

ABSTRACT
Bacteria have two pathways to restart stalled replication forks caused by environmental stresses, error-prone translesion DNA synthesis (TLS) catalyzed by TLS polymerase and error-free template switching catalyzed by RecA, and their competition on the arrested fork affects bacterial SOS mutagenesis. DnaE2 is an error-prone TLS polymerase, and its functions require ImuA and ImuB. Here, we investigated the transcription of imuA, imuB, and dnaE2 in UV-C-irradiated Myxococcus xanthus and found that the induction of imuA occurred significantly earlier than that of the other two genes. Mutant analysis showed that unlike that of imuB or dnaE2, the deletion of imuA significantly delayed bacterial regrowth and slightly reduced the bacterial mutation frequency and UV resistance. Transcriptomic analysis revealed that the absence of ImuA released the expression of some known SOS genes, including recA1, recA2, imuB, and dnaE2. Yeast two-hybrid and pulldown analyses proved that ImuA interacts physically with RecA1 besides ImuB. Protein activity analysis indicated that ImuA had no DNA-binding activity but inhibited the DNA-binding and recombinase activity of RecA1. These findings indicate the new role of ImuA in SOS mutagenesis; that is, ImuA inhibits the recombinase activity of RecA1, thereby facilitating SOS mutagenesis in M. xanthus.
IMPORTANCE DnaE2 is responsible for bacterial SOS mutagenesis in nearly one-third of sequenced bacterial strains. However, its mechanism, especially the function of one of its accessory proteins, ImuA, is still unclear. Here, we report that M. xanthus ImuA could affect SOS mutagenesis by inhibiting the recombinase activity of RecA1, which helps to explain the mechanism of DnaE2-dependent TLS and the selection of the two restart pathways to repair the stalled replication fork.
KEYWORDS: DnaE2, ImuA, Myxococcus xanthus, SOS mutagenesis, replication restart
INTRODUCTION
DNA lesions caused by endogenous or environmental stresses, including damaged bases or base modifications, apurinic (AP) or apyrimidinic sites, DNA gaps, pyrimidine dimers, intrastrand and interstrand cross-links (ICLs), and DNA-protein complexes (DPCs), can easily block DNA replication, resulting in the collapse of the replication fork, breaks in DNA strands, and even cell death (1). Bacteria possess two pathways (also named postreplication repair pathways in some publications) to reinitiate the stalled replication fork: translesion DNA synthesis (TLS) and template switching (see Fig. S1 in the supplemental material). Template switching is mediated by RecA-based homologous recombination and is error free, while TLS is inherently error prone since it involves bypassing the DNA lesion by inserting a nucleotide opposite the lesion. TLS is the main source of SOS mutagenesis, which opportunistically leads to adaptive evolution in response to environmental stress (2–5).
Escherichia coli contains three different TLS polymerases, polymerase II (Pol II), polymerase IV (Pol IV), and polymerase V (Pol V), which are induced by the SOS response (2, 6, 7). Compared with Pol II and Pol IV, Pol V catalyzes DNA synthesis with the lowest fidelity and is the main enzyme for bacterial SOS mutagenesis (6, 8, 9). However, only approximately one-third (1,925 strains) of the sequenced bacteria (6,107 strains [KEGG data in February 2021]) encode Pol V. In recent years, the DnaE2 (ImuC in some publications) protein has been proven to play a key role in SOS mutagenesis (10–16), found in about 1/3 of the sequenced bacteria (2,062 strains of 6,107 strains). DnaE2 is a homolog of the DnaE1 subunit of replicative DNA polymerase III. Unlike DnaE1, DnaE2 does not bind to the ε subunit (3′-to-5′ exonuclease activity) of DNA polymerase III, and therefore, the DNA synthesis catalyzed by DnaE2 has no proofreading function, resulting in a high mutation frequency (17). Additionally, DnaE2 lacks the C-terminal domain (CTD) to bind to the τ subunit, which acts as a loading subunit to assemble DnaE onto the replication fork (12, 17). Thus, DnaE2-catalyzed TLS requires the assistance of other proteins, such as ImuA and ImuB (16, 17).
The imuA and imuB genes, usually located in the SOS-regulated imuA-imuB-dnaE2 operon, are required for DnaE2 mutagenesis (10, 11, 13, 14). imuA or imuB mutants of Caulobacter crescentus and Mycobacterium tuberculosis showed a significant reduction in the SOS-induced mutation frequency, similar to that of the dnaE2 mutant (14, 16). ImuB retains a β-clamp-binding motif (354QLPLWG359) and thus can interact with the β-clamp. Warner et al. speculated that the interaction between ImuB and the β-clamp mediates the assembly of DnaE2 to the replication fork, leading to DnaE2-induced mutation (16).
ImuA is an inactive RecA-like protein (14, 16). RecA has been reported to be involved in the error-prone TLS pathway catalyzed by Pol V by forming an active mutasome (Pol V-RecA-ATP) (18). However, RecA was not involved in DnaE2-mediated error-prone TLS (11). As mentioned above, ImuA was involved in DnaE2-catalyzed TLS, but ImuA did not interact directly with DnaE2 (16). Therefore, the role of ImuA in DnaE2-mediated TLS does not imitate the mechanism of RecA in the Pol V-RecA mutasome but remains unclear.
Myxococcus xanthus DK1622 is the model strain of myxobacteria, and its genome size is >9 Mbp, with a large number of DNA repeats, such as duplicate recA genes (19–21). Recently, we showed that the expression level of RecA1 (MXAN_1441) with recombinase activity is very low in M. xanthus DK1622 cells (22), which might limit the template-switching events at stalled forks. M. xanthus has a dnaE2 gene (MXAN_3982)-encoded error-prone DNA polymerase for DNA replication (23). Because recombination-mediated template switching in M. xanthus is weak, the DnaE2-catalyzed TLS pathway plays a more important role in restarting replication forks, and so M. xanthus is an ideal strain for studying DnaE2-catalyzed TLS. Here, we study the function of ImuA in M. xanthus and found that it interacts directly with RecA1 and probably functions as a mediator to balance the template-switching and TLS pathways.
RESULTS
Induction of imuA by UV-C irradiation occurs earlier than that of imuB and dnaE2.
The imuA, imuB, and dnaE2 genes, adjacently located either in an operon or not, are SOS-regulated genes and can be induced by DNA-damaging agents such as UV-C light (254 nm) (10, 13, 14, 16). In M. xanthus DK1622, imuA and imuB are adjacent, and dnaE2 is located downstream, separated from the two genes by a 6.57-kb fragment containing seven cistrons (Fig. 1A). The three genes each possess an independent promoter region and thus are transcribed independently (Fig. 1B). Although the promoters of the three genes did not have the same sequence as the M. xanthus SOS box, sequence alignment showed that similar SOS box-like sequences were found in the promoter region of dnaE2 or imuB but not imuA. In UV-C-irradiated M. xanthus cells, the expressions of imuA, imuB, and dnaE2 genes were induced with different induction times and strengths (Fig. 1C). Under low-dose UV-C radiation treatment (5 J/m2), imuA transcription was upregulated 1 h after irradiation, dnaE2 responded to irradiation 2 h later, and imuB was not induced. When treated with high-dose radiation, the transcriptions of imuA, imuB, and dnaE2 were all significantly upregulated: imuA was induced 1 h after irradiation, while imuB and dnaE2 were induced 2 h later. Notably, the relative transcription level of imuA was higher than those of imuB and dnaE2 in response to UV-C irradiation with a high or low dose.
FIG 1.

Gene location and transcription analysis of imuA, imuB, and dnaE2 in M. xanthus DK1622. (A) Schematic gene location and promoter alignment. The RNA polymerase-binding regions (−10 and −35) are marked in red, and the SOS box regions are underlined in blue. The promoters of recA1, recA2, and lexA are also provided for comparison. The SOS box consensus sequence here has been reported in a previous publication (41). (B) PCR amplification of the imuA-imuB intergenic region. Primers used for PCR amplification are indicated above the gel map. Total RNA was extracted from M. xanthus and reverse transcribed into cDNA for PCR. Genomic DNA was used as a control. MW, molecular weight. (C) Analysis of induction of imuA, imuB, and dnaE2 with UV-C irradiation. The induction of recA1 is used as a control. DK1622 cells were exposed to UV-C irradiation at a dose of 5 or 15 J/m2 using a UV cross-linking machine. After irradiation, the samples were postincubated at 30°C for different times (from 0 to 7 h), and the total RNAs were then extracted from cells for RT-PCR. Relative gene expression was quantified by the comparative CT (2−ΔΔCT) method. Error bars represent means ± standard errors of the means (SEM) (n = 3) (P < 0.05).
Compared with the imuB or dnaE2 mutant, the imuA mutant has a long lag period, a high mutation frequency, and high radiation resistance.
To investigate the functions of the three genes, we constructed deletion mutants of imuA and imuB (named IA and IB) (see Fig. S2A in the supplemental material). The deletion of dnaE2 (YL1601) (23), imuB, or recA1 (22) did not significantly affect bacterial growth, but the imuA mutant showed a delay of about 12 h before the exponential growth phase, although its maximum growth rate (μmax) did not change significantly (Fig. 2A). The analysis of survival against UV-C radiation showed that the absence of imuA slightly decreased the survival rate of M. xanthus cells with an increase of the radiation dose, and the absence of imuB or dnaE2 had a more significant effect (Fig. 2B).
FIG 2.

Growth, mutability, and survival analyses of the imuA, imuB, and dnaE2 mutants. (A) Growth analysis of the imuA, imuB, and dnaE2 mutants. M. xanthus DK1622 and the SOS gene recA1 mutant were used as controls. Strains were treated with 15 J/m2 UV-C and then shaken at 30°C for different times to measure the OD600 values of the culture. DK, wild-type M. xanthus DK1622; IA, imuA mutant; IB, imuB mutant; YL1601, dnaE2 mutant. The μmax value was calculated as μmax = (ΔlnOD600) Δt−1 = 0.071 (h−1), where t is the time in hours and OD600 is the value determined at time t. (B) Survival analysis with different doses of UV-C irradiation. (C) Mutability analysis. A total of 5 × 109 cells (irradiated with UV-C) were spread on CYE agar containing 40 μg/ml nalidixic acid, and the resistant clones were counted to represent the mutation ability. Error bars indicate the standard errors from six independent experiments.
SOS mutagenesis was analyzed by calculating the target site mutation frequency of nalidixic acid of M. xanthus (23, 24). Without UV radiation, the mutation frequency of wild-type M. xanthus cells was approximately 3 × 10−8, in line with a previous report (23), and UV-C irradiation doubled the mutation frequency of M. xanthus (Fig. 2C). The deletion of dnaE2 or imuB significantly reduced the mutation frequency of this bacterium, whether it was irradiated or not, which suggested that ImuB and DnaE2 are involved in spontaneous mutagenesis. The deletion of imuA also decreased the mutation frequency, but it was not as obvious as that of imuB or dnaE2. In particular, the mutation frequency of the imuA mutant still retained UV inducibility (Fig. 2C), which suggested that ImuA plays a minor role in UV-induced mutagenesis.
ImuA interacts with ImuB and RecA1.
To understand the potential roles of imuA, the transcriptomes of the imuA-deleted and -overexpressing strains were compared with that of the wild-type M. xanthus strain. A total of 260 differentially expressed genes (DEGs) were identified (Tables S1 and S2), and among them, 135 known DEGs were functionally classified into genetic information processing (63 genes), metabolism (47 genes), environmental information processing (23 genes), and cellular processes (2 genes) (Fig. S3). Notably, the DEGs belonging to genetic information processing were mainly inhibited by ImuA, including the known SOS-induced genes (recA1, recA2, recF, recR, dnaE2, and imuB). ImuA promoted the gene expression of various metabolic pathways (Fig. S3B) but clearly inhibited the biosynthesis of other secondary metabolites (Fig. S3C). These outcomes suggested that ImuA was associated with a bacterial SOS response that was involved in the induction of secondary metabolites, DNA repair, and mutagenesis (25–27).
Based on the above-described transcriptome analysis, the yeast two-hybrid system was used to screen the ImuA-interacting proteins from the proteins near the stalling replication fork, including recombination proteins (RecA, RecF, RecO, and RecR), TLS proteins (ImuB and DnaE2), replication fork-binding proteins (DnaE1 and DnaN), single-stranded-DNA-binding protein (SSB), and the known SOS response regulator (LexA). The results showed that ImuA interacted significantly with ImuB and the recombinase RecA1 (Fig. S4). Further protein-protein interaction analysis among the three SOS mutagenesis proteins (ImuA, ImuB, and DnaE2) and two recombinases (RecA1 and RecA2) proved that ImuA could interact with RecA1 and ImuB but not DnaE2. RecA1 interacted with all the other four proteins (ImuA, ImuB, DnaE2, and RecA2), and RecA2 interacted only with RecA1 (Fig. 3A). These outcomes suggested that RecA1 might be involved in DnaE2’s TLS process.
FIG 3.
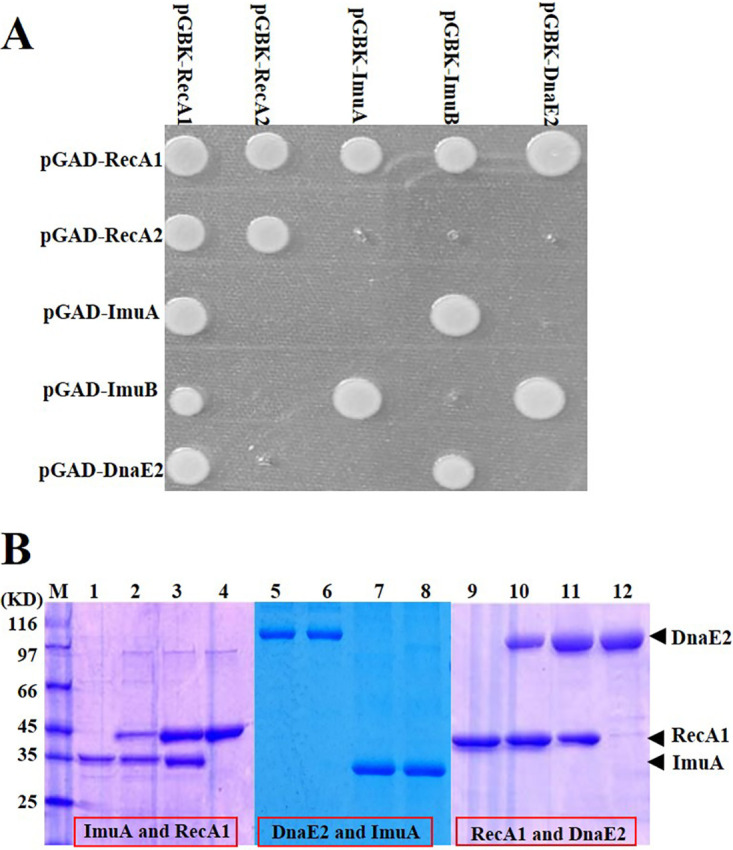
Protein interaction analysis. (A) Yeast two-hybrid analysis of pairwise interactions between RecA1, RecA2, ImuA, ImuB, and DnaE2. The genes of these proteins were linked to plasmids pGBKT7 and pGADT7, and the recombination plasmids were pairwise transformed into yeast AH109 cells to inspect protein interactions on a plate with 3-AT. (B) His pulldown analysis. Lanes: M, molecular marker; 1, His-ImuA; 2, His-ImuA and RecA1; 3, His-RecA1 and ImuA; 4, His-RecA1; 5, His-DnaE2; 6, His-DnaE2 and ImuA; 7, His-ImuA and DnaE2; 8, His-ImuA; 9, His-RecA1; 10, His-RecA1 and DnaE2; 11, His-DnaE2 and RecA1; 12, His-DnaE2.
We further expressed the RecA1, ImuA, and DnaE2 proteins with and without a His tag to verify their interactions using a His tag pulldown analysis. According to SDS-PAGE (Fig. 3B), His-tagged proteins, i.e., His-ImuA (lanes 1 and 8), His-RecA1 (lanes 4 and 9), and His-DnaE2 (lanes 5 and 12), bound to the nickel column. Besides, RecA1 and ImuA proteins in combination with His-ImuA (Fig. 3B, lane 2) and His-RecA1 (lane 3) also bound to the nickel column, respectively. Similarly, the RecA1 and DnaE2 proteins interacted with each other (Fig. 3B, lanes 10 and 11), but ImuA and DnaE2 did not interact (lanes 6 and 7).
ImuA inhibits the DNA-binding and recombinase activities of RecA1.
The RecA monomers aggregate on DNA to form nucleoprotein filaments and contain two polymerization motifs (PMs) in the N terminus and the core ATPase domain (core-PM) (28, 29). As an analog of RecA, ImuA has only one conserved core-PM site in its core ATPase domain and thus could not form RecA-like filaments theoretically (Fig. 4A and Fig. S5). Protein-protein docking prediction showed that ImuA could bind to the RecA filament through its core-PM site (Fig. S5D). Therefore, ImuA has the potential to participate in the RecA filament through the core-PM site, preventing RecA filament formation and extension (Fig. S5E).
FIG 4.

ImuA function analysis. (A) Comparison sketch of ImuA proteins. RecA, E. coli RecA; MxImuA, M. xanthus ImuA; CcImuA, C. crescentus ImuA; MtImuA, M. tuberculosis ImuA; PM, polymerization motif; WA, Walker A motif; WB, Walker B motif; L1 and L2, DNA-binding loops 1 and 2; NTD, N-terminal domain; aa, amino acids. (B) DNA-binding ability. RecA1 was used as a positive control, and bovine serum albumin (BSA) was used as a negative control. (C) Ability of ImuA to bind to RecA1-coated single-stranded DNA (ssDNA). DNA fixed on Sepharose resin was prebound with excess RecA1 protein. ImuA was added to the RecA1-coated ssDNA. SDS-PAGE was used to detect the unbound ImuA protein in the flowthrough (FT) and ImuA/RecA proteins bound to resin (R). (D) Effects of increased concentrations of ImuA on the binding of RecA1 to DNA. (E) Effects of ImuA on the recombinase activity of RecA1. jmDNA, joint molecular DNA; dsDNA, double-stranded DNA.
The ImuA protein lacks the known DNA-binding regions, including the DNA-binding loops and the conserved N terminus of RecA (Fig. 4A and Fig. S5). In the in vitro binding of ImuA to DNA, either single-stranded DNA (ssDNA) or double-stranded DNA (dsDNA) incubated with RecA1 lagged significantly, while DNA bands with ImuA or bovine serum protein (negative control) showed no change compared with the blank (no protein added) (Fig. 4B). To test whether ImuA could indirectly bind to DNA through RecA1 protein, 12 μg of the ImuA protein (∼0.4 nmol) was incubated with 200 ng of ssDNA coated with RecA1 (Fig. 4C). The addition of excessive ImuA did not destroy the RecA filament, and the RecA protein was not dissociated clearly (Fig. 4C, lane 4), but a small amount of ImuA was bound to the resin (lane 5). These findings suggested that ImuA interacted with DNA through RecA1.
A gradient concentration of ImuA proteins was added to the binding reaction solution containing RecA1 and DNA, and the DNA-protein complex was detected using agarose gel electrophoresis (Fig. 4D). RecA bound ssDNA or dsDNA to form nucleoprotein filaments (Fig. 4D, lane 2 or lane 7). The molecular weight and quantity of RecA nucleoprotein filaments decreased (Fig. 4D, lanes 3 to 5 or lanes 8 to 10) with increasing ImuA concentrations. This result indicated that ImuA has the ability to inhibit the formation of RecA filaments, probably inhibiting filament extension by interacting with RecA1.
RecA is a bacterial DNA homologous recombinase that forms a nucleoprotein filament on DNA to catalyze DNA recombination (28, 29). RecA1 catalyzed the recombination of circular ssDNA with homologous linear dsDNA, forming a lagging joined DNA band on the electropherogram, while the addition of ImuA to the reaction system significantly reduced this activity (Fig. 4E). Thus, ImuA inhibited RecA-mediated recombination, which was in line with the above-described DNA-binding results that ImuA inhibited RecA binding to ssDNA to form nucleoprotein filaments and further inhibited DNA recombination.
Effect of recA1 deletion or overexpression on mutation frequencies of DK1622 and IA strains.
To verify the effect of RecA1 on the SOS mutagenesis of M. xanthus, we deleted or overexpressed recA1 in the DK1622 and IA strains, respectively, and analyzed their relative mutation frequencies (Fig. 5).
FIG 5.
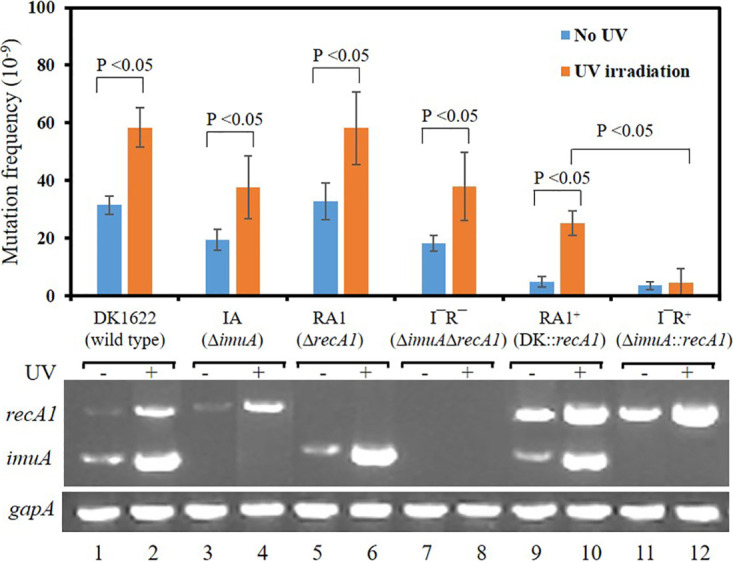
Effect of the deletion or overexpression of recA1 on the mutation frequencies of the DK1622 and IA strains. The relative expression of imuA or recA1 determined by RT-PCR is listed at the bottom. Expression of the gapA gene was used as a quantitative control of cDNA. DK1622 (wild type), M. xanthus DK1622; IA (ΔimuA), imuA deletion mutant; RA1 (ΔrecA1), recA1 deletion mutant; I− R− (ΔimuA ΔrecA1), imuA-recA1 double-deletion mutant; RA1+ (DK1622::recA1), recA1 overexpression strain; I− R+ (ΔimuA::recA1), recA1 overexpression in IA. Error bars indicate the standard deviations (SD) from four independent experiments.
In the wild type (DK1622), the relative expression level of recA1 is lower, and the recA1 deletion (RA1) has no significant effect on the mutation frequency (Fig. 5, lanes 5 and 6), which showed that the SOS mutagenesis of M. xanthus does not need the participation of RecA1. In addition, the imuA-recA1 double-deletion mutant (L− R−) showed a mutation frequency similar to that of the imuA deletion mutant (IA), further proving that the low expression level of recA1 had little effect on the mutation frequency.
In recA1 overexpression strains (RA1+ and I− R+), recA1 showed higher expression levels (Fig. 5, lanes 9 and 10), and the mutation frequencies of the two strains were significantly repressed, regardless of UV radiation, which indicated that RecA1 could inhibit bacterial mutation. Compared with the RA1+ strain, the overexpression of the recA1 gene in the imuA mutant (I− R+) more clearly showed inhibition of bacterial mutation and fully lost the UV inducibility of the mutation frequency, similar to that in the imuB-dnaE2 mutant. The results show that imuA participates in SOS mutagenesis by inhibiting RecA1 in M. xanthus.
DISCUSSION
The sequenced bacterial genomes in the KEGG database were queried for imuA, imuB, and dnaE2 sequences. A total of 2,062 genomes containing dnaE2 appeared in the search, of which 1,249 genomes belong to Proteobacteria, 632 belong to Actinobacteria, and the remaining 181 belong to Bacteroidetes, Chloroflexi, Deinococcus-Thermus, Firmicutes, and so on (see Fig. S6A in the supplemental material). Out of the 2,062 dnaE2-containing genomes, 1,793 contain an imuB gene (86.95% coexistence rate), suggesting a functional correlation between ImuB and DnaE2. Furthermore, out of the 1,793 genomes with dnaE2 and imuB, 1,234 contain a recA analog (defined as imuA) in front of imuB. The coexistence ratio of imuA and dnaE2 is approximately 59.84% of the total strains containing the dnaE2 gene (Fig. S6B). Although the coexistence ratio of imuA is lower than that of imuB, the presence of ImuA is important for SOS mutagenesis catalyzed by DnaE2. For example, both Streptomyces coelicolor and M. tuberculosis belong to the actinobacteria, and both encode the DnaE2 protein. S. coelicolor lacks the imuA gene, and its DnaE2 cannot function in SOS mutagenesis (15), while M. tuberculosis encodes ImuA, which plays an important role in SOS mutagenesis catalyzed by DnaE2 (16).
M. xanthusimuA, imuB, and dnaE2 are all SOS genes, which can be induced by UV radiation, but their expression is LexA/RecA independent (30). The fact that RecA1 does not regulate the expression of imuA, imuB, and dnaE is beneficial for studying the role of RecA1 in DnaE2-catalyzed SOS mutagenesis and discovering the interaction between ImuA and RecA1. In addition, the expression level of RecA1 is very low in M. xanthus (22, 31), which may lead to the disadvantage of RecA1-catalyzed error-free template switching in competition with DnaE2-catalyzed error-prone TLS, and thus, the inhibition of ImuA on RecA1 activity has less of an effect on the mutation frequency. This may be the reason why the effect of the imuA deletion on the mutation frequency is not as obvious as that of the imuB or dnaE2 deletion (Fig. 2) and is supported by the fact that the deletion of recA1 has little effect on the bacterial mutation frequency (Fig. 5). The overexpression of recA1 significantly inhibited the mutation frequency in both irradiated and nonirradiated cells, and the deletion of imuA in the recA1 overexpression strain showed significant inhibition of SOS mutagenesis, similar to the imuA mutation in other bacteria (10–14).
The recA gene is highly conserved in the genomes with the dnaE2 gene (Table S3). A total of 2,051 out of 2,062 genomes containing dnaE2 genes also contained the recA gene (KEGG data in February 2021). Inevitable competition exists between RecA-catalyzed template switching and DnaE2-catalyzed TLS on the stalled replication fork. In E. coli, RecA-mediated homologous recombination and the Pol V-mediated TLS process compete at the stalled replication fork (32). To prevent homologous recombination, Pol V and RecA, located in the 3′ terminus of the RecA filament, formed an active mutasome and catalyzed RecA filaments to decompose from the template in the 3′-to-5′ direction in the presence of SSB (33). In DnaE2-catalyzed TLS, ImuA, a RecA-like protein, probably undertook the function of inhibiting RecA recombination by binding to the 3′ termini of RecA filaments.
In addition to inhibiting RecA1, ImuA could interact with ImuB (Fig. S3), which has also been confirmed in M. tuberculosis, presumably required for the assembly of ImuB-DnaE2 onto DNA (16). So M. xanthus ImuA may play a role in the initial stage of replication fork restart repair, choosing one of the two restart pathways to repair the stalling replication fork (Fig. 6). In detail, DNA damage blocks the replication process, exposing ssDNA as a DNA damage signal to activate stalled fork repair. RecA protein was bound to the ssDNA at the stalled replication fork to form nucleoprotein filaments to initiate the template-switching pathway. The binding of ImuA to the RecA filament hindered the recombination ability of RecA, thus inhibiting template switching. Meanwhile, the interaction of ImuA and ImuB may promote the assembly of the ImuB-DnaE2 dimer on the DNA replication fork. As ImuB-DnaE2 binds to DNA, RecA filaments disaggregate, release RecA away from DNA, and initiate the TLS pathway.
FIG 6.

Hypothetical model of ImuA function in template switching and TLS in M. xanthus. Bacteria have two pathways to rescue stalled replication forks, error-prone TLS and error-free template switching. When replication encounters a DNA damage lesion and is blocked, ssDNA is exposed as a DNA damage signal to activate the SOS response. The SOS genes, including recA1, imuA, imuB, and dnaE2, are induced. RecA protein binds to the ssDNA at the stalled replication fork to form nucleoprotein filaments and initiates the template-switching pathway. At the same time, DnaE2 approaches the stalled replication fork to initiate the TLS pathway. The combination of ImuA and RecA hinders the extension of RecA nucleoprotein filaments and the recombinase activity of RecA, thus inhibiting template switching. ImuB guides DnaE2 to assemble on the replication fork by binding to ImuA/β-clamp to start TLS. In the absence of ImuA, RecA binds to the exposed ssDNA to form a filament, which promotes error-free template switching.
ImuB is a homolog of the UmuC subunit of Pol V and has the potential to bind to RecA, ImuA, and the β-clamp (17, 28). The binding of ImuB to RecA/ImuA was probably through its C-terminal tail (similar to the RecA N-terminal [RecA-NT] site) binding to the core ATPase domain (core-PM site) of RecA/ImuA, similar to the pattern of RecA filaments (34). Thus, ImuB has the potential to detach the RecA/ImuA protein from the filaments in the 3′-to-5′ direction by competitive binding to the core-PM site of RecA/ImuA (Fig. 6), and the interactions among ImuA, ImuB, and RecA1, as well as their further interaction with DnaE2 or other subunits of DNA polymerase, demand an in-depth investigation to reveal SOS mutagenesis catalyzed by DnaE2.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Details of bacterial strains, plasmids, and oligonucleotides used in this study are provided in Tables 1 and 2. The M. xanthus strains were cultivated in Casitone-based Casitone-yeast extract (CYE) medium for growth assays, and E. coli strains were routinely incubated on Luria-Bertani (LB) medium. M. xanthus strains were incubated at 30°C, and E. coli strains were incubated at 37°C. Solid or liquid medium was supplemented with 33 μg/ml of kanamycin (Kan) or 100 μg/ml of ampicillin (Amp) as required.
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Genotype and/or description | Reference or source |
|---|---|---|
| Strains | ||
| M. xanthus | ||
| DK1622 | Wild-type strain | 21 |
| IA | DK1622 (ΔimuA) | This study |
| IA+ | DK1622::pSW3991 (imuA with its promoter integrated at the attB site) | This study |
| IB | DK1622 (ΔimuB) | This study |
| YL1601 | DK1622 (ΔdnaE2) | 23 |
| E. coli | ||
| DH5α(λpir) | F− 80dlacZΔM15 Δ(lacZYA-argF)U169 deoR recA1 endA1 hsdR17 (rK− mK+) phoA supE44 λ− thi-1 gyrA96 relA1 | TaKaRa |
| BL21(DE3) | Expression strain; λ(DE3 [lacI lacUV5-T7 gene 1 ind1 sam7 nin5]) [malB+] K-12(λS) | This study |
| Saccharomyces cerevisiae | ||
| AH109 | MATatrp1-901 leu2-3,112 ura3-52 his3-200 gal4Δ gal80Δ LYS2::GAL1UAS-GAL1TATA-HIS3 MEL1 GAL2UAS-GAL2TATA-ADE2 URA3::MEL1UAS-MEL1TATA-lacZ | Clontech |
| Plasmids | ||
| pBJ113 | Gene replacement vector with galK cassette; Kanr | 35 |
| pBJ3991 | Upstream and downstream homologous arms of imuA inserted into the EcoRI/HindIII site of pBJ113; Kanr | This study |
| pGBK-T7 | Yeast two-hybrid vector; Kanr | Clontech |
| pGAD-T7 | Yeast two-hybrid vector; Ampr | Clontech |
| pSWU19 | Site-specific integration vector with Mx8 attP integration site; Kanr | 36 |
| pSW3991 | imuA with its upstream 247-bp promoter sequence, inserted into the XbaI/EcoRI sites of pSWU19; Kanr | This study |
| pET3a | E. coli expression vector; Ampr | Novagen |
| pET15b | E. coli expression vector; Ampr | Novagen |
TABLE 2.
Primers used in this study
| Primer | Sequence (5′–3′) | |
|---|---|---|
| Gene knockout | ||
| MXAN_3991_UF | CGGGAAGTCCAGCACCACGC | |
| MXAN_3991_UR | ACGGATCCAACGGGCTGGGCATCCTGGG | |
| MXAN_3991_DF | TTGGATCGCTCATCTCGCCTCCTCGCC | |
| MXAN_3991_DR | CGTGACGGAGCCGCTGGTGT | |
| MXAN_3990_UF | CTCGTGAATGGTGGGTGACGGTGC | |
| MXAN_3990_UR | AAGGATCCGGCTGGGCGGGGAGTGGTGGT | |
| MXAN_3990_DF | TTGGATCCTGCCTTCAGCGCCGAGGTGGA | |
| MXAN_3990_DR | GTGGGCTGCTCCTGCTGCTGACTT | |
| MXAN_1441_UF | ATGGATCCGCCGCCGCCACTGCCTTCA | |
| MXAN_1441_UR | GTATCCACACCCGTCACTTCC | |
| MXAN_1441_DF | GACCGCACGGGGCTCTTCAAT | |
| MXAN_1441_DR | ATGGATCCTAGACGGAGGACGCCAACAC | |
| Gene transcription | ||
| Fa | GGCTGCTCCTGCTGCTGACT | |
| Ra | CGGCAGGGCCGCGTCCAGTC | |
| Fb | TGGCCTTCGCCTCCACCTCG | |
| Rb | GTATTCCATGGGCCGCCTCG | |
| Gene overexpression | ||
| MXAN_3991_OF | CCGAATTCGACAAGAAGGTGGAAGTGAAGG | |
| MXAN_3991_OR | ATTCTAGAGAAGCGGGTGAGGTGCAGATA | |
| pilAp-up | TTCCCGGGCTGGCGAACTACTTCCTGTCC | |
| pilAp-down | CTTCTCCGCCAGCTTGCTCATGGGGGTCCTCAGAGAAGGTTG | |
| MXAN_1441_OF | CAACCTTCTCTGAGGACCCCCATGAGCAAGCTGGCGGAGAAG | |
| MXAN_1441_OR | AAGGTACCCGGTCAAGCTGGACGTGTT | |
| Protein expression | ||
| MXAN_1441_PF | AGGGTACCAAAACCACCGCCGAGGATGAC | |
| MXAN_1441_PR | AATCTAGACACTGCCTTCAGCTTCTCCGC | |
| MXAN_3982_PF | AAGGTACCAGAGCTGCCACGCGGTGGGAG | |
| MXAN_3982_PR | ATTCTAGAGCGGCAGACCAGCTCGGCGTA | |
| MXAN_3991_PF | TTGGTACCGCAGCGGAGCCCTTCTTCTT | |
| MXAN_3991_PR | TTTCTAGACCCCTGCCCCGCTCCCACTCG | |
| RT-PCR | ||
| RTPCR_3982_F | ATCATATGGACTACGCCGAGCTGGT | |
| RTPCR_3982_R | ACGGGGTGCGGCTCATCCTGGGCG | |
| RTPCR_3991_F | GTGGGCTGCTCCTGCTGCTGA | |
| RTPCR_3991_R | GGCGTGCCGTCCTCCGTGTCCT | |
| RTPCR_3990_F | TTCAGCGTGAAGGTGAGCCG | |
| RTPCR_3990_R | CGCTGGAAGAGGTGTTGGAGGA | |
| RTPCR_5350_F | GCACGAGCACGTCAGCCTCT | |
| RTPCR_5350_F | AGGCGCGTGATTTCATCGAG | |
| 3-kbp dsDNA | ||
| FM13 | ACTATTACCCCCTCTGGCAAAACT | |
| RM13 | GAAACCAATCAATAATCGGCTGTC | |
Gene mutation and overexpression in M. xanthus.
Markerless deletion mutation was carried out to knock out the imuA or imuB gene in M. xanthus DK1622 using the pBJ113 plasmid. The plasmid contained a kanamycin resistance cassette for the first round of screening and a galK gene for negative screening (35, 36). To avoid a potential effect on downstream gene expression, the middle sequence of the imuA gene was deleted, and 9 bp from the N terminus and 93 bp from the C terminus were retained (see Fig. S2 in the supplemental material). Similarly, the middle sequence of imuB was deleted, and 54 bp of the N terminus and 99 bp of the C terminus were kept (Fig. S2). Briefly, up- and downstream homologous arms were amplified using primers (Table 2) and ligated using the BamHI site. The fragment was inserted into the EcoRI/HindIII site of pBJ113. The resulting plasmid was introduced into M. xanthus strains via an electroporation method (1.25 kV, 200 W, 25 mF, and 1-mm cuvette gap). The second round of screening was performed on CYE plates containing 1% galactose (Sigma). The imuA mutant (named IA) and the imuB mutant (named IB) were identified and verified by PCR amplification and sequencing (Fig. S2).
To overexpress the imuA gene, the whole imuA gene, along with its promoter, was amplified and cloned into the M. xanthus integrative vector pSWU19 (37). The recombinant plasmid was transformed into M. xanthus DK1622 cells. Individual kanamycin-resistant clones were selected and validated using PCR amplification. We also transformed the empty vector into the M. xanthus wild-type strain as a control.
To analyze the effect of the imuA deletion on the mutation frequency under the genetic background of recA1 deletion or overexpression, we further deleted or overexpressed recA1 in DK1622 and IA, respectively. The recA1 deletion and overexpression strains were constructed as described above, using the primers listed in Table 2. Deletion of the recA1 gene was carried out using the pBJ113 plasmid according to the protocol described above. To overexpress the recA1 gene, the recA1 gene was ligated under the control of a pilA promoter, cloned into the XbaI-KpnI sites of the pSWU19 vector, and transformed into M. xanthus strains (DK1622 and IA). The resulting strains were identified by PCR and sequencing.
Restriction enzymes, DNA ligase, and other enzymes were purchased from Thermo Scientific (Shanghai, China) and used according to the manufacturer’s recommendations. All DNA products were validated using DNA sequencing.
Growth, irradiation, and mutation frequency assays.
M. xanthus cells in exponential phase were used to determine cell growth and mutation frequency. For UV-C irradiation, 15 ml of the cell suspension in 130-mm glass petri dishes was stirred gently with a magnetic rod and irradiated with UV-C rays (254 nm) at room temperature. Next, the cells were diluted in fresh medium and incubated at 30°C. The density of the cells was measured every 12 h to generate growth curves.
The mutation frequency assay was conducted by screening nalidixic acid-resistant strains as described previously (24). Briefly, cells in logarithmic phase were mock irradiated or irradiated with 15 J/m2 UV-C. After a 4-h acclimation period, approximately 5 × 109 M. xanthus cells were placed on CYE agar containing 40 μg/ml nalidixic acid and incubated for 5 days. Nalidixic acid-resistant candidates were counted, and the mutation frequency was calculated using the following formula: mutation frequency = nalidixic acid-resistant clones/5 × 109.
RNA extraction, RT-PCR, and RNA sequencing.
M. xanthus cells in the logarithmic phase were UV-C irradiated at doses of 0, 5, and 15 J/m2, and these cells were then diluted in fresh medium and incubated for 0 to 7 h. Total RNA from these cells was extracted using RNAiso Plus reagent (TaKaRa, Beijing, China) according to the manufacturer’s protocol. The RNA concentration was quantified using a Nanodrop 2000 instrument (Thermo Fisher Scientific, USA). The cDNA was synthesized using a PrimeScript reverse transcription (RT) reagent kit (TaKaRa, Beijing, China). Genomic DNA (gDNA) in the RNA sample was removed with gDNA Eraser (TaKaRa, Beijing, China). First-strand cDNA was synthesized by using the PrimeScript RT enzyme according to the manufacturer’s instructions with random primers.
The resulting cDNA was diluted 5-fold before performing RT-PCR. The primers designed for the imuA, dnaE2, recA1, and recA2 genes are listed in Table 2. RT-PCR was accomplished using the SYBR premix ExTaq kit (TaKaRa, China) on an ABI StepOnePlus real-time PCR system (Thermo Fisher Scientific, USA) with the following program: 3 min at 95°C, followed by 40 cycles of 30 s at 95°C, 30 s at 55°C, and 15 s at 72°C. The relative quantification of mRNAs of interest was performed by the comparative threshold cycle (CT) (2−ΔΔCT) method, using the gapA (glyceraldehyde-3-phosphate dehydrogenase) gene as an endogenous control, as described previously (23). The mock-treated samples (0 J/m2) were used as a control.
RNA sequencing (RNA-seq) was conducted by a sequencing company (Vazyme, Nanjing, China). Briefly, purified double-stranded cDNA was end repaired, A tailed, and ligated to the sequencing linker. After PCR amplification, the cDNA was sequenced on the Illumina HiSeq 4000 platform to generate >30 million reads for each sample. Three replicates were sequenced for each sample, and the sequencing reads were processed using HTSeq software (Illumina, CA, USA). All the upregulated and downregulated genes were obtained by comparison with the control, and their gene functions were explored using database annotations such as Gene Ontology (GO) and KEGG.
Expression, purification, and pulldown analyses of ImuA, DnaE2, and RecA1.
Genes imuA, recA1, and dnaE2 were cloned into E. coli expression vectors pET15b and pET3a, respectively. Next, these recombinant plasmids were transformed into Escherichia coli BL21(DE3) to express proteins. For ImuA and RecA1 proteins, the strains were cultured to an optical density at 600 nm (OD600) of approximately 0.5 at 37°C, and protein expression was induced with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) for 4 h. For DnaE2 protein expression, the strains were cultured at 30°C until the OD600 reached 0.5, and IPTG was added to a final concentration of 1 mM to induce protein expression for 6 h at 30°C. Cells were harvested and sonicated to prepare the crude extract. 6×His-tagged proteins were purified from the crude extract using Ni-nitrilotriacetic acid (NTA) agarose columns (Qiagen) according to the manufacturer’s instructions, and no 6×His-tagged proteins were purified by salting out and ion-exchange chromatography.
In the His pulldown analysis, purified His-tagged proteins were first incubated with Ni-NTA agarose beads. Subsequently, the beads were extensively washed and incubated with no His-tag-labeled protein. The proteins bound to the beads were eluted with 150 mM imidazole and separated by SDS-PAGE.
Electrophoretic mobility shift assay.
To test the DNA-binding ability, electrophoretic mobility shift assays were performed in reaction mixtures containing 25 mM Tris-HCl (pH 7.0), 50 mM NaCl, 4% glycerol, 1 mM dithiothreitol (DTT), 10 mM MgCl2, 1.5 mM ATP, M13 circle ssDNA or 3-kb dsDNA (derived from M13), and protein (DNA and protein concentrations depicted in the figures). The solution was incubated at 32°C for 20 min and electrophoresed on a 1% agarose gel for 2 h at 2 V cm−1 at 4°C. This gel was stained with ethidium bromide. This experiment was performed three times, and a representative blot is shown.
DNA strand exchange reactions.
The RecA-dependent DNA strand exchange reaction was carried out as described previously (38) between M13 circular ssDNA and the linear dsDNA (3-kbp fragment derived from M13). The reactions were carried out at 30°C in strand exchange buffer (SEB) containing 25 mM Tris-HCl (pH 7.0), 50 mM NaCl, 4% glycerol, 1 mM DTT, 10 mM MgCl2, and an ATP-regenerating system (10 U/ml of pyruvate kinase and 3.3 mM phosphoenolpyruvate). After preincubation of ssDNA with RecA1 protein at 30°C for 5 min, the reaction mixture was supplemented with 1.5 mM ATP and ImuA, and incubation was continued for 5 min. Linear dsDNA was added to start the DNA strand exchange reactions. The reactions were stopped by the addition of 5 μl of gel loading buffer (0.125% bromophenol blue, 25 mM EDTA, 25% glycerol, and 5% SDS) to the reaction mixture. Furthermore, these samples were electrophoresed in an 0.8% agarose gel with Tris-acetate EDTA (TAE) buffer.
Yeast two-hybrid assay.
The yeast two-hybrid phenotypic assay to evaluate protein interactions was performed as described previously (39). Genes imuA, imuB, dnaE2, dnaE1, recA1, recA2, recO, recR, dnaN, lexA, and ssb were cloned into the pGBKT7 and pGADT7 plasmids. These plasmids were transformed into yeast AH109 cells, and interacting phenotypes were screened for the ability to grow on selective medium with 3-amino-1,2,4-triazole (3-AT). These experiments were repeated three times.
Analysis of binding of ImuA to RecA-coated ssDNA.
M13mp18 ssDNA was immobilized on CNBr-activated Sepharose 4B (GE), as described previously (40). Twenty microliters of the agarose beads containing 200 ng M13 plus strand (∼0.6 nmol) was mixed with 14.4 μg RecA1 protein (∼0.4 nmol) in 200 μl of SEB buffer for 5 min at 30°C by constant tapping. Theoretically, 1 RecA molecule binds to 3 nucleotides, and 14.4 μg RecA1 is twice the amount of RecA required to theoretically bind 200 ng of DNA. The beads were captured by centrifugation at low speed and washed twice with SEB buffer to remove unbound proteins. Next, 12 μg ImuA protein was loaded onto the RecA-DNA-coated beads, and incubation was continued at 30°C for 5 min. The unbound protein was washed out with SEB buffer. The DNA-binding proteins on beads were cut using ultrasonic treatment, collected by centrifugation, and separated by SDS-PAGE.
Sequence analysis.
Genes were retrieved from the sequenced bacterial genomes in the KEGG database, and the gene loci were downloaded from the KEGG Genome map database (https://www.genome.jp). The nucleotide sequences of imuA, imuB, and dnaE2 were obtained from GenBank, and their promoters were determined by sequence annotations and further software analysis using the Berkeley Neural Network Promoter Prediction tool (http://www.fruitfly.org/seq_tools/promoter.html).
Protein domain analysis was performed on a Pfam-based PRODOM database using multiple-sequence alignments, and the three-dimensional structure was constructed using Swiss-Model (https://swissmodel.expasy.org) and visualized using PyMOL (version 1.20; https://pymol.org).
Data availability.
All data sets generated for this study are included in the manuscript and the supplemental material. The raw sequencing data generated from this study have been deposited in the NCBI SRA under the accession number SRP312127.
ACKNOWLEDGMENTS
This work was financially supported by funding from the National Natural Science Foundation of China (NSFC) (numbers 31670076 and 31471183), the National Key Research and Development Programs of China (numbers 2018YFA0900400 and 2018YFA0901704), the Key Program of the Shandong Natural Science Foundation (number ZR2016QZ002), and Special Investigation on Scientific and Technological Basic Resources (number 2017FY100302) to Y.L. and by from the Special National Project on the Investigation of Basic Resources of China (number 2019FY100700) and the Key Research and Developmental Program of Shandong Province (number 2019JZZY020308) to D.S.
We declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Supplemental material is available online only.
Contributor Information
Duohong Sheng, Email: dhsheng@sdu.edu.cn.
Yuezhong Li, Email: lilab@sdu.edu.cn.
Maia Kivisaar, University of Tartu.
REFERENCES
- 1.Conti B, Smogorzewska A. 2020. Mechanisms of direct replication restart at stressed replisomes. DNA Repair (Amst) 95:102947. 10.1016/j.dnarep.2020.102947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Agashe D. 2017. The road not taken: could stress-specific mutations lead to different evolutionary paths? PLoS Biol 15:e2002862. 10.1371/journal.pbio.2002862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hershberg R. 2015. Mutation—the engine of evolution: studying mutation and its role in the evolution of bacteria. Cold Spring Harb Perspect Biol 7:a018077. 10.1101/cshperspect.a018077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lovett ST. 2017. Template-switching during replication fork repair in bacteria. DNA Repair (Amst) 56:118–128. 10.1016/j.dnarep.2017.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shee C, Gibson JL, Darrow MC, Gonzalez C, Rosenberg SM. 2011. Impact of a stress-inducible switch to mutagenic repair of DNA breaks on mutation in Escherichia coli. Proc Natl Acad Sci U S A 108:13659–13664. 10.1073/pnas.1104681108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goodman MF, Woodgate R. 2013. Translesion DNA polymerases. Cold Spring Harb Perspect Biol 5:a010363. 10.1101/cshperspect.a010363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schlacher K, Goodman MF. 2007. Lessons from 50 years of SOS DNA-damage-induced mutagenesis. Nat Rev Mol Cell Biol 8:587–594. 10.1038/nrm2198. [DOI] [PubMed] [Google Scholar]
- 8.Fuchs RP, Fujii S, Wagner J. 2004. Properties and functions of Escherichia coli Pol IV and Pol V. Adv Protein Chem 69:229–264. 10.1016/S0065-3233(04)69008-5. [DOI] [PubMed] [Google Scholar]
- 9.Jaszczur M, Bertram JG, Robinson A, van Oijen AM, Woodgate R, Cox MM, Goodman MF. 2016. Mutations for worse or better: low-fidelity DNA synthesis by SOS DNA polymerase V is a tightly regulated double-edged sword. Biochemistry 55:2309–2318. 10.1021/acs.biochem.6b00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abella M, Erill I, Jara M, Mazón G, Campoy S, Barbé J. 2004. Widespread distribution of a lexA-regulated DNA damage-inducible multiple gene cassette in the Proteobacteria phylum. Mol Microbiol 54:212–222. 10.1111/j.1365-2958.2004.04260.x. [DOI] [PubMed] [Google Scholar]
- 11.Alves IR, Lima-Noronha MA, Silva LG, Fernández-Silva FS, Freitas ALD, Marques MV, Galhardo RS. 2017. Effect of SOS-induced levels of imuABC on spontaneous and damage-induced mutagenesis in Caulobacter crescentus. DNA Repair (Amst) 59:20–26. 10.1016/j.dnarep.2017.09.003. [DOI] [PubMed] [Google Scholar]
- 12.Boshoff HI, Reed MB, Barry CE, Mizrahi V. 2003. DnaE2 polymerase contributes to in vivo survival and the emergence of drug resistance in Mycobacterium tuberculosis. Cell 113:183–193. 10.1016/s0092-8674(03)00270-8. [DOI] [PubMed] [Google Scholar]
- 13.Erill I, Campoy S, Mazon G, Barbé J. 2006. Dispersal and regulation of an adaptive mutagenesis cassette in the bacteria domain. Nucleic Acids Res 34:66–77. 10.1093/nar/gkj412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Galhardo RS, Rocha RP, Marques MV, Menck CF. 2005. An SOS-regulated operon involved in damage-inducible mutagenesis in Caulobacter crescentus. Nucleic Acids Res 33:2603–2614. 10.1093/nar/gki551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsai HH, Shu HW, Yang CC, Chen CW. 2012. Translesion-synthesis DNA polymerases participate in replication of the telomeres in Streptomyces. Nucleic Acids Res 40:1118–1130. 10.1093/nar/gkr856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Warner DF, Ndwandwe DE, Abrahams GL, Kana BD, Machowski EE, Venclovas C, Mizrahi V. 2010. Essential roles for imuA′- and imuB-encoded accessory factors in DnaE2-dependent mutagenesis in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 107:13093–13098. 10.1073/pnas.1002614107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Timinskas K, Balvočiūtė M, Timinskas A, Venclovas Č. 2014. Comprehensive analysis of DNA polymerase III a subunits and their homologs in bacterial genomes. Nucleic Acids Res 42:1393–1413. 10.1093/nar/gkt900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jiang Q, Karata K, Woodgate R, Cox MM, Goodman MF. 2009. The active form of DNA polymerase V is UmuD′(2) C-RecA-ATP. Nature 460:359–363. 10.1038/nature08178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen X-J, Han K, Feng J, Zhuo L, Li Y-J, Li Y-Z. 2016. The complete genome sequence and analysis of a plasmid-bearing myxobacterial strain Myxococcus fulvus 124B02 (M 206081). Stand Genomic Sci 11:1. 10.1186/s40793-015-0121-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goldman BS, Nierman WC, Kaiser D, Slater SC, Durkin AS, Eisen JA, Ronning CM, Barbazuk WB, Blanchard M, Field C, Halling C, Hinkle G, Iartchuk O, Kim HS, Mackenzie C, Madupu R, Miller N, Shvartsbeyn A, Sullivan SA, Vaudin M, Wiegand R, Kaplan HB. 2006. Evolution of sensory complexity recorded in a myxobacterial genome. Proc Natl Acad Sci U S A 103:15200–15205. 10.1073/pnas.0607335103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaiser D. 1979. Social gliding is correlated with the presence of pili in Myxococcus xanthus. Proc Natl Acad Sci U S A 76:5952–5956. 10.1073/pnas.76.11.5952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sheng DH, Wang YX, Qiu M, Zhao JY, Yue XJ, Li YZ. 2020. Functional division between the RecA1 and RecA2 proteins in Myxococcus xanthus. Front Microbiol 11:140. 10.3389/fmicb.2020.00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peng R, Chen JH, Feng WW, Zhang Z, Yin J, Li ZS, Li YZ. 2017. Error-prone DnaE2 balances the genome mutation rates in Myxococcus xanthus DK1622. Front Microbiol 8:122. 10.3389/fmicb.2017.00122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tzeng L, Ellis TN, Singer M. 2006. DNA replication during aggregation phase is essential for Myxococcus xanthus development. J Bacteriol 188:2774–2779. 10.1128/JB.188.8.2774-2779.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baharoglu Z, Mazel D. 2014. SOS, the formidable strategy of bacteria against aggressions. FEMS Microbiol Rev 38:1126–1145. 10.1111/1574-6976.12077. [DOI] [PubMed] [Google Scholar]
- 26.Kreuzer KN. 2013. DNA damage responses in prokaryotes: regulating gene expression, modulating growth patterns, and manipulating replication forks. Cold Spring Harb Perspect Biol 5:a012674. 10.1101/cshperspect.a012674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weissman KJ, Müller R. 2010. Myxobacterial secondary metabolites: bioactivities and modes-of-action. Nat Prod Rep 27:1276–1295. 10.1039/c001260m. [DOI] [PubMed] [Google Scholar]
- 28.Del Val E, Nasser W, Abaibou H, Reverchon S. 2019. RecA and DNA recombination: a review of molecular mechanisms. Biochem Soc Trans 47:1511–1531. 10.1042/BST20190558. [DOI] [PubMed] [Google Scholar]
- 29.Leite WC, Galvão CW, Saab SC, Iulek J, Etto RM, Steffens MB, Chitteni-Pattu S, Stanage T, Keck JL, Cox MM. 2016. Structural and functional studies of H. seropedicae RecA protein—insights into the polymerization of RecA protein as nucleoprotein filament. PLoS One 11:e0159871. 10.1371/journal.pone.0159871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sheng D-H, Wang Y, Wu S-G, Duan R-X, Li Y-Z. 24May2021. The regulation of LexA on UV induced SOS response in Myxococcus xanthus based on transcriptome analysis. J Microbiol Biotechnol 10.4014/jmb.2103.03047. [DOI] [PMC free article] [PubMed]
- 31.Norioka N, Hsu MY, Inouye S, Inouye M. 1995. Two recA genes in Myxococcus xanthus. J Bacteriol 177:4179–4182. 10.1128/jb.177.14.4179-4182.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Naiman K, Pagès V, Fuchs RP. 2016. A defect in homologous recombination leads to increased translesion synthesis in E. coli. Nucleic Acids Res 44:7691–7699. 10.1093/nar/gkw488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pham P, Bertram JG, O’Donnell M, Woodgate R, Goodman MF. 2001. A model for SOS-lesion-targeted mutations in Escherichia coli. Nature 409:366–370. 10.1038/35053116. [DOI] [PubMed] [Google Scholar]
- 34.Timinskas K, Venclovas Č. 2019. New insights into the structures and interactions of bacterial Y-family DNA polymerases. Nucleic Acids Res 47:4393–4405. 10.1093/nar/gkz198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Julien B, Kaiser AD, Garza A. 2000. Spatial control of cell differentiation in Myxococcus xanthus. Proc Natl Acad Sci U S A 97:9098–9103. 10.1073/pnas.97.16.9098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ueki T, Inouye S, Inouye M. 1996. Positive-negative KG cassettes for construction of multi-gene deletions using a single drug marker. Gene 183:153–157. 10.1016/s0378-1119(96)00546-x. [DOI] [PubMed] [Google Scholar]
- 37.Wu SS, Kaiser D. 1997. Regulation of expression of the pilA gene in Myxococcus xanthus. J Bacteriol 179:7748–7758. 10.1128/jb.179.24.7748-7758.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shan Q, Cox MM, Inman RB. 1996. DNA strand exchange promoted by RecA K72R. Two reaction phases with different Mg2+ requirements. J Biol Chem 271:5712–5724. 10.1074/jbc.271.10.5712. [DOI] [PubMed] [Google Scholar]
- 39.Fields S, Sternglanz R. 1994. The two-hybrid system: an assay for protein-protein interactions. Trends Genet 10:286–292. 10.1016/0168-9525(90)90012-u. [DOI] [PubMed] [Google Scholar]
- 40.Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. 1999. Short protocols in molecular biology, 3rd ed. John Wiley & Sons, New York, NY. [Google Scholar]
- 41.Campoy S, Fontes M, Padmanabhan S, Cortés P, Llagostera M, Barbé J. 2003. LexA-independent DNA damage-mediated induction of gene expression in Myxococcus xanthus. Mol Microbiol 49:769–781. 10.1046/j.1365-2958.2003.03592.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures S1 to S6, Tables S1 to S3. Download AEM.00919-21-s0001.pdf, PDF file, 1.6 MB (1.6MB, pdf)
Data Availability Statement
All data sets generated for this study are included in the manuscript and the supplemental material. The raw sequencing data generated from this study have been deposited in the NCBI SRA under the accession number SRP312127.