Cl (6.23 mg, 0.02 mmol) in dd-water (1.0 mL) was added 2MByOH (4 μL, 0.04 mmol), resulting in a rapid color change to green. After stirring for 2 h at room temperature, the solvent and other volatiles were removed under high vacuum, affording [{99Tc(O)O2(tacn)}2(2MByOH)]Cl2 in quantitative yield. IR [cm–1]: 3456s, 3412s, 3120m, 2991w, 2913w, 2845w, 2050w, 1637s, 1619s, 1541w, 1488w, 1455w, 1423w, 1381w, 1356w, 1286w, 1264w, 1230w, 1174w, 1110w, 1064m, 1014m, 967s, 931m, 847w, 837m, 802w, 746w, 716w, 676w, 621w, 601w, 565w, 525w, 467w, 436w. 1H NMR (500 MHz, D2O): δ 8.11 (s, CH isomer 1, 1 H), 7.58 (s, CH isomer 2, 1 H), 3.77–2.20 (m, tacn, 36 H), 1.60 (s, CH3 isomer 1, 6 H), 1.45 (s, CH3 isomer 2, 3 H), 1.24 (s, CH3 isomer 2, 3 H). 13C NMR (125 MHz, D2O): δ 129.21 (O2CRR′, 1 C), 123.94 (CH isomer 2, 1 C), 120.07 (CH isomer 1, 1 C), 57.94–45.21 (tacn, 6 C), 28.02 (CH3 isomer 2, 1 C), 26.98 (CH3 isomer 1, 2 C), 25.07 (CH3 isomer 2, 1 C). See Scheme 3 for a definition of isomers 1 and 2.
Crystals of [{99Tc(O)O2(tacn)}2(2MByOH)]Br2 suitable for single-crystal X-ray diffraction analysis were obtained by slow evaporation of an aqueous solution of the product in the presence of excess KBr.
X-ray Structure Analysis
Crystallographic data were collected at 183(2) K with Mo Kα radiation (λ = 0.7107 Å) monochromatized with graphite on an Oxford Diffraction Xcalibur system with a Ruby detector. Suitable crystals were covered with oil (Infineum V8512, formerly known as Paratone N), mounted atop a glass fiber, and immediately transferred to the diffractometer. The CrysAlisPro(43) program suite was used for data collection, semiempirical absorption correction, and data analysis. The structure was solved with direct methods using SIR97(44) and refined by full-matrix least-squares methods on F2 with SHELXL-2018(45) using the Olex2 GUI.46 The refinement was done with anisotropic thermal parameters for all non-H atoms, unless otherwise indicated. The positions of the H atoms were calculated using the “riding atom” option in SHELXL-2018. More details on data collection and structure calculations are given in Table 1 and in the crystallographic information file.
Computational Methods
The majority of DFT calculations (including full geometry optimizations in the presence of a solvent) were carried out with the ADF 2018 program system.47 Relativistic effects were taken into account with the ZORA48 method, applied both as a scalar correction and with spin–orbit coupling at the two-component level. A parallel set of calculations were carried out with the same basis set but with a nonrelativistic Hamiltonian. Specially optimized all-electron ZORA STO-TZ2P basis sets were used throughout. A variety of exchange-correlation functionals were tested, including OLYP,49,50 B3LYP,51,52 PBE0,53,54 and OPBE0.55 The potential influence of dispersion corrections was examined, and, in general, they did not make a significant difference. Our results therefore generally refer to the pristine functionals. Zero-point energy and thermal corrections (vibrational, rotational, and translational) were made to the electronic energies in the calculation of the thermodynamic parameters. Enthalpies (H) and Gibbs free energies (G) were calculated from
| 1 |
| 2 |
| 3 |
where U is the gas-phase thermodynamic energy, Eel the total electronic energy, and Enuc the nuclear internal energy (sum of the vibrational, rotational, and translational energies and the zero-point energy correction); R is the ideal gas constant, T the temperature, and S the entropy. S was calculated from the temperature-dependent partition function in ADF at 298.15 K. Solvent effects were taken into account with COSMO (conductor-like screening model),56−58 as implemented59 in ADF. The type of cavity used is Esurf,60 and the solvent used was MeCN (eps = 37.5; Rad = 2.76).
The Gaussian 16 program system61 was used for the PBE-D26263 calculations. The basis set was 6-311G(d,p) on all nonmetallic atoms and LANL2DZ with an ECP augmented with one f-polarization function on Re (0.869) and Tc (1.134). The polarizable continuum model (PCM)64 as its integral equation formalism variant (IEFPCM)65 was used for solvent (MeCN) calculations in Gaussian.
Acknowledgments
This work was supported by Grant PZ00P2_126414 of the Swiss National Science Foundation, Grant 262229 of the Research Council of Norway, and Grants 129270 and 132504 of the South African National Research Foundation to J.C.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.1c00995.
Selected spectra, additional crystallographic details, and optimized DFT coordinates (PDF)
Accession Codes
CCDC 2071332 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no competing financial interest.
Supplementary Material
References
- Abram U.; Alberto R. Technetium and rhenium: coordination chemistry and nuclear medical applications. J. Braz. Chem. Soc. 2006, 17, 1486–1500. 10.1590/S0103-50532006000800004. [DOI] [Google Scholar]
- Eckelman W. C. Unparalleled Contribution of Technetium-99m to Medicine Over 5 Decades. J. Am. Coll. Cardiol. Cardiovasc. Imaging 2009, 2, 364–368. 10.1016/j.jcmg.2008.12.013. [DOI] [PubMed] [Google Scholar]
- Jürgens S.; Herrmann W. A.; Kühn F. E. Rhenium and technetium based radiopharmaceuticals: Development and recent advances. J. Organomet. Chem. 2014, 751, 83–89. 10.1016/j.jorganchem.2013.07.042. [DOI] [Google Scholar]
- Papagiannopoulou D. Technetium-99m radiochemistry for pharmaceutical applications. J. Labelled Compd. Radiopharm. 2017, 60, 502–520. 10.1002/jlcr.3531. [DOI] [PubMed] [Google Scholar]
- Pearlstein R. M.; Davison A. Alkene—glycol interconversion with technetium and rhenium oxo complexes. Polyhedron 1988, 7, 1981–1989. 10.1016/S0277-5387(00)80713-5. [DOI] [Google Scholar]
- Tooyama Y.; Braband H.; Spingler B.; Abram U.; Alberto R. High-Valent Technetium Complexes with the [99TcO3]+ Core from in situ Prepared Mixed Anhydrides of [99TcO4]− and Their Reactivities. Inorg. Chem. 2008, 47, 257–264. 10.1021/ic701908q. [DOI] [PubMed] [Google Scholar]
- Braband H.; Tooyama Y.; Fox T.; Alberto R. Syntheses of High-Valent fac-[99mTcO3]+ Complexes and [3 + 2] Cycloadditions with Alkenes in Water as a Direct Labelling Strategy. Chem. - Eur. J. 2009, 15, 633–638. 10.1002/chem.200801757. [DOI] [PubMed] [Google Scholar]
- Braband H.; Tooyama Y.; Fox T.; Simms R.; Forbes J.; Valliant J. F.; Alberto R. fac-[TcO3(tacn)]+: A Versatile Precursor for the Labelling of Pharmacophores, Amino Acids and Carbohydrates through a New Ligand-Centred Labelling Strategy. Chem. - Eur. J. 2011, 17, 12967–12974. 10.1002/chem.201101275. [DOI] [PubMed] [Google Scholar]
- Ghosh A. Electronic Structure of Corrole Derivatives: Insights from Molecular Structures, Spectroscopy, Electrochemistry, and Quantum Chemical Calculations. Chem. Rev. 2017, 117, 3798–3881. 10.1021/acs.chemrev.6b00590. [DOI] [PubMed] [Google Scholar]
- Alemayehu A. B.; McCormick L. J.; Vazquez-Lima H.; Ghosh A. Relativistic Effects on a Metal–Metal Bond: Osmium Corrole Dimers. Inorg. Chem. 2019, 58, 2798–2806. 10.1021/acs.inorgchem.8b03391. [DOI] [PubMed] [Google Scholar]
- Alemayehu A. B.; Thomas K. E.; Einrem R. F.; Ghosh A.. The Story of 5d Metallocorroles: From Metal-Ligand Misfits to New Building Blocks for Cancer Phototherapeutics. Acc. Chem. Res. 2021, Article ASAP. 10.1021/acs.accounts.1c00290. [DOI] [PMC free article] [PubMed]
- Alemayehu A. B.; Vazquez-Lima H.; McCormick L. J.; Ghosh A. Relativistic effects in metallocorroles: comparison of molybdenum and tungsten biscorroles. Chem. Commun. 2017, 53, 5830–5833. 10.1039/C7CC01549F. [DOI] [PubMed] [Google Scholar]
- Alemayehu A. B.; Vazquez-Lima H.; Gagnon K. J.; Ghosh A. Tungsten Biscorroles: New Chiral Sandwich Compounds. Chem. - Eur. J. 2016, 22, 6914–6920. 10.1002/chem.201504848. [DOI] [PubMed] [Google Scholar]
- Einrem R. F.; Braband H.; Fox T.; Vazquez-Lima H.; Alberto R.; Ghosh A. Synthesis and Molecular Structure of 99Tc Corroles. Chem. - Eur. J. 2016, 22, 18747–18751. 10.1002/chem.201605015. [DOI] [PubMed] [Google Scholar]
- Einrem R. F.; Gagnon K. J.; Alemayehu A. B.; Ghosh A. Metal-Ligand Misfits: Facile Access to Rhenium-Oxo Corroles by Oxidative Metalation. Chem. - Eur. J. 2016, 22, 517–520. 10.1002/chem.201504307. [DOI] [PubMed] [Google Scholar]
- Alemayehu A. B.; Vazquez-Lima H.; Gagnon K. J.; Ghosh A. Stepwise Deoxygenation of Nitrite as a Route to Two Families of Ruthenium Corroles: Group 8 Periodic Trends and Relativistic Effects. Inorg. Chem. 2017, 56, 5285–5294. 10.1021/acs.inorgchem.7b00377. [DOI] [PubMed] [Google Scholar]
- Alemayehu A. B.; Gagnon K. J.; Terner J.; Ghosh A. Oxidative Metalation as a Route to Size-Mismatched Macrocyclic Complexes: Osmium Corroles. Angew. Chem., Int. Ed. 2014, 53, 14411–14414. 10.1002/anie.201405890. [DOI] [PubMed] [Google Scholar]
- Thomas K. E.; Vazquez-Lima H.; Fang Y.; Song Y.; Gagnon K. J.; Beavers C. M.; Kadish K. M.; Ghosh A. Ligand Noninnocence in Coinage Metal Corroles: A Silver Knife-Edge. Chem. - Eur. J. 2015, 21, 16839–16847. 10.1002/chem.201502150. [DOI] [PubMed] [Google Scholar]
- Thomas K. E.; Alemayehu A. B.; Conradie J.; Beavers C. M.; Ghosh A. Synthesis and Molecular Structure of Gold Triarylcorroles. Inorg. Chem. 2011, 50, 12844–12851. 10.1021/ic202023r. [DOI] [PubMed] [Google Scholar]
- Metal oxidation states are omitted henceforth, unless essential for the purposes of clarity.
- Gorin D. J.; Toste F. D. Relativistic effects in homogeneous gold catalysis. Nature 2007, 446, 395–403. 10.1038/nature05592. [DOI] [PubMed] [Google Scholar]
- Demissie T. B.; Garabato B. D.; Ruud K.; Kozlowski P. M. Mercury Methylation by Cobalt Corrinoids: Relativistic Effects Dictate the Reaction Mechanism. Angew. Chem., Int. Ed. 2016, 55, 11503–11506. 10.1002/anie.201606001. [DOI] [PubMed] [Google Scholar]
- Takashima C.; Ikabata Y.; Kurita H.; Takano H.; Shibata T.; Nakai H. Relativistic Effect on Homogeneous Catalytic Reaction by Cationic Iridium Catalysts. J. Comput. Chem., Jpn. 2019, 18, 136–138. 10.2477/jccj.2019-0021. [DOI] [Google Scholar]
- Dirac P. A. M. Quantum Mechanics of Many-Electron Systems. Proc. R. Soc. London A 1929, 123, 714–733. 10.1098/rspa.1929.0094. [DOI] [Google Scholar]
- Pitzer K. S. Relativistic effects on chemical properties. Acc. Chem. Res. 1979, 12, 271–76. 10.1021/ar50140a001. [DOI] [Google Scholar]
- Pyykkö P.; Desclaux J. P. Relativity and the periodic system of elements. Acc. Chem. Res. 1979, 12, 276–281. 10.1021/ar50140a002. [DOI] [Google Scholar]
- Pyykkö P. Relativistic Effects in Chemistry: More Common Than You Thought. Annu. Rev. Phys. Chem. 2012, 63, 45–64. 10.1146/annurev-physchem-032511-143755. [DOI] [PubMed] [Google Scholar]
- Mews J.-M.; Schwerdtfeger P. Exclusively Relativistic: Periodic Trends in the Melting and Boiling Points of Group 12. Angew. Chem., Int. Ed. 2021, 60, 7703–7709. 10.1002/anie.202100486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyykkö P. Theoretical chemistry of gold. Angew. Chem., Int. Ed. 2004, 43, 4412–4456. 10.1002/anie.200300624. [DOI] [PubMed] [Google Scholar]
- Ghosh A.; Conradie J. The Valence States of Copernicium and Flerovium. Eur. Eur. J. Inorg. Chem. 2016, 2016, 2989–2992. 10.1002/ejic.201600146. [DOI] [Google Scholar]
- Conradie J.; Ghosh A. The Blue–Violet Color of Pentamethylbismuth: A Visible Spin-Orbit Effect. ChemistryOpen 2017, 6, 15–17. 10.1002/open.201600131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demissie T. B.; Conradie J.; Vazquez-Lima H.; Ruud K.; Ghosh A. Rare and Nonexistent Nitrosyls: Periodic Trends and Relativistic Effects in Ruthenium and Osmium Porphyrin-Based {MNO}7 Complexes. ACS Omega 2018, 3, 10513–10516. 10.1021/acsomega.8b01434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pershina V.; Iliaš M. Carbonyl compounds of Tc, Re, and Bh: Electronic structure, bonding, and volatility. J. Chem. Phys. 2018, 149, 204306. 10.1063/1.5055066. [DOI] [PubMed] [Google Scholar]
- Conry R. R.; Mayer J. M. Oxygen atom transfer reactions of cationic rhenium(III), rhenium(V), and rhenium(VII) triazacyclononane complexes. Inorg. Chem. 1990, 29, 4862–4867. 10.1021/ic00349a010. [DOI] [Google Scholar]
- Braband H.; Abram U. Technetium Complexes with Triazacyclononane. Inorg. Chem. 2006, 45, 6589–6591. 10.1021/ic060569m. [DOI] [PubMed] [Google Scholar]
- The integrals for the glycolato CH proton signals at 8.11 and 7.58 ppm showed a ratio of 2:1 for the two diastereomers. Also, 13C–1H correlation spectra along with one-dimensional nuclear Overhauser effect experiments led to the assignment of signals to the different diastereomers. Furthermore, the two CH3 groups of isomer 2 are diastereotopic and yield separate signals at 1.45 and 1.24 ppm.
- Middleditch M.; Anderson J. C.; Blake A. J.; Wilson C. A Series of [3 + 2] Cycloaddition Products from the Reaction of Rhenium Oxo Complexes with Diphenyl Ketene. Inorg. Chem. 2007, 46, 2797–2804. 10.1021/ic062290b. [DOI] [PubMed] [Google Scholar]
- Hopmann K. How Accurate is DFT for Iridium-Mediated Chemistry?. Organometallics 2016, 35, 3795–3807. 10.1021/acs.organomet.6b00377. [DOI] [Google Scholar]
- Nadeem Q.; Meola G.; Braband H.; Bolliger R.; Blacque O.; Hernández-Valdés D.; Alberto R. Angew. Chem., Int. Ed. 2020, 59, 1197–1200. 10.1002/anie.201912994. [DOI] [PubMed] [Google Scholar]
- Braband H.; Tooyama Y.; Fox T.; Alberto R. Syntheses of High-Valent fac-[99mTcO3]+ Complexes and [3 + 2] Cycloadditions with Alkenes in Water as a Direct Labelling Strategy. Chem. - Eur. J. 2009, 15, 633–638. 10.1002/chem.200801757. [DOI] [PubMed] [Google Scholar]
- Herrmann W. A.; Roesky P. W.; Kühn F. E.; Scherer W.; Kleine M. Heterolyse von Re2O7: Erzeugung und Stabilisierung des Kations [ReO3]+. Angew. Chem. 1993, 105, 1768–1770. 10.1002/ange.19931051207. [DOI] [Google Scholar]
- Wieghardt K.; Pomp C.; Nuber B.; Weiss J. Syntheses of [LRe(CO)3]+ and [LRe(NO)(CO)2]2+ and their oxidative decarbonylation product [LReO3]+. Crystal structure of [LReO3]Cl (L = 1,4,7-triazacyclononane). Inorg. Chem. 1986, 25, 1659–1661. 10.1021/ic00230a027. [DOI] [Google Scholar]
- CrysAlisPro Software System; version 171.32; Oxford Diffraction Ltd.: Oxford, U.K..
- Altomare A.; Burla M. C.; Camalli M.; Cascarano G. L.; Giacovazzo C.; Guagliardi A.; Moliterni A. G. G.; Polidori G.; Spagna R. SIR97: A new tool for crystal structure determination and refinement. J. Appl. Crystallogr. 1999, 32, 115–119. 10.1107/S0021889898007717. [DOI] [Google Scholar]
- Sheldrick G. M. Crystal Structure Refinement with SHELXL. Acta Crystallogr., Sect. C: Struct. Chem. 2015, C71, 3–8. 10.1107/S2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolomanov O. V.; Bourhis L. J.; Gildea R. J.; Howard J. A. K.; Puschmann H. OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339. 10.1107/S0021889808042726. [DOI] [Google Scholar]
- te Velde G.; Bickelhaupt F. M.; Baerends E. J.; Fonseca Guerra C.; van Gisbergen S. J. A.; Snijders J. G.; Ziegler T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. 10.1002/jcc.1056. [DOI] [Google Scholar]
- van Lenthe E.; Baerends E. J.; Snijders J. G. Relativistic regular two-component Hamiltonians. J. Chem. Phys. 1993, 99, 4597. 10.1063/1.466059. [DOI] [Google Scholar]
- Handy N. C.; Cohen A. J. Left-right correlation energy. Mol. Phys. 2001, 99, 403–412. 10.1080/00268970010018431. [DOI] [Google Scholar]
- Lee C.; Yang W.; Parr R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron-density. Phys. Rev. B: Condens. Matter Mater. Phys. 1988, 37, 785–789. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- Becke A. D. Density-functional exchange-energy approximation with correct asymptotic behaviour. Phys. Rev. A: At., Mol., Opt. Phys. 1988, 38, 3098–3100. 10.1103/PhysRevA.38.3098. [DOI] [PubMed] [Google Scholar]
- Miehlich B.; Savin A.; Stoll H.; Preuss H. Results Obtained with the Correlation Energy Density Functionals of Becke and Lee, Yang and Parr. Chem. Phys. Lett. 1989, 157, 200–206. 10.1016/0009-2614(89)87234-3. [DOI] [Google Scholar]
- Perdew J. P.; Ernzerhof M.; Burke K. Rationale for mixing exact exchange with density functional approximations. J. Chem. Phys. 1996, 105, 9982–9985. 10.1063/1.472933. [DOI] [Google Scholar]
- Adamo C.; Barone V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. 10.1063/1.478522. [DOI] [Google Scholar]
- Swart M.; Ehlers A. W.; Lammertsma K. Performance of the OPBE exchange-correlation functional. Mol. Phys. 2004, 102, 2467–2474. 10.1080/0026897042000275017. [DOI] [Google Scholar]
- Klamt A.; Schüürmann G. COSMO: A New Approach to Dielectric Screening in Solvents with Explicit Expressions for the Screening Energy and Its Gradient. J. Chem. Soc., Perkin Trans. 2 1993, 799–805. 10.1039/P29930000799. [DOI] [Google Scholar]
- Klamt A. Conductor-like Screening Model for Real Solvents: A New Approach to the Quantitative Calculation of Solvation Phenomena. J. Phys. Chem. 1995, 99, 2224–2235. 10.1021/j100007a062. [DOI] [Google Scholar]
- Klamt A.; Jonas V. Treatment of the outlying charge in continuum solvation models. J. Chem. Phys. 1996, 105, 9972–9981. 10.1063/1.472829. [DOI] [Google Scholar]
- Pye C. C.; Ziegler T. An implementation of the conductor-like screening model of solvation within the Amsterdam density functional package. Theor. Chem. Acc. 1999, 101, 396–408. 10.1007/s002140050457. [DOI] [Google Scholar]
- Pascual-Ahuir J. L.; Silla E.; Tuñon I. GEPOL: An improved description of molecular surfaces. III. A new algorithm for the computation of a solvent-excluding surface. J. Comput. Chem. 1994, 15, 1127–1138. 10.1002/jcc.540151009. [DOI] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Petersson G. A.; Nakatsuji H.; Li X.; Caricato M.; Marenich A. V.; Bloino J.; Janesko B. G.; Gomperts R.; Mennucci B.; Hratchian H. P.; Ortiz J. V.; Izmaylov A. F.; Sonnenberg J. L.; Williams-Young D.; Ding F.; Lipparini F.; Egidi F.; Goings J.; Peng B.; Petrone A.; Henderson T.; Ranasinghe D.; Zakrzewski V. G.; Gao J.; Rega N.; Zheng G.; Liang W.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Throssell K.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M. J.; Heyd J. J.; Brothers E. N.; Kudin K. N.; Staroverov V. N.; Keith T. A.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A. P.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Millam J. M.; Klene M.; Adamo C.; Cammi R.; Ochterski J. W.; Martin R. L.; Morokuma K.; Farkas O.; Foresman J. B.; Fox D. J.. Gaussian 16, revision C.01; Gaussian, Inc.: Wallingford, CT, 2016. [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]; Errata:; Phys. Rev. Lett. 1997, 78, 1396 - 1396.10.1103/PhysRevLett.78.1396
- Grimme S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. 10.1002/jcc.20495. [DOI] [PubMed] [Google Scholar]
- Marenich A. V.; Cramer C. J.; Truhlar D. G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
- Skyner R. E.; Mcdonagh J. L.; Groom C. R.; van Mourik T.; Mitchell J. B. O. A review of methods for the calculation of solution free energies and the modelling of systems in solution. Phys. Chem. Chem. Phys. 2015, 17, 6174–6191. 10.1039/C5CP00288E. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.