

Abstract
Simple Summary
Cellular metabolism including lipid metabolism is often altered in cancer cells. To keep up with the high energetic demand of cancer cells lipid metabolism is increased. Thus, mutations, changes in gene expression or other alterations of lipid metabolism genes are often seen in cancer cells. In this report we characterized DGAT2, a gene required for triacylglycerol synthesis and cell membrane structure using the Catalogue of Somatic Mutations in Cancers (COSMIC). We identified a hotspot mutation at D222V that may affect enzyme activity in cancer cells. Additionally, we find that DGAT2 mutations in cancer cells are distinguishable from a conserved mutation that is linked to Axonal Charcot-Marie-Tooth disease, an inherited condition leading to muscle degeneration. This suggests that DGAT2 mutations and alterations in cancer cells are specific to drive cellular transformation and immortalization.
Abstract
DGAT2 is a transmembrane protein encoded by the DGAT2 gene that functions in lipid metabolism, triacylglycerol synthesis, and lipid droplet regulation. Cancer cells exhibit altered lipid metabolism and mutations in DGAT2 may contribute to this state. Using data from the Catalogue of Somatic Mutations in Cancer (COSMIC), we analyzed all cancer genetic DGAT2 alterations, including mutations, copy number variations and gene expression. We find that several DGAT2 mutations fall within the catalytic site of the enzyme. Using the Variant Effect Scoring Tool (VEST), we identify multiple mutations with a high likelihood of contributing to cellular transformation. We also found that D222V is a mutation hotspot neighboring a previously discovered Y223H mutation that causes Axonal Charcot-Marie-Tooth disease. Remarkably, Y223H has not been detected in cancers, suggesting that it is inhibitory to cancer progression. We also identify several single nucleotide polymorphisms (SNP) with high VEST scores, indicating that certain alleles in human populations have a pathogenic predisposition. Most mutations do not correlate with a change in gene expression, nor is gene expression dependent on high allele copy number. However, we did identify eight alleles with high expression levels, suggesting that at least in certain cases, the excess DGAT2 gene product is not inhibitory to cellular proliferation. This work uncovers unknown functions of DGAT2 in cancers and suggests that its role may be more complex than previously appreciated.
Keywords: lipid metabolism, lipid droplet, lipid storage, DNA damage, mutation, gene expression, cancer
1. Introduction
Lipids play many important roles in the cell, including energy storage, structure, signaling, gene regulation, and metabolism [1]. Although much is known about the reprogramming of lipid metabolism by cancer cells, such as upregulated fatty acid oxidation and cholesterol accumulation [2], the roles of many lipid regulators involved in specific genomic instability processes have only recently been extensively characterized [3,4,5]. One major category of lipids is nonmembrane lipids, which are primarily neutral lipids such as fatty acids (FAs) and derivatives such as triacylglycerol. Triacylglycerols are often found sequestered into lipid droplets (LDs) and form the pool of free FAs for mitochondrial beta oxidation [6].
Triacylglycerol biosynthesis follows either the glycerol phosphate pathway or the monoacylglycerol pathway [7]. Within both pathways, the acyl CoA diacylglycerol acyltransferase enzymes DGAT1 and DGAT2 catalyze the conversion of diacylglycerol to triacylglycerol. Both enzymes are found on the surface of the ER membrane, with DGAT1 on the luminal side and DGAT2 facing the cytoplasmic side [8].
While DGAT1 has additional functions, DGAT2 is specific to triacylglycerol synthesis. It catalyzes a covalent bond between de novo synthesized activated fatty acids (FA-CoA) and diacylglycerol [9]. DGAT2 is of crucial importance to cell membrane stability. Knocking out DGAT2 in mice is lethal because it leads to impaired skin permeability and dehydration [8]. DGAT2 mutations are also associated with physiological alterations like Charcot-Marie-Tooth disease in humans, compromising muscle structure and function [10].
Lipid droplets are primarily composed of triacylglycerols and other neutral lipids [11]. These lipids are enclosed by a phospholipid monolayer, creating a droplet that can store nonpolar molecules in the aqueous cytosol [12]. Lipid droplets have a variety of functions, including preventing cellular damage by sequestering lipotoxic molecules, providing FAs for energy production, and maintaining ER and membrane homeostasis [13].
Lipid droplet metabolism involves the balance between lipolysis, lipophagy, and lipogenesis. Lipolysis releases FAs from lipid droplets through a series of sequential enzymes that break down triacylglycerols [13]. In adipose tissues, the products are released into the bloodstream, while in nonadipose tissues, the FAs are used for beta oxidation [14].
Lipid droplets are also degraded in autophagic compartments by lytic enzymes [14]. An overabundance of free FAs causes an increase in lipid droplet synthesis, which protects the cell from damaging lipotoxic effects [13]. This protective role and the ability of LDs to efficiently transfer FAs to the mitochondria ensure that LD biogenesis occurs even during periods of nutrient deprivation, using autophagy of structural lipids [13].
Cancer cells exhibit the Warburg effect in which glycolytic products are diverted away from the Krebs Cycle [15]. Instead of undergoing normal cellular respiration when oxygen is present, cancer metabolism prioritizes fermentation. This increases glutamine metabolism, lactate production, and gluconeogenesis, a process that results in elevated lipogenic precursors such as citrate. Consequently, cancer cells show greater lipid synthesis to meet the increased energetic demands of tumors [16]. This metabolic shift is characterized by de novo synthesis of FAs, which can be converted to triacylglycerol by DGAT2 and stored in lipid droplets. Many cancers have been found to display accumulation of lipid droplets, including those affecting brain, lung, renal, and skin tissues [13].
DGAT2 mutations in cancers are common but have not yet been thoroughly investigated. In this study, we carried out a pan-cancer analysis of DGAT2 mutations. Using data from the Catalogue of Somatic Mutations in Cancers (COSMIC) [17], we analyzed the mutational landscape of DGAT2 and characterized pathogenic mutations associated with changes in DGAT2 function that likely contribute to cellular transformation.
2. Materials and Methods
COSMIC is a repository for somatic mutations and genome alterations in cancers. It combines data from the NIH Cancer Genome Atlas (TCGA) project, the International Cancer Genome Consortium (ICGC), the Cell Lines Project, as well as other independent studies [17]. The COSMIC data is manually curated by experts and often includes PubMed IDs of studies which we used for references in this report. An Excel file with all DGAT2 COSMIC mutations was downloaded from https://cancer.sanger.ac.uk/cosmic (version 92, accessed on 29 October 2020) and statistically analyzed to reveal mutation frequencies that may affect the functional role of DGAT2 in cancer metabolism. The copy number variation file was generated using the Copy Number Analysis (CONAN) tool. Expression data of DGAT2 was also downloaded from the same site.
Protein alignments of the DGAT2 isoforms were generated using COBALT (https://www.ncbi.nlm.nih.gov/tools/cobalt/re_cobalt.cgi, accessed on 4 March 2021) as described previously [18,19]. The lollipop figures were made as reported earlier [20,21].
The Cancer-Related Analysis of Variants Toolkit (CRAVAT) software was employed to determine the likelihood of mutations being drivers or passengers and was used as previously described [22,23].
All statistical analyses and graphs were done using SPSS® version 17. The figures were created in Photoshop®.
3. Results and Discussion
3.1. DGAT2 Isoforms and Conservation
The DGAT2 gene contains eight introns and nine exons [24]. Two accepted transcript variants and protein isoforms and several alternatively spliced isoforms have been reported (Table 1). The major transcript reported on COSMIC (accessed in October 2020) is NM_032564.5 (ENST00000228027.11), corresponding to isoform 1. However, an isoform 2, as well as a predicted transcript X1, are also listed on NCBI. All amino acid annotations/mutations listed in this report use isoform 1. An alignment of the major DGAT2 isoforms shows that a central region is conserved among all of them (Supplementary Figure S1). Previous studies have also shown that DGAT2 is ubiquitous in both plants and animals [24,25]. A comprehensive study of 117 sequences from 70 organisms showed that DGAT2 is more conserved than DGAT1 [26]. An alignment of several animal sequences shows lower conservation in the first 40 amino acids (Supplementary Figure S1, Table S1). Additionally, human Isoform X1 appears to be the major transcript in several other species suggesting that the N-terminus of the protein is more plastic and often functionally dispensable.
Table 1.
DGAT2 Transcripts and isoforms.
| 1 Transcript | Transcript Length (Nucleotides) | 2 Protein Isoforms | Protein Size (Amino Acids) | UniProt Identifier |
|---|---|---|---|---|
| NM_032564.5 | 2407 | 1 | 388 | Q96PD7-1 |
| NM_001253891.2 | 2278 | 2 | 345 | Q96PD7-2 |
| XM_011545304 | 2156 | X1 | 358 | N/A |
| - | - | 3 | 295 | S4R3S3 |
| - | - | 4 | 160 | S4R383 |
| - | - | 5 | 112 | S4R3Z3 |
| - | - | 6 | 113 | S4R449 |
1 The variants are from NCBI. 2 The isoforms 1 and 2 are accepted isoforms from NCBI. Isoform X1 is predicted. Isoforms 3–6 are possible isoforms from UniProt.
3.2. DGAT2 Mutation Distribution in Human Cancers
The COSMIC database reports a total of 398 DGAT2 mutations detected in 21 different cancers (Figure 1A,B and Supplementary Table S2). The majority of the mutations within DGAT2 are intronic, substitution/missense, and synonymous (Figure 1A). Non-sense mutations also represent frameshifts resulting from insertions and deletions introducing a stop codon before the endogenous stop codon. The highest frequency of missense mutations was observed in skin tissue samples (frequency (f) = 21). Other tissues showing elevated mutation frequencies include the lungs (f = 16), large intestines (f = 16), and endometrium (f = 15) (Figure 1B). The cancer type for some mutations is not specified. Consistent with the high mutation frequencies seen in epithelial tissues, the most common cancer type associated with DGAT2 mutations was carcinoma, followed by malignant melanoma (Figure 1C). This result also correlates with a high prevalence of cancer in skin, lung, large intestine, and endometrial tissues (Figure 1D).
Figure 1.

Distribution of DGAT2 mutations in cancer samples. (A) Frequency of mutation types in all cancer samples reported on COSMIC. “Intronic” mutations also include 5′ and 3′ UTRs. (B) Tissue distribution of missense, non-sense and indel mutations. Synonymous, intronic and UTR mutations were excluded from this analysis as they are not likely to change the function of the gene. The panel follows the same color coding as in (A). (C) Distribution of missense, non-sense and InDel mutations by cancer histology. (D) Distribution of cancer histology by tissue. This panel follows the same color coding as in (C). (E) Frequency of missense, non-sense and InDel pathogenic mutations. (F) Scatter graph to show tissue correlation with the FATMHH score. The distribution of the FATHMM score was also graphed as a boxplot for easy visualization of median and quartiles. Probability values represent Pearson’s two-tailed significance.
3.3. Mutation Pathogenicity Is Correlated with Older Age
COSMIC reports a FATHMM score that indicates the pathogenicity of a mutation [27]. Scores range between 0 and 1 and mutations with a score of 0.7 and higher are predicted to be pathogenic. A graph of all FATMHH scores of DGAT2 CDS mutations (excluding intronic and 5′ and 3′ UTRs) shows that most of the missense, non-sense, and InDel mutations were associated with high pathogenicity scores (Figure 1E). Pathogenic mutations were identified in all cancers, but esophagus, large intestine, lung, and skin tissue mutations were characterized by clusters of high pathogenic scores (Figure 1F). This heavy burden of pathogenic mutations within the coding sequence is expected considering the high level of DGAT2 sequence conservation. Consistent with the general lack of sequence conservation in the N-terminus, the average FATMHH score for mutations within the first 39 amino acids is low, but variability is high (0.48 ± 0.37). This suggests that mutations in certain key residues are highly pathogenic while others are not. Predictably, the pathogenic score for all other residues is substantial (0.77 ± 0.3).
Genomic instability is a defining feature of cancer cells and a driving force behind tumor evolution [28]. It is also an established hallmark of aging [29]. The accumulation of genetic damage both results from and contributes to the aging process [30]. COSMIC reports patient age at the time of tissue extraction and sequencing for most samples. Thus, we investigated any correlation between pathogenic mutations and patient age.
DGAT2 mutations linked to elevated pathogenicity were observed in samples from patients whose ages were near the post-reproductive period or in the senescent stage of life (Figure 2 and Supplementary Figure S2). For instance, endometrium samples were taken from patients in the 40–50 age range (Figure 2A). Patients with reported ages of 50–55 years contributed samples with prevalent DGAT2 mutations in large intestine and skin tissues, while stomach mutations originated primarily in samples from septuagenarians. Thyroid cancers mostly revealed DGAT2 mutations in specimens from younger individuals. Similarly, in gliomas, DGAT2 mutations were mainly detected in subjects around the age of 40. The significance of these observations is not immediately clear due to the limitations of small sample sizes for these tissues.
Figure 2.

Correlation of patient age with mutation pathogenicity. (A) Age and tissue distribution of mutations. (B) Age and histology distribution of mutations. Only tissues for which at least five samples were reported were included in (A) and (B). Complete data are reported in Figure S2. For A and B, circles represent outliners and asterisks extreme values. (C) Scatter graph to show the correlation of FATMHH pathogenicity score and age. (D) Scatter graph to show the distribution of FATMHH pathogenicity score by cancer histology.
Carcinomas and malignant melanomas were linked to DGAT2 mutations in samples from patients in the 50–70 age group (Figure 2B). These results agree with the expected increase of cancer prevalence observed during senescence. Intriguingly, the young age of patients whose samples were linked to gliomas (~40 years) may indicate a set point in mid-maturity when alterations to DGAT2 can result from and contribute to oncogenic states in the brain. However, these age data are interpreted with some caution because it is not clear from COSMIC whether the age reported in the database occurs at diagnosis or following patient death. Moreover, COSMIC data does not reveal age of cancer onset, which limits potential inferences to relative comparisons with published data on the subject.
The high pathogenicity scores of DGAT2 mutations in carcinoma samples (above 0.9625) and in many tissue types, notably skin and esophagus, suggests that the DGAT2 mutations in these tissues may be critical for cellular immortalization or cancer progression. Shifts in lipid metabolism are an established hallmark of cancer [2]. Thus, given the crucial involvement of DGAT2 in lipid regulation, pathogenic mutations in this gene may contribute to shifts in lipid metabolism that are implicated in promotion of cancer. Our findings on DGAT2 mutation pathogenicity in carcinomas support previous research on hepatocellular carcinomas (HCC). DGAT2 was found to be downregulated in HCC, while increased DGAT2 expression repressed tumor growth [31]. The DGAT2 mutational landscape includes alterations that may decrease or inhibit its enzymatic activity, resulting in hypomorphic DGAT2 in the cell. This would create a similar effect to that of DGAT2 downregulation observed in hepatocellular carcinomas and suggests an important role of DGAT2 functional insufficiency in cancer progression.
To further understand the contribution of DGAT2 mutations to carcinogenesis, we used the Cancer-Related Analysis of Variants Toolkit (CRAVAT) software which predicts the potential of a mutation to be driver or pathogenic [22,32,33,34]. All COSMIC-reported mutations (including noncoding/intronic) were processed using this analysis. Because of the small data set, we extracted only mutations with p-values lower than 0.05 and false discovery rates (FDR) lower than 0.01 (Supplementary Table S3).
Our analysis reveals that none of the DGAT2 mutations (coding or noncoding) are drivers. This was an expected outcome because, to our knowledge, this gene has not been characterized as a driver gene. However, certain mutations including D222V (see Section 3.3) were pathogenic within statistical significance (Table 2). Notably, all pathogenic mutations fall within the DGAT2 catalytic site, and they do not correlate with any tissue specificity. None of the intronic or 5′ and 3′ UTR mutations are pathogenic. We also observed that high pathogenicity correlates well with specimens from patients older than 40 years of age (Figure 2C). In general, however, carcinomas and melanomas, which are exceedingly represented in samples from older patients, are statistically enriched in higher levels of pathogenic mutations (Figure 2D).
Table 2.
DGAT2 alleles with significant VEST values.
| 1 Mutation | 2 VEST p-Value | 2 VEST FDR | Tissue | 3 dbSNP |
|---|---|---|---|---|
| G212C | 0.00071000 | 0.05 | CNS | rs777087960 |
| H163R | 0.00071000 | 0.05 | Endometrium | |
| R259H | 0.00091000 | 0.05 | Large Intestine | |
| E243Q | 0.00121000 | 0.05 | Upper Aer Dig | |
| F314S | 0.00152000 | 0.05 | Upper Aer Dig | |
| P329S | 0.00182000 | 0.05 | Endometrium | |
| F262V | 0.00182000 | 0.05 | Pancreas | |
| G261C | 0.00253000 | 0.05 | Lung | |
| G197D | 0.00283000 | 0.05 | Endometrium | rs369680804 |
| S278F | 0.00283000 | 0.05 | Soft Tissue | |
| G318R | 0.00314000 | 0.05 | Kidney | |
| L267R | 0.00314000 | 0.05 | Liver | |
| G164S | 0.00536000 | 0.05 | Haem and Lymph | |
| P215H | 0.00607000 | 0.05 | Endometrium | |
| G120D | 0.008 | 0.10 | Skin | |
| R205K | 0.0082 | 0.10 | Lung | |
| A241V | 0.01002000 | 0.10 | Lung | rs766238005 |
| L245V | 0.01113000 | 0.10 | Lung | |
| W100L | 0.01214000 | 0.10 | Skin | |
| N155S | 0.01295000 | 0.10 | Skin | |
| G270R | 0.01326000 | 0.10 | Lung | |
| G167D | 0.01427000 | 0.10 | Prostate | |
| P141S | 0.01488000 | 0.10 | Lung | |
| S244C | 0.01528000 | 0.10 | Liver | |
| K146N | 0.0164 | 0.10 | Large Intestine | |
| R218W | 0.01791000 | 0.10 | Skin | rs528376420 |
| A310T | 0.01893000 | 0.10 | CNS | rs761761542 |
| W126R | 0.02054000 | 0.10 | Stomach | |
| P215S | 0.02146000 | 0.10 | Skin | |
| D222V | 0.02257000 | 0.10 | Kidney, Large intestine | |
| N228Y | 0.0248 | 0.10 | Stomach | |
| S294F | 0.0249 | 0.10 | Skin | |
| R297Q | 0.02844000 | 0.10 | Haem and Lymph | rs140793537 |
| R297 * | 0.03186000 | 0.10 | Stomach | rs771080849 |
| Y139 * | 0.03273000 | 0.10 | Esophagus | |
| R189L | 0.0339 | 0.10 | Skin | |
| R137 * | 0.03489000 | 0.10 | Skin | rs572486802 |
4 Diagram with mutation distribution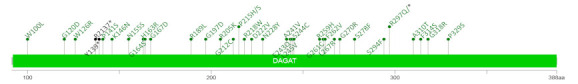
| ||||
1 Only those mutations with significant VEST values are shown. Mutations are organized by p-value significance. 2 Significance was chosen for mutations with p-value < 0.05 and FDR < 0.1. 3 NCBI Identifiers for mutations that have single nucleotide polymorphisms. 4 Green labels represent point mutations and black labels represent truncations.
Multiple DGAT2 mutations have previously been identified as single nucleotide polymorphisms (SNPs) by COSMIC as well as NCBI (Supplementary Table S3), including eight that the CRAVAT algorithm identifies as pathogenic (Table 2). NCBI does not list clinical significance for any of the eight SNPs nor could we find any literature describing the phenotypes of these alleles. However, one publication does report several intergenic SNPs associated with decreased DGAT2 expression, which promotes predisposition to prostate cancer [35]. We investigated expression data for DGAT2 mutations (see Section 3.7) and indeed, one of the alleles (R218W, rs528376420) appears to show increased expression levels, though the results are highly variable and not statistically significant under our analysis.
Taken together, these data show that certain DGAT2 mutations are likely to promote cellular transformation. Moreover, certain SNPs in human populations may predispose individuals to cancer development. However, the clinical significance of these SNPs remains to be further investigated.
3.4. Identification of a Mutation Hotspot in DGAT2
DGAT2 is a transmembrane protein with two domains embedded in the phospholipid bilayer on the N-terminal half [7,36] (Figure 3A). Two other domains towards the C-terminus are also embedded but do not cross the entire membrane. The C-terminus of DGAT2 is necessary for the catalytic function of the enzyme [37]. Several conserved domains have been identified within the C-terminus: the membrane-binding domain (MBD) from residues 156 to 199 interacts with the ER membrane, and the lipid droplet (LD) targeting domain from 293–309, interacts with lipid droplets. These findings are consistent with a recent report that identified three cysteine residues in the C-terminus at 172, 214, and 312 that are essential for DGAT2 catalytic activity [9]. The catalytic domain of the enzyme spans between amino acids 93 and 387. Two conserved consensus sequences HPHG (conserved in DGAT family proteins) [36] and FLXLXXXn (conserved in lipid metabolizing enzymes) [38] have also been identified. This structure of the protein is inferred from mouse data and simulated models [7,36] because, to our knowledge, it has not yet been determined in humans.
Figure 3.

Mutation distribution over the open reading frame of DGAT2. (A) Cartoon of DGAT2 protein structure adapted from [7]. Residue numbers are inferred from the mouse data. The protein is predicted to cross the ER membrane once between amino acids 66–115 and bind (but not cross) the membrane two more times between 156–119 and 293–309. The amino acid sequence HPHG is conserved in the DGAT2 family proteins. The FLXLXXXn is a consensus sequence for lipid metabolizing proteins. (B) Frequency distribution of mutations that fall over the CDS of DGAT2. A D222B hotspot is identified in kidney cancers.
A visual inspection of COSMIC mutations shows that they are distributed throughout the entire region of the coding sequence. However, the Kolmogorov–Smirnov test for uniformity reveals a statistically nonuniform distribution (p = 0.007). Indeed, we identified a D222V hotspot which appears primarily in renal carcinomas (Figure 3B) [39]. Remarkably, a recent report characterized a mutation in the neighboring residue (Y223H) in a family with Axonal Charcot-Marie-Tooth disease [10]. The authors propose that this mutation explains the autosomal dominant inheritance of the disease. Phenotypes associated with p.Y223H in CMT include decreased serum TG levels and decreased axonal branching. Both the D222 and Y223 residues are highly conserved in animals (Supplementary Figure S1).
However, a mutation at Y223 does not appear in cancers reported on COSMIC. Overexpression of the DGAT2 Y223H mutant inhibited cellular proliferation [10], so it is possible that it negatively affects cancer progression and is therefore selected against. An analysis of mutation preferences in cancer cells shows that Y to H (tyrosine to histidine) mutations are twice as likely to appear in cancers than D to V (aspartate to valine) [40], which would suggest that the Y223H mutation should be more likely than D222V. Y to H mutations also have a higher evolution tendency than D to V [41]. Thus, this strongly indicates that mutation of the Y223 residue affects the function of the protein in a manner that is inhibitory to cancer development.
3.5. Mutational Landscape
We next investigated the distribution of DGAT2 mutations by mutation type (Figure 4). Several non-sense mutations, including three frameshifts that introduce a stop codon were identified (Figure 4A). These mutations appear in multiple cancer types: hematopoietic and lymphoid (A242Gfs*2), urinary tract (E15*), lung (W40*, E283*, E291*), endometrium (H163Pfs*81), skin (Q143*, R137*), stomach (R297*), large intestine (R317*) and esophagus (Y139*). For one, the tissue is not specified (G280EFs*9). Mutations that appear early in the sequence are predicted to have a more drastic effect because they truncate larger regions of the protein. Non-sense mutations (p.E15*, p.W40*, p.R137*, p.Y139*, and p.Q143*) may truncate the protein before the MBD and LD targeting domains. Additionally, mutations (p.E283*, p.E291*, and p.R297*) may result in an incomplete LD targeting domain. A frameshift insertion (H163Pfs*81) within the MBD segment, an insertion before the LD domain (A242Gfs*2), and a deletion (G280Efs*9) could also impact the normal function of DGAT2. Unfortunately, COSMIC does not provide information on zygosity of these mutations, so it is unclear if one or both alleles of the gene are affected. Therefore, in this analysis, we cannot rule out changes to DGAT2 function that stem from haploinsufficiency.
Figure 4.

Distribution of DGAT mutations by mutation type. Shown are non-sense (blue) and frameshift (red) (A), missense (B) and synonymous (C). Only mutations that appear more than twice are labeled. Complete data is shown in Supplementary Table S2.
Missense mutations may also impact the catalytic activity in the C-terminus. As previously mentioned, the p.Y223H mutation contributes to the human disease Charcot-Marie-Tooth disease (CMT) [10]. Of the cancer samples found in COSMIC, the highest frequency missense mutation was p.D222V, occurring just before the defining CMT residue and within the same well-conserved N-acetyltransferase superfamily (NAT-SF) domain (Figure 4B). Several other mutations with a frequency ≥2 appear within the catalytic subunit of the enzyme and within or near the MBD or LD domains (Figure 4B). Some substitutions such as W126R/S, G270R, G293S, A310T, P345H and P380L cause a change from polar to nonpolar residues or vice versa. This observation indicates potential changes in structure that could impact the enzymatic function of the protein.
Synonymous mutations do not alter protein sequence but may affect mRNA stability or codon usage during translation [42]. Thus, they can affect gene expression. A graph of synonymous mutations shows a complete absence in the middle of the protein between amino acids 77–189 (Figure 4C). The reason for this is unclear but suggests that nucleotide alterations may not be tolerated in that region.
3.6. DGAT2 Expression Levels in Cancer Tissues
Gene expression levels are available for certain TCGA samples. We retrieved DGAT2 expression levels for the various cohorts listed on COSMIC (Supplementary Figure S3). The data is represented as a Z-score, which is internally normalized to control [43]. If there is no change in gene expression compared to control the Z-value is 0. Z-values greater than +2 or lower than −2 represent over-expression or under-expression, respectively. Our analysis of the various cohorts shows that all are skewed towards over-expressed DGAT2, suggesting that certain individuals within the groups have high expression levels of the gene. This does not appear to be cancer specific. We found this paradoxical since several studies have shown that DGAT2 overexpression inhibits cell proliferation [10,31,35].
To understand whether DGAT2 mutations affect gene expression levels, we extracted Z-values for all TCGA samples (Supplementary Table S4). We find that most mutation values are within the normal range. We identified 10 mutations that were over-expressed (Table 3).
Table 3.
Expression levels of select DGAT2 mutations.
| Sample ID 1 | Sample Name | Mutation | Mutation Z-Score | Sample Z-Sore Range 2 |
|---|---|---|---|---|
| COSU540 | Cecum carcinoma | p.G120D | 2.221 | 0.30 ± 4.68 |
| COSU413 3 | Bladder urothelial carcinoma | p.P275= | 2.335 | 0.19 ± 1.19 |
| COSU418 | Lung squamous cell carcinoma | p.P141S | 2.583 | 0.07 ± 1.18 |
| COSU419 | Uterine corpus endometrial carcinoma | p.R189W | 2.706 | 0.22 ± 1.19 |
| COSU540 | Cecum carcinoma | p.R218W | 3.292 | 0.30±4.68 |
| COSU418 | Lung squamous cell carcinoma | p.W40* | 3.368 | 0.07 ± 1.18 |
| COSU435 | Prostate adenocarcinoma | p.G167D | 3.647 | 0.05 ± 1.05 |
| COSU377 | Acute myeloid leukemia | c.*27A>G | 4.978 | 0.0040 ± 0.98 |
| COSU414 | Breast invasive carcinoma | p.I94V | 5.195 | 0.140 ± 2.34 |
| COSU419 | Uterine corpus endometrial carcinoma | c.*556A>T | 6.743 | 0.22 ± 1.19 |
1See Supplementary Figure S3B for tissue information on Sample ID. 2 See Supplementary Figure S3A for extended information on Z-score range. 3 Red samples are statistically significant.
Because Z-values are internally controlled, expression data of individual samples may only be compared within the cohort. When compared with the cohort range, eight out of the 10 mutations showed increased expression levels (Table 3). We searched the literature to see if expression levels for the 10 mutations were previously characterized. TCGA P141S expression levels were determined in lung squamous cell carcinoma. Additionally, a study on BRAFV600 melanoma patients who received vemurafenib and dabrafenib chemotherapy also identified the mutation [44]. However, the melanoma study did not determine expression levels. Four different mutations at the R189 residues have been identified: R189=, R189L and R189Q [45] in the skin; and R189W in the endometrium. Only the TCGA data lists expression levels for W40*, G167D, I94V and for the two noncoding mutations *27A>G and *556A>T. These analyses indicate that most DGAT2 mutations do not affect gene expression.
3.7. Copy Number Variations of DGAT2 Alleles
We next used the Copy Number Analysis (CONAN) tool to investigate whether cancers are characterized by variations in DGAT2 copy numbers. Indeed, we find that most cancer tissues feature an increased copy number of DGAT2, including some samples with substantial increases (e.g., breast and ovary) (Figure 5A). NCBI reports the highest DGAT2 expression levels in fat tissue followed by liver and skin. The other tissues are characterized by low gene expression [46]. A higher copy number may allow higher gene expression in certain cancers. Nonetheless, an increased copy number does not appear to correlate with either the type of cancer or with tissues where normal expression is high.
Figure 5.

Copy number variation of DGAT2 alleles. (A) Copy number extracted from COSMIC with the CONAN tool. (B) Distribution of DGAT2 amplification (AMP), homozygous deletion (HD) and loss of heterozygosity (LOH) in CONAN samples. (C) Correlation of copy number with expression level (Z-value).
To investigate if copy number translates to higher expression, we extracted Z-values for samples derived from CONAN (Supplementary Table S5). Our analysis shows that there is no correlation between copy number and gene expression (Figure 5C). Remarkably, gene expression is highest when copy number is 10–20. Further experimental work is necessary to reveal the functional implications of this observation. A smaller subset of samples is characterized by homozygous deletion, which suggests that in those contexts, DGAT2 is not essential for cell proliferation or cancer progression.
4. Conclusions
Analyzing the mutational landscape of more regulators such as DGAT2 increases our understanding of deregulated lipid metabolism in cancer. Our analysis identified several things about mutated DGAT2 in cancer tissues. First, mutations distribute evenly throughout the CDS sequence. This is an expected observation given the high conservation of DGAT2 sequence and function. Second, DGAT2 mutations are found in all cancer types suggesting that deregulated gene activity is not cancer specific. Third, we identify pathogenic mutations and the likelihood that certain SNPs may predispose individuals to cancer. Fourth, expression levels are not associated with cancer type or allele copy number. Taken together, these data suggest that the role of deregulated DGAT2 is context-dependent across cancers and is associated with structural alterations in conserved functional domains (Figure 6). Future examination of mutations in other lipid regulators will help determine the impact of deregulated lipid metabolism on cancer progression and facilitate a probabilistic assessment of cancer predisposition following post-biopsy sequencing.
Figure 6.

Model of the functional consequence in cancers linked to DGAT2 mutations. An initial DNA damage event on the sequence of DGAT2 occurs through various mechanisms. DGAT2 pathogenic mutations become fixed in cells that adapt and thrive in specific tissue environments. As the population of DGAT2 mutant cells outcompetes its wild-type counterparts, lipid metabolism changes due to altered function in the DGAT2 protein and associated lipid regulators. Cancer-promoting conditions select for DGAT2 deregulation to contribute to cellular transformation.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/biology10080714/s1, Figure S1: Sequence alignment of DGAT2 from various animal species, Figure S2: Mutation distribution by age, Figure S3: DGAT2 cohorts expression levels, Table S1: DGAT2 sequences used for the alignment in Supplementary Figure S1, Table S2: DGAT2 mutations reported on COSMIC separated by cancer type, Table S3: CRAVAT analyses, Table S4: Expression data for TCGA samples, Table S5: DGAT2 expression profiles in copy number variants.
Author Contributions
Conceptualization, W.E. and R.C.P.; writing-original draft preparation, M.G., W.E., and R.C.P.; writing-review and editing: M.G., H.B., R.W., and M.V.; formal analysis: W.E. and R.C.P.; investigation: M.G. and A.P.; supervision: W.E and R.C.P.; funding acquisition: W.E and R.C.P. All authors have read and agreed to the published version of the manuscript.
Funding
Funding for WE, MG, HB, RW, and MV was provided by the Biology Department Research Fund and the College of Arts and Sciences Undergraduate Research Program at Xavier University. Funding for RCP and AP was provided by The Ohio State University Comprehensive Cancer Center Pelotonia Fund.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data used in this study i available on COSMIC (https://cancer.sanger.ac.uk/cosmic, accessed on 29 October 2020).
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Shyu P., Wong X.F.A., Crasta K., Thibault G. Dropping in on lipid droplets: Insights into cellular stress and cancer. Biosci. Rep. 2018;38 doi: 10.1042/BSR20180764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang W., Bai L., Li W., Cui J. The Lipid Metabolic Landscape of Cancers and New Therapeutic Perspectives. Front. Oncol. 2020;10 doi: 10.3389/fonc.2020.605154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Palian B.M., Rohira A.D., Johnson S.A.S., He L., Zheng N., Dubeau L., Stiles B.L., Johnson D.L. Maf1 Is a Novel Target of PTEN and PI3K Signaling That Negatively Regulates Oncogenesis and Lipid Metabolism. PLoS Genet. 2014;10:e1004789. doi: 10.1371/journal.pgen.1004789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shetty M., Noguchi C., Wilson S., Martinez E., Shiozaki K., Sell C., Mell J.C., Noguchi E. Maf1-dependent transcriptional regulation of tRNAs prevents genomic instability and is associated with extended lifespan. Aging Cell. 2020;19:e13068. doi: 10.1111/acel.13068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hammerquist A.M., Escorcia W., Curran S.P. Maf1 regulates intracellular lipid homeostasis in response to DNA damage response activation. Mol. Biol. Cell. 2021;32:1086–1093. doi: 10.1091/mbc.E20-06-0378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhatt-Wessel B., Jordan T.W., Miller J.H., Peng L. Role of DGAT enzymes in triacylglycerol metabolism. Arch. Biochem. Biophys. 2018;655:1–11. doi: 10.1016/j.abb.2018.08.001. [DOI] [PubMed] [Google Scholar]
- 7.Yen C.L.E., Stone S.J., Koliwad S., Harris C., Farese R.V. DGAT enzymes and triacylglycerol biosynthesis. J. Lipid Res. 2008;49:2283–2301. doi: 10.1194/jlr.R800018-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chitraju C., Walther T.C., Jr., Farese R.V. The triglyceride synthesis enzymes DGAT1 and DGAT2 have distinct and overlapping functions in adipocytes. J. Lipid Res. 2019;60:1112–1120. doi: 10.1194/jlr.m093112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jung S., Choi M., Choi K., Bin Kwon E., Kang M., Kim D.-E., Jeong H., Kim J., Kim J.H., Kim M.O., et al. Inactivation of human DGAT2 by oxidative stress on cysteine residues. PLoS ONE. 2017;12:e0181076. doi: 10.1371/journal.pone.0181076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bin Hong Y., Kang J., Kim J.H., Lee J., Kwak G., Hyun Y.S., Nam S.H., Hong H.D., Choi Y.-R., Jung S.-C., et al. DGAT2Mutation in a Family with Autosomal-Dominant Early-Onset Axonal Charcot-Marie-Tooth Disease. Hum. Mutat. 2016;37:473–480. doi: 10.1002/humu.22959. [DOI] [PubMed] [Google Scholar]
- 11.Meyers A., Weiskittel T.M., Dalhaimer P. Lipid Droplets: Formation to Breakdown. Lipids. 2017;52:465–475. doi: 10.1007/s11745-017-4263-0. [DOI] [PubMed] [Google Scholar]
- 12.Khor V.K., Shen W.-J., Kraemer F. Lipid droplet metabolism. Curr. Opin. Clin. Nutr. Metab. Care. 2013;16:632–637. doi: 10.1097/MCO.0b013e3283651106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Petan T., Jarc E., Jusović M. Lipid Droplets in Cancer: Guardians of Fat in a Stressful World. Mol. 2018;23:1941. doi: 10.3390/molecules23081941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang C.-W. Lipid droplets, lipophagy, and beyond. Biochim. Biophys. Acta (BBA)—Mol. Cell Biol. Lipids. 2016;1861:793–805. doi: 10.1016/j.bbalip.2015.12.010. [DOI] [PubMed] [Google Scholar]
- 15.Warburg O. On the Origin of Cancer Cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 16.Currie E., Schulze A., Zechner R., Walther T., Farese R.V. Cellular Fatty Acid Metabolism and Cancer. Cell Metab. 2013;18:153–161. doi: 10.1016/j.cmet.2013.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tate J.G., Bamford S., Jubb H.C., Sondka Z., Beare D.M., Bindal N., Boutselakis H., Cole C.G., Creatore C., Dawson E., et al. COSMIC: The Catalogue of Somatic Mutations In Cancer. Nucleic Acids Res. 2019;47:D941–D947. doi: 10.1093/nar/gky1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nelakurti D.D., Pappula A.L., Rajasekaran S., Miles W.O., Petreaca R.C. Comprehensive Analysis of MEN1 Mutations and Their Role in Cancer. Cancers. 2020;12:2616. doi: 10.3390/cancers12092616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McPherson M.T., Holub A.S., Husbands A.Y., Petreaca R.C. Mutation Spectra of the MRN (MRE11, RAD50, NBS1/NBN) Break Sensor in Cancer Cells. Cancers. 2020;12:3794. doi: 10.3390/cancers12123794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jay J.J., Brouwer C. Lollipops in the Clinic: Information Dense Mutation Plots for Precision Medicine. PLoS ONE. 2016;11:e0160519. doi: 10.1371/journal.pone.0160519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang W., Wang C., Zhang X. Mutplot: An easy-to-use online tool for plotting complex mutation data with flexibility. PLoS ONE. 2019;14:e0215838. doi: 10.1371/journal.pone.0215838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Douville C., Carter H., Kim R., Niknafs N., Diekhans M., Stenson P.D., Cooper D.N., Ryan M., Karchin R. CRAVAT: Cancer-related analysis of variants toolkit. Bioinform. 2013;29:647–648. doi: 10.1093/bioinformatics/btt017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Masica D.L., Douville C., Tokheim C., Bhattacharya R., Kim R., Moad K., Ryan M.C., Karchin R. CRAVAT 4: Cancer-Related Analysis of Variants Toolkit. Cancer Res. 2017;77:e35–e38. doi: 10.1158/0008-5472.CAN-17-0338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Turchetto-Zolet A.C., Maraschin F.S., de Morais G.L., Cagliari A., Andrade C.M., Margis-Pinheiro M., Margis R. Evolutionary view of acyl-CoA diacylglycerol acyltransferase (DGAT), a key enzyme in neutral lipid biosynthesis. BMC Evol. Biol. 2011;11:263. doi: 10.1186/1471-2148-11-263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Turchetto-Zolet A.C., Christoff A.P., Kulcheski F., Loss-Morais G., Margis R., Margis-Pinheiro M. Diversity and evolution of plant diacylglycerol acyltransferase (DGATs) unveiled by phylogenetic, gene structure and expression analyses. Genet. Mol. Biol. 2016;39:524–538. doi: 10.1590/1678-4685-gmb-2016-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cao H. Structure-Function Analysis of Diacylglycerol Acyltransferase Sequences from 70 Organisms. BMC Res. Notes. 2011;4:249. doi: 10.1186/1756-0500-4-249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rogers M.F., Shihab H.A., Mort M.E., Cooper D.N., Gaunt T., Campbell C. FATHMM-XF: Accurate prediction of pathogenic point mutations via extended features. Bioinform. 2018;34:511–513. doi: 10.1093/bioinformatics/btx536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee J.-K., Choi Y.-L., Kwon M., Park P.J. Mechanisms and Consequences of Cancer Genome Instability: Lessons from Genome Sequencing Studies. Annu. Rev. Pathol. Mech. Dis. 2016;11:283–312. doi: 10.1146/annurev-pathol-012615-044446. [DOI] [PubMed] [Google Scholar]
- 29.López-Otín C., Blasco M.A., Partridge L., Serrano M., Kroemer G. The Hallmarks of Aging. Cell. 2013;153:1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Z., Zhang Z., Ren Y., Wang Y., Fang J., Yue H., Ma S., Guan F. Aging and age-related diseases: From mechanisms to therapeutic strategies. Biogerontology. 2021;22:165–187. doi: 10.1007/s10522-021-09910-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Y., Li T., Jin Y., Shen J. Dgat2 reduces hepatocellular carcinoma malignancy via downregulation of cell cycle-related gene expression. Biomed. Pharmacother. 2019;115:108950. doi: 10.1016/j.biopha.2019.108950. [DOI] [PubMed] [Google Scholar]
- 32.Carter H., Chen S., Isik L., Tyekucheva S., Velculescu V., Kinzler K.W., Vogelstein B., Karchin R. Cancer-Specific High-Throughput Annotation of Somatic Mutations: Computational Prediction of Driver Missense Mutations. Cancer Res. 2009;69:6660–6667. doi: 10.1158/0008-5472.CAN-09-1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Douville C., Masica D.L., Stenson P.D., Cooper D.N., Gygax D.M., Kim R., Ryan M., Karchin R. Assessing the Pathogenicity of Insertion and Deletion Variants with the Variant Effect Scoring Tool (VEST-Indel) Hum. Mutat. 2016;37:28–35. doi: 10.1002/humu.22911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carter H., Douville C., Stenson P.D., Cooper D.N., Karchin R. Identifying Mendelian disease genes with the Variant Effect Scoring Tool. BMC Genom. 2013;14:S3. doi: 10.1186/1471-2164-14-S3-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nurminen R., Rantapero T., Wong S.C., Fischer D., Lehtonen R.J., Tammela T.L., Nykter M., Visakorpi T., Wahlfors T., Schleutker J. Expressional profiling of prostate cancer risk SNPs at 11q135 identifiesDGAT2as a new target gene. Genes Chromosom. Cancer. 2016;55:661–673. doi: 10.1002/gcc.22368. [DOI] [PubMed] [Google Scholar]
- 36.Stone S.J., Levin M.C., Farese R.V., Jr. Membrane topology and identification of key functional amino acid residues of murine acyl-CoA:diacylglycerol acyltransferase-2. J. Biol. Chem. 2006;281:40273–40282. doi: 10.1074/jbc.M607986200. [DOI] [PubMed] [Google Scholar]
- 37.McFie P.J., Banman S.L., Kary S., Stone S.J. Murine Diacylglycerol Acyltransferase-2 (DGAT2) Can Catalyze Triacylglycerol Synthesis and Promote Lipid Droplet Formation Independent of Its Localization to the Endoplasmic Reticulum. J. Biol. Chem. 2011;286:28235–28246. doi: 10.1074/jbc.M111.256008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alam M., Gilham D., Vance D.E., Lehner R. Mutation of F417 but not of L418 or L420 in the lipid binding domain decreases the activity of triacylglycerol hydrolase. J. Lipid Res. 2006;47:375–383. doi: 10.1194/jlr.M500344-JLR200. [DOI] [PubMed] [Google Scholar]
- 39.Gerlinger M., Horswell S., Larkin J., Rowan A.J., Salm M., Varela I., Fisher R., McGranahan N., Matthews N., Santos C.R., et al. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat. Genet. 2014;46:225–233. doi: 10.1038/ng.2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anoosha P., Sakthivel R., Gromiha M.M. Exploring preferred amino acid mutations in cancer genes: Applications to identify potential drug targets. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2016;1862:155–165. doi: 10.1016/j.bbadis.2015.11.006. [DOI] [PubMed] [Google Scholar]
- 41.Creixell P., Schoof E.M., Tan C.S., Linding R. Mutational properties of amino acid residues: Implications for evolvability of phosphorylatable residues. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2012;367:2584–2593. doi: 10.1098/rstb.2012.0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brule C.E., Grayhack E.J. Synonymous Codons: Choose Wisely for Expression. Trends Genet. 2017;33:283–297. doi: 10.1016/j.tig.2017.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cheadle C., Vawter M.P., Freed W.J., Becker K.G. Analysis of Microarray Data Using Z Score Transformation. J. Mol. Diagn. 2003;5:73–81. doi: 10.1016/S1525-1578(10)60455-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Van Allen E.M., Wagle N., Sucker A., Treacy D.J., Johannessen C.M., Goetz E.M., Place C.S., Taylor-Weiner A., Whittaker S., Kryukov G.V., et al. Faculty Opinions recommendation of The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Fac. Opin.—Post-Publ. Peer Rev. Biomed. Lit. 2015;4:94–109. doi: 10.3410/f.718184868.793510330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pickering C.R., Zhou J.H., Lee J.J., Drummond J.A., Peng S.A., Saade R.E., Tsai K.Y., Curry J.L., Tetzlaff M.T., Lai S.Y., et al. Mutational Landscape of Aggressive Cutaneous Squamous Cell Carcinoma. Clin. Cancer Res. 2014;20:6582–6592. doi: 10.1158/1078-0432.CCR-14-1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fagerberg L., Hallström B.M., Oksvold P., Kampf C., Djureinovic D., Odeberg J., Habuka M., Tahmasebpoor S., Danielsson A., Edlund K., et al. Analysis of the Human Tissue-specific Expression by Genome-wide Integration of Transcriptomics and Antibody-based Proteomics. Mol. Cell. Proteom. 2014;13:397–406. doi: 10.1074/mcp.M113.035600. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data used in this study i available on COSMIC (https://cancer.sanger.ac.uk/cosmic, accessed on 29 October 2020).