

Abstract
Oxidative stress-induced neuron apoptosis plays a crucial role in the early brain injury (EBI) after subarachnoid hemorrhage (SAH). Kisspeptin has been reported as antioxidant to reduce oxidative stress-induced neuronal cell death through G protein-coupled receptor 54 (GPR54). The goal of this study was to determine the neuroprotection of the Kisspeptin/GRP54 signaling pathway against EBI after SAH. Two hundred and ninety-two Sprague Dawley male rats were used and SAH was induced by the endovascular perforation. Exogenous Kisspeptin 54 (KP54) was delivered intranasally. Small interfering ribonucleic acid (siRNA) for endogenous KISS1, a selective GPR54 antagonist kisspeptin 234, or β-arrestin 2 siRNA for ARRB2 (a functional adaptor of GPR54) were administered intracerebroventricularly. Post-SAH evaluations included neurobehavioral tests, SAH grade, Western blot, immunofluorescence, Fluoro-Jade C, TUNEL, and Nissl staining. The results showed that endogenous KISS1 knockdown aggravated but exogenous KP54 (1.0 nmol/kg) treatment attenuated neurological deficits, brain oxidative stress, and neuronal apoptosis at 24 h after SAH. The benefits of KP54 persisted to 28 days after SAH, which significantly improved cognitive function in SAH rats. The GPR54 blockade or the ARRB2 knockout offset the neuroprotective effects of KP54 in SAH rats. In conclusion, our results suggested that administration of KP54 attenuated oxidative stress, neuronal apoptosis and neurobehavioral impairments through GPR54/ARRB2/AKT/GSK3β signaling pathway after SAH in rat. Thus, KP54 may provide an effective treatment strategy for SAH patients.
Keywords: Subarachnoid hemorrhage, oxidative stress, neuronal apoptosis, kisspeptin 54, GPR 54, rat
1. Introduction
Subarachnoid hemorrhage (SAH) is a clinical syndrome caused by the rupture of intracranial diseased blood vessels with sudden bleeding into the subarachnoid space [1]. Accounting for about 10% of acute strokes, SAH is a severe cerebrovascular disease [2]. Early brain injury (EBI) within 72 hours is considered to be the main cause of high mortality and delayed neurological damage [3, 4] after SAH. Emerging evidence showed that oxidative stress-induced neuron apoptosis played a crucial role in the pathogenesis of EBI after SAH [5]. Therefore, targeting oxidative stress and subsequent neuronal apoptosis would be an effective way to improve the overall outcomes of SAH.
Kisspeptins (KPs), neuropeptides encoded by KISS1 gene, play multiple functions by binding with orphan G protein-coupled receptor 54 (GPR54) [6]. The KP preproprotein is a 145 amino acid protein that can be enzymatically cleaved into 54 amino acids (KP54), 14 amino acids (KP14), 13 amino acids (KP13) and 10 amino acids (KP10) in length, respectively [7]. Sharing a c-terminal sequence with conserved 10 amino acids region, these fragments have the similar binding affinity and efficacy to GPR54 receptor [8]. KPs and GPR54 were expressed in the central nervous systems [9] and cardio-cerebrovascular systems [6]. Endogenous KPs/GPR54 signaling played a critical role in the regulation of metastasis [10], pubertal development [11] and reproductive function through the hypothalamic-pituitary-gonadal axis [12]. Exogenous KPs treatments have been shown to act as antioxidant and attenuate oxidative damages of liver and brain in rat models [13, 14]. KPs treatment reduced the neuronal death by increasing the activity of superoxide dismutase (SOD) and catalase after oxidative brain injury [14]. KPs bind to GPR54 that subsequently activates the phosphatidylinositol 3-kinase/AKT (PI3K/AKT) signaling pathway in neurons [15]. The activation of PI3K/AKT/glycogen synthase kinase-3β (GSK3β) signaling pathway was reported to decrease oxidative stress [16, 17] and inhibit caspase-3-dependent apoptosis in a rat model of cerebral ischemia-reperfusion injury [18]. A functional adaptor of GPR54, β-arrestin 2 (ARRB2) [19], served as a stimulator of PI3K/AKT signaling and decreased the cell apoptosis and caspase-3 activation by increasing the levels of phosphorylated-AKT (pAKT) and phosphorylated-GSK3β (pGSK3β) in cancer cells [20]. The protein levels of pGSK3β elevated in brain tissues after SAH [21], which reduced oxidative stress by enhancing endogenous antioxidant mechanism [22]. Collectively, the activation GPR54/ARRB2/AKT/GSK3β signaling pathway may serve as a potential anti-oxidative stress and anti-apoptotic strategy in the treatment of SAH.
KP54 and KP10 are new drugs approved by U.S. Food and Drug Administration (FDA) for treating disorders of reproductive system. They are currently investigated in the phase 1 or 2 of clinical trials (KP54, ClinicalTrials.gov Identifier: NCT02081924, NCT01667406; KP10, ClinicalTrials.gov Identifier: NCT00914823). Compared with KP10, KP54 shows its stronger blood-brain barrier penetration and longer-lasting pharmacokinetics in animal studies [23]. Recent preclinical studies showed that KPs treatment were beneficial to Alzheimer’s disease [24] and oxidative stress-induced brain injury [14]. However, the neuroprotective effects of KP54 and the underlying mechanisms of protection have not been investigated in the setting of SAH. In the light of previous studies, we therefore hypothesized that KP54 would attenuate oxidative stress and neuronal apoptosis of EBI through GPR54/ARRB2/AKT/GS3Kβ pathway after SAH in rats.
2. Materials and Methods
This study was approved by the Institutional Animal Care and Use Committee (IACUC) of Loma Linda University (LLU, No. 8170018). All the experiment procedures were performed in accordance with the National Institutes of Health Guidelines for the Use of Animals in Neuroscience Research and ARRIVE guidelines (Animal Research: Reporting of In Vivo Experiments).
2.1. Animals
Adult male Sprague-Dawley (SD) rats (weight 280 – 320 g) were purchased from Harlan Laboratories (Indianapolis, Indiana) and housed in the Animal Care Facility of LLU with 12-h light/dark cycle.
2.2. SAH model
SAH was induced using a modified endovascular perforation method as previously described [25]. Briefly, the isoflurane-anesthetized rat was intubated and ventilated (Harvard Apparatus, Holliston, MA, USA) in a supine position. Following a middle line incision in the neck, the left common carotid artery, internal carotid artery (ICA) and external carotid artery (ECA) were exposed by the blunt separation. A 4.0 cm-long monofilament nylon thread (4–0) with sharpened tip was put into the left ICA through the ECA stump and was advanced further to puncture the bifurcation of the anterior and the middle cerebral arteries. The sham-operated rats underwent the identical surgical procedures without arterial perforation. At end of surgery, the rat was extubated and monitored closely in a separate cage until it recovered from anesthesia. When rats were sacrificed at different time points, SAH grade was evaluated blindly by two independent investigators using the previously described scoring standard [26]. The SAH grade (0 – 18) was the sum of the grading scores in 6 brain regions based on the amounts of subarachnoid blood clot. Due to lacking of significant neurological deficits, rats with an insufficient SAH (SAH grade ≤ 8) were excluded as previously reported [27].
2.3. Drug administration
2.3.1. Intranasal administration
KP54 (a selective agonist of GPR54) was diluted in phosphate-buffered saline (PBS, 0.01M). PBS (vehicle) or KP54 was administered via the intranasal (i.n) route at 1h after SAH as previously described [28]. Briefly, rats were placed in the supine position under anesthesia with 3% isoflurane. A total volume of 10 μl PBS or 10 μl KP54 of three different doses (0.1 nmol/kg, 1.0 nmol/kg, or 10.0 nmol/kg) were administered with 5 μl/ one nostril and alternated every 5 min.
2.3.2. Intracerebroventricular injection
A volume of 5 μl (500 pmol) KISS1 small interfering ribonucleic acid (siRNA) (Thermo Fisher Scientific, MA, USA), ARRB2 siRNA (Santa Cruz Biotechnology, TX, USA) or scrambled siRNA (Thermo Fisher Scientific, MA, USA) were diluted with the 10 μL Entranster-in vivo RNA transfection reagent (Engreen Biosystem, Beijing, China). The solution was mixed and incubated for 15 min at room temperature. Finally, the total volume of 15 μL Entranster-in vivo–siRNA mixture was administered via the intracerebroventricular (i.c.v) route at 48h before SAH. Kisspeptin 234 (KP234), a selective GPR54 antagonist (No.3881, Tocris Bioscience Inc., MN, USA) was diluted in normal saline (NS). NS (5.0 μl) or KP234 (1.0 nmol in 5.0 μl) were administered via the i.c.v route at 1h before SAH. As previously described [28], rats were placed in a stereotaxic apparatus under 3% isoflurane anesthesia. The needle of a Hamilton syringe (Microliter701, Hamilton Company, NV, USA) was inserted into the left lateral ventricle (1.0 mm caudal and 1.5 mm lateral to the bregma, 3.5 mm in depth) through a burr hole on the skull. The drugs were administrated at a speed of 0.5 μl/min using an infusion pump (Quintessential Stereotaxic Injector, Stoelting Co., IL, USA). Upon the completion of injection, the needle was kept in the same place for another 10 min to prevent leakage, after which it was removed slowly within 5 min. The burr hole was sealed by bone wax and the incision was sutured.
2.4. Experimental design
Five separate experiments were performed in this study (Fig. 1). Rats were randomly assigned to each experimental group. The excluded rats (died during surgery or mild SAH grading) were replaced to ensure the same numbers of rats/group for statistical analysis.
Fig. 1.
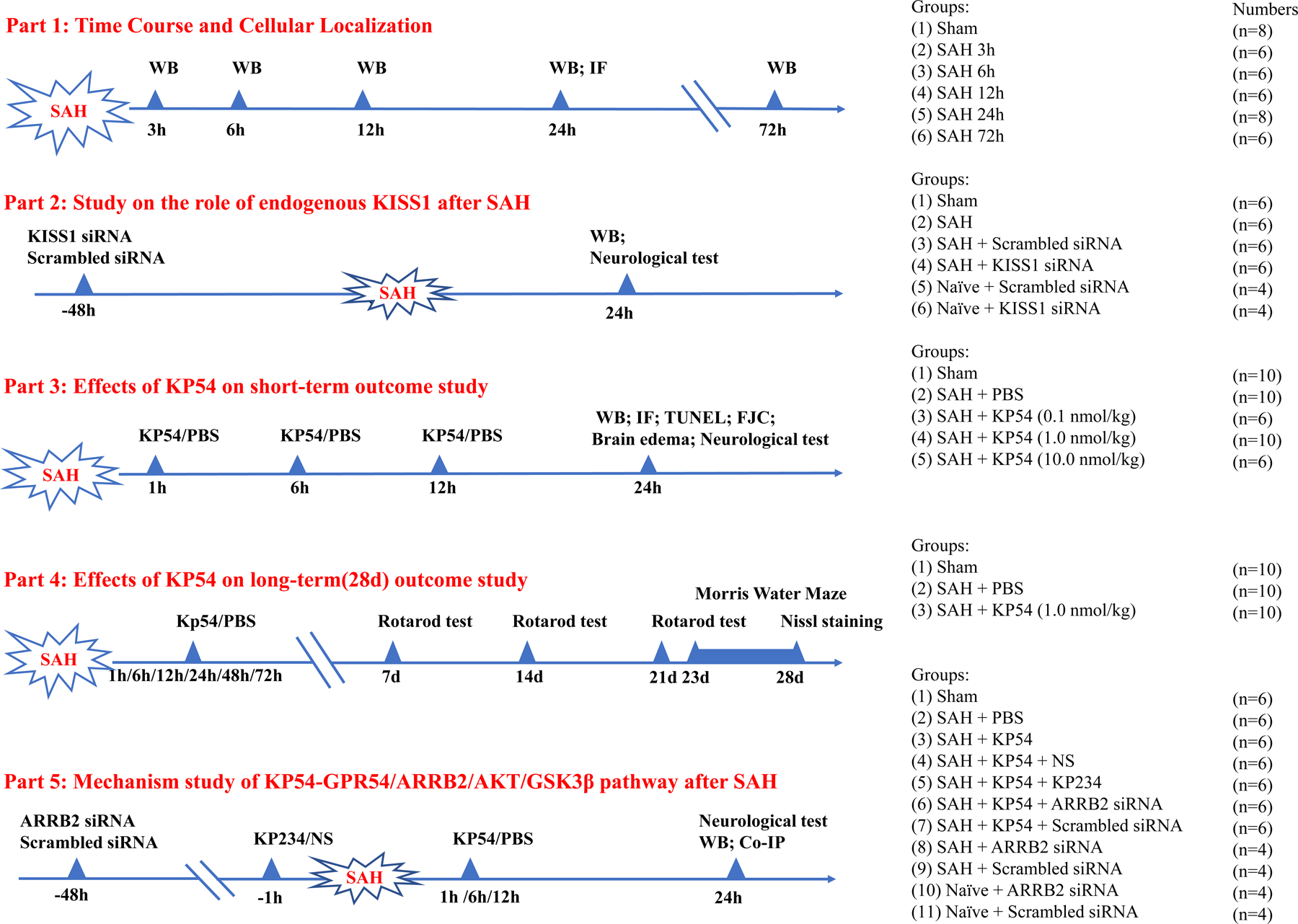
Experimental design and animal groups.
2.4.1. Experiment 1. Time-course and cellular localization.
This experiment was to determine the endogenous protein levels of KISS1, GPR54, and ARRB2 in the sham rats and SAH rats. The 40 rats were randomly divided into 6 groups including Sham (n = 8), SAH-3h (n = 6), SAH-6h (n = 6), SAH-12h (n = 6), SAH-24h (n = 8) and SAH-72h (n = 6). Western blot was performed to determine the protein levels of KISS1, GPR54 and ARRB2 in the ipsilateral (left) brain hemisphere (n = 6 per group). Cellular localization of GRP54 or ARRB2 was evaluated by the double immunofluorescence staining in the sham and SAH-24h group (n = 2 per group). GPR54 or ARRB2 was co-stained with the neuron marker of neuronal nuclei (NeuN), the microglial marker of calcium-binding adaptor molecule1 (Iba-1) or the astrocyte marker of glial fibrillary acidic protein (GFAP).
2.4.2. Experiment 2. Role of endogenous KISS1 in the oxidative stress and neuronal apoptosis 24 h after SAH.
This experiment was to evaluate the effect of endogenous KISS1 knockdown on neurobehavioral outcome, oxidative stress and neuronal apoptosis after SAH.
First, a set of 8 rats were randomized to two groups of naïve + KISS1 siRNA, naïve + scrambled siRNA (n = 4 per group) for validating the knock down efficiency of KISS1 siRNA. The naïve group were healthy rats and did not undergo any surgical procedures. KISS1 siRNA or scrambled siRNA was injected via i.c.v at 72 h before the brain tissues were collected in naïve rats.
Another set of 24 rats were randomized into four groups (n = 6 per group): Sham, SAH, SAH + scrambled siRNA, SAH + KISS1 siRNA. KISS1 siRNA or scrambled siRNA was injected via i.c.v at 48 h before SAH induction. Neurobehavioral tests (modified Garcia test and beam balance test) and western blot assessments ( KISS1, 4HNE, HO1, Bcl2 and BAX) were conducted at 24 h after SAH.
2.4.3. Experiment 3. The effect of KP54 treatment on short-term outcomes 24 h after SAH.
This experiment was to evaluate the effects of GPR54 activation with exogenous KP54 treatment (SCP0186, Sigma-Aldrich Inc., MO, USA) on short-term outcome after SAH. A total of 30 rats were randomly assigned to five groups (n = 6 per group): Sham, SAH + PBS, SAH + KP54 0.1 nmol/kg, SAH + KP54 1.0 nmol/kg, SAH + KP54 10.0 nmol/kg. PBS or KP54 was administered at 1st h, 6th h, 12th h, and 24th h after SAH. Neurological scores (modified Garcia test and beam balance test) and SAH grade were assessed at 24 h after SAH. Based on the results of neurological scores, the dose of 1.0 nmol/kg was determined as the best dosage of KP54 and was used for western blot, immunohistochemistry staining and the mechanistic experiments. To evaluate oxidative stress injury and neuronal apoptosis, an additional 12 rats were used and randomized to groups of Sham, SAH + PBS, and SAH + KP54 (1.0 nmol/kg) groups (n = 4 per group). Immunofluorescence staining of 8-hydroxydeoxyguanosine (8-OHdG) and mitochondrial superoxide (Mitosox), Fluoro-Jade C (FJC) staining, terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining and western blot were performed at 24 h after SAH.
2.4.4. Experiment 4. The effect of KP54 treatment on long-term outcomes after SAH.
This experiment was to evaluate the treatment effects of KP54 on long-term neurological outcomes after SAH. Thirty rats were randomly assigned to three groups (n = 10 per group): Sham, SAH + PBS, SAH + KP54 (1.0 nmol/kg). PBS or KP54 were administered intranasally at 1st h, 6th h, 12th h, 24th h,48th h and 72nd h after SAH. The Rotarod test was conducted on the 7th day, 14th day and 21st day after SAH. The Morris water maze test was performed between days 23 and 28 after SAH, after which all the rats were sacrificed. Nissl staining was used to evaluate the neuronal degeneration within the ipsilateral hippocampus.
2.4.5. Experiment 5. The mechanisms underlying neuroprotective effects of KP54 treatment
To explore the signaling pathway of KP54 mediated GPR54/ARRB2/AKT/GSK3β activation after SAH, a total of 50 rats were randomly assigned to five groups: Sham (n = 10), SAH + PBS (n = 10), SAH + KP54 (n = 10), SAH + KP54+ NS (n = 6), and SAH + KP54 + KP234 (n = 6). NS or KP234 (GPR 54 inhibitor) were administered i.c.v at 1h before SAH. PBS or KP54 was administered intranasally at 1st h, 6th h, 12th h, and 24th h after SAH. At 24 h after SAH, neurobehavioral scores and SAH grade were evaluated. Western blot was performed to assess the protein levels of GRP54, ARRB2, AKT and GSK3β in the ipsilateral brain tissues at 24 h after SAH.
To determine the knock down efficiency of ARRB2 siRNA, additional 16 rats were randomized into four groups (n=4 per group): naïve + ARRB2 siRNA, naïve + scrambled siRNA, SAH + ARRB2 siRNA, SAH+ scrambled siRNA. ARRB2 siRNA or scrambled siRNA was injected via i.c.v at 48 h before SAH induction in SAH rats or 72 h before scarification in naïve rats. To further access the role of ARRB2 in KP54 treatment-mediated GRP54 activation, another set of rats were randomized to two groups (n=6 per group) of SAH + KP54 + ARRB2 siRNA and SAH + KP54 + scrambled siRNA. Western blot was performed to assess the protein levels of ARRB2, AKT and GSK3β in the ipsilateral brain tissues at 24 h after SAH. Immunoprecipitation was performed to evaluate the relationship between ARRB2 and GPR 54 specifically.
2.5. Neurobehavioral tests
Neurobehavioral tests were performed by an investigator blind from the experimental group’s information.
2.5.1. Short-term neurobehavioral outcomes at 24h after SAH
Modified Garcia scale (score = 0 – 18) was used to assess sensorimotor function including hemiplegia, motor performance deficits and abnormal postures. Beam balance test (score = 0 – 4) was used to assess complex movements and coordination, whose score was averaged from three repeated tests. The higher score indicated the better performance in the neurobehavioral tests.
2.5.2. Long-term neurobehavioral outcomes
To access the balance and coordination function, rotarod test was performed at 7th day, 14th day, 21st day after SAH. Briefly, the rats were placed on the rotarod (Columbus Instruments, OH, USA) and started to run at a speed of 5 or 10 revolutions per minute (RPM) with an acceleration of 2 RPM every 5 seconds. The final duration that the rats stayed on the rotarod was recorded and averaged from the five repeated tests.
To evaluate the spatial learning memory and cognitive function, Morris water maze test was performed from the 23rd day to 28th day after SAH as previously described [28]. The following data were recorded by the EthoVision video tracking system (Noldus, VA, USA): 1) the time that rats reached the submerged platform for 5 consecutive days; 2) the time that rats spent in the southwest quadrant (where the platform was used to locate) on the last day of 60-s probe trial; 3) Swimming speed and distance as well as escape latency.
2.6. Immunofluorescence staining
Under deep anesthesia, the rats were transcardially perfused with 0.1 M PBS (4°C, 200 ml) followed by 10% formalin (4°C , 200 ml). The brain were harvested and fixed in 10% formalin for 24 h, after which they were dehydrated in 30% sucrose solution at 4 °C until saturation. The brain tissues were embedded in OCT compound (Scigen Scientific Gardena, CA, USA) and frozen in liquid nitrogen. The brain samples were cut into 10-µm coronal sections using a Leica CM1860 cryostat microtome (Leica Biosystems, Nussloch, Germany) and mounted on microscope slides (Genesee Scientific, CA, USA). Immunofluorescence staining was performed as described previously [28]. Briefly, the brain slices were incubated with 0.3% Triton X-100 (Sigma-Aldrich Inc., MO, USA) for 30 min and 5% normal donkey serum for 2 h at room temperature. The primary antibodies used for overnight incubation at 4 °C were rabbit anti-GPR54 (1:100, bs-2501R, Bioss, MA, USA), mouse anti-NeuN (marker of neuron, 1:100, ab104224, Abcam, MA, USA), mouse anti-ARRB2 (1:50, sc-13140, Santa Cruz, TX, USA), rabbit anti-NeuN (1:100, ab177487, Abcam, MA, USA), goat anti-Iba-1 (marker of microglia, 1:100, ab5076, Abcam, MA, USA), goat anti-GFAP (marker of astrocyte, 1:100, ab53554, Abcam, MA, USA). On the second day, the brain slices were washed with PBS (0.1 M) and incubated with the corresponding fluorescence-conjugated secondary antibodies (1:100, Jackson ImmunoResearch, PA, USA) at room temperature for 1 h. The cell nuclei were stained by 4′,6-diamidino-2-phenylindole (DAPI, Sigma-Aldrich Inc., MO, USA). Finally, the brain slices were covered with coverslip and observed under a fluorescence microscope (DMi8, Leica Microsystems, Wetzlar, Germany).
2.7. Measurement of oxidative stress
Immunohistochemistry staining were used to evaluate the oxidative stress-induced DNA damage (8-OHdG) and mitochondrial superoxide (Mitosox) in the brain. Freshly prepared frozen 10μm brain slices were placed on the microscope slides (Genesee Scientific, CA, USA). The slides were first immersed in the pH 6.0 antigen retrieval solution and heated in microwave oven for 15 min to expose the antigen. Secondly, the sections were blocked with endogenous peroxidase for 10 min using 3% H2O2 followed by the incubation of 8-OHdG antibody (1:200, ab62623, Abcam, MA, USA) or Mitosox antibody (1:1000, M36008, Thermo Fisher, CA, USA) at room temperature. The slides were observed under the microscope under magnification of 200x for Mitosox staining or 400x for 8-OHdG. The fluorescence intensity was averaged from four randomly selected brain regions within ipsilateral entorhinal cortex using ImageJ 1.52p (National Institutes of Health, MD, USA).
2.8. Assessment of neuronal damage
2.8.1. FJC staining
Neuronal degeneration was evaluated using FJC staining (modified FJC Ready-to-Dilute Staining Kit, Biosensis, CA, USA) at 24 h after SAH according to the manufacture’s instruction. Under fluorescence microscope (DMi8, Leica Microsystems, Wetzlar, Germany), FJC positive cells were counted within ipsilateral entorhinal cortex under 200x magnification and averaged from four randomly selected brain regions within ipsilateral entorhinal cortex.
2.8.2. TUNEL staining
Neuronal death was evaluated using TUNEL staining (situ Apoptosis Detection Kit, 12156792910, Roche, MO, USA) at 24 h after SAH according to the manufacturer’s instructions. Under a fluorescence microscope (DMi8, Leica Microsystems, Wetzlar, Germany), TUNEL-positive neurons were counted within ipsilateral entorhinal cortex under 200x magnification and averaged from four brain slices.
2.8.3. Nissl staining
Hippocampus neuronal damage was evaluated using Nissl staining at 28th day after SAH. The brain sample was cut into 20-µm coronal sections using a Leica CM1860 cryostat microtome (Leica Biosystems, Nussloch, Germany) and mounted on microscope slides (Genesee Scientific, CA, USA). Nissl staining was performed using 0.5% crystal violet as described previously [29]. Under a light microscope, the surviving neurons within the hippocampal cornu ammonis (CA)1, CA3, and dentate gyrus (DG) per brain slice were counted at 400× magnification. Ni-positive neurons were counted within ipsilateral entorhinal cortex under 200x magnification and averaged from four brain slices.
2.9. Western blot analysis
Western blot was performed as previously described [29]. Briefly, the rats were transcardially perfused with 0.1 M PBS (pre-cold, 200 ml) under deep anesthesia. The ipsilateral (left) hemisphere was harvested and homogenized in RIPA lysis buffer (Santa Cruz Biotechnology, TX, USA). The supernatant was collected after centrifuging at 14,000 rpm for 30 min at 4 °C. The protein concentrations were estimated using the DC Protein Assay kit (Bio-Rad, CA, USA) and the Genesys 10S UV-Vis spectrophotometer (Thermo Fisher Scientific, MA, USA). A total of 40 µg proteins were loaded in each well, separated though sodium dodecyl sulfate polyacrylamide gel electrophoresis (10% or 12.5%), transferred onto nitrocellulose membranes (0.2 µm or 0.45 µm) and incubated at 4 °C for 12 h with primary antibodies as follows: rabbit anti-KISS1 (1:300, ab19028, Abcam, MA, USA), rabbit anti-GPR54 (1:500, bs-2501R, Bioss, MA, USA), mouse anti-ARRB2 (1:500, sc-13140, Santa Cruz, TX, USA), rabbit anti-PI3K (1:1000; #4228, Cell Signaling Technology, MA, USA); rabbit anti-phosphorylated-AKT (1:1000; #9271, Cell Signaling Technology, MA, USA); rabbit anti-AKT (1:1000; #9272, Cell Signaling Technology, MA, USA), mouse anti-phosphorylated-GSK3β (1:1000, sc-373800, Santa Cruz, TX, USA), mouse anti-GSK3β (1:1000, sc-377213, Santa Cruz, TX, USA), rabbit anti-4HNE (1:1000, ab46545, Abcam, MA, USA), rabbit anti-HO-1 (1:30000, ab68477, Abcam, MA, USA), rabbit anti-Bcl2 (1:1000, ab59348, Abcam, MA, USA), rabbit anti-BAX (1:4000, ab182734; Abcam, MA, USA), rabbit anti-cleaved caspase 3 (CC3) (1:800; #9661, Cell Signaling Technology, MA, USA); mouse anti-β-actin (1:1000, sc-47778, Santa Cruz, TX, USA). The membranes were incubated with HRP-conjugated rabbit (1:5000, Millipore Corporation, MA, USA) or mouse (1:3000, Santa Cruz, TX, USA) secondary antibodies for 1 h and visualized using enhanced chemiluminescence (ECL) reagent kit (Genesee Scientific, CA, USA) on the imaging system (Model 4000, Bio-Rad, CA, USA). The β-actin was used as an internal control. The data were quantified with optical methods using the ImageJ 1.52p (National Institutes of Health, MD, USA).
2.10. Co-immunoprecipitation
Co-immunoprecipitation (Co-IP) was performed to detect the protein interaction between GPR54 and ARRB2. Briefly, protein A/G Sepharose beads (50 µl, ab193262, Abcam, MA, USA) were added into the protein lysate (200 µl) and incubated with either 2 µg mouse anti-ARRB2 (sc-13140, Santa Cruz, TX, USA), rabbit anti-GPR54 (bs-2501R, Bioss, MA, USA), or normal mouse IgG (sc-2025, Santa Cruz, TX, USA) at 4 °C overnight. The beads were gently rinsed with PBS (pre-cold, 0.01 M), collected by centrifuging at 14,000 rpm at 4 °C for 30 min., The beads were resuspended in 200 μl loading buffer and heated at 95 °C for 10 min. The western blot assessments and data analysis were subsequent performed as described above.
2.11. Statistical analysis
The data were expressed as mean ± standard deviation (SD). Statistical analysis was performed using GraphPad Prism 8 (GraphPad Software, CA, USA). One-way or two-way ANOVA followed by Tukey’s post hoc test was used for comparisons among multiple groups. A p-value less than 0.05 was considered statistically significant.
3. Results
3.1. Mortality and SAH grades
As shown in Table 1, a total of 292 male rats were used in this present study. Among those, there were 40 sham rats, 236 SAH rats, and 16 naïve rats. A total of 45 SAH rats were excluded from the experiment due to the mild SAH grade (SAH grade ≤ 8, n = 11) or death during surgery (n = 34). The overall mortality was 17.8% (34/191) in SAH group, 0% (0/40) in sham group, and 0% (0/16) in the naïve group, respectively. The highest mortality (33.3%) occurred in the SAH + KISS1 siRNA, SAH + KP54 (10.0 nmol/kg), and long-term SAH + PBS groups. However, no significant difference was found in mortality among all the SAH groups (p > 0.05). Blood clots were mainly distributed around the Willis and ventral brain stem in SAH rats, but rarely seen in the shams (Fig. 2A). No significant difference of the SAH grades was found among SAH groups (Fig. 2B).
Table 1.
Summary of animal numbers and mortality.
| Groups | Mortality | Excluded |
|---|---|---|
| Part 1: Time Course and Cellular Localization | ||
| Sham (n=8) | 0.0% (0/8) | 0 |
| SAH (3h, 6h, 12h, 24h, 72h) (n=32) | 12.5% (5/40) | 3 |
| Part 2: Study on the role of endogenous KISS1 after SAH | ||
| Sham (n=6) | 0.0% (0/6) | 0 |
| SAH (n=6) | 12.5% (1/8) | 1 |
| SAH + scrambled siRNA (n=6) | 12.5% (1/8) | 1 |
| SAH + KISS1 siRNA (n=6) | 33.3% (3/9) | 0 |
| Naïve + scrambled siRNA (n=4) | 0.0% (0/4) | 0 |
| Naïve + KISS1 siRNA (n=4) | 0.0% (0/4) | 0 |
| Part 3: Effects of KP54 on short-term outcome study | ||
| Sham (n=10) | 0.0% (0/10) | 0 |
| SAH + PBS (n=10) | 15.4% (2/13) | 1 |
| SAH + KP54 (0.1 nmol/kg) (n=6) | 22.2% (2/9) | 1 |
| SAH + KP54 (1.0 nmol/kg) (n=10) | 15.4% (2/13) | 1 |
| SAH + KP54 (10.0 nmol/kg) (n=6) | 33.3% (3/9) | 0 |
| Part 4: Effects of KP54 on long-term(28d) outcome study | ||
| Sham (n=10) | 0.0% (0/10) | 0 |
| SAH + PBS (n=10) | 33.3% (5/15) | 0 |
| SAH + KP54 (1.0 nmol/kg) (n=10) | 16.7% (2/12) | 0 |
| Part 5: Mechanism study of KP54/GPR54/ARRB2/AKT/GSK3β pathway after SAH | ||
| Sham (n=6) | 0.0% (0/6) | 0 |
| SAH + PBS (n=6) | 12.5% (1/8) | 1 |
| SAH + KP54 (1.0 nmol/kg) (n=6) | 14.3% (1/7) | 0 |
| SAH + KP54 + NS (n=6) | 12.5% (1/8) | 1 |
| SAH + KP54 + KP234 (n=6) | 25.0% (2/8) | 0 |
| SAH + KP54 + scrambled siRNA (n=6) | 0.0% (0/6) | 0 |
| SAH + KP54 + ARRB2 siRNA (n=6) | 14.3% (1/7) | 1 |
| Naïve + ARRB2 siRNA (n=4) | 0.0% (0/4) | 0 |
| Naïve + scrambled siRNA (n=4) | 0.0% (0/4) | 0 |
| SAH + ARRB2 siRNA (n=4) | 20.0% (1/5) | 0 |
| SAH + scrambled siRNA (n=4) | 0.0% (0/4) | 0 |
| Total | ||
| Naïve | 0.0% (0/16) | 0 |
| Sham | 0.0% (0/40) | 0 |
| SAH | 17.8% (34/191) | 11 |
Fig. 2. The temporal expressions of KISS1, GPR54 and ARRB2 within ipsilateral brain hemisphere after subarachnoid hemorrhage (SAH).
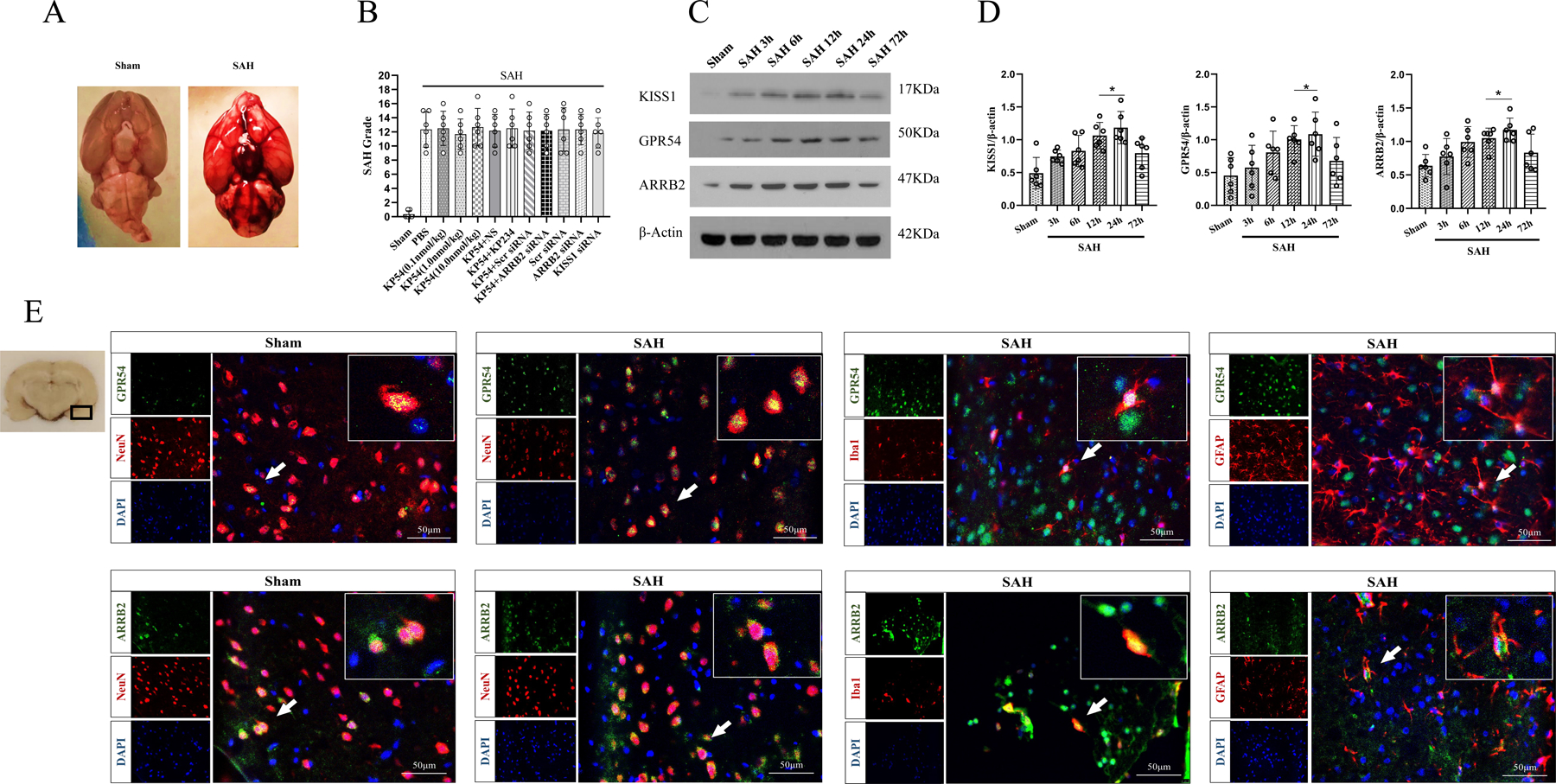
(A) Representative pictures showed that subarachnoid blood clots mainly presented around the circle of Willis in the rat brain at 24 h after SAH; (B) SAH grading scores of all SAH groups. (C) Representative western blot bands of KISS1, GPR54 and ARRB2; (D) Time course and densitometric quantification of KISS1, GPR54 and ARRB2. *p < 0.05 vs. Sham group. Data was represented as mean ± SD, n = 6 per group, one-way ANOVA Tukey test was used for the comparison between sham and SAH groups. (E) Immunofluorescence staining of GPR54 or ARRB2 with neurons (NeuN, red), microglia (Iba-1, red) and astrocytes (GFAP, red) at 24 h after SAH. The small black box in the coronal brain slice (left of top panel) indicated the area where microphotograph was taken. Scale bar = 50μm, n = 2 for each group.
3.4. Time course of endogenous KISS1, GPR54 and ARRB2 expressions in ipsilateral hemisphere after SAH
The western blot showed that endogenous protein levels of KISS1, GPR54 and ARRB2 started increasing at 3 h and peaked at 24 h after SAH, compared with sham group (P < 0.05, Fig. 2C, 2D). Double immunofluorescence staining showed that GPR54 or ARRB2 co-localized with NeuN positive neurons, Iba-1 positive microglia, and GFAP positive astrocytes, respectively (Fig. 2E).
3.5. Endogenous KISS1 knockdown exacerbated short-term outcomes at 24 h after SAH
To evaluate the role of endogenous KISS1 in oxidative stress and apoptosis after SAH, KISS1 siRNA was administered at 48 h before SAH induction. Compared with sham group, there were significant higher the brain expressions of KISS1, 4HNE, HO1 and BAX but lower Bcl2 in SAH rats at 24 h after SAH. KISS1 siNRA downregulated the KISS1 and Bcl2 expressions but increased 4HNE and BAX expressions in SAH rats compared to no-treated or scramble siRNA treated SAH rats. (P < 0.01, Fig. 3A, 3B). Modified Garcia and beam balance scores were significantly lower in SAH rats than shams (P < 0.05, Fig. 3C). KISS1 knockdown by KISS1 siRNA exacerbated neurobehavioral deficits compared to SAH and SAH + scrambled siRNA groups (P < 0.05, Fig. 3C). Western blot showed that the protein levels of KISS1 were significantly decreased in KISS1 siRNA treated either naïve or SAH rats (P < 0.01, Fig. 3D, 3E), suggesting knock down efficiency of KISS1 siRNA.
Fig. 3. Endogenous KISS1 knockdown exacerbated short-term outcomes at 24 h after SAH.
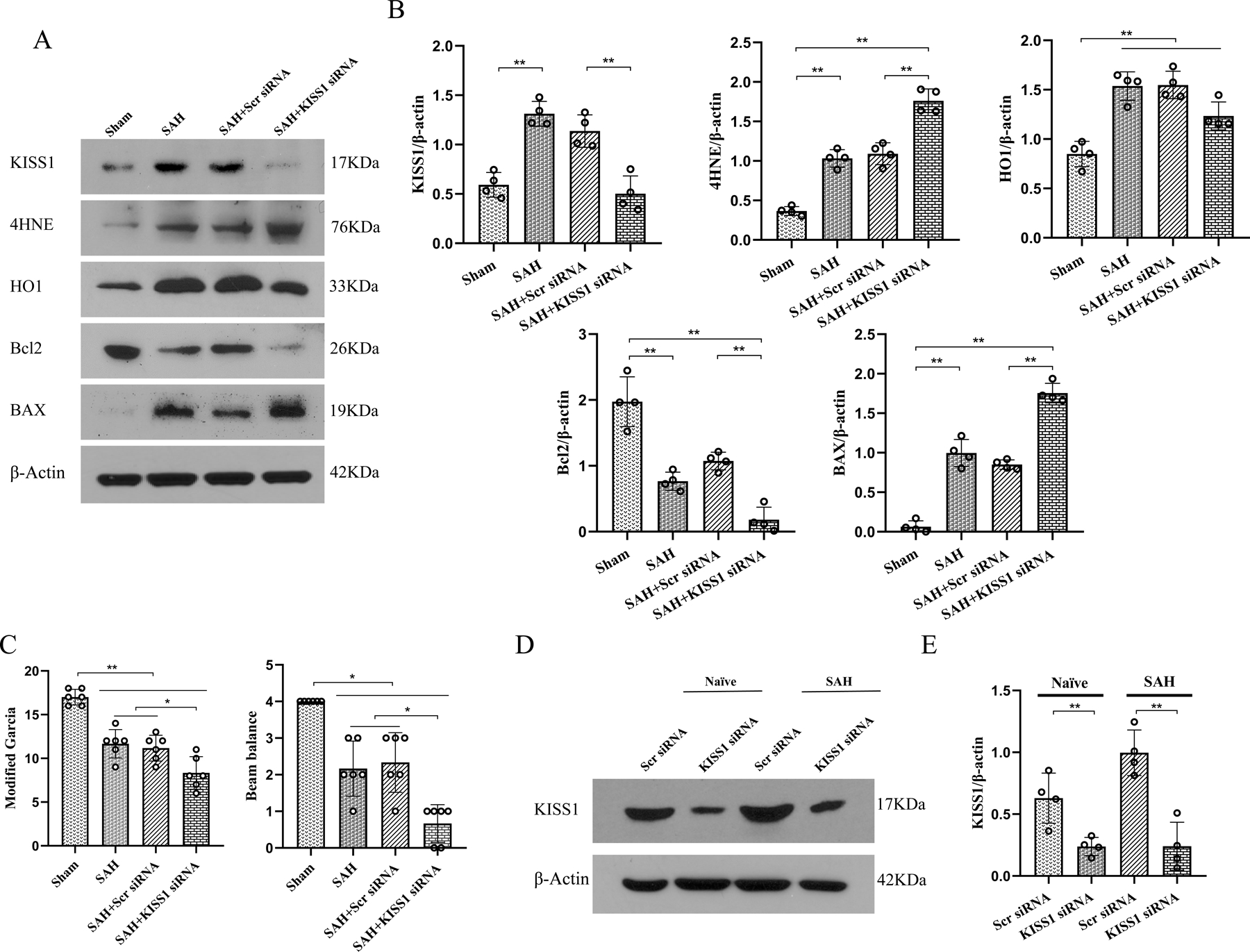
(A) Representative western blot bands of KISS1, 4HNE, HO1,Bcl2, and BAX; (B) Densitometric quantification of KISS1, 4HNE, HO1, Bcl2, and BAX at 24 h after SAH; (C) KISS1 siRNA knockdown significantly aggravated the neurological deficits, resulting in the lower modified Garcia score and beam balance score in SAH rats than SAH alone or scramble siRNA-treated SAH; (D, E) Representative western blot bands and densitometric quantification of KISS1 demonstrated the knockdown efficacy of KISS1 siRNA on brain KISS protein levels in naïve and SAH rats, *P < 0.05, **P < 0.01. Data was represented as mean ± SD. n = 4–10/group. One-way ANOVA, Tukey’s post hoc test.
3.6. KP54 treatment improved short-term neurobehavioral deficits and ipsilateral hemisphere oxidative stress injury at 24 h after SAH.
There were significant neurological impairments in the SAH rats at 24 h after SAH (Fig. 4 A and B). KP54 treatment at the dose of 1.0 nmol/kg, but not 0.1 nmol/kg or 10.0 nmol/kg, significantly improved the SAH-induced neurological deficits accessed by the modified Garcia test (Fig. 4A) and beam balance test (Fig. 4B). Based on the neurological results, the KP 54 dose of 1.0 nmol/kg was used as the best dose in the subsequent studies. Western blot showed that the brain expression levels of 4HNE (P < 0.01) and HO1 (P < 0.05) were significantly increased in SAH + PBS group compared with sham group at 24 h after SAH. KP54 treatment significantly reduced the elevation of 4HNE (P < 0.05) but further increased the expression of HO1 (P < 0.05) in SAH rats compared with SAH + PBS group (Fig. 4C, 4D).
Fig. 4. KP54 treatment attenuated the neurobehavioral deficits and oxidative stress at 24 h after SAH.
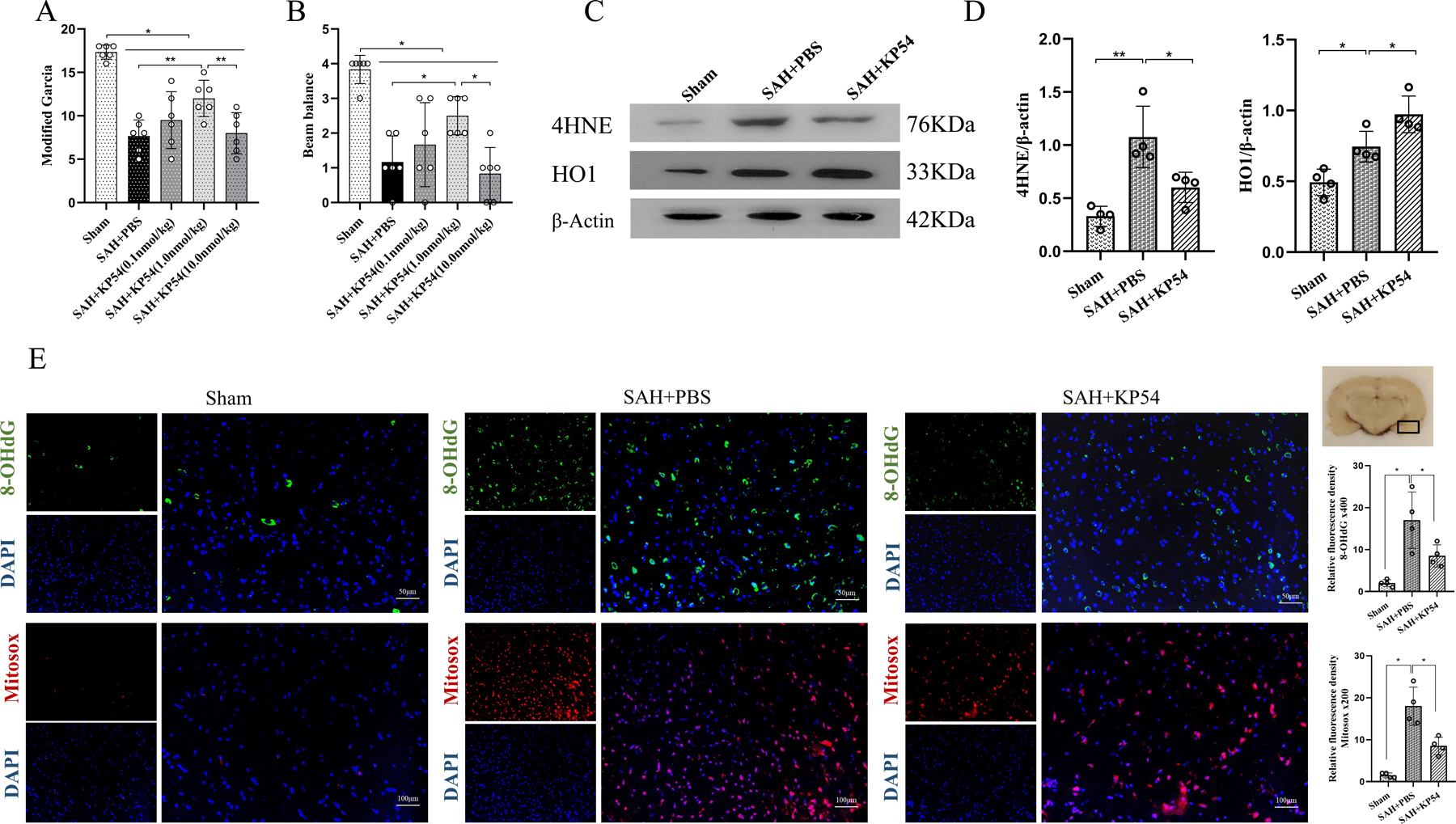
(A) Modified Garcia score at 24 h after SAH; (B) Beam balance score at 24 h after SAH; (C) Representative western blot bands of the brain oxidative stress markers at 24 h after SAH; (D) Densitometric quantification for the brain oxidative stress markers at 24 h after SAH; (E) Representative microphotographs and quantitative analysis for 8-OHdG and mitosox staining in the ipsilateral brain cortex. The small black rectangular box in the coronal brain slice (the right top panel) indicates the area for staining quantification. Scale bar = 50 or 100 μm. *P < 0.05, **P < 0.01. Data were represented as mean ± SD, n = 4 or 6 per group, one-way ANOVA Tukey test.
The 8-OHdG and Mitosox staining were for the evaluation of oxidative stress injury in ipsilateral brain tissue at 24 h after SAH. There were significantly increases in 8-OHdG and Mitosox positive neurons in the SAH + PBS group, which were reversed by KP54 treatment at the dose of 1.0 nmol/kg (SAH + KP54) (P < 0.05, Fig. 4E).
3.7. KP 54 treatment reduced neuronal degeneration and neuronal apoptosis at 24 h after SAH
At 24 h after SAH, western blot showed that apoptotic proteins level of BAX (P < 0.05) and CC3 (P < 0.01) were significantly increased within the ipsilateral hemisphere, while anti-apoptotic Bcl2 were significantly decreased in SAH + PBS group compared with sham group (P < 0.05). KP54 treatment significantly reduced the levels of BAX and CC3 but increased the expression of Bcl2 in SAH + KP54 compared with SAH + PBS group (P < 0.05, Fig. 5A, 5B).
Fig. 5. KP54 treatment attenuated the neuronal damage at 24 h after SAH.
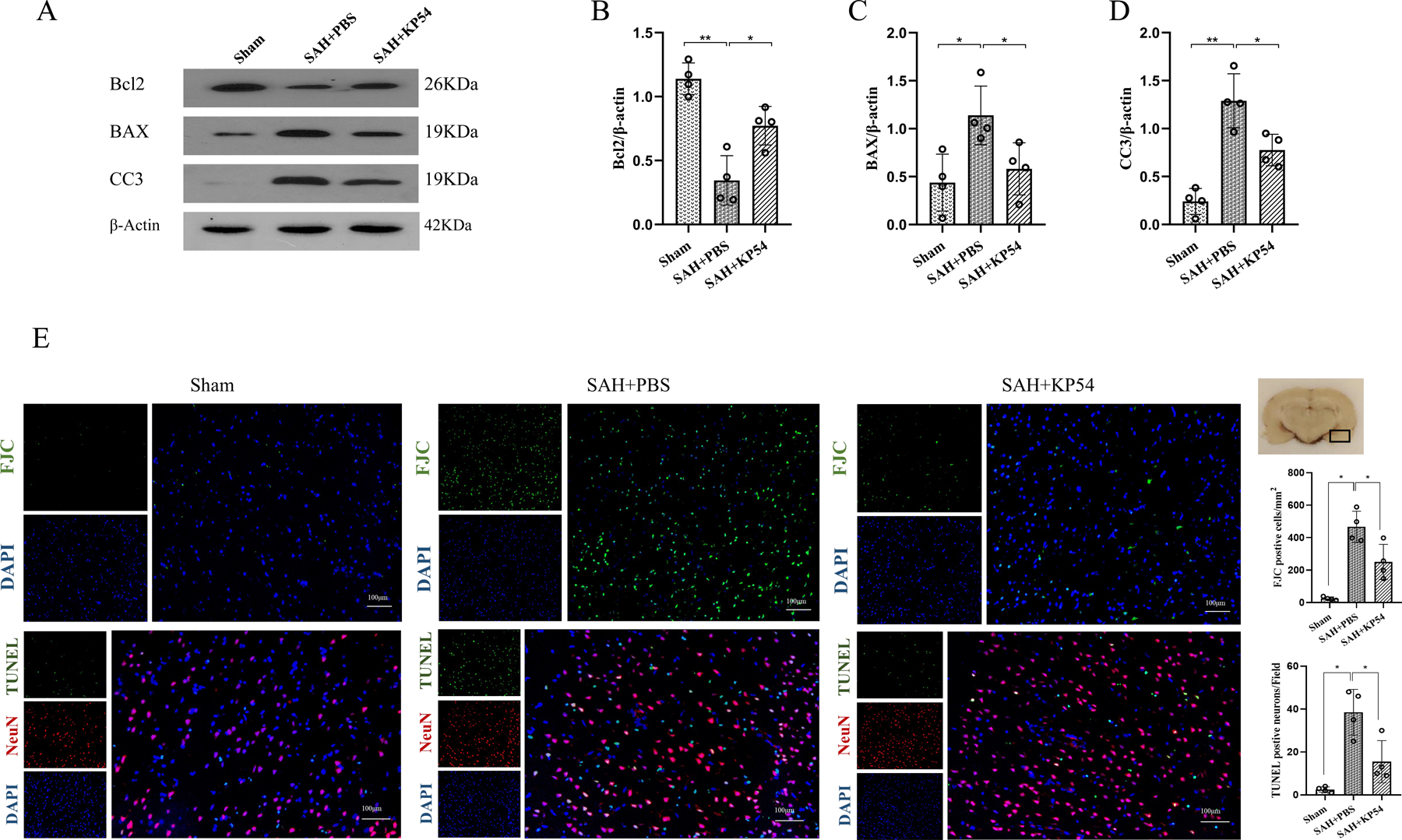
(A) Representative western blot bands of the brain apoptosis markers at 24 h after SAH; (B-D) Densitometric quantification for the apoptosis markers in brain at 24 h after SAH; (E) Representative microphotographs and quantitative analysis for FJC and TUNEL staining in the ipsilateral cortex of rat brain. The small black rectangular box in the coronal brain slice (the right top panel) indicates the area for staining quantification. Scale bar = 100 μm. *P < 0.05, **P < 0.01. Data were represented as mean ± SD, n = 4 per group, one-way ANOVA Tukey test.
FJC staining were used for assessing neuronal degeneration and TUNEL staining were performed for the evaluating neuronal death. At 24 h after SAH, there were significantly greater numbers of FJC-positive cells and TUNEL-positive neurons in SAH + PBS group than sham group at 24 h after SAH. These SAH-induced neuronal damages were significantly reduced by KP54 treatment at the dose of 1.0 nmol/kg (P < 0.05, Fig. 5C).
3.8. KP54 treatment improved long-term neurological deficits and reduced neuronal degeneration at 28 d after SAH.
In Rotarod tests of 5 RPM and 10 RPM, SAH rats were associated with significant shorter falling latency than shams at 1, 2, 3 weeks after SAH (P < 0.01, Fig. 6A, 6B). Such impaired neurological performances were significantly improved by KP54 treatment in SAH rats (P < 0.01, Fig. 6A, 6B).
Fig. 6. KP54 treatment improved the long-term outcomes at 28d after SAH.
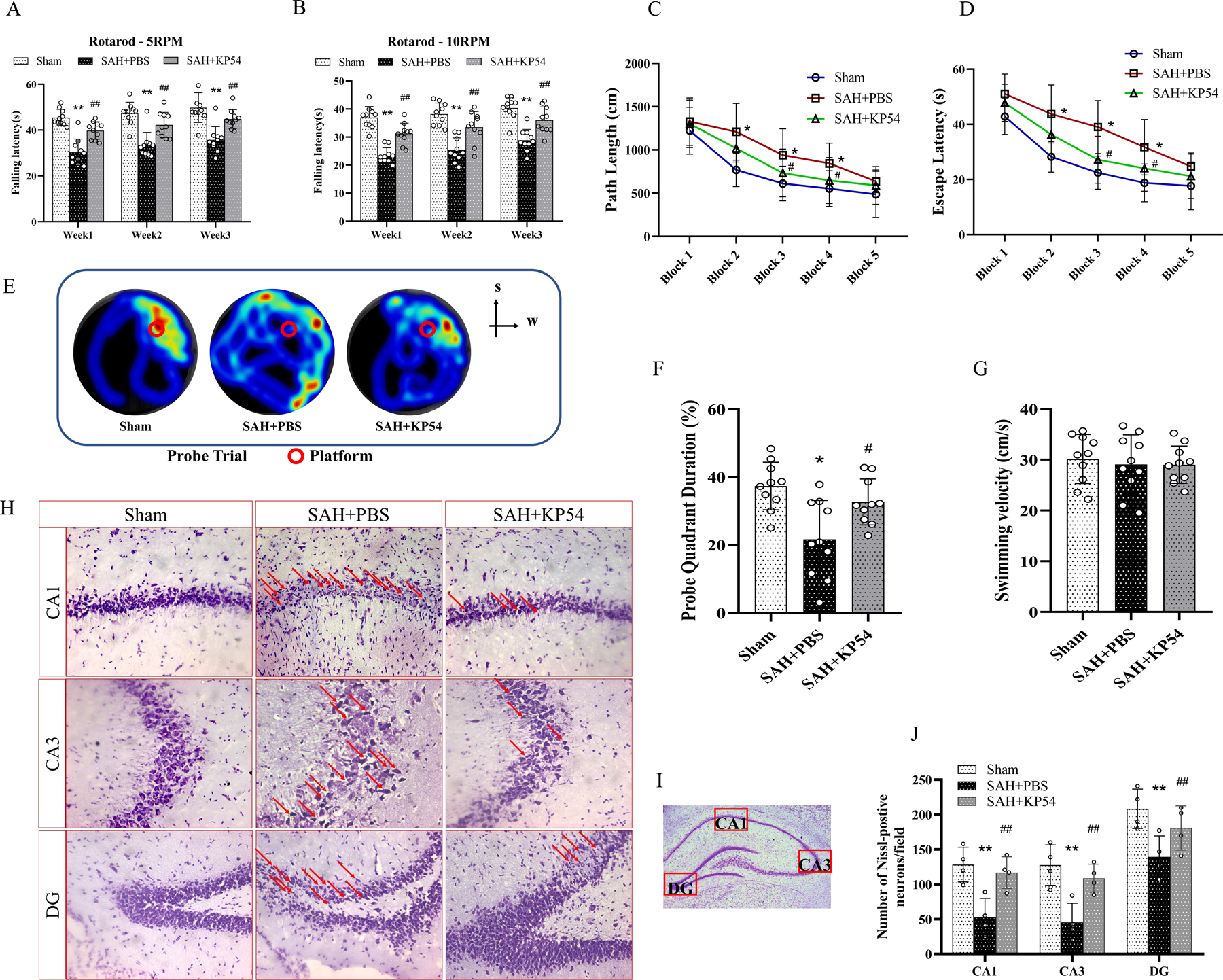
(A, B) Rotarod tests of 5rpm and 10rpm at every week; (C, D) Escape latency and swim distance of water maze test at 28d,; (E) Representative heat map of probe test showed that KP54 treated SAH rats spent more time in target quadrant; (F) Quantification of the probe quadrant duration in the probe trial,; (G) Swimming velocities of different groups in probe trial; (H) Representative microphotographs of Nissl staining within the hippocampal CA1, CA3 and DG regions showed the damaged neurons (red arrows); (I) Representative microphotograph of Nissl staining showed the location of CA1, CA3 and DG regions within the left hippocampus; (J) Quantifications of the surviving neuron within in hippocampal CA1, CA3 and DG regions, *P < 0.05, **P < 0.01 vs. Sham group; #P < 0.05, ##P < 0.01 vs. SAH + PBS group. Data represent with mean ± SD, n = 4 or 10 per group. Two-way ANOVA-Tukey for repeated measures of Rotarod and water mazes. One-way ANOVA-Turkey for measurements in probe trial and Nissl staining.
In Morris water maze test, both swim distance and escape latency to reach the platform were significantly longer in the SAH rats rats than shams at all testing days (P < 0.05, Fig. 6C, 6D). Such neurological deficits were attenuated by KP54 treatment on the 3rd and 4th day of Morris water maze test (P < 0.05, Fig. 6C. 6D). In the probe quadrant trials on 6th day of Morris water maze test, the time spent in the southwest quadrant (platform was used placed) was calculated to assess the spatial memory and learning ability. The SAH rats spent a notably less time in the target quadrant than shams, which was significantly increased in KP54-treated SAH rats (P < 0.05, Fig. 6F). The swimming speeds were not significant among all three groups, suggesting comparable motor functions (Fig. 6G).
Nissl staining showed that the morphological abnormalities of hippocampal neurons with atrophic cell body and concentrated nucleus within the ipsilateral hippocampus at 28 d after SAH (Fig. 6H). KP54 treatment significantly attenuated SAH-induced neuronal injury in CA1 (P < 0.01), CA3 (P < 0.01), and DG regions (P < 0.01, Fig. 6J).
3.9. KP234 and ARRB2 siRNA abolished the protective effects of KP54 treatment on short-term neurobehavioral outcomes at 24 h after SAH.
KP234 was i.c.v administrated at 1 h before SAH to inhibit the binding of KPs/GPR54. ARRB2 siRNA knockdown was i.c.v administrated at 48 h before SAH induction to investigate the role of ARRB2 upon KP54-mediated GRP 54 activation. Either GPR54 antagonism with KP234 or ARRB2 siRNA knockdown reversed the neurobehavioral benefits of KP54 at 24 h after SAH (P < 0.05, Fig.7A, 7B). Western blot showed that the protein levels of ARRB2 in ARRB2 siRNA-treated naïve and SAH were significantly decreased at 24 h after SAH (P < 0.01, Fig. 7C, 7D), suggesting the knock down efficiency of ARRB2 siRNA. Modified Garcia and beam balance scores were significantly lower in SAH rats than Naïve groups (P < 0.01, Fig. 7E, 7F). ARRB2 knockdown by siRNA further exacerbated neurobehavioral performances compared to SAH + scrambled siRNA group (P < 0.05, Fig.7E, 7F).
Fig. 7. Neuroprotection of KP54 depended on GPR54/ARRB2 at 24 h after SAH.
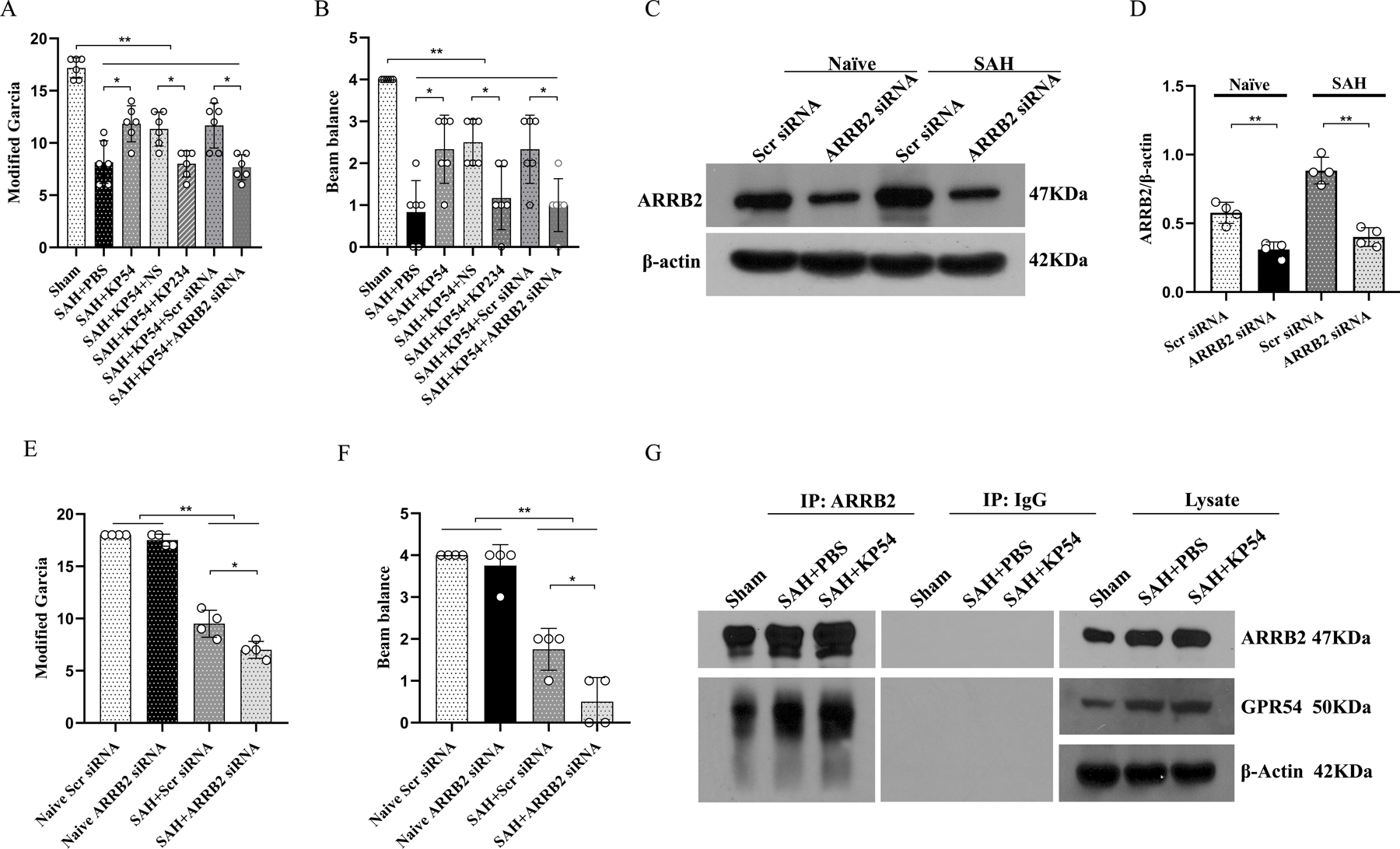
(A, B) GPR54 inhibitor KP234 and ARRB2siRNA knockdown reversed the neurobehavioral benefits of KP54 treatment at 24 h after SAH. (C, D) Representative western blot bands and densitometric quantification of ARRB2 demonstrated the efficacy of ARRB2 siRNA knockdown in naïve and SAH rats. (E, F) ARRB2 knockdown significantly aggravated the neurological deficits in SAH rats compared to scramble siRNA-treated SAH rats; (G) The effects of KP54 on the GPR54/ARRB2 interaction at 24 h after SAH. *P < 0.05, **P < 0.01. Data were represented as mean ± SD, n=4–6 per group, One-way ANOVA-Tukey.
Co-immunoprecipitation (Co-IP) for suggested that the GPR54 interacted with ARRB2 in both shams and SAH rats. KP54 treatment significantly enhanced the GPR54/ARRB2 interaction in SAH rats (Fig. 7G).
3.10. KP54 treatment attenuated oxidative stress and neuronal apoptosis via activating GPR54/ARRB2/AKT/GSK3β signaling pathway at 24 h after SAH.
As shown in Fig. 8, the brain expressions of KISS1, GPR54, ARRB2, 4HNE, HO1, BAX, CC3 were significantly increased (P < 0.05) within ipsilateral hemisphere, while the Bcl2 was notably decreased at 24 h after SAH compared with the shams (P < 0.05). KP54 treatment further increased the protein levels of KISS1, GPR54, ARRB2, PI3K, pAKT, pGSK3β, HO1and Bcl2 in brain tissues, while decreased the brain expressions of 4HNE, BAX, and CC3 in the SAH rats when compared with the SAH + PBS group (P < 0.05). GPR54 antagonism with KP234 significantly revered the effects of KP54 on protein levels of GPR54, ARRB2, PI3K, pAKT, pGSK3β, HO1, Bcl2, 4HNE, BAX and CC3 in the KP54-treated SAH rats (P < 0.05). ConsistentlyARRB2 knockdown using ARRB2 siRNA significantly reversed the KP54 effects on brain protein levels of ARRB2, PI3K, pAKT, pGSK3β, Bcl2, HO1 4HNE, BAX, and CC3 in SAH rats at 24 h after SAH (P < 0.05).
Fig. 8. KP54 treatment attenuated oxidative stress and neuron apoptosis via activating GPR54/ARRB2/AKT/GSK3β pathway after SAH.
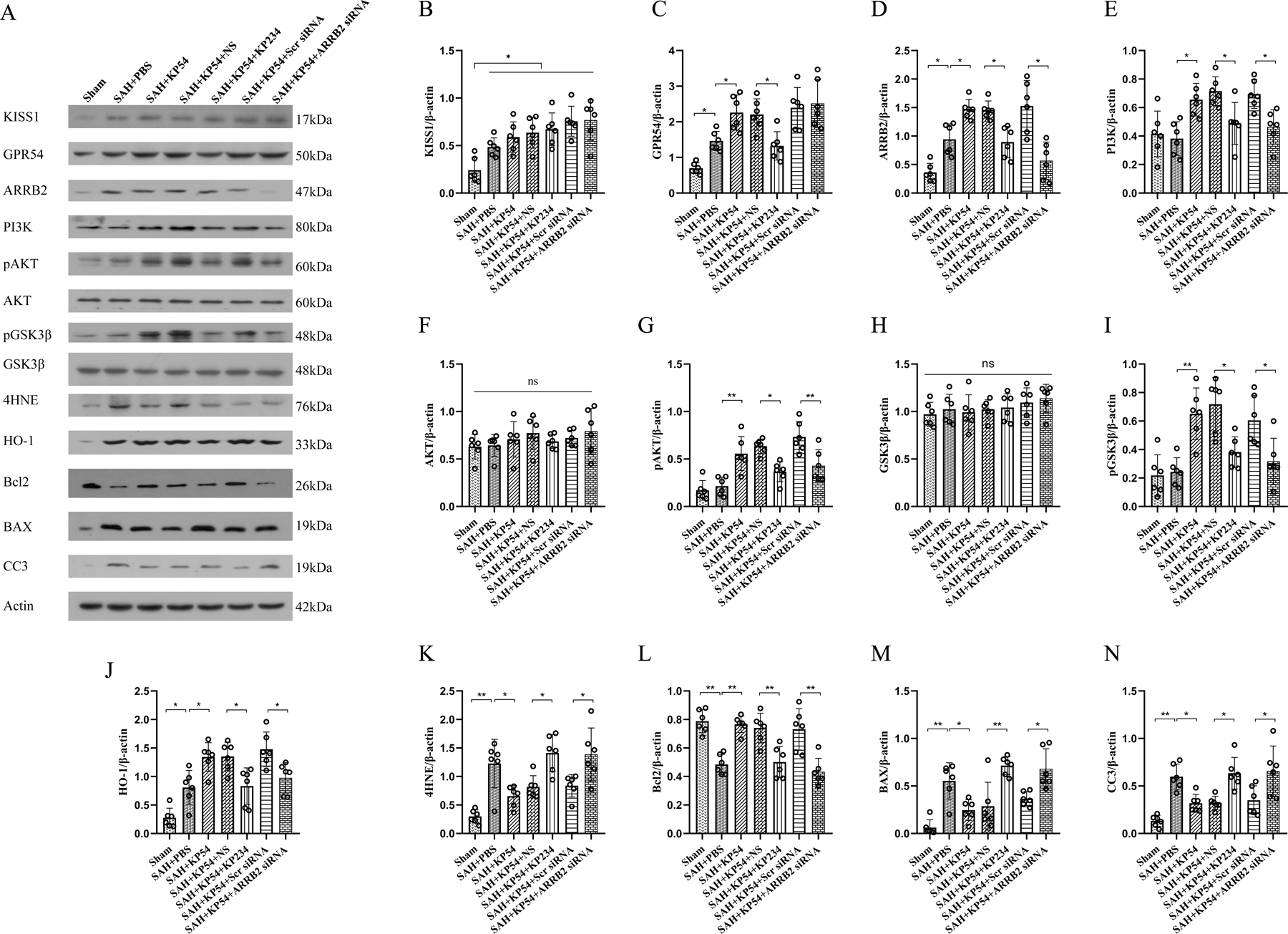
(A) Representative western blot bands; (B-N) Densitometric quantification of KISS1, GPR54, ARRB2, PI3K, pAKT, AKT, pGSK3β, GSK3β, 4HNE, HO-1, Bcl2, BAX, and CC3 in the ipsilateral brain hemisphere at 24 h after SAH. *P < 0.05, **P < 0.01. Data were represented as mean ± SD, n = 6 per group. One-way ANOVA-Tukey.
4.0. Discussion
In the present study, we explored the neuroprotective effects of KP54 against oxidative stress and neuronal apoptosis of EBI and the potential mechanisms involving the GPR54/ARRB2/AKT/GSK3β signaling pathway in a rat model of SAH (Fig. 9). The results showed: (1) the endogenous protein levels of KISS1, GPR54 and ARRB2 were increased and peaked within ipisilateral brain tissues at 24 h after SAH. GPR54 or ARRB2 was colocalized with neurons, astrocytes and microglia. (2) KISS1 siRNA knockdown aggravated neurological deficits and the brain expressions of markers for the oxidative stress and neuronal apoptosis in rats 24 h after SAH. (3) Intranasal administration of KP54 (1.0 nmol/kg) attenuated oxidative stress and neuronal apoptosis in SAH rats, and significantly improved the neurological deficits and neuronal degeneration in both 24 and 28 d after SAH. (4) Mechanistically, KP54 treatment increased the protein levels of GPR54, ARRB2, PI3K, pAKT, pGSK3β, HO1, Bcl2, and decreased the levels of 4HNE, BAX, CC3 in the ipsilateral hemisphere at 24 h after SAH. The GPR54 antagonism or the ARRB2 siRNA knockout partially abolished the beneficial effects of KP54 on neurological dysfunctions, the oxidative stress injury and neuronal apoptosis. Taken together, our results suggested that KP54 attenuated oxidative stress and neuronal apoptosis as well as the neurobehavioral deficits though activating GPR54/ARRB2/AKT/GSK3β pathway after SAH in rats.
Fig. 9.
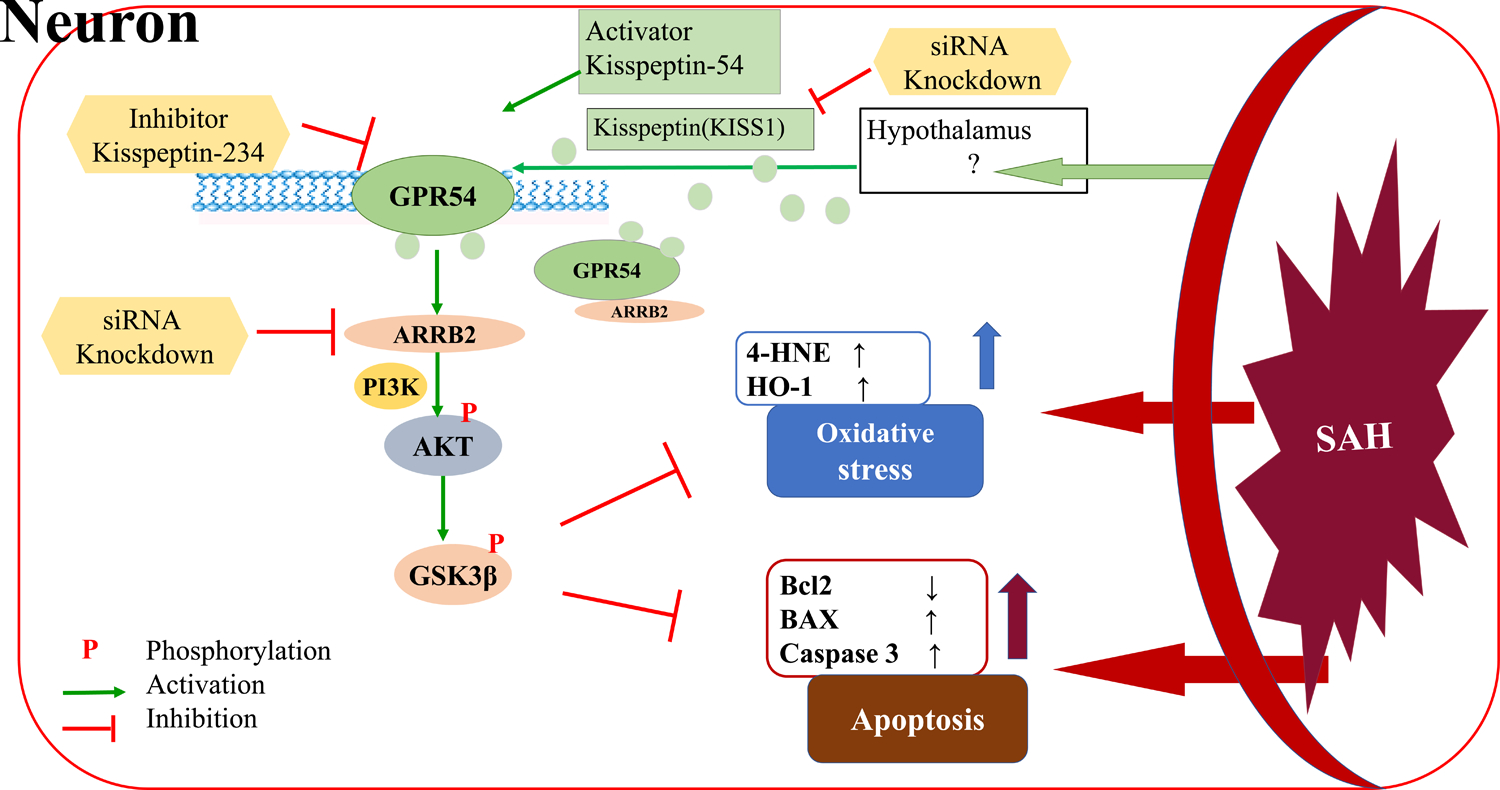
The graphic abstract. The KP54 attenuated oxidative stress and neuronal apoptosis via the activation of GPR54/ARRB2/PI3K/AKT/GSK3β signaling pathway after SAH.
KPs and the receptor GPR54 express in the hypothalamus, pituitary gland, hippocampus, cortex, and cardio vasculature [30, 31]. Exogenous KPs treatment reduced amyloid-β peptide-induced oxidative damage and neurotoxicity in Alzheimer’s disease [24]. ARRB2 protein level was upregulated and peaked at 24h after cerebral ischemia/reperfusion injury and ARRB2 deficiency exacerbated stroke outcomes [32]. In current study, our results showed that endogenous KISS1, GPR54 and ARRB2 were significantly increased at 24 h after SAH. The endogenous KISS1 knockdown by KISS1 siRNA aggravated neurological deficits at 24 h after SAH. Such detrimental effects were accompanying with the increased protein markers of oxidative stress and neuronal apoptosis in the ipsilateral brain tissues. Thus, the temporal elevation of KISS1/GPR54/ARRB2 protein expressions may be an endogenous neuroprotective mechanism upregulated after SAH. However, such upregulation was not sufficient to counteract the overall brain injury in SAH rats.
Previous studies showed that oxidative stress injury and neuron apoptosis were involved in the entire pathogenesis of EBI after SAH [33, 34]. After SAH, blood enters the subarachnoid space. The rapid degradation of hemoglobin enhances the oxidative reactions and inhibits antioxidant system, leading to the increases in a variety of reactive oxygen species, such as superoxide anions, hydroxyl radicals, hydrogen peroxide, etc.[33, 35]. As antioxidants, KPs treatment increased SOD activity and reduced ischemia-reperfusion injury in ovary and uterus [36]. It was also able to increase SOD levels in the liver tissue of young male rats [13]. Akkaya et al reported [14] that delayed KPs administration reduced neuronal cell death by upregulating SOD activity and decreasing malondialdehyde level after oxidative stress-induced brain injury in rats. In current study, our results for the first time, demonstrated that intranasal administration of KP54 at dose of 1.0 nmol/kg reduced oxidative stress injury and neuronal apoptosis in a GPR54 dependent manner at 24 h after SAH. In addition, KPs have been shown to regulate neuronal activity [37] and enhance brain responses to olfactory and visual cues of attraction in men [38]. The activation of KPs/GPR54 pathway mitigated amyloid-beta–induced memory impairment in mice [39, 40]. Similarly, we found that KP54 treatment significantly reduced short- and long-term neurologic deficits and improved the memory/ spatial learning after SAH.
The dose-dependent effects of KPs had been previously reported regarding endothelial cell migration and proliferation [41]. Low concentration increased but high concentration inhibited cell migration and proliferation [41, 42]. A high dose of KPs (50 nmol/day) could acutely induce testicular degeneration in adult male rats [43]. Other studies showed that a very high concentration (10 μmol) of KPs could promote vasoconstriction and induce atherosclerotic plaque progression and instability [44]. In this study, we selected three dosages based on the previous publication [23]. The doses of 1.0 nmol/kg improved the short-term and long-term neurological deficits after SAH, but the higher dose of 10.0 nmol/kg exacerbated neurological outcomes. The results suggest that the appropriate dosage is necessary to ensure the benefits of KP54 in treatment of SAH. KP54 is an FDA approved treatment for in vitro fertilization that is currently under clinical investigation. It would be feasible to apply KP54 for SAH patients, however, the optimal dose needs to be verified to avoid any side effects.
ARRB2, an adapter of GPR54, played an important role in the regulation of GPR54 downstream signaling [19]. Recent study suggested that GPR54 actively regulated the downstream extracellular regulated protein kinases pathways through an interdependent manner with ARRB2 [45]. Our results consistently demonstrated that there was a protein-protein interaction between GPR54 and ARRB2 in shans and SAH brains at 24 h after SAH, which was further enhanced by KP54 treatment. When GPR54 was antagonized by KP234, the brain expression of ARRB2 was reduced. Indeed, ARRB2 itself was suggested as a promising therapeutic target for stroke [46]. It mitigated cerebral ischemia/reperfusion injury [32] and protected HEK293 cells from oxidative stress-induced apoptosis [47]. In different models, GPR54 and ARRB2 both were shown to activate the PI3K/AKT signaling pathway [15, 20]. However, it remains unclear whether GPR54 regulated the downstream PI3K/AKT pathway through ARRB2. Although GSK3β might play dual roles in the process of nerve cell apoptosis [48]. Nevertheless, we found the GSK3β is the downstream protein of kisspeptin-mediated anti-apoptotic signaling pathway. The activation of PI3K/AKT/GSK3β signaling pathway was anti-oxidant in Alzheimer’s disease [49] and anti-apoptosis in SAH rats [21]. Our results indicated that KP54 treatment increased the brain expressions of GPR54, ARRB2, pAKT and pGSK3β, but reduced the brain expressions of oxidative stress-related proteins and neuronal apoptosis at 24h after SAH. Mechanistically, the pretreatment of GPR54 inhibitor KP234 or ARRB2 siRNA reversed the neurobehavioral benefits and anti-oxidative stress/ neuronal apoptosis effects of KP54 treatment. Immunoprecipitation assessments revealed that GPR54 interacted with ARRB2 in response to KP54 treatment and further activating the downstream PI3K/AKT/GSK3β signaling pathway. Collectively, our results suggested that KP54 attenuated oxidative stress and neuronal apoptosis through GPR54/ARRB2/AKT/GSK3β pathway after SAH in rat.
There were several limitations in this study. Firstly, the GPR54 and ARRB2 were also expressed in microglia and astrocyte cells. KPs/GPR54 signaling were associated with the resistance to matrix metalloproteinases 9 [50] and inflammation [51]. Therefore, we could not exclude the contribution of anti-inflammatory effects and blood-brain barrier protection of KP54 to the improved post-SAH neurological outcomes. Secondly, other downstream signaling pathway of GPR54/ARRB2 other than PI3K/GSK3β need further investigation in the setting of SAH. Thirdly, our study only evaluated the neuroprotective effects of KPs/GPR54 in male rats. Hypothalamic KPs/GPR54 activation could increase gonadotrophin secretion in mammals and estrogen has been shown to be protective against SAH [52]. Therefore, the effects of KPs/GRP54 signaling activation in female rats need to be further elucidated after SAH.
In conclusion, intranasal administration of KP54 attenuated oxidative stress and neuronal apoptosis through activating GPR54/ARRB2/AKT/GSK3β pathway after SAH in rat. Thus, KP54 may serve as a potential new treatment for SAH patients.
Highlights.
the endogenous protein levels of KISS1, GPR54 and ARRB2 were increased and peaked within ipsilateral brain tissues at 24 h after SAH.
KISS1 siRNA knockdown aggravated neurological deficits, oxidative stress and neuronal apoptosis in rats 24 h after SAH.
KP54 attenuated oxidative stress and neuronal apoptosis as well as the neurobehavioral deficits though activating GPR54/ARRB2/AKT/GSK3β pathway after SAH in rats.
Acknowledgments
This study was supported by the grants from the National Institutes of Health (NS081740 and NS082184) to John H. Zhang and the Ningbo Health Branding Subject Fund (PPXK2018–04) to Yi Huang.
Abbreviations:
- SAH
subarachnoid hemorrhage
- EBI
early brain injury
- siRNA
small interfering ribonucleic acid
- WB
western blot
- IF
Immunofluorescence staining
- KP54
kisspeptin 54
- GPR54
G protein-coupled receptor 54
- ARRB2
β-arrestin 2
- SOD
superoxide dismutase
- PI3K
phosphatidylinositol 3-kinase
- GSK3β
glycogen synthase kinase-3β
- IACUC
Institutional Animal Care and Use Committee
- PBS
phosphate buffered saline
- NS
normal saline
- SD
Sprague-Dawley
- i.c.v
intracerebroventricular
- 8-OHdG
8-hydroxydeoxyguanosine
- Mitosox
mitochondrial superoxide
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end labeling
- FJC
Fluoro-Jade C
- h
hour
- d
day
- ICA
internal carotid artery
- ECA
external carotid artery
- Co-IP
Co-immunoprecipitation
- KP234
kisspeptin 234
- NeuN
neuronal nuclei
- GFAP
glial fibrillary acidic protein
- Iba-1
calcium-binding adaptor molecule1
- CC3
cleaved caspase 3
- CA
cornu ammonis
- DG
dentate gyrus
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
The authors declare that they have no conflict of interest.
Reference
- [1].Neifert SN, Chapman EK, Martini ML, Shuman WH, Schupper AJ, Oermann EK, Mocco J, Macdonald RL, Aneurysmal Subarachnoid Hemorrhage: the Last Decade, Transl Stroke Res (2020). [DOI] [PubMed] [Google Scholar]
- [2].Toth G, Cerejo R, Intracranial aneurysms: Review of current science and management, Vasc Med 23(3) (2018) 276–288. [DOI] [PubMed] [Google Scholar]
- [3].Fujii M, Yan J, Rolland WB, Soejima Y, Caner B, Zhang JH, Early brain injury, an evolving frontier in subarachnoid hemorrhage research, Transl Stroke Res 4(4) (2013) 432–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kamp MA, Steiger HJ, van Lieshout JH, Experimental Aneurysmal Subarachnoid Hemorrhage: Tiding Over, Transl Stroke Res 11(1) (2020) 1–3. [DOI] [PubMed] [Google Scholar]
- [5].Fumoto T, Naraoka M, Katagai T, Li Y, Shimamura N, Ohkuma H, The Role of Oxidative Stress in Microvascular Disturbances after Experimental Subarachnoid Hemorrhage, Transl Stroke Res 10(6) (2019) 684–694. [DOI] [PubMed] [Google Scholar]
- [6].Mead EJ, Maguire JJ, Kuc RE, Davenport AP, Kisspeptins: a multifunctional peptide system with a role in reproduction, cancer and the cardiovascular system, Br J Pharmacol 151(8) (2007) 1143–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Pasquier J, Kamech N, Lafont AG, Vaudry H, Rousseau K, Dufour S, Molecular evolution of GPCRs: Kisspeptin/kisspeptin receptors, J Mol Endocrinol 52(3) (2014) T101–17. [DOI] [PubMed] [Google Scholar]
- [8].Kotani M, Detheux M, Vandenbogaerde A, Communi D, Vanderwinden JM, Le Poul E, Brezillon S, Tyldesley R, Suarez-Huerta N, Vandeput F, Blanpain C, Schiffmann SN, Vassart G, Parmentier M, The metastasis suppressor gene KiSS-1 encodes kisspeptins, the natural ligands of the orphan G protein-coupled receptor GPR54, J Biol Chem 276(37) (2001) 34631–6. [DOI] [PubMed] [Google Scholar]
- [9].Navarro VM, Metabolic regulation of kisspeptin - the link between energy balance and reproduction, Nat Rev Endocrinol 16(8) (2020) 407–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Murphy KG, Kisspeptins: regulators of metastasis and the hypothalamic-pituitary-gonadal axis, J Neuroendocrinol 17(8) (2005) 519–25. [DOI] [PubMed] [Google Scholar]
- [11].Abreu AP, Kaiser UB, Pubertal development and regulation, Lancet Diabetes Endocrinol 4(3) (2016) 254–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Dhillo WS, Chaudhri OB, Patterson M, Thompson EL, Murphy KG, Badman MK, McGowan BM, Amber V, Patel S, Ghatei MA, Bloom SR, Kisspeptin-54 stimulates the hypothalamic-pituitary gonadal axis in human males, J Clin Endocrinol Metab 90(12) (2005) 6609–15. [DOI] [PubMed] [Google Scholar]
- [13].Aydin M, Oktar S, Yonden Z, Ozturk OH, Yilmaz B, Direct and indirect effects of kisspeptin on liver oxidant and antioxidant systems in young male rats, Cell Biochem Funct 28(4) (2010) 293–9. [DOI] [PubMed] [Google Scholar]
- [14].Akkaya H, Kilic E, Dinc SE, Yilmaz B, Postacute effects of kisspeptin-10 on neuronal injury induced by L-methionine in rats, J Biochem Mol Toxicol 28(8) (2014) 373–7. [DOI] [PubMed] [Google Scholar]
- [15].Castano JP, Martinez-Fuentes AJ, Gutierrez-Pascual E, Vaudry H, Tena-Sempere M, Malagon MM, Intracellular signaling pathways activated by kisspeptins through GPR54: do multiple signals underlie function diversity?, Peptides 30(1) (2009) 10–5. [DOI] [PubMed] [Google Scholar]
- [16].Fu J, Sun H, Wei H, Dong M, Zhang Y, Xu W, Fang Y, Zhao J, Astaxanthin alleviates spinal cord ischemia-reperfusion injury via activation of PI3K/Akt/GSK-3beta pathway in rats, J Orthop Surg Res 15(1) (2020) 275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Arslan F, Lai RC, Smeets MB, Akeroyd L, Choo A, Aguor EN, Timmers L, van Rijen HV, Doevendans PA, Pasterkamp G, Lim SK, de Kleijn DP, Mesenchymal stem cell-derived exosomes increase ATP levels, decrease oxidative stress and activate PI3K/Akt pathway to enhance myocardial viability and prevent adverse remodeling after myocardial ischemia/reperfusion injury, Stem Cell Res 10(3) (2013) 301–12. [DOI] [PubMed] [Google Scholar]
- [18].Chen L, Xiang Y, Kong L, Zhang X, Sun B, Wei X, Liu H, Hydroxysafflor yellow A protects against cerebral ischemia-reperfusion injury by anti-apoptotic effect through PI3K/Akt/GSK3beta pathway in rat, Neurochem Res 38(11) (2013) 2268–75. [DOI] [PubMed] [Google Scholar]
- [19].Pampillo M, Camuso N, Taylor JE, Szereszewski JM, Ahow MR, Zajac M, Millar RP, Bhattacharya M, Babwah AV, Regulation of GPR54 signaling by GRK2 and {beta}-arrestin, Mol Endocrinol 23(12) (2009) 2060–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Sun X, Zhang Y, Wang J, Wei L, Li H, Hanley G, Zhao M, Li Y, Yin D, Beta-arrestin 2 modulates resveratrol-induced apoptosis and regulation of Akt/GSK3ss pathways, Biochim Biophys Acta 1800(9) (2010) 912–8. [DOI] [PubMed] [Google Scholar]
- [21].Ma J, Wang Z, Liu C, Shen H, Chen Z, Yin J, Zuo G, Duan X, Li H, Chen G, Pramipexole-Induced Hypothermia Reduces Early Brain Injury via PI3K/AKT/GSK3beta pathway in Subarachnoid Hemorrhage rats, Sci Rep 6 (2016) 23817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Duan J, Cui J, Yang Z, Guo C, Cao J, Xi M, Weng Y, Yin Y, Wang Y, Wei G, Qiao B, Wen A, Neuroprotective effect of Apelin 13 on ischemic stroke by activating AMPK/GSK-3beta/Nrf2 signaling, J Neuroinflammation 16(1) (2019) 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].d’Anglemont de Tassigny X, Jayasena CN, Murphy KG, Dhillo WS, Colledge WH, Mechanistic insights into the more potent effect of KP-54 compared to KP-10 in vivo, PLoS One 12(5) (2017) e0176821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Milton NG, Chilumuri A, Rocha-Ferreira E, Nercessian AN, Ashioti M, Kisspeptin prevention of amyloid-beta peptide neurotoxicity in vitro, ACS Chem Neurosci 3(9) (2012) 706–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Duris K, Manaenko A, Suzuki H, Rolland W, Tang J, Zhang JH, Sampling of CSF via the Cisterna Magna and Blood Collection via the Heart Affects Brain Water Content in a Rat SAH Model, Transl Stroke Res 2(2) (2011) 232–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Sugawara T, Ayer R, Jadhav V, Zhang JH, A new grading system evaluating bleeding scale in filament perforation subarachnoid hemorrhage rat model, J Neurosci Methods 167(2) (2008) 327–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zhan Y, Chen C, Suzuki H, Hu Q, Zhi X, Zhang JH, Hydrogen gas ameliorates oxidative stress in early brain injury after subarachnoid hemorrhage in rats, Crit Care Med 40(4) (2012) 1291–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Fang Y, Shi H, Ren R, Huang L, Okada T, Lenahan C, Gamdzyk M, Travis ZD, Lu Q, Tang L, Huang Y, Zhou K, Tang J, Zhang J, Zhang JH, Pituitary Adenylate Cyclase-Activating Polypeptide Attenuates Brain Edema by Protecting Blood-Brain Barrier and Glymphatic System After Subarachnoid Hemorrhage in Rats, Neurotherapeutics (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Okada T, Enkhjargal B, Travis ZD, Ocak U, Tang J, Suzuki H, Zhang JH, FGF-2 Attenuates Neuronal Apoptosis via FGFR3/PI3k/Akt Signaling Pathway After Subarachnoid Hemorrhage, Mol Neurobiol 56(12) (2019) 8203–8219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wolfe A, Hussain MA, The Emerging Role(s) for Kisspeptin in Metabolism in Mammals, Front Endocrinol (Lausanne) 9 (2018) 184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kirby HR, Maguire JJ, Colledge WH, Davenport AP, International Union of Basic and Clinical Pharmacology. LXXVII. Kisspeptin receptor nomenclature, distribution, and function, Pharmacol Rev 62(4) (2010) 565–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Chen L, Kong L, Wei X, Wang Y, Wang B, Zhang X, Sun J, Liu H, beta-arrestin 2 negatively regulates NOD2 signalling pathway through association with TRAF6 in microglia after cerebral ischaemia/reperfusion injury, J Cell Mol Med 23(5) (2019) 3325–3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ayer RE, Zhang JH, Oxidative stress in subarachnoid haemorrhage: significance in acute brain injury and vasospasm, Acta Neurochir Suppl 104 (2008) 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Gaetani P, Pasqualin A, Rodriguez y Baena R, Borasio E, Marzatico F, Oxidative stress in the human brain after subarachnoid hemorrhage, J Neurosurg 89(5) (1998) 748–54. [DOI] [PubMed] [Google Scholar]
- [35].Yang Y, Chen S, Zhang JM, The Updated Role of Oxidative Stress in Subarachnoid Hemorrhage, Curr Drug Deliv 14(6) (2017) 832–842. [DOI] [PubMed] [Google Scholar]
- [36].Aslan M, Erkanli Senturk G, Akkaya H, Sahin S, Yilmaz B, The effect of oxytocin and Kisspeptin-10 in ovary and uterus of ischemia-reperfusion injured rats, Taiwan J Obstet Gynecol 56(4) (2017) 456–462. [DOI] [PubMed] [Google Scholar]
- [37].Liu X, Herbison AE, Kisspeptin Regulation of Neuronal Activity throughout the Central Nervous System, Endocrinol Metab (Seoul) 31(2) (2016) 193–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Yang L, Demetriou L, Wall MB, Mills EG, Zargaran D, Sykes M, Prague JK, Abbara A, Owen BM, Bassett PA, Rabiner EA, Comninos AN, Dhillo WS, Kisspeptin enhances brain responses to olfactory and visual cues of attraction in men, JCI Insight 5(3) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Jiang JH, He Z, Peng YL, Jin WD, Wang Z, Han RW, Chang M, Wang R, Kisspeptin-13 enhances memory and mitigates memory impairment induced by Abeta1–42 in mice novel object and object location recognition tasks, Neurobiol Learn Mem 123 (2015) 187–95. [DOI] [PubMed] [Google Scholar]
- [40].Ebrahimi Khonacha S, Janahmadi M, Motamedi F, Kisspeptin-13 Improves Spatial Memory Consolidation and Retrieval against Amyloid-beta Pathology, Iran J Pharm Res 18(Suppl1) (2019) 169–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Golzar F, Javanmard SH, The effects of kisspeptin-10 on migration and proliferation of endothelial cell, Adv Biomed Res 4 (2015) 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Olbrich T, Ziegler E, Turk G, Schubert A, Emons G, Grundker C, Kisspeptin-10 inhibits bone-directed migration of GPR54-positive breast cancer cells: Evidence for a dose-window effect, Gynecol Oncol 119(3) (2010) 571–8. [DOI] [PubMed] [Google Scholar]
- [43].Thompson EL, Amber V, Stamp GW, Patterson M, Curtis AE, Cooke JH, Appleby GF, Dhillo WS, Ghatei MA, Bloom SR, Murphy KG, Kisspeptin-54 at high doses acutely induces testicular degeneration in adult male rats via central mechanisms, Br J Pharmacol 156(4) (2009) 609–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Sato K, Shirai R, Hontani M, Shinooka R, Hasegawa A, Kichise T, Yamashita T, Yoshizawa H, Watanabe R, Matsuyama TA, Ishibashi-Ueda H, Koba S, Kobayashi Y, Hirano T, Watanabe T, Potent Vasoconstrictor Kisspeptin-10 Induces Atherosclerotic Plaque Progression and Instability: Reversal by its Receptor GPR54 Antagonist, J Am Heart Assoc 6(4) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Szereszewski JM, Pampillo M, Ahow MR, Offermanns S, Bhattacharya M, Babwah AV, GPR54 regulates ERK1/2 activity and hypothalamic gene expression in a Galpha(q/11) and beta-arrestin-dependent manner, PLoS One 5(9) (2010) e12964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Wang H, Deng QW, Peng AN, Xing FL, Zuo L, Li S, Gu ZT, Yan FL, beta-arrestin2 functions as a key regulator in the sympathetic-triggered immunodepression after stroke, J Neuroinflammation 15(1) (2018) 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Zhang Z, Hao J, Zhao Z, Ben P, Fang F, Shi L, Gao Y, Liu J, Wen C, Luo L, Yin Z, beta-Arrestins facilitate ubiquitin-dependent degradation of apoptosis signal-regulating kinase 1 (ASK1) and attenuate H2O2-induced apoptosis, Cell Signal 21(7) (2009) 1195–206. [DOI] [PubMed] [Google Scholar]
- [48].Jacobs KM, Bhave SR, Ferraro DJ, Jaboin JJ, Hallahan DE, Thotala D, GSK-3beta: A Bifunctional Role in Cell Death Pathways, Int J Cell Biol 2012 (2012) 930710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Ali T, Kim T, Rehman SU, Khan MS, Amin FU, Khan M, Ikram M, Kim MO, Natural Dietary Supplementation of Anthocyanins via PI3K/Akt/Nrf2/HO-1 Pathways Mitigate Oxidative Stress, Neurodegeneration, and Memory Impairment in a Mouse Model of Alzheimer’s Disease, Mol Neurobiol 55(7) (2018) 6076–6093. [DOI] [PubMed] [Google Scholar]
- [50].Tomita K, Oishi S, Ohno H, Peiper SC, Fujii N, Development of novel G-protein-coupled receptor 54 agonists with resistance to degradation by matrix metalloproteinase, J Med Chem 51(23) (2008) 7645–9. [DOI] [PubMed] [Google Scholar]
- [51].Mi WL, Mao-Ying QL, Liu Q, Wang XW, Li X, Wang YQ, Wu GC, The distribution of kisspeptin and its receptor GPR54 in rat dorsal root ganglion and up-regulation of its expression after CFA injection, Brain Res Bull 78(4–5) (2009) 254–60. [DOI] [PubMed] [Google Scholar]
- [52].Tabuchi S, Relationship between Postmenopausal Estrogen Deficiency and Aneurysmal Subarachnoid Hemorrhage, Behav Neurol 2015 (2015) 720141. [DOI] [PMC free article] [PubMed] [Google Scholar]