

Abstract
Many germ cells are eliminated during development, long before oogenesis or spermatogenesis. In mouse foetal testes, the majority of germ cell apoptosis coincides with the onset of male differentiation, suggesting coordination of these processes. We studied foetal germ cell fates and discovered that both apoptosis and differentiation initiate in clonally-related clusters. Lineage tracing confirmed that germ cells die as clones independent of intercellular bridges, suggesting that shared intrinsic properties are apoptotic determinants. We identified transcriptional heterogeneity among foetal germ cells that included an apoptosis-susceptible population characterized by failure to differentiate, whereas successful differentiation to prospermatogonia occurred through expression of epigenetically-regulated genes including LINE1. Our results indicate that foetal germ cell fate is based upon discrete, cell-heritable identities. Elevated DNA methylation in the apoptosis-susceptible subpopulation supports our hypothesis that earlier errors in germ cell epigenetic reprogramming derail differentiation in cellular progeny, leading to foetal apoptotic selection that ultimately improves gamete quality.
The transmission of genetic information through the gametes is a critical bottleneck for evolutionary selection. Most organisms produce gametes in excessive quantities, particularly in males. Gamete wastage is regarded as an optimization of parental energy investment, success of fertilization, and embryo fitness1. The precursors of mature gametes in the embryo similarly undergo overproduction and elimination. Importantly, only those precursors that survive will later contribute to sexual reproduction, so selection in the embryonic period can be highly influential in shaping the gametic, and genetic, pool.
Primordial germ cells (PGCs) are established at gastrulation in mammals2.Their subsequent development entails a lengthy migration3 and interaction with diverse niches that regulate PGC proliferation and motility4. Concurrently, PGCs undergo extensive epigenetic reprogramming5. Following migration to the gonads, PGCs respond to external signals by initiating sex-specific differentiation: mitotic arrest and male gene expression in the foetal testis or meiosis in the foetal ovary6,7. The ability of PGCs to respond appropriately to these developmental challenges determines their fitness to continue onward toward gametogenesis.
Apoptosis is a conserved feature of PGC development linked by phenotypes to reproductive fitness8. In mice, PGCs failing migration to the gonads succumb to apoptosis, prompted by the absence of growth factors in ectopic locations9. Blocking apoptosis increases the number of total and ectopic PGCs, resulting in male sterility10,11 or increased frequency of defects in oocytes12. During sex differentiation in the foetal testis from E13.5 to E17.5, germ cells are subjected to a stereotyped wave of death13,14 which depends on the Bax-mediated intrinsic but not the extrinsic apoptotic pathway10, and occurs in the absence of exogenous insults. As studies of Bax mutant mice have focused on postnatal defects in spermatogenesis, less is known about the foetal apoptotic wave and how the fates of individual PGCs are determined.
While apoptosis in the germline prevents participation in gametogenesis and genome propagation, other processes such as differentiation and proliferation positively select individual germ cells. Accumulated FGF pathway mutations in human testes confer a selective advantage in survival and proliferation to spermatogonial stem cells15. Studies with tetrachimeric mice suggest that selection during male germ cell development prunes the founder population for adult spermatogenesis16. The divergence in male and female development may explain differences in the selection landscape of developing foetal germ cells: although the metabolic cost to producing oocytes is higher, their individual capacity to contribute to offspring is more limited compared to that of self-renewing spermatogonial stem cells.
These studies, together with male sterility in Bax mutants, motivated our investigation of developmentally-scheduled apoptosis in the foetal testis to determine how eliminated PGCs differ from survivors. We found that PGC apoptosis acts clonally and yet autonomously to eliminate related PGCs with aberrant differentiation. We identified distinct PGC subpopulations with divergent fates of differentiation to prospermatogonia versus susceptibility to apoptosis due to contrasting expression and methylation of epigenetically-regulated genes. Our findings demonstrate that differences in epigenetic reprogramming in PGC ancestors can underlie clonal heterogeneity in survival and male sex differentiation. These results argue that apoptosis during male germline development functions as a quality control mechanism to promote efficient differentiation and ultimately fitter gametes.
RESULTS
Germ Cell Apoptosis is Spatially Clustered in FoetalTestes
To map dying germ cells during the wave of apoptosis, we employed 3D imaging in intact foetal testes. Wholemount immunostaining against a germ cell nuclear marker, TRA98, resolved individual PGCs. Using the late-stage apoptotic marker cleaved-PARP (cPARP), we observed an acute increase in PGC death from E12.5 through E14.5, consistent with previous characterizations (Extended Data Figure 1a)14. Although variable by background17 (Extended Data Figure 1b), the apoptotic frequency in PGCs in this window surpasses other embryonic tissues18 and E11.5 PGCs19 by an order of magnitude. Apoptotic PGCs in the testis from E12.5–14.5 were localized nonrandomly in clusters (Figure 1a, Supplemental Movie 1), as verified by Ripley’s K-function (Figure 1b). Spatial bias in apoptosis was not observed at a tissue-wide level as apoptotic clusters themselves were randomly distributed throughout the testis (Extended Data Figure 1c). Further investigation of the immediate environment of apoptotic clusters did not show correlation with distance or ratio between PGCs and supportive Sertoli cells (Extended Data Figure 1d–f) that promote postnatal PGC survival20. Thus, the local environment does not strongly contribute to PGC apoptosis.
Figure 1. Germ cell apoptosis is spatially clustered in foetal testes.
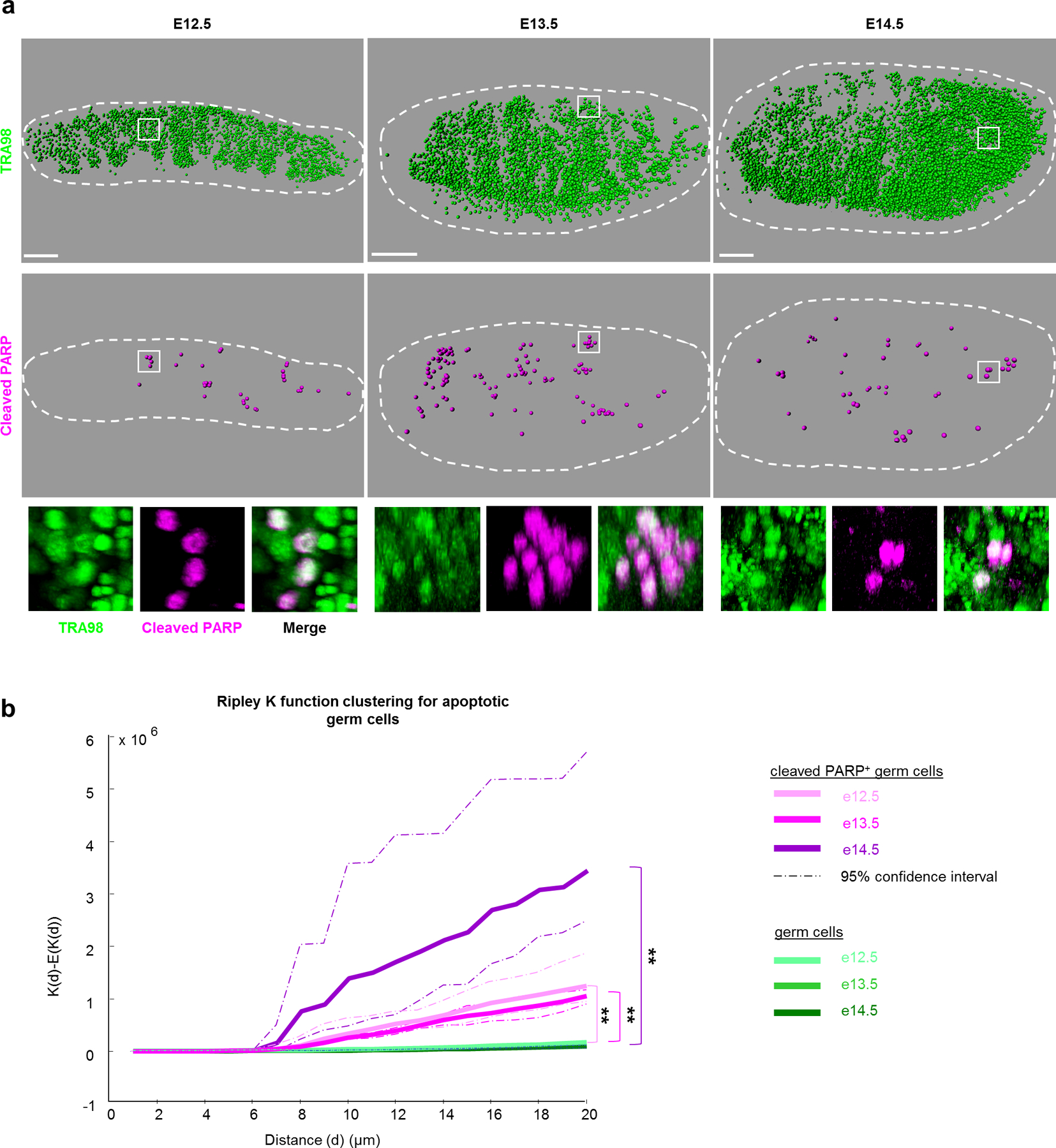
a, Wholemount imaging for germ cell nuclei and apoptosis enables spatial reconstructions of germ cell distributions in foetal testes during apoptotic period from E12.5-E14.5. Germ cells were identified by nuclear marker TRA98 and individually mapped in three-dimensional space. Apoptosis within this germ cell population was detected by cleaved PARP. Scale bar = 100μm. Bottom: Higher magnification examples of apoptotic germ cell clusters from wholemount imaging. b, Ripley K-factor analysis of spatial clustering of cleaved PARP+ germ cells from E12.5–E14.5 testes compared to the extent of clustering of all germ cells (TRA98+), n=3 animals for each timepoint **: p=0 for each comparison as calculated by RipleyGUI using a between-treatments sum of squares (see Methods).
Clustered apoptosis could arise from phylogenetically-conserved and specialized intercellular bridges between PGCs formed by incomplete cytokinesis beginning at E10.521. Intercellular bridges permit sharing of cytoplasmic components22 and could convey apoptotic signals between connected cells. To determine the contribution of bridges to apoptotic clustering, we examined the distribution of apoptotic PGCs in Tex14−/− mice lacking intercellular bridges23. The frequency of PGC apoptosis in Tex14 did not differ from that of wild-type littermates (Extended Data Figure 1g). Importantly, clusters of apoptotic PGCs were still detected in mutant testes (Figure 2a) and we statistically confirmed their clustered distribution (Figure 2b), although the average cluster size was smaller (Extended Data Figure 1h). That apoptotic clustering persists in Tex14−/− indicates that apoptotic potential is maintained in nearby disconnected cells. As cytoplasm is not shared between Tex14−/− PGCs, diffusible apoptotic signals are therefore not necessary for the observed apoptotic clustering. This contrasts with clonal apoptosis in Drosophila spermatogonia that requires intra-cyst cytoplasmic sharing24. It is likely that bridges in murine male PGCs coordinate some degree of timing, however this result raises the possibility that shared apoptotic potential among adjacent PGCs is cell autonomous.
Figure 2. Clustered germ cell apoptosis does not require cytoplasmic sharing.

a, Apoptotic germ cell distributions in Tex14−/− mutants lacking intercellular bridges among germ cells compared to littermate wild-type controls. Scale bar = 100μm. b, Spatial clustering of apoptotic germ cells in Tex14 compared to complete spatial randomness (CSR). Tex14+/+, n=3 animals;Tex14+/−, n=2 animals, Tex14−/− n=4 animals. CSR determined by 1,000 simulated random distributions. †† p=0.1716. p values were calculated by RipleyGUI using a between-treatments sum of squares (see Methods). c, Experimental design for clonal labelling of germ cells as well as interrogation of intercellular bridge function within labelled clones inTex14 mutants. d, Comparison of production or absence of bicoloured clones in E13.5 Rainbow testes on a Tex14 background. Left: Section view from wholemount imaging of a clone simultaneously expressing mCherry and mCerulean in Rainbow; Tex14+/+ where intercellular bridges are intact. Right: Disconnected clonal structure and absence of multicoloured clones in Tex14−/−. Scale bar = 20μm. e, Model for bicoloured clone formation in Tex14+/+ but not Tex14−/− through germ cell intercellular bridge-dependent cytoplasmic sharing when recombination is induced in multi-cell cysts.
Multicolour Labelling Reveals Clonal Germ Cell Apoptosis
To investigate lineage relationship among clustered apoptotic PGCs, we utilized multicolour reporter systems. By inducibly and permanently labelling multiple PGC clones with distinct colours in the same testis, we could compare behaviours in neighbouring, differently-labelled clones. Rosa26-Confetti25 or Rosa26-Rainbow26 mice were crossed to Pou5f1-CreERT227 and pulsed once with Tamoxifen at E10.5 to induce recombination and fluorophore expression exclusively in PGCs by E11–11.5, subsequent to migration (Figure 2c). By E13.5, distinctly-coloured PGC clones grew to a mean size of 8 (Supplemental Movie 2) and were consistent in size with previously described cysts25. At higher doses of Tamoxifen, we observed a low frequency of bicoloured clones (Figure 2d), which likely arose from separate recombination events in an interconnected 2-cell cyst (Figure 2e). By contrast, when multicolour labelling was performed on a Tex14−/− background, bicoloured clones were not observed and clones were more interdigitated than wild-type (Figure 2d, Supplemental Movie 3,4), confirming that cytoplasmic sharing is dependent upon intercellular bridges.
Throughout the apoptotic wave, we observed that individual clusters of lineage-marked cPARP+ PGCs (Figure 3a) shared the same colour (Figure 3b) and that apoptotic cells lay within the boundary occupied by a single clone (Extended Data Figure 2). We categorized the distribution of apoptosis in clonally-labelled local clusters and found it was strictly monoclonal (Figure 3c), even when differently-labelled clones were adjacent and interspersed. The absence of polyclonal apoptosis in a local cluster strongly argues against an extrinsic basis for apoptosis, including secreted pro-apoptotic factors. Instead, these results suggest that intrinsic, clonally-shared properties predispose subpopulations of PGCs toward apoptosis. To investigate if such pro-apoptotic potential is not only clonally shared but also cell-autonomous, we disrupted cytoplasmic sharing using Tex14−/− on our multicoloured clonal-labelling background. Apoptosis still occurred clonally despite constituent cells lacking bridges (Extended Data Figure 3a,b), indicating that apoptotic potential is maintained in individual cells and not reliant upon diffusible signals.
Figure 3: Multicolour clonal labelling reveals clonal apoptosis during the foetal apoptotic wave.
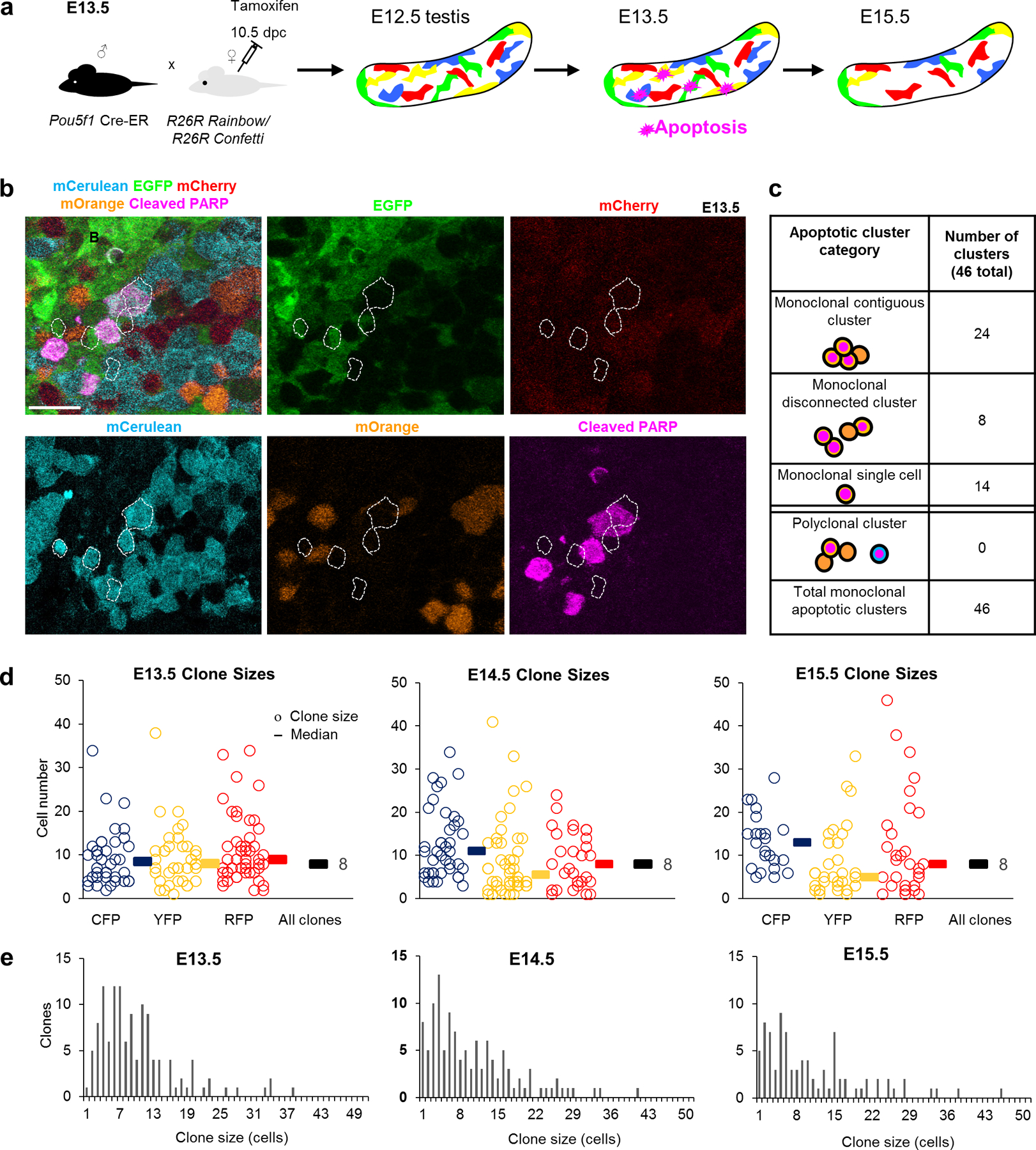
a, Experimental design for labelling of germ cells with multicolour reporters Confetti or Rainbow to determine the clonality of apoptosis. b, Apoptotic cluster in E13.5 Pou5f1-CreERT2; R26Rainbow testes containing cleaved-PARP+ germ cells strictly within a labelled (mCerulean) clone. Scale bar = 30μm. c, Clustered Confetti or Rainbow apoptotic germ cells categorized by cluster organization and clonal label. d, Clone size measurements during the apoptotic wave in Confetti testes (3 animals). Each clone is represented by a circle with the corresponding colour. Median clone sizes are shown for each colour as well as for all colours combined (black bars, median clone size value adjacent). e, Histogram displaying clone size distributions throughout apoptotic wave. One-way ANOVA for all 3 stages: p=0.806248.
Although many PGC clones were entirely or partially cPARP-positive, our detection of apoptosis was limited by the transience of cPARP in dying cells and the rapid subsequent cellular breakdown. To determine whether apoptosis cumulatively eliminates all cells in clones or only a subset, we compared the size of clones across development: at E13.5 following PGC mitotic arrest28 and at E15.5 concluding the apoptotic wave. A simple model of clonal dynamics during apoptosis predicts that partial death of a clone would manifest as a decline in average size of clones. However, we observed a consistent mean clone size of 8 from E13.5 until E15.5 past the conclusion of the apoptotic wave (Figure 3d,e). This concurs with single-colour labelling of one clone per testis23, demonstrating the scalability of a multicolour approach. Together, constant clone size and confined clonal apoptosis argue that PGCs in the foetal testis are eliminated based upon mitotically-heritable properties. Furthermore, distinct survival by clone suggests that the PGC population contains significantly heterogeneous subpopulations.
Single-cell RNA Sequencing Identifies an Apoptosis-Poised Germ Cell Subpopulation
To identify the basis of the observed heterogeneity in PGC fates, we performed single-cell RNA-sequencing (scRNA-seq) of purified wild-type PGCs from E13.5 testes during peak apoptosis (Figure 4a). scRNA-seq can reveal diverse cell states within the population that would be masked in bulk analysis. Oct4ΔPE-GFP+ PGCs purified from E13.5 testes yielded 2,556 transcriptomes, which clustered into 7 distinct subpopulations characterized by markers (Supplemental Table 1).
Figure 4: Single cell RNA sequencing of male germ cells identifies an apoptotically-poised subpopulation.

a, Isolation of E13.5 male germ cells for single cell RNA-sequencing. Transcriptional heterogeneity and clustering organise E13.5 germ cells into 7 populations as represented by t-distributed stochastic neighbour embedding (t-SNE). b, Average expression of apoptosis-related genes in each of the seven germ cell states, including Bcl2-family pro-apoptotic (Bad, Bax, Bim, Bid) and anti-apoptotic (Bcl-X) genes. Expression from n=2,556 single cells. c, Immunofluorescence staining for coexpression of p53 and apoptosis (cleaved-PARP) in E13.5 testis sections. Scale bar=30μm. Right: larger magnification of boxed area, scale bar = 10μm. d, Gene ontology of transcriptional markers upregulated or downregulated in the Trp53-high germ cell state 6 compared to all other clustered populations. Expression from n=2,556 single cells. p-values determined by hypergeometric test. e, Expression heatmap for genes related to germ cell death and differentiation in apoptosis-poised (AP) germ cells (subpopulation 6) versus apoptosis-resistant male-differentiating (MD) germ cells (subpopulation 3).
Most E13.5 PGCs maintained high levels of the pro-apoptotic gene Bax (Figure 4b, Extended Data Figure 4a), consistent with prior observations9. Subpopulation 6 was distinguished by Trp53 as well as downstream targets (Extended Data Figure 4a). In blood and early embryogenesis, elevated expression of Trp53 and its protein product P53 confers disadvantage in cell competition29,30. Given the established role of p53 in apoptosis31, we examined other pro-apoptotic genes (Figure 4b). Subpopulation 6 expressed the highest levels of transcripts associated with PGC apoptosis including Bax , Bim and Bad32 as well as the lowest level of anti-apoptotic Bcl-X33. Immunostaining revealed levels of P53 to be heterogeneous among PGCs and elevated in those undergoing apoptosis in clusters (Figure 4c), consistent with clonal expression. We termed subpopulation 6 ‘apoptosis-poised’ (AP), which was supported by the similar 9.5% frequency in the scRNA-seq dataset compared to 2–8% of cPARP+ germ cells previously detected at E13.5 (Figure 1a, Extended Data Figure 1b, Extended Data Figure 4b). Specific elevation of Pecam1, Epcam, Fragilis, and cKit indicated that AP-germ cells are not simply losing PGC identity, nor dying from neglect or loss of adhesion (Extended Data Figure 4d).
Apoptosis-Poised Germ Cells Exhibit Aberrant Male Differentiation
Gene ontology analysis of upregulated genes specific to AP-germ cells found enrichment for cell death and stress pathways (Figure 4d). Conversely, genes downregulated in AP-germ cells represented pathways involved in PGC differentiation. Although most PGC subpopulations at E13.5 exhibited high levels of pro-apoptosis transcripts, in agreement with bulk analyses33, we found subpopulation 3 to be uniquely depleted of apoptosis and AP-germ cell-associated markers (Figure 4b).
Male differentiation further distinguished subpopulation 3 from AP-germ cells. The latter were defined by expression of Rhox family genes (Figure 4e), which are expressed in PGCs but decrease sharply after E12.5 in males34 in association with differentiation to prospermatogonia. Persistent Rhox levels in E13.5 AP-germ cells could therefore indicate a developmentally delayed state. This hypothesis was bolstered by elevation of Nodal signalling-responsive components including Lefty2 in the AP subpopulation (Figure 4e). Nodal signalling is transiently active in E12.5 male germ cells but must be extinguished for male differentiation to proceed efficiently; negative feedback quickly shuts Nodal signalling down by E13.535. Increased Lefty2 in AP-germ cells relative to all other E13.5 subpopulations therefore reflects a more sex-undifferentiated identity. In contrast, transcripts involved in male sex differentiation, including Nanos236 and piRNA-regulating genes37, were lowest in AP-germ cells but highest in subpopulation 3, hence designated the male-differentiated (MD) state (Figure 4e).
Further supporting their reciprocal states, the AP-germ cell marker Trp53 and MD-marker Nanos2 were inversely distributed in the entire E13.5 single-cell RNA-seq dataset (Figure 5a), which was validated by RNA in-situ hybridization (Figure 5b). Nanos3 expression was also reciprocal between MD and AP-subpopulations; this suppressor of germ cell apoptosis38 was highest in MD-germ cells (Extended Data Figure 4c), consistent with a dichotomy between apoptosis and differentiation.
Figure 5: AP germ cells exhibit aberrant male sex differentiation at E13.5.
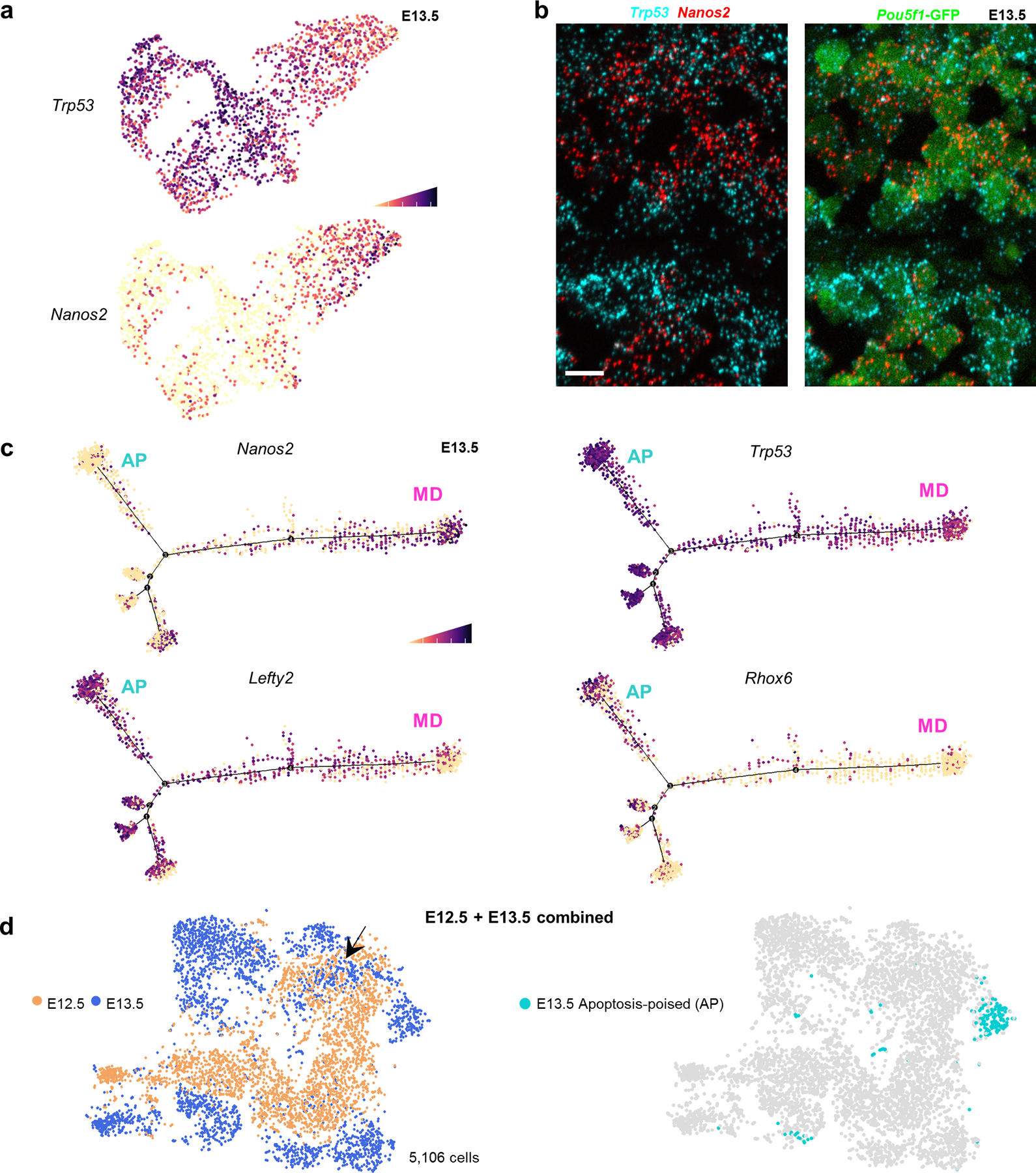
a,Trp53 expression in E13.5 single cells compared to differentiation marker Nanos2. Cell shading is derived from single cell expression values normalized to global expression for all genes and log-transformed via Seurat. b, Expression of Trp53 and Nanos2 mRNA in E13.5 testis sections by RNAscope. Scale bar =10μm. Image representative of 2 biological replicates. c, Pseudotime distribution of E13.5 single cells with expression for markers of male-differentiating (MD) germ cells (Nanos2) versus markers of an undifferentiated (Rhox6, Lefty2) or apoptotic state (Trp53) that together characterize the apoptosis-poised population (AP). d, t-SNE plot of combined E12.5 and E13.5 populations with age identified by colour. Arrow: Overlapping E13.5 subset with E12.5 germ cells. Right: Apoptosis-poised (AP) population separately identified on the combined E12.5-E13.5 tSNE distribution.
While higher Trp53 expression is associated with AP-germ cells, we noted that p53−/− germ cells were increased for expression of AP-marker LEFTY1/2 at E13.5 (Extended Data Figure 5a,b). This increase in sex-undifferentiated germ cells at E13.5 over wild-type suggests that p53 promotes male differentiation. The contrasting association of p53 with both male differentiation and an AP-state likely reflects the complex role of p53 in regulating multiple cellular processes and indicates that p53 expression is not deterministic for AP identity.
AP-Germ Cells Deviate from an Inferred Trajectory of Normal Male Differentiation
We used pseudotime analysis to place AP-germ cells in a differentiation context. The inferred trajectory clustered Nanos2-high cells at one extreme, suggesting that these are the most advanced male-differentiated germ cells, or prospermatogonia (Figure 5c). Corresponding absence of Lefty2 is consistent with MD identity. The remaining cells on the trajectory that were Nanos2-positive expressed lower Lefty2, suggesting that they maintained some degree of undifferentiated germ cell character and were likely transitioning from E12.5 to E13.5.
By contrast, the branch most devoid of Nanos2 (Figure 5c) exhibited uniquely high Rhox6/9, as well as elevated Trp53 and Lefty2, most consistent with the AP-germ cell state. To test the possibility that Nanos2-low PGCs represented an earlier state of differentiation, we performed scRNAseq on E12.5 male PGCs, computationally pooled with E13.5 dataset, and batch-corrected by timepoint identity. Clustering revealed that the majority of E12.5 germ cells were transcriptionally distinct from E13.5 and expressed appropriate stage-specific developmental markers (Figure 5d, Extended Data Figure 6a,b). Maturation markers such as Nanos2 were entirely absent at E12.5, whereas markers of a sex-undifferentiated state – Lefty and pro-apoptotic transcripts – were uniformly high. A subset of E13.5 PGCs overlapped with a subset at E12.5 and likely represents an intermediate state with slightly delayed differentiation. However, AP-germ cells were not associated with this overlapping population and instead remained transcriptionally distinct from all E12.5 cells. This distinctness suggests that AP-germ cells resemble neither sex-differentiated E13.5 cells nor undifferentiated E12.5-E13.5 cells, but instead deviate toward apoptosis (Extended Data Figure 7)
Aberrantly Differentiated PGCs Are Retained in the Absence of Apoptosis
Expression of developmentally-inappropriate genes in the AP subpopulation suggests that the wave of scheduled apoptosis eliminates PGCs that fail to differentiate properly and further predicts that mis-differentiated germ cells bearing hallmarks of the AP subset would persist in the absence of apoptosis. We examined Bax−/− mice at E15.5, after the foetal apoptotic wave would have removed germ cells destined to die. In normal male PGC development, expression of AP-marker Lefty2 is nearly ubiquitous at E12.5 (Figure 6a) but decreases to less than one percent by E15.5 when apoptosis concludes and germ cells complete male differentiation. However, we found that LEFTY1/2 was four-fold more frequent in Bax−/− male germ cells at E15.5 compared to littermate controls (Figure 6b,e). Similarly, germ cells expressing the AP marker, P53, were increased in Bax−/− (Figure 6c).
Figure 6: Aberrantly differentiated germ cells are retained in the absence of apoptosis.

a, Percent Lefty expression in germ cells during male differentiation from E12.5-E14.5. n=2 animals for each timepoint. Data are presented as mean values. b, Percent LEFTY+ expression in Bax−/− germ cells at E15.5. n=6 Bax+/+ animals, n=4 Bax+/− animals, n=11 Bax−/− animals. Data are presented as mean values ± SD. ** = p=0.041., two-tailed t-test. c, Percent P53+ expression in Bax−/− germ cells at E15.5. n=6 animals for each genotype. Data are presented as mean values ± SD. ** = p=0.0265, two-tailed t-test. d, Immunofluorescent detection of MAEL in Bax−/− testes at E15.5. MAEL-low and LEFTY-positive cluster of cells are outlined. Scale bar = 15μm. e, Immunofluorescent detection of LEFTY expression in germ cells from Bax+/− versus littermate Bax−/− testes at E15.5. LEFTY-positive clusters are outlined. White arrowheads highlight LEFTY-positive germ cell groups. Scale bar = 50μm.
LEFTY1/2+ PGCs in Bax−/− also exhibited lower expression of MD markers such as MAEL (Figure 6d). Persistent LEFTY1/2+, Maelstrom-negative germ cells in the absence of apoptosis suggests that these immature, non-differentiated germ cells are ordinarily removed during the apoptotic wave. Additionally, these LEFTY1/2+ germ cells retained at E15.5 in Bax−/− are distributed in a clustered manner typical of clonal expression (Figure 6e); indeed, clonality of LEFTY1/2+ clusters was verified in wild-type (Extended Data Figure 8). Given that foetal germ cell apoptosis is clonal, this observed pattern in Bax−/− is suggestive of clonal survival of AP-germ cells through E15.5.
Epigenetically-regulated Genes Underlie Germ Cell Heterogeneity
The clonality of germ cell apoptosis and accompanying mis-differentiation argues that heterogeneity between PGCs is stably cell-heritable. Although spontaneous genetic mutations are a potential source of de novo heritable heterogeneity, the mutation rate in mice39 is too low to account for the observed frequency of PGC apoptosis. Cell-heritable changes could otherwise arise from epi-mutations transmitted to daughter cells through proliferation. Genome-wide DNA demethylation of PGCs between E7.5 through E12.540 provides a significant opportunity for the occurrence and propagation of epi-mutations to clones.
The importance of accurate DNA demethylation for PGC fate is suggested by disrupted expression of germline reprogramming-responsive (GRR) genes involved in sex differentiation in Tet1 mutants41. Additionally, deletion of Dnmt1 in PGCs causes precocious demethylation and male differentiation42. Because male differentiation is driven by genes regulated by promoter methylation, expression of these genes can indicate the fidelity of epigenetic reprogramming. A significant number of GRRs and Dnmt1-regulated genes overlapped with MD markers (Figure 7a, Supplemental Table 2). This overlap confirms the prospermatogonial identity of MD-germ cells and suggests their epigenetic state is permissive for male differentiation. Conversely, markers for the AP-germ cell state significantly included genes upregulated in E14.5 Tet1-KO male germ cells, which supports epigenetic reprogramming defects as a basis for AP identity (Figure 7a, Supplemental Table 2).
Figure 7: Epigenetic differences distinguish AP and MD germ cell subpopulations.
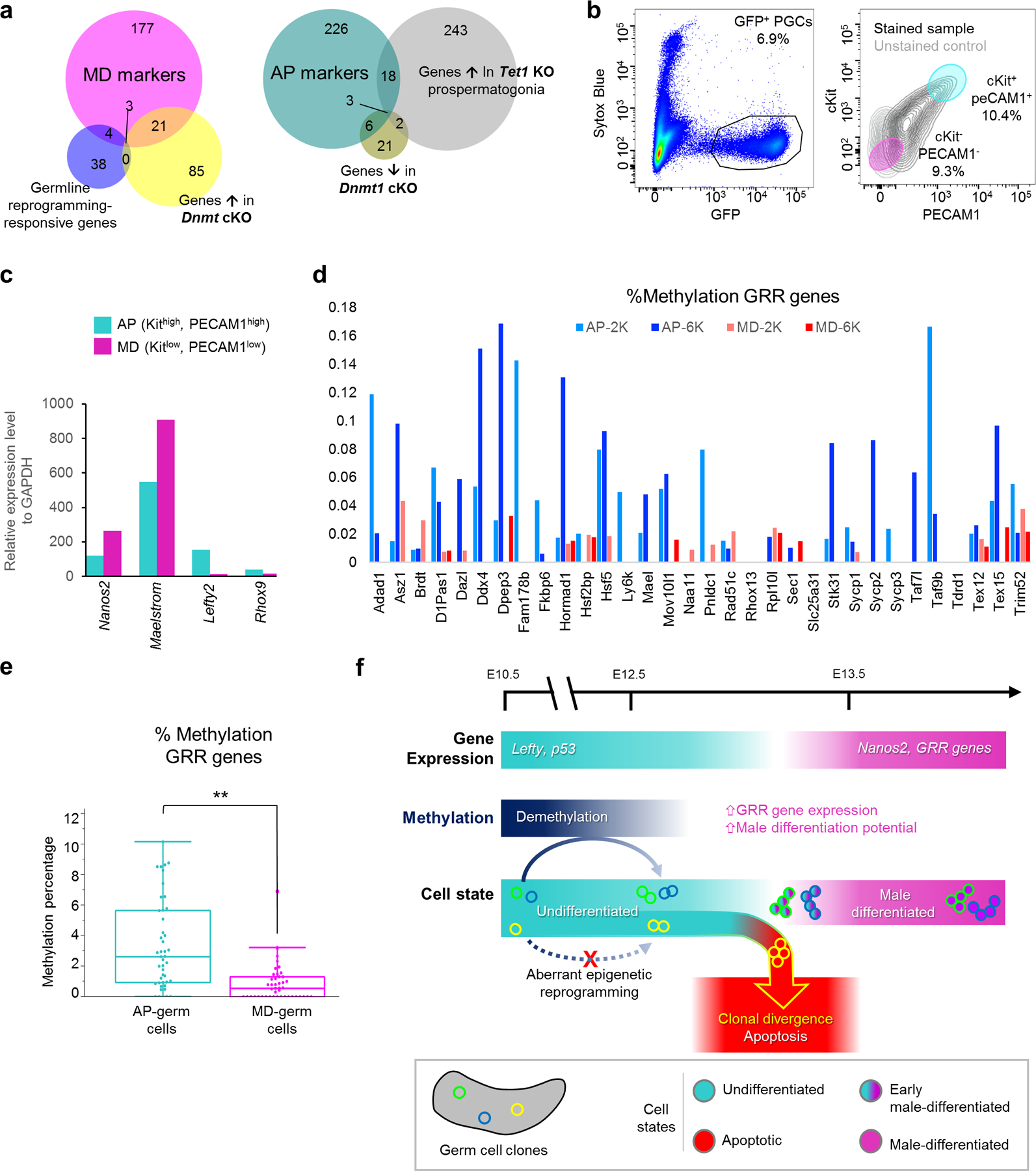
a, Overlap of epigenetically-regulated genes with MD and AP markers. Numbers in Venn diagram represent genes for each relationship among sets. MD overlap with GRR: p=1.69e-7; with Dnmt1 cKO: p=1.41e-26. AP overlap with Tet1 KO: p=2.66e-12; with Dnmt1 cKO: p=2.22e-11, one-tailed Fisher exact test. b, FACS purification scheme to isolate AP and MD populations from wild-type Pou5f1ΔPE-GFP E13.5 germ cells following selection for live cells (Sytox Blue-negative) and GFP expression. Based on scRNA-seq cluster markers, AP-germ cells were distinguished as Kithigh, PECAM1high while MD-germ cells were Kitlow, PECAM1low. c, qPCR for expression of AP and MD markers in sorted cell populations, normalized to GAPDH. QPCR performed on n=16,300 cells for MD population and n=11,700 cells for AP. d, Methylation of individual GRR genes in sorted AP versus MD cells from sequencing of two runs of n=2,000 (AP-2K, MD-2K) and n=6,000 (AP-6K, MD-6K) cells of each respective population. e, Methylation at all GRR loci in sorted AP versus MD cells both sequencing runs combined, **: p=2.13e-05, two-sided Wilcoxon test. n=8,000 cells per state from 2 combined runs. Data are presented as median values with first and third quartiles. Whiskers represent minimum and maximums with single outlier excluded in MD. f, Model for clonal origin of heterogeneous differentiation leading to clonal apoptosis or survival. Normal male differentiation involves downregulation of genes such as Lefty and p53 while upregulating Nanos2 and GRR genes. Male differentiation is facilitated by demethylation, particularly at GRRs, which is heritable and can produce divergent clonal outcomes. Aberrant epigenetic reprogramming disrupts a clone’s differentiation capacity, resulting in the apoptotic elimination of affected clones.
To validate that AP and MD-germ cells differed epigenetically, we isolated both populations using surface markers that distinguished each population (Figure 7b). We confirmed by quantitative PCR that purified AP and MD populations reciprocally expressed their respective markers (Figure 7c), including epigenetically-regulated genes such as Mael that were deficient in the AP population. This result indicates that the Maelstrom-negative cells retained in Bax−/− (Figure 6d) are likely AP-germ cells. Bisulfite sequencing on sorted AP and MD populations revealed that AP-germ cells were significantly hypermethylated compared to MD-germ cells (Extended Data Figure 9a). Importantly, this AP hypermethylation was most pronounced at GRRs (Figure 6d,e), confirming that AP-germ cells are epigenetically restricted from proceeding to a pro-differentiation state. The heritability of epimutations or aberrant reprogramming can epigenetically direct germ cells to this AP state, thereby producing divergent clonal fates (Figure 7f).
LINE-1 Is Associated with Male Differentiation and Successful Epigenetic Reprogramming
Epigenetic reprogramming in PGC development also de-represses the expression of transposable elements (TE)43. LINE-1s are the predominant active TE expressed in germ cells and their expression dynamics mirror that of GRRs during male differentiation. LINE1 product ORF1p was absent at E12.5 but expressed in nearly all germ cells by E15.5 (Extended Data Figure 9b). ORF1p+ clusters of PGCs first appeared at E13.5, reminiscent of cPARP+ clones. To ascertain if ORF1p expression is similarly clonal, we measured levels on a Pou5f1-CreERT2;R26R-Confetti background at E13.5 (Figure 8a). Within each clone, constituent cells expressed similar levels of ORF1p, demonstrating that the ORF1p-expressing state is stably heritable among related PGCs (Figure 8b) but clonally heterogeneous across all PGCs.
Figure 8: LINE1 expression is clonally heterogeneous and associated with male differentiation.

a, LINE1 expression detected by ORF1p antibody with clonal label. Individual Confetti-labelled clones delineated by dashed lines. White arrowheads: ORF1p-high clones, open arrowheads: ORF1p-low clones. Scale bar = 20μm. b, Quantification of ORF1p immunofluorescence in individual cells of clones. Each column represents a clone and circles are individual cells. Mean intensity per clone denoted by bar. c, Single cell repeat element RNA-seq identifying LINE1 L1MdA as a marker of a MD-like Nanos2-high germ cell subpopulation. Violin plots display cell densities as shape and individual cells as dots. The number of cells for each cluster are detailed in source data. d, Methylation at L1Md compared between sorted AP and MD populations. Data represent two separate sequencing runs on 2,000 and 6,000 cells per population. n=2,000 cells each, AP 2K, MD 2K. n=6,000 cells each, AP 6K, MD 6k. Whiskers represent minimum and maximum values. e, Quantification of ORF1p-negative germ cells in Bax−/− at E15.5. n=2 animals, Bax+/− ; n=3 animals, Bax−/−. f, Reciprocal ORF1p expression to immature differentiation marker LEFTY in E15.5 germ cells retained on apoptosis-deficient Bax−/− background. Dashed lines indicate ORF1p-negative cluster. Scale bar = 20μm.
In light of the clonally-variable expression of ORF1p, we considered whether the inability to repress TEs could be the basis for male germ cell elimination, which has been shown in foetal oocytes44. However, we found no correlation between ORF1p-high clones and apoptosis in the testis (Extended Data Figure 9c).
Alignment of scRNA data to a transposon reference genome45 confirmed heterogeneity of TE transcripts at E13.5. Although few TEs marked any subpopulations (Extended Data Figure 10a–d), the youngest and most active LINE-1, L1MdA, was elevated in germ cells that highly express MD-marker genes such as Nanos2 (Figure 8c). L1MdA is known to resemble GRRs in its demethylation and expression dynamics42, and we confirmed that L1Md loci were hypomethylated in MD cells (Figure 8d). Compared to other MD and AP markers, L1MdA was less strongly reciprocal (Extended Data Figure 10b), which suggests it is less dynamically expressed than genes orchestrating male differentiation such as GRRs.
Since LINE-1 turns on through epigenetic de-silencing during male differentiation, and AP-germ cells are hypermethylated at L1Md loci (Figure 8d), we predicted that the epigenetic landscape in AP-germ cells restricts LINE-1 expression. In Bax−/− testes at E15.5, we observed a 5-fold increase in the percent of germ cells that fail to express ORF1p (Figure 8e). We confirmed these were AP-germ cells based on aberrantly-sustained LEFTY expression (Figure 8f). Furthermore, ORF1p-negative germ cells in Bax−/− were also organized in clusters, suggesting that this differentiation defect is a clonally-heritable property. These results argue that reciprocal expression of LEFTY and ORF1p defines two divergent PGC fates: inappropriately sustained LEFTY expression is associated with aberrant differentiation and apoptosis (AP-germ cells), while ORF1p expression reflects progression to a male-differentiated (MD-germ cell state) that survives.
DISCUSSION
Successful reproduction and genetic transmission depend on the appropriate sex differentiation of germ cells. In this study, we find that PGCs are heterogenous but clonal in their decisions to undergo differentiation or apoptosis. We identify an apoptosis-poised state distinguished by sexually-undifferentiated gene expression that is retained in mutants without apoptosis. We find that germ cells most advanced in male sex differentiation downregulate pro-apoptosis genes, exist in clones, and retain an opposite epigenetic state from AP-germ cells. Together our results suggest that variations in demethylation are cell-heritable through division and deterministic (Figure 7f). Here, we posit that clonally heterogeneous epigenetic states likely arise from stochastic variability in demethylation. At critical loci, such as GRRs that are highly influential for facilitating a significant state change through male differentiation, even subtle epigenetic variation can be amplified to produce vastly different fates. Male sex differentiation is the litmus test of earlier epigenetic reprogramming and foetal germ cell apoptosis effectively enriches the quality of the spermatogonial progenitor pool by eliminating those incompetent to differentiate. Clonal apoptosis has been observed in vertebrate brain development46, suggesting that the coordination of apoptosis and differentiation is an important developmental paradigm to corral cellular populations toward the appropriate endpoint.
The elimination of aberrantly differentiated PGC clones may be beneficial toward ensuring reproductive success. Adult Bax−/− males are infertile and demonstrate meiotic errors postnatally10,11, but here we show that germ cell defects are already apparent much earlier in the foetal period. At E13.5, AP-germ cells are distinguished by aberrantly elevated Nodal signalling and inappropriate expression of immaturity markers such as Lefty and Rhox6/9. AP-germ cells still fail to reach the MD-germ cell state even when spared from elimination and afforded a longer differentiation window in Bax mutants. The continued failure to differentiate suggests that initial defects in AP-germ cells are not merely a transient delay, but persist with unabated Nodal signalling. Nodal is associated with pluripotency in germ cells37 and Nodal-high cells have increased tumorigenic potential47. High Lefty and Nodal expression is found in carcinoma in situ tumour stem cells that produce testicular germ cell tumours (TGCT)48. Apoptosis may therefore function to remove PGCs with aberrantly persistent Nodal signalling to prevent the initiation of TGCTs upon defective differentiation. While TGCTs were not reported in Bax mice, the tumorigenic effects of inhibiting germ cell quality control may be more evident on a TGCT-prone background49.
The association of LINE-1 with MD-germ cells was surprising considering the concurrent upregulation of piRNA-biogenesis genes such as Mili and Mael. While LINE-1 is ultimately repressed by piRNAs in adult germ cells, its foetal expression could be beneficial to differentiation or to priming the piRNA system. Fetal piRNA biogenesis involves a ping-pong amplification cycle in which sense transcripts from active transposons guide piRNA amplification and direct methylation at TEs for more durable repression50. Elevated LINE-1 in MD-germ cells suggests that this population would be first to initiate ping-pong piRNA biogenesis and subsequently maintain lower levels of LINE1 throughout adulthood. Hence, the survival advantage of MD-germ cells may enrich for germ cells with more robust piRNA production and improved long-term TE suppression. In human foetal germ cells, elevated TE expression has also been observed in an advanced male subpopulation46. Advanced human foetal germ cells that also express relatively higher piRNA genes demonstrate later repression of TEs, consistent with active PIWI-piRNA silencing of TEs following ping-pong amplification. Future investigations that follow the earliest differentiating germ cells (such as MD-germ cells) beyond the foetal period can determine how primacy in male differentiation improves TE regulation and genomic integrity.
Methods
Mice
For timed pregnancies, female mice were set up with individual males and checked daily for seminal plugs each morning. Date of plug detection was considered E0.5. For WT embryo collection, CD1 females were mated to Oct4-ΔPE-GFPSzabo males (MGI: 4835542). For clonal labeling, R26R-ConfettiSnippert (MGI:104735) and R26R-RainbowRinkevich mice (gift from I. Weissman, Stanford University) were outcrossed onto CD1 to generate mixed background homozygous females and then crossed to heterozygous Pou5f1-cre/EsiGreder (MGI:5049897) males for Tamoxifen-inducible germ-cell specific labeling after e8.5. Tex14tm1Zuk (Tex14−/−) mice (MGI: 3623684) were a gift from M. Matzuk (Baylor College of Medicine). B6.129S2-Trp53tm1Tyj/J(Trp53−/−) mice (MGI: 1857263) were obtained from Jackson Laboratory. All animal work was approved by the University of California, San Francisco Institutional Animal Care Use Committee (IACUC). Mice were maintained at 25°C, 40% relative humidity with a 12h:12h light:dark cycle.
Wholemount and Section Imaging
Tissues were fixed in 4% PFA for 2h, washed with PBS, and blocked with 2% BSA, 0.1% Triton X-100 in PBS for 3 hours. Primary antibodies incubation was performed in 0.2% BSA, 0.1% Triton X-100 in PBS for 2 or more days at 4°C, followed by washing with 0.1% Triton X-100 in PBS. Primary antibodies used were Tra98, (Abcam ab82527), 1:200; cleaved-PARP Alexa 647-conjugated (BD Biosciences F21–852), 1:20; cleaved-PARP(Cell Signaling 9544), 1:100; P53 (Cell Signaling 2524S), 1:100.; AMH (Santa Cruz sc-6886), 1:50; LEFTY, (R&D AF746), 1:100; MAEL (Abcam ab216324), 1:500; ORF1p (Abcam ab216324), 1:500.
Secondary antibody incubation was performed in 0.2% BSA, 0.2% BSA, 0.1% Triton X-100 in PBS. Tissues were washed with PbS and dehydrated through a 25%, 50%, 75%, 100%, 100% methanol series. Tissues were cleared with a 2:1 benzyl benzoate:benzyl alcohol (BABB) solution and imaged in BABB with a 10x/0.4 dry HCX PL APO CS objective on a Leica SP8 upright confocal microscope with LAS X software. Confocal images were processed and analyzed with Imaris and Volocity for quantitative measurements and object detection.
For section immunofluorescence, tissues were fixed in 4% PFA for 2h, washed with PBS, and dehydrated overnight in 30% sucrose at 4°C. Tissues were embedded in OCT and flash-frozen and stored at −80°C. Thick cryosections were cut at 25um and 50um; otherwise, sections were cut at 8um thickness and affixed to Superfrost Plus slides (Fisher Scientific). Sections were washed with PBS and incubated overnight at 4°C with primary antibody in 5% donkey serum, 0.5% Triton X-100. Sections were washed with PBS and incubated with secondary antibody for 1h at room temperature. Slides were mounted with Vectashield and imaged on a SP5 Leica confocal microscope. For antibodies requiring antigen retrieval, sections were immersed in 10mM sodium citrate and heated until boiling. Sections were washed with PBS and stained with primary antibody as described.
RNA in situ hybridization
Testes were prepared for section immunofluorescence and sectioned at 5μm. RNA was detected using the RNAscope Multiplex Fluorescent kit v2 with probes against mouse Mm-Tp53-C2 and Mm-Nanos2-C1. Tissue sections were pretreated with RNAscope Protease III for 6 minutes at 37°C and incubated in hydrogen peroxide for 10 minutes at 37°C. Probes were incubated for 2 hours followed by the standard Multiplex Fluorescent v2 assay. Probes were detected by Opal dyes 570/650 (Perkin Elmer). For subsequent antibody-based immunofluorescence, sections were prepared using the described section immunofluorescence protocol.
Spatial statistical analysis
Wholemount tissues were stained for makers of apoptosis, GCs, and nuclei. Objects were identified using the Cells module in Imaris (Bitplane) to determine the three-dimensional coordinates of each object centroid. Spatial analysis of clustering was based on the Ripley K function using the RipleyGUI51 platform in Matlab. K-function scores were calculated to evaluate deviation (K(t) − E[K(t)]) from an expected random distribution, CSR, which was simulated independently 100 times for each spatial distribution analyzed. The relative degree of clustering for apoptotic GCs versus all GC distributions, or across time points, was tested with the between-treatments sum of squares (BTSS) and compared to a 95% confidence interval for the BTSS value of the null hypothesis (two-tailed). The null hypothesis assumes two sets have interchangeable distributions and is tested by 5,000 random resamplings from both sets with replacement to generate an accumulated probability distribution from the resamplings, which is then compared to the BTSS values for the real sets to generate a p-value for significance.
Clonal analysis
R26R-Confetti and R26R-Rainbow female mice were mated with Pou5f1-cre/Esr males and intraperitoneally injected with Tamoxifen (Sigma, 20mg/ml dissolved in sunflower seed oil) at E10.5. Tamoxifen dosage was scaled to the pregnant female’s weight and adjusted to produce distinguishable coloured populations (1.25mg and 2.5mg/40g female for Confetti and Rainbow, respectively). Clonally labeled gonads were dissected and fixed for wholemount staining or section immunofluorescence as described.
Due to the prevalent fragmentation of germ cell clones, we first established the maximum distance within which similarly colored germ cells were considered to be clonal. To generate clonal density conditions under which single discrete clones can be readily identified for measuring this clonal dispersion distance, we administered a low (0.5mg/40g female weight) dose of Tamoxifen to Confetti and Rainbow pregnant females, which produced at most one germ cell clone per E13.5 testis. We measured the distance between nearest-neighbor cells from two different fragments to determine the dispersion between fragments and took the largest of these nearest-neighbor distances among all fragment pairs as the maximum potential dispersion of clonal fragmentation. In evaluating multiple single clones, we found the dispersion between fragments to be less than 50μm, as demonstrated in a representative clone in Extended Data Figure 2b. This distance was corroborated with measurements from secondary analysis of published single clone fragmentation in similarly aged testes21. For clonal identification of apoptotic clusters in Rainbow and Confetti, we considered similarly coloured PARP+ cells to be clonal if they laid within this 50μm distance.
To clear tissues and preserve endogenous fluorescence for wholemount imaging, tissues were washed with PBS following secondary antibody incubation and placed in Scale CUBIC Reagent 152i overnight. Cleared tissues were imaged in Scale CUBIC Reagent 1 on a white-light Leica SP8 confocal microscope. Excitation for CFP was with a 458nm laser line; GFP and YFP, 514nm white-light; RFP, 561 white-light. Fluorescence was collected for CFP between 465–495nm, airy 1.5; GfP and YFP, 521–555nm; RFP, 565–590nm.
Clonal populations were analyzed using the Cell module on Imaris to identify individual cells of a clone and quantify clone size. Clones were detected by CFP, YFP, or RFP intensity with a threshold set at 2 standard deviations below the median intensity value. Intensity was measured over the cell body with a 1.2um background filter and a 5um minimum cell diameter. Individual cells were separated using a 7um estimated cell diameter.
Single cell RNA seq
For E12.5 and E13.5 male germ cell collection, WT testes from E12.5 and E13.5 timed matings were collected together. Testes were dissected in cold PBS and non-gonadal tissue removed. Testes were digested in 0.25% trypsin/EDTA at 37°C for 20 minutes with trituration every 10 minutes, followed by the addition of 1mg/ml DNAse and further digestion for 10 minutes. An equal volume of fetal bovine serum was added to halt digestion and the digest was strained through a single-cell filter. Dead cells were labeled with Sytox Blue and live germ cells were obtained by sorting on GFP+, Sytox− into 0.04% BSA. 2,517 and 2,606 cells were recovered for E12.5 and E13.5 timepoints, respectively. Cells were processed for 10X sequencing by the UCSF Institute for Human Genetics. Cell by gene matrices were obtained by alignment with mouse genome assembly mm10 and repeat assembly mm10_rmsk_TE53 (M. Hammell lab, Cold Spring Harbor Laboratory) and performing CellRanger analysis on 10x reads.
Single cell expression data was analyzed using Seurat to identify differentially expressed genes and perform principal component analysis. Statistically significant principal components (n=11, p<0.05) were used to cluster cells in an unsupervised manner. Differentially expressed genes by cluster (germ cell state) were identified by receiver operating characteristic (ROC) test, bimodal test, and Wilcoxon rank sum test. Significance cutoffs were AUC>0.6, ROC test and p<0.05 for bimodal and Wilcoxon rank sum test. A p<0.05 for Wilcoxon rank sum test was used to identify markers that were significant, positive classifiers of a cluster. Fold changes and normalized gene expression was expressed in natural log space as a default output of Seurat. For fold change calculations pertaining to cluster markers, the mean expression across all cells of one cluster was compared to the mean expression of all other cells in non-log space, and then the fold difference was expressed in natural log space.
Clusters were visualized by t-distribute stochastic neighbor embedding (t-SNE) plot generated in Seurat. Expression of individual genes across all cells were determined by using the LogNormalize in Seurat to normalize gene expression in each cell by total expression and log-transforming the result. These expression data were plotted on t-SNEs using the FeaturePlot function in Seurat with expression values represented by a colour gradient.
Gene ontology analysis was performed using biomarker lists for each clustered population identified by Seurat using GSEA molecular signature database analysis (MSigDB) on the C5 GO:Biological Process collection54,55. Biomarkers were analyzed for statistical overrepresentation in biological process categories by hypergeometric test and semantically sorted using ReviGO.
For pseudotime analysis, E13.5 GC expression data was analyzed using Monocle. We used the unsupervised dpFeature procedure to identify the top 1000 highly variable genes among clusters for constructing the differentiation trajectory. Cells were clustered with a rho of 15 and delta of 8.
For multimodal single-cell repeat element RNA-seq, pre-processing quality control based on repeat element expression removed more cells than the quality control performed for the initial non-repeat RNA-seq analysis, which was based only on expression of non-repeat elements. Consequently, the filtered cells were matched for both repeat and non-repeat datasets before performing downstream analysis. We directed the clustering algorithm to produce a similar number of clusters (7) to the initial non-repeat RNA-seq analysis. This produced slightly different cluster identities than the initial analysis but we verified the resulting cluster identities by AP and MD marker expression to relate them back to the initial clustering performed non-repeat elements only. Clustering was also performed using repeat elements only and the resulting clusters assessed for AP and MD marker expression.
Whole Genome Bisulfite Sequencing by Post-Bisulfite Adapter Tagging
For AP and MD germ cell collection, CD1 females were mated with Oct4-ΔPE-GFPSzabo males. Testes from E13.5 timed matings were dissected and digested in 0.25% trypsin/EDTA at 37°C for 20 minutes with trituration every 10 minutes, followed by the addition of 1mg/ml DNAse and further digestion for 10 minutes. An equal volume of fetal bovine serum was added to halt digestion. Cells were treated with Fc block (Biolegend 101319) for 10 min on ice, followed by incubation with 1ug/1M cells in 100ul PE-PECAM1 (Biolegend 102407) and 1ug/1M cells in 100ul APC-c-Kit (Biolegend 105812) on ice for 30 min. Dead cells were excluded with Sytox Blue. AP germ cells were obtained by sorting on GFP+ PE-PECAM1high APC-c-Kithigh, whereas MD GCs were sorted on GFP+ PE-PECAM1low APC-c-Kitlow. Flow cytometry data was analyzed using FlowJo (v10.7).
Sorted cells were pelleted and lysed using EZ DNA Methylation-DirectTM Kit (Zymo D5020). 60 fg of unmethylated phage Lamba (Promega D1521) was spiked into each sample to assess bisulfite conversion rate. Bisulfite conversion and library preparation was carried out with the Pico Methyl-Seq Library Prep Kit (Zymo D5455). We performed purification steps described in sections 3, 4, and 5 of the manufacturer’s protocol using a 5:1 ratio of Binding Buffer:Sample. The library was amplified through 9 PCR cycles (see section 4 of the manufacturer protocol). Concentration and purity of each library was assessed by Nanodrop, and the quality of the libraries was evaluated using Agilent BioAnalyzer High Sensitivity DNA Chips.
Libraries were sequenced on NextSeq 500 High Output and NovaSeq 6000 S1 flow cells yielding to 75-bp, and 50-bp paired-end reads, respectively. A total of two libraries per cell population (AP vs MD) were sequenced.
Bisulfite Sequencing Analysis
Raw sequence reads were trimmed by 10 nt and filtered by removing reads with a mean Phred quality score of less than 20. Adapters were removed using cutadapt (v1.12). We used Bismark (v0.14.3) to align the filtered sequences to the mouse genome assembly GRC38/mm10 with the non_directional parameter and using Bowtie (v1.2) within Bismark. Only one read pair was kept when multiple mates aligned to the same genomic location. CpG methylation was calculated at each CpG locus by counting the number of methylated versus unmethylated reads. Bisulfite conversion rate was estimated by calculating the percentage of unmethylated counts for CpGs on the phage lambda genome.
To assess CpG methylation at GRR promoters, we calculated the average weighted methylation score for each GRR promoter. The weighted methylation score takes into account variable coverage, and it is calculated by summing the methylated counts and the unmethylated counts of all CpGs in a region; the methylation percentage is then derived from these summed counts. This calculates the average methylation of multiple CpGs but adds more weight to the CpGs with more coverage. For GRR promoters, we used CpGs in the region from 1,000 nt upstream of the TSS to 500 nt downstream of the TSS, as defined by the GENCODE annotation (vM24). For L1_Md elements, we used CpGs within the repeat regions, as defined by the repeat masker annotation. DNA methylation levels were visualized as box and whisker plots.
We compared methylation levels in AP and MD cell populations by calculating p-values with the two-sided paired Wilcoxon test.
Statistics and Reproducibility
All images are representative of a minimum of three biological replicates unless otherwise indicated. Statistical calculations were obtained using built-in functions in Seurat, Ripley GUI, and MSigDB. For all other calculations, Paired t-tests, ANOVA, and hypergeometric tests were performed using Microsoft Excel or GraphPad Prism 7.
Data Availability
Data availability. scRNA-seq and BS-seq data that support the findings of this study have been deposited in the Gene Expression Omnibus (GEO) under accession codes GSE119045 and GSE155122. All other data supporting the findings of this study are available from the corresponding author on reasonable request. Gene ontology analysis on MSigDB computed overlaps with GO:BP (https://www.gsea-msigdb.org/gsea/msigdb/genesets.jsp?collection=GO:BP).
Code Availability
Code for the scRNAseq analysis is available at https://github.com/dnucsf/NatureCellBiology2020.
Extended Data
Extended Data Fig. 1. Apoptotic clusters are randomly distributed throughout fetal testes.

a, Percent apoptotic cleaved-PARP+ germ cells in wholemount testes on a mixed genetic background during the apoptotic wave. n=4 animals. Data presented as mean values ± SD. Dashed line denotes reported average apoptotic index in developing fetal tissues21. b, Percent cleaved-PARP+ cells on apoptosis-sensitive C57/Bl6 background20 at peak apoptosis (E13.5). c, Distribution of apoptotic clusters measured for spatial bias. 3D coordinates of each cluster’s centroid were evaluated by Ripley’s K-function. These were compared against simulated distributions for an equivalent number of points. Representative plots depict each simulated distribution type (right). Cluster coordinates from n=5 animals.**: p=0, †: p=0.10622, between-treatments sum of squares. d, E13.5 testis section stained for Sertoli cells (AMH), germ cells (TRA98), and apoptosis (cleaved-PARP). Apoptotic cords are defined as cords containing any apoptotic germ cells (red outline) in contrast to non-apoptotic cords (white outline). Scale bar = 20μm. e, Ratio of total germ cells to Sertoli cells in apoptotic and non-apoptotic cords, p=0.584, two-tailed t-test. Data points represent individual cords pooled from sections from 4 animals. n =15 non-apoptotic cords and n = 11 apoptotic cords. Data presented as mean values ± SD. f, Average numbers of Sertoli cells in direct contact with each germ cell in apoptotic and non-apoptotic cords. Within apoptotic cords, apoptotic (PARP+) and non-apoptotic (PARP-) germ cells were separately considered. p=0.737154, one-way ANOVA. Data points represent individual germ cells (n) analyzed in sections from 4 animals. Total cell counts per category are detailed in source data. Data presented as mean values ± SD. g, Quantification of apoptotic germ cells in E13.5 Tex14 mutants. p=0.4491, two-tailed t-test. n=4 animals per genotype. Data presented as mean values ± SD. h, Apoptotic cluster size by cell number in Tex14 E13.5. p=0.0073, two-tailed T-test. Data points (n) are available in source data and represent individual apoptotic clusters analyzed from 2 animals (Tex14+/+) and 3 animals (Tex14−/−). Data presented as mean values ± SD.
Extended Data Fig. 2. Clonal boundary determination for fragmented apoptotic clusters.
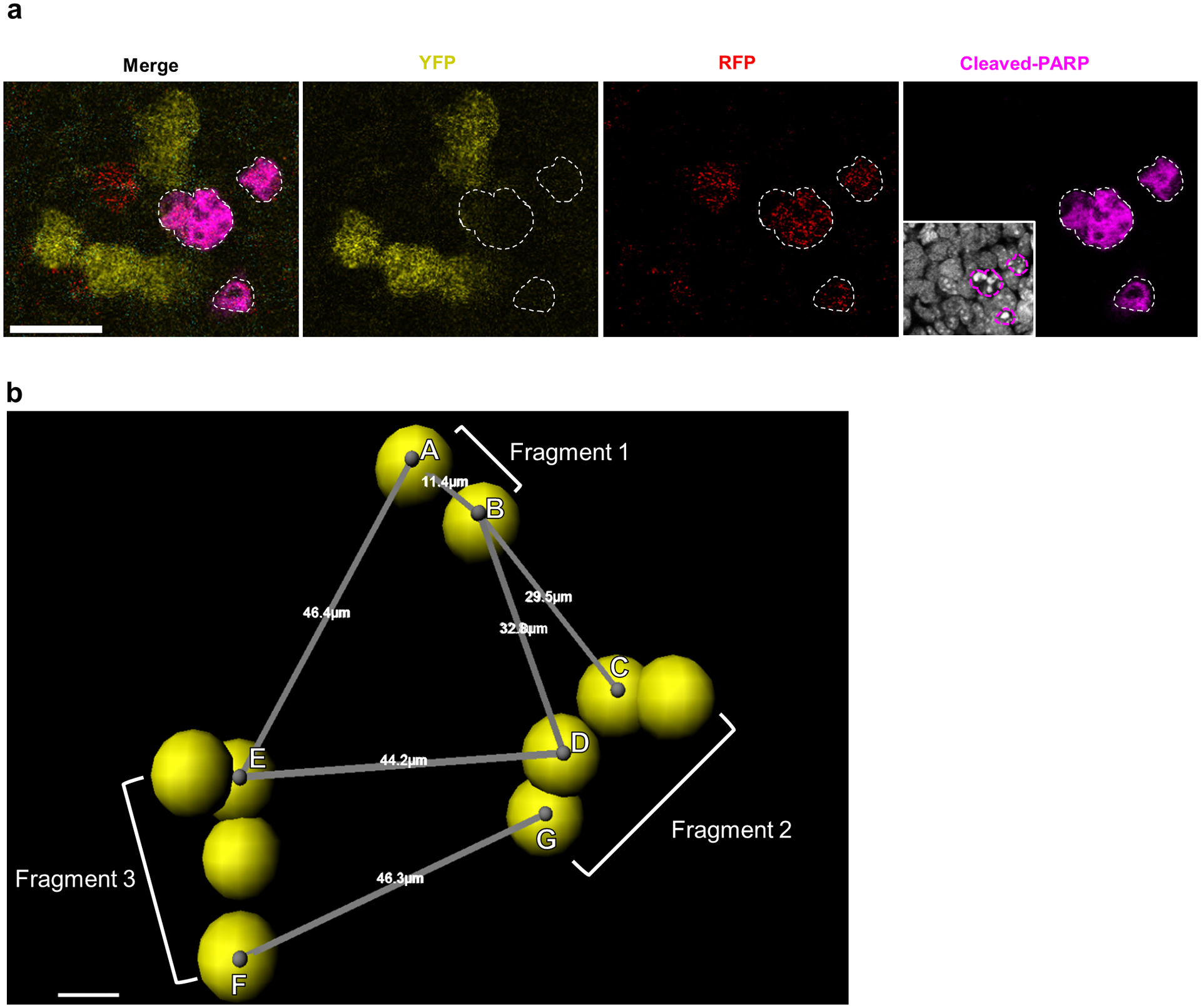
a, Detection of clonal apoptosis in Confetti labeled germ cells at E13.5. Apoptotic germ cells identified by cleaved-PARP staining (white outlines). Scale bar = 20um. 5 animals. Inset: Hoechst staining. b, Confetti -labeled germ cells imaged in wholemount and segmented using the Imaris Cell module displayed fragmentation by E13.5, in accordance with published findings21. We identified three-dimensional coordinates for centroids (grey dots) of each cell (represented by yellow spheres) within a clone to measure the centroid-to-centroid distances between nearest-neighbor germ cells belonging to different fragments. In depicted clone, spans nearest-neighbor cells between Fragments 1 and 2 while spans nearest-neighbor cells from Fragments 3 and 2. We took the largest of these nearest-neighbor distances from all fragments pairs to represent the maximum potential dispersion between fragments of a single clone. Scale bar = 5um.
Extended Data Fig. 3. Clonal apoptotic clusters are maintained in the absence of intercellular bridges.

a, Cleaved-PARP staining for apoptosis in wholemount E13.5 Tex14−/− testes on a R26R Rainbow clonally-labeled background reveals mCherry apoptotic clone (outlined in yellow). Scale bar = 10μm. Rotated view of same clones confirming that constituent cells of mCherry clone are disconnected. Nearby mCerulean clone is negative for apoptosis. b, Extended view of entire wholemount E13.5 testis. Scale bar = 100μm.
Extended Data Fig. 4. Single cell-RNAseq analysis of E13.5 male germ cells.

a, Average expression of p53-activated genes by germ cell subpopulation. Expression from n=2,556 single cells. b, Subpopulation size in cell number and as a percentage of total germ cells analyzed for single-cell RNA sequencing at E13.5. c, Average pro-survival gene Nanos3 expression by germ cell subpopulation. Expression from n=2,556 single cells. d, Average expression of prosurvival genes Kit, adhesion factor Epcam, and PGC identity gene Ifitm3 by germ cell subpopulation. Expression from n=2,556 single cells.
Extended Data Fig. 5. LEFTY1/2+ cells are increased in p53−/− testes by E13.5 compared to p53+/−.

a, Immunofluorescence detection of LEFTY1/2 in sections from E13.5 p53+/− and p53−/− testes. Scale bar = 50μm. 2 animals per genotype. b, Quantification of percent LEFTY1/2+ germ cells from E13.5 p53+/− and p53−/− testes. n=2 animals per genotype.
Extended Data Fig. 6. E12.5 and E13.5 germ cells remain transcriptionally distinct in merged analysis.
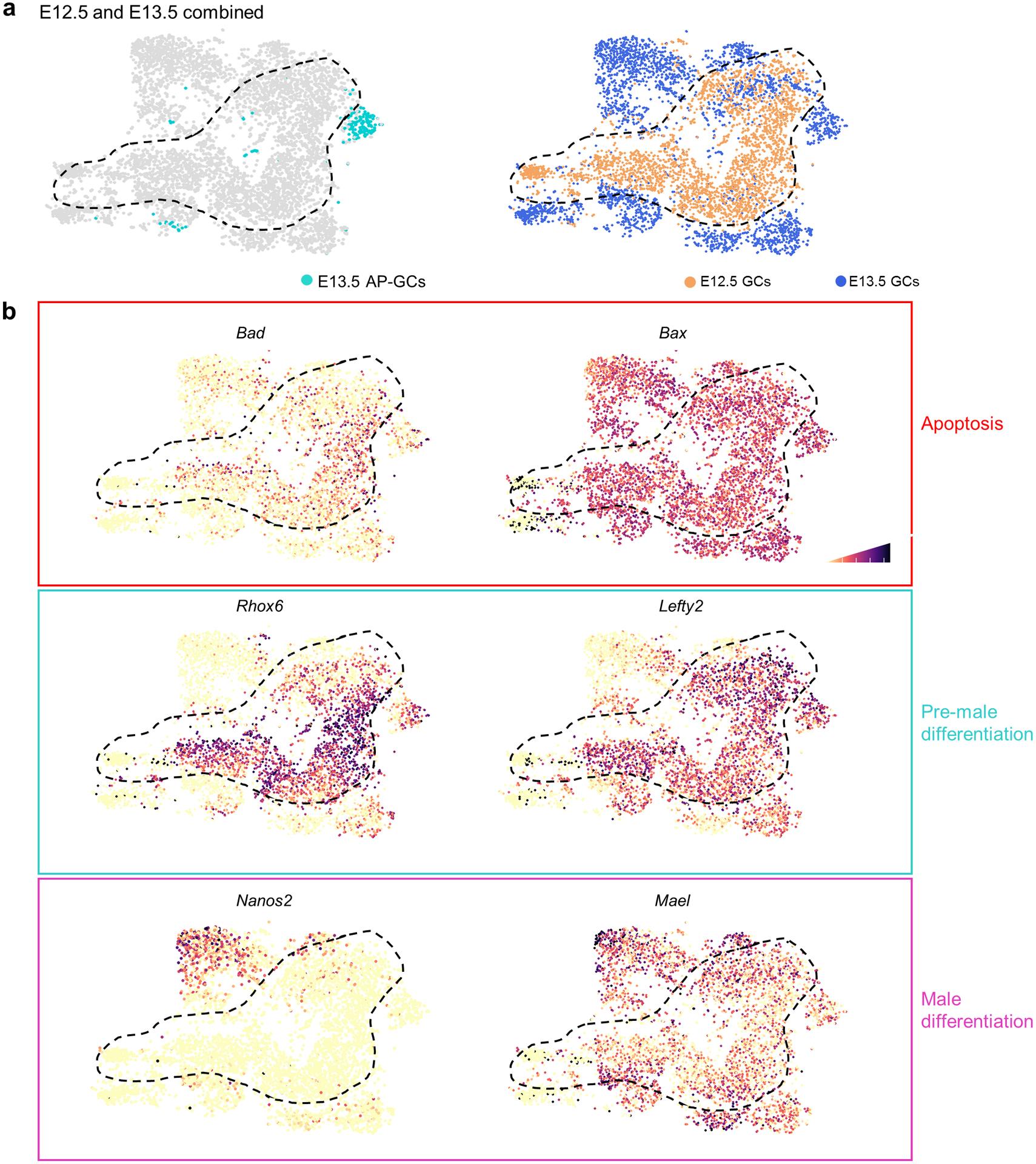
a, Merged dataset with E12.5 and E13.5 germ cells after transcriptional clustering. E13.5 AP-germ cell identities colored while E12.5 boundary is denoted by dashed black line. t-SNE represents 5,106 cells. b, t-SNE plots showing expression of genes (Bad, Bax), male differentiation genes (Nanos2, Mael), and apoptotic genes associated with a pre-male differentiation population (Rhox6, Lefty2). Cell shading is derived from single cell expression values normalized to global expression for all genes and log-transformed via Seurat.
Extended Data Fig. 7. Model for coordination of clonal apoptosis with male differentiation.

Clonal variation in differentiation potential results in divergent clonal fates: germ cells normally activate male differentiation programs marked by Nanos2 but a subset of clones aberrantly maintain an undifferentiated state characterized by expression of pro-apoptotic genes that ultimately results in clonal death.
Extended Data Fig. 8. LEFTY expression is clonally heterogeneous.

LEFTY expression in E13.5 Confetti testis, LEFTY-high cells (open arrowheads) and LEFTY-low cells (white arrowheads) with clone boundaries indicated by dashed outlines. Scale bar = 20μm
Extended Data Fig. 9. Population-specific expression of epigenetically-regulated genes including LINE1 during male differentiation.
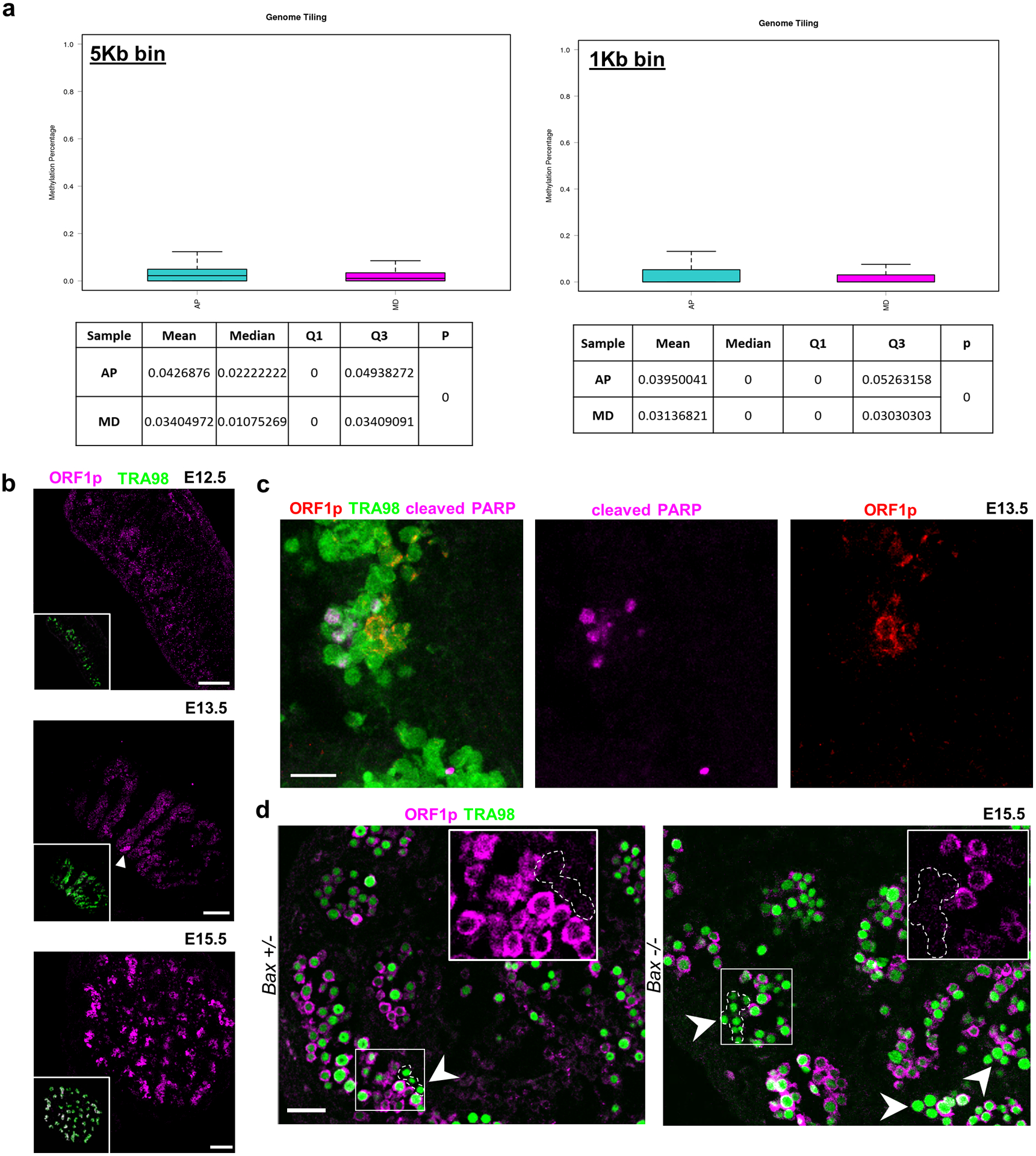
a, Whole-genome bisulfite sequencing comparison of sorted AP and MD populations for relative methylation percentage. Data are presented as median values with first and third quartiles. Whiskers represent minimum and maximums. Sequencing was performed on 2 separate runs of n=2,000 and n=6,000 cells each for both populations. b, ORF1p expression during apoptotic wave. ORF1p is expressed in small clusters of germ cells at E13.5 (white arrowhead) and progresses toward homogenous expression by E15.5. Scale bar = 100μm. c, ORF1p and cleaved PARP staining in E13.5 wild type sections. Scale bar = 25μm. d, ORF1p-negative germ cell clusters in E15.5 Bax−/− compared to Bax+/−. Arrowheads denote ORF1p-negative cells. Insets: Higher-magnification image of ORF1p-negative cluster compared to ORF1p-positive neighboring germ cells. Scale bar = 50μm.
Extended Data Fig. 10. Differentially expressed repeat elements identify distinct subpopulations of E13.5 germ cells.

a, Abridged table of repeat element markers for subpopulations defined by scRNA-seq clustering based on expression of non-repetitive elements. p value calculated by two-sided Wilcoxon rank sum test. Expression from n=2,043 cells. Full table of markers available in source data. b, Expression of MD marker Nanos2, AP marker Rhox6, and LINE1 members across clusters identified by expression of non-repetitive elements. Predominant expression of Nanos2 in cluster 4 is characteristic of MD population. Predominant expression of AP marker Rhox6 in cluster 5 is characteristic of AP population. Violin plots display cell densities as shape and individual cells as dots. The number of cells (n) for each cluster are detailed in source data. c, Abridged table of repeat element markers for subpopulations defined by scRNA-seq based only on expression of repetitive elements. P value calculated by two-sided Wilcoxon rank sum test. Expression from n=2,043 cells. Full table of markers available in source data. d, Expression of MD marker Nanos2, AP marker Rhox6, and LINE1 members across clusters identified only by expression of repeat elements. Violin plots display cell densities as shape and individual cells as dots. The number of cells (n) for each cluster are detailed in source data.
Supplementary Material
Supplementary Table 1. Transcriptional markers that distinguish each single germ cell state compared to all others were identified among differentially expressed genes in an E13.5 germ cell population by the two-tailed Wilcoxon rank sum test-based FindMarkers operation in Seurat, which provides a p-value that is further adjusted by bonferroni correction using all genes in the dataset.
Supplementary Table 2. Comparison of markers for MD-germ cells with GRRs and genes upregulated in Dnmt1-cKO male germ cells42. Comparison of markers for AP-germ cells with genes upregulated in Tet1-KO43 male germ cells and genes downregulated in Dnmt1-cKO. Adjusted p-values for MD- and AP-markers were generated by Wilcoxon rank sum test with bonferroni correction. Statistics for markers from Dnmt1-cKO and Tet1-KO were obtained from referenced studies.
Wholemount visualization of apoptotic clusters of PGCs at E13.5. Whole E13.5 testis with cell nuclei identified by Hoechst and PGCs by Tra98. Apoptosis is identified by cleaved-PARP and rotated views highlight the spatial distribution of apoptotic PGCs. Magnified views show a representative apoptotic germ cell cluster.
PGC proliferation is clonal. E13.5 PGCs are clonally labeled using Rainbow and cells in late G2/M are marked by phosphor-histone H3. Magnified view shows two separate groups of phosphohistone H3+ cells that are clonal (mOrange clone and a GFPclone).
Multicolor clone labeling reveals shared fluorophores within germ cell cysts. 3-dimensional perspective of whole E13.5 Rainbow;Tex14+/+;Pou5f1Cre-ER testis with bicolored clone (mCerulean and mCherry) outlined as a surface, followed by individual channels confirming bicolored labeling.
Tex14 mutant testes differ in clone structure and do not form bicolored clones. 3-dimensional perspective of whole E13.5 Rainbow; Tex14−/−; Pou5f1Cre-ER testis with strictly single-colored clones as a consequence of lacking intercellular bridges.
Footnotes
Competing Interests
The authors declare no competing interests.
REFERENCES
- 1.Lehtonen J & Parker GA Gamete competition, gamete limitation, and the evolution of the two sexes. Mol. Hum. Reprod 20, 1161–1168 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Gardner RL & Rossant J Investigation of the fate of 4–5 day post-coitum mouse inner cell mass cells by blastocyst injection. J. Embryol. Exp. Morphol 52, 141–152 (1979). [PubMed] [Google Scholar]
- 3.Anderson R, Copeland TK, Schöler H, Heasman J & Wylie C The onset of germ cell migration in the mouse embryo. Mech. Dev 91, 61–68 (2000). [DOI] [PubMed] [Google Scholar]
- 4.Cantú AV, Altshuler-Keylin S & Laird DJ Discrete somatic niches coordinate proliferation and migration of primordial germ cells via Wnt signaling. J Cell Biol 214, 215–229 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saitou M, Kagiwada S & Kurimoto K Epigenetic reprogramming in mouse pre-implantation development and primordial germ cells. Development 139, 15–31 (2012). [DOI] [PubMed] [Google Scholar]
- 6.Menke DB, Koubova J & Page DC Sexual differentiation of germ cells in XX mouse gonads occurs in an anterior-to-posterior wave. Dev. Biol 262, 303–312 (2003). [DOI] [PubMed] [Google Scholar]
- 7.Ohta K et al. Male differentiation of germ cells induced by embryonic age-specific Sertoli cells in mice. Biol. Reprod 86, 112 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aitken RJ, Findlay JK, Hutt KJ & Kerr JB Apoptosis in the germ line. Reproduction 141, 139–150 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Runyan C et al. Steel factor controls midline cell death of primordial germ cells and is essential for their normal proliferation and migration. Development 133, 4861–4869 (2006). [DOI] [PubMed] [Google Scholar]
- 10.Knudson CM, Tung KS, Tourtellotte WG, Brown GA & Korsmeyer SJ Bax-deficient mice with lymphoid hyperplasia and male germ cell death. Science 270, 96–99 (1995). [DOI] [PubMed] [Google Scholar]
- 11.Rodriguez I, Ody C, Araki K, Garcia I & Vassalli P An early and massive wave of germinal cell apoptosis is required for the development of functional spermatogenesis. EMBO J. 16, 2262–2270 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perez GI, Knudson CM, Leykin L, Korsmeyer SJ & Tilly JL Apoptosis-associated signaling pathways are required for chemotherapy-mediated female germ cell destruction. Nat. Med 3, 1228–1232 (1997). [DOI] [PubMed] [Google Scholar]
- 13.Coucouvanis EC, Sherwood SW, Carswell-Crumpton C, Spack EG & Jones PP Evidence That the Mechanism of Prenatal Germ Cell Death in the Mouse Is Apoptosis. Exp. Cell Res 209, 238–247 (1993). [DOI] [PubMed] [Google Scholar]
- 14.Wang RA, Nakane PK & Koji T Autonomous cell death of mouse male germ cells during fetal and postnatal period. Biol. Reprod 58, 1250–1256 (1998). [DOI] [PubMed] [Google Scholar]
- 15.Goriely A, McVean GAT, Röjmyr M, Ingemarsson B & Wilkie AOM Evidence for Selective Advantage of Pathogenic FGFR2 Mutations in the Male Germ Line. Science 301, 643–646 (2003). [DOI] [PubMed] [Google Scholar]
- 16.Ueno H, Turnbull BB & Weissman IL Two-step oligoclonal development of male germ cells. Proc. Natl. Acad. Sci 106, 175–180 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cook MS, Munger SC, Nadeau JH & Capel B Regulation of male germ cell cycle arrest and differentiation by DND1 is modulated by genetic background. Development 138, 23–32 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Foley JGD & Bard JBL Apoptosis in the cortex of the developing mouse kidney. J. Anat 201, 477–484 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Laird DJ, Altshuler-Keylin S, Kissner MD, Zhou X & Anderson KV Ror2 Enhances Polarity and Directional Migration of Primordial Germ Cells. PLOS Genet. 7, e1002428 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mendis SHS, Meachem SJ, Sarraj MA & Loveland KL Activin A Balances Sertoli and Germ Cell Proliferation in the Fetal Mouse Testis. Biol. Reprod 84, 379–391 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Lei L & Spradling AC Mouse primordial germ cells produce cysts that partially fragment prior to meiosis. Development 140, 2075–2081 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ventelä S, Toppari J & Parvinen M Intercellular Organelle Traffic through Cytoplasmic Bridges in Early Spermatids of the Rat: Mechanisms of Haploid Gene Product Sharing. Mol. Biol. Cell 14, 2768–2780 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Greenbaum MP et al. TEX14 is essential for intercellular bridges and fertility in male mice. Proc. Natl. Acad. Sci 103, 4982–4987 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu KL & Yamashita YM Germ cell connectivity enhances cell death in response to DNA damage in the Drosophila testis. eLife 6, e27960 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Snippert HJ et al. Intestinal Crypt Homeostasis Results from Neutral Competition between Symmetrically Dividing Lgr5 Stem Cells. Cell 143, 134–144 [DOI] [PubMed] [Google Scholar]
- 26.Rinkevich Y, Lindau P, Ueno H, Longaker MT & Weissman IL Germ-layer and lineage-restricted stem/progenitors regenerate the mouse digit tip. Nature 476, 409–413 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Greder LV et al. Brief Report: Analysis of Endogenous Oct4 Activation during Induced Pluripotent Stem Cell Reprogramming Using an Inducible Oct4 Lineage Label. STEM CELLS 30, 2596–2601 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Western PS, Miles DC, van den Bergen JA, Burton M & Sinclair AH Dynamic regulation of mitotic arrest in fetal male germ cells. Stem Cells Dayt. Ohio 26, 339–347 (2008). [DOI] [PubMed] [Google Scholar]
- 29.Bondar T & Medzhitov R p53-mediated hematopoietic stem and progenitor cell competition. Cell Stem Cell 6, 309–322 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bowling S et al. P53 and mTOR signalling determine fitness selection through cell competition during early mouse embryonic development. Nat. Commun 9, 1763 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haupt S, Berger M, Goldberg Z & Haupt Y Apoptosis - the p53 network. J. Cell Sci 116, 4077–4085 (2003). [DOI] [PubMed] [Google Scholar]
- 32.de Felici MD et al. Bcl-2 and Bax regulation of apoptosis in germ cells during prenatal oogenesis in the mouse embryo. Cell Death Differ. 6, 908–915 (1999). [DOI] [PubMed] [Google Scholar]
- 33.Rucker EB et al. Bcl-x and Bax Regulate Mouse Primordial Germ Cell Survival and Apoptosis during Embryogenesis. Mol. Endocrinol 14, 1038–1052 (2000). [DOI] [PubMed] [Google Scholar]
- 34.Maclean JA et al. Rhox: a new homeobox gene cluster. Cell 120, 369–382 (2005). [DOI] [PubMed] [Google Scholar]
- 35.Spiller CM et al. Endogenous Nodal signaling regulates germ cell potency during mammalian testis development. Development 139, 4123–4132 (2012). [DOI] [PubMed] [Google Scholar]
- 36.Saba R, Kato Y & Saga Y NANOS2 promotes male germ cell development independent of meiosis suppression. Dev. Biol 385, 32–40 (2014). [DOI] [PubMed] [Google Scholar]
- 37.Molaro A et al. Two waves of de novo methylation during mouse germ cell development. Genes Dev. 28, 1544–1549 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Suzuki A & Saga Y Nanos2 suppresses meiosis and promotes male germ cell differentiation. Genes Dev. 22, 430–435 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Milholland B et al. Differences between germline and somatic mutation rates in humans and mice. Nat. Commun 8, 15183 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seisenberger S et al. The Dynamics of Genome-wide DNA Methylation Reprogramming in Mouse Primordial Germ Cells. Mol. Cell 48, 849–862 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hill PWS et al. Epigenetic reprogramming enables the transition from primordial germ cell to gonocyte. Nature 555, 392–396 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hargan-Calvopina J et al. Stage-Specific Demethylation in Primordial Germ Cells Safeguards against Precocious Differentiation. Dev. Cell 39, 75–86 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hajkova P et al. Epigenetic reprogramming in mouse primordial germ cells. Mech. Dev 117, 15–23 (2002). [DOI] [PubMed] [Google Scholar]
- 44.Malki S, van der Heijden GW, O’Donnell KA, Martin SL & Bortvin A A Role for Retrotransposon LINE-1 in Fetal Oocyte Attrition in Mice. Dev. Cell 29, 521–533 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reznik B et al. Heterogeneity of transposon expression and activation of the repressive network in human fetal germ cells. Development dev.171157 (2019). doi: 10.1242/dev.171157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brockway NL et al. Multicolour lineage tracing using in vivo time-lapse imaging reveals coordinated death of clonally related cells in the developing vertebrate brain. Dev. Biol 453, 130–140 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Watanabe K et al. Cripto-1 Is a Cell Surface Marker for a Tumorigenic, Undifferentiated Subpopulation in Human Embryonal Carcinoma Cells. STEM CELLS 28, 1303–1314 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Spiller CM, Bowles J & Koopman P Nodal/Cripto signaling in fetal male germ cell development: implications for testicular germ cell tumors. Int. J. Dev. Biol 57, 211–219 (2013). [DOI] [PubMed] [Google Scholar]
- 49.Dawson EP et al. Delayed male germ cell sex-specification permits transition into embryonal carcinoma cells with features of primed pluripotency. Development 145, dev156612 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Aravin AA et al. A piRNA pathway primed by individual transposons is linked to de novo DNA methylation in mice. Mol. Cell 31, 785–799 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hansson K et al. Ripleygui: software for analyzing spatial patterns in 3d cell distributions. Frontiers in Neuroinformatics. 7, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Susaki EA et al. Whole-brain imaging with single-cell resolution using chemical cocktails and computational analysis. Cell 157, 726–739 (2014). [DOI] [PubMed] [Google Scholar]
- 53.Jin Y et al. TEtranscripts: A package for including transposable elements in differential expression analysis of RNA-seq datasets. Bioinformatics 31, 3593–3599 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liberzon A et al. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Subramanaian A et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. Transcriptional markers that distinguish each single germ cell state compared to all others were identified among differentially expressed genes in an E13.5 germ cell population by the two-tailed Wilcoxon rank sum test-based FindMarkers operation in Seurat, which provides a p-value that is further adjusted by bonferroni correction using all genes in the dataset.
Supplementary Table 2. Comparison of markers for MD-germ cells with GRRs and genes upregulated in Dnmt1-cKO male germ cells42. Comparison of markers for AP-germ cells with genes upregulated in Tet1-KO43 male germ cells and genes downregulated in Dnmt1-cKO. Adjusted p-values for MD- and AP-markers were generated by Wilcoxon rank sum test with bonferroni correction. Statistics for markers from Dnmt1-cKO and Tet1-KO were obtained from referenced studies.
Wholemount visualization of apoptotic clusters of PGCs at E13.5. Whole E13.5 testis with cell nuclei identified by Hoechst and PGCs by Tra98. Apoptosis is identified by cleaved-PARP and rotated views highlight the spatial distribution of apoptotic PGCs. Magnified views show a representative apoptotic germ cell cluster.
PGC proliferation is clonal. E13.5 PGCs are clonally labeled using Rainbow and cells in late G2/M are marked by phosphor-histone H3. Magnified view shows two separate groups of phosphohistone H3+ cells that are clonal (mOrange clone and a GFPclone).
Multicolor clone labeling reveals shared fluorophores within germ cell cysts. 3-dimensional perspective of whole E13.5 Rainbow;Tex14+/+;Pou5f1Cre-ER testis with bicolored clone (mCerulean and mCherry) outlined as a surface, followed by individual channels confirming bicolored labeling.
Tex14 mutant testes differ in clone structure and do not form bicolored clones. 3-dimensional perspective of whole E13.5 Rainbow; Tex14−/−; Pou5f1Cre-ER testis with strictly single-colored clones as a consequence of lacking intercellular bridges.
Data Availability Statement
Data availability. scRNA-seq and BS-seq data that support the findings of this study have been deposited in the Gene Expression Omnibus (GEO) under accession codes GSE119045 and GSE155122. All other data supporting the findings of this study are available from the corresponding author on reasonable request. Gene ontology analysis on MSigDB computed overlaps with GO:BP (https://www.gsea-msigdb.org/gsea/msigdb/genesets.jsp?collection=GO:BP).