

Abstract
Proton transfer reactions (PTR) have emerged as a powerful tool for the study of intact proteins. When coupled with m/z-selective kinetic excitation, such as parallel ion parking (PIP), one can exert exquisite control over rates of reaction with a high degree of specificity. This allows one to “concentrate”, in the gas-phase, nearly all the signal from an intact protein charge state envelope into a single charge state improving signal-to-noise by 10x or more. While this approach has been previously reported, here we show that implementing these technologies on a 21 T FT-ICR MS provides tremendous advantage for intact protein analysis. Advanced strategies for performing PTR with PIP were developed to complement this unique instrument, including subjecting all analyte ions entering the mass spectrometer to PTR and PIP. This experiment, which we call “PTR-MS1-PIP”, generates a pseudo-MS1 spectrum derived from ions that are exposed to PTR reagent and PIP waveforms, but have not undergone any prior true mass filtering or ion isolation. The result is an extremely rapid and significant improvement in spectral signal-to-noise (S:N) of intact proteins. This permits observation of many more proteoforms and reduces ion injection periods for subsequent tandem mass spectrometry characterization. Additionally, the product ion parking waveform has been optimized to enhance the proton transfer reaction rate without compromise to parking efficiency. We demonstrate that this process, called “rapid park”, can improve reaction rates by 5–10x and explore critical factors discovered to influence this process. Finally, we demonstrate how coupling PTR-MS1 and rapid park provides a ten-fold reduction in ion injection time, improving the rate of tandem MS sequencing.
Graphical Abstract
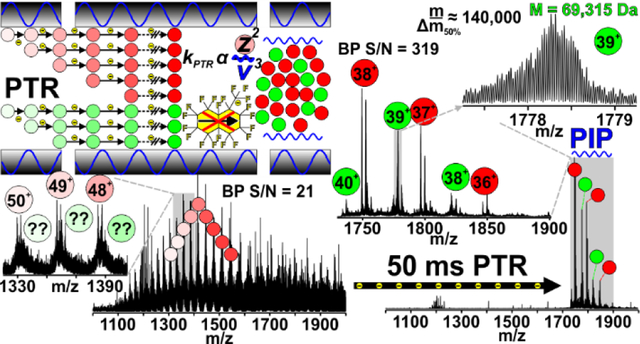
Gas-phase reactions have been reliable analytical tools for mass spectrometrists through the ages[1–3]. These reactions occur ubiquitously during ionization in the source region (APCI[4], APPI, ESI[5], etc) and while ions traverse/reside in the various hardware elements of all mass spectrometry (MS) systems. An example of the latter includes abstraction of protons by residual water molecules in the vacuum chamber. Often these reactions are hardly noticeable due to their intrinsically slow rates, or they are deliberately minimized to protect instrument performance. However, ion-neutral and ion-ion reactions can be harnessed deliberately within the framework of any mass spectrometer where ions have the proper residence time and are sufficiently mixed to permit reaction, including ion cyclotron resonance (ICR) cells (Penning trap)[6], radio-frequency (RF) multipole storage devices (Paul trap)[7, 8], or even beam-type devices[9]. During storage, reagents are introduced to promote formation of desired gas-phase product ion(s). For example, ion-ion reactions such as electron transfer dissociation (ETD)[7] or proton transfer reactions (PTR)[10] involve reactions with reagent anions to produce charge-reduced product ions. Many pioneers have demonstrated that these reactions provide tremendous analytical value[11], especially for protein sequencing. This has led to increased commercial availability of implementations of ion-ion reaction capabilities[12–14].
Intact or “top-down” protein analysis has been a long-term science driver for the field of analytical MS because it addresses the shortcomings of bottom-up protein analysis[15]. In principle, top-down can provide direct insight into biologically relevant proteoforms[16] including identification via accurate mass and primary amino acid structure determination via tandem MS sequencing. Identification and site-localization of post-translational modifications[17] (PTMs), and stoichiometric interrogation of native protein complexes[18] have been demonstrated. Quantification is also achievable on the proteoform level with top-down MS approaches[19]. However, analysis of intact proteins is far more challenging than enzymatically produced peptide fragments[20]. Part of the reason for this difficulty is that ion signal is divided among isotopologues, charge states, proteoforms, adducts, etc.[21, 22] Division in ion signal reduces observed dynamic range and increases the ion accumulation period required for obtaining high quality tandem MS spectra.
PTR involving multiply charged precursor ions generated by ESI of proteins was first introduced by Stephenson and McLuckey as a means of separating mixtures of overlapping charge state distributions prior to low-resolution quadrupole ion trap mass analysis[10, 23]. While extensive charge reduction can greatly simplify intact and product ion spectra[24–27] of proteins and lessen the requirement for high resolving power, eventually products of PTR are charge-neutralized, or fall outside the available m/z-range of the mass analyzer. This spurred development of ion parking[28] and parallel ion parking[29] (PIP) to m/z-selectively halt ion-ion reaction kinetics. PTR coupled with PIP simplifies spectra by concentrating ion signal into fewer channels, thereby simultaneously increasing signal-to-noise (S:N).
The past two decades have seen seismic shifts in the capabilities of Fourier transform mass spectrometry (FTMS) instrumentation that have, in turn, catalyzed advances in top-down proteomics. Robust implementations of PTR, which was commercially released by Thermo Fisher Scientific in 2019, and PIP on today’s FTMS instrumentation could overcome many of the challenges associated with top-down proteomics[19, 30]. In 2019, Ugrin et. al. demonstrated coupling PTR with PIP on a modified Orbitrap Elite™ for identification of intact E. coli ribosomal proteins[31]. In the same year Huguet et. al. demonstrated targeted use of PTR for improved detection and intact mass determination of large (>30 kDa) proteins derived from P. aeruginosa[14]. More recently Kline et. al. utilized PTR to reduce congestion in MS2 and MS3 spectra of large intact proteins[32]. These reports illustrate how appropriate use of gas-phase ion chemistry and careful ion manipulation can dramatically improve intact protein analysis.
Here we report implementation of PTR and PIP on a custom 21 tesla (T) Fourier transform ion cyclotron resonance (FT-ICR) MS instrument[33]. In experiments employing multiple activation stages, we show how performing ETD followed by PTR alleviates “extreme” spectral congestion to improve sequence coverage of a 50 kDa protein (protein A/G) compared to ETD alone. Advanced strategies for PTR coupled with PIP that best exploit the unique instrument configuration of the 21 T FT-ICR MS system are described. This involved development of pseudo-MS1 methodology, in which all ions electrosprayed into the mass spectrometer are exposed to PTR reagent (with no prior isolation; PTR-MS1) and subjected to PIP. This approach, which we term, “PTR-MS1-PIP”, achieves high S:N improvement through concentration of all observed protein charge, and eliminates unnecessary ion-handling/manipulation overhead, saving appreciable analysis time. In proof-of-principle experiments with apomyoglobin, PTR-MS1-PIP elicited a 10-fold gain in S:N. Single- transient acquisitions yielded isotopic resolution of protein A/G (50.5 kDa) and exo-Klenow fragment (68 kDa) in magnitude mode, and exhibited a >10-fold improvement in S:N over the most abundant charge state observed in normal MS1 acquisitions. In an LC-MS analysis of MCF-7 (H. sapiens) cell lysate proteins 75% more proteoforms were observed (2472 w/ PTR-MS1-PIP vs. 1404 Normal MS1), along with an increase in the average molecular weight (MW) of the proteins detected, and significant gains in the number of >30 kDa proteoforms observed.
In efforts to optimize the PIP waveform, we discovered that, under certain conditions, the PTR rate could be accelerated dramatically (~5–10x) over previously reported efforts[31]. This effect, which we refer to as “rapid park”, saves valuable analysis time without compromise to parking efficiency. Additionally, improvement in S:N directly translates to a reduction in precursor ion injection period for subsequent tandem MS. Ion injection periods represent a significant fraction of the total analysis time. Also, when ion injection periods are long or “maxed out” poor proteoform identification rates result. PTR-MS1-PIP followed by tandem MS with CID yielded 10x lower ion injection periods for apomyoglobin and equivalent or better sequence coverage across a variety of charge states compared with normal MS1 followed by CID tandem MS.
Experimental Section:
Sample Preparation
Pierce™ Intact Protein Standard Mix, and recombinant protein A/G were purchased from Thermo Fisher Scientific (Waltham, MA). Equine myoglobin was purchased from Sigma Aldrich (St. Louis, MO). These were reconstituted in water and aliquots of myoglobin and protein A/G were diluted to a final concentration of 1 μM in a solution containing 50/50 methanol and 0.1% acetic acid (v/v) for direct infusion. An aliquot of the standard mix was diluted with solvent A (see Liquid Chromatography) to a final concentration of 20 ng/μL for LC-MS.
Pellets of 1×107 MCF-7 cells were thawed on ice and suspended in ten volumes of lysis buffer (4% SDS, 100 mM Tris pH 7.5, 10 mM DTT, 10 mM sodium butyrate and 1X Thermo Halt Protease and Phosphatase Inhibitor Cocktail). Cell pellets were lysed by heating at 95 °C for 10 minutes, vortexing every 2 minutes. Cellular debris was removed by centrifugation at 20,000 × g for 20 minutes. Acetone protein precipitation was performed on the supernatant and the resulting protein pellet was suspended in 150 μL of 1 % SDS for quantification via bicinchoninic acid (BCA) assay. Size-based separation of approximately 400 μg of each sample into 12 fractions was performed using a GELFrEE 8100 fractionation station with a 10% GELFrEE cartridge (Expedeon) following the manufacturer’s recommended procedure. Methanol-chloroform precipitation[34] was performed on each fraction to remove SDS, and each pellet was reconstituted in 35 μL of solvent A (0.3% formic acid, and 5% acetonitrile in water with % expressed as v/v). Fractions 3 and 4, containing primarily 10–30 kDa proteins, were subjected to LC-tandem MS as outlined below.
Instrumentation
All data presented herein was collected on an in-house constructed 21 T FT-ICR MS system. This hybrid dual-cell linear RF ion trap 21 T FT-ICR MS system has been previously described[33]. The linear RF ion trap is a modified form of a Velos Pro[35] (Thermo Fisher Scientific, San Jose, CA). The Velos Pro is equipped with a commercial Orbitrap™ Fusion[36] API inlet/front-end ETD (FETD) reagent ion source (Thermo Fisher Scientific). The glow discharge FETD source enables simultaneous generation of reagent ions for both ETD and PTR ion-ion reactions[8]. An external multipole storage device (MSD)[37], situated between the velos dual linear RF ion trap (ITMS) and the ICR cell (FTMS) allows for multiple ion fills to be collected prior to FTMS acquisition. This device contains an axial field for sequestration of ions from one end of the MSD to the other. Ions are delivered via a controlled ejection process with an auxillary RF pseudopotential as described by Kaiser et al.[38]
The FETD source was further modified to introduce perfluoromethyldecalin (PFMD, m/z 512) reagent anions, and the ion trap control language (ITCL) modified to include the ability to perform PTR as described by Ugrin et. al[24]. Significant additional code development was required to enable both PTR-MS1 and rapid ion parking. To enable PTR-MS1 a single stage of PTR activation was added to the scan matrix for both the normal and AGC matrices. All activation parameters necessary for this step were stored temporarily in the MSn matrix variables responsible for the first stage of MSn (see Figures S1–S4 for long-term stability of this operation mode). Rapid ion parking was enabled by developing a new algorithm to build custom auxiliary waveforms directly within the ITCL using existing hardware with physical modification to optics or circuit boards.
Liquid Chromatography
For each injection, 2–4 μL sample aliquots were loaded onto an in-house-fabricated 360 μm o.d. × 150 μm i.d. fused-silica microcapillary trap column packed 2.5 cm with PLRP-S resin (5 μm particle, 1000 Å pore, Agilent Technologies, Palo Alto, CA). The nano-HPLC system (ACQUITY M-Class, Waters, Milford, MA) was operated at 2.5 μL/min for loading/trapping, and the sample was washed with 95% A for 10 min. Separation was achieved on an in-house-fabricated 360 μm o.d. × 75 μm i.d. fused-silica microcapillary analytical column packed with PLRP-S to 17.5 cm length. For intact protein standards, samples were gradient eluted at 0.3 μL/min with a solvent composition of 5–35% B in 5 min, 35–55% B in 30 min, 55–75% B in 5 min, 75% B wash step for 5 min, and a 5% B re-equilibration step in 15 min (60 min total length). For MCF7 cell lysates, the gradient utilized a solvent composition of 5–15 %B in 5 min, 15–55 %B in 80 min, 55–75 %B in 5 min, and a 5 %B re-equilibration step in 15 min (105 min total length). Following separation, samples were directly ionized by microelectrospray ionization using a 15 μm fused-silica PicoTip (New Objective, Woburn, MA) emitter packed 3 mm with PLRP-S resin. A UWPR nanospray source was utilized for application of the ionization voltage, fixturing the column, and providing fine adjustment for the ESI emitter (http://proteomicsresource.washington.edu/protocols05/nsisource.php).
Mass Spectrometry
For all experiments, the source voltage was biased at 2.5 kV, and the heated capillary temperature was 325 °C. Spectra were collected in reduced profile mode (with the exception of protein A/G data which utilized full profile mode) with the ICR mass analyzer. Direct infusion of myoglobin and protein A/G was performed at 0.5 μL/min with a laser-pulled (Sutter P-2000, Novato, CA) fused silica capillary emitter tip. Myoglobin, and protein A/G spectra were taken as the sum of 10, and 50 transients, respectively.
For LC-MS experiments, the instrument was operated with Xcalibur software (Thermo Fisher Scientific). For standards, 50 ng total protein were injected, and “normal” (denaturing-ESI) MS1 spectral acquisition was toggled with PTR-MS1-PIP spectral acquisition. For MCF7 experiments, 4 μL of each reconstituted fraction (F3 and 4) were injected twice; once to acquire only normal MS1 spectra; once to acquire only PTR-MS1-PIP spectra.
All LC-MS spectra were acquired from 600–2500 m/z, with 5E5 MS1 AGC target, and were taken as a single transient for standards, and as the sum of 4 transients for MCF7 samples. Transient durations were 1.524 s and 0.762 s (corresponding to 1.2 M and 600 k resolving power at m/z 200) for standards and MCF7 samples, respectively. The 21 T FT-ICR system at NHMFL routinely produces results with resolving powers at ~100% the Nyquist limit for a transient processed via FFT in magnitude mode. For PTR-MS1-PIP experiments, a PIP waveform was applied to the x-rods of the high-pressure cell of the dual linear ion trap during PTR. The waveform included frequency components corresponding to m/z 1750–2750, with normalized activation amplitude of 0.11. The waveform also included a reagent activation window (centered at m/z 512, 20 Th window), where the amplitude was lowered to 0.02[31]. The Mathieu q of the reagent was set to 0.55. PTR reagent AGC was 5E5, and the entire precursor ion population (no isolation) was exposed to reagent for 150 ms.
Data Handling
All data were processed in .raw file format (Thermo Fisher Scientific) in reduced profile mode (noise baseline subtracted). For standards, results were obtained by visual inspection of the raw data with Xcalibur™ Qual Browser software (Thermo Fisher Scientific). Additionally, to determine sequence coverage, spectra were deconvolved with the QualBrowser-embedded Xtract algorithm and fragment ions matched to sequences using ProSight Lite[39] with a product ion mass tolerance of +/− 10 ppm. All protein A/G fragments were manually validated or assigned.
For MCF7, raw files were m/z-to-mass deconvolved with Thermo Protein Deconvolution 4.0 software (Thermo Fisher Scientific) to obtain a count of observed proteoform masses. Xtract parameters were set as follows: 70% fit factor, minimum S/N 2, 10% remainder threshold, 2 minimum detected charge states, and charge range of +5 to +50. A sliding window of 0.5 minutes and 50% offset was used to deconvolute the retention time range of 0 – 110 minutes. Deconvolved masses were further aggregated with Proteoform Suite version 0.3.4 (https://github.com/smith-chem-wisc/ProteoformSuite/releases) utilizing merging mass tolerances of 3 ppm, 1 min retention time tolerance, and allowing for misassignment of monoisotopic masses by up to two isotopologues.
Results and Discussion:
The development of PTR was well ahead of the technology required to take full advantage analytically. Since then, PTR has been employed to alleviate issues associated with extreme spectral congestion[14, 24, 40, 41] in top-down proteomics by reducing overlap of isotopic peak clusters. For example, ETD is highly effective for intact protein sequence analysis[42], however, resultant fragment ions are often clustered around the precursor within a narrow m/z-range. Application of an additional stage of activation by PTR (ETD-PTR) serves to distribute ETD-product ions more evenly throughout the usable m/z-range of the instrument[43]. Figure 1 shows ETD tandem MS spectra of the [M+50H]50+ charge state of recombinant protein A/G (50.4 kDa) without (Fig. 1A) and with an additional PTR activation stage (Fig. 1B). These spectra were generated from the same number of precursor ions (5E6 cumulative ion target). The insets illustrate the qualitative difference in spectral complexity over a 100 m/z domain. Manually validated fragments matched to the sequence with ProSight Lite following automated spectral deconvolution with the Xtract algorithm yielded 12% sequence coverage with ETD-only vs. 39% from the ETD-PTR spectrum (See Figure S5 for fragment ion maps). Additionally, PTR provides dramatic S:N improvements that originate from the dependence of the rate of charge reduction on precursor charge state. Higher charged fragments are preferentially charge-reduced, and fragment ion current is collapsed into fewer individual charge states as the reaction progresses. This also simplifies the resulting mass spectrum for more straight forward data interpretation (either manually or algorithmically).
FIGURE 1.
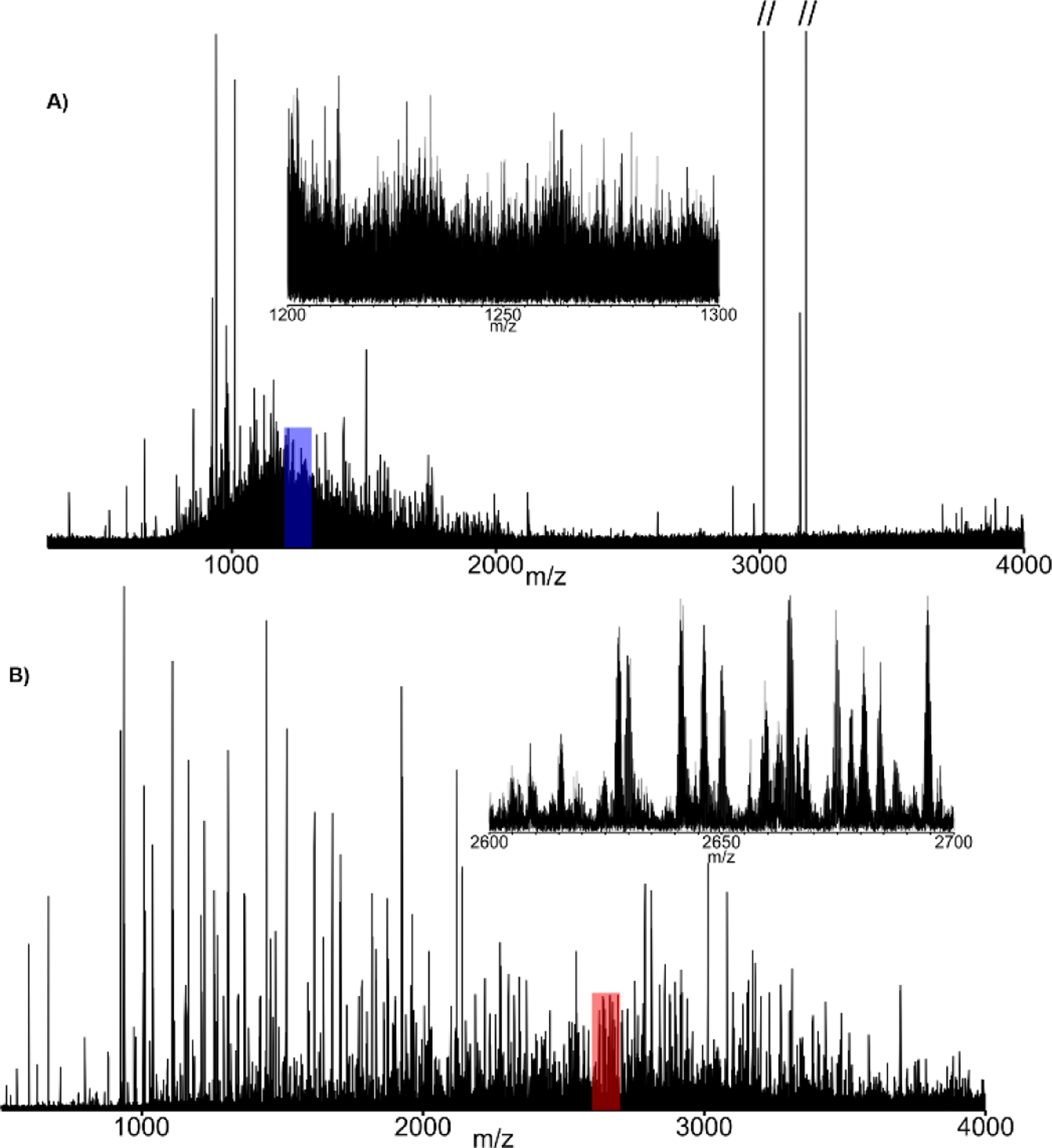
Tandem MS spectra of protein AG [M+50H]50+ (m/z 1010.2) utilizing both ETD alone (A) and ETD followed by a PTR (B). These results illustrate the importance of relieving “spectral congestion”, even with ultra-high resolution FT-ICR MS (see Figure S1 for identified fragment ion maps).
By pairing PTR with parallel ion parking (PTR-PIP), ion signal that is normally dispersed across a wide m/z domain in the form of a protein charge envelope can be concentrated into nearly a single protein charge state. Implementation of PTR-PIP for intact mass analysis as described by Ugrin et al.[31] employed wide (400 Th) linear IT precursor isolation prior to PTR-PIP. While this approach is quite successful, precursor isolation limits S:N improvement, as well as the ability to quantitatively interrogate PTR-PIP data, as more charge states of some protein precursors may be isolated and subjected to PTR-PIP than others. Additionally, ion isolation prior to PTR-PIP requires valuable analysis time. The overhead associated with the unnecessary steps is magnified when multiple fills are performed prior to mass analysis to build ion population, improve S:N, or compensate for reduced ITMS space charge capacity. Executing the steps required to accumulate and isolate these large m/z regions requires ~30–50 ms, but this time must be multiplied by the number of ion fills employed to minimize ITMS space charge effects, and improve S:N. This can result in hundreds of milliseconds worth of unnecessary overhead per spectrum. Here we demonstrate an approach called PTR-MS1 in which all ions entering the mass spectrometer are exposed to the PTR reagent without any pre-processing/manipulation. The resulting spectrum can be thought of as a “pseudo-MS1” spectrum. While this approach is entirely decoupled from the application of PIP, it is most effective when PTR-MS1 is coupled with PIP (PTR-MS1-PIP). In Figure 2, an example is given where normal MS1 and PTR-MS1-PIP conditions are used to analyze an intact protein standard mixture by LC-MS. The spectra were acquired during the apex of the elution from each of two high-MW recombinant proteins: protein A/G (50 kDa) and exo-Klenow fragment (68 kDa). Base peak S:N was improved by a factor of ~13x for protein A/G, and by ~18x for exo-Klenow fragment. Insets within each mass spectrum provide an enhanced view of each isotopically resolved envelope used to calculate S:N. These data are the result of single transient acquisitions in magnitude mode which represents a significant milestone in FTMS technology. While high MW isotopic resolution has been achieved with both ICR and Orbitrap technology, extensive transient averaging is required on the order of 100’s to 1000’s of summed transients to achieve a similar result. The S:N improvement imparted by PTR-MS1-PIP, including the ability to isotopically resolve high MW proteins in single transients, provides a major benefit to LC-MS analysis of intact proteins.
FIGURE 2.
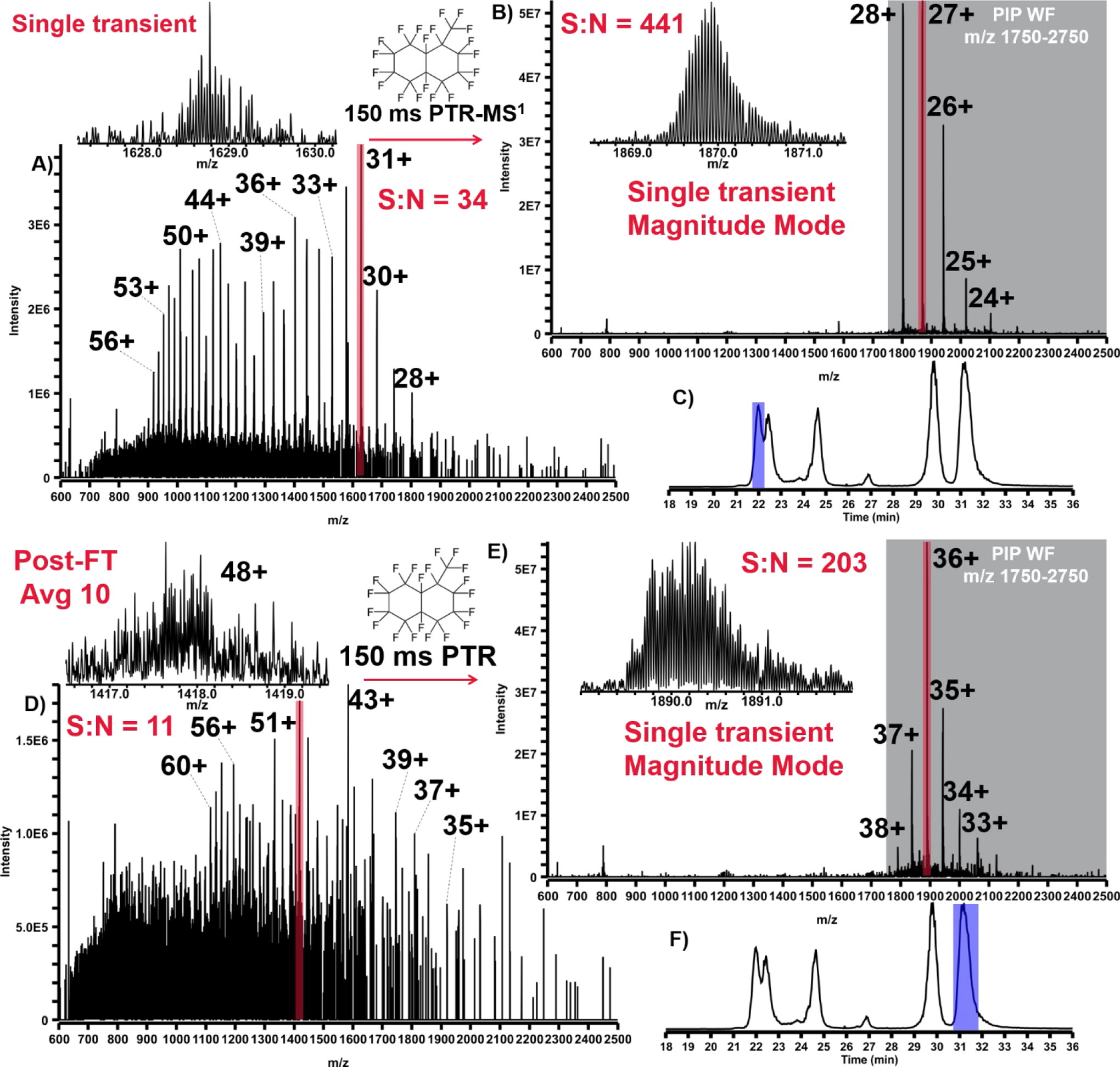
LC-MS analysis of intact protein standard mixture with spectra taken from the apex of elution profiles for both protein AG (50 kDa) and exo-Klenow fragment (68 kDa). A) Spectrum of protein AG under normal MS1 conditions with the most abundant charge state highlighted in red. B) Spectrum of protein AG under PTR-MS1-PIP conditions with the most abundant charge state highlighted in red and the range of m/z intended for PIP shaded in grey. C) LC-MS total-ion chromatogram for the analysis with the region over which protein AG elutes highlighted in blue. Figures 2D, E, & F the same respectively for the exo-Klenow fragment.
PTR-MS1-PIP technology was used to analyze proteins recovered from H. sapiens MCF7 (breast cancer) cell lysate, prefractionated by GELFrEE[44]. Due to sample availability limitations, our analysis focused on fractions 3 and 4. We often observed many more proteoforms of a given protein from these samples using PTR-MS1-PIP (by sometimes 3x or more) compared to normal MS1. Figure 3 presents one such example in which a proteoform family isotopic peak envelope is detected under both normal MS1 (Fig. 3A) and PTR-MS1-PIP (Fig. 3B) conditions. Using the QualBrowser embedded Xtract algorithm (Fig. 3C&D), 5 co-eluting proteoforms were accurately deconvolved from the normal MS1 spectrum vs. 15 co-eluting proteoforms for PTR-MS1-PIP spectrum. For the two samples analyzed, the total number of proteoforms detected following automated peak deconvolution and chromatographic clustering[45] improved by >%75 (Figure 4) through the use of PTR-MS1-PIP (1404 vs. 2472 proteoforms; normal MS1 vs. PTR-MS1-PIP). Other important aspects of the analysis that improved through the use of PTR-MS1-PIP include the average MW of the proteoforms detected (15.6 kDa vs. 20.4 kDa; normal MS1 vs. PTR-MS1-PIP) and the number of proteoforms >30 kDa (2 vs. 133; normal MS1 vs. PTR-MS1-PIP). Thousands of proteoforms were detectable throughout the duration of the analytical gradient, which indicates co-elution of proteoforms occurs ubiquitously throughout the HPLC seperation. We attribute the improvement in detected number of proteoforms to reflect performance improvement from PTR-MS1-PIP especially for co-eluting protein species. The next logical step is to interrogate these species using tandem MS for sequence information and PTM localization, however, at the time of these experiments tandem MS based upon PTR-MS1-PIP precursor ion acquisitions required further development of instrument control software.
FIGURE 3.
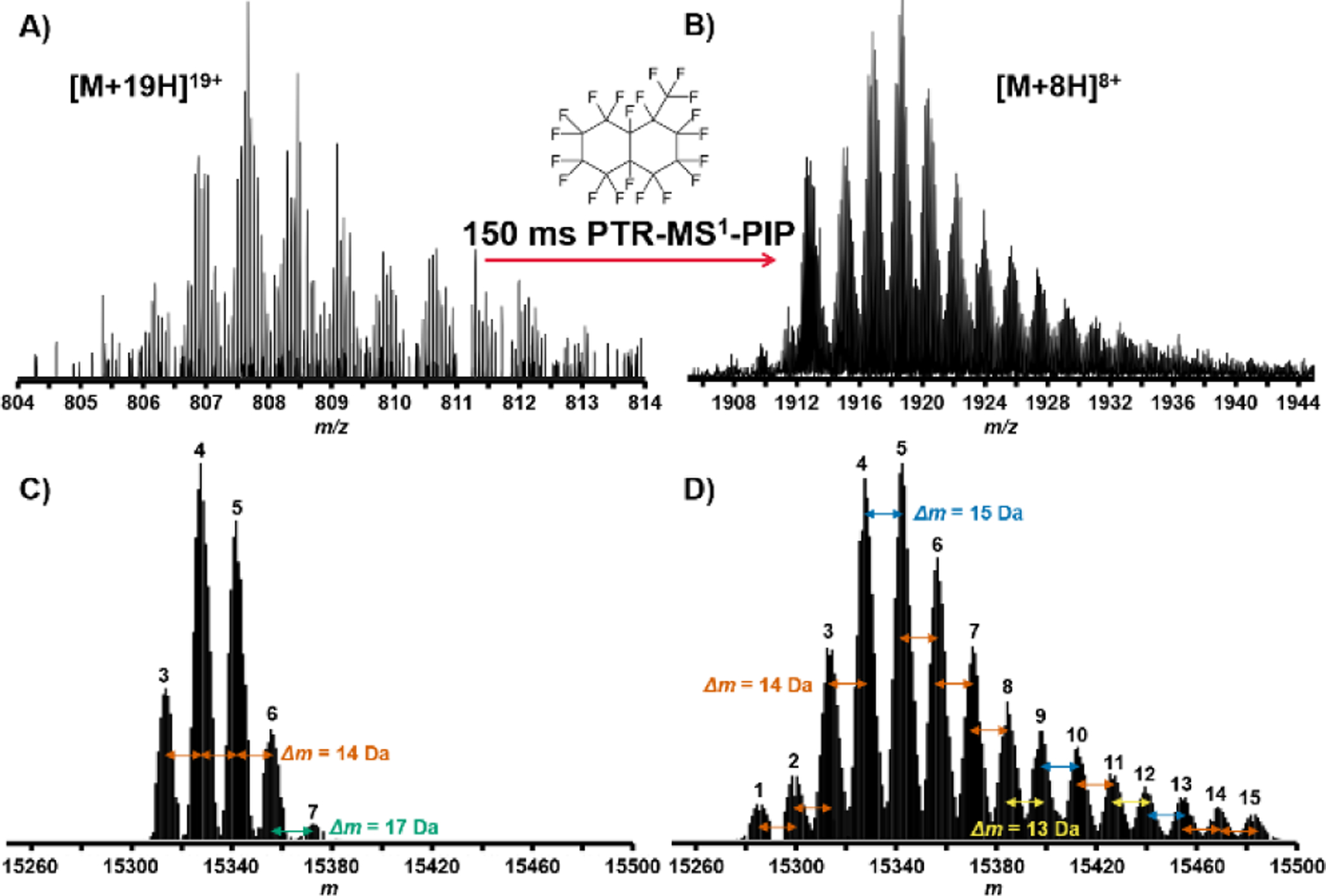
LC-MS analysis of whole cell lysate proteins from H. Sapiens MCF7 cells. A) Region of mass spectrum taken with normal MS1 conditions containing isotopic peak clusters for a protein exhibiting a “family” of associated proteoforms. B) The same region measured under PTR-MS1-PIP conditions shows a more defined set of associated proteoforms from improved S:N. C) Deconvolution of the peak clusters shown in the normal MS1 spectrum yielded 5 proteoforms. D) Deconvolution of the respective peak clusters of the PTR-MS1-PIP spectrum yielded 15 proteoforms. Xtract-determined monoisotopic mass differences (Δm, Da) among proteoforms are indicated.
FIGURE 4.
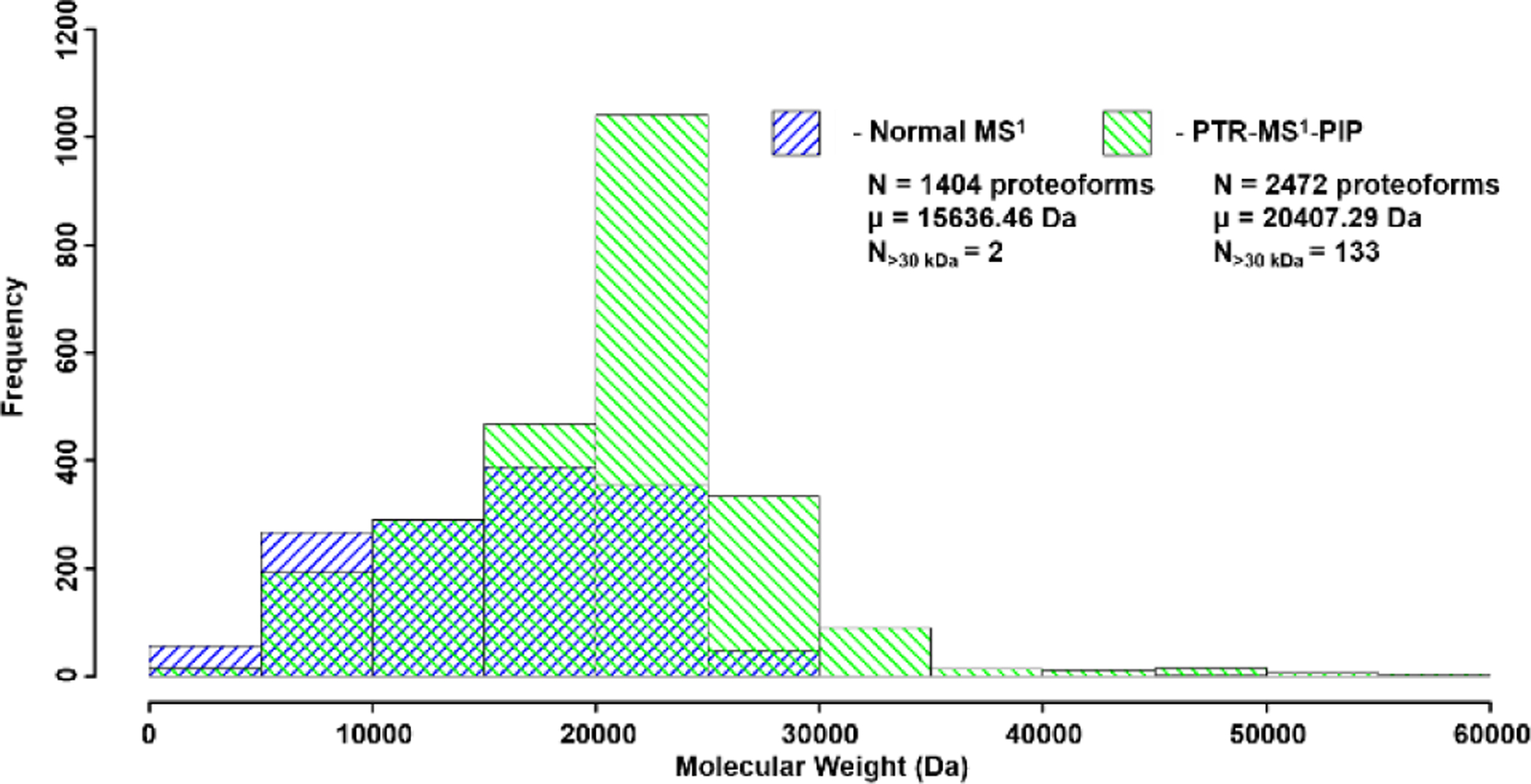
Histogram of MW for all detected proteoforms from the LC-MS analysis of GELFrEE fractions 3 and 4 of whole cell lysate proteins derived from H. sapiens MCF7 cells. PTR-MS1-PIP improved the total number of detected proteoforms, especially those of larger MW.
The PIP waveforms utilized thus far in this study mirrored the waveform frequency and amplitude composition utilized by Ugrin et. al[31], which demonstrated that kinetic excitation of the reagent anion, in addition to desired charge-reduced precursor ions, enabled far more efficient ion parking in the ITMS, albeit at a reduced reaction rate. Here, parking efficiency is defined by the following relationship:
| Equation(1) |
The abundance (A) of the target charge state divided by the abundance of all observed charge states following PTR-PIP. An unfortunate side effect of PTR is that precursor charge is consumed through each successive generation of proton transfer. For example, if an [M+21H]21+ precursor were isolated and subjected to PTR-PIP, with the [M+14H]14+ being the desired charge-reduced product, 1/3 of the total charge will be consumed. This presents an upper bound for the yield of the reaction products to be 2/3 the total starting charge (or abundance) prior to PTR. This consumption of charge is unfortunate because the charge capacity of the ion trap cannot be fully exercised, which limits S:N improvement potential when performing PTR-PIP. However, even with this limitation, 10x gain in S:N over normal MS1 data is often obtainable with our implementation on the 21 T FT-ICR MS at NHMFL (see Figure 2).
Here, under these conditions, high parking efficiency was achieved when computed with Eqn. 1, however, the PTR rate is relatively slow (k = ~50 sec−1). With this PIP waveform and reaction rate, measured with apomyoglobin as the analyte, 120 ms is required to convert a purely isolated [M+21H]21+ to the [M+14H]14+ with ~94% parking efficiency (see Figures S6A & S7A). While this experiment was contrived to study reaction rates and to monitor product ion conversion for each successive reaction generation, it represents a lower limit for overall reaction rates possible during PTR-MS1-PIP type experiments. Taken alone, 120 ms is a relatively short period of time. However, if multiple ion fills are employed to maximize S:N in the FTMS and minimize space charge effects during ion manipulations within the ITMS (including PTR and ETD), the required PTR duration can strain the chromatographic-compatibility of the analysis. For example, if 10 ion fills are used with this PIP waveform, 1.2 sec. duration has been spent simply charge-reducing and parking ions. This represents a significant fraction of total analysis time and fundamentally limits the total number of acquisitions possible per unit time. LC-MS experiments under both targeted or discovery approaches benefit from higher data acquisition repetition rates. Therefore, optimization of the PIP waveform was undertaken to ensure the fastest possible PTR kinetics, and highest spectral acquisition rates possible when PTR-MS1-PIP is utilized at 21 T.
During experiments in which the amplitude of the frequency components responsible for parking the reagent ion and apomyoglobin product ions were optimized, rapid ion parking was discovered. When keeping the product ion parking amplitude constant while iteratively increasing the amplitude utilized for resonant excitation of the reagent anion, the PTR reaction rate decreased until a local minimum in reaction rate was achieved. This minimum also represents the point at which maximum parking efficiency is achieved and is consistent with observations by Ugrin et. al. When the resonant excitation amplitude was increased, collision-activated dissociation of the reagent anion ensues to a small degree, with the predominant reagent fragment ion exhibiting loss of F (seeFigure S8). The radical electron is preserved and the fragment is still a capable PTR reagent. However, the loss of F places this species (m/z 493) outside the frequency range of the PIP waveform for the PTR reagent (20 Th centered at m/z 512), allowing radial excursion of this reactive species to relax back to the thermalized steady state near r=0 over the course of ~1–2 ms[31] period (see Figure S9). This relaxation in radius and velocity of the reagent anion fragments promotes a concomitant increase in the rate of PTR (see Eqn. 2).
| Equation (2) |
McLuckey and Stephenson derived Eqn. 2 to explain dependencies observed during ion-ion reactions, where k is the rate constant, ν is the magnitude of the differential velocity between reacting species, z is the integer charge state of the reactants, e is the elementary charge, and μ is the two bodied reduced mass of the reactants[46]. The rate is inversely proportional to the cube of the differential velocity of the reacting ion clouds. Therefore, when the PFMD reagent anion is activated and undergoes loss of F, it is no longer being driven by the PIP WF and its effective rate of charge reduction is accelerated relative to intact reagent anion. Thus, the overall rate constant for the process becomes a linear combination of the k for the original reagent anion and k’ for the reagent anion fragment. While the process that leads to rapid ion parking is not yet fully understood, we hypothesize that under the appropriate reagent anion activation conditions, this reactive anion fragment is generated in “just-the-right” abundance to increase the overall PTR rate without excessive charge-reduction that would impact parking efficiency (see Figure S6B & S7B). The amplitude of the frequency components used for kinetic excitation of the reagent anion must be carefully controlled to induce this condition (see Figure S10). The onset and rate at which these PTR-active reagent fragment ions are formed from the greater population of reagent anions has been found to be critically important (see Figure S11). When amplitudes are increased further beyond that which induces rapid ion parking, resultant spectra resemble those in which anion excitation was not employed (i.e. parking efficiency is dramatically reduced). Further investigation into the mechanism which enables rapid ion parking is ongoing, however, this initial report is meant to describe this phenomenon and its impact on performance. Additionally, rapid ion parking was successfully implemented on a second FT-ICR instrument operating at 14.5 T with nearly identical front-end hardware. If rapid ion parking proves to be enabled by progressively altering the reactivity of the reagent anion, it is expected that this process can also be initiated by developing a PIP WF with a time-dependent reagent amplitude component. In this mode, one could modulate the kinetic excitation of the reagent anion slowly, during the PTR period, to maintain PTR rate and parking efficiency.
In addition to increasing PTR kinetics (see Figure S12) without compromise to parking efficiency, the principal performance impact of rapid ion parking is evident from the following proof-of-principle experiments. In Figure 5A & B, a normal spectrum of apomyoglobin without PTR-MS1 is shown and can be directly compared against a spectrum in which PTR-MS1-PIP with rapid ion parking WF was enabled. In this experiment, only 20 ms PTR reaction was required to achieve a 10x boost in S:N (vs. 120 ms without rapid ion parking). The total additional time required to conduct PTR-MS1-PIP with rapid ion parking is 70 ms; this overhead time includes a discrete AGC ITMS acquisition, in order to inform the correct number of charges for the subsequent FTMS acquisition (AGC target of 1×106 charges). When these ion populations are subject to tandem MS characterization with CID, the primary advantage of rapid ion parking is realized in precursor ion accumulation time. Figure 5A & C shows a single charge state, [M+13H]13+, of apomyoglobin isolated (shown in red) and subjected to CID under normal MS1 conditions. Here we performed multiple fragment ion fills where each of four fills of the MSD was derived from 2 × 105 precursor charges, requiring 171.07 ms to accumulate per fill (total ion injection time = 684.28 ms). With PTR-MS1-PIP and rapid ion parking prior to precursor isolation, one can pre-concentrate nearly all of the denaturing-ESI charge state distribution into the desired charge state targeted for tandem MS characterization. In doing so, a dramatic reduction in the ion injection period is observed. In Figure 5B & D, the same charge state ([M+13H]13+) of apomyoglobin has been interrogated by CID tandem MS, however, prior to ion isolation, PTR-MS1-PIP with rapid ion parking was performed. The ion injection times for the same AGC target used in Fig. 5C were decreased by >10x (5.09 ms per ion fill or 20.36 ms total ion injection period). The tandem MS spectra shown in Fig. 5C & D have nearly indistinguishable features including S:N and dynamic range. When these spectra are mined for sequence informative b/y fragment ions with ProSight Lite, nearly identical sequence coverage is obtained (36% Normal MS1 vs. 39% PTR-MS1-PIP with rapid ion parking).
FIGURE 5.
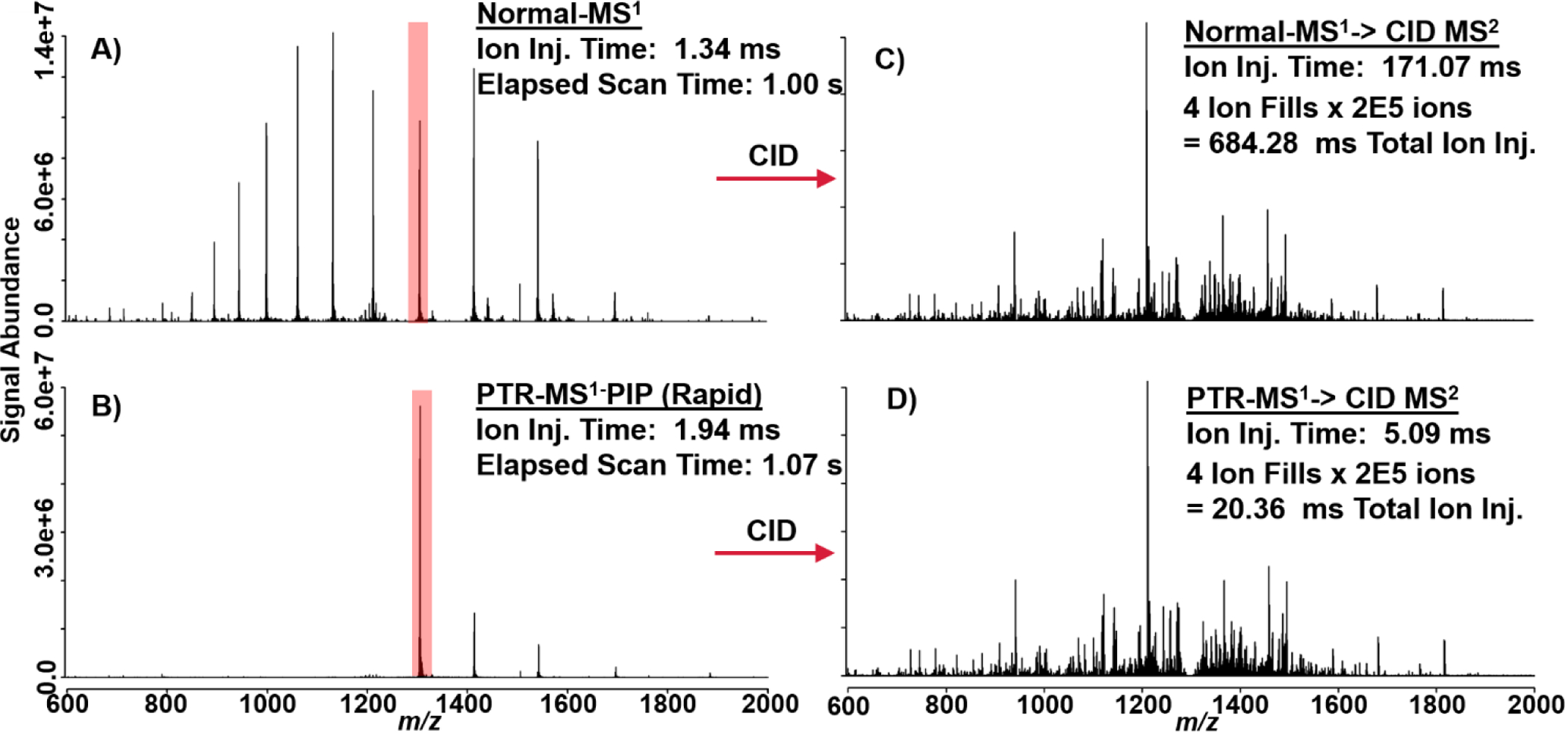
These spectra illustrate proof-of-principal behind the combination of PTR-MS1, PIP, and rapid ion parking prior to tandem MS analysis. A) Apomyoglobin mass spectrum under normal MS1 and B) PTR-MS1-PIP with rapid ion parking. The [M+13H]13+ is highlighted in red to indicate its isolation and tandem MS by CID. C) CID spectrum generated from isolation of the [M+13H]13+ under normal MS1 conditions and D) PTR-MS1-PIP with rapid ion parking prior to ion isolation and CID. CID tandem MS spectra are nearly indistinguishable, were generated from the same number of charges (AGC ion target), but precursor ion accumulation times were far lower for the PTR-MS1-PIP condition.
The reduction in ion injection period afforded by PTR-MS1-PIP with rapid ion parking dramatically improves the tandem MS duty cycle. In Figure 6, charge states 14–9+ were interrogated with CID tandem MS with and without PTR-MS1-PIP, and ion injection periods and sequence coverage from the resultant spectra were compared. From these data, a 10-fold reduction in the ion injection period was observed for all charge states (Fig. 6A). Sequence coverage for each charge state was similar or better with the PTR-MS1-PIP rapid ion parking approach, especially for the lower charge states studied (Fig. 6B). This is likely the beneficial effect of having converted all the higher charge states of apomyoglobin into the charge state of interest through PTR-MS1-PIP. We hypothesize that residual gas-phase structure is retained from their previous charge state, or randomized refolding following the loss of charge[47] provides access to a unique set of fragments not attainable through CID of precursors of the same charge states produced by denaturing-ESI alone.
FIGURE 6.
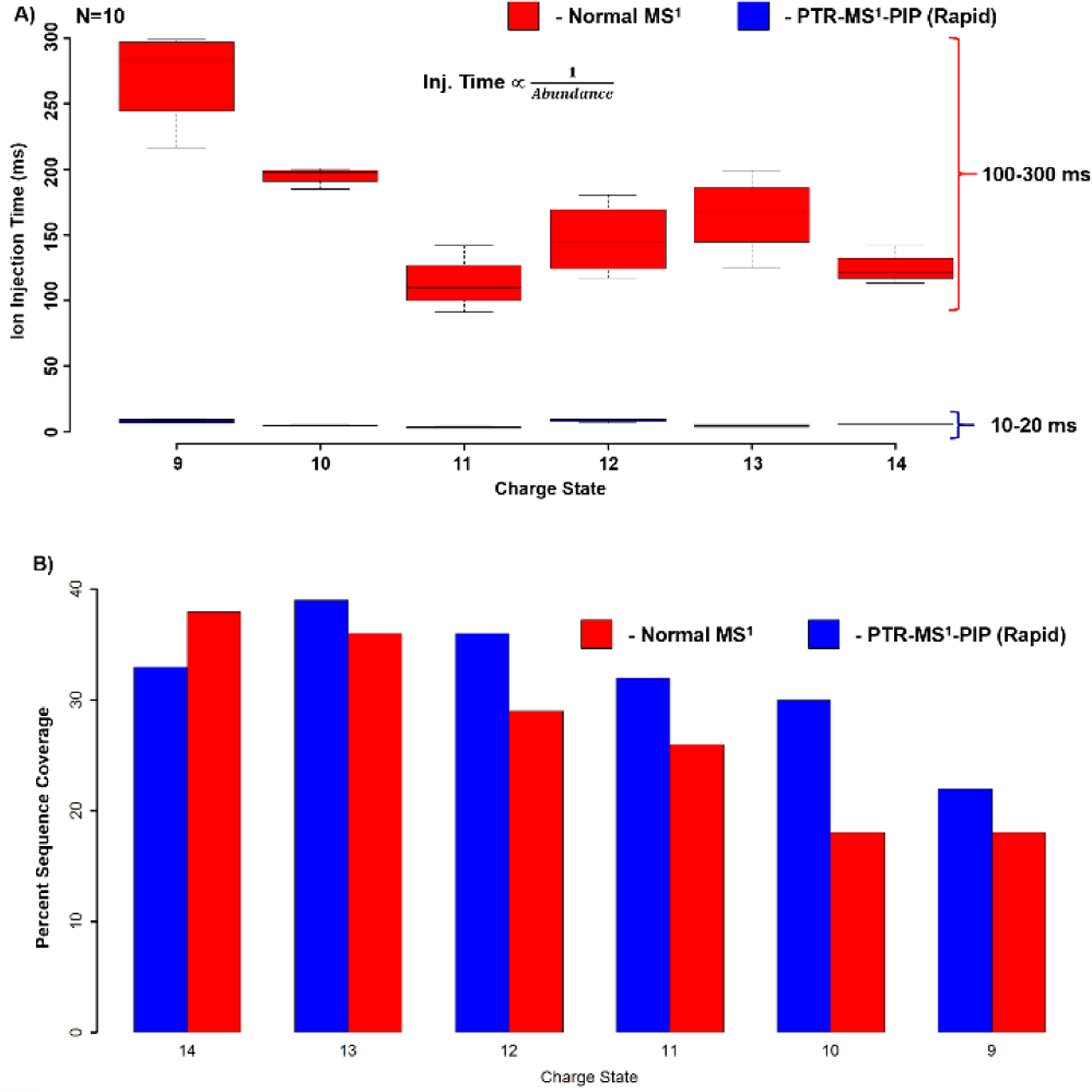
A) Box- and- whisker plot of ion injection periods required for tandem MS isolation and fragmentation of various apomyoglobin charge states under normal MS1 and PTR-MS1-PIP with rapid ion parking conditions (N=10). Median injection period is represented by the line through each box, box edges represent the upper and lower quantile, and the whiskers represent the most extreme datapoints. B) Bar plot indicating the sequence coverage observed from automated fragment ion assignment using ProSight Lite.
Conclusions:
The pairing of PTR-PIP technology with 21 T FT-ICR MS provides significant analytical advantage to intact protein analysis. PTR-MS1-PIP technology provides over 10-fold improvement in S:N compared with conventional precursor acquisitions. The greatest benefit is realized with high MW proteins, especially when their signal is spread among a vast number of PTM’s and charge states. Many more proteoforms were observed in MCF7 cell analysis compared with conventional precursor ion acquisition. Rapid ion parking improves the PTR rates such that use of PTR-MS1-PIP imparts negligible overhead in the total acquisition time (~70 ms for apomyoglobin). The observed 10-fold gain in S:N translates to 10-fold reduction in ion injection periods for apomyoglobin, while maintaining tandem MS sequence coverage. These proof-of-principle experiments reveal a promising avenue toward improvement of both targeted and discovery-based analysis of intact proteins. Further development is required for incorporation of these approaches into automated data-dependent acquisition workflows. These technological developments will undoubtedly enable deeper top-down proteome coverage by enabling detection of lower-abundance proteoforms, and more rapid tandem MS sequencing for greater proteoform identification rates. In principle, PTR-MS1, PIP, and rapid ion parking could be implemented on existing commercial instruments for similar performance improvement.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Thermo Fisher Scientific Research and Development (San Jose, CA) for providing reagent ion source hardware. The Hunt laboratory gratefully acknowledges National Institutes of Health grant GM-037537. We gratefully acknowledge support from the National Science Foundation Division of Chemistry through DMR-1157490, DMR-1644779, and the State of Florida.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
PTR_PIP_21T_Supplemental_verFinal.pdf has been included with a set of additional figures to support the main text.
All raw data and metadata associated with this manuscript can be found at DOI: 10.17605/OSF.IO/TAQN7.
REFERENCES
- 1.Brodbelt JS Analytical applications of ion-molecule reactions. Mass Spectrom.Rev 1997, 16, 91–110. [Google Scholar]
- 2.Green MK; Lebrilla CB Ion-molecule reactions as probes of gas-phase structures of peptides and proteins. Mass Spectrom.Rev 1997, 16, 53–71. [DOI] [PubMed] [Google Scholar]
- 3.Prentice BM; McLuckey SA Gas-phase ion/ion reactions of peptides and proteins: acid/base, redox, and covalent chemistries. Chem.Commun.(Camb) 2013, 49, 947–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carroll DI; Dzidic I; Stillwell RN; Horning MG; Horning EC Subpicogram detection system for gas phase analysis based upon atmospheric pressure ionization (API) mass spectrometry. Anal.Chem 1974, 46, 706–710. [Google Scholar]
- 5.Whitehouse CM; Dreyer RN; Yamashita M; Fenn JB Electrospray interface for liquid chromatographs and mass spectrometers. Anal.Chem 1985, 57, 675–679. [DOI] [PubMed] [Google Scholar]
- 6.Nibbering NMM Gas-phase ion/molecule reactions as studied by Fourier transform ion cyclotron resonance. Acc.Chem.Res 1990, 23, 279–285. [Google Scholar]
- 7.Syka JE; Coon JJ; Schroeder MJ; Shabanowitz J; Hunt DF Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc.Natl.Acad.Sci.U.S.A 2004, 101, 9528–9533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Earley L; Anderson LC; Bai DL; Mullen C; Syka JE; English AM; Dunyach JJ; Stafford GC; Shabanowitz J; Hunt DF; Compton PD Front-end electron transfer dissociation: a new ionization source. Anal.Chem 2013, 85, 8385–8390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Williams JP; Brown JM; Campuzano I; Sadler PJ Identifying drug metallation sites on peptides using electron transfer dissociation (ETD), collision induced dissociation (CID) and ion mobility-mass spectrometry (IM-MS). Chem.Commun 2010, 46, 5458–5460. [DOI] [PubMed] [Google Scholar]
- 10.Stephenson JL; McLuckey SA Ion/Ion Reactions in the Gas Phase: Proton Transfer Reactions Involving Multiply-Charged Proteins. J.Am.Chem.Soc 1996, 118, 7390–7397. [Google Scholar]
- 11.Riley NM; Coon JJ The Role of Electron Transfer Dissociation in Modern Proteomics. Anal.Chem 2018, 90, 40–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McAlister GC; Phanstiel D; Good DM; Berggren WT; Coon JJ Implementation of Electron-Transfer Dissociation on a Hybrid Linear Ion Trap–Orbitrap Mass Spectrometer. Anal.Chem 2007, 79, 3525–3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaplan DA; Hartmer R; Speir JP; Stoermer C; Gumerov D; Easterling ML; Brekenfeld A; Kim T; Laukien F; Park MA Electron transfer dissociation in the hexapole collision cell of a hybrid quadrupole-hexapole Fourier transform ion cyclotron resonance mass spectrometer. Rapid Commun.Mass Spectrom 2008, 22, 271–278. [DOI] [PubMed] [Google Scholar]
- 14.Huguet R; Mullen C; Srzentic K; Greer JB; Fellers RT; Zabrouskov V; Syka JEP; Kelleher NL; Fornelli L Proton Transfer Charge Reduction Enables High-Throughput Top-Down Analysis of Large Proteoforms. Anal.Chem 2019, 91, 15732–15739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chait BT Mass Spectrometry: Bottom-Up or Top-Down? Science 2006, 314, 65. [DOI] [PubMed] [Google Scholar]
- 16.Smith LM; Kelleher NL; Linial M; Goodlett D; Langridge-Smith P; Ah Goo Y; Safford G; Bonilla* L; Kruppa G; Zubarev R; Rontree J; Chamot-Rooke J; Garavelli J; Heck A; Loo J; Penque D; Hornshaw M; Hendrickson C; Pasa-Tolic L; Borchers C; Chan D; Young* N; Agar J; Masselon C; Gross* M; McLafferty F; Tsybin Y; Ge Y; Sanders* I; Langridge J; Whitelegge* J; Marshall A; The Consortium for Top, Down Proteomics. Proteoform: a single term describing protein complexity. Nature Methods 2013, 10, 186–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Siuti N; Kelleher NL Decoding protein modifications using top-down mass spectrometry. Nature methods 2007, 4, 817–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Skinner OS; Haverland NA; Fornelli L; Melani RD; Do Vale LHF; Seckler HS; Doubleday PF; Schachner LF; Srzentić K; Kelleher NL; Compton PD Top-down characterization of endogenous protein complexes with native proteomics. Nature chemical biology 2018, 14, 36–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schaffer LV; Millikin RJ; Miller RM; Anderson LC; Fellers RT; Ge Y; Kelleher NL; LeDuc RD; Liu X; Payne SH; Sun L; Thomas PM; Tucholski T; Wang Z; Wu S; Wu Z; Yu D; Shortreed MR; Smith LM Identification and Quantification of Proteoforms by Mass Spectrometry. Proteomics 2019, 19, 1800361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aebersold R; Mann M Mass spectrometry-based proteomics. Nature 2003, 422, 198–207. [DOI] [PubMed] [Google Scholar]
- 21.Riley NM; Mullen C; Weisbrod CR; Sharma S; Senko MW; Zabrouskov V; Westphall MS; Syka JE; Coon JJ Enhanced Dissociation of Intact Proteins with High Capacity Electron Transfer Dissociation. J.Am.Soc.Mass Spectrom 2016, 27, 520–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Compton PD; Kelleher NL; Gunawardena J Estimating the Distribution of Protein Post-Translational Modification States by Mass Spectrometry. J.Proteome Res 2018, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stephenson JL; McLuckey SA Ion/ion proton transfer reactions for protein mixture analysis. Anal.Chem 1996, 68, 4026–4032. [DOI] [PubMed] [Google Scholar]
- 24.Stephenson JL; McLuckey SA Simplification of product ion spectra derived from multiply charged parent ions via ion/ion chemistry. Anal.Chem 1998, 70, 3533–3544. [DOI] [PubMed] [Google Scholar]
- 25.Coon JJ; Ueberheide B; Syka JE; Dryhurst DD; Ausio J; Shabanowitz J; Hunt DF Protein identification using sequential ion/ion reactions and tandem mass spectrometry. Proc.Natl.Acad.Sci.U.S.A 2005, 102, 9463–9468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Holden DD; McGee WM; Brodbelt JS Integration of Ultraviolet Photodissociation with Proton Transfer Reactions and Ion Parking for Analysis of Intact Proteins. Anal.Chem 2016, 88, 1008–1016. [DOI] [PubMed] [Google Scholar]
- 27.Sanders JD; Mullen C; Watts E; Holden DD; Syka JEP; Schwartz JC; Brodbelt JS Enhanced Sequence Coverage of Large Proteins by Combining Ultraviolet Photodissociation with Proton Transfer Reactions. Anal.Chem 2020, 92, 1041–1049. [DOI] [PubMed] [Google Scholar]
- 28.McLuckey SA; Reid GE; Wells JM Ion parking during ion/ion reactions in electrodynamic ion traps. Anal.Chem 2002, 74, 336–346. [DOI] [PubMed] [Google Scholar]
- 29.Chrisman PA; Pitteri SJ; McLuckey SA Parallel ion parking of protein mixtures. Anal.Chem 2006, 78, 310–316. [DOI] [PubMed] [Google Scholar]
- 30.Chen B; Brown KA; Lin Z; Ge Y Top-Down Proteomics: Ready for Prime Time? Anal.Chem 2018, 90, 110–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ugrin SA; English AM; Syka JEP; Bai DL; Anderson LC; Shabanowitz J; Hunt DF Ion-Ion Proton Transfer and Parallel Ion Parking for the Analysis of Mixtures of Intact Proteins on a Modified Orbitrap Mass Analyzer. J.Am.Soc.Mass Spectrom 2019, 30, 2163–2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kline JT; Mullen C; Durbin KR; Oates RN; Huguet R; Syka JEP; Fornelli L Sequential Ion-Ion Reactions for Enhanced Gas-Phase Sequencing of Large Intact Proteins in a Tribrid Orbitrap Mass Spectrometer. J.Am.Soc.Mass Spectrom 2021, [DOI] [PubMed] [Google Scholar]
- 33.Hendrickson CL; Quinn JP; Kaiser NK; Smith DF; Blakney GT; Chen T; Marshall AG; Weisbrod CR; Beu SC 21 Tesla Fourier Transform Ion Cyclotron Resonance Mass Spectrometer: A National Resource for Ultrahigh Resolution Mass Analysis. J.Am.Soc.Mass Spectrom 2015, 26, 1626–1632. [DOI] [PubMed] [Google Scholar]
- 34.Wessel D; Flugge UI A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal.Biochem 1984, 138, 141–143. [DOI] [PubMed] [Google Scholar]
- 35.Second TP; Blethrow JD; Schwartz JC; Merrihew GE; MacCoss MJ; Swaney DL; Russell JD; Coon JJ; Zabrouskov V Dual-pressure linear ion trap mass spectrometer improving the analysis of complex protein mixtures. Anal.Chem 2009, 81, 7757–7765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Senko MW; Remes PM; Canterbury JD; Mathur R; Song Q; Eliuk SM; Mullen C; Earley L; Hardman M; Blethrow JD; Bui H; Specht A; Lange O; Denisov E; Makarov A; Horning S; Zabrouskov V Novel parallelized quadrupole/linear ion trap/Orbitrap tribrid mass spectrometer improving proteome coverage and peptide identification rates. Anal.Chem 2013, 85, 11710–11714. [DOI] [PubMed] [Google Scholar]
- 37.Wilcox BE; Hendrickson CL; Marshall AG Improved ion extraction from a linear octopole ion trap: SIMION analysis and experimental demonstration. J.Am.Soc.Mass Spectrom 2002, 13, 1304–1312. [DOI] [PubMed] [Google Scholar]
- 38.Kaiser NK; Savory JJ; Hendrickson CL Controlled ion ejection from an external trap for extended m/z range in FT-ICR mass spectrometry. J.Am.Soc.Mass Spectrom 2014, 25, 943–949. [DOI] [PubMed] [Google Scholar]
- 39.Fellers RT; Greer JB; Early BP; Yu X; LeDuc RD; Kelleher NL; Thomas PM ProSight Lite: graphical software to analyze top-down mass spectrometry data. Proteomics 2015, 15, 1235–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McLuckey SA; Goeringer DE Ion/molecule reactions for improved effective mass resolution in electrospray mass spectrometry. Anal.Chem 1995, 67, 2493–2497. [DOI] [PubMed] [Google Scholar]
- 41.Anderson LC; English AM; Wang W; Bai DL; Shabanowitz J; Hunt DF Protein derivatization and sequential ion/ion reactions to enhance sequence coverage produced by electron transfer dissociation mass spectrometry. Int.J.Mass.Spectrom 2015, 377, 617–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weisbrod CR; Kaiser NK; Syka JEP; Early L; Mullen C; Dunyach JJ; English AM; Anderson LC; Blakney GT; Shabanowitz J; Hendrickson CL; Marshall AG; Hunt DF Front-End Electron Transfer Dissociation Coupled to a 21 Tesla FT-ICR Mass Spectrometer for Intact Protein Sequence Analysis. J.Am.Soc.Mass Spectrom 2017, 28, 1787–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Anderson LC; Karch KR; Ugrin SA; Coradin M; English AM; Sidoli S; Shabanowitz J; Garcia BA; Hunt DF Analyses of Histone Proteoforms Using Front-end Electron Transfer Dissociation-enabled Orbitrap Instruments. Mol Cell Proteomics 2016, 15, 975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tran JC; Doucette AA Gel-eluted liquid fraction entrapment electrophoresis: an electrophoretic method for broad molecular weight range proteome separation. Anal.Chem 2008, 80, 1568–1573. [DOI] [PubMed] [Google Scholar]
- 45.Cesnik AJ; Shortreed MR; Schaffer LV; Knoener RA; Frey BL; Scalf M; Solntsev SK; Dai Y; Gasch AP; Smith LM Proteoform Suite: Software for Constructing, Quantifying, and Visualizing Proteoform Families. J.Proteome Res 2018, 17, 568–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McLuckey SA; Stephenson JL; Asano KG Ion/ion proton-transfer kinetics: implications for analysis of ions derived from electrospray of protein mixtures. Anal.Chem 1998, 70, 1198–1202. [DOI] [PubMed] [Google Scholar]
- 47.Laszlo KJ; Munger EB; Bush MF Folding of Protein Ions in the Gas Phase after Cation-to-Anion Proton-Transfer Reactions. J.Am.Chem.Soc 2016, 138, 9581–9588. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.