

Abstract
In the multi-hit model of carcinogenesis, a precancerous state often precedes overt malignancy. Identification of these states has been of great interest as they allow for early identification of at-risk individuals before the appearance of a future cancer. One such condition has recently been described for blood cancers: Clonal Hematopoiesis of Indeterminate Potential (CHIP). Recent research advances have elucidated the risk of progression of CHIP to myeloid malignancies, its potential as a precursor for non-myeloid blood cancers, and its association with non-hematological cancers. Understanding the evolution of CHIP to hematological malignancy may help identify CHIP carriers at high risk of transformation and lead to the development of targeted therapies that can be deployed preemptively.
Introduction
Hematopoietic stem cells (HSCs) reside at the apex of a constantly active hematopoietic system. To fill this role, these cells are uniquely capable of either self-renewal or initiating a clonal lineage of progenitors capable of producing about one trillion blood cells daily [1]. Inherent to the regular cellular divisions HSCs undergo are endogenous stressors such as reactive oxygen species or stochastic DNA replication errors that result in somatic mutations [2]. Additionally, as these tissues age certain somatic mutation classes increase in frequency due to spontaneous deamination of 5-methylcytosine to thymine and incorrect nonhomologous end joining of DNA double-strand breaks [2,3]. Thus, with about 200,000 HSCs each acquiring about one exonic mutation per decade via these various processes, 1.4 million protein-coding mutations will be distributed across one’s HSC pool by the age of 70 [4•,5]. Importantly, as the top of the hematopoietic hierarchy, any mutations in the founding HSC will propagate to its clonal descendants. While many of these mutations will be neutral passenger mutations, just a single driver mutation that confers a fitness advantage may be sufficient to promote clonal expansion of the mutated HSC’s progeny. This condition may be referred to as clonal hematopoiesis (CH). Whether these mutations might be precursors to frank malignancy was unknown until recently. Here, we will review recent advances in our understanding of CH and its association to malignancy.
CHIP and Myeloid Cancers
Over the last decade, the advent of accessible next-generation sequencing technologies has enabled the identification of recurrent drivers of hematological cancers [6–8]. In 2014, three groups examined whole exome sequencing (WES) data from peripheral blood DNA in ∼30,000 people unselected for hematological phenotypes. Remarkably, they found a cancer-associated mutation in more than 10% of those age 70 or older [9–11], most frequently loss-of-function mutations in genes DNMT3A and TET2, which are also commonly mutated in acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS). The prevalence of CH, therefore, greatly exceeds the prevalence of frank hematological cancers (Figure 1A-B). The presence of these mutations was associated with a 10-fold increased relative risk of hematological cancers, while the absolute risk of progression to overt malignancy was 0.5–1% per year. These studies complemented earlier work on patients with AML who were found to frequently have HSCs with mutations in DNMT3A or TET2 at remission [12,13]. The functional consequences of these and other common mutations in AML and MDS have been extensively reviewed elsewhere [14–16]. In order to distinguish the presence of CH due to a cancer driver mutation in the absence of a malignancy from frank malignancy (which is also considered a clonal hematopoietic state), the term clonal hematopoiesis of indeterminate potential (CHIP) was coined [17]. Variant allele fraction (VAF), which is a measure of the clone size, of at least 2% was proposed for the definition of CHIP. A cutoff was a practical necessity because, if sequenced deeply enough, we anticipated that nearly everyone would harbor a very small CH clone, a hypothesis that has subsequently been validated [18]. If these very small clones are ubiquitous, then comparisons of outcomes between carriers and non-carriers of CH lack meaning or utility. A VAF of 2% was chosen based on the fact that the studies prior to 2015 had not examined the consequences of harboring clones smaller than this size. However, the exact cutoff for this practical definition of CHIP may be modified as new data emerges. In several recent studies, larger clones have been associated with an increased risk of pathological consequences such as cancer and cardiovascular disease, but very small clones appear to not be associated with such risk [9,19,20••,21••,22]. Further investigation will be required to determine if there is a clinically significant threshold for risk of transformation, which may differ by driver gene.
Figure 1. Aging increases risk for clonal hematopoiesis and hematological malignancies.
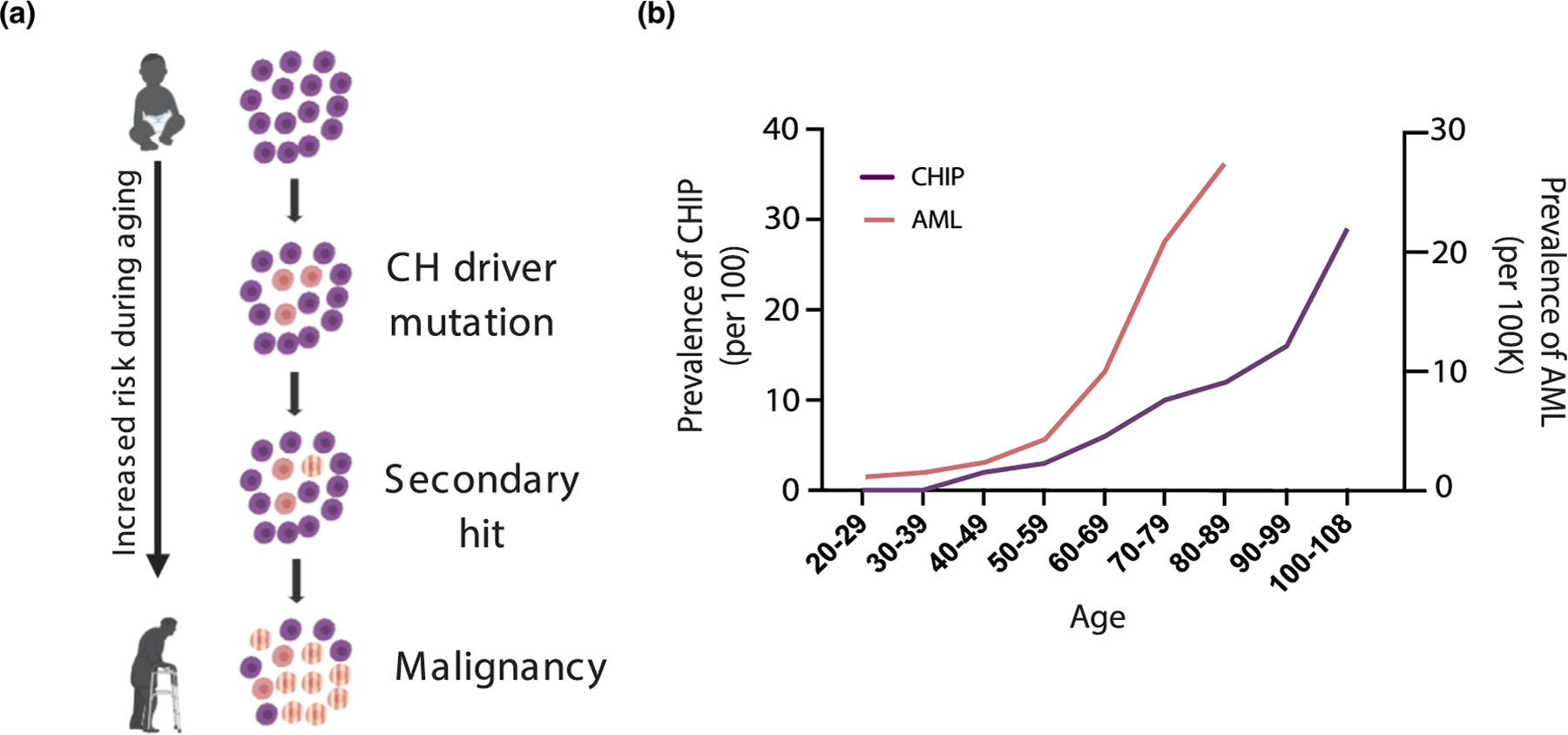
A) Representation of CH driver mutations as a function of aging. After clonal expansion, CH clones may acquire additional secondary mutations that induce malignant transformation. B) Prevalence of CHIP and AML as a function of age. Data for the prevalence of CHIP were adopted from studies found in Jaiswal et al. 2014 [9]. Prevalence of AML among patients in the US were reported using Surveillance Research Program, National Cancer Institute SEER*Explorer (seer.cancer.gov/explorer) for AML patients diagnosed between 2013–2017 [57].
These initial studies were powered to be able to show that CHIP as a whole was associated with cancer risk, but did not have enough people who developed hematological cancers to determine risk at the level of the individual driver mutations. More recently, studies have examined the risk of progression of CH (including CHIP with VAF > 2% as well as clones below this size, which would not be considered CH) to acute myeloid leukemia (AML) in case-control studies within large cohorts of individuals with peripheral blood samples collected before the onset of disease [20••,21••]. According to these studies, CH mutations can be found in approximately three-quarters of the AML patients before diagnosis and are presumed to be founder mutations for the subsequent cancer [20••]. These studies also found factors that were predictive of AML development. One such factor was VAF, which was also found to be associated with malignancy development in a prior study [19]. CHIP carriers who went on to develop AML had not only larger clones, but also a different mutational profile compared to CHIP carriers who never developed AML. CHIP carriers who did not develop AML tended to harbor mutations such as TET2 and DNMT3A, while those who progressed to AML had a bigger representation of mutations such as TP53, spliceosome mutations (SF3B1, SRSF2, U2AF1), and in one study IDH1/IDH2 mutations (Figure 2) [20••,21••]. However, there was no difference in clonal expansion rates between pre-AML and non-leukemic CHIP carriers [21••]. Not all AMLs have a demonstrable antecedent CHIP phase, especially those which occur in younger populations where CHIP is less frequent. It has been observed that approximately 75% of younger patients have cytogenetic rearrangements or other chromosomal structural changes that could act as drivers of the disease [7,23,24]. In contrast, nearly 50% of adult AML patients, mostly comprised of elderly individuals, do not have any abnormal karyotype status and only harbor point mutations that commonly occur in AML [23,24]. This suggests that CHIP may become a more important factor in the development of AML relative to cytogenetic rearrangements as individuals age.
Figure 2. Risk factors for progression of CHIP to malignancies.
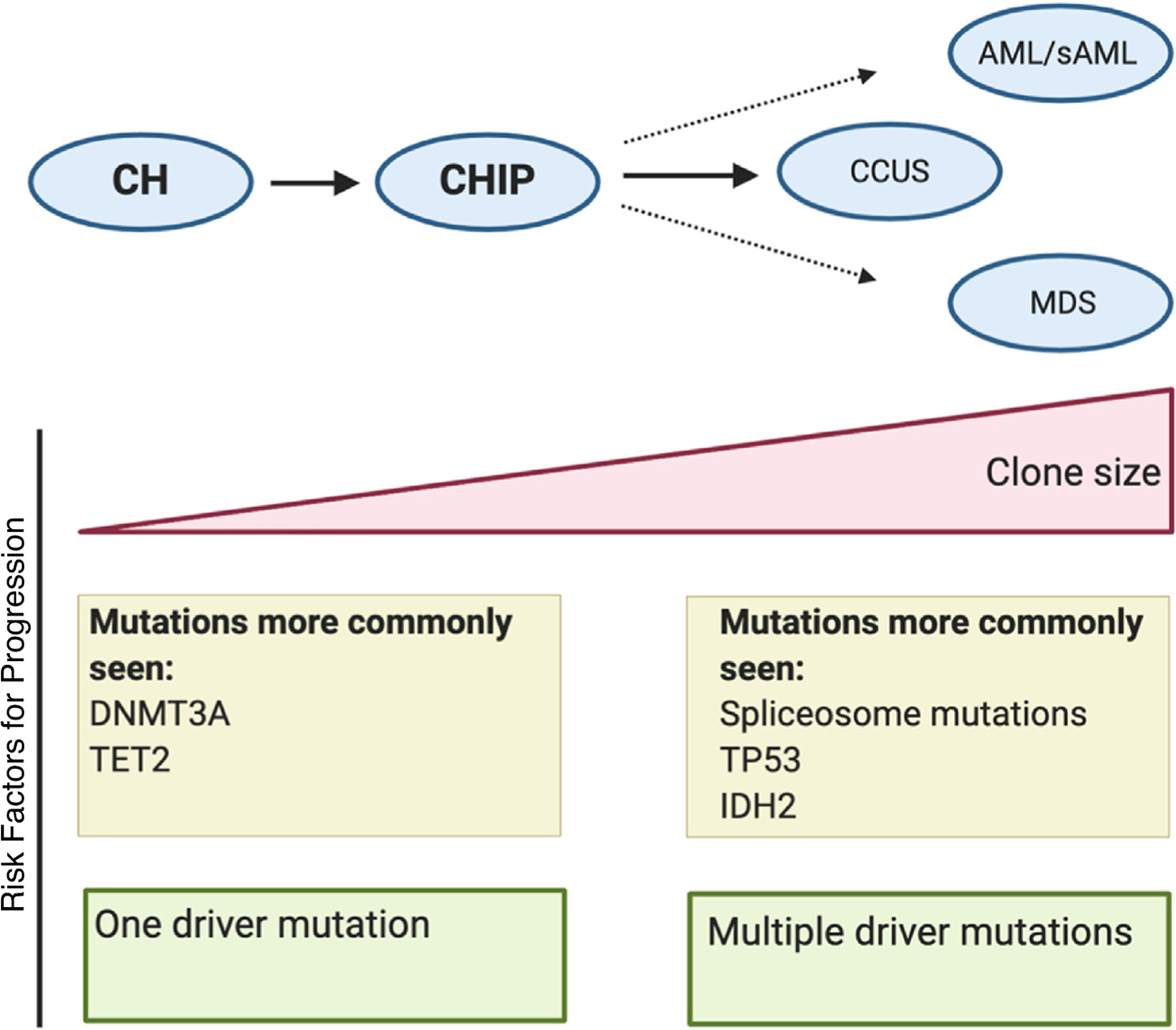
CHIP is derived from clonal hematopoiesis (CH) once clonal mutated cells carry a VAF of at least 2% and harbor at least one mutation commonly found in hematological malignancies. CHIP may progress to clonal cytopenia, MDS, or AML in some people. Several features distinguish non-malignant carriers of CHIP from those at high-risk of transformation. Those who progress to MDS or AML are more likely to have larger VAFs in comparison to control groups. Patients who go on to develop malignancy also typically harbor a different mutational profile and carry multiple driver mutations in comparison to those who are non-malignant carriers.
So far, there have been limited studies on the transformation of CHIP to MDS. However, many of the most commonly mutated genes seen in CHIP are also commonly mutated in MDS [8], therefore, it is expected that CHIP is also a pre-malignant state for MDS. Idiopathic cytopenia of undetermined significance (ICUS) is defined by the presence of unexplained cytopenias in patients that do not meet criteria for myeloid neoplasms such as MDS and AML. Approximately 35% of individuals diagnosed with ICUS [25,26] have detectable CHIP mutations, which is referred to as clonal cytopenia of undetermined significance (CCUS). This frequency is substantially higher than in populations without known cytopenias, suggesting that in some cases the clones may be causally responsible for low blood counts. Indeed, one study found that some individuals with CCUS have similar outcomes as those with low risk MDS [27]. Certain features were more predictive of MDS development in these patients, for example, VAF ≥ 10% or harboring 2 or more mutations. The presence of SF3B1, SRSF2, U2AF1, JAK2, and RUNX1 mutations alone had high positive predictive value for the risk of transforming to myeloid neoplasms, but the presence of TET2, DNMT3A or ASXL1 mutations were only strongly predictive in the presence of a co-mutation with another gene, which suggests synergy between certain genes to drive malignant transformation [27]. In sum, these studies suggest that mutational patterns may be more indicative of outcome in patients with CCUS than morphological criteria.
The myeloproliferative neoplasms (MPNs), polycythemia vera and essential thrombocythemia, are largely driven by mutations that lead to JAK/STAT pathway activation [28]. The most commonly mutated gene in MPNs is JAK2, which is also one of the most commonly mutated genes in CHIP. Based on mouse models of the JAK2 V617 F mutation and the lack of other driver mutations in many patients with MPN, it has been presumed that the mutation is sufficient for development of an MPN phenotype in humans [29–31]. However, the prevalence of large JAK2 mutant clones in unselected populations of middle-aged individuals is approximately 1 in 1000 [32•], which is much higher than the prevalence of MPNs. The factors which influence the progression of MPN from JAK2 mutated CHIP remain unknown.
CHIP and Other Blood Cancers
In addition to predisposing carriers to myeloid malignancies, CHIP mutations have been associated with some lymphomas.
Several studies have demonstrated a strong association between certain CHIP mutations and T-cell lymphomas, particularly angioimmunoblastic T cell lymphoma (AITL), peripheral T cell lymphoma not otherwise specified (PCTL-NOS), and adult T cell leukemia/lymphoma (ATLL). TET2 mutations have been found in 33–100% of AITL cases, 20%–60% of PTCL-NOS cases, and 8–14% of ATLL cases [33–38]. DNMT3A mutations have also been identified in 23–33% of AITL cases, 12%–28% of PTCL-NOS cases, and 2% of ATLL cases [34,37,39]. In addition to being a frequent finding in T-cell lymphomas, there is also evidence of a CHIP state preceding the development of these cancers. In one study, a patient with AITL was shown to have had a founding heterozygous TET2 mutation present in all bone marrow cells [34]. This mutation was followed by two different TET2 mutations in two smaller subclones leading to biallelic inactivation of TET2 in the majority of the hematopoietic cells. This was subsequently followed by an ASXL1 mutation, and a RHOA mutation which led to the development of AITL [39,40]. Another study performed DNA sequencing of in vitro derived CD34+ single cell colonies to demonstrate that CHIP status predated transformation to T-cell lymphoma. One patient acquired a TET2 mutation in a bone marrow HSC clone, which was followed by several additional mutations that were ultimately only found in the tumor population [33]. Similar to the previous case study, acquisition of a CHIP mutation in the bone marrow predated and possibly predisposed carriers to a subsequent peripheral T-cell lymphoma carrying the original CHIP mutation.
Amongst B-cell lymphomas, TET2 mutations have been reported in 5% of mantle cell lymphoma and 0–12% of diffuse large B-cell lymphoma (DLBCL) cases [33,41–43]. Furthermore, a mouse model demonstrated that the loss of Tet2 predisposes to malignant transformation of germinal center B-cells into DLBCL, which was accompanied by widespread alteration of methylation at enhancer elements [40]. Thus, CHIP may also be a precursor state for B-cell malignancies with TET2 mutations.
CHIP in solid tumors
Multiple studies have shown that CHIP mutations are more common in patients with lymphoid cancers and solid cancers compared to the general population [44–46]. This seems to be driven by selection for specific mutations, likely due to the iatrogenic pressures seen in the cancer setting. In patients with B-cell lymphoma, there was an increased prevalence of DNA damage response (DDR) mutations, primarily PPM1D and TP53, compared to cancer-free CHIP carriers, likely due to selection from therapy-related genotoxic stress. These DDR mutations were strongly associated with poorer late overall survival, inferior event-free survival, and significantly increased incidence of therapy-related myeloid neoplasms (tMN) following autologous stem-cell transplantation (ASCT) for lymphoma[47–49]. Common CHIP mutations, such as DNMT3A, TET2, and ASXL1, did not associate with survival outcomes of patients following chemotherapy and ASCT compared to non-CHIP carriers in one study [47], while they did in another [48].
CHIP may also be of concern for patients receiving treatments for non-hematological cancers, as the presence of CHIP was associated with increased risk for developing tMN in solid tumor patients [50••]. In this study, prospective analysis demonstrated that mutations seen in the pre-malignant CHIP phase formed the dominant clone in the tMN [50••]. While previous work had shown that mutations in TP53 and PPM1D were strongly associated with prior cancer therapy, more recent studies have shown that prior radiation therapy and chemotherapy with platinum and topoisomerase II inhibitors place patients at particularly high risk for these mutations [44,50••].
Similar to what has been seen in the healthy population, the presence of CHIP is associated with increased mortality rate in patients with solid cancers. The excess mortality seen in these patients is most often due to relapse of their initial neoplasm rather than to development of tMN derived from the mutated hematopoietic clone [50••]. It is uncertain why this is the case, but it is plausible that this may be a causal association. It has been reported that loss of Tet2 switches tumor-associated macrophages (TAMs) into a proinflammatory, M1-like phenotype from an immunosuppressive, M2-like phenotype in a mouse model of melanoma [51]. The pro-inflammatory cytokine interleukin-1 beta (IL-1B) has been shown to be upregulated in murine macrophages and the plasma of humans who have TET2 mutations [19,32•,52]. In humans, treatment with an antibody against IL-1B reduced the number of lung cancers that developed in an exploratory analysis of the CANTOS trial [53]. Thus, one hypothesis for adverse outcomes in cancer patients with CHIP is a pro-tumorigenic, inflammatory microenvironment due to infiltration by mutated immune cells. Indeed, it has been observed that patients with breast cancer have leukocytes within the tumor microenvironment that harbor CHIP-associated mutations [54].
Recently, cell-free DNA (cfDNA) analysis has been used for the early detection of solid cancers. Perhaps unsurprisingly, many variants found in cfDNA were shown to be from CH, and this was especially problematic for JAK2 and TP53 mutations [55]. A subsequent study using high-depth sequencing of cfDNA and matched blood cell DNA determined that the majority of mutations that were detected in cfDNA were canonical CH mutations, which led the authors to conclude that matched white blood cell samples can help to filter mutations that may be due to CH [56]. Thus, while early detection of solid tumors using cfDNA remains promising, its utility may be limited by confounding due to CH.
Outlook
Clonal expansions of hematopoietic cells are very common in aging humans. While CHIP is clearly causally linked to subsequent hematologic cancers, it is currently not recommended to screen for these mutations in the general population. This is primarily due to 1) imprecise ability to predict the likelihood of progression to malignancy and 2) lack of low-risk therapeutic options in those at risk for transformation. For the former, we foresee the development of novel biomarkers, which, when combined with known parameters such as mutated driver gene(s), VAF, and clinical parameters, may offer more precise risk stratification. For the latter, a greater mechanistic understanding of the causes of clonal expansion of the size of the clone is necessary to rationally design therapeutic interventions that prevent progression to frank malignancy. Despite the lack of actionability at the present moment, clonal hematopoiesis is an exemplar of a pre-malignancy that will continue to yield important insights into cancer biology.
Acknowledgements
This work was supported by awards to S.J from Burroughs Wellcome Foundation, Ludwig Cancer Center, American Society of Hematology, and N.I.H. (1DP2HL157540). S.R.M. is supported by training grant 2T32HL098049 from N.I.H. Figures used templates derived from Biorender.com.
Footnotes
Declaration of competing interest
S.J. reports consulting income from Novartis and Roche Genentech related to this topic.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Liggett LA, Sankaran VG: Unraveling Hematopoiesis through the Lens of Genomics. Cell 2020, 182:1384–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beerman I, Seita J, Inlay MA, Weissman IL, Rossi DJ: Quiescent hematopoietic stem cells accumulate DNA damage during aging that is repaired upon entry into cell cycle. Cell Stem Cell 2014, 15:37–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL et al. : Signatures of mutational processes in human cancer. Nature 2013, 500:415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.•. Lee-Six H, Obro NF, Shepherd MS, Grossmann S, Dawson K, Belmonte M, Osborne RJ, Huntly BJP, Martincorena I, Anderson E et al. : Population dynamics of normal human blood inferred from somatic mutations. Nature 2018 This study used clonal evolutionary histories of hematopoietic stem cells and their descendents to estimate the number of HSCs in adult humans.
- 5.Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, Wartman LD, Lamprecht TL, Liu F, Xia J et al. : The origin and evolution of mutations in acute myeloid leukemia. Cell 2012, 150:264–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bejar R, Stevenson K, Abdel-Wahab O, Galili N, Nilsson B, Garcia-Manero G, Kantarjian H, Raza A, Levine RL, Neuberg D et al. : Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med 2011, 364:2496–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cancer Genome Atlas Research N, Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A, Hoadley K, Triche TJ Jr, Laird PW et al. : Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 2013, 368:2059–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Papaemmanuil E, Gerstung M, Malcovati L, Tauro S, Gundem G, Van Loo P, Yoon CJ, Ellis P, Wedge DC, Pellagatti A et al. : Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013, 122:3616–3627quiz 3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A et al. : Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 2014, 371:2488–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Genovese G, Kahler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, Chambert K, Mick E, Neale BM, Fromer M et al. : Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 2014, 371:2477–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, McMichael JF, Schmidt HK, Yellapantula V, Miller CA et al. : Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med 2014, 20:1472–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jan M, Snyder TM, Corces-Zimmerman MR, Vyas P, Weissman IL, Quake SR, Majeti R: Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci Transl Med 2012, 4:149ra118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shlush LI, Zandi S, Mitchell A, Chen WC, Brandwein JM, Gupta V, Kennedy JA, Schimmer AD, Schuh AC, Yee KW et al. : Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 2014, 506:328–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sperling AS, Gibson CJ, Ebert BL: The genetics of myelodysplastic syndrome: from clonal haematopoiesis to secondary leukaemia. Nat Rev Cancer 2017, 17:5–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bowman RL, Busque L, Levine RL: Clonal Hematopoiesis and Evolution to Hematopoietic Malignancies. Cell Stem Cell 2018, 22:157–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jaiswal S, Ebert BL: Clonal hematopoiesis in human aging and disease. Science 2019, 366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Steensma DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP, Ebert BL: Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015, 126:9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Young AL, Challen GA, Birmann BM, Druley TE: Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat Commun 2016, 7:12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, McConkey M, Gupta N, Gabriel S, Ardissino D et al. : Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N Engl J Med 2017, 377:111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.••. Abelson S, Collord G, Ng SWK, Weissbrod O, Mendelson Cohen N, Niemeyer E, Barda N, Zuzarte PC, Heisler L, Sundaravadanam Y et al. : Prediction of acute myeloid leukaemia risk in healthy individuals. Nature 2018, 559:400–404 These studies performed case-control analyses to determine the risk of progression to AML from CH, and identified VAF and driver gene as the most predictive factors for transformation.
- 21.••. Desai P, Mencia-Trinchant N, Savenkov O, Simon MS, Cheang G, Lee S, Samuel M, Ritchie EK, Guzman ML, Ballman KV et al. : Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat Med 2018, 24:1015–1023 These studies performed case-control analyses to determine the risk of progression to AML from CH, and identified VAF and driver gene as the most predictive factors for transformation.
- 22.Bick AG, Pirruccello JP, Griffin GK, Gupta N, Gabriel S, Saleheen D, Libby P, Kathiresan S, Natarajan P: Genetic IL-6 Signaling Deficiency Attenuates Cardiovascular Risk in Clonal Hematopoiesis. Circulation 2019. [DOI] [PMC free article] [PubMed]
- 23.Mrozek K, Heerema NA, Bloomfield CD: Cytogenetics in acute leukemia. Blood Rev 2004, 18:115–136. [DOI] [PubMed] [Google Scholar]
- 24.Mrozek K, Marcucci G, Nicolet D, Maharry KS, Becker H, Whitman SP, Metzeler KH, Schwind S, Wu YZ, Kohlschmidt J et al. : Prognostic significance of the European LeukemiaNet standardized system for reporting cytogenetic and molecular alterations in adults with acute myeloid leukemia. J Clin Oncol 2012, 30:4515–4523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kwok B, Hall JM, Witte JS, Xu Y, Reddy P, Lin K, Flamholz R, Dabbas B, Yung A, Al-Hafidh J et al. : MDS-associated somatic mutations and clonal hematopoiesis are common in idiopathic cytopenias of undetermined significance. Blood 2015, 126:2355–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cargo CA, Rowbotham N, Evans PA, Barrans SL, Bowen DT, Crouch S, Jack AS: Targeted sequencing identifies patients with preclinical MDS at high risk of disease progression. Blood 2015, 126:2362–2365. [DOI] [PubMed] [Google Scholar]
- 27.Malcovati L, Galli A, Travaglino E, Ambaglio I, Rizzo E, Molteni E, Elena C, Ferretti VV, Catricala S, Bono E et al. : Clinical significance of somatic mutation in unexplained blood cytopenia. Blood 2017, 129:3371–3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nangalia J, Massie CE, Baxter EJ, Nice FL, Gundem G, Wedge DC, Avezov E, Li J, Kollmann K, Kent DG et al. : Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med 2013, 369:2391–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Akada H, Yan D, Zou H, Fiering S, Hutchison RE, Mohi MG: Conditional expression of heterozygous or homozygous Jak2V617F from its endogenous promoter induces a polycythemia vera-like disease. Blood 2010, 115:3589–3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mullally A, Lane SW, Ball B, Megerdichian C, Okabe R, Al-Shahrour F, Paktinat M, Haydu JE, Housman E, Lord AM et al. : Physiological Jak2V617F expression causes a lethal myeloproliferative neoplasm with differential effects on hematopoietic stem and progenitor cells. Cancer Cell 2010, 17:584–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marty C, Lacout C, Martin A, Hasan S, Jacquot S, Birling MC, Vainchenker W, Villeval JL: Myeloproliferative neoplasm induced by constitutive expression of JAK2V617F in knock-in mice. Blood 2010, 116:783–787. [DOI] [PubMed] [Google Scholar]
- 32.•. Bick AG, Weinstock JS, Nandakumar SK, Fulco CP, Bao EL, Zekavat SM, Szeto MD, Liao X, Leventhal MJ, Nasser J et al. : Inherited causes of clonal haematopoiesis in 97,691 whole genomes. Nature 2020, 586:763–768 Analysis of clonal hematopoiesis in ∼100,000 participants from the TOPMed consortium found associations with circulating cytokines and germline polymorphisms.
- 33.Quivoron C, Couronne L, Della Valle V, Lopez CK, Plo I, Wagner-Ballon O, Do Cruzeiro M, Delhommeau F, Arnulf B, Stern MH et al. : TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell 2011, 20:25–38. [DOI] [PubMed] [Google Scholar]
- 34.Sakata-Yanagimoto M, Enami T, Yoshida K, Shiraishi Y, Ishii R, Miyake Y, Muto H, Tsuyama N, Sato-Otsubo A, Okuno Y et al. : Somatic RHOA mutation in angioimmunoblastic T cell lymphoma. Nat Genet 2014, 46:171–175. [DOI] [PubMed] [Google Scholar]
- 35.Lemonnier F, Couronne L, Parrens M, Jais JP, Travert M, Lamant L, Tournillac O, Rousset T, Fabiani B, Cairns RA et al. : Recurrent TET2 mutations in peripheral T-cell lymphomas correlate with TFH-like features and adverse clinical parameters. Blood 2012, 120:1466–1469. [DOI] [PubMed] [Google Scholar]
- 36.Odejide O, Weigert O, Lane AA, Toscano D, Lunning MA, Kopp N, Kim S, van Bodegom D, Bolla S, Schatz JH et al. : A targeted mutational landscape of angioimmunoblastic T-cell lymphoma. Blood 2014, 123:1293–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kataoka K, Nagata Y, Kitanaka A, Shiraishi Y, Shimamura T, Yasunaga J, Totoki Y, Chiba K, Sato-Otsubo A, Nagae G et al. : Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat Genet 2015, 47:1304–1315. [DOI] [PubMed] [Google Scholar]
- 38.Shimoda K, Shide K, Kameda T, Hidaka T, Kubuki Y, Kamiunten A, Sekine M, Akizuki K, Shimoda H, Yamaji T et al. : TET2 Mutation in Adult T-Cell Leukemia/Lymphoma. J Clin Exp Hematop 2015, 55:145–149. [DOI] [PubMed] [Google Scholar]
- 39.Cairns RA, Iqbal J, Lemonnier F, Kucuk C, de Leval L, Jais JP, Parrens M, Martin A, Xerri L, Brousset P et al. : IDH2 mutations are frequent in angioimmunoblastic T-cell lymphoma. Blood 2012, 119:1901–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dominguez PM, Ghamlouch H, Rosikiewicz W, Kumar P, Beguelin W, Fontan L, Rivas MA, Pawlikowska P, Armand M, Mouly E et al. : TET2 Deficiency Causes Germinal Center Hyperplasia, Impairs Plasma Cell Differentiation, and Promotes B-cell Lymphomagenesis. Cancer Discov 2018, 8:1632–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD, Johnson NA, Severson TM, Chiu R, Field M et al. : Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 2011, 476:298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Asmar F, Punj V, Christensen J, Pedersen MT, Pedersen A, Nielsen AB, Hother C, Ralfkiaer U, Brown P, Ralfkiaer E et al. : Genome-wide profiling identifies a DNA methylation signature that associates with TET2 mutations in diffuse large B-cell lymphoma. Haematologica 2013, 98:1912–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kubuki Y, Yamaji T, Hidaka T, Kameda T, Shide K, Sekine M, Kamiunten A, Akizuki K, Shimoda H, Tahira Y et al. : TET2 mutation in diffuse large B-cell lymphoma. J Clin Exp Hematop 2017, 56:145–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Coombs CC, Zehir A, Devlin SM, Kishtagari A, Syed A, Jonsson P, Hyman DM, Solit DB, Robson ME, Baselga J et al. : Therapy-Related Clonal Hematopoiesis in Patients with Non-hematologic Cancers Is Common and Associated with Adverse Clinical Outcomes. Cell stem cell 2017, 21:374–382.e374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gillis NK, Ball M, Zhang Q, Ma Z, Zhao Y, Yoder SJ, Balasis ME, Mesa TE, Sallman DA, Lancet JE et al. : Clonal haemopoiesis and therapy-related myeloid malignancies in elderly patients: a proof-of-concept, case-control study. Lancet Oncol 2017, 18:112–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ptashkin RN, Mandelker DL, Coombs CC, Bolton K, Yelskaya Z, Hyman DM, Solit DB, Baselga J, Arcila ME, Ladanyi M et al. : Prevalence of Clonal Hematopoiesis Mutations in Tumor-Only Clinical Genomic Profiling of Solid Tumors. JAMA Oncol 2018, 4:1589–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Husby S, Favero F, Nielsen C, Sørensen BS, Bæch J, Grell K, Hansen JW, Rodriguez-Gonzalez FG, Haastrup EK, Fischer-Nielsen A et al. : Clinical impact of clonal hematopoiesis in patients with lymphoma undergoing ASCT: a national population-based cohort study. Leukemia 2020:1–13. [DOI] [PubMed]
- 48.Gibson CJ, Lindsley RC, Tchekmedyian V, Mar BG, Shi J, Jaiswal S, Bosworth A, Francisco L, He J, Bansal A et al. : Clonal Hematopoiesis Associated With Adverse Outcomes After Autologous Stem-Cell Transplantation for Lymphoma. J Clin Oncol 2017, 35:1598–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Takahashi K, Wang F, Kantarjian H, Doss D, Khanna K, Thompson E, Zhao L, Patel K, Neelapu S, Gumbs C et al. : Preleukaemic clonal haemopoiesis and risk of therapy-related myeloid neoplasms: a case-control study. Lancet Oncol 2017, 18:100–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.••. Bolton KL, Ptashkin RN, Gao T, Braunstein L, Devlin SM, Kelly D, Patel M, Berthon A, Syed A, Yabe M et al. : Cancer therapy shapes the fitness landscape of clonal hematopoiesis. Nat Genet 2020, 52:1219–1226 This study identified specific oncologic therapies associated with clonal hematopoiesis in patients with solid tumors at a large cancer center.
- 51.Pan W, Zhu S, Qu K, Meeth K, Cheng J, He K, Ma H, Liao Y, Wen X, Roden C et al. : The DNA Methylcytosine Dioxygenase Tet2 Sustains Immunosuppressive Function of Tumor-Infiltrating Myeloid Cells to Promote Melanoma Progression. Immunity 2017, 47:284–297.e285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, Wu C-L, Sano S, Muralidharan S, Rius C et al. : Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science (New York, N.Y.) 2017, 355:842–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD et al. : Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med 2017, 377:1119–1131. [DOI] [PubMed] [Google Scholar]
- 54.Kleppe M, Comen E, Wen HY, Bastian L, Blum B, Rapaport FT, Keller M, Granot Z, Socci N, Viale A et al. : Somatic mutations in leukocytes infiltrating primary breast cancers. npj Breast Cancer 2015, 1:15005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hu Y, Ulrich BC, Supplee J, Kuang Y, Lizotte PH, Feeney NB, Guibert NM, Awad MM, Wong KK, Janne PA et al. : False-Positive Plasma Genotyping Due to Clonal Hematopoiesis. Clin Cancer Res 2018, 24:4437–4443. [DOI] [PubMed] [Google Scholar]
- 56.Razavi P, Li BT, Brown DN, Jung B, Hubbell E, Shen R, Abida W, Juluru K, De Bruijn I, Hou C et al. : High-intensity sequencing reveals the sources of plasma circulating cell-free DNA variants. Nature Medicine 2019. [DOI] [PMC free article] [PubMed]
- 57.Surveillance E, and End Results (SEER) Program. Edited by; 2013–2017.