

Abstract
The Gram-positive bacterium Listeria monocytogenes is the causative agent of the foodborne disease listeriosis, one of the deadliest bacterial infections known. In order to cause disease, L. monocytogenes must properly coordinate its metabolic and virulence programs in response to rapidly changing environments within the host. However, the mechanisms by which L. monocytogenes senses and adapts to the many stressors encountered as it transits through the gastrointestinal (GI) tract and disseminates to peripheral organs are not well understood. In this study, we investigated the role of the redox-responsive transcriptional regulator Rex in L. monocytogenes growth and pathogenesis. Rex is a conserved canonical transcriptional repressor that monitors the intracellular redox state of the cell by sensing the ratio of reduced and oxidized nicotinamide adenine dinucleotides (NADH and NAD+, respectively). Here, we demonstrated that L. monocytogenes Rex represses fermentative metabolism and is therefore required for optimal growth in the presence of oxygen. We also show that in vitro, Rex represses the production of virulence factors required for survival and invasion of the GI tract, as a strain lacking rex was more resistant to acidified bile and invaded host cells better than wild type. Consistent with these results, Rex was dispensable for colonizing the GI tract and disseminating to peripheral organs in an oral listeriosis model of infection. However, Rex-dependent regulation was required for colonizing the spleen and liver, and L. monocytogenes lacking the Rex repressor were nearly sterilized from the gallbladder. Taken together, these results demonstrated that Rex functions as a repressor of fermentative metabolism and suggests a role for Rex-dependent regulation in L. monocytogenes pathogenesis. Importantly, the gallbladder is the bacterial reservoir during listeriosis, and our data suggest redox sensing and Rex-dependent regulation are necessary for bacterial survival and replication in this organ.
Author summary
Listeriosis is a foodborne illness caused by Listeria monocytogenes and is one of the deadliest bacterial infections known, with a mortality rate of up to 30%. Following ingestion of contaminated food, L. monocytogenes disseminates from the gastrointestinal (GI) tract to peripheral organs, including the spleen, liver, and gallbladder. In this work, we investigated the role of the redox-responsive regulator Rex in L. monocytogenes growth and pathogenesis. We demonstrated that alleviation of Rex repression coordinates expression of genes necessary in the GI tract during infection, including fermentative metabolism, bile resistance, and invasion of host cells. Accordingly, Rex was dispensable for colonizing the GI tract of mice during an oral listeriosis infection. Interestingly, Rex-dependent regulation was required for bacterial replication in the spleen, liver, and gallbladder. Taken together, our results demonstrate that Rex-mediated redox sensing and transcriptional regulation are important for L. monocytogenes metabolic adaptation and virulence.
Introduction
To successfully colonize different niches, bacteria must be able to rapidly sense and respond to environmental changes. The Gram-positive bacterium Listeria monocytogenes is an excellent example of this adaptability. As a saprophyte and intracellular pathogen, L. monocytogenes coordinates its metabolic and virulence programs to transition from life in nature to the mammalian host where it causes the foodborne disease listeriosis. Following ingestion of contaminated foods by the host, L. monocytogenes contends with acid stress in the stomach and acidic bile in the small intestine before descending to the cecum where it traverses the intestinal barrier [1]. Traveling via the lymph or blood, L. monocytogenes disseminates to the spleen and liver where it replicates intracellularly. The intracellular lifecycle requires L. monocytogenes to quickly escape the oxidizing vacuolar compartment to replicate in the highly reducing environment of the cytosol and then spread cell-to-cell [2–5]. From the liver, the bacteria enter the gallbladder and replicate extracellularly to very high densities and then reseed the intestinal tract upon bile secretion [6–8]. Bile itself is antimicrobial, acting as a detergent that disrupts bacterial membranes and denatures proteins [9]. Although L. monocytogenes virulence determinants have been investigated for decades, the vast majority of studies injected the bacteria intravenously rather than infecting mice through the natural foodborne route [10, 11]. Therefore, the mechanisms by which L. monocytogenes senses and adapts to the many stressors of the host environment during oral infection are not well understood.
Given its ability to replicate in diverse environmental niches, it is critical for L. monocytogenes to appropriately modify its metabolism in response to the changing extracellular surroundings. Reduced and oxidized nicotinamide adenine dinucleotides (NADH and NAD+, respectively) play important roles in many biological processes and are therefore key molecules for sensing the intracellular redox state [12]. For example, during aerobic respiration the NADH:NAD+ ratio is kept low as NADH is oxidized to NAD+ by the electron transport chain (ETC). In hypoxic environments or when the ETC is inhibited, NADH levels become elevated and NAD+ is no longer available to fuel carbon oxidation for growth. Therefore, the NADH:NAD+ ratio is the primary indicator of the metabolic state of a cell.
The transcriptional repressor Rex monitors the intracellular redox state of the cell by directly sensing the NADH:NAD+ ratio and repressing target genes when this ratio is low [13, 14]. An increase in relative NADH abundance following reduced respiration results in Rex dissociating from DNA and derepression of target genes [15–18]. Rex is widely conserved across Gram-positive bacteria and while there is considerable variability in the identity of Rex-dependent genes among organisms, Rex generally functions to regulate metabolic pathways involved in NAD+-regeneration, such as fermentation [13, 14].
L. monocytogenes encodes a Rex protein that shares 65% and 56% identity with homologues in Bacillus subtilis and Staphylococcus aureus [19]. We hypothesized L. monocytogenes Rex may be important during infection to sense the changing environment and regulate metabolic pathways accordingly. In this study, we identified Rex-dependent transcriptional changes in L. monocytogenes and demonstrated a role for Rex regulation during oral listeriosis.
Results
Transcriptomics identifies Rex-regulated genes
To investigate the role of Rex in L. monocytogenes, we generated a Δrex mutant via allelic exchange and analyzed the Rex-dependent transcriptome under standard growth conditions. RNA sequencing (RNA-seq) was performed on RNA harvested from mid-log and stationary phase cultures of wild type (wt) and Δrex strains grown aerobically in brain heart infusion (BHI) broth (Tables A-D in S1 Text). We did not observe dramatic growth phase-dependent differences in Rex-dependent regulation so here we focus on the stationary phase results for simplicity. In the Δrex mutant, 196 transcripts were significantly increased in abundance at least two-fold (p < 0.01), indicating these genes are repressed by Rex during aerobic growth (Table 1 and Table A in S1 Text). Some of the most dramatically increased transcripts were involved in fermentative metabolism, including those encoding alcohol dehydrogenase (lap), pyruvate formate lyase (pflA and pflBC), and lactate dehydrogenase (ldhA) (Table 1). Unexpectedly, transcripts encoding virulence factors involved in bile resistance (bsh, bile salt hydrolase) and host cell invasion (inlAB, internalin A and B) were in greater abundance in the Δrex mutant, indicating Rex-dependent regulation may impact virulence. The RNA-seq results were validated via quantitative RT-PCR (qPCR) by measuring the expression of 5 genes in wt and Δrex during stationary phase aerobic growth. We also measured gene expression in the rex complemented strain, in which rex was expressed from its native promoter at an ectopic site in the chromosome (Δrex p-rex). In the Δrex mutant, all 5 transcripts were increased in abundance compared to the wt and Δrex p-rex strains (S1 Fig), consistent with the RNA-seq results.
Table 1. Rex-repressed genes-of-interest.
| 10403S | EGD-e | Gene | Function | Fold change in Δrex |
|---|---|---|---|---|
| LMRG_01332 | lmo1634 | lap | bifunctional acetaldehyde-CoA/alcohol dehydrogenase | 342.30 |
| LMRG_00859 | lmo1407 | pflC | pyruvate formate-lyase 1-activating enzyme | 88.30 |
| LMRG_00858 | lmo1406 | pflB | formate acetyltransferase | 59.84 |
| LMRG_00046 | lmo0355 | frdA | fumarate reductase flavoprotein subunit | 85.24 |
| LMRG_01064 | lmo1917 | pflA | formate acetyltransferase | 77.91 |
| LMRG_01979 | lmo2717 | cydB | cytochrome d ubiquinol oxidase subunit II | 19.05 |
| LMRG_01980 | lmo2716 | cydC | ABC transporter | 17.62 |
| LMRG_01981 | lmo2715 | cydD | ABC transporter | 15.76 |
| LMRG_01978 | lmo2718 | cydA | cytochrome bd-I oxidase subunit I | 13.99 |
| LMRG_01659 | lmo2173 | - | sigma-54-dependent transcriptional regulator | 16.62 |
| LMRG_00127 | lmo0434 | inlB | internalin B | 10.97 |
| LMRG_00126 | lmo0433 | inlA | internalin A | 10.26 |
| LMRG_01801 | lmo2447 | - | Rgg/GadR/MutR family transcriptional regulator | 10.76 |
| LMRG_02012 | lmo0912 | - | formate transporter | 10.71 |
| LMRG_01217 | lmo2067 | bsh | bile acid hydrolase | 5.57 |
| LMRG_02632 | lmo0210 | ldhA | lactate dehydrogenase | 3.30 |
Highlighted genes are predicted to be in an operon [20].
In silico promoter analysis of genes in the 10403S genome exhibiting Rex-dependent regulation was performed to determine potential Rex binding sites using the Bacillus subtilis Rex consensus sequence [13]. Allowing up to 3-mismatches with the B. subtilis consensus sequence, we identified potential Rex binding sites in the promoter regions of 48 genes and/or operons repressed by Rex (Table E in S1 Text). Specifically, we identified putative Rex binding sites upstream of lap, pflBC, and pflA, indicating Rex likely represses fermentative metabolism directly. Rex binding sites were also predicted upstream of bsh and inlAB, further suggesting direct involvement of Rex in virulence gene regulation.
In the absence of rex, 110 transcripts were less abundant during aerobic growth, indicating the presence of Rex is required to fully activate these genes (Table C in S1 Text). As Rex is a canonical transcriptional repressor, we hypothesize these changes are due to indirect effects. Indeed, promoter analysis did not identify any putative Rex binding sites in the promoters of genes activated in the presence of Rex, suggesting these changes are likely due to indirect Rex-dependent regulation.
Fermentative metabolism is repressed by Rex
Transcriptional analysis indicated that Rex-mediated repression functions to down-regulate fermentative metabolism during aerobic growth (Fig 1A). To verify the role of Rex in regulating metabolism, we first assessed growth of the wt and Δrex strains during both aerobic and anaerobic growth. A small, but significant, growth defect was observed for rex-deficient L. monocytogenes, beginning 4 hours post-inoculation into aerobic shaking flasks (Fig 1B). This defect was not due to a change in glucose uptake, as wt and Δrex consumed glucose similarly (Fig 1C). In contrast, the Δrex strain exhibited no growth defect when grown anaerobically (S2A Fig), demonstrating Rex-dependent repression is alleviated during wt anaerobic growth.
Fig 1. Fermentative metabolism is repressed by Rex during aerobic growth.

A. Model of aerobic and anaerobic central metabolic pathways in L. monocytogenes. Enzymes encoded by genes repressed by Rex are denoted in purple text. Underlined metabolic end-products were those differentially produced by Δrex compared to wt during aerobic growth. LdhA, lactate dehydrogenase; PflABC, pyruvate formate lyase; Lap, alcohol dehydrogenase; ATP, adenosine triphosphate; NAD(H), nicotinamide adenine dinucleotide; ETC, electron transport chain; TCA, tricarboxylic acid cycle. B. Aerobic growth of wt, Δrex, and the complemented strain (Δrex p-rex) measured by optical density (OD600). C-F. Glucose and extracellular metabolites were quantified 4 hours post-inoculation in aerobic (black, blue, and light blue bars) and anaerobic (grey and purple bars) cultures. Concentrations of glucose (C), lactate (D), formate (E), and acetate (F) were determined and normalized to OD600. G. Relative intracellular ATP concentration was measured at 4 hours during aerobic (black, blue, and light blue bars) and anaerobic (grey and purple bars) growth. Data are the means and standard error of the mean (SEM) of three independent experiments in all panels with the exception of panel G. Here, anaerobic samples are the means and SEMs of two independent experiments. p values were calculated using a heteroscedastic Student’s t test. * p < 0.05; ** p < 0.01; **** p < 0.0001; n.s. p > 0.05. In all panels, no significant difference was found between wt and Δrex p-rex. In C-G, no significant difference was found between the wt and Δrex strains grown in anaerobic conditions.
To clarify the effect of Rex regulation on L. monocytogenes aerobic growth, extracellular metabolites were quantified 4 hours post-inoculation. The Δrex mutant secreted approximately 90% more lactate and ~55% more formate than wt and the complemented strain (Fig 1D and 1E). This was accompanied by a concomitant decrease in the primary aerobic by-product acetate (Fig 1F). These metabolite changes confirmed the transcriptional analysis showing increased transcript abundance of ldhA, pflA, and pflBC in the absence of rex and demonstrated that carbon-flux is being directed primarily towards lactate fermentation (Fig 1A). When grown anaerobically, both the wt and Δrex strains produced more lactate and formate, demonstrating a switch to fermentative metabolism in the absence of oxygen (Fig 1D and 1E). We were unable to determine if the increased expression of lap in Δrex impacted ethanol production under aerobic conditions, as this volatile metabolite evaporates in a shaking flask and could not be reliably quantified. However, ethanol was detected when bacteria were grown anaerobically in a sealed tube. During mid-log growth, the wt and Δrex strains produced similar amounts of ethanol (S2F Fig). However, following glucose depletion and entry into stationary phase at 6 hours (S2G Fig), the Δrex mutant made significantly more ethanol than wt (S2H Fig). These results were not unexpected, as lap expression was increased in the Δrex mutant following 7 hours of anaerobic growth (S1B Fig), indicating that Rex functions to repress lap transcription when bacteria enter stationary phase.
Results from the extracellular metabolite analysis led us to hypothesize that the aerobic growth defect exhibited by the Δrex strain is a result of aberrant carbon flux through fermentation. This would result in decreased intracellular ATP stores compared to wt aerobic growth which generates ATP through oxidative phosphorylation and the ETC (Fig 1A). To test this hypothesis, we measured ATP concentrations 4 hours post-inoculation when the growth defect of Δrex becomes apparent. Indeed, the rex-deficient strain had 50% the amount of ATP compared to wt and the complemented strain in aerobic conditions (Fig 1G). Similarly, strains grown anaerobically had ~50% the amount of ATP compared to wt growing aerobically, further demonstrating the Δrex strain is undergoing fermentation during aerobic growth (Fig 1G). As previously stated, glucose consumption and extracellular metabolite profiles were similar between the wt and Δrex strains when incubated anaerobically, demonstrating that Rex-mediated repression is normally alleviated in this growth environment (Figs 1C–1G and S2). Taken together, these data indicate that L. monocytogenes Rex functions to repress fermentative metabolism in the presence of oxygen and a strain lacking rex is impaired for aerobic growth as a result of altered carbon-flux and decreased ATP production.
L. monocytogenes Δrex is more resistant to acidified bile in vitro
In addition to metabolic genes, the transcriptional profile revealed that Rex represses virulence determinants necessary in the host gastrointestinal (GI) tract, including bile salt hydrolase (bsh) and internalins A and B (inlAB). Bsh detoxifies bile, which is encountered during transit through the GI tract and colonization of the gallbladder [21–23]. To investigate the role of Rex in bile resistance, we first generated L. monocytogenes strains lacking bsh or both rex and bsh, and assessed their survival following a 24-hour exposure to porcine bile in BHI. L. monocytogenes colonizing the gallbladder would be exposed to bile at neutral pH [9], which we found to have no effect on the survival of any bacterial strain tested (Fig 2). These results are consistent with published reports demonstrating that L. monocytogenes bsh is not required for survival in neutral bile [22, 23]. In contrast, L. monocytogenes in the GI tract encounters acidified bile following its release from the gallbladder into the low pH environment of the duodenum [9, 24]. Acidified bile was highly bactericidal, resulting in a 2.5-log reduction in wt survival 24 hours post-inoculation (Fig 2). This killing was dependent on bile, as all strains grew equally well in acidified BHI lacking bile (S3 Fig). L. monocytogenes lacking bsh was even more sensitive to acidified bile, exhibiting a 4-log decrease in survival (Fig 2). As predicted, the Δrex mutant was more resistant to the toxic effects of acidified bile, exhibiting only a ~9-fold reduction in CFU. Trans-complementation of rex returned the susceptibility to similar levels as wt (Δrex p-rex). Moreover, the ΔrexΔbsh double mutant displayed similar susceptibility as the Δbsh mutant (Fig 2), indicating the increased survival of the Δrex mutant is dependent upon bsh expression. These results demonstrated that L. monocytogenes lacking the Rex repressor are more resistant to acidified bile due to increased bsh expression.
Fig 2. Alleviation of Rex repression promotes bacterial survival in acidified bile.
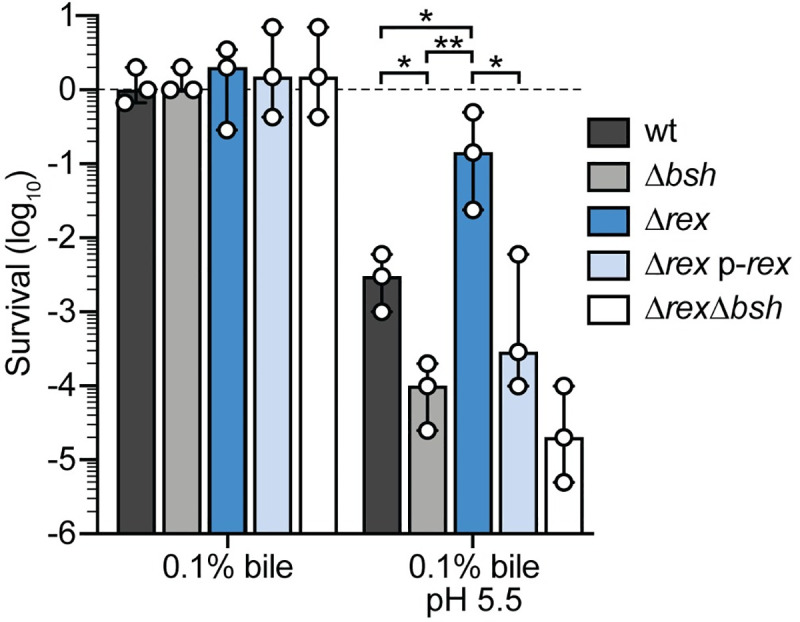
Survival of wt (black), Δbsh (grey), Δrex (blue), Δrex p-rex (light blue) and ΔrexΔbsh (white) normalized to the initial inoculum (dashed line = 1) and expressed as log-transformed CFU per mL of culture. Strains were evaluated for survival 24 hours post-inoculation in BHI supplemented with bile or acidified BHI supplemented with bile under aerobic conditions. Data are the means and ranges of three independent experiments. p values were calculated using a heteroscedastic Student’s t test. * p < 0.05; ** p < 0.01; n.s. p > 0.05. No significant difference was found between the Δbsh and ΔrexΔbsh strains.
The role of Rex in the intracellular lifecycle of L. monocytogenes
The intracellular lifecycle of L. monocytogenes begins with entry into host cells by phagocytosis or bacterial-mediated invasion, followed by replication within the cytosol, and cell-to-cell spread via actin polymerization [2]. RNA-seq revealed that transcripts encoding InlA and InlB were increased in the Δrex mutant, leading to the hypothesis that Rex regulates invasion of non-phagocytic cells via receptor-mediated endocytosis. Specifically, InlA and InlB mediate invasion of epithelial cells and hepatocytes by engaging the host receptors E-cadherin and Met, respectively [5, 25]. To investigate the effects of increased inlAB transcription in the Δrex mutant, we measured bacterial invasion of Caco-2 human intestinal epithelial cells and Huh7 human hepatocytes. To measure invasion, cells were treated with gentamicin 1 hour post-infection and intracellular bacteria were enumerated 2 hours post-infection. We observed a significant 41% increase in invasion of Caco-2 cells by the Δrex strain, which could be reduced back to wt levels with ectopic expression of rex (Fig 3A). This increase in Δrex was completely dependent on inlAB expression, as invasion was not significantly different between the ΔinlAB and ΔrexΔinlAB strains. Similarly, Δrex exhibited increased invasion of Huh7 cells in an InlAB-dependent manner (Fig 3B). Together, these results demonstrated that increased inlAB transcription in L. monocytogenes Δrex results in increased invasion of human intestinal epithelial cells and human hepatocytes in vitro.
Fig 3. Alleviation of Rex repression promotes bacterial invasion of Caco-2 epithelial cells and Huh7 hepatocytes.

The ability of L. monocytogenes strains to invade Caco-2 epithelial cells (A) and Huh7 hepatocytes (B) was evaluated. Bacterial invasion was measured 2 hours post-infection and 1 hour after adding gentamicin to kill extracellular bacteria. Invasion was normalized to the initial inocula and is reported as a percentage of wt. In panel A, data are the means and SEMs of 5 independent experiments performed in duplicate. In panel B, data are the means and SEMs of 3 independent experiments performed in duplicate. p values were calculated using a heteroscedastic Student’s t test. * p < 0.05; ** p < 0.01; *** p < 0.001. No statistical significance was found between strains lacking inlAB.
After invading host cells via receptor-mediated endocytosis or phagocytosis, L. monocytogenes replicates intracellularly and spreads cell-to-cell using actin-based motility [2–5]. To investigate the role of Rex regulation in these facets of pathogenesis, we first measured intracellular growth in several relevant cell types. We found that the Δrex strain replicated intracellularly at the same rate as wt in activated bone marrow-derived macrophages (BMMs), Huh7 human hepatocytes, and TIB73 murine hepatocytes (Fig 4A–4C). These results suggested that Rex-mediated repression is not required for intracellular growth and therefore, deleting the Rex repressor had no effect on intracellular replication.
Fig 4. Intracellular growth and cell-to-cell spread are not impaired in Δrex.

A-C. Intracellular growth kinetics of wt and Δrex in IFNγ-activated BMMs (A), Huh7 cells (B), and TIB73 cells (C). D. Plaque area in L2 fibroblasts and TIB73 hepatocytes, measured as a percentage of wt. Data in panels A and D are the means and SEMs of three independent experiments. Data in panels B and C are the means and SEMs of two independent experiments performed in duplicate. In all panels, p values were calculated using a heteroscedastic Student’s t test. * p < 0.05; n.s. p > 0.05.
Next, cell-to-cell spread was evaluated via plaque assays in which a monolayer of cells is infected and both intracellular growth and intercellular spread are measured over 3 days [26]. The Δrex mutant formed plaques ~10% larger than those formed by wt L. monocytogenes in L2 murine fibroblasts and formed plaques ~5% smaller in TIB73 hepatocytes (Fig 4D), indicating that Rex regulation is not required for cell-to-cell spread. Taken together, these data demonstrated that L. monocytogenes lacking the Rex repressor has an advantage at early stages of infection, as Δrex displayed increased invasion of host cells. However, Rex is dispensable for intracellular growth and intercellular spread in all cell types analyzed.
Rex is required for virulence in a murine oral model of infection
We hypothesized that during oral infection of L. monocytogenes, Rex-mediated repression is alleviated due to the hypoxic environment of the GI tract. Derepression of Rex target genes would not only up-regulate fermentative metabolism for energy production in this environment but would also increase transcription of virulence factors required for successful infection of the GI tract. To test the ability of Δrex to survive in the murine GI tract, 6- to 8-week old female BALB/c mice were orally infected with 108 CFU of wt, Δrex, or Δrex p-rex and housed in cages with elevated wire bottoms to limit reinoculation by coprophagy [27, 28]. Prior to infection, mice were treated with streptomycin for 48 hours to increase susceptibility to oral L. monocytogenes infection [8, 27, 28]. Changes in body weight were recorded throughout the infection as a global measurement of disease severity [8]. Mice infected with wt and Δrex p-rex L. monocytogenes lost ~12% of their initial body weight throughout the 4 day infection (Fig 5A). In contrast, mice infected with Δrex lost only ~3% of their initial weight 3 days following infection and returned to their initial weight by 4 days post-infection (Fig 5A). These results indicated mice infected with Δrex experienced less severe disease than mice infected with either wt or the complemented strain following oral infection.
Fig 5. Rex is required for full virulence in an oral listeriosis model.

Female BALB/c mice were orally infected with 108 CFU of wt (black open squares), Δrex (blue circles), or Δrex p-rex (blue open triangles) and the number of bacteria present in each tissue was determined at 1 and 4 days post-infection. A. The body weights of the mice over time, reported as a percentage of body weight prior to infection. For wt and Δrex, data are the means and SEMs of n = 30 (day 1), n = 20 (day 2), n = 15 (day 3), and n = 10 (day 4). For Δrex p-rex, data represent the means and SEMs of n = 10 (day 1) and n = 5 (days 2–4). Significance between wt and Δrex is denoted by *, while # indicates significance between Δrex and Δrex p-rex. B-H. Mice were sacrificed each day and organs were harvested to enumerate bacterial burden. Each symbol represents an individual mouse (n = 10 per group for wt and Δrex and n = 5 per group for Δrex p-rex) and the solid lines indicate the geometric means. Dashed lines indicate the limit of detection (l.o.d.). Data are combined from two independent experiments for the wt and Δrex strains. Results are expressed as log-transformed CFU per organ or per gram of feces. p values were calculated using a heteroscedastic Student’s t test. * or # p < 0.05; ## p < 0.01; *** p < 0.001; **** p < 0.0001.
To determine bacterial burden, organs and feces were harvested, homogenized, and plated. Specifically within the GI tract, we analyzed the small intestinal tissue, intestinal contents, cecum, and feces. Similar bacterial loads were observed between all strains in the GI tract and feces throughout the infection, indicating that Rex-mediated transcriptional repression is dispensable for the GI phase of infection, as predicted (Fig 5B–5E). Rex was also not required for dissemination from the GI tract to internal organs, as evidenced by the similar bacterial burdens between all strains in the spleen, liver, and gallbladder 1 day post-infection (Fig 5F–5H). However, by day 4 of the infection, we observed a significant attenuation of Δrex compared to wt. Bacterial burdens in the spleens and livers of mice infected with Δrex were decreased by approximately 1-log compared to the wt strain (Fig 5F and 5G). The most dramatic attenuation was observed in the gallbladder, with Δrex decreased approximately 5-logs compared to wt 4 days post-infection (Fig 5H). Importantly, virulence could be restored back to wt levels in all three organs when mice were infected with the rex complemented strain (Fig 5F–5H). We also assessed bacterial burdens in mice infected with either the wt or Δrex strains over the course of a 4-day infection in two additional independent experiments and found similar results in all organs (S4 and S5 Figs). Taken together, these results confirmed our hypothesis that Rex-dependent repression is not required for colonization and invasion of the GI tract during oral infection, as a Δrex mutant was able to colonize and disseminate similar to wt. These results suggest that Rex is normally derepressed in this anaerobic environment. In addition, the infection studies revealed that Δrex is able to disseminate to internal organs in the early stages of infection. However, Rex-dependent regulation was required for replication in the spleen, liver, and gallbladder after oral infection.
Discussion
In this study, we investigated the role of the redox-responsive transcriptional regulator Rex in L. monocytogenes. Transcriptional and in silico promoter analyses identified dozens of genes likely to be directly repressed by Rex in vitro. We demonstrated that derepression of Rex target genes induces fermentative metabolism, resulting in decreased ATP production and impaired aerobic growth of L. monocytogenes lacking rex. We also present evidence that the presence of Rex impacts virulence factor production in vitro. These studies revealed that Δrex is more resistant to acidified bile in a Bsh-dependent manner and that over-expression of inlAB in the Δrex mutant leads to increased invasion of host cells. In vivo experiments demonstrated that Rex is dispensable for colonizing the GI tract and disseminating to peripheral organs in an oral listeriosis model of infection. However, Rex was required for colonization of the spleen, liver, and gallbladder. This in vivo attenuation was not a result of impaired intracellular replication or cell-to-cell spread, as the Δrex mutant performed similar to wt in cell culture assays of infection. Taken together, our results indicate an important role for redox sensing and Rex-mediated transcriptional repression during L. monocytogenes infection.
Transcriptome analysis revealed that L. monocytogenes Rex regulates metabolic pathways similarly to what has been described in other Gram-positive bacteria [13, 15–17]. Specifically, fermentative metabolic pathways were the most significantly changed in the Δrex mutant. We identified putative Rex binding sites in the promoters of 48 Rex-repressed genes and/or operons, including those involved in fermentation and virulence, suggesting Rex likely binds and directly represses these genes. In contrast, genes activated by Rex lacked a Rex-binding site and we hypothesize they are indirectly regulated. Further protein-DNA binding analysis is needed to elucidate the direct regulon of L. monocytogenes Rex.
The results herein demonstrate that L. monocytogenes Rex functions to repress fermentation during aerobic growth in order to maximize energy generation. We found the Δrex mutant over-expressed genes necessary for fermentative metabolism (lap, ldhA, and pflBC/pflA) and accordingly, produced more lactate and formate than wt when replicating aerobically. While acetate is the major by-product generated by wt L. monocytogenes during aerobic growth [29, 30], we observed a concomitant decrease in acetate production by Δrex. Together, these results suggest that in the absence of Rex repression, there is an increased metabolic flux from pyruvate towards lactate and away from acetate production. The increased LdhA activity to produce lactate funnels NADH away from the ETC, resulting in less ATP generation by respiration. Indeed, the L. monocytogenes Δrex strain exhibited an aerobic growth defect and produced half as much ATP as wt. The metabolic and growth phenotypes were ameliorated during anaerobic growth when Rex-mediated repression is relieved and fermentation is required for growth. Taken together, these results demonstrated that L. monocytogenes Rex is necessary to repress fermentative metabolism in the presence of oxygen in order to efficiently produce ATP.
Our transcriptional results suggested a role for L. monocytogenes Rex in regulating production of virulence factors necessary during oral listeriosis. Specifically, alleviation of Rex-mediated repression increased expression of genes encoding the bile detoxifying enzyme Bsh and the internalin proteins InlA and InlB. Within the GI tract, L. monocytogenes encounters acidified bile that can disrupt bacterial membranes, dissociate membrane proteins, and induce DNA damage and oxidative stress [9, 31]. Bsh detoxifies conjugated bile acids and contributes to bacterial survival in the GI tract, which is evidenced by the wide distribution of homologous enzymes among commensal gut bacteria [21, 22, 32]. Also within the GI tract, L. monocytogenes invades intestinal epithelial cells and disseminates to peripheral organs. InlA and InlB mediate invasion of non-phagocytic cells by engaging the host cell receptors E-cadherin and Met, respectively [5, 25, 33]. We demonstrated that rex-deficient L. monocytogenes were significantly more resistant to acidified bile stress and were better able to invade intestinal epithelial cells in vitro. It is important to note that while these in vitro experiments confirmed transcriptome analyses, it is unlikely that increased transcription of inlAB in the Δrex mutant had any effect on this strain during the oral infection in mice. While InlAB-mediated invasion is required for invasion of non-phagocytes in cell culture, it has been reported that neither of these adhesins are required for successful oral infection in the mouse listeriosis model [28, 34, 35]. Together, our in vitro results suggest that alleviation of Rex-mediated repression coordinates expression of genes necessary in the GI tract during infection, including fermentative metabolism, bile resistance, and invasion of host cells.
Following invasion of intestinal epithelial cells, L. monocytogenes disseminates via the lymph and blood to the spleen and liver where it replicates intracellularly and spreads cell-to-cell without entering the extracellular space [4]. We found that Rex was dispensable for intracellular replication in activated bone marrow-derived macrophages and hepatocytes. Furthermore, L. monocytogenes Δrex grew and spread cell-to-cell at the same rate as wt in both fibroblasts and hepatocytes. Combined, our in vitro results suggested that Rex is dispensable for the intracellular lifecycle and implied that Rex repression is alleviated in the GI tract during oral infection and this results in the upregulation of anaerobic metabolism and virulence factors.
To investigate the role of L. monocytogenes Rex in pathogenesis, we took advantage of a recently optimized oral listeriosis model of murine infection [8, 28]. As predicted from our in vitro results, we found that Rex was completely dispensable for colonizing the GI tract, suggesting that in wt L. monocytogenes, Rex repression is fully relieved in this hypoxic environment. Further, within the first 24 hours of infection, Δrex disseminated to the spleen, liver, and gallbladder similarly to wt. However, the Δrex mutant was attenuated overall, as mice infected with this strain lost significantly less body weight than mice infected with wt L. monocytogenes. Four days post-infection, we observed a 10-fold decrease in bacterial burden in the spleens and livers of mice infected with Δrex compared to wt-infected mice. Surprisingly, Δrex was attenuated ~5-logs in the gallbladders 4 days post-infection. This phenotype was quite dramatic, as only 2 of the 13 mice infected with Δrex harbored more than 10 CFU in the gallbladders when all three infection experiments were combined. This was in stark contrast to mice infected with the wt or complemented strain, which each harbored approximately 106 CFU per gallbladder at this time point.
A handful of other studies have identified L. monocytogenes mutants defective in colonizing the gallbladder, however these mutants were also more sensitive to bile stress in vitro or lacked known virulence factors [8, 23, 36–39]. In contrast, the Δrex strain was insensitive to neutral bile, more resistant to acidified bile stress, and was not impaired for intracellular growth or intercellular spread. Other than bile stress, not much is known about impediments to bacterial proliferation in the gallbladder, despite its importance to L. monocytogenes pathogenesis. Early during infection, a few bacteria are released from lysed hepatocytes and transit through the common bile duct to colonize the lumen of the gallbladder where they replicate to very high densities [6–8]. After a meal, the gallbladder contracts and delivers bile along with a bolus of L. monocytogenes back into the small intestine where it can reseed the intestinal tract [40]. Thus, the gallbladder quickly becomes the primary reservoir of L. monocytogenes during infection [7]. Ongoing work is aimed at identifying the stressors present in the gallbladder that inhibit Δrex replication in this organ, which may have more broad implications for other bacterial pathogens that replicate in the gallbladder, such as Salmonella spp [41].
Redox homeostasis and bacterial pathogenesis are intricately tied, although the mechanisms are not entirely understood [42]. Rex-regulated metabolic pathways have been indirectly implicated in virulence in other pathogens. S. aureus Rex controls expression of lactate dehydrogenase, which is essential for bacterial survival when exposed to nitric oxide produced by phagocytes [17, 43]. Similarly, Clostridium difficile Rex regulates butyrate production, which induces toxin synthesis during gut colonization [44]. In contrast, we identified putative Rex-binding sites in the promoters of inlAB and bsh, suggesting that L. monocytogenes Rex directly regulates these virulence factors. Interestingly, bsh and inlAB are positively regulated by the master virulence regulator PrfA and the stress-responsive alternative sigma factor SigB and are induced following exposure to bile and acidic conditions [21, 45–47]. This work demonstrated that bsh and inlAB are also induced under anaerobic conditions when Rex repression is alleviated. We predict these regulatory factors sense distinct or potentially overlapping environmental signals and converge on these virulence factors for appropriate and efficient regulation. Future research will investigate the crosstalk between these transcriptional regulators during pathogenesis and the variable redox environments encountered by L. monocytogenes during infection.
Overall, this work suggests a model in which L. monocytogenes Rex-mediated repression is alleviated in the anaerobic environment of the GI tract, thus upregulating fermentative metabolism and virulence factor production. However, following dissemination to internal organs, Rex is required to regulate factors critical for survival within the gallbladder. As the primary reservoir of L. monocytogenes during infection, identifying the factors required for survival and replication in the gallbladder is imperative for understanding L. monocytogenes pathogenesis.
Materials and methods
Ethics statement
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All protocols were reviewed and approved by the Animal Care and Use Committee at the University of Washington (Protocol 4410–01).
Bacterial strains and culture conditions
L. monocytogenes mutants were derived from wild type strain 10403S [48, 49] and cultured in brain heart infusion (BHI) broth at 37°C with shaking (220 rpm), unless otherwise stated. Antibiotics (purchased from Sigma Aldrich) were used at the following concentrations: streptomycin, 200 μg/mL; chloramphenicol, 10 μg/mL (Escherichia coli) and 7.5 μg/mL (L. monocytogenes); and carbenicillin, 100 μg/mL. Porcine bile (Sigma Aldrich) was dissolved in sterile BHI with streptomycin to ensure sterility. In cases where pH adjustments of media were carried out, 1N HCl was used and the pH was determined using VWR sympHony benchtop pH meter. L. monocytogenes strains are listed in Table F in S1 Text and E. coli strains are listed in Table G in S1 Text. Plasmids were introduced in E. coli via chemical competence and heat-shock and introduced into L. monocytogenes wt via trans-conjugation from E. coli SM10 [50].
Cell lines
Huh7 and Caco-2 are cancer cell lines derived from human males with hepatocellular carcinoma and colon adenocarcinoma, respectively. TIB73 is a spontaneously immortalized hepatocyte cell line from a normal BALB/c embryo liver. Huh7, Caco-2, and TIB73 cell lines were obtained from Joshua Woodward (University of Washington) [51]. L2 fibroblasts were described previously [52]. Cell lines were grown at 37°C in 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) with 10% heat-inactivated fetal bovine serum (FBS; 20% for Caco-2 cells) and supplemented with sodium pyruvate (2 mM) and L-glutamine (1 mM). For passaging, cells were maintained in Pen-Strep (100 U/ml) but were plated in antibiotic-free media for infections. Initial infection of TIB73 cells was carried out in DMEM with 0.1% FBS and replaced with 10% FBS in DMEM during gentamicin treatment.
Vector construction and cloning
To construct the Δrex mutant, ~700 bp regions upstream and downstream of rex (LMRG_01223) were PCR amplified using L. monocytogenes 10403S genomic DNA as a template. PCR products were restriction digested and ligated into pKSV7-oriT [53]. The plasmid pKSV7xΔbsh was constructed via Gibson assembly using the NEBuilder HiFi DNA assembly master mix. Regions ~1000 bp upstream and downstream of bsh were amplified with linker regions identical to those flanking a ccdB toxin cassette in pKSV7x [54]. pKSV7x was PCR amplified and DpnI treated. The linearized vector and insert PCR products were combined in the NEB master mix and ligated according to manufacturer instructions. pKSV7Δrex and pKSV7xΔbsh were transformed into E. coli and sequences were confirmed by Sanger DNA sequencing. Plasmids with the mutant Δrex and Δbsh alleles were introduced into L. monocytogenes via trans-conjugation and integrated into the chromosome. Colonies were purified on selective nutrient agar and subsequently cured of the plasmid by conventional methods [52]. Allelic exchange was confirmed by PCR. To construct the ΔrexΔbsh mutant, pKSV7Δrex was trans-conjugated into L. monocytogenes Δbsh and integrated into the chromosome as described above.
To construct the ΔrexΔinlAB mutant, the mutant rex region was amplified from pKSV7Δrex, restriction digested, and ligated into the pLIM1 plasmid containing a PheS* counterselection marker (provided as a generous gift from Arne Rietsch, Case Western Reserve University). Sequences were confirmed by Sanger DNA sequencing. The plasmid was introduced into L. monocytogenes ΔinlAB via trans-conjugation and integrated into the chromosome as previously described [52, 55]. Briefly, transconjugants were selected by growing on BHI containing streptomycin and chloramphenicol at 30°C for 24 hours. A colony from this plate was re-streaked onto a fresh plate and incubated at 42°C for 24–48 hours. A colony was re-steaked and grown at 42°C two additional times to ensure integration of the pLIM1Δrex plasmid into the chromosome. One colony was inoculated into BHI broth and grown overnight at 30°C. The culture was diluted 10−4 and 100 μl was plated on BHI agar supplemented with p-chloro-phenylalanine (18 mM) and incubated at 37°C overnight. Colonies that grew on the counterselection plates were validated to be chloramphenicol-sensitive and confirmed by PCR.
Ectopic expression of genes in L. monocytogenes was carried out using pPL2 integration plasmids [56]. The plasmid for complementing Δrex was constructed by PCR amplifying rex along with its predicted native promoter using L. monocytogenes 10403S genomic DNA as a template. Sequences were confirmed by Sanger DNA sequencing. The constructed pPL2 plasmid was trans-conjugated into L. monocytogenes Δrex and L. monocytogenes ΔrexΔinlAB integration was confirmed by antibiotic resistance.
RNA isolation
Nucleic acids were purified from bacteria harvested from broth culture as previously described [57]. Briefly, bacteria were grown overnight in BHI shaking at 37°C and normalized to an optical density at 600 nm (OD600) of 0.02 into 25 mL BHI. After 4 and 7 hours of aerobic growth at 37°C shaking, bacteria were mixed 1:1 with ice-cold methanol, pelleted, and stored at -80°C. Bacteria were lysed in phenol:chloroform containing 1% SDS by bead beating with 0.1 mm diameter silica/zirconium beads. Nucleic acids were precipitated from the aqueous fraction overnight at -20°C in ethanol containing sodium acetate (150 mM, pH 5.2). Precipitated nucleic acids were washed with ethanol and treated with TURBO DNase per manufacturer’s specification (Life Technologies Corporation).
Transcriptomics
Ribosomal RNA was removed from total RNA samples using the Ribo-Zero rRNA Removal kit, according to manufacturer’s recommendations (Illumina, Inc., San Diego, CA, USA). Depleted samples were analyzed and sequenced by the Genomics & Bioinformatics Shared Resources at Fred Hutchinson Cancer Research Center as previously described [58]. Results were evaluated using CLC Genomics Workbench (Qiagen) and transcripts that were changed >2-fold (p < .01) were included in our analysis. In addition, the data were technically validated by measuring expression of 6 genes via quantitative RT-PCR (qPCR) and a correlation was confirmed (R2 = .92). In Table 1, we included genes of interest and those known to be regulated by Rex in other organisms.
Quantitative RT-PCR of bacteria transcripts
Transcript analysis was performed as previously described [52]. Briefly, for the measurement of aerobic transcripts overnight cultures were normalized to an OD600 of 0.02 in 25 mL BHI in 250 mL flasks and were incubated with shaking at 37°C for 7 hours. For measurement of anaerobic transcripts, filter sterilized BHI broth was degassed overnight in an anaerobic chamber. Media was transferred to 16 x 125 mm Hungate Anaerobic Tubes (Chemglass Life Sciences) inside the anaerobic chamber and the tubes were autoclaved. The OD600 was normalized to an OD600 of 0.02 in 10 mL BHI in the Hungate tubes and incubated at 37°C for 7 hours. Following 7 hours of both aerobic and anaerobic growth, RNA was harvested as described above with the exception of DNase treatment, which was carried out using Thermo Scientific DNaseI (Thermo Scientific) according to the manufacturer’s instructions. The synthesis of cDNA was carried out using an iScript cDNA synthesis kit (Bio-Rad). Quantitative RT-PCR was performed on cDNA with the iTaq universal SYBR green supermix (Bio-Rad).
Growth curves
For aerobic growth in broth, the cultures were normalized to an OD600 of 0.02 in 25 mL BHI in 250 mL flasks and were incubated with shaking at 37°C. The OD600 was measured every hour. For anaerobic growth in broth, filter sterilized BHI broth was degassed overnight in an anaerobic chamber. Media was transferred to 16 x 125 mm Hungate Anaerobic Tubes (Chemglass Life Sciences) inside the anaerobic chamber and the tubes were autoclaved. The OD600 was normalized to an OD600 of 0.02 in 10 mL BHI in the Hungate tubes and incubated at 37°C, with OD600 measurements every hour.
Measurement of bacterial metabolites
Bacteria from aerobic and anaerobic cultures were collected (1 mL aliquots) after 4 hours of growth and centrifuged at 13,000 x g for 2 min. The supernatants were removed, sterile filtered, and stored at -20°C until use. Extracellular metabolites were determined using Roche Yellow Line Kits (R-Biopharm), according to the manufacturer’s recommendation. Intracellular ATP concentrations were determined using a BacTiter-Glo kit (Promega) according to the manufacturer’s protocol and normalized to optical units.
Bile sensitivity assays
Overnight cultures were diluted 1:200 into BHI pH 5.5, BHI supplemented with 0.1% porcine bile, or BHI pH 5.5 supplemented with 0.1% porcine bile. Aerobic cultures were incubated for 24 hours at 37°C shaking, followed by serial dilutions and plating on BHI agar to enumerate CFU.
Bacterial invasion assays
Caco-2 or Huh7 cells were seeded 2.0 x 105 cells per well in 24-well plates and washed twice in sterile PBS just prior to infection. Bacterial cultures were incubated overnight in BHI broth at 30°C static and then washed twice with sterile PBS and resuspended in cell culture media. Bacteria were added to cell monolayers at a multiplicity of infection (MOI) of 10 for Caco-2 cells and 20 for Huh7 cells. To measure bacterial invasion, monolayers were washed twice with PBS after 1 hour of infection and incubated with cell culture media containing gentamicin (50 μg/mL) for 1 hour. Monolayers were washed with PBS twice and lysed with 0.1% Triton X-100 and internalized bacteria were enumerated following plating on BHI agar [25, 59].
Intracellular growth curves
Growth curves in bone marrow-derived macrophages (BMMs) were performed as previously described [58], with the following modifications. Briefly, BMMs were harvested as previously reported [60] and seeded at a concentration of 6 x 105 cells per well in a 24-well plate the day before infection. BMMs were activated by incubating the monolayer with recombinant murine IFNg (100 ng/mL, PeproTech) overnight and during infection. Overnight bacterial cultures incubated at 30°C statically were washed twice with PBS and resuspended in warmed BMM media [52]. BMMs were washed twice with PBS and infected at an MOI of 0.1. Thirty minutes post-infection, cells were washed twice with PBS and BMM media containing gentamicin (50 μg/mL) was added to each well. To measure bacterial growth, cells were lysed by addition of 250 μL cold PBS with 0.1% Triton X-100 and incubated for 5 min at room temperature, followed by serial dilutions and plating on BHI agar to enumerate CFU.
Growth curves in Huh7 and TIB73 cells were performed as previously described [51]. Briefly, Huh7 and TIB73 cells were seeded at a concentration of 2.0 x 105 cells per well in 24-well plates the day before infection. Overnight bacterial cultures incubated at 30°C statically were washed twice with sterile PBS and resuspended in cell culture media. Huh7 and TIB73 cells were infected at an MOI of 20 or 50, respectively. Sixty minutes post-infection, cells were washed twice with PBS and cell culture media containing gentamicin (50 μg/mL) was added to each well. To measure bacterial growth, cells were lysed by addition of 250 μL cold PBS with 0.1% Triton X-100 and incubated for 5 min at room temperature, followed by serial dilutions and plating on BHI agar to enumerate CFU. Experiments were performed with technical replicates and repeated two times.
Plaque assays
Plaque assays were performed as previously described [26, 52]. Briefly, 6-well plates were seeded with L2 fibroblasts or TIB73 cells at a density of 1.2 x 106 and 1.5 x 106, respectively. Bacterial cultures were incubated overnight at 30°C in BHI broth and were then diluted in sterile PBS (1:10 for L2 infections; 1:2 for TIB73 infections). L2 fibroblasts and TIB73 cells were infected with 5 μL or 10 μL of diluted bacteria, respectively. 1 hour post-infection, cells were washed twice with sterile PBS and agarose overlays containing DMEM and gentamicin were added to the wells. 2 days post-infection, cells were stained with neutral red dye and incubated overnight. Plaques were imaged 72 hours post-infection and plaque area was quantified using Image J software [61].
Mice
Female BALB/c mice were purchased from The Jackson Laboratory at 5 weeks of age and used in experiments when they were 6–7 weeks old. All mice were maintained under specific-pathogen-free conditions at the University of Washington South Lake Union animal facility. All protocols were reviewed and approved by the Animal Care and Use Committee at the University of Washington (Protocol 4410–01).
Oral murine infection
Infections were performed as previously described [8, 28, 62–64]. Groups of 3 or 5 mice were placed in cages with wire flooring raised 1 inch to prevent coprophagy, and streptomycin (5 mg/mL) was added to drinking water 48 hours prior to infection. Food and water were removed 16 hours prior to infection to initiate overnight fasting. L. monocytogenes cultures were grown overnight in BHI broth at 30°C static. The cultures were diluted 1:10 into fresh BHI broth and grown at 37°C shaking for 2 hours. Bacteria were diluted in PBS and mice were fed 108 bacteria in 20 μL via pipette. The inocula were plated and enumerated after infection to ensure consistent dosage between strains. Food and water were returned to cages following infection and mice were euthanized at 1, 2, 3, and 4 days post-infection. Livers, spleens, and feces were harvested and homogenized in 0.1% NP-40. Gallbladders were harvested and ruptured in 1 mL of 0.1% NP-40 with a sterile stick. The cecum sections were emptied, flushed with sterile PBS, and homogenized in 0.1% NP-40 buffer. The small intestines were cut lengthwise with sterile forceps and flushed with sterile PBS. Intestinal contents were resuspended in PBS and intestinal tissue was homogenized in 0.1% NP-40. All organs were serial diluted in PBS and plated on LB agar to enumerate CFU.
Supporting information
Gene expression measured by quantitative RT-PCR following 7 hours of aerobic (A) and anaerobic (B) growth in the wt, Δrex, and Δrex p-rex strains. Data are graphed as the fold change over wt (wt = 1). In both panels, data are the means and SEMs of three independent experiments. Student’s unpaired t test was used to compare fold changes between the Δrex and wt strains and between the Δrex and Δrex p-rex strains (n.s., p > 0.05; *, p < 0.05; **, p < 0.01; ****, p < 0.0001). Data were not statistically significant between wt and Δrex p-rex.
(TIF)
A. Anaerobic growth of wt and Δrex strains, measured by OD600. B-F. Supernatants were sampled at 4 hours during anaerobic growth. Concentrations of glucose (B), lactate (C), formate (D), acetate (E), and ethanol (F) were determined and normalized to OD600. G. Concentration of glucose was measured in the supernatant over time. H. Concentration of ethanol in the supernatant over time, normalized to the OD600 I. Relative intracellular ATP concentration was measured at 4 hours. In panels A-H, data are the means and SEMs of three independent experiments. Data in panel I is the mean and SEM of 2 independent experiments. A heteroscedastic Student’s unpaired t test was used to compare results from wt and Δrex (n.s., p > 0.05; ****, p < 0.0001).
(TIF)
Growth of wt (black), Δbsh (grey), Δrex (blue), and ΔrexΔbsh (white) normalized to the initial inoculum (dashed line = 1). Strains were evaluated 24 hours post-inoculation in acidified BHI grown aerobically. Data are the means and range of three independent experiments. Strains were not significantly different (heteroscedastic Student’s t test; p > 0.05).
(TIF)
Female BALB/c mice were orally infected with 108 CFU of wt (black squares) or Δrex (blue circles) and the number of bacteria present in each tissue was determined over time. A. The body weights of the mice over time, reported as a percentage of body weight prior to infection. Data are the means and SEMs of n = 20 (day 1), n = 15 (day 2), n = 10 (day 3) and n = 5 (day 4). B-H. Mice were sacrificed each day and organs were harvested to enumerate bacterial burden. Each symbol represents an individual mouse (n = 5 per group), and the solid lines indicate the geometric means. Dashed lines indicate the limit of detection (l.o.d.). Results are expressed as log-transformed CFU per organ or per gram of feces. p values were calculated using a heteroscedastic Student’s t test. * p < 0.05; ** p < 0.01; *** p < 0.001.
(TIF)
Female BALB/c mice were orally infected with 108 CFU of wt (black squares) or Δrex (blue circles) and the number of bacteria present in each tissue was determined over time. A-F. Mice were sacrificed on 1, 2, and 4 days post-infection and organs were harvested to enumerate bacterial burden. Panel A includes small intestinal tissue only; bacterial burden in the intestinal contents was not evaluated. Each symbol represents an individual mouse (n = 3 per group) and the solid lines indicate the geometric means. Dashed lines indicate the limit of detection (l.o.d.). Results are expressed as log-transformed CFU per organ or per gram of feces. p values were calculated using a heteroscedastic Student’s t test. * p < 0.05; *** p < 0.001.
(TIF)
Table A in S1 Text. All Rex repressed genes during stationary phase. Table B in S1 Text All Rex repressed genes during mid-log phase. Table C in S1 Text All transcripts less abundant in Δrex during stationary phase. Table D in S1 Text All transcripts less abundant in Δrex during mid-log phase. Table E in S1 Text Predicted Rex binding sites in the 10403S genome. Table F in S1 Text L. monocytogenes strains used in this study. Table G in S1 Text E. coli strains used in this study.
(DOCX)
Acknowledgments
The authors would like to thank Arne Rietsch (Case Western Reserve University) for the pLIM plasmid and Steve Libby (University of Washington) for technical assistance.
Data Availability
All relevant data are within the manuscript and its Supporting Information files.
Funding Statement
Research in the Reniere Lab is funded by NIH grant R01 AI132356 (MLR). C.R.H. is funded by NIH grant T32AI055396. R.C.G. is funded by NIH grant 5T32AI055396. This work used the Genomics and Bioinformatics Shared Resources at Fred Hutchinson Cancer Research Center which is partially funded from Cancer Center grant NCI 5P30CA015704-43. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Vázquez-Boland JA, Kuhn M, Berche P, Chakraborty T, Domínguez-Bernal G, Goebel W, et al. Listeria Pathogenesis and Molecular Virulence Determinants. Clin Microbiol Rev. 2001;14: 584–640. doi: 10.1128/CMR.14.3.584-640.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tilney, Lewis G. P Daniel A. Actin filaments and the growth, movement, and spread of the intracellular bacterial parasite, Listeria monocytogenes. J Cell Biol. 1989;109: 1597–1608. doi: 10.1083/jcb.109.4.1597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schnupf P, Portnoy DA. Listeriolysin O: a phagosome-specific lysin. Microbes and Infection. 2007;9: 1176–1187. doi: 10.1016/j.micinf.2007.05.005 [DOI] [PubMed] [Google Scholar]
- 4.Freitag NE, Port GC, Miner MD. Listeria monocytogenes—from saprophyte to intracellular pathogen. Nat Rev Microbiol. 2009;7: 623–628. doi: 10.1038/nrmicro2171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Radoshevich L, Cossart P. Listeria monocytogenes: towards a complete picture of its physiology and pathogenesis. Nat Rev Microbiol. 2018;16: 32–46. doi: 10.1038/nrmicro.2017.126 [DOI] [PubMed] [Google Scholar]
- 6.Hardy J. Extracellular Replication of Listeria monocytogenes in the Murine Gall Bladder. Science. 2004;303: 851–853. doi: 10.1126/science.1092712 [DOI] [PubMed] [Google Scholar]
- 7.Zhang T, Abel S, Abel zur Wiesch P, Sasabe J, Davis BM, Higgins DE, et al. Deciphering the landscape of host barriers to Listeria monocytogenes infection. Proc Natl Acad Sci. 2017;114: 6334–6339. doi: 10.1073/pnas.1702077114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Louie A, Zhang T, Becattini S, Waldor MK, Portnoy DA. A Multiorgan Trafficking Circuit Provides Purifying Selection of Listeria monocytogenes Virulence Genes. mBio. 2019;10. doi: 10.1128/mBio.02948-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Begley M, Gahan CGM, Hill C. The interaction between bacteria and bile. FEMS Microbiol Rev. 2005;29: 625–651. doi: 10.1016/j.femsre.2004.09.003 [DOI] [PubMed] [Google Scholar]
- 10.Pitts MG, D’Orazio SEF. A Comparison of Oral and Intravenous Mouse Models of Listeriosis. Pathogens. 2018;7: 13. doi: 10.3390/pathogens7010013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.D’Orazio SEF. Animal models for oral transmission of Listeria monocytogenes. Front Cell Infect Microbiol. 2014;4. doi: 10.3389/fcimb.2014.00004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mesquita I, Varela P, Belinha A, Gaifem J, Laforge M, Vergnes B, et al. Exploring NAD+ metabolism in host–pathogen interactions. Cell Mol Life Sci. 2016;73: 1225–1236. doi: 10.1007/s00018-015-2119-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ravcheev DA, Li X, Latif H, Zengler K, Leyn SA, Korostelev YD, et al. Transcriptional Regulation of Central Carbon and Energy Metabolism in Bacteria by Redox-Responsive Repressor Rex. J Bacteriol. 2012;194: 1145–1157. doi: 10.1128/JB.06412-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Richardson AR, Somerville GA, Sonenshein AL. Regulating the Intersection of Metabolism and Pathogenesis in Gram-positive Bacteria. Microbiol Spectr. 2015;3. doi: 10.1128/microbiolspec.MBP-0004-2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brekasis D, Paget MS. A novel sensor of NADH/NAD+ redox poise in Streptomyces coelicolor A3(2). EMBO J. 2003;22: 4856–4865. doi: 10.1093/emboj/cdg453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang E, Bauer MC, Rogstam A, Linse S, Logan DT, von Wachenfeldt C. Structure and functional properties of the Bacillus subtilis transcriptional repressor Rex. Mol Microbiol. 2008;69: 466–478. doi: 10.1111/j.1365-2958.2008.06295.x [DOI] [PubMed] [Google Scholar]
- 17.Pagels M, Fuchs S, Pané-Farré J, Kohler C, Menschner L, Hecker M, et al. Redox sensing by a Rex-family repressor is involved in the regulation of anaerobic gene expression in Staphylococcus aureus. Mol Microbiol. 2010;76: 1142–1161. doi: 10.1111/j.1365-2958.2010.07105.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gyan S, Shiohira Y, Sato I, Takeuchi M, Sato T. Regulatory Loop between Redox Sensing of the NADH/NAD+ Ratio by Rex (YdiH) and Oxidation of NADH by NADH Dehydrogenase Ndh in Bacillus subtilis. J Bacteriol. 2006;188: 7062–7071. doi: 10.1128/JB.00601-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ruhland BR, Reniere ML. Sense and sensor ability: redox-responsive regulators in Listeria monocytogenes. Current Opinion in Microbiology. 2019;47: 20–25. doi: 10.1016/j.mib.2018.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wurtzel O, Sesto N, Mellin JR, Karunker I, Edelheit S, Bécavin C, et al. Comparative transcriptomics of pathogenic and non-pathogenic Listeria species. Mol Syst Biol. 2012;8: 583. doi: 10.1038/msb.2012.11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dussurget O, Cabanes D, Dehoux P, Lecuit M, the European Listeria Genome Consortium, Buchrieser C, et al. Listeria monocytogenes bile salt hydrolase is a PrfA-regulated virulence factor involved in the intestinal and hepatic phases of listeriosis. Mol Microbiol. 2002;45: 1095–1106. doi: 10.1046/j.1365-2958.2002.03080.x [DOI] [PubMed] [Google Scholar]
- 22.Begley M, Sleator RD, Gahan CGM, Hill C. Contribution of Three Bile-Associated Loci, bsh, pva, and btlB, to Gastrointestinal Persistence and Bile Tolerance of Listeria monocytogenes. Infect Immun. 2005;73: 894–904. doi: 10.1128/IAI.73.2.894-904.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dowd GC, Joyce SA, Hill C, Gahan CGM. Investigation of the Mechanisms by Which Listeria monocytogenes Grows in Porcine Gallbladder Bile. Infect Immun. 2011;79: 369–379. doi: 10.1128/IAI.00330-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kochan TJ, Shoshiev MS, Hastie JL, Somers MJ, Plotnick YM, Gutierrez-Munoz DF, et al. Germinant Synergy Facilitates Clostridium difficile Spore Germination under Physiological Conditions. mSphere. 2018;3: e00335–18, /msphere/3/5/mSphere335-18.atom. doi: 10.1128/mSphere.00335-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pizarro-Cerdá J, Kühbacher A, Cossart P. Entry of Listeria monocytogenes in Mammalian Epithelial Cells: An Updated View. Cold Spring Harb Perspect Med. 2012;2. doi: 10.1101/cshperspect.a010009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun AN, Camilli A, Portnoy DA. Isolation of Listeria monocytogenes small-plaque mutants defective for intracellular growth and cell-to-cell spread. Infect Immun. 1990;58: 3770–3778. doi: 10.1128/iai.58.11.3770-3778.1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Becattini S, Littmann ER, Carter RA, Kim SG, Morjaria SM, Ling L, et al. Commensal microbes provide first line defense against Listeria monocytogenes infection. J Exp Med. 2017;214: 1973–1989. doi: 10.1084/jem.20170495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ghanem ENB, Jones GS, Myers-Morales T, Patil PD, Hidayatullah AN, D’Orazio SEF. InlA Promotes Dissemination of Listeria monocytogenes to the Mesenteric Lymph Nodes during Food Borne Infection of Mice. PLoS Pathog. 2012;8: e1003015. doi: 10.1371/journal.ppat.1003015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Romick TL, Fleming HP, McFeeters RF. Aerobic and anaerobic metabolism of Listeria monocytogenes in defined glucose medium. Appl Environ Microbiol. 1996;62: 304–307. doi: 10.1128/aem.62.1.304-307.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Müller-Herbst S, Wüstner S, Mühlig A, Eder D, M. Fuchs T, Held C, et al. Identification of genes essential for anaerobic growth of Listeria monocytogenes. Microbiology. 2014;160: 752–765. doi: 10.1099/mic.0.075242-0 [DOI] [PubMed] [Google Scholar]
- 31.Gahan CGM, Hill C. Gastrointestinal phase of Listeria monocytogenes infection. Journal of Applied Microbiology. 2005;98: 1345–1353. doi: 10.1111/j.1365-2672.2005.02559.x [DOI] [PubMed] [Google Scholar]
- 32.Jones BV, Begley M, Hill C, Gahan CGM, Marchesi JR. Functional and comparative metagenomic analysis of bile salt hydrolase activity in the human gut microbiome. Proc Natl Acad Sci. 2008;105: 13580–13585. doi: 10.1073/pnas.0804437105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lamond NM, McMullen PD, Paramasvaran D, Visvahabrathy L, Eallanardo SJ, Maheswhari A, et al. Cardiotropic isolates of Listeria monocytogenes with enhanced vertical transmission dependent upon the bacterial surface protein InlB. Infect Immun. 2020; IAI.00321-20, iai;IAI.00321-20v1. doi: 10.1128/IAI.00321-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lecuit M, Dramsi S, Gottardi C, Fedor-Chaiken M, Gumbiner B, Cossart P. A single amino acid in E-cadherin responsible for host specificity towards the human pathogen Listeria monocytogenes. EMBO J. 1999;18: 3956–3963. doi: 10.1093/emboj/18.14.3956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gregory SH, Sagnimeni AJ, Wing EJ. Expression of the inlAB operon by Listeria monocytogenes is not required for entry into hepatic cells in vivo. Infect Immun. 1996;64: 3983–3986. doi: 10.1128/iai.64.10.3983-3986.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Faith NG, Kathariou S, Neudeck BL, Luchansky JB, Czuprynski CJ. A P60 mutant of Listeria monocytogenes is impaired in its ability to cause infection in intragastrically inoculated mice. Microbial Pathogenesis. 2007;42: 237–241. doi: 10.1016/j.micpath.2007.01.004 [DOI] [PubMed] [Google Scholar]
- 37.Quillin SJ, Schwartz KT, Leber JH. The novel Listeria monocytogenes bile sensor BrtA controls expression of the cholic acid efflux pump MdrT. Mol Microbiol. 2011;81: 129–142. doi: 10.1111/j.1365-2958.2011.07683.x [DOI] [PubMed] [Google Scholar]
- 38.Chen L-H, Köseoğlu VK, Güvener ZT, Myers-Morales T, Reed JM, D’Orazio SEF, et al. Cyclic di-GMP-dependent Signaling Pathways in the Pathogenic Firmicute Listeria monocytogenes. PLoS Pathog. 2014;10. doi: 10.1371/journal.ppat.1004301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Faith NG, Kim J-W, Azizoglu R, Kathariou S, Czuprynski C. Purine biosynthesis mutants (purA and purB) of serotype 4b Listeria monocytogenes are severely attenuated for systemic infection in intragastrically inoculated A/J Mice. Foodborne Pathog Dis. 2012;9: 480–486. doi: 10.1089/fpd.2011.1013 [DOI] [PubMed] [Google Scholar]
- 40.Hardy J, Margolis JJ, Contag CH. Induced Biliary Excretion of Listeria monocytogenes. Infect Immun. 2006;74: 1819–1827. doi: 10.1128/IAI.74.3.1819-1827.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gonzalez-Escobedo G, Marshall JM, Gunn JS. Chronic and acute infection of the gall bladder by Salmonella Typhi: understanding the carrier state. Nat Rev Microbiol. 2011;9: 9–14. doi: 10.1038/nrmicro2490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reniere ML. Reduce, Induce, Thrive: Bacterial Redox Sensing during Pathogenesis. J Bacteriol. 2018;200. doi: 10.1128/JB.00128-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Richardson AR, Libby SJ, Fang FC. A Nitric Oxide-Inducible Lactate Dehydrogenase Enables Staphylococcus aureus to Resist Innate Immunity. Science. 2008;319: 1672–1676. doi: 10.1126/science.1155207 [DOI] [PubMed] [Google Scholar]
- 44.Bouillaut L, Dubois T, Francis MB, Daou N, Monot M, Sorg JA, et al. Role of the global regulator Rex in control of NAD+-regeneration in Clostridioides (Clostridium) difficile. Mol Microbiol. 2019;111: 1671–1688. doi: 10.1111/mmi.14245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kazmierczak MJ, Mithoe SC, Boor KJ, Wiedmann M. Listeria monocytogenes σB Regulates Stress Response and Virulence Functions. J Bacteriol. 2003;185: 5722–5734. doi: 10.1128/JB.185.19.5722-5734.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scortti M, Monzó HJ, Lacharme-Lora L, Lewis DA, Vázquez-Boland JA. The PrfA virulence regulon. Microbes and Infection. 2007;9: 1196–1207. doi: 10.1016/j.micinf.2007.05.007 [DOI] [PubMed] [Google Scholar]
- 47.Guariglia-Oropeza V, Orsi RH, Guldimann C, Wiedmann M, Boor KJ. The Listeria monocytogenes Bile Stimulon under Acidic Conditions Is Characterized by Strain-Specific Patterns and the Upregulation of Motility, Cell Wall Modification Functions, and the PrfA Regulon. Front Microbiol. 2018;9: 120. doi: 10.3389/fmicb.2018.00120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bishop DK, Hinrichs DJ. Adoptive transfer of immunity to Listeria monocytogenes. The influence of in vitro stimulation on lymphocyte subset requirements. J Immunol. 1987;139: 2005–2009. [PubMed] [Google Scholar]
- 49.Bécavin C, Bouchier C, Lechat P, Archambaud C, Creno S, Gouin E, et al. Comparison of Widely Used Listeria monocytogenes Strains EGD, 10403S, and EGD-e Highlights Genomic Differences Underlying Variations in Pathogenicity. mBio. 2014;5: e00969–14. doi: 10.1128/mBio.00969-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Simon R, Priefer U, Puhler A. A Broad Host Range Mobilization System for in vivo Genetic Engineering: Transposon Mutagenesis in Gram-Negative Bacteria. Nat Biotechnol. 1983;1: 784–791. doi: 10.1038/nbt1183-784 [DOI] [Google Scholar]
- 51.McFarland AP, Burke TP, Carletti AA, Glover RC, Tabakh H, Welch MD, et al. RECON-Dependent Inflammation in Hepatocytes Enhances Listeria monocytogenes Cell-to-Cell Spread. mBio. 2018;9: e00526–18, /mbio/9/3/mBio.00526-18.atom. doi: 10.1128/mBio.00526-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reniere ML, Whiteley AT, Portnoy DA. An In Vivo Selection Identifies Listeria monocytogenes Genes Required to Sense the Intracellular Environment and Activate Virulence Factor Expression. PLoS Pathog. 2016;12. doi: 10.1371/journal.ppat.1005741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Camilli A, Tilney LG, Portnoy DA. Dual roles of plcA in Listeria monocytogenes pathogenesis. Mol Microbiol. 1993;8: 143–157. doi: 10.1111/j.1365-2958.1993.tb01211.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Whiteley AT, Ruhland BR, Edrozo MB, Reniere ML. A Redox-Responsive Transcription Factor Is Critical for Pathogenesis and Aerobic Growth of Listeria monocytogenes. Infect Immun. 2017;85. doi: 10.1128/IAI.00978-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Argov T, Rabinovich L, Sigal N, Herskovits AA. An Effective Counterselection System for Listeria monocytogenes and Its Use To Characterize the Monocin Genomic Region of Strain 10403S. Appl Environ Microbiol. 2017;83: e02927-16, e02927-16. doi: 10.1128/AEM.02927-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lauer P, Chow MYN, Loessner MJ, Portnoy DA, Calendar R. Construction, Characterization, and Use of Two Listeria monocytogenes Site-Specific Phage Integration Vectors. J Bacteriol. 2002;184: 4177–4186. doi: 10.1128/JB.184.15.4177-4186.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Reniere ML, Whiteley AT, Hamilton KL, John SM, Lauer P, Brennan RG, et al. Glutathione activates virulence gene expression of an intracellular pathogen. Nature. 2015;517: 170–173. doi: 10.1038/nature14029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cesinger MR, Thomason MK, Edrozo MB, Halsey CR, Reniere ML. Listeria monocytogenes SpxA1 is a global regulator required to activate genes encoding catalase and heme biosynthesis enzymes for aerobic growth. Mol Microbiol. 2020;114: 230–243. doi: 10.1111/mmi.14508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Burkholder KM, Bhunia AK. Listeria monocytogenes Uses Listeria Adhesion Protein (LAP) To Promote Bacterial Transepithelial Translocation and Induces Expression of LAP Receptor Hsp60. Infect Immun. 2010;78: 5062–5073. doi: 10.1128/IAI.00516-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sauer J-D, Sotelo-Troha K, von Moltke J, Monroe KM, Rae CS, Brubaker SW, et al. The N -Ethyl- N -Nitrosourea-Induced Goldenticket Mouse Mutant Reveals an Essential Function of Sting in the In Vivo Interferon Response to Listeria monocytogenes and Cyclic Dinucleotides. Infect Immun. 2011;79: 688–694. doi: 10.1128/IAI.00999-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of Image Analysis. Nat Methods. 2012;9: 671–675. doi: 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bou Ghanem EN, Myers-Morales T, Jones GS, D’Orazio SEF. Oral Transmission of Listeria monocytogenes in Mice via Ingestion of Contaminated Food. JoVE. 2013; 50381. doi: 10.3791/50381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jones GS, Bussell KM, Myers-Morales T, Fieldhouse AM, Bou Ghanem EN, D’Orazio SEF. Intracellular Listeria monocytogenes Comprises a Minimal but Vital Fraction of the Intestinal Burden following Foodborne Infection. Infect Immun. 2015;83: 3146–3156. doi: 10.1128/IAI.00503-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jones GS D ’Orazio SEF. Monocytes are the predominate cell type associated with Listeria monocytogenes in the gut but they do not serve as an intracellular growth niche. J Immunol. 2017;198: 2796–2804. doi: 10.4049/jimmunol.1602076 [DOI] [PMC free article] [PubMed] [Google Scholar]