

Abstract
Significance: Physiological concentrations of nitric oxide (NO•) and related reactive nitrogen species (RNS) mediate multiple signaling pathways in the nervous system. During inflammaging (chronic low-grade inflammation associated with aging) and in neurodegenerative diseases, excessive RNS contribute to synaptic and neuronal loss. “NO signaling” in both health and disease is largely mediated through protein S-nitrosylation (SNO), a redox-based posttranslational modification with “NO” (possibly in the form of nitrosonium cation [NO+]) reacting with cysteine thiol (or, more properly, thiolate anion [R-S−]).
Recent Advances: Emerging evidence suggests that S-nitrosylation occurs predominantly via transnitros(yl)ation. Mechanistically, the reaction involves thiolate anion, as a nucleophile, performing a reversible nucleophilic attack on a nitroso nitrogen to form an SNO-protein adduct. Prior studies identified transnitrosylation reactions between glyceraldehyde-3-phosphate dehydrogenase (GAPDH)-nuclear proteins, thioredoxin-caspase-3, and X-linked inhibitor of apoptosis (XIAP)-caspase-3. Recently, we discovered that enzymes previously thought to act in completely disparate biochemical pathways can transnitrosylate one another during inflammaging in an unexpected manner to mediate neurodegeneration. Accordingly, we reported a concerted tricomponent transnitrosylation network from Uch-L1-to-Cdk5-to-Drp1 that mediates synaptic damage in Alzheimer's disease.
Critical Issues: Transnitrosylation represents a critical chemical mechanism for transduction of redox-mediated events to distinct subsets of proteins. Although thousands of thiol-containing proteins undergo S-nitrosylation, how transnitrosylation regulates a myriad of neuronal attributes is just now being uncovered. In this review, we highlight recent progress in the study of the chemical biology of transnitrosylation between proteins as a mechanism of disease.
Future Directions: We discuss future areas of study of protein transnitrosylation that link our understanding of aging, inflammation, and neurodegenerative diseases. Antioxid. Redox Signal. 35, 531–550.
Keywords: S-nitrosylation, transnitrosylation, neurodegenerative diseases, nitric oxide
Introduction
At physiological or basal levels, nitric oxide (NO•)-related species support many aspects of brain function, including neuronal development, synaptic plasticity, and neuronal survival. In contrast, mounting evidence suggests that aged brains, like the rest of body, undergo inflammaging, the low-grade chronic inflammation associated with advanced age, in part, characterized by increased levels of reactive oxygen species and reactive nitrogen species (ROS/RNS), including superoxide anion (O2•−) and NO• (8, 20, 134). This increase in ROS/RNS results in oxidative and nitrosative stress, contributing to neuropathological processes, such as protein misfolding, mitochondrial dysfunction, neuroinflammation, synaptic injury, and neuronal loss.
Notably, cellular synthesis of NO-related species can lead to activation of multiple cellular signaling pathways, which modulate numerous pathophysiological responses. For example, Snyder and colleagues initially identified NO• synthase (NOS) in neurons (17, 18). Subsequent studies revealed the presence of three distinct subtypes of NOS, namely, NOS1 (or neuronal NOS [nNOS]), NOS2 (or inducible NOS [iNOS]), and NOS3 (or endothelial NOS), catalyzing NO• production from l-arginine (97, 132). The activity of NOS1 and NOS3 relies on intracellular calcium/calmodulin levels, whereas induction of NOS2 expression by inflammatory stimuli produces neurotoxic amounts of NO• in a calcium-independent manner. Interestingly, while nearly all eucaryotes can produce NO• from NOS, the roundworm Caenorhabditis elegans lacks its own NOS. In this case, bacteria that the worms naturally consume synthesize NO-related species in the gut flora of the nematode, influencing thermotolerance and C. elegans life span (51).
Initial studies on the biological effects of NO• identified cyclic guanosine monophosphate (cGMP) as an important mediator in eliciting physiological responses, such as smooth muscle relaxation and anticoagulation (60, 100). Later studies, however, revealed a ubiquitous, cGMP-independent mechanism for NO• bioactivity in which an NO-related group (possibly in the form of nitrosonium cation, NO+) covalently reacts with a nucleophilic thiol group (i.e., thiolate anion, RS−) on target proteins to form an S-nitrosothiol. As noted by Smith and Marletta (148), free NO+ in solution would very quickly react with water to produce NO2−, and so the reaction would have to represent a concerted transfer. In this context, transition metals, such as copper and iron, present in metalloproteins have been proposed to contribute to the formation of S-nitrosothiols via promotion of a one-electron oxidation of NO•, thereby balancing the electrons in the reaction mechanism (47, 58, 82). For example, it is possible that iron could contribute under some conditions because Fe3+–NO• would possess a significant Fe2+–NO+ character (148). In addition, another pathway for cellular S-nitrosothiol formation could involve direct reaction between thiyl radical (RS•) and NO• radical (158). These and other mechanistic details have been well summarized in a previous excellent review (148).
Mechanism notwithstanding, this redox-based posttranslational modification (PTM), termed protein S-nitrosylation (SNO), is now well-recognized as a major contributor to both the physiological and pathophysiological activities of “NO” (1, 5, 7, 107, 138, 150). Although the field generally uses the terms protein S-nitrosylation and transnitrosylation (as used here), because a nitroso group is in general being transferred (rather than a nitrosyl group, which is formally an NO• radical), chemically the proper terms are S-nitrosation and transnitrosation (148). Note that other forms of RNS, such as peroxynitrite (ONOO−), can induce disulfide bond formation or nitration of protein tyrosine, forming 3-nitrotyrosine that can contribute to pathology (61).
Early Work on S-Nitrosothiol Formation That Regulates Protein Function, Termed Protein S-Nitrosylation
Lipton's group (86), subsequently in collaboration with Stamler and colleagues (90), demonstrated that one neuroprotective mechanism of “NO signaling” involved negative feedback onto the N-methyl-d-aspartate (NMDA)-type glutamate receptor (NMDAR), which is physically tethered to nNOS, via S-nitrosylation to inhibit receptor overactivation. The overall effect is to suppress excitotoxic neuronal damage. Notably, site-directed mutagenesis studies revealed that S-nitrosylation (SNO) of the NMDAR is predominantly mediated through reaction with at least five cysteine (Cys) residues: Cys399 on the GluN2A subunit, Cys744 and Cys798 on the GluN1 subunit, and Cys87 and Cys320 on the GluN2A subunit (and potentially with analogous cysteine residues at positions 79 and 308 on GluN2B) (31).
Intriguingly, under ambient air conditions, the two pairs of cysteine residues (i.e., Cys744 and Cys798 on the GluN1 subunit and Cys87 and Cys320 on the GluN2A subunit) can form either disulfide bonds or free thiols, depending on the redox microenvironment. Moreover, X-ray crystallographic studies confirmed the presence of S-nitrosothiol at Cys744/798 on GluN1 if an “NO donor” was added after reduction of the disulfide (157). In addition, relative hypoxia, as seen in a normal brain (15–40 torr), produced a less oxidizing environment than ambient air conditions, favoring the presence of at least some free thiol groups over disulfide formation. However, during pathological hypoxia (<8–10 torr), the further increase in free thiol groups at Cys744 and Cys798 on the GluN1 subunit facilitated a greater degree of reaction with NO-related species (157). Our electrophysiological recording data showed that conditions favoring S-nitrosothiol formation at Cys744 and/or Cys798 on the GluN1 subunit sensitized Cys399 on Glu2A to undergo S-nitrosylation. Finally, structural modeling analysis revealed that the conformational changes precipitated by S-nitrosylation of GluN2A at Cys399 resulted in enhanced binding of glutamate to the clam-shell-shaped ligand-binding domain and of Zn2+ to a second clam shell domain on the NMDAR, leading to receptor desensitization and subsequently closure of the ion channel (91). Importantly, with the development of the biotin-switch assay, Snyder and colleagues found that the NMDAR was S-nitrosylated in intact brain tissue, implying that this reaction occurs in vivo and not merely under artificial tissue culture or in vitro conditions (63, 64). Hence, S-nitrosylation of the NMDAR at available free thiols (or more properly, thiolate anions) is critical for the inhibitory effects of NO-related species on this receptor, preventing excessive activity associated with excitotoxicity.
Since the initial discovery of S-nitrosylation of the NMDAR, it has been speculated that as many as 3000 proteins can be potentially S-nitrosylated (141). Notably, depending on the target protein or site of modification, S-nitrosylation can activate or inhibit protein function, and can contribute to either the neuroprotective or neurotoxic effects of NO• in the nervous system. A similar range of activities has been reported for protein S-nitrosylation in many other organs, including the circulatory and cardiovascular systems, kidney, pancreas, lungs, and elsewhere. As discussed below, the determinants of selective and specific S-nitrosothiol formation in an in vivo environment are influenced by the cellular localization and compartmentalization of the target protein, the local redox milieux, pH, and availability of transition metals, as well as the presence of an S-nitrosylation motif or partial motif near the target thiol (59, 107). In this review, we highlight signaling pathways affecting neuronal function via protein S-nitrosylation with a focus on protein–protein transnitrosylation.
Specificity and Stability of Protein S-Nitrosylation
While a majority of cellular proteins contain multiple cysteine residues, under certain conditions S-nitrosylation can occur on specific free thiols/thiolates to form S-nitrosylated proteins (SNO-proteins) (59, 86, 90, 107, 152). Such selective S-nitrosylation can be achieved, for example, when target proteins are located adjacent to the NOS. Close proximity to the source of NO• production allows exposure of SNO target proteins to high concentrations of S-nitrosylating species, increasing the probability of S-nitrosothiol formation (107, 148, 152). For example, the direct interaction of nNOS with the NMDAR via PSD-95 facilitates SNO-NMDAR and SNO-PSD-95 formation after activation of nNOS by calcium influx through the NMDAR-associated ion channel (86, 90, 107). Another important cellular attribute that promotes protein S-nitrosylation is a hydrophobic environment, as seen in lipid membranes (or possibly within a particular protein structure). In this case, hydrophobicity is known to increase the stability of S-nitrosylating species; thus, S-nitrosylation of proteins localized in lipid membranes occurs more efficiently (152).
Another important mechanism implicated in targeting specific cysteine residue(s) for S-nitrosylation involves the presence of a consensus acid-base motif near a critical cysteine residue (39, 104, 151), although such motifs have been criticized and other considerations have been shown to be equally important (148). We and our colleagues initially discovered that charged acidic and/or basic side chains adjacent to the target cysteine influence thiol pKa and SNO stability, thereby increasing the susceptibility of the sulfhydryl or thiolate to form an S-nitrosothiol (151). More recent structure studies indicated that the acidic or basic residue is generally located within 8 Å of SNO sites (96). In addition, in some cases, the acid-base motif contributes to selective S-nitrosylation of target sites via facilitation of transnitrosylation, for example, by S-nitrosoglutathione (GSNO, a physiological NO donor); this probably occurs via increasing protein–protein interaction, as discussed below (96).
The biological effects of S-nitrosylation can be counteracted by denitrosylation reactions. For example, enzymes such as thioredoxin (Trx) and Trx-related proteins, protein-disulfide isomerase (PDI), SNO-CoA reductase, and class III alcohol dehydrogenase (also known as ADH5 or GSNO reductase [GSNOR]) have been reported to catalyze the removal of SNO from target proteins (152), but an effect of GSNOR directly on S-nitrosylated proteins has been refuted (148). Moreover, considering catalytic reactions, however, the use of the words S-nitrosylase and denitrosylase to reflect enzymatic action has been used somewhat loosely in the literature for protein S-nitrosylation and denitrosylation. Specifically, for enzymatic activity, a catalyst generally follows Michaelis–Menten kinetics, affects kcat/Km, and increases the reaction rates over spontaneous reaction rates. This type of analysis has not been achieved for most proposed S-nitrosylases or denitrosylases; in fact, initial studies by Marletta and colleagues may represent the first example demonstrating catalytic transnitrosation/denitrosation reactions, in this case mediated by Trx (12, 102, 103, 148).
Mechanism notwithstanding, our group and others have recently discovered that an NO-related species can be transferred between proteins via protein–protein transnitrosylation (25, 30, 68, 76, 102, 106, 108, 122, 140). In this reaction, the donor protein, providing an NO-related moiety to another protein, is selectively denitrosylated, whereas the recipient protein, accepting an NO-related group from the donor protein, is specifically S-nitrosylated. Although further investigations are required to reveal the precise chemistry behind the protein–protein transnitrosylation reactions, transnitrosylation has now been recognized as a major basis for transduction of SNO-mediated cell signaling pathways, as delineated below (68, 76, 105, 140). It remains to be demonstrated definitively whether at least some of these transnitrosylation reactions indeed represent catalytic processes as opposed to a noncatalytic homogeneous chemical reaction involving thiolate anion, as a nucleophile, performing a reversible nucleophilic attack on the nitroso nitrogen to form an SNO-protein adduct.
Protein S-nitrosylation is generally regarded as a labile modification (146), although SNO can be stabilized via amortization of electrophilicity in the outer pi molecular orbital by the presence of a tryptophan or tyrosine ring (91). Some of the pathophysiological effects following SNO can be sustained through further oxidation of the same cysteine residue (24, 49). For example, S-nitrosylation can lead to more stable disulfide bond formation between vicinal cysteines (10, 90, 176). In addition, S-nitrosylation often causes conformational changes to the target proteins. These alterations in the tertiary structure may render the same thiol group more susceptible to subsequent oxidative modification. Accordingly, the remaining free thiol undergoes facile reaction with ROS, yielding sulfenic acid adducts (-SOH) or hyperoxidation products such as sulfinic acid (-SO2H) or irreversible sulfonic acid (-SO3H) (49).
Protein–Protein Transnitrosylation
Under physiological conditions, NO-related species can react with thiol (or more properly thiolate) containing low-molecular-weight compounds, such as cysteine and glutathione, leading to the formation of S-nitrosocysteine (CysNO or SNOC) and GSNO, respectively. SNOC or GSNO then acts as an endogenous (S)NO donor, if the chemical milieux and redox potential permit, to produce S-nitrosylated proteins (59). For mechanistic considerations of the reaction, see Smith and Marletta (148). Evidence from our laboratory and others now suggests that protein–protein transnitrosylation also occurs in a broad range of organisms, including bacteria, plants, and vertebrates (25, 30, 68, 76, 102, 106, 108, 122, 140). Such transnitrosylation reactions generally require that the donor and recipient proteins are present in the sample protein complex, allowing these proteins to remain in close contact for transnitrosylation. Moreover, in some cases, the interaction of the two proteins can lead to conformational changes, possibly affecting the redox potential of the key cysteine residue(s). In part, the apparent difference in the redox potentials of the two proteins determines the direction of “NO” (and electron) transfer. Specifically, during protein–protein transnitrosylation, a SNO-protein with a higher redox potential will acquire an electron and donate “NO” as NO+, while the recipient protein with a lower redox potential will accept the NO+ group at a cysteine thiolate anion, thus losing an electron. Accordingly, protein–protein transnitrosylation can mediate not only selective SNO-formation but also S-nitrosylation-dependent intracellular signaling pathways.
Chemically, transnitrosylation also represents the reaction mechanism from some small-molecular-weight “NO donors” such as nitroprusside to cysteine thiolate, as first shown for the NMDAR (59, 86, 90, 107, 152). Perhaps the first evidence for protein–protein transnitrosylation was reported for S-nitrosylated hemoglobin (SNO-Hb) and the anion exchanger 1 (AE1) (118). S-Nitrosylation of Hb occurs at Cys93 of the Hb β chain. In peripheral tissues, the NO group has been reported to be released from SNO-Hb and exported out of erythrocytes to trigger blood vessel relaxation, modulating blood flow to maximize oxygen delivery (70). Mechanistically, it was reported that the low oxygen environment in peripheral tissues induces the binding of SNO-Hb to the cytoplasmic domain of the membrane protein AE1. The interaction of Hb and AE1 reportedly initiates transnitrosylation from Hb to AE1, followed by extracellular liberation of NO• from AE1 localized at the erythrocyte membrane (118, 149).
Subsequently, several research groups discovered additional protein–protein transnitrosylation reactions, involving Trx, caspases, X-linked inhibitor of apoptosis (XIAP), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (14, 76, 102, 108). More recently, studies identified multiprotein complexes that mediate protein–protein transnitrosylation (68, 140), such as a heterotrimeric protein complex composed of iNOS, S100A8, and S100A9 in immune cells, and a multiprotein complex, containing hybrid cluster protein (Hcp) and nitrate reductase NarGHI, in Escherichia coli. Very recently, our group demonstrated that S-nitrosylated ubiquitin carboxy-terminal hydrolase L1 (Uch-L1) activates a transnitrosylation network involving Cdk5 and dynamin-related protein 1 (Drp1) (106). In summary, transnitrosylation has emerged as an important mechanism to transduce S-nitrosylation-dependent signaling. Accordingly, we review here our current understanding of protein–protein transnitrosylation reactions in physiological and pathophysiological processes in the nervous system.
Transnitrosylation Involving Trx and Trx-Related Proteins
Trx1 is a 12 kDa globular protein ubiquitously expressed essentially in all living cells (88). Trx was first isolated and characterized from E. coli as an electron donor for ribonucleotide reductase (RNR), the rate-limiting enzyme in DNA synthesis (83). Two Trx proteins exist in mammalian cells: Trx1 in the cytosol and Trx2 in mitochondria. Mice with homozygous knockout (KO) of Trx1 or Trx2 genes show early embryonic lethality, consistent with the notion that Trx activity is critical for cell survival (99).
Both Trx1 and Trx2 belong to the Trx superfamily, featuring the well-known Trx fold, consisting of a central core of five β-strands surrounded by four α-helices (42). Five Cys residues are present in human Trx1, including two cysteine residues at the highly conserved active site (-Trp-Cys-Gly-Pro-Cys-). Besides the active-site cysteines, three additional cysteines (Cys62, Cys69, and Cys73) exist in Trx1. Cys62 and Cys69 can form an intramolecular disulfide bond in response to oxidative stress (56, 170), whereas Cys73 protrudes from the surface and contributes to intermolecular disulfide bond formation, including homodimerization of Trx1 (126, 172).
Trx proteins are major protein disulfide oxidoreductases in mammalian cells. Besides RNR, substrates for Trx proteins include methionine sulfoxide reductase and peroxiredoxins (Prx). In the case of Prx, a typical 2-Cys Prx functions as a homodimer, reacting with and thus removing excess H2O2 and thereby regulating intracellular redox status (22, 128). Mechanistically, the N-terminal active-site Cys in one of the subunits is easily oxidized by H2O2 to sulfenic acid (Cys-SOH), which then reacts with the C-terminal Cys of the other subunit to generate a disulfide bond. The disulfide bridge can be reduced by Trx proteins through a disulfide exchange mechanism. The resulting oxidized Trx can then be reduced by Trx reductase (TrxR), with NADPH as the ultimate electron donor for the system (94). Accordingly, the Trx/Prx protein cascade represents a critical antioxidant enzyme system in mammalian cells.
Under certain conditions, Trx proteins have also been reported to catalyze denitrosylation of both low-molecular-weight thiol and protein thiol groups (153, 178). In addition to Trx proteins, several enzymes have been shown to catabolize GSNO, including alcohol dehydrogenase class III (ADH, also known as GSNOR), carbonyl reductase, PDI, glutaredoxin (Grx), TrxR, and Trx-related proteins such as TRP-14 (66, 109, 114, 127, 147). Among these, PDI, Grx, Trx, and Trx-related proteins all belong to the Trx-fold superfamily, comprised a Trx-fold and CXXC active-site motif, arguing that this structure is important and effective in thiol denitrosylation.
Trx proteins can also directly denitrosylate other protein S-nitrosothiols, the most well-studied case being S-nityrosylated caspase-3. Caspase-3 belongs to a family of cysteinyl aspartate-specific proteases that serve as central regulators of apoptosis (46). Caspase-3 and other caspases are synthesized as inactive procaspases, which are cleaved and activated in response to apoptotic signals either from mitochondrial (intrinsic pathway) or extracellular (extrinsic pathway) stimuli. Both of these apoptotic pathways converge on caspase-3 (34). NO-related species can S-nitrosylate procaspase-3 at its active-site cysteine (Cys163) to inhibit its activity, and thereby protect the cell from apoptosis (Fig. 1A) (95, 101, 131, 159). Interestingly, studies also showed that activation of the cell surface death receptor Fas stimulates caspase-3 activity not only because of the cleavage of caspase-3 but also via selective denitrosylation of the cleaved form of caspase-3 (95). These findings suggest that protein S-nitrosylation and denitrosylation play important regulatory roles in apoptotic signaling.
FIG. 1.

Trx mediates protein transnitrosylation and denitrosylation reactions. (A) Trx that is oxidized or reduced at its active site catalyzes transnitrosylation reactions through the nonactive-site cysteine Cys62 or Cys73. Under nitrosative conditions, Trx is S-nitrosylated at Cys62 or Cys73, which in turn can serve as an “NO donor” to transnitrosylate a thiol/thiolate group in other proteins such as caspase-3 and Trx itself. (B) Trx can also catalyze the transnitrosylation and denitrosylation of the same protein, such as caspase-3, although the forward rate reaction for S-nitrosylation has been reported to predominate (102, 103). Trx-mediated denitrosylation of the active-site cysteine of caspase-3 promotes activation of caspase-3, and thus it is proapoptotic. Transnitrosylation of the same cysteine of caspase-3 by Trx (Cys73) is antiapoptotic because it attenuates caspase-3 activation. Trx, thioredoxin.
Subsequent studies showed that both Trx1 and Trx2 facilitate denitrosylation of caspase-3 in specific cellular environments. For example, under some conditions, Trx1 appears to actively denitrosylate cytosolic caspase-3, maintaining low levels of S-nitrosylated caspase-3. Upon stimulation of Fas, Trx2-mediated denitrosylation of caspase-3 takes place possibly at the mitochondrial outer membrane. This denitrosylation of mitochondria-associated caspase-3 contributes to caspase-3 activation (Fig. 1B) (14). Considering the fact that Trx2 contains Cys only at the active site, this active-site cysteine most likely participiates in the denitrosylation reaction. Two possible mechanisms have been proposed for the reaction. The first mechanism involves the formation of an intermolecular disulfide bond between Cys32 of Trx1 and the active-site cysteine of caspase-3 (Cys163), leading to the release of NO•. Subsequently, Cys35 of Trx1 attacks the intermolecular disulfide bond and forms a new intramolecular disulfide within Trx1, releasing free caspase-3. An alternative pathway starts with transnitrosylation from Cys163 of caspase-3 to Cys32 of Trx1, which results in SNO-free caspase-3 and S-nitrosylated Trx1 (153). Given that S-nitrsoylation of vicinal cysteines can facilitate disulfide formation, this reaction on Trx1 is followed by formation of an intramolecular disulfide bond between Cys32 and Cys35 on Trx1 with concomitant release of NO•. Interestingly, caspase-3 can also serve as the (S)NO donor for other proteins such as XIAP via transnitrosylation (108). This reaction is discussed in detail later in this review.
Trx1 can reportedly also be S-nitrosylated at its nonactive-site cysteines, including Cys62, Cys69, and Cys73 (12, 52, 56, 171). A mass spectrometry (MS) study using HeLa cells revealed that S-nitrosylation of Trx1(Cys73) occurs only when its two active-site cysteine residues are in disulfide form (177). Another study using recombinant Trx1 showed that incubation of fully reduced Trx1 with GSNO leads to S-nitrosylation at Cys69 and Cys73 as well as disulfide bond formation between the active-site cysteines (56). Tannenbaum and Marletta further showed that SNO-Trx formation depends on the redox state of the active-site cysteines (Fig. 1A) (12). Specifically, when the Trx1 active site is oxidized, S-nitrosylation occurs primarily at Cys73, with a secondary SNO site at Cys69, whereas fully reduced Trx1 is selectively S-nitrosylated at Cys62. Considering the fact that under physiological conditions most Trx1 cysteine residues appear to be in reduced form and that oxidative/nitrosative stress increases oxidation of Trx1 (170), SNO-Trx1 may regulate separate signaling pathways under different cellular redox environments.
Notably, Trx1 nitrosylated at Cys62 or Cys73 can participate in additional transnitrosylatyion reactions. Thus, unlike active-site Trx1 Cys, SNO-Cys62 or SNO-Cys73 has been shown to S-nitrosylate other substrate proteins (12, 102, 103). An MS approach identified ∼40 such proteins. These substrate proteins include Prx1, caspase-3, thioredoxin interacting protein (TXNIP), and heat shock protein 90-β (HSP90-β) (177). Among these possible targets, the transnitrosylation reaction from Trx1(SNO-Cys73) to caspase-3 is the best characterized to date, and has been shown to occur at the caspase-3 active-site cysteine (Cys163) both in vitro and in intact cells (102, 103).
In addition, using a biochemical assay for caspase-3 activity, Marletta's group has demonstrated that the rate constant (kcat) for SNO-caspase-3 formation from Trx1(SNO-Cys73) is 120-fold faster than from GSNO, consistent with the notion that Trx1(SNO-Cys73) acts as an enzymatic transnitrosylase toward caspase-3 (102, 103). Although NO can be transferred back to Trx1 from SNO-caspase-3, the rate of SNO-caspase-3 denitrosylation/reactivation proceeds at a much slower rate under the conditions studied, suggesting that the S-nitrosothiol is more stable on the active-site cysteine of caspase-3 (Cys163) due to its greater nucleophilicity.
Transnitrosylase and denitrosylase activities of Trx proteins have also been found in lower organisms such as the mushroom Inonotus obliquus (189). Similar to human Trx1, the nonactive-site cysteine of mushrorom Trx can mediate transnitrosylation reactions. Three isoforms of Trx are present in I. obliquus: Trx1–Trx3. Both Trx1 and Trx3 have an extra nonactive-site cysteine and exhibit the ability to transnitrosylate GSNOR in the mushroom. In contrast, mushroom Trx2 only has the active-site cysteine and lacks transnitrosylation activity toward GSNOR. Hence, these studies showed the importance of the nonactive-site cysteine of Trx proteins in transnitrosylation reactions. Further studies are warranted to elucidate the role of Trx transnitrosylate activity in the mammalian nervous system.
PDI is another member of the Trx-fold superfamily that can chemically reduce both low-molecular mass, thiol-containing molecules (e.g., GSNO) and protein S-nitrosothiols (123, 147, 183). PDI was initially identified in the lumen of the endoplasmic reticulum, where it functions as a molecular chaperone and a disulfide isomerase to assist nascent protein folding. Although at lower levels, PDI was subsequently found in other cellular compartments, such as the cytosol and on the external surface of the cell membrane (4). The latter cell surface PDI (csPDI) has drawn special attention in SNO signaling. For example, csPDI denitrosylates extracellular GSNO as well as S-nitrosylated proteins (e.g., serum albumin) through a mechanism involving the active-site cysteines of PDI. NO-related species thus accumulated at the membrane may react with O2 to produce the nitrosylating agent N2O3; this schema has been proposed to transduce the biological effects of NO-related species into the intracellular compartment (123, 130, 183). Intriguingly SNO-PDI formed in erythrocytes has been reported to facilitate the efflux of S-nitrosothiols into the blood and the eventual transfer of SNO to endothelial cells, inducing vasodilation (72). Hence, PDI is involved in both cellular release and entry of NO-related species.
In addition, our group has shown that aberrant S-nitrosylation of PDI occurs in human brains in several neurodegenerative disorders, including Alzheimer's disease (AD), Parkinson's disease (PD), and amyotrophic lateral sclerosis (ALS). The SNO-PDI thus formed abrogates the normal protein-folding activity of PDI, thus contributing to protein misfolding and the consequent neuronal damage in these neurodegenerative diseases (67, 165).
Transnitrosylation of XIAP by Caspases
Caspases involved in apoptosis are classified into initiator caspases (caspases-8, -9, and -10) and effector caspases (caspase-3, -6, and -7). During normal embryogenesis, the basal activity of caspases is essential for neurodevelopment; in contrast, excessively activated caspases contribute to neuronal loss in neurodegenerative diseases (37, 46, 108). Activation of the intrinsic apoptotic pathway typically involves a two-step mechanism in which the initiator caspase-9 is activated via dimerization at the apoptosome after which it processes executioner caspase-3 and caspase-7 for activation. In opposition to this mechanism of cell death, inhibitor of apoptosis proteins (IAPs) block the apoptotic events through direct interaction with caspases. For example, XIAP, thought to be the most potent IAP, binds to caspase-3, caspase-7, and caspase-9 through its baculovirus IAP repeat (BIR) domains and their flanking regions, suppressing the proteolytic activity of caspases. Specifically, a flanking region near the BIR2 domain selectively targets active caspase-3 and caspase-7, whereas the activity of capsase-9 is inhibited via the BIR3 domain of XIAP (37, 145). In addition, the RING finger domain in XIAP is responsible for ubiquitin E3 ligase activity toward caspases, further decreasing caspase activity via proteasomal degradation of caspases.
Our group has demonstrated that the excessive production of NO that occurs in neurodegenerative disorders contributes, in part, to apoptotic neuronal loss via S-nitrosylation of XIAP (forming SNO-XIAP) (108, 164). Mechanistically, S-nitrosylation of XIAP occurs at a Cys in the RING domain that perturbs its conformation. This in turn leads to a decrease in E3 ligase activity and thus accumulation of active caspases. Supporting the premise that S-nitrosylated XIAP contributes to caspase-dependent neuronal loss, expression of non-nitrosylatable mutant XIAP protected neurons from NMDA-induced excitotoxicity (108). In addition, our groups and others have demonstrated that many other RING-type ubiquitin E3 ligases (e.g., parkin, cIAP1, CHIP, and RNF213) are targets of S-nitrosylation (6, 32, 92, 112, 129, 155, 182, 184), suggesting that S-nitrosylation could widely affect this type of enzyme in both PD and tauopathy models. For example, S-nitrosylation of the E3 ligase RNF213 leads to increased levels of NFAT-1 and FILAMIN-A, which amplify noncanonical Wnt/Ca2+ signaling and contributes to pathology in the P301S mouse model of tauopathy (6).
While SNO-XIAP exhibits proapoptotic properties, NO• is also known to show neuroprotective activity, in part, via S-nitrosylation-mediated inhibition of caspases (14, 95, 159). Upon activation of cell death cascades, selective denitrosylation of caspases removes SNO-mediated inhibition of the caspases (95, 159). Along these lines, we found that S-nitrosylated caspase-3 can transnitrosylate XIAP. In this context, the transfer of an NO-related group from caspase-3 to XIAP represents a proapoptotic event for two reasons. This transnitrosylation results in denitrosylation of caspase-3, thus restoring caspase-3 activity, and S-nitrosylation of XIAP, effectively decreasing its antiapoptotic activity (Fig. 2). As additional evidence for this pathway, transnitrosylation does not occur from caspase-3 to an XIAP mutant that lacks the ability to bind to caspase-3, consistent with the notion that transnitrosylation from caspase-3 to XIAP requires direct interaction of the two enzymes.
FIG. 2.

SNO-Caspase-mediated transnitrosylation of XIAP contributes to caspase-dependent cell death. Through direct binding to caspases, XIAP antagonizes caspase activity. XIAP also serves as a ubiquitin E3 ligase that targets caspases for proteasomal degradation. Several caspases are known to be S-nitrosylated under basal conditions, inhibiting their proapoptotic protease activity. However, during apoptotic cell death, transnitrosylation from constitutively S-nitrosylated caspases (e.g., caspase-3 and caspase-9) to XIAP occurs. The resulting formation of S-nitrosylated XIAP (forming SNO-XIAP) at a cysteine residue in the RING domain decreases XIAP ubiquitination activity. This diminishes caspase degradation, leading to an increase in caspase activity, thus promoting caspase-dependent neuronal cell death. SNO, S-nitrosylation; XIAP, X-linked inhibitor of apoptosis.
Furthermore, we devised a quantitative method based on the Nernst equation to assess redox immunoblots to determine if the transnitrosylation reaction is thermodynamically favored, assuming the reaction goes to steady state, as might be expected in a chronic disease. The Nernst equation quantifies the net electromotive force for electron movement between a redox pair, in this case the oxidized (or S-nitrosylated) and reduced forms of the proteins, as quantified on the immunoblots, involved in the reaction. This allows us to calculate the associated Gibbs free energy associated with the reaction, and we thus found that transnitrosylation from caspase-3 to XIAP is the favored direction of this chemical reaction (rather than transferring NO from XIAP to caspase-3). In addition, we found evidence from biotin-switch assays for SNO-XIAP in human postmortem brains from patients with AD, Lewy body dementia (LBD), and Huntington's disease (HD). Collectively, these findings are consistent with the notion that transfer of NO-related species from caspase-3 to XIAP takes place in vivo in these neurodegenerative diseases (108).
Similar to these results with caspase-3, Zhang and colleagues recently showed that transnitrosylation from caspase-9 to XIAP occurs in models of cerebral ischemia (186). As support for transnitrosylation from caspase-9 to XIAP in vivo, the group found that cerebral ischemia/reperfusion causes S-nitrosylation of XIAP and denitrosylation of (pro-)caspase-9 simultaneously. In addition, inhibiting caspase-9/XIAP complex formation using pharmacological approaches, prevented ischemia/reperfusion from increasing SNO-XIAP and decreasing SNO-caspase-9 levels. Thus, these findings are consistent with the concept that direct association of caspase-9 and XIAP triggers transnitrosylation between these two proteins. Notably, Trx activity appeared to augment SNO-XIAP formation after ischemia/reperfusion, although additional studies will be needed to determine how Trx facilitates the transfer of NO-related species from caspase-9 to XIAP. Another open question concerns the fact that despite evidence for simultaneous denitrosylation of procaspase-9 and S-nitrosylation of XIAP, XIAP only binds to the processed (cleaved) form of capase-9. Hence, further studies will be required to characterize the role of XIAP in procaspase-9 denitrosylation. One possibility is that an as-yet unknown third protein participates in transnitrosylation from caspase-9 to XIAP. In addition, since both SNO-caspase-3 and SNO-caspase-9 are capable of transnitrosylating XIAP, it will be important to test if SNO-caspase-7 can also transnitrosylate XIAP.
GAPDH-Mediated Transnitrosylation
GAPDH was initially discovered as a metabolic enzyme in the glycolysis cascade but then turned out to be a multitasking protein (163). For example, GAPDH is known to regulate gene expression and cell death pathways, in part, via formation of SNO-GAPDH (54, 76). Depending on the stimulus, S-nitrosylation of GAPDH can occur at different cysteine residues. In particular, lipopolysaccharide and other apoptotic stimuli induce S-nitrosylation of GAPDH at its active-site cysteine (Cys152 in human and Cys150 in mouse and rat). In contrast, oxidatively modified low-density lipoprotein (LDLox) plus interferon (IFN)-γ induce SNO-GAPDH at Cys247 (69). As would be expected, S-nitrosylation of GAPDH at the active-site cysteine abolishes its enzyme activity (54), while formation of GAPDH (SNO-Cys247) enhances proteasomal degradation of ribosomal protein L13a, leading to defective translation (69).
Moreover, although GAPDH itself does not contain a nuclear localization signal (NLS), S-nitrosylation of GAPDH at the active site promotes its binding to the E3 ubiquitin ligase Siah1, which possesses an NLS. This binding partner thus allows SNO-GAPDH to translocate into the nucleus in a SNO-GAPDH/Siah1 complex, contributing to signaling pathways associated with various cellular functions, including dendritic outgrowth and neuronal cell death (133). Siah1 has a wide range of substrates in the nucleus (Fig. 3). However, the high turnover rate of Siah1 restricts its enzymatic function in the nucleus. Binding to GAPDH slows the Siah1 turnover rate, thus accelerating degradation of its nuclear targets (54). As a consequence, Siah1-mediated degradation of histone-methylating enzyme suppressor of variegation 3–9 homologue 1 (SUV39H1) is increased, leading to a decrease in methylation of lysine 9 on histone H3 and promoting dendritic outgrowth (133). Apart from stabilizing Siah1, GAPDH can also bind p300/CBP (CREB binding protein) in the nucleus to facilitate expression of p300/CBP target genes, such as p53, thereby promoting cell death (135).
FIG. 3.

GAPDH-mediated transnitrosylation. S-Nitrosylation of GAPDH promotes its binding to Siah1, facilitating translocation into the nucleus as a SNO-GAPDH/Siah1 complex. Complex formation stabilizes Siah1 protein, thus enhancing degradation of its nuclear targets. SNO-GAPDH can also bind and activate p300/CBP in the nucleus. Moreover, SNO-GAPDH can mediate transnitrosylation of other nuclear proteins, such as SIRT1, HDAC2, and DNAPK, altering their function. DNAPK, DNA-activated protein kinase; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; HDAC2, histone deacetylase 2; SIRT1, sirtuin 1.
In addition, SNO-GAPDH facilitates protein–protein transnitrosylation in the nucleus. Transnitrosylation targets of GAPDH include deacetylating enzyme sirtuin 1 (SIRT1), histone deacetylase 2 (HDAC2), DNA activated protein kinase (DNAPK), and B23/nucleophosmin (Fig. 3) (76, 85). In fact, GAPDH has proven to be a better NO donor toward SIRT1 than GSNO. S-Nitrosylation of SIRT1 abrogates its deacetylation activity, leading to increased acetylation of PGC1α and thus a decrease in its transcriptional activity (76). Moreover, SNO-GAPDH-dependent inactivation of SIRT1 facilitates pathological acetylation of tau, possibly contributing to tau aggregation in AD (136). Since SNO-GAPDH formation has been found in models of other neurodegenerative diseases (e.g., PD and HD), the potential pathophysiological role of SNO-GAPDH-mediated transnitrosylation in these and other neurological conditions remains to be explored.
In addition to its role in the nucleus, SNO-GAPDH appears to mediate protein–protein transnitrosylation in mitochondria (75). In this case, SNO-GAPDH enters the mitochondrial matrix, possibly due to its putative mitochondrial import sequence, thus increasing S-nitrosylation of mitochondrial proteins such as HSP60 and VDAC1 through transnitrosylation reactions. Further studies are needed to examine how SNO-GAPDH-initiated mitochondrial transnitrosylation might regulate mitochondrial function.
Transnitrosylation from SNO-DJ-1 to Phosphatase and Tensin Homologue
Deletion or mutation of the DJ-1 (PARK7) gene has been linked to familial autosomal recessive early-onset PD and in rare cases adult PD (16). In addition, virtually all postmortem brains with sporadic PD or AD that have been studied to date contain excessively oxidized or S-nitrosylated DJ-1, which disrupts its neuroprotective action (9, 29). DJ-1 appears to be a multifunctional neuroprotective protein, but its exact mechanism(s) of action remains unclear (9, 15). The most well-characterized function of DJ-1 is its antioxidative property mediated by three redox-sensitive cysteine residues (Cys46, Cys53, and Cys106) (21, 143, 190). Of these three cysteine residues, Cys106 is most sensitive to oxidation, often forming a cysteine sulfinic acid (SO2H) (21, 120, 175). Moreover, Cys106 can be further oxidized to sulfonic acid (SO3H), irreversibly inactivating DJ-1 activity (190). Supporting this notion, mutation of Cys106 abolishes the antioxidant properties of DJ-1 (21, 156). In addition, while Cys46 and Cys53 of DJ-1 are susceptible to S-nitrosylation by artificially high concentrations of “NO donor” (>0.25 mM GSNO) in vitro and in cell-based systems (62), our group found that only Cys106 is S-nitrosylated under physiological conditions. Specifically, increasing endogenous NO via nNOS activation or the addition of low concentrations of “NO donor” (≤25 μM SNOC) produced S-nitrosylation only at DJ-1(C106) (30). Moreover, when we and our colleagues solved the crystal structure of DJ-1 at a 1.5 Å resolution/pH 4.6 in the presence of NO, the predominant S-nitrosylation site was at Cys106 (30). Taken together, these findings are consistent with the notion that S-nitrosylation of DJ-1 occurs predominantly at Cys106 (30).
In addition, our group previously found that phosphatase and tensin homologue (PTEN) is S-nitrosylated by endogenous NO after activation of nNOS or in response to low concentrations of exogenous NO donor (≤25 μM SNOC). S-Nitrosylation of PTEN decreases its phosphatase activity (79, 110). PTEN is a well-known tumor suppressor that is mutated in many forms of cancer (87), negatively regulating phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt) activity. In the central nervous system (CNS), PTEN activity is critical for diverse functions such as neuronal differentiation and synaptogenesis (11, 48, 81), neuronal plasticity (23), and axonal branching (41, 116). PTEN activity is regulated by several PTMs, including phosphorylation, oxidation, and S-nitrosylation. For example, phosphorylation at the C-terminal tail region of PTEN (i.e., Ser380, Thr382, or Thr383) decreases its phosphatase activity (162, 167). In addition, oxidation of PTEN at Cys124 inactivates its inhibitory functions through formation of an intramolecular disulfide bond between Cys71 and Cys124 (80). Formation of this disulfide results in activation of the PI3K/Akt signaling pathway (179). We and our colleagues found that PTEN can be S-nitrosylated, predominantly at Cys83, but in some circumstances also at Cys71 and Cys124 (79, 110). By mutating these cysteine residues, the resulting non-nitrosylatable PTEN mutant completely abolished S-nitrosothiol formation and polyubiquitination of PTEN, suggesting that degradation of PTEN is at least, in part, dependent on its S-nitrosylation (79, 110). Increased levels of SNO-PTEN have been observed not only in human postmortem brains with AD and PD but also in the penumbra of ischemic mouse brains, outside of the region of maximal damage. One possibility is that SNO-PTEN may represent an early neuroprotective event in the disease process, activating the PI3K/Akt signaling pathway to promote neuronal cell survival (79, 110, 119).
Prior evidence had suggested that DJ-1 inhibits enzymatic activity of PTEN via direct binding to PTEN (3, 73, 181). Since we had found that both DJ-1 and PTEN were S-nitrosylated under physiological conditions, we postulated that DJ-1(SNO-Cys106) might transnitrosylate PTEN (Fig. 4). We subsequently found considerable evidence for this hypothesis. For example, knockdown of cytosolic DJ-1 decreases SNO-PTEN formation and thereby increases PTEN enzymatic activity, consistent with the notion that transnitrosylation proceeds from DJ-1 to PTEN in intact cells (30, 105). SNO-DJ-1-mediated transnitrosylation of PTEN would thus stimulate the prosurvival of PI3K/Akt signaling pathway (79, 110). Recently, another group reported that SNO-DJ-1 also mediates S-nitrosylation of parkin, which maintains mitochondrial function and integrity to protect neurons from cell death (113). These authors found that S-nitrosylation of parkin is impaired in DJ-1 KO cells, whereas overexpression of wild-type DJ-1, but not mutant DJ-1(C106S), preserves SNO-parkin levels in DJ-1 KO cells. While these results are consistent with the notion that parkin might be a target for transnitrosylation by SNO-DJ-1 (113), further work will be needed to determine if this is indeed a direct reaction.
FIG. 4.
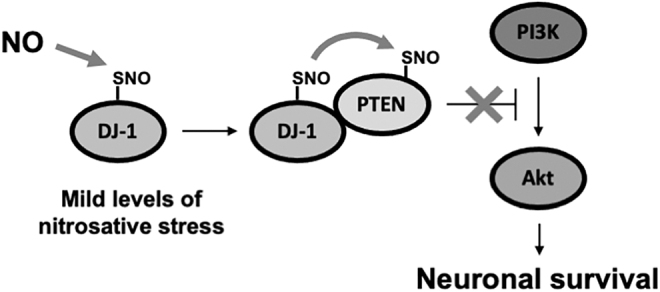
Schema of SNO-DJ-1 contributing to neuronal survival via transnitrosylation of PTEN. SNO-DJ-1 inhibits PTEN activity via transnitrosylation (forming SNO-PTEN). SNO-PTEN prevents PTEN from antagonizing the action of PI3K and thus activates the PI3K/Akt pathway. Akt, protein kinase B; PI3K, phosphoinositide 3-kinase; PTEN, phosphatase and tensin homologue.
Transnitrosylation of Drp1 by SNO-Cdk5
Cdk5, a member of the cyclin-dependent kinase (Cdk) family, is a neuronal specific kinase and has emerged as a crucial regulator of cell survival, axonal guidance, neuronal migration, and synaptic density (74, 180, 188). The kinase activity of Cdk5 is dictated by its temporal and spatial expression as well as the intracellular localization of Cdk5 regulators, such as p39, p35, and p25 (the cleaved form of p35) (38, 187). Dysregulated Cdk5 kinase activity contributes to the pathology of several neurodegenerative diseases, including AD, PD, ALS, and HD (115, 117, 121).
Emerging evidence demonstrates that S-nitrosylation of Cdk5 occurs at Cys83 and Cys157 (44, 122, 188) and that S-nitrosylation increases Cdk5 kinase activity (122). Arguably the best correlate to cognitive decline in human with AD is the degree of synaptic loss in the brain (160), and Cdk5 is known to regulate the synaptic number and excitatory synaptic transmission (45, 74). Along these lines, our group observed significantly greater levels of SNO-Cdk5 in human postmortem AD brains compared with control brains. Moreover, we showed that SNO-Cdk5 contributes to amyloid β (Aβ)- or NMDAR-mediated dendritic spine loss, which is rescued by expression of non-nitrosylatable mutant Cdk5 (122). These findings are consistent with the notion that increased Cdk5 kinase activity via aberrant S-nitrosylation may be a critical mechanism of cognitive impairment in AD. In many neurodegenerative diseases including AD, mitochondrial dysfunction, with consequent bioenergetic compromise and synaptic loss, contributes to cognitive dysfunction (57, 125). Aberrant Cdk5 activity is associated with mitochondrial dysfunction (154, 173), suggesting that SNO-Cdk5-dependent synaptic loss may be mediated through mitochondrial abnormalities.
Mitochondria are dynamic organelles consistently undergoing fusion and fission throughout the cell. These mitochondrial dynamics are important for both mitochondrial inheritance and the maintenance of mitochondrial function (174). Dysfunction in mitochondrial dynamics is implicated in age-associated neurodegenerative diseases (2, 36, 84, 169, 191). Among the mitochondrial fusion/fission-regulating proteins, our group discovered that Drp1, which encodes a dynamin-like GTPase and facilitates mitochondrial fission, is S-nitrosylated at Cys644 in human AD brains (27). In healthy neurons, mitochondria display an elongated filamentous morphology along its neurites, whereas fragmented mitochondria are observed in neurons exposed to Aβ oligomers, which are known to induce nitrosative stress. In this context, NO contributes to increased mitochondrial fission via upregulation of Drp1 GTPase activity (13). Dimerization/oligomerization of Drp1 is known to enhance its GTPase activity. Taking these findings into account, we found that S-nitrosylation increases both Drp1 dimerization and GTPase activity, resulting in excessive mitochondrial fragmentation and cristae damage. This results in bioenergetic compromise and synaptic loss. In contrast, expression of non-nitrosylatable mutant Drp1(C644S) prevents Aβ-induced mitochondrial fragmentation and synaptic loss (27). These findings support the notion that SNO-Drp1 manifests excessive GTPase activity, inducing abnormal mitochondrial fragmentation, compromising bioenergetics, and thereby impairing synaptic structure and function in AD.
Emerging evidence suggests that Cdk5 directly regulates Drp1 activity and thus mitochondrial dynamics, and various mechanisms of action for this have been suggested, including PTM, for example, phosphorylation and S-nitrosylation (2, 26, 50, 65, 122). Interestingly, our group demonstrated that transfer of an NO group from SNO-Cdk5 to Drp1 via transnitrosylation contributes to NMDA/Aβ-mediated synaptic spine loss. Moreover, this transnitrosylation reaction is thermodynamically favored in the intact cells (105). Although we and others also found that SNO-Cdk5 can potentially phosphorylate Drp1 (50, 65), Aβ-mediated synaptic loss was significantly rescued by NOS inhibitors, non-nitrosylatable mutant Drp1 (27), or non-nitrosylatable mutant Cdk5 (122). These findings support the notion that transnitrosylation of Drp1 by SNO-Cdk5 might be the predominant neuropathological event leading to mitochondrial dysfunction during the pathogenesis of AD. In the next section, we consider recent discoveries elucidating the upstream events leading to Cdk5-to-Drp1 transnitrosylation in AD.
Multitransnitrosylation Cascade from Uch-L1 to Cdk1 to Drp1
Uch-L1 is one of the most abundant proteins in the brain (1%–2% of the total soluble proteins) and has been characterized as a deubiquitinating enzyme (142). Uch-L1 dysfunction is associated with neurodegenerative diseases. For example, downregulation, redox-dependent PTMs, and genetic mutations of Uch-L1 have all been observed in human AD and PD brains (19, 28, 77); however, the molecular mechanism of Uch-L1-mediated neurodegeneration has remained unknown until very recently. Emerging evidence showed that S-nitrosylation of Uch-L1 at Cys90, Cys152, and Cys220 occurs in cells exposed to high concentrations of GSNO, accompanied by decreased ubiquitin binding affinity through conformational changes and decreased deubiquitinating activity (77). In addition, circular dichroism spectroscopy revealed significant structural changes in Uch-L1 when incubated with increasing concentrations of GSNO, but the triple cysteine mutant Uch-L1(C90A/C152A/C220A) was resistant to these GSNO-induced conformational changes (77). Moreover, it was reported that SNO-Uch-L1 underwent α-syn fibrillization, promoting cytosolic aggregation, as seen in Lewy bodies of PD and LBD (77).
In contrast, in our hands, we found that Uch-L1 is predominately S-nitrosylated in vivo only at Cys152 in AD transgenic mouse brains (106). Moreover, the crystal structure of Uch-L1 (35) revealed that a full S-nitrosylation motif (comprising Glu7 and Arg153) is present in the vicinity of Cys152 but not in the other cysteine residues previously reported to be nitrosylated. These data are consistent with the notion that Cys152 is the predominant target of S-nitrosylation in Uch-L1 (68, 151). We further showed that expression of non-nitrosylatable mutant Uch-L1(C152A) is sufficient to abrogate the effect of NO on Uch-L1 catalytic activity and ubiquitin binding (106). S-Nitrosylation of Uch-L1 also induces synaptic spine loss both in vitro and in vivo in cell-based and animal models of AD. Importantly, compared with control human brains, the relative increases in SNO-Uch-L1 levels in human AD brains, mouse models of AD, and cell-based neuronal models exposed to Aβ oligomers are all comparable (106), consistent with the notion that a pathophysiologically relevant amount of SNO-Uch-L1 is present in our AD model systems. This fact provides confidence that the conclusions made about SNO-Uch-L1 from the model systems are germane to human AD.
As discussed above, Aβ-induced transnitrosylation from SNO-Cdk5 to Drp1 results in mitochondrial fragmentation, bioenergetic compromise, and consequent synaptic loss. We recently discovered that this pathway is triggered upstream initially by transnitrosylation from SNO-Uch-L1 to Cdk5. Interestingly, direct binding of Uch-L1 to Cdk5 was already known to enhance Cdk5 kinase activity in a Uch-L1 activity-independent manner (71). Our group then demonstrated that Uch-L1 can transnitrosylate Cdk5 in vitro and in intact cell systems (106). Remarkably, we found a cascade of concerted transnitrosylation from SNO-UchL-1 to SNO-Cdk5 to SNO-Drp1 (Fig. 5). In a critical experiment, we found that depletion of Cdk5 attenuates SNO-Drp1 formation in the presence of SNO-Uch-L1; this finding is consistent with the notion that transfer of nitrosothiol from Uch-L1 to Drp1 requires Cdk5 (106). Moreover, in an in vitro transnitrosylation assay, SNO-Cdk5 does not transnitrosylate Uch-L1 but does transnitrosylate Drp1 in the same reaction, suggesting the concerted nature of the reaction schema (106, 122). Moreover, we show that the concerted transnitrosylation from SNO-Uch-L1 to Cdk5 to Drp1 is both kinetically and thermodynamically favored (106). Assuming the reaction reaches steady state, as might be reasonably expected in an age-related neurodegenerative disease, the Nernst equation and the associated Gibb's free energy for transnitrosylation from SNO-UchL1 to Cdk5 produce a value of nearly −20 kJ/mol, indicating that the reaction is highly thermodynamically favorable (106). A similar analysis shows that subsequent transnitrosylation from SNO-Cdk5 to Drp1 is also energetically favorable, consistent with the concerted action of this pathway. Hence, our findings argue that Cdk5 is transnitrosylated from Uch-L1, and then, in turn, serves as an NO donor, to transnitrosylate Drp1. Our evidence suggests that this triple transnitrosylation cascade represents a pathogenic event in AD because lentiviral infection with non-nitrosylatable mutant Uch-L1(C152A) constructs significantly preserves synapses in transgenic mouse models of AD (106).
FIG. 5.

Schematic modeling of multiple transnitrosylation steps from Uch-L1 to Cdk5 to Drp1 in the pathogenesis of AD. In AD, oligomerized Aβ, other misfolded proteins, aging, and neuroinflammation can all increase intracellular NO• levels in neurons. The increase in NO• occurs via extrasynaptic (e)NMDAR-mediated nNOS activation in neurons and iNOS activation in glia cells. Excessive elevation of NO• causes aberrant mitochondrial fragmentation, resulting in bioenergetic compromise with consequent neuronal damage and synaptic loss. This loss of synapses contributes to cognitive decline in AD. The aberrant transnitrosylation pathway that triggers the loss of synapses involves transfer of NO+ from SNO-Uch-L1 to Cdk5 to Drp1. Accounting for the mitochondrial damage, the formation of SNO-Drp1 activates its GTPase activity and increases mitochondrial fragmentation. This multistep transnitrosylation cascade represents a non-canonical signaling pathway distinct from the known enzymatic functions of Uch-L1, Cdk5, and Drp1. Aβ, amyloid β; AD, Alzheimer's disease; Cdk, cyclin-dependent kinase; Drp1, dynamin-related protein 1; iNOS, inducible nitric oxide synthase; NMDAR, N-methyl-d-aspartate-type glutamate receptor; nNOS, neuronal nitric oxide synthase; NO•, nitric oxide; Uch-L1, ubiquitin carboxy-terminal hydrolase L1.
In summary, this new work shows that mechanistically distinct enzymes, that is, a kinase, a guanosine triphosphatase, and a ubiquitin protein hydrolase, which function in disparate biochemical pathways, can also act in concert to mediate a series of redox reactions. Each enzyme manifests a second, noncanonical function—transnitrosylation—triggering a pathological biochemical cascade in AD. The resulting series of transnitrosylation reactions contributes to synaptic loss, the major pathological correlate to cognitive decline in AD. This concept of “noncanonical” or “hidden” transnitrosylation networks may represent a set of biochemical reactions not currently enveloped in standard bioinformatic packages such as Gene Ontology (GO) enrichment or KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway analysis, and thus, this finding could change our concept of pathway analysis and selection pressure on such cascades. Given this network operates in the postreproductive period associated with aging, at a time of increasing susceptibility to neurodegenerative disorders, natural selection pressure may be lessened on this aberrant alternative activity. In the future, identification of additional noncanonical transnitrosylation networks using MS-based approaches should be explored and may well lead to new therapeutic targets for neurodegenerative disorders.
Transnitrosylation Pathways in Non-CNS Tissues and Other Organisms
In the aged brain, protein–protein transnitrosylation mediates both protective and destructive pathways depending on the signaling cascades affected by the reactions. In other tissues and in nonmammalian organisms, recent studies have identified additional transnitrosylation events, including the Hcp in E. coli and S100A8/S100A9 in mouse bone marrow-derived macrophages exposed to IFN-γ and LDLox (68, 140). Here we review the roles of these proteins involved in transnitrosylation. We also evaluate the potential influence of these studies on redox reactions relevant to the field of neuroscience.
Hcp-dependent transnitrosylation in E. coli
Activation of nitrate reductase (NarGHI) in E. coli growing anaerobically in nitrate results in an increase in protein S-nitrosylation, although the underlying mechanism for the generation of the S-nitrosylating species in these bacteria remains unclear (139). Notably, S-nitrosylation of the transcription factor OxyR under anaerobic conditions induces expression of a distinct subset of the genes regulating cell growth, metabolism, and nitrosative stress (139).
Using this bacterial model system, Stamler's group revealed that a majority of the SNO-proteins formed in E. coli are primarily due to transnitrosylation from Hcp (140) (Fig. 6A). E. coli Hcp is a hybrid protein containing a [2Fe-2S] cluster and a [4Fe-2S-2O] cluster (166). Its expression is induced during anaerobic respiration without oxygen and with nitrate (139). Iron/sulfur clusters in a protein are thought to mediate one-electron oxidation of NO• to NO+, facilitating oxidative modification of thiolate anion (S−) by NO+ and thereby contributing to the generation of S-nitrosothiol on the protein (82). Along these lines, the iron/sulfur centers of Hcp are reported to catalyze auto-S-nitrosylation of Hcp at a cysteine residue coordinating an Fe atom. In addition, Hcp forms a large multiprotein complex, including NarGHI, OxyR, and a series of proteins mediating transnitrosylation, such as GAPDH and LpdA (dihydrolipoyl dehydrogenase). Accordingly, SNO-Hcp serves as an anchoring protein that facilitates transfer of an NO group from Hcp to its interacting partners, conveying SNO-based signals to downstream proteins. Importantly, Hcp-mediated transnitrosylation pathways not only regulate metabolism and motility but also confer protection against nitrosative stress, consistent with the notion that these redox reactions are physiologically important (140). Thus, the Hcp interactome represents a redox machinery that triggers S-nitrosylation-dependent signaling in facultative anaerobic bacteria.
FIG. 6.

Transnitrosylation in Escherichia coli and immune cells. (A) Schematic of Hcp-mediated transnitrosylation. Nitrate reductase (NarGHI) contributes to production of NO-related species in E. coli during anaerobic respiration using nitrate. The iron/sulfur cluster of Hcp may promotes NO• oxidation to NO+, facilitating formation of an SNO at an Fe-coordinating cysteine residue. Transnitrosylation from SNO-Hcp to interacting proteins, such as OxyR, GAPDH, and LpdA, regulates cellular metabolism and nitrosative stress in E. coli. (B) S100A9-mediated transnitrosylation in myeloid cells. Inflammatory stimuli induce iNOS expression, followed by assembly of a protein complex consisting of iNOS, S100A8, and S100A9. iNOS-driven S-nitrosylation of S100A9 at Cys3 (forming SNO-S100A9) transfers NO-related species to target proteins recruited to the complex by S100A8. These target proteins include GAPDH, annexin V, ezrin, moesin, and vimentin. The transnitrosylation events result in dysregulation of the GAIT (IFN-γ-activated inhibitor of translation) complex, contributing to prolonged expression of inflammatory genes. Hcp, hybrid cluster protein; IFN, interferon.
Importantly, transition metal-catalyzed protein S-nitrosylation has also been widely reported in mammalian systems (98), as exemplified by hemoglobin, matrix metalloproteinases, and NOS isoforms (49, 93, 118, 124). Therefore, in the mammalian brain, it will be interesting to examine if metalloproteins, especially those with iron/sulfur centers, support transnitrosylation pathways during inflammaging and neurodegenerative processes.
S100A8/A9-mediated transnitrosylation in inflammation
S100A8 and S100A9 proteins belong to the large S100 protein family, functioning not only as intracellular Ca2+ sensors but also as extracellular ligands for cell surface receptors such as toll-like receptor 4 and receptor for advanced glycosylation end products. S100A8 and S100A9 are abundantly expressed in myeloid lineage cells, including neutrophils, monocytes, and mast cells, forming stable hetero- or homodimers (168). S100A8 and S100A9 each contain a single cysteine residue. The thiol groups of these cysteines are highly sensitive to ROS-mediated oxidative modifications and RNS-mediated S-nitrosylation (55, 68, 89, 111). For example, oxidative stress triggers disulfide linkage between S100A8 molecules, forming homodimers, and decreases S100A8 activity as an extracellular chemotactic factor that stimulates the recruitment of myeloid cells to inflammatory sites (55). Moreover, S-nitrosylated S100A8 mediates some of the anti-inflammatory properties of NO, at least, in part, via inhibition of mast cell degranulation (89). Intriguingly, transnitrosylation from S100A8 to hemoglobin occurs in vitro (89), suggesting that S100A8 can participate in transfer of an NO group to other proteins in a cellular context. In addition, ROS-mediated oxidation of S100A9 at Cys3 leads to mTORC1 activation, thus contributing to proliferation of cancer stem-like cells (111). However, the pathophysiological role of S-nitrosylated S100A9 remains unclear.
Jia and colleagues have shown that the S100A8/A9 heterodimer binds to iNOS, directing selective S-nitrosylation of other interacting proteins and eliciting an anti-inflammatory response (68) (Fig. 6B). Specifically, in peripheral human myeloid cells, inflammatory stimuli promote complex formation among iNOS, S100A8/9 heterodimers, and target proteins to be S-nitrosylated, including GAPDH. Within this protein complex, iNOS-derived NO-related species initially S-nitrosylate S100A9 at Cys3. In addition, S100A8 can interact with a target protein, for example, GAPDH, annexin V, ezrin, or vimentin, thereby facilitating transnitrosylation from S100A9 to these interacting proteins. The study further identified a consensus motif (I/L-X-C-X-X-D/E) for iNOS/S100A8/S100A9-dependent S-nitrosylation, in which a nearby hydrophobic residue (I or L) and acidic amino acid (D or E) increase the nucleophilicity of the target cysteine.
In the CNS, S100A8 and S100A are normally expressed in microglia and to some extent in astrocytes and neurons, while increased expression is associated with neurodegenerative disorders such as AD (33). For example, both S100A8 and S100A9 are present within neurofibrillary tangles (144), promote γ- and β-secretase activity to increase Aβ levels (78), and directly interact and coaggregate with Aβ peptide (185). However, whether iNOS/S100A8/S100A9-mediated transnitrosylation influences the pathophysiology of AD or other neurological disorders remains unknown. Future studies will be needed to decipher the role of S100A9-mediated transnitrosylation in neuroinflammation during AD pathogenesis.
Concluding Remarks and Future Directions
Depending on the quantity and site of production, RNS can trigger either physiological functions, such as synaptic transmission, neuronal plasticity, and cell survival, or pathological phenotypes, including protein misfolding, synaptic damage, and cognitive decline. Subserving these events, NO-dependent protein-S-nitrosylation leads to activation or inhibition of multiple pathways in the brain. Furthermore, protein–protein transnitrosylation has emerged as an important signaling mechanism, transducing the biological effects of NO to distinct downstream proteins not only in human brains but in most if not all tissues and organisms. Hence, S-nitrosylation/transnitrosylation must be tightly regulated to ensure proper physiological function; aberrant activation of these pathways can contribute to the pathogenesis of many types of diseases, including neurodegenerative disorders, cancer, and autoimmune diseases. Moreover, these findings suggest that elucidation of additional transnitrosylation networks that operate under either physiological or pathological conditions could open up new avenues of exploration for potential therapeutic targets to prevent NO•-associated and inflammatory neurodegeneration in aged brains. Further studies are needed that utilize MS-based proteomics for identification of additional transnitrosylation networks as well as biochemical approaches to support or refute an enzymatic process that potentially underlies each transnitrosylation reaction.
Moreover, the primary source(s) of NO• that contributes to aberrant protein S-nitrosylation pathways in vivo remains unclear. Hence, additional work will be needed to determine which NOS (e.g., iNOS or nNOS) provides NO• in this regard. In addition, what cell types (i.e., neurons, astrocytes, oligodendrocytes, and microglia) are involved in RNS production in vivo and how various transnitrosylation networks contribute to aberrant S-nitrosylation of specific proteins all remain to be elucidated.
MS methods to enhance the field of S-nitrosylation research
Progress in biochemistry has long depended on the development of advanced analytical and physical methods of analysis. The mammalian brain contains 10–20,000 proteins, and further variations based on PTMs and the number of cysteines that can be modified may yield a number an order of magnitude larger than that. In the early 1990s, S-nitrosylation was discovered as a significant component of NO chemistry, a PTM of proteins, and a significant factor in signaling and toxicity (90). In most cases, the instability of the –SNO group was a significant block to the characterization of protein identity and the specific cysteines that had been modified. Slowly it became known that the nitrosylation process was both thermodynamically and kinetically driven with many essential components, and that the process was reversible (denitrosylation), and transferable, for example, to other proteins. In 2001, a dramatic turn of events emerged in the publication of Jaffrey and Snyder (63, 64) of a method that involved selective cysteine blocking, denitrosylation of -SNO with ascorbate to the thiol (90), and substitution of the newly generated thiol with an entity (e.g., biotin) that could be selectively captured for further proteomic identification. The method, called “biotin switch,” rapidly generated hundreds of citations with protein identities important to the health of brain and other organ systems.
The biotin-switch assay was so successful that it stimulated research to advance the rate and capacity for protein identification and cysteine-site identification. A major advance appeared in 2006 from the Gross laboratory (53) under the acronym SNOSID (SNO site Identification). SNOSID is an extension of the biotin-switch method in which the captured proteins are trypsinized before capture to yield the SNO-cysteine peptides. Using this approach, the laboratory was able to identify 68 proteins and associated cysteine-SNO sites from rat cerebellum. Another step toward improvement came from Stamler and colleagues with resin-assisted capture of SNO-proteins. This method substitutes thiopropyl sepharose for biotin/avidin to reduce the required steps for enrichment of SNO-proteins. Since this technique combines thiol labeling and protein enrichment into one step, it avoids additional processes that could affect background labeling and thus sensitivity such as removal of excess biotinylating agent and pulldown by streptavidin (43, 161). Shortly thereafter the Ischiropoulos laboratory greatly improved the recovery of SNO-peptides and proteins through covalent binding of the cysteine sulfur with phenylmercury, which has no reactivity documented for disulfides, sulfinic or sulfonic acids, S-glutathionylated, S-alkylated, or S-sulfhydrated cysteine residues (40). This enabled structural profiling of sites in proximity to SNO-cysteines and opened the field to possibilities of predicting these sites (39).
The search for even better methods continued. Two problems continued to plague the biotin-switch method. False positives from incomplete blocking of naked cysteines, and false negatives from incomplete denitrosylation with ascorbate and subsequent biotinylation of SNO-sites. False positives are by far the worst problem because it introduces misinformation that will confound subsequent systems analysis. A solution to the false-positive problem would be to develop a reagent that would react with and couple to the SNO-site, and this was accomplished by the Tannenbaum laboratory in 2013 (137) and then applied to mouse models of AD (6, 138), autism (5), and arsenic toxicity (7). In each case, hundreds to thousands of SNO-proteins were identified in both control and treated mouse frontal cortex.
Once we enter the era of big data with protein S-nitrosylation, we will need the tools of systems biology to explain what the data mean. The tools we have now include genomic platforms, GO, pathway analysis, and STRING for protein interactions. In the future, we will have to develop methods in artificial intelligence and machine learning to help develop models and hypotheses incorporating all of this new information on the S-nitrosoproteome in health and disease.
Acknowledgment
We thank Scott R. McKercher of The Scripps Research Institute for critical reading of the article.
Abbreviations Used
- Aβ
amyloid β
- AD
Alzheimer's disease
- AE1
anion exchanger 1
- Akt
protein kinase B
- ALS
amyotrophic lateral sclerosis
- BIR
baculovirus IAP repeat
- Cdk
cyclin-dependent kinase
- cGMP
cyclic guanosine monophosphate
- CNS
central nervous system
- csPDI
cell surface PDI
- Cys
cysteine
- DNAPK
DNA-activated protein kinase
- Drp1
dynamin-related protein 1
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GO
Gene Ontology
- Grx
glutaredoxin
- GSNO
S-nitrosoglutathione
- GSNOR
GSNO reductase
- Hcp
hybrid cluster protein
- HD
Huntington's disease
- HDAC2
histone deacetylase 2
- HSP
heat shock protein
- IAP
inhibitor of apoptosis protein
- IFN
interferon
- iNOS
inducible NOS
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- KO
knockout
- LBD
Lewy body dementia
- LDLox
oxidatively modified low-density lipoprotein
- MS
mass spectrometry
- NFAT-1
nuclear factor of activated T cells 1
- NMDAR
N-methyl-d-aspartate-type glutamate receptor
- nNOS
neuronal NOS
- NO•
nitric oxide
- NO+
nitrosonium cation
- NOS
NO synthase
- O2•−
superoxide anion
- PD
Parkinson's disease
- PDI
protein-disulfide isomerase
- PGC1α
proliferator-activated receptor-γ coactivator 1α
- PI3K
phosphoinositide 3-kinase
- Prx
peroxiredoxins
- PTEN
phosphatase and tensin homologue
- PTM
posttranslational modification
- RNR
ribonucleotide reductase
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- RS−
thiolate anion
- RS•
thiyl radical
- SIRT1
sirtuin 1
- SNOC (or CysNO)
S-nitrosocysteine
- SNO-protein
S-nitrosylated protein
- SNOSID
SNO site Identification
- TRP-14
Trx-related protein 14
- Trx
thioredoxin
- TrxR
Trx reductase
- Uch-L1
ubiquitin carboxy-terminal hydrolase L1
- XIAP
X-linked inhibitor of apoptosis
Authors' Contributions
T.N., C.O., and X.Z. prepared a draft article; S.R.T. and S.A.L. edited and finalized the article; T.N., C.O., X.Z., S.R.T., and S.A.L. approved the final version of the article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported, in part, by NIH grants RF1 AG057409, R01 DA048882, R01 NS086890, P30 NS076411, P01 ES016738, and DP1 DA041722 (to Stuart A. Lipton), R01 AG056259 (to Stuart A. Lipton and Steven R. Tannenbaum), R01 AG061845 (to Tomohiro Nakamura); by an award from the California Tobacco-Related Disease Research Program (TDRP 27IR-0010 to Stuart A. Lipton); by a Distinguished Investigator Award from the Brain & Behavior Research Foundation (to Stuart A. Lipton); by a New Investigator Award from the Alzheimer's Association (to Tomohiro Nakamura); by the Michael J. Fox Foundation (to Stuart A. Lipton and Tomohiro Nakamura); and by the Step Family Foundation (to Stuart A. Lipton).
References
- 1.Ahern GP, Klyachko VA, and Jackson MB. cGMP and S-nitrosylation: two routes for modulation of neuronal excitability by NO. Trends Neurosci 25: 510–517, 2002 [DOI] [PubMed] [Google Scholar]
- 2.Alexander C, Votruba M, Pesch UE, Thiselton DL, Mayer S, Moore A, Rodriguez M, Kellner U, Leo-Kottler B, Auburger G, Bhattacharya SS, and Wissinger B. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet 26: 211–215, 2000 [DOI] [PubMed] [Google Scholar]
- 3.Aleyasin H, Rousseaux MW, Marcogliese PC, Hewitt SJ, Irrcher I, Joselin AP, Parsanejad M, Kim RH, Rizzu P, Callaghan SM, Slack RS, Mak TW, and Park DS. DJ-1 protects the nigrostriatal axis from the neurotoxin MPTP by modulation of the AKT pathway. Proc Natl Acad Sci U S A 107: 3186–3191, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ali Khan H and Mutus B.. Protein disulfide isomerase a multifunctional protein with multiple physiological roles. Front Chem 2: 70, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Amal H, Barak B, Bhat V, Gong G, Joughin BA, Wang X, Wishnok JS, Feng G, and Tannenbaum SR. Shank3 mutation in a mouse model of autism leads to changes in the S-nitroso-proteome and affects key proteins involved in vesicle release and synaptic function. Mol Psychiatry 25: 1835–1848, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Amal H, Gong G, Gjoneska E, Lewis SM, Wishnok JS, Tsai LH, and Tannenbaum SR. S-nitrosylation of E3 ubiquitin-protein ligase RNF213 alters non-canonical Wnt/Ca +2 signaling in the P301S mouse model of tauopathy. Transl Psychiatry 9: 44, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Amal H, Gong G, Yang H, Joughin BA, Wang X, Knutson CG, Kartawy M, Khaliulin I, Wishnok JS, and Tannenbaum SR. Low doses of arsenic in a mouse model of human exposure and in neuronal culture lead to S-nitrosylation of synaptic proteins and apoptosis via nitric oxide. Int J Mol Sci 21: 3948, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Andersen JK.Oxidative stress in neurodegeneration: cause or consequence? Nat Med 10 Suppl: S18–S25, 2004 [DOI] [PubMed] [Google Scholar]
- 9.Ariga H, Takahashi-Niki K, Kato I, Maita H, Niki T, and Iguchi-Ariga SM. Neuroprotective function of DJ-1 in Parkinson's disease. Oxid Med Cell Longev 2013: 683920, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arnelle DR and Stamler JS. NO+, NO, and NO- donation by S-nitrosothiols: implications for regulation of physiological functions by S-nitrosylation and acceleration of disulfide formation. Arch Biochem Biophys 318: 279–285, 1995 [DOI] [PubMed] [Google Scholar]
- 11.Backman SA, Stambolic V, Suzuki A, Haight J, Elia A, Pretorius J, Tsao MS, Shannon P, Bolon B, Ivy GO, and Mak TW. Deletion of PTEN in mouse brain causes seizures, ataxia and defects in soma size resembling Lhermitte-Duclos disease. Nat Genet 29: 396–403, 2001 [DOI] [PubMed] [Google Scholar]
- 12.Barglow KT, Knutson CG, Wishnok JS, Tannenbaum SR, and Marletta MA. Site-specific and redox-controlled S-nitrosation of thioredoxin. Proc Natl Acad Sci U S A 108: E600–E606, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barsoum MJ, Yuan H, Gerencser AA, Liot G, Kushnareva Y, Graber S, Kovacs I, Lee WD, Waggoner J, Cui J, White AD, Bossy B, Martinou JC, Youle RJ, Lipton SA, Ellisman MH, Perkins GA, and Bossy-Wetzel E. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J 25: 3900–3911, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Benhar M, Forrester MT, Hess DT, and Stamler JS. Regulated protein denitrosylation by cytosolic and mitochondrial thioredoxins. Science 320: 1050–1054, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Biosa A, Sandrelli F, Beltramini M, Greggio E, Bubacco L, and Bisaglia M. Recent findings on the physiological function of DJ-1: beyond Parkinson's disease. Neurobiol Dis 108: 65–72, 2017 [DOI] [PubMed] [Google Scholar]
- 16.Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, Dekker MC, Squitieri F, Ibanez P, Joosse M, van Dongen JW, Vanacore N, van Swieten JC, Brice A, Meco G, van Duijn CM, Oostra BA, and Heutink P. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299: 256–259, 2003 [DOI] [PubMed] [Google Scholar]
- 17.Bredt DS, Hwang PM, and Snyder SH. Localization of nitric oxide synthase indicating a neural role for nitric oxide. Nature 347: 768–770, 1990 [DOI] [PubMed] [Google Scholar]
- 18.Bredt DS and Snyder SH. Isolation of nitric oxide synthetase, a calmodulin-requiring enzyme. Proc Natl Acad Sci U S A 87: 682–685, 1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Butterfield DA, Gnjec A, Poon HF, Castegna A, Pierce WM, Klein JB, and Martins RN. Redox proteomics identification of oxidatively modified brain proteins in inherited Alzheimer's disease: an initial assessment. J Alzheimers Dis 10: 391–397, 2006 [DOI] [PubMed] [Google Scholar]
- 20.Calabrese V, Mancuso C, Calvani M, Rizzarelli E, Butterfield DA, and Stella AM. Nitric oxide in the central nervous system: neuroprotection versus neurotoxicity. Nat Rev Neurosci 8: 766–775, 2007 [DOI] [PubMed] [Google Scholar]
- 21.Canet-Aviles RM, Wilson MA, Miller DW, Ahmad R, McLendon C, Bandyopadhyay S, Baptista MJ, Ringe D, Petsko GA, and Cookson MR. The Parkinson's disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. Proc Natl Acad Sci U S A 101: 9103–9108, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chae HZ, Chung SJ, and Rhee SG. Thioredoxin-dependent peroxide reductase from yeast. J Biol Chem 269: 27670–27678, 1994 [PubMed] [Google Scholar]
- 23.Chang N, El-Hayek YH, Gomez E, and Wan Q. Phosphatase PTEN in neuronal injury and brain disorders. Trends Neurosci 30: 581–586, 2007 [DOI] [PubMed] [Google Scholar]
- 24.Chang TS, Jeong W, Woo HA, Lee SM, Park S, and Rhee SG. Characterization of mammalian sulfiredoxin and its reactivation of hyperoxidized peroxiredoxin through reduction of cysteine sulfinic acid in the active site to cysteine. J Biol Chem 279: 50994–51001, 2004 [DOI] [PubMed] [Google Scholar]
- 25.Chen L, Wu R, Feng J, Feng T, Wang C, Hu J, Zhan N, Li Y, Ma X, Ren B, Zhang J, Song CP, Li J, Zhou JM, and Zuo J. Transnitrosylation mediated by the non-canonical catalase ROG1 regulates nitric oxide signaling in plants. Dev Cell 53: 444–457 e5, 2020 [DOI] [PubMed] [Google Scholar]
- 26.Cho B, Cho HM, Kim HJ, Jeong J, Park SK, Hwang EM, Park JY, Kim WR, Kim H, and Sun W. CDK5-dependent inhibitory phosphorylation of Drp1 during neuronal maturation. Exp Mol Med 46: e105, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cho DH, Nakamura T, Fang J, Cieplak P, Godzik A, Gu Z, and Lipton SA. S-nitrosylation of Drp1 mediates β-amyloid-related mitochondrial fission and neuronal injury. Science 324: 102–105, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Choi J, Levey AI, Weintraub ST, Rees HD, Gearing M, Chin LS, and Li L. Oxidative modifications and down-regulation of ubiquitin carboxyl-terminal hydrolase L1 associated with idiopathic Parkinson's and Alzheimer's diseases. J Biol Chem 279: 13256–13264, 2004 [DOI] [PubMed] [Google Scholar]
- 29.Choi J, Sullards MC, Olzmann JA, Rees HD, Weintraub ST, Bostwick DE, Gearing M, Levey AI, Chin LS, and Li L. Oxidative damage of DJ-1 is linked to sporadic Parkinson and Alzheimer diseases. J Biol Chem 281: 10816–10824, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Choi MS, Nakamura T, Cho SJ, Han X, Holland EA, Qu J, Petsko GA, Yates JR, 3rd, Liddington RC, and Lipton SA. Transnitrosylation from DJ-1 to PTEN attenuates neuronal cell death in Parkinson's disease models. J Neurosci 34: 15123–15131, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Choi YB, Tenneti L, Le DA, Ortiz J, Bai G, Chen HS, and Lipton SA. Molecular basis of NMDA receptor-coupled ion channel modulation by S-nitrosylation. Nat Neurosci 3: 15–21, 2000 [DOI] [PubMed] [Google Scholar]
- 32.Chung KK, Thomas B, Li X, Pletnikova O, Troncoso JC, Marsh L, Dawson VL, and Dawson TM. S-nitrosylation of parkin regulates ubiquitination and compromises parkin's protective function. Science 304: 1328–1331, 2004 [DOI] [PubMed] [Google Scholar]
- 33.Cristovao JS and Gomes CM. S100 proteins in Alzheimer's disease. Front Neurosci 13: 463, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.D'Amelio M, Cavallucci V, and Cecconi F. Neuronal caspase-3 signaling: not only cell death. Cell Death Differ 17: 1104–1114, 2010 [DOI] [PubMed] [Google Scholar]
- 35.Das C, Hoang QQ, Kreinbring CA, Luchansky SJ, Meray RK, Ray SS, Lansbury PT, Ringe D, and Petsko GA. Structural basis for conformational plasticity of the Parkinson's disease-associated ubiquitin hydrolase UCH-L1. Proc Natl Acad Sci U S A 103: 4675–4680, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Delettre C, Lenaers G, Griffoin JM, Gigarel N, Lorenzo C, Belenguer P, Pelloquin L, Grosgeorge J, Turc-Carel C, Perret E, Astarie-Dequeker C, Lasquellec L, Arnaud B, Ducommun B, Kaplan J, and Hamel CP. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet 26: 207–210, 2000 [DOI] [PubMed] [Google Scholar]
- 37.Denault JB, Eckelman BP, Shin H, Pop C, and Salvesen GS. Caspase 3 attenuates XIAP (X-linked inhibitor of apoptosis protein)-mediated inhibition of caspase 9. Biochem J 405: 11–19, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dhavan R and Tsai LH. A decade of CDK5. Nat Rev Mol Cell Biol 2: 749–759, 2001 [DOI] [PubMed] [Google Scholar]
- 39.Doulias PT, Greene JL, Greco TM, Tenopoulou M, Seeholzer SH, Dunbrack RL, and Ischiropoulos H. Structural profiling of endogenous S-nitrosocysteine residues reveals unique features that accommodate diverse mechanisms for protein S-nitrosylation. Proc Natl Acad Sci U S A 107: 16958–16963, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Doulias PT, Raju K, Greene JL, Tenopoulou M, and Ischiropoulos H. Mass spectrometry-based identification of S-nitrosocysteine in vivo using organic mercury assisted enrichment. Methods 62: 165–170, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Drinjakovic J, Jung H, Campbell DS, Strochlic L, Dwivedy A, and Holt CE. E3 ligase Nedd4 promotes axon branching by downregulating PTEN. Neuron 65: 341–357, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Forman-Kay JD, Clore GM, Wingfield PT, and Gronenborn AM. High-resolution three-dimensional structure of reduced recombinant human thioredoxin in solution. Biochemistry 30: 2685–2698, 1991 [DOI] [PubMed] [Google Scholar]
- 43.Forrester MT, Thompson JW, Foster MW, Nogueira L, Moseley MA, and Stamler JS. Proteomic analysis of S-nitrosylation and denitrosylation by resin-assisted capture. Nat Biotechnol 27: 557–559, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Foster MW, Forrester MT, and Stamler JS. A protein microarray-based analysis of S-nitrosylation. Proc Natl Acad Sci U S A 106: 18948–18953, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fu WY, Chen Y, Sahin M, Zhao XS, Shi L, Bikoff JB, Lai KO, Yung WH, Fu AK, Greenberg ME, and Ip NY. Cdk5 regulates EphA4-mediated dendritic spine retraction through an ephexin1-dependent mechanism. Nat Neurosci 10: 67–76, 2007 [DOI] [PubMed] [Google Scholar]
- 46.Fuentes-Prior P and Salvesen GS. The protein structures that shape caspase activity, specificity, activation and inhibition. Biochem J 384: 201–232, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gaston BM, Carver J, Doctor A, and Palmer LA. S-nitrosylation signaling in cell biology. Mol Interv 3: 253–263, 2003 [DOI] [PubMed] [Google Scholar]
- 48.Groszer M, Erickson R, Scripture-Adams DD, Lesche R, Trumpp A, Zack JA, Kornblum HI, Liu X, and Wu H. Negative regulation of neural stem/progenitor cell proliferation by the Pten tumor suppressor gene in vivo. Science 294: 2186–2189, 2001 [DOI] [PubMed] [Google Scholar]
- 49.Gu Z, Kaul M, Yan B, Kridel SJ, Cui J, Strongin A, Smith JW, Liddington RC, and Lipton SA. S-nitrosylation of matrix metalloproteinases: signaling pathway to neuronal cell death. Science 297: 1186–1190, 2002 [DOI] [PubMed] [Google Scholar]
- 50.Guo MY, Shang L, Hu YY, Jiang LP, Wan YY, Zhou QQ, Zhang K, Liao HF, Yi JL, and Han XJ. The role of Cdk5-mediated Drp1 phosphorylation in Aβ1–42 induced mitochondrial fission and neuronal apoptosis. J Cell Biochem 119: 4815–4825, 2018 [DOI] [PubMed] [Google Scholar]
- 51.Gusarov I, Gautier L, Smolentseva O, Shamovsky I, Eremina S, Mironov A, and Nudler E. Bacterial nitric oxide extends the lifespan of C. elegans. Cell 152: 818–830, 2013 [DOI] [PubMed] [Google Scholar]
- 52.Haendeler J, Hoffmann J, Tischler V, Berk BC, Zeiher AM, and Dimmeler S. Redox regulatory and anti-apoptotic functions of thioredoxin depend on S-nitrosylation at cysteine 69. Nat Cell Biol 4: 743–749, 2002 [DOI] [PubMed] [Google Scholar]
- 53.Hao G, Derakhshan B, Shi L, Campagne F, and Gross SS. SNOSID, a proteomic method for identification of cysteine S-nitrosylation sites in complex protein mixtures. Proc Natl Acad Sci U S A 103: 1012–1017, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hara MR, Agrawal N, Kim SF, Cascio MB, Fujimuro M, Ozeki Y, Takahashi M, Cheah JH, Tankou SK, Hester LD, Ferris CD, Hayward SD, Snyder SH, and Sawa A. S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat Cell Biol 7: 665–674, 2005 [DOI] [PubMed] [Google Scholar]
- 55.Harrison CA, Raftery MJ, Walsh J, Alewood P, Iismaa SE, Thliveris S, and Geczy CL. Oxidation regulates the inflammatory properties of the murine S100 protein S100A8. J Biol Chem 274: 8561–8569, 1999 [DOI] [PubMed] [Google Scholar]
- 56.Hashemy SI and Holmgren A. Regulation of the catalytic activity and structure of human thioredoxin 1 via oxidation and S-nitrosylation of cysteine residues. J Biol Chem 283: 21890–21898, 2008 [DOI] [PubMed] [Google Scholar]
- 57.Hashimoto M, Rockenstein E, Crews L, and Masliah E. Role of protein aggregation in mitochondrial dysfunction and neurodegeneration in Alzheimer's and Parkinson's diseases. Neuromolecular Med 4: 21–36, 2003 [DOI] [PubMed] [Google Scholar]
- 58.Heinrich TA, da Silva RS, Miranda KM, Switzer CH, Wink DA, and Fukuto JM. Biological nitric oxide signalling: chemistry and terminology. Br J Pharmacol 169: 1417–1429, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hess DT, Matsumoto A, Kim SO, Marshall HE, and Stamler JS. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol 6: 150–166, 2005 [DOI] [PubMed] [Google Scholar]
- 60.Hobbs AJ, and Ignarro LJ. Nitric oxide-cyclic GMP signal transduction system. Methods Enzymol 269: 134–148, 1996 [DOI] [PubMed] [Google Scholar]
- 61.Ischiropoulos H, Zhu L, and Beckman JS. Peroxynitrite formation from macrophage-derived nitric oxide. Arch Biochem Biophys 298: 446–451, 1992 [DOI] [PubMed] [Google Scholar]
- 62.Ito G, Ariga H, Nakagawa Y, and Iwatsubo T. Roles of distinct cysteine residues in S-nitrosylation and dimerization of DJ-1. Biochem Biophys Res Commun 339: 667–672, 2006 [DOI] [PubMed] [Google Scholar]
- 63.Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, and Snyder SH. Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat Cell Biol 3: 193–197, 2001 [DOI] [PubMed] [Google Scholar]
- 64.Jaffrey SR and Snyder SH. The biotin switch method for the detection of S-nitrosylated proteins. Sci STKE 2001: pl1, 2001 [DOI] [PubMed] [Google Scholar]
- 65.Jahani-Asl A, Huang E, Irrcher I, Rashidian J, Ishihara N, Lagace DC, Slack RS, and Park DS. CDK5 phosphorylates DRP1 and drives mitochondrial defects in NMDA-induced neuronal death. Hum Mol Genet 24: 4573–4583, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jensen DE, Belka GK, and Du Bois GC. S-nitrosoglutathione is a substrate for rat alcohol dehydrogenase class III isoenzyme. Biochem J 331: 659–668, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jeon GS, Nakamura T, Lee JS, Choi WJ, Ahn SW, Lee KW, Sung JJ, and Lipton SA. Potential effect of S-nitrosylated protein disulfide isomerase on mutant SOD1 aggregation and neuronal cell death in amyotrophic lateral sclerosis. Mol Neurobiol 49: 796–807, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jia J, Arif A, Terenzi F, Willard B, Plow EF, Hazen SL, and Fox PL. Target-selective protein S-nitrosylation by sequence motif recognition. Cell 159: 623–634, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jia J, Arif A, Willard B, Smith JD, Stuehr DJ, Hazen SL, and Fox PL. Protection of extraribosomal RPL13a by GAPDH and dysregulation by S-nitrosylation. Mol Cell 47: 656–663, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jia L, Bonaventura C, Bonaventura J, and Stamler JS. S-nitrosohaemoglobin: a dynamic activity of blood involved in vascular control. Nature 380: 221–226, 1996 [DOI] [PubMed] [Google Scholar]
- 71.Kabuta T, Mitsui T, Takahashi M, Fujiwara Y, Kabuta C, Konya C, Tsuchiya Y, Hatanaka Y, Uchida K, Hohjoh H, and Wada K. Ubiquitin C-terminal hydrolase L1 (UCH-L1) acts as a novel potentiator of cyclin-dependent kinases to enhance cell proliferation independently of its hydrolase activity. J Biol Chem 288: 12615–12626, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kallakunta VM, Slama-Schwok A, and Mutus B. Protein disulfide isomerase may facilitate the efflux of nitrite derived S-nitrosothiols from red blood cells. Redox Biol 1: 373–380, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kim RH, Peters M, Jang Y, Shi W, Pintilie M, Fletcher GC, DeLuca C, Liepa J, Zhou L, Snow B, Binari RC, Manoukian AS, Bray MR, Liu FF, Tsao MS, and Mak TW. DJ-1, a novel regulator of the tumor suppressor PTEN. Cancer Cell 7: 263–273, 2005 [DOI] [PubMed] [Google Scholar]
- 74.Kim Y, Sung JY, Ceglia I, Lee KW, Ahn JH, Halford JM, Kim AM, Kwak SP, Park JB, Ho Ryu S, Schenck A, Bardoni B, Scott JD, Nairn AC, and Greengard P. Phosphorylation of WAVE1 regulates actin polymerization and dendritic spine morphology. Nature 442: 814–817, 2006 [DOI] [PubMed] [Google Scholar]
- 75.Kohr MJ, Murphy E, and Steenbergen C. Glyceraldehyde-3-phosphate dehydrogenase acts as a mitochondrial trans-S-nitrosylase in the heart. PLoS One 9: e111448, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kornberg MD, Sen N, Hara MR, Juluri KR, Nguyen JV, Snowman AM, Law L, Hester LD, and Snyder SH. GAPDH mediates nitrosylation of nuclear proteins. Nat Cell Biol 12: 1094–1100, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kumar R, Jangir DK, Verma G, Shekhar S, Hanpude P, Kumar S, Kumari R, Singh N, Sarovar Bhavesh N, Ranjan Jana N, and Kanti Maiti T. S-nitrosylation of UCHL1 induces its structural instability and promotes α-synuclein aggregation. Sci Rep 7: 44558, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kummer MP, Vogl T, Axt D, Griep A, Vieira-Saecker A, Jessen F, Gelpi E, Roth J, and Heneka MT. Mrp14 deficiency ameliorates amyloid β burden by increasing microglial phagocytosis and modulation of amyloid precursor protein processing. J Neurosci 32: 17824–17829, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kwak YD, Ma T, Diao S, Zhang X, Chen Y, Hsu J, Lipton SA, Masliah E, Xu H, and Liao FF. NO signaling and S-nitrosylation regulate PTEN inhibition in neurodegeneration. Mol Neurodegener 5: 49, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kwon J, Lee SR, Yang KS, Ahn Y, Kim YJ, Stadtman ER, and Rhee SG. Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proc Natl Acad Sci U S A 101: 16419–16424, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lachyankar MB, Sultana N, Schonhoff CM, Mitra P, Poluha W, Lambert S, Quesenberry PJ, Litofsky NS, Recht LD, Nabi R, Miller SJ, Ohta S, Neel BG, and Ross AH. A role for nuclear PTEN in neuronal differentiation. J Neurosci 20: 1404–1413, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lancaster JR Jr.How are nitrosothiols formed de novo in vivo? Arch Biochem Biophys 617: 137–144, 2017 [DOI] [PubMed] [Google Scholar]
- 83.Laurent TC, Moore EC, and Reichard P. Enzymatic synthesis of deoxyribonucleotides. IV. Isolation and characterization of thioredoxin, the hydrogen donor from Escherichia coli B. J Biol Chem 239: 3436–3444, 1964 [PubMed] [Google Scholar]
- 84.Lawson VH, Graham BV, and Flanigan KM. Clinical and electrophysiologic features of CMT2A with mutations in the mitofusin 2 gene. Neurology 65: 197–204, 2005 [DOI] [PubMed] [Google Scholar]
- 85.Lee SB, Kim CK, Lee KH, and Ahn JY. S-nitrosylation of B23/nucleophosmin by GAPDH protects cells from the SIAH1-GAPDH death cascade. J Cell Biol 199: 65–76, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lei SZ, Pan ZH, Aggarwal SK, Chen HS, Hartman J, Sucher NJ, and Lipton SA. Effect of nitric oxide production on the redox modulatory site of the NMDA receptor-channel complex. Neuron 8: 1087–1099, 1992 [DOI] [PubMed] [Google Scholar]
- 87.Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, Bigner SH, Giovanella BC, Ittmann M, Tycko B, Hibshoosh H, Wigler MH, and Parsons R. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 275: 1943–1947, 1997 [DOI] [PubMed] [Google Scholar]
- 88.Lillig CH and Holmgren A. Thioredoxin and related molecules—from biology to health and disease. Antioxid Redox Signal 9: 25–47, 2007 [DOI] [PubMed] [Google Scholar]
- 89.Lim SY, Raftery M, Cai H, Hsu K, Yan WX, Hseih HL, Watts RN, Richardson D, Thomas S, Perry M, and Geczy CL. S-nitrosylated S100A8: novel anti-inflammatory properties. J Immunol 181: 5627–5636, 2008 [DOI] [PubMed] [Google Scholar]
- 90.Lipton SA, Choi YB, Pan ZH, Lei SZ, Chen HS, Sucher NJ, Loscalzo J, Singel DJ, and Stamler JS. A redox-based mechanism for the neuroprotective and neurodestructive effects of nitric oxide and related nitroso-compounds. Nature 364: 626–632, 1993 [DOI] [PubMed] [Google Scholar]
- 91.Lipton SA, Choi YB, Takahashi H, Zhang D, Li W, Godzik A, and Bankston LA. Cysteine regulation of protein function—as exemplified by NMDA-receptor modulation. Trends Neurosci 25: 474–480, 2002 [DOI] [PubMed] [Google Scholar]
- 92.Lipton SA, Nakamura T, Yao D, Shi ZQ, Uehara T, and Gu Z. Comment on “S-nitrosylation of parkin regulates ubiquitination and compromises parkin's protective function”. Science 308: 1870, 2005; author reply 1870 [DOI] [PubMed] [Google Scholar]
- 93.Luchsinger BP, Rich EN, Gow AJ, Williams EM, Stamler JS, and Singel DJ. Routes to S-nitroso-hemoglobin formation with heme redox and preferential reactivity in the β subunits. Proc Natl Acad Sci U S A 100: 461–466, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Luthman M and Holmgren A. Rat liver thioredoxin and thioredoxin reductase: purification and characterization. Biochemistry 21: 6628–6633, 1982 [DOI] [PubMed] [Google Scholar]
- 95.Mannick JB, Hausladen A, Liu L, Hess DT, Zeng M, Miao QX, Kane LS, Gow AJ, and Stamler JS. Fas-induced caspase denitrosylation. Science 284: 651–654, 1999 [DOI] [PubMed] [Google Scholar]
- 96.Marino SM and Gladyshev VN. Structural analysis of cysteine S-nitrosylation: a modified acid-based motif and the emerging role of trans-nitrosylation. J Mol Biol 395: 844–859, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Marletta MA, Yoon PS, Iyengar R, Leaf CD, and Wishnok JS. Macrophage oxidation of L-arginine to nitrite and nitrate: nitric oxide is an intermediate. Biochemistry 27: 8706–8711, 1988 [DOI] [PubMed] [Google Scholar]
- 98.Marozkina N and Gaston B. An update on thiol signaling: S-nitrosothiols, hydrogen sulfide and a putative role for thionitrous acid. Antioxidants (Basel) 9, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Matsui M, Oshima M, Oshima H, Takaku K, Maruyama T, Yodoi J, and Taketo MM. Early embryonic lethality caused by targeted disruption of the mouse thioredoxin gene. Dev Biol 178: 179–185, 1996 [DOI] [PubMed] [Google Scholar]
- 100.McDonald LJ, and Murad F. Nitric oxide and cGMP signaling. Adv Pharmacol 34: 263–275, 1995 [DOI] [PubMed] [Google Scholar]
- 101.Melino G, Bernassola F, Knight RA, Corasaniti MT, Nistico G, and Finazzi-Agro A. S-nitrosylation regulates apoptosis. Nature 388: 432–433, 1997 [DOI] [PubMed] [Google Scholar]
- 102.Mitchell DA and Marletta MA. Thioredoxin catalyzes the S-nitrosation of the caspase-3 active site cysteine. Nat Chem Biol 1: 154–158, 2005 [DOI] [PubMed] [Google Scholar]
- 103.Mitchell DA, Morton SU, Fernhoff NB, and Marletta MA. Thioredoxin is required for S-nitrosation of procaspase-3 and the inhibition of apoptosis in Jurkat cells. Proc Natl Acad Sci U S A 104: 11609–11614, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mnatsakanyan R, Markoutsa S, Walbrunn K, Roos A, Verhelst SHL, and Zahedi RP. Proteome-wide detection of S-nitrosylation targets and motifs using bioorthogonal cleavable-linker-based enrichment and switch technique. Nat Commun 10: 2195, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Nakamura T and Lipton SA. Emerging role of protein-protein transnitrosylation in cell signaling pathways. Antioxid Redox Signal 18: 239–249, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nakamura T, Oh CK, Liao L, Zhang X, Lopez KM, Gibbs D, Deal AK, Scott HR, Spencer B, Masliah E, Rissman RA, Yates JR, 3rd, and Lipton SA. Non-canonical transnitrosylation network contributes to synapse loss in Alzheimer's disease. Science 371: eaaw0843, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Nakamura T, Tu S, Akhtar MW, Sunico CR, Okamoto S, and Lipton SA. Aberrant protein s-nitrosylation in neurodegenerative diseases. Neuron 78: 596–614, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nakamura T, Wang L, Wong CC, Scott FL, Eckelman BP, Han X, Tzitzilonis C, Meng F, Gu Z, Holland EA, Clemente AT, Okamoto S, Salvesen GS, Riek R, Yates JR, 3rd, and Lipton SA. Transnitrosylation of XIAP regulates caspase-dependent neuronal cell death. Mol Cell 39: 184–195, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Nikitovic D and Holmgren A. S-nitrosoglutathione is cleaved by the thioredoxin system with liberation of glutathione and redox regulating nitric oxide. J Biol Chem 271: 19180–19185, 1996 [DOI] [PubMed] [Google Scholar]
- 110.Numajiri N, Takasawa K, Nishiya T, Tanaka H, Ohno K, Hayakawa W, Asada M, Matsuda H, Azumi K, Kamata H, Nakamura T, Hara H, Minami M, Lipton SA, and Uehara T. On-off system for PI3-kinase-Akt signaling through S-nitrosylation of phosphatase with sequence homology to tensin (PTEN). Proc Natl Acad Sci U S A 108: 10349–10354, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ohata H, Shiokawa D, Obata Y, Sato A, Sakai H, Fukami M, Hara W, Taniguchi H, Ono M, Nakagama H, and Okamoto K. NOX1-dependent mTORC1 activation via S100A9 oxidation in cancer stem-like cells leads to colon cancer progression. Cell Rep 28: 1282–1295.e8, 2019 [DOI] [PubMed] [Google Scholar]
- 112.Ozawa K, Komatsubara AT, Nishimura Y, Sawada T, Kawafune H, Tsumoto H, Tsuji Y, Zhao J, Kyotani Y, Tanaka T, Takahashi R, and Yoshizumi M. S-nitrosylation regulates mitochondrial quality control via activation of parkin. Sci Rep 3: 2202, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ozawa K, Tsumoto H, Miura Y, Yamaguchi J, Iguchi-Ariga SMM, Sakuma T, Yamamoto T, and Uchiyama Y. DJ-1 is indispensable for the S-nitrosylation of parkin, which maintains function of mitochondria. Sci Rep 10: 4377, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Pader I, Sengupta R, Cebula M, Xu J, Lundberg JO, Holmgren A, Johansson K, and Arner ES. Thioredoxin-related protein of 14 kDa is an efficient L-cystine reductase and S-denitrosylase. Proc Natl Acad Sci U S A 111: 6964–6969, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Paoletti P, Vila I, Rife M, Lizcano JM, Alberch J, and Gines S. Dopaminergic and glutamatergic signaling crosstalk in Huntington's disease neurodegeneration: the role of p25/cyclin-dependent kinase 5. J Neurosci 28: 10090–10101, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Park KK, Liu K, Hu Y, Smith PD, Wang C, Cai B, Xu B, Connolly L, Kramvis I, Sahin M, and He Z. Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science 322: 963–966, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, and Tsai LH. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 402: 615–622, 1999 [DOI] [PubMed] [Google Scholar]
- 118.Pawloski JR, Hess DT, and Stamler JS. Export by red blood cells of nitric oxide bioactivity. Nature 409: 622–626, 2001 [DOI] [PubMed] [Google Scholar]
- 119.Pei DS, Sun YF, and Song YJ. S-nitrosylation of PTEN Invovled in ischemic brain injury in rat hippocampal CA1 region. Neurochem Res 34: 1507–1512, 2009 [DOI] [PubMed] [Google Scholar]
- 120.Premkumar L, Dobaczewska MK, and Riedl SJ. Identification of an artificial peptide motif that binds and stabilizes reduced human DJ-1. J Struct Biol 176: 414–418, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Qu D, Rashidian J, Mount MP, Aleyasin H, Parsanejad M, Lira A, Haque E, Zhang Y, Callaghan S, Daigle M, Rousseaux MW, Slack RS, Albert PR, Vincent I, Woulfe JM, and Park DS. Role of Cdk5-mediated phosphorylation of Prx2 in MPTP toxicity and Parkinson's disease. Neuron 55: 37–52, 2007 [DOI] [PubMed] [Google Scholar]
- 122.Qu J, Nakamura T, Cao G, Holland EA, McKercher SR, and Lipton SA. S-nitrosylation activates Cdk5 and contributes to synaptic spine loss induced by β-amyloid peptide. Proc Natl Acad Sci U S A 108: 14330–14335, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ramachandran N, Root P, Jiang XM, Hogg PJ, and Mutus B. Mechanism of transfer of NO from extracellular S-nitrosothiols into the cytosol by cell-surface protein disulfide isomerase. Proc Natl Acad Sci U S A 98: 9539–9544, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ravi K, Brennan LA, Levic S, Ross PA, and Black SM. S-nitrosylation of endothelial nitric oxide synthase is associated with monomerization and decreased enzyme activity. Proc Natl Acad Sci U S A 101: 2619–2624, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Reddy PH and Beal MF. Amyloid β, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer's disease. Trends Mol Med 14: 45–53, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ren X, Björnstedt M, Shen B, Ericson ML, and Holmgren A. Mutagenesis of structural half-cystine residues in human thioredoxin and effects on the regulation of activity by selenodiglutathione. Biochemistry 32: 9701–9708, 1993 [DOI] [PubMed] [Google Scholar]
- 127.Ren X, Sengupta R, Lu J, Lundberg JO, and Holmgren A. Characterization of mammalian glutaredoxin isoforms as S-denitrosylases. FEBS Lett 593: 1799–1806, 2019 [DOI] [PubMed] [Google Scholar]
- 128.Rhee SG.Cell signaling. H2O2, a necessary evil for cell signaling. Science 312: 1882–1883, 2006 [DOI] [PubMed] [Google Scholar]
- 129.Romagny S, Bouaouiche S, Lucchi G, Ducoroy P, Bertoldo JB, Terenzi H, Bettaieb A, and Plenchette S. S-nitrosylation of cIAP1 switches cancer cell fate from TNFα/TNFR1-mediated cell survival to cell death. Cancer Res 78: 1948–1957, 2018 [DOI] [PubMed] [Google Scholar]
- 130.Root P, Sliskovic I, and Mutus B. Platelet cell-surface protein disulphide-isomerase mediated S-nitrosoglutathione consumption. Biochem J 382: 575–580, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Rossig L, Fichtlscherer B, Breitschopf K, Haendeler J, Zeiher AM, Mulsch A, and Dimmeler S. Nitric oxide inhibits caspase-3 by S-nitrosation in vivo. J Biol Chem 274: 6823–6826, 1999 [DOI] [PubMed] [Google Scholar]
- 132.Sakuma I, Stuehr DJ, Gross SS, Nathan C, and Levi R. Identification of arginine as a precursor of endothelium-derived relaxing factor. Proc Natl Acad Sci U S A 85: 8664–8667, 1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Sawa A, Khan AA, Hester LD, and Snyder SH. Glyceraldehyde-3-phosphate dehydrogenase: nuclear translocation participates in neuronal and nonneuronal cell death. Proc Natl Acad Sci U S A 94: 11669–11674, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sbodio JI, Snyder SH, and Paul BD. Redox mechanisms in neurodegeneration: from disease outcomes to therapeutic opportunities. Antioxid Redox Signal 30: 1450–1499, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Sen N, Hara MR, Kornberg MD, Cascio MB, Bae BI, Shahani N, Thomas B, Dawson TM, Dawson VL, Snyder SH, and Sawa A. Nitric oxide-induced nuclear GAPDH activates p300/CBP and mediates apoptosis. Nat Cell Biol 10: 866–873, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Sen T, Saha P, and Sen N. Nitrosylation of GAPDH augments pathological tau acetylation upon exposure to amyloid-β. Sci Signal 11: eaao6765, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Seneviratne U, Godoy LC, Wishnok JS, Wogan GN, and Tannenbaum SR. Mechanism-based triarylphosphine-ester probes for capture of endogenous RSNOs. J Am Chem Soc 135: 7693–7704, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Seneviratne U, Nott A, Bhat VB, Ravindra KC, Wishnok JS, Tsai LH, and Tannenbaum SR. S-nitrosation of proteins relevant to Alzheimer's disease during early stages of neurodegeneration. Proc Natl Acad Sci U S A 113: 4152–4157, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Seth D, Hausladen A, Wang YJ, and Stamler JS. Endogenous protein S-nitrosylation in E. coli: regulation by OxyR. Science 336: 470–473, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Seth D, Hess DT, Hausladen A, Wang L, Wang YJ, and Stamler JS. A multiplex enzymatic machinery for cellular protein S-nitrosylation. Mol Cell 69: 451–464 e6, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Seth D and Stamler JS. The SNO-proteome: causation and classifications. Curr Opin Chem Biol 15: 129–136, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Setsuie R and Wada K. The functions of UCH-L1 and its relation to neurodegenerative diseases. Neurochem Int 51: 105–111, 2007 [DOI] [PubMed] [Google Scholar]
- 143.Shendelman S, Jonason A, Martinat C, Leete T, and Abeliovich A. DJ-1 is a redox-dependent molecular chaperone that inhibits α-synuclein aggregate formation. PLoS Biol 2: e362, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Shepherd CE, Goyette J, Utter V, Rahimi F, Yang Z, Geczy CL, and Halliday GM. Inflammatory S100A9 and S100A12 proteins in Alzheimer's disease. Neurobiol Aging 27: 1554–1563, 2006 [DOI] [PubMed] [Google Scholar]
- 145.Shiozaki EN, Chai J, Rigotti DJ, Riedl SJ, Li P, Srinivasula SM, Alnemri ES, Fairman R, and Shi Y. Mechanism of XIAP-mediated inhibition of caspase-9. Mol Cell 11: 519–527, 2003 [DOI] [PubMed] [Google Scholar]
- 146.Singh SP, Wishnok JS, Keshive M, Deen WM, and Tannenbaum SR. The chemistry of the S-nitrosoglutathione/glutathione system. Proc Natl Acad Sci U S A 93: 14428–14433, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Sliskovic I, Raturi A, and Mutus B. Characterization of the S-denitrosation activity of protein disulfide isomerase. J Biol Chem 280: 8733–8741, 2005 [DOI] [PubMed] [Google Scholar]
- 148.Smith BC and Marletta MA. Mechanisms of S-nitrosothiol formation and selectivity in nitric oxide signaling. Curr Opin Chem Biol 16: 498–506, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Stamler JS, Jia L, Eu JP, McMahon TJ, Demchenko IT, Bonaventura J, Gernert K, and Piantadosi CA. Blood flow regulation by S-nitrosohemoglobin in the physiological oxygen gradient. Science 276: 2034–2037, 1997 [DOI] [PubMed] [Google Scholar]
- 150.Stamler JS, Lamas S, and Fang FC. Nitrosylation. the prototypic redox-based signaling mechanism. Cell 106: 675–683, 2001 [DOI] [PubMed] [Google Scholar]
- 151.Stamler JS, Toone EJ, Lipton SA, and Sucher NJ. (S)NO signals: translocation, regulation, and a consensus motif. Neuron 18: 691–696, 1997 [DOI] [PubMed] [Google Scholar]
- 152.Stomberski CT, Hess DT, and Stamler JS. Protein S-nitrosylation: determinants of specificity and enzymatic regulation of S-nitrosothiol-based signaling. Antioxid Redox Signal 30: 1331–1351, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Stoyanovsky DA, Tyurina YY, Tyurin VA, Anand D, Mandavia DN, Gius D, Ivanova J, Pitt B, Billiar TR, and Kagan VE. Thioredoxin and lipoic acid catalyze the denitrosation of low molecular weight and protein S-nitrosothiols. J Am Chem Soc 127: 15815–15823, 2005 [DOI] [PubMed] [Google Scholar]
- 154.Sun KH, de Pablo Y, Vincent F, and Shah K. Deregulated Cdk5 promotes oxidative stress and mitochondrial dysfunction. J Neurochem 107: 265–278, 2008 [DOI] [PubMed] [Google Scholar]
- 155.Sunico CR, Nakamura T, Rockenstein E, Mante M, Adame A, Chan SF, Newmeyer TF, Masliah E, Nakanishi N, and Lipton SA. S-nitrosylation of parkin as a novel regulator of p53-mediated neuronal cell death in sporadic Parkinson's disease. Mol Neurodegener 8: 29, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Taira T, Saito Y, Niki T, Iguchi-Ariga SM, Takahashi K, and Ariga H. DJ-1 has a role in antioxidative stress to prevent cell death. EMBO Rep 5: 213–218, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Takahashi H, Shin Y, Cho SJ, Zago WM, Nakamura T, Gu Z, Ma Y, Furukawa H, Liddington R, Zhang D, Tong G, Chen HS, and Lipton SA. Hypoxia enhances S-nitrosylation-mediated NMDA receptor inhibition via a thiol oxygen sensor motif. Neuron 53: 53–64, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Tannenbaum SR and White FM. Regulation and specificity of S-nitrosylation and denitrosylation. ACS Chem Biol 1: 615–618, 2006 [DOI] [PubMed] [Google Scholar]
- 159.Tenneti L, D'Emilia DM, and Lipton SA. Suppression of neuronal apoptosis by S-nitrosylation of caspases. Neurosci Lett 236: 139–142, 1997 [DOI] [PubMed] [Google Scholar]
- 160.Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, and Katzman R. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 30: 572–580, 1991 [DOI] [PubMed] [Google Scholar]
- 161.Thompson JW, Forrester MT, Moseley MA, and Foster MW. Solid-phase capture for the detection and relative quantification of S-nitrosoproteins by mass spectrometry. Methods 62: 130–137, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Tolkacheva T, Boddapati M, Sanfiz A, Tsuchida K, Kimmelman AC, and Chan AM. Regulation of PTEN binding to MAGI-2 by two putative phosphorylation sites at threonine 382 and 383. Cancer Res 61: 4985–4989, 2001 [PubMed] [Google Scholar]
- 163.Tristan C, Shahani N, Sedlak TW, and Sawa A. The diverse functions of GAPDH: views from different subcellular compartments. Cell Signal 23: 317–323, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Tsang AH, Lee YI, Ko HS, Savitt JM, Pletnikova O, Troncoso JC, Dawson VL, Dawson TM, and Chung KK. S-nitrosylation of XIAP compromises neuronal survival in Parkinson's disease. Proc Natl Acad Sci U S A 106: 4900–4905, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Uehara T, Nakamura T, Yao D, Shi ZQ, Gu Z, Ma Y, Masliah E, Nomura Y, and Lipton SA. S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature 441: 513–517, 2006 [DOI] [PubMed] [Google Scholar]
- 166.van den Berg WA, Hagen WR, and van Dongen WM. The hybrid-cluster protein (‘prismane protein’) from Escherichia coli. Characterization of the hybrid-cluster protein, redox properties of the [2Fe-2S] and [4Fe-2S-2O] clusters and identification of an associated NADH oxidoreductase containing FAD and [2Fe-2S]. Eur J Biochem 267: 666–676, 2000 [DOI] [PubMed] [Google Scholar]
- 167.Vazquez F, Grossman SR, Takahashi Y, Rokas MV, Nakamura N, and Sellers WR. Phosphorylation of the PTEN tail acts as an inhibitory switch by preventing its recruitment into a protein complex. J Biol Chem 276: 48627–48630, 2001 [DOI] [PubMed] [Google Scholar]
- 168.Wang S, Song R, Wang Z, Jing Z, Wang S, and Ma J. S100A8/A9 in inflammation. Front Immunol 9: 1298, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Waterham HR, Koster J, van Roermund CW, Mooyer PA, Wanders RJ, and Leonard JV. A lethal defect of mitochondrial and peroxisomal fission. N Engl J Med 356: 1736–1741, 2007 [DOI] [PubMed] [Google Scholar]
- 170.Watson WH, Pohl J, Montfort WR, Stuchlik O, Reed MS, Powis G, and Jones DP. Redox potential of human thioredoxin 1 and identification of a second dithiol/disulfide motif. J Biol Chem 278: 33408–33415, 2003 [DOI] [PubMed] [Google Scholar]
- 171.Weichsel A, Brailey JL, and Montfort WR. Buried S-nitrosocysteine revealed in crystal structures of human thioredoxin. Biochemistry 46: 1219–1227, 2007 [DOI] [PubMed] [Google Scholar]
- 172.Weichsel A, Gasdaska JR, Powis G, and Montfort WR. Crystal structures of reduced, oxidized, and mutated human thioredoxins: evidence for a regulatory homodimer. Structure 4: 735–751, 1996 [DOI] [PubMed] [Google Scholar]
- 173.Weishaupt JH, Kussmaul L, Grotsch P, Heckel A, Rohde G, Romig H, Bahr M, and Gillardon F. Inhibition of CDK5 is protective in necrotic and apoptotic paradigms of neuronal cell death and prevents mitochondrial dysfunction. Mol Cell Neurosci 24: 489–502, 2003 [DOI] [PubMed] [Google Scholar]
- 174.Westermann B.Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol 11: 872–884, 2010 [DOI] [PubMed] [Google Scholar]
- 175.Wilson MA, Collins JL, Hod Y, Ringe D, and Petsko GA. The 1.1-A resolution crystal structure of DJ-1, the protein mutated in autosomal recessive early onset Parkinson's disease. Proc Natl Acad Sci U S A 100: 9256–9261, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Wolhuter K, Whitwell HJ, Switzer CH, Burgoyne JR, Timms JF, and Eaton P. Evidence against stable protein S-nitrosylation as a widespread mechanism of post-translational regulation. Mol Cell 69: 438–450 e5, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Wu C, Liu T, Chen W, Oka S, Fu C, Jain MR, Parrott AM, Baykal AT, Sadoshima J, and Li H. Redox regulatory mechanism of transnitrosylation by thioredoxin. Mol Cell Proteomics 9: 2262–2275, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Wu C, Parrott AM, Liu T, Beuve A, and Li H. Functional proteomics approaches for the identification of transnitrosylase and denitrosylase targets. Methods 62: 151–160, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Wu KL, Wu CA, Wu CW, Chan SH, Chang AY, and Chan JY. Redox-sensitive oxidation and phosphorylation of PTEN contribute to enhanced activation of PI3K/Akt signaling in rostral ventrolateral medulla and neurogenic hypertension in spontaneously hypertensive rats. Antioxid Redox Signal 18: 36–50, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Xie Z, Sanada K, Samuels BA, Shih H, and Tsai LH. Serine 732 phosphorylation of FAK by Cdk5 is important for microtubule organization, nuclear movement, and neuronal migration. Cell 114: 469–482, 2003 [DOI] [PubMed] [Google Scholar]
- 181.Yang Y, Gehrke S, Haque ME, Imai Y, Kosek J, Yang L, Beal MF, Nishimura I, Wakamatsu K, Ito S, Takahashi R, and Lu B. Inactivation of Drosophila DJ-1 leads to impairments of oxidative stress response and phosphatidylinositol 3-kinase/Akt signaling. Proc Natl Acad Sci U S A 102: 13670–13675, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Yao D, Gu Z, Nakamura T, Shi ZQ, Ma Y, Gaston B, Palmer LA, Rockenstein EM, Zhang Z, Masliah E, Uehara T, and Lipton SA. Nitrosative stress linked to sporadic Parkinson's disease: S-nitrosylation of parkin regulates its E3 ubiquitin ligase activity. Proc Natl Acad Sci U S A 101: 10810–10814, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Zai A, Rudd MA, Scribner AW, and Loscalzo J. Cell-surface protein disulfide isomerase catalyzes transnitrosation and regulates intracellular transfer of nitric oxide. J Clin Invest 103: 393–399, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Zaman K, Knight J, Hussain F, Cao R, Estabrooks SK, Altawallbeh G, Holloway K, Jafri A, Sawczak V, Li Y, Getsy P, Sun F, Raffay T, Cotton C, Brodsky JL, Periasamy A, Lewis SJ, and Gaston B. S-nitrosylation of CHIP enhances F508Del-CFTR maturation. Am J Respir Cell Mol Biol 61: 765–775, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Zhang C, Liu Y, Gilthorpe J, and van der Maarel JR. MRP14 (S100A9) protein interacts with Alzheimer β-amyloid peptide and induces its fibrillization. PLoS One 7: e32953, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Zhang D, Zhao N, Ma B, Wang Y, Zhang G, Yan X, Hu S, and Xu T. Procaspase-9 induces its cleavage by transnitrosylating XIAP via the Thioredoxin system during cerebral ischemia-reperfusion in rats. Sci Rep 6: 24203, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Zhang P, Fu WY, Fu AK, and Ip NY. S-nitrosylation-dependent proteasomal degradation restrains Cdk5 activity to regulate hippocampal synaptic strength. Nat Commun 6: 8665, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Zhang P, Yu PC, Tsang AH, Chen Y, Fu AK, Fu WY, Chung KK, and Ip NY. S-nitrosylation of cyclin-dependent kinase 5 (cdk5) regulates its kinase activity and dendrite growth during neuronal development. J Neurosci 30: 14366–14370, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Zhao Y, He M, Ding J, Xi Q, Loake GJ, and Zheng W. Regulation of anticancer styrylpyrone biosynthesis in the medicinal mushroom Inonotus obliquus requires thioredoxin mediated transnitrosylation of S-nitrosoglutathione reductase. Sci Rep 6: 37601, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Zhou W, Zhu M, Wilson MA, Petsko GA, and Fink AL. The oxidation state of DJ-1 regulates its chaperone activity toward α-synuclein. J Mol Biol 356: 1036–1048, 2006 [DOI] [PubMed] [Google Scholar]
- 191.Zuchner S, Mersiyanova IV, Muglia M, Bissar-Tadmouri N, Rochelle J, Dadali EL, Zappia M, Nelis E, Patitucci A, Senderek J, Parman Y, Evgrafov O, Jonghe PD, Takahashi Y, Tsuji S, Pericak-Vance MA, Quattrone A, Battaloglu E, Polyakov AV, Timmerman V, Schroder JM, and Vance JM. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet 36: 449–451, 2004 [DOI] [PubMed] [Google Scholar]