

Abstract
Barley (Hordeum vulgare) is the fourth most cultivated crop in the world in terms of production volume, and it is also the most important raw material of the malting and brewing industries. Barley belongs to the grass (Poaceae) family and plays an important role in food security and food safety for both humans and livestock. With the global population set to reach 9.7 billion by 2050, but with less available and/or suitable land for agriculture, the use of biotechnology tools in breeding programs are of considerable importance in the quest to meet the growing food gap. Proteomics as a member of the “omics” technologies has become popular for the investigation of proteins in cereal crops and particularly barley and its related products such as malt and beer. This technology has been applied to study how proteins in barley respond to adverse environmental conditions including abiotic and/or biotic stresses, how they are impacted during food processing including malting and brewing, and the presence of proteins implicated in celiac disease. Moreover, proteomics can be used in the future to inform breeding programs that aim to enhance the nutritional value and broaden the application of this crop in new food and beverage products. Mass spectrometry analysis is a valuable tool that, along with genomics and transcriptomics, can inform plant breeding strategies that aim to produce superior barley varieties. In this review, recent studies employing both qualitative and quantitative mass spectrometry approaches are explored with a focus on their application in cultivation, manufacturing, processing, quality, and the safety of barley and its related products.
Keywords: barley, plant proteomics, mass spectrometry, plant breeding
Introduction
Hordeum vulgare (barley) is among one of the first domesticated cereal crops derived from its wild relative Hordeum spontaneum with domestication occurring approximately 11 000 years ago in the Fertile Crescent.1 Barley has become the fourth most important member of the cereal grain family with a total production of 158 million metric tons worldwide in 2019.2 Barley belongs to the grass (Poaceae) family, and it plays an important role in food security and food safety for both humans and livestock. It has been malted for beer and whisky and also has been used as a food product due to its health benefits, such as lowering blood cholesterol,3 improving regulation of blood sugar,4 and modulating gut microbiota.5,6 Barley grain is a rich source of carbohydrate (including dietary fiber), protein, E group vitamins such as tocopherols and tocotrienols, and B group vitamins such as thiamine, riboflavin, and niacin.7−9 The global barley market is expected to grow from 19.98 billion USD in 2017 to 25.18 billion USD in 2022.10 Its application can be categorized into two main sectors: (1) food and beverages; and (2) other applications such as animal feed, cosmetic products, pharmaceuticals, biofuels, and nutraceuticals.11−14 It has been used in the diets of many countries worldwide such as the Middle East, Russia, Poland, Tibet, Japan, North African countries, and India. It can be in meals such as soups, stews, casseroles, different kinds of pastas, noodles, and bakeries to produce flat bread and pastries.15,16 According to Roman texts, barley has been used as a staple for gladiators, and interestingly they were known as hordearii which means “barley men”. Hordearii followed a particular diet that consisted of barley and beans to gain weight and also provide subcutaneous fat with the aim of having more protection during battle.17 Different methods of processing barley for food products include pearling, malting, grinding or roller milling, flaking, and extruding,16 and the main products of barley after this processing include dehulled barley, malt, barley flour, pearl barley, and pot barley.13
Concerns about nutrition (health facts) and safety (origin, allergy) of food are increasingly affecting consumers’ choices. Specific medical conditions or dietary preferences have led to free-from diets such as gluten-free and dairy-free, or vegetarian/vegan and more recently plant-based diets. Consumers are also searching for foods that contain healthy ingredients, often fortified with minerals, vitamins, or bioactive molecules. Plant-based proteins have received considerable attention in recent years, and owing to soluble fiber such as β-glucan and high protein content barley has attracted significant attention. In addition, barley glutelin proteins have demonstrated oil-binding capacities and considerable emulsifying stability, while barley hordein showed good foaming properties which provide opportunities for value-added food applications for barley protein.14−18 The impact of these proteins on the final product will be discussed later in this paper. The use of barley as a high-quality protein ingredient in confectionary products has shown the potential to improve product flavor, texture, and storage stability.14,19 There is a high demand for foods that contain high-quality protein or that are free from specific proteins. This demand coupled with constraints on both land and resources requires further optimization of food systems, for instance, by meeting crop yield potentials. To achieve this goal, sustainable agriculture needs to adopt modern biotechnological tools of which proteomics is one of the key technologies that can support crop breeding programs.
The proteome refers to the whole set of expressed proteins in a cell, tissue, or organism at a specific time and condition, and the term proteomics describes the global identification and characterization of the protein complement of a biological sample.20 The results obtained from proteomics can be beneficial in many different research fields, for instance, to provide information about detection of diagnostic markers and alteration of expression patterns in response to different signals to understand pathogenicity mechanisms.21−23 In agriculture and food studies, proteomics is applied for the identification and characterization of proteins and to elucidate their function and interactions. Functional analysis of proteins is often achieved by qualitative and quantitative measurements of plant tissues at specific developmental or physiological stages.24−26
Quantitative mass spectrometry (MS)-based proteomics can be used to provide information on the grain proteome, that is, all proteins expressed within the edible part of cereal crops. Genomics and transcriptomics studies focus on the genes present in a genome and their expression, while proteomics defines the qualitative and quantitative composition of expressed proteins as well as how the expressed levels change under different environmental conditions. In addition to protein abundance, the presence of protein modifications can be informative from both understanding biological processes (i.e., signaling) or chemical alterations (i.e., food processing).27,28
There have been several valuable reviews covering different aspects of crops, particularly in barley which the reader can refer to for further information.24−37 In recent years, advances in MS-based tools and methods have resulted in increasing application of proteomics to barley employing a range of different techniques. This review aims to give an overview of barley proteomics studies and to highlight the application of mass spectrometry to different parts of the value chain from cultivation to processing and final product quality and safety. These research studies range from qualitative to quantitative approaches as described in the subsequent sections.
Proteins in Barley, Malt, and Beer
Major Proteins in Barley
Barley produces structurally diverse proteins that play fundamental roles in plant development, cellular renewal, nutrient uptake and transport, and biotic and abiotic stress responses.38 In the mature barley grain, 8–15% of the total dry weight consists of protein; however, the level varies depending on the genetic background, environmental conditions, and nitrogen availability.39
Although the Osborne solubility-based classification of barley proteins has been an enormous contribution to cereal protein studies, modern proteomics enables and requires a more systematic categorization.40 Therefore, barley proteins can be categorized by more practical classification according to their molecular functions: (1) storage proteins; (2) metabolic and structural proteins; and (3) protective proteins (Figure 1).41
Figure 1.
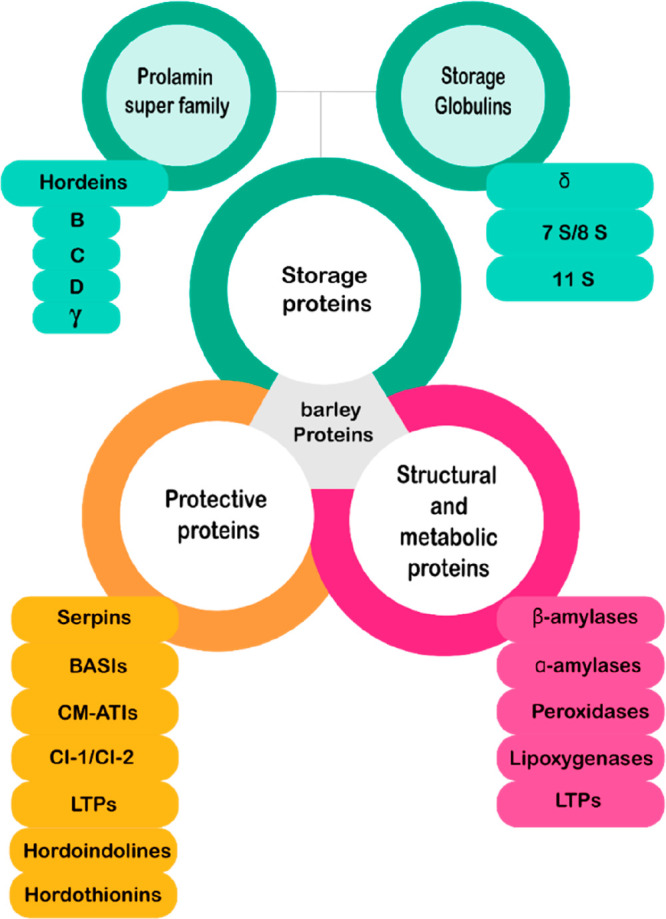
Barley protein classification. Abbreviations used: BASI, barley amylase/subtilisin inhibitor; CM-ATIs, chloroform methanol-extractable α-amylase/trypsin inhibitors; CI, chymotrypsin inhibitors; LTPs, lipid transfer proteins. Abbreviations: BASI - barley amylase/subtilisin inhibitor; CM-ATI - chloroform methanol soluble α-amylase/trypsin inhibitors; CI - chymotrypsin inhibitors; LTPs, lipid transfer proteins; serpins - serine protease inhibitors. Figure was adapted from Gubatz and Shewry.39 Used with permission. Copyright 2010 Wiley.
Storage Proteins
Storage proteins provide energy and are considered as a source of nitrogen, sulfur, and carbon to fuel germination and the growth of the seedlings. Barley storage proteins include proteins from both the prolamin superfamily and storage globulins. The grain prolamins are present in the tribe Triticeae including wheat (gliadins and glutenins), barley (hordeins), rye (secalins), and other grains such as corn (zeins), sorghum (kafirins), and oats (avenins). Prolamin proteins share a conserved pattern of cysteine residues, and they are classified into sulfur-rich, sulfur-poor, and high molecular weight prolamins.42 On the basis of recent evidence, the avenin-like proteins in wheat, that are also present in barley and share amino acid sequence homology with hordeins, are also involved in stress responses including abiotic (drought) and biotic (Fusarium head blight) stresses.43 The hordeins are the dominant proteins in the endosperm, and they comprise ∼55% of the total grain protein.44,45 They are rich in proline and glutamine residues, hence the term prolamin.46 Hordeins are classified into four subgroups according to their molecular weight: the D-hordeins with an approximate size of 105 kDa, the sulfur-poor C-hordeins of size 55–65 kDa, the B-hordeins of size ∼50 kDa, and the sulfur-rich γ-hordeins of size 35–45 kDa.42,47−49 The storage globulins of the barley grain are present in the embryo, endosperm, and the aleurone layer. These proteins include 7–8S globulins, which can be found in both the aleurone layer and the embryo, and the 11–12S globulins, which are found exclusively in the endosperm.50 Seed storage proteins are produced at a specific stage of seed development in the endosperm (in cereals), they accumulate in organelles known as protein bodies, and fractions of storage proteins show polymorphism between genotypes.51,52
Metabolic and Structural Proteins
Metabolic and structural proteins are diverse and have different properties, and they may have other roles than metabolic or structural activities. In barley they include (1) enzymes such as β-amylases, α-amylases, peroxidases (Prx), and lipoxygenases (LOX); and (2) small sulfur-rich proteins such as nonspecific lipid transfer proteins (ns-LTPs).39 Amylases are hydrolytic enzymes that degrade starch, a major energy reserve of barley seed, during germination into sugars and oligosaccharides. Amylases are important to the malting and brewing process involved in fermentable sugar production during mashing.53 β-Amylases are different in resting and germinated seeds. During grain development, β-amylases are synthesized, and a portion of the enzyme becomes insoluble during maturation and desiccation. The presence and abundance of β-amylases play a crucial role in the mashing process.46,54
Peroxidase enzymes, of which barley seed-specific peroxidase 1 (BSSP1) and barley peroxidase 1 (BSP1) have been already identified,55 oxidize a wide range of substrates in the barley during grain filling and germination. Lipoxygenases are involved in metabolic processes and catalyze the synthesis of xylipins, compounds derived from polyunsaturated fatty acids.56 In barley, they are present in three isoforms: (1) LOX-1 is present in quiescent grains; (2) LOX-2 is a germination-associated isoform; and (3) LOX-3 isoform expression has been detected only after germination and is similar to that of LOX-2, and it has several roles in brewing such as causing stale flavor in beer as a result of degradation and oxidation of polyunsaturated fatty acids during the malting and mashing processes.57
The barley seed proteome includes LTPs which have been implicated in several biological processes including developmental processes, metabolic and protective roles.58,59
Protective Proteins
Seeds are a rich source of proteins and nutrients and as such are subject to different biotic stresses such as attack by pests and pathogens. Several grain proteins play protective roles, and their accumulation can be increased under these situations. In barley, the protective proteins include enzyme inhibitors such as serine protease inhibitors (serpin), α-amylases, and trypsin inhibitors (ATI). ATIs are composed of three subgroups of (1) chloroform–methanol soluble proteins (CMATIs); (2) dimeric ATIs; and (3) monomeric ATIs. Additionally, barley amylase/subtilisin inhibitor (BASI), chymotrypsin inhibitors (CI) Cl-1/Cl-2, LTPs, hordothionins, hordoindolines, and defensins also belong here.29,39,46,60
Like other cereals, barley contains protein inhibitors that can act against α-amylases and proteases from pathogens and pests. Serpins inhibit chymotrypsin-like enzymes from insects and pathogens; in the developing barley grain, two isoforms, BS24 and BS27, are expressed and are suggested to be present in the endosperm and aleurone.61,62 ATIs have roles in grain filling and maturation, and many can be selectively extracted by chloroform/methanol (CM), and as such they are termed CM-proteins.63,64 In barley, CMa, CMb, CMd inhibit α-amylases, while CMc and CMe were observed to have inhibitor activity against trypsin.63,64 BASI is another protein that inhibits both subtilisin and amylase-2 enzymes during premature sprouting; this inhibitor is a member of the Kunitz-type protein inhibitor family and is an abundant protein of the endosperm and the aleurone layers of the mature seed.65
Chymotrypsin inhibitors of barley include CI-1 and CI-2. They lack cysteine residues and hence disulfide bonds, and they belong to a family of proteins including the potato inhibitor I and the leech inhibitor elgin.66 Another abundant protein group in barley aleurone layers is the nonspecific lipid transfer protein (ns-LTP) family. They are involved in the plant defense mechanisms to biotic and abiotic stresses and have protective roles in the assembly of extracellular hydrophobic polymers. These proteins survive during malting and brewing.39 Hordoindolines are reported to be present in the mature barley endosperm as two isoforms of a and b proteins, with hordoindoline b being the major isoform in the mature barley endosperm, which also survives in the malting and mashing stages.67 Hordothionins are cysteine-rich proteins and barley grain composed of two forms of thionins as α- and β-hordothionins. They inhibit the growth of pathogens such as fungi and bacteria, and they survive during brewing process.62 Plant defensins are the final group of protective proteins. They appear to be among the most widespread antifungal peptides in plants. There are two types of defensins in barley termed γ- and ω-hordothionins which are sulfur-rich proteins, and they share homology to α- and β-hordothionins.68
Major Proteins in Malt
During the malting process, proteases play an essential role in the partial degradation of storage proteins to yield peptides and amino acids which are important contributors to wort and beer quality. Several studies have investigated the role of proteins and their modifications in the malting and brewing processes, and they indicated the role of proteins in the final beer foam, haze stability, and flavor.69−73 During the malting process, enzymes such as α-amylases, β-amylases, limit dextrinases, α-glucosidases, β-glucanases, and more than 40 proteases are present and carry out their functions.61,74,75
Upon commencement of germination, α-amylase expression is increased, and it acts to cleave α-(1–4)-glycosidic bonds in starch. This enzyme has three forms, α-amylase I, II, and III, and they appear at different stages of germination.76 The actions of β-amylase liberate maltose by cleaving nonreducing ends of amylose and amylopectin, which is the most abundant sugar produced during the mashing stage. The resulting maltose serves as a source of energy for yeast during fermentation.77 This enzyme has three forms (Sd1, Sd2, and Sd3), and they vary in their thermostability. As Swanston and Molina-Cano (2001) indicated, this characteristic depends on the barley variety; they also reported that for the studied genotypes, there was a small, but highly significant, effect of the environment on the proportion of total β-amylase that was water-soluble.54 Limit dextrinase (LD) is another enzyme that hydrolyzes α-(1–6)-glycosidic bonds in amylopectin and branched dextrin, and it can be in three forms of active, inactive, or bound during germination.77 According to Huang et al. (2016), the activity of LD is increased during germination, and during mashing, it continues to convert dextrins into linear maltosachharides.78 Mashing temperature and pH are known to affect the activity of LD.77−80
The enzyme α-glucosidase is synthesized in the aleurone layer and embryo tissue during germination, and its activity increases in the presence of gibberellic acid (GA) as are other enzymes in the barley grain. Alpha-glucosidase catalyzes the α-glycosidic bonds in oligosaccharides and glucans to produce glucose.81 Its efficiency depends on critical parameters such as the type of the substrate (oligosaccharides or starch polymers), temperature, and pH.82
During germination, the enzyme β-glucanase catalyzes the hydrolysis of β-glucan. Studies have shown it has two isoenzymes EI and EII.83 The degradation of β-glucan, a major component of the barley cell wall, affects the malt quality. The enzymes β-glucanase and xylanase are considered important because high amounts of β-glucan and arabinoxylan in the final beer are considered a negative factor, impacting viscosity and the filtering process.83,84
The last group of enzymes is the proteases, which break the proteins into peptides and amino acids to provide a nitrogen source for the seedling. They are categorized into four classes according to their active-site residues in barley: cysteine-, serine-, aspartic-, and metalloproteases.85 The classes are differentiated by the catalytic mechanisms of the enzyme and the chemicals that inhibit their activity. Cysteine proteases contain the amino acid cysteine at their active centers, and they are the main proteases involved in the germination. Serine proteases possess a serine residue in their active site, aspartic proteases have two asparagine-residues in the active center and a conserved three-dimensional structure, and metalloproteases use a metal ion in their catalytic reactions.85,86 Jones and Marinac (2000) demonstrated that by increasing the temperature during mashing proteases were inactivated, and most of the proteolytic activity occurred during malting and mashing.85 Dormant barley grain possesses less proteolytic activity, but during malting, this activity increases.30,86
Major Proteins in Beer
Beer is one of the most consumed beverages around the world. Beer quality is affected by the proteins that remain intact after malting, mashing, and brewing. During malting, proteases partially hydrolyze storage proteins into free amino acids and soluble peptides, and glucanases and xylanases hydrolyze the endosperm cell wall substrates. Poor hydrolysis of beta-glucans and arabinoxylans results in runoff and filtration and haze issues in the final beer. Hydrolyzed proteins may play positive roles such as delivering body and mouthfeel or foam formation; or negative roles, such as haze formation.
Beer foam stability, flavor, mouthfeel, and haze formation are considered important characteristics in beer production.83−87 Therefore, numerous studies identified and characterized proteins in beer that influence these traits. Among those proteins, ns-LTPs and the serpin protein Z have been shown to have major effects, which will be explained in the following sections.88
Nonspecific LTPs are polypeptides characterized by an eight-cysteine motif, and as the name suggests, play a role in transferring lipids within plant membranes.89 Although their specific role is still unknown, they are known as pathogenesis-related (PR) proteins and play a role in defense mechanisms under biotic and abiotic stresses.90 They are small cysteine-rich proteins, and they are classified into two types of nsLTP1 (9 kDa) and nsLTP2 (7 kDa) according to their molecular size. There is also a modern classification of LTPs including five major (LTP1, LTP2, LTPc, LTPd, and LTPg) and five minor types with fewer members (LTPe, LTPf, LTPh, LTPj, and LTPk), this classification is based on the position of the conserved intron, the identity of the amino acid sequence, and the spacing between the cysteine residues.91
In barley seeds, the LTPs are deposited in the aleurone layer and persist in beer. LTP1 was found to be a surface-active protein that is modified and accumulates in beer foam during brewing.92 LTP1 is not only protease-resistant but also stable under high temperatures. During mashing, barley grain is subjected to long-term high temperatures, during which conversion of starch to monosaccharides occurs, and proteins are glycated. The interaction of d-glucose with free amine groups leads to a product called a Schiff base, and it is modified to form a more stable compound known as an Amadori product.93 LTP1 glycation inhibits its unfolding and accumulation during the boiling step. In a study undertaken by Jegou et al. (2000), LTP1 in its unfolded state was shown to affect beer quality with unfolding observed to take place after wort boiling.94 High temperature during boiling (103–110 °C) increases protein precipitation and reduces the level of LTP1 in beer; therefore, lower wort boiling temperatures near 96 °C maintains the LTP1 level in beer.95,96 In brewing, LTP1 stabilizes beer foam by binding foam stabilizing lipids, and a high amount of LTP1 can prevent the formation of stale flavor in beer because it binds to certain intermediate compounds, such as α-ketol 9-hydroxy-10-oxo-12 (z)-octadecenoic acid.97
Another protein that particularly affects beer flavor is lipoxygenase. This enzyme catalyzes the oxygenation of polyunsaturated fatty acids (PUFAs).98 In barley seed, LOX-1 is present in the germ, and it carries out the oxidative degradation of PUFAs to produce compounds that influence beer flavor.99 Linoleic acid, one of the lipids in malt, undergoes oxidation by LOX-1 and as a result produces a compound called 2(E)-nonenal, which causes stale flavor in long storage beers.100 Therefore, malting cultivars that have low LOX-1 activity are desirable.71,101
Serpins are another protein family that impacts beer quality. They were first found to be active as serine protease inhibitors, but they have other functions. All of the serpin types share three β-sheets, 8–9 α-helices, and a semiconserved reactive center loop domain in their secondary structures.102 Their regulatory function relates to the control of cell death by inhibiting endogenous proteinases. In cereals, they have a defensive role by inhibiting the chymotrypsin-like enzymes of pests and pathogens.39
Protein Z, a member of serpin family, contains at least four antigenically identical molecular forms with different isoelectric points and molecular masses near 40 kDa.103 Protein Z comprises two cysteines and 20 lysine residues per monomer, and it is also rich in hydrophobic residues.104 The glycation of protein Z is commenced from the early stages of malting.105 The quantity of protein Z is positively correlated with beer foam stability. The addition of purified protein Z from barley malt into the finished beer was shown to enhance the beer foam stability.71
Two isoforms, protein Z4 and Z7, are present both in free and bound forms, and these two share an approximately 70% sequence homology.89 Protein Z4 has high elasticity and surface viscosity, and according to Evans et al. (1999) when malt is less modified the effect of protein Z in foam stability is lower.89 When modification is increased, the impact of the protein Z on the foam stability is noted to be enhanced. The amount of protein Z in the final beer has been observed to be dependent on nitrogen fertilization rates.106
As previously mentioned, hordeins are barley storage proteins that are grouped into four families: B-hordeins (30–45 kDa), C-hordeins (45–75 kDa), D-hordeins (105 kDa), and γ-hordeins (35–40 kDa). Hordeins like other barley proteins are subjected to chemical (Maillard reaction, hydrogen bond formation) and enzymatic (proteolysis) modifications that mainly occur during malting and mashing. These proline-rich proteins are considered as the main cause of haze formation in beer, that is related to protein–polyphenol interaction.107,108 In this interaction, proline is involved in the binding site of protein to polyphenols. Numerous studies demonstrated that hordeins are mainly responsible for chill haze formation.106,109 It is also reported that polypeptides derived from hordein influence beer foam since they are concentrated in beer foam fraction.110 Aside from influencing the quality of the beer, hordeins are also known to trigger gluten sensitivities and autoimmune disorders termed celiac disease (CD). Among the barley hordeins, B- and C-hordeins contain higher numbers of immunogenic peptides which are implicated in CD. Therefore, much work has focused on those proteins by using quantitative proteomic techniques to ensure the safety and quality of produced barley and its related products.111−113
Proteomic Methods Used for Barley Analysis
Proteome characterization provides a path to understanding barley biochemistry and is of fundamental importance to improve productivity for sustainable agriculture, future food security, and resource conservation, especially under changing climate conditions.38 Proteomics is applied for the identification and characterization of proteins to elucidate their function and interactions. Additionally, understanding the modifications that proteins undergo and their interactions within the cell is also critical. Functional analysis of proteins is often achieved by qualitative and quantitative measurements of plant tissue at specific developmental or physiological stages.24,25 In an effort to achieve maximum coverage and resolution, proteomics studies benefit from the use of different but complementary technologies and approaches. There are crucial stages in proteomic research, including preparation, separation, identification, and quantitation of proteins in a sample. According to the aim of the study, the selection and application of approaches for each stage may be different.28
Gel-Based Proteomics
Generally, gel electrophoresis techniques can be identified according to the dimension of the gel system and the labeling procedure for visualization of proteins. Two-dimensional gel electrophoresis (2-DE) is a form of gel electrophoresis technique which was introduced by O’Farrell and Klose in 1975.114−117 In 2-DE, proteins are separated according to their isoelectric points in the first dimension, and in the second dimension, they are separated according to their molecular mass through SDS-PAGE.116 In these methods, the basis of gel staining is detection of proteins by visual inspection. Typically, a protein-specific dye-binding chemical reaction is conducted in proteins within the gel. A photograph of the stained gel is used for more investigation. By technology development, a UV light box or scanners have been used for documentation of the stained gel. There are a number of staining methods that are used including Coomassie Brilliant Blue, silver, fluorescent dye, zinc, and functional group staining.116
Difference gel electrophoresis (DIGE) is one of the powerful comparative techniques in proteomics in which samples are labeled with cyanine dyes prior to electrophoresis, and the efficiency of this method is based on natural or modified differences in charge between individual polypeptide chains and dissimilarities of their molecular size under native or denatured conditions. There are some advantages of using this method including being applicable for large-scale proteomic studies, direct protein visualization, and fluorescent labeling with highly sensitive dyes; however, there are some disadvantages which limit usage of this method such as restriction in separation of complex protein mixtures, cross-contamination of individual protein spots for highly abundant polypeptides, and under-representing some protein species (extremely low/high pI, highly hydrophobic proteins) in 2-DE gels.116,117
Early research studies in barley proteomics implemented the 2-DE technique to study the barley leaf proteome to discriminate and characterize cultivars based on the obtained spot patterns.117 Although gel-based techniques were successful in identifying barley proteins, they do have some limitations such as incomplete separation of the entire proteome in a complex sample, wherein large abundant proteins mask the low abundant ones so they cannot be detected. Therefore, other analytical techniques such as different chromatography techniques have been applied for separation of proteins and subsequent MS analyses to identify proteins.
To identify and characterize proteins, there are two fundamental MS-based approaches which are termed “top-down” and “bottom-up” wherein most barley proteomics studies use a bottom-up proteomics workflow (Figure 2).
Figure 2.

Bottom-up proteomic workflows in MS-based proteomics.
Bottom-Up Proteomics
Bottom-up proteomics or the peptide-centric approach is a common strategy that can be performed through different methods depending on the goal of the research. In this strategy, proteins are extracted and digested by a protease such as trypsin. This produces peptides that are subsequently separated before MS analysis. The peptides are then analyzed and detected within the mass spectrometer to determine their mass and are commonly fragmented within the mass spectrometer to yield MS/MS spectra that reveal the mass of the product ions (or fragments) that are subsequently used to identify the peptides.
There are three different acquisition modes that are commonly used to acquire mass spectra in proteomics studies: (1) data-dependent acquisition (DDA); (2) quantitative acquisition; and (3) data-independent acquisition (DIA) (Table 1).118−120 These are discussed in the section below.
Table 1. Comparison of Three Different Data Acquisition Methods Employed in Bottom-Up Proteomicsa.
| bottom-up
proteomics acquisition methods | ||
|---|---|---|
| data-dependent acquisition (DDA) | quantitative MS/MS | data-independent acquisition (DIA) |
| data analysis is easy | number of peptides that can be quantified in each injection is limited (10s–100s) | generates highly reproducible data |
| number of most abundant ions for fragmentation should be defined | data analysis is easy | able to quantify 10000s of peptides |
| lower reproducibility | generates highly reproducible data | information should be defined including mass range, precursor ion window width and number of MS/MS scans |
| generates product ion spectra of peptides for either identification or as SWATH-MS ion library | highly specific and sensitive | requires creation of spectral library by DDA |
| often used as a prerequisite for targeted MS method development | requires optimization of method for target peptides | data analysis and interpretation is more complex with specific software |
Abbreviations: SWATH - sequential window acquisition of all theoretical fragmentation spectra.
Bottom-up acquisition methods have seen great application in the grain science area in recent years due to technical innovation and optimization of techniques that have increased the depth of coverage and provided more accurate information. In the following sections, the application of three advanced acquisition methods in barley proteomics will be explained (Table 2).
Table 2. Proteomic Studies in Barleya.
| plant material | technique | number of identified spots/proteins | quantitation | ref |
|---|---|---|---|---|
| leaf | 2-DE | 29 | (117) | |
| seed and malt | 2-DE and MS | 27 for seeds, 3 for malt | (121) | |
| seed | 2-DE and MS | 19 | (122) | |
| seed and malt | 2-DE and MS | 62 in total | (123) | |
| seed | 2-DE and MS | 250 | yes | (124) |
| seed, malt, and beer | 2-DE and LC-MS/MS | 40 for seed, 41 for malt, 30 for beer | (125) | |
| seed | 2-DE and MS | 48 | (126) | |
| seed | 2-DE and MS | 48 | (127) | |
| malted beer | 2-DE and LC-MS/MS | 85 | (128) | |
| wort | 2-DE and MS | 63 | (129) | |
| seed, germinated grain, green malt and malt | 2-DE, MALDI-TOF/TOF MS | 6 (focus on hordeins) | yes, iTRAQ | (130) |
| beer | LC-MS/MS | 33 | yes, emPAI | (131) |
| flour, wort, and beer | LC-MS/MS | 144 in flour, 27 in wort, 79 in beer | yes, MRM | (111) |
| barley and malt | 2-DE, MALDI-TOF/TOF MS | 12 in barley, 9 in malt | (132) | |
| seeds and breakfast products | LC-MS/MS | 96 | yes, MRM | (133) |
| seed | 2-DE and MS | 23 | (134) | |
| leaf | 2-DE and LC-MS/MS | 9258 | yes, intensity | (135) |
| grain | 2-DE, LC-MS/MS | 136 DE spots | (136) | |
| grain | 2-DE, LC-MS/MS | 63 DE proteins | (137) | |
| seeds | LC-MS/MS | 1168 | yes, SPC | (50) |
| seeds | LC-MS/MS | focus on prolamin oxidation | yes, MRM | (138) |
| malt | LC-MS/MS | 1418 | yes, SPC | (139) |
| wort and beer | LC-MS/MS | 210 | yes, SWATH | (73) |
| seed | LC-MS/MS | 220 | yes, SWATH | (140) |
| spent grain | 2-DE, LC-MS/MS | 1346 | yes, intensity | (141) |
| leaf | LC-MS/MS | 1800 | yes, TMT | (142) |
| wort | LC-MS/MS | 87 | yes, SWATH | (143) |
| grain | LC-MS/MS | 1483 | (144) | |
| seeds | LC-MS/MS | 6 (focus on ATIs) | yes, MRM | (145) |
| grain | 2-DE, LC-MS/MS | hordein accumulation | (49) | |
| malt rootlet | LC-MS/MS | 2111 | (146) | |
| wort | 2-DE, LC-MS/MS | protein Z4 and Z7 analysis | (147) | |
| leaf | LC-MS/MS | 6921 | yes, DIA | (148) |
| grain | LC-MS/MS | 1907 | yes, SWATH | (149) |
Abbreviations: ATIs - α-amylase trypsin inhibitors, 2-DE - two2-dimensional electrophoresis, emPAI - exponentially modified protein abundance index; iTRAQ - isobaric tags for relative and absolute quantitation; LC - liquid chromatography; MALDI - matrix-assisted laser desorption ionization; MRM - multiple reaction monitoring; MS - mass spectrometry; SPC - spectral counting; SWATH - sequential Window Acquisition of all Theoretical fragmentation spectra; TMT - tandem mass tag, TOF - time-of-flight.
Data-Dependent Acquisition
In discovery proteomics by DDA methods, all ions which coelute at a specific time in the chromatogram result in a mass spectrum. The instrument then switches to acquiring product ion mass spectra. The precursor ions are selected from the previous survey scan and are sequentially isolated and fragmented.150 This approach is used to identify the maximum number of proteins in the sample; however, it often results in repeated identification of peptides derived from high abundant proteins and is limited by the stochastic nature of ion sampling.151
In an initial analysis of barley seed and malt, Østergaard et al. (2002) by using DDA acquisition method of MALDI-TOF MS demonstrated that in seed protein extracts, α-amylase/trypsin inhibitor was one of the proteins that caused variation between barley cultivars. Moreover, in malt extracts, multiple forms of the α-amylase isozyme 2 were identified according to varying spot patterns of the cultivars.121 Tissue-specific studies performed on barley grain by the DDA technique revealed that although the starchy endosperm comprises nearly 85% of a seed’s dry weight, it includes less than 50% of soluble proteins, and interestingly, the aleurone layer and embryo showed a significant contribution in the number of identified proteins using 2-DE.152
Concerning analysis of proteins both in barley grain and the corresponding malt, Bak-Jensen et al.123 identified an increased number of proteins by implementing tandem MS wherein the proteins identified were involved in glycolysis, pathogen defense, nutrient storage, protein folding, detoxification, and nitrogen metabolism. Further research explored the environmental impact on grain filling, such as how nitrogen availability can influence seed proteome changes.153
Subsequently, discovery proteomics by DDA has been applied to study the early stages of barley grain development and protein changes in malt. For instance, Perrocheau et al. (2005) explored the barley proteome changes during the malting and brewing processes. In this study employing gel electrophoresis combined with MS, 40 proteins were identified from barley grain, 41 proteins from malt, and 30 proteins from beer. They reported that most of the heat-stable proteins during brewing are disulfide-rich proteins, and these are involved in defense mechanisms and include protein Z, amylase-protease inhibitors, and LTPs (LTP1 and LTP2).125
Another significant study focused on the proteome changes of aleurone, embryo, and endosperm across the time frame of germination. Late embryogenesis abundant (LEA) proteins, ABA-induced proteins, a HSP70 fragment, and a β-type proteasome subunit showed alteration in abundance during the early stages of germination, while the pattern of redox-related proteins altered at the end of germination.126
A proteomic study of barley genetic diversity used a proteome map that was integrated for chromosome 1H, 2H, 3H, 5H, and 7H, and the results indicated that more than 60 protein spots showed variation between cultivars, including peroxidases, serpins, and proteins with unknown functions. MS data confirmed that single nucleotide polymorphisms (SNP) in the coding gene region can change the function of proteins and represent a connection between a cultivar’s genome, proteome, and phenotype.154 Further studies used MS-based proteomics to address barley quality improvement for beer production. This was accomplished by constructing a beer proteome map, which showed eight families of barley proteins that included protein Z (Z4, Z7), BDAI-1, CMb, LTP1, TAI, BTI-CMe, and subtilisin-chymotrypsin inhibitor CI-1B.128 Progress and achievement in genome sequencing and annotation of the barley cultivar Morex had a huge impact on the implementation of modern MS methods to investigate the proteome of barley and its products.155 Comparative proteome analysis is often applied to barley. For instance, using 2-DE and MS, a feed barley cultivar and a malting barley cultivar were subjected to comparative study with the purpose of identifying protein markers, which have the potential to affect the grain protein composition and quality. The results identified 23 proteins, and malting quality was suggested to be characterized by an accumulation of a serpin protein, α-amylase/trypsin inhibitor CMb, and α-amylase inhibitor BDAI-1.134 Another comparative analysis explored flag leaves of near-isogenic late- and early senescing barley germplasm by applying both gel-based and gel-free quantitative techniques, wherein >9000 proteins were reported with pathogenesis-related proteins, membrane and intracellular receptors and coreceptors, involving enzymes in attacking pathogen cell walls and DNA repair enzymes up-regulated in an early senescing line. Additionally, a link between early senescence and up-regulated defense functions was observed.135
So-called “shotgun” approaches have been more common in barley grain proteomic studies in recent years. A comparison of two-rowed and six-rowed cultivars was conducted to provide a comprehensive characterization of barley seed proteome by shotgun proteomics. This study could identified 1168 proteins, and among these proteins, specifically hordoindolines were differentiated. It was reported that the type of hordoindoline proteins may cause seed hardness differences between two cultivars. Differences in protein profiles of cultivars were suggested to be utilized for investigation of important complex traits such as mating quality.50 Later, using gel-free shotgun proteomics changes during different stages of malting were explored, and results revealed more than 1400 identified proteins during different stages of malting. These proteins were associated with carbohydrate metabolism and enzyme regulation which offer potential targets for breeding with the aim of improving malting quality. Moreover, this research confirmed that most of the proteins necessary for seed germination are synthesized during later stages of seed maturation.139 In a recent study, the same label-free shotgun approach was applied to investigate barley rootlets, a byproduct of malting process, and as a result 800 proteins were identified. Gene ontology (GO) enrichment analysis of barley rootlets highlighted the enrichment of primary metabolism-related terms including glycolysis/gluconeogenesis, TCA cycle, and pentose phosphate shunt that involved in sugar production enrichment. Furthermore, GO term analysis for molecular function and cellular component identified the translation process as a key feature in barley rootlet proteome. This study also revealed that pathways that are involved in stress responses such as ascorbate-glutathione pathways were significantly enriched due to a steeping regime that seeds undergo during the malting process.146
A recent study compared different buffer compositions commonly used in cereal grain protein extraction to assess the extraction efficiency experiments and was performed by means of shotgun proteomics. A total of 1497 proteins were identified from two barley varieties (Hindmarsh and Commander) using an optimized extraction protocol, and the results revealed 272 (18.2%) commonly extracted proteins by three experimented extraction methods including Tris-HCl, urea, and isopropyl alcohol/dithiothreitol (IPA/DTT). As demonstrated in the results, Tris-HCl and urea-based buffers extracted a maximum number of proteins of all functional classes from barley compared to IPA/DTT.144
As it can be seen in the barley proteomics literature, results obtained through discovery proteomics laid the foundation for developing targeted proteomics methods and caused the discovery proteomics to improve to hypothesis-based approaches, which are explained in the following.
Quantitative Mass Spectrometry
MS-based quantitation can be classified into two types according to the goal of the study: relative and absolute quantitation. Alternatively, they can be classified according to the technology used: label-based and label-free.156
The label-based quantitation method is based on the comparison between samples through labeling and detecting them according to specific changes in size, and this method includes three kinds of labeling: metabolic, chemical, and enzymatic labeling.157
Isobaric tags for relative and absolute quantification (iTRAQ) is one of the stable isotope labeling approaches which has been implemented for quantitative proteomic analysis. In barley studies, this technique was implemented to investigate the effect of the malting process on hordein composition, and results showed that the majority of the hordein components in barley grain are present in all stages of the malting process and the amount of hordeins was reported to be decreased during malting; specifically, C-hordein decreased by 65%. This technology has a high level of sensitivity and specificity, and it is considered as a high through-put method which can be used for quantifying proteins across wide ranges of MW and pI; however, it is time-consuming and expensive.157
Label-free quantitation is an easy and cost-effective method for relative quantitation of proteins that does not require expensive reagents for labeling. In this method, samples are injected independently in MS, and quantitation can be done at the MS scan level by measuring the area under the curve (AUC) or signal intensity through extracted ion chromatograms (XICs). It can be performed at the MS/MS scan level by counting the number of peptide-to-spectrum matches (PSMs) for each protein, termed spectral counting. The advantage of this method is that an unlimited number of samples can be measured and compared, and it has relatively high quantitative proteome coverage. Yet, the main disadvantage of this method is a higher variation that can result from individual preparation of each sample.157 There are some solutions to reduce the variation such as training of personnel, application of robotics, and experimental design. Moreover, the application of internal standards can decrease the variability that arises from instrument responses.158
Targeted MS methods aim to detect and quantify protein targets of interest often in complex protein mixtures. Selected reaction monitoring (SRM) or multiple reaction monitoring (MRM) is a targeted scan type commonly employed on triple quadrupole (QQQ) mass spectrometers. The development of targeted methods requires prior knowledge of the precursor/product ion pairs.159
In MRM, by filtering out all other ions, one can dramatically increase the sensitivity to specific transitions. Quantitation by MRM can be achieved by relative and absolute approaches but also by label-based and label-free workflows.160 Since 2012, studies have increasingly employed quantitative methods in addition to qualitative identification. For example, a shotgun proteome analysis of beer by means of HPLC-ESI-MS/MS demonstrated the presence of low-molecular-weight beer components (hordein-derived peptides), potentially harmful to people with CD.131 Application of targeted MS methods such as MRM has facilitated proteomic studies that have increased sensitivity with greater accuracy.160 This technique has now seen widespread application in plant proteomics studies, including barley. For instance, targeted approaches were used to identify B-, C-, D-, and γ-hordeins in the wort and beer samples. In this study, researchers identified 144 proteins in flour, 27 protein in wort, and 79 proteins in beer with the majority of them nonspecific LTP1 and α-amylase trypsin inhibitors. They also identified degradation of C-hordein products in wort, but these C-hordein fragments were not detected after the brewing process.111 Additionally, LC-MS/MS was used to develop a scheduled MRM method using nine barley-specific peptides that enabled the detection of barley contamination as a means to provide food safety assurance in gluten-free food production.133 Concerning immunogenic proteins of barley, it is known that degradation of cereal prolamin proteins and peptides can reduce their toxicities for celiac disease (CD) patients; in a study, the MRM method was established to detect and quantify proline oxidation fragments in barley. Additionally fragmentation, aggregation and side chain modifications were identified, and free thiol loss, carbonyl formation, and dityrosine formation were among those modifications. The result of this study reported that the immunoreactivity of the oxidized hordein isolate was considerably decreased in all metal-catalyzed oxidation systems.138 In an LC–MS discovery proteomics analysis of barley cultivars, extraction efficiency of three buffers were investigated,144 and a targeted MRM quantitation method was used with a focus on 6 α-amylase trypsin inhibitors across 12 barley cultivars. The research indicated that a relative targeted quantitation approach by MRM can be used for identifying and quantifying ATIs involved in autoimmune responses, in order to develop barley lines with a low amount of immune responsive ATIs.145
Quantitation of targeted peptides in label-free approach relies on the direct evaluation of mass spectrometry signal intensities of naturally occurring peptides contained in a sample.160 Quantitation by MRM is ideally suited for projects that involve quantitation of low-abundant proteins and peptides with maximal accuracy.161
Data-Independent Acquisition
An alternative approach that does not rely on the preselection of target proteins and their peptide derivatives is called data-independent acquisition (DIA) MS. In DIA-MS, precursor ions are sampled across a defined mass range, and all those precursor ions are subjected to simultaneous fragmentation resulting in the generation of mosaic MS/MS spectra. DIA-MS can be implemented in a range of different mass spectrometers, but generally, these approaches help to increase the detection of low-abundant and isobaric peptides, and as a result increased the identification of low-abundant proteins.162 One variant of DIA is termed Sequential Window Acquisition of all THeoretical fragmentation spectra (SWATH).163
The SWATH method mostly depends on the peptide spectral library, which is required to be established in advance by shotgun proteomics. It is worth mentioning that SWATH-MS can quantify an unlimited number of target peptides as long as they have been previously observed by shotgun MS.164,165 Therefore, a “discovery” data-dependent experiment followed by data-independent quantitation by SWATH is a suitable choice when reproducible and accurate quantitation is among the main goals of the project.164−168,182,183
Application of novel methods such as SWATH-MS has enabled investigation of the beer proteome. For instance, one study used a global untargeted SWATH analysis during beer production revealing protein modifications by protease digestion, glycation, or oxidation during the processing steps. They suggested that heat and high concentrations of protein catalyze glycation and oxidation modification, and the result is reduced sugars present in wort. These sugars are critical contributors to the color and flavor of beer. The SWATH-MS results for sweet wort, hopped wort, and beer were compared and demonstrated a difference between boiling and fermentation stages, and protein abundance of high molecular weight proteins were decreased during the boil, while hydrophobic proteins with high grand average hydrophobicity (GRAVY) scores were reduced in abundance during fermentation. This study showed the opportunities that modern MS-based techniques can offer for investigating and understanding the brewing process. The authors suggested that SWATH-MS methods can be used for exploring beer biochemistry to improve beer quality.73 The implemented SWATH-MS workflow was applied to investigate the variability of barley seed to explore the difference of barley variety and the burden of fungal disease at the proteome level. Obtained results of this study demonstrated that the abundance of several proteins across the investigated diseases and locations were significantly affected by disease burden. Among those proteins, oxalate oxidase 2 (OXO2) abundance was significantly increased under pathogen infection. The importance of this protein is due to its role in plant stress responses and production of hydrogen peroxide in the apoplast. This study highlighted the differences between barley varieties with application in feed or malting. Those malting varieties showed higher levels of proteins involved in starch synthesis and beer quality than those varieties used for feed. The results of this study showed the potential of using the SWATH-MS workflow for quality control purposes of barley products.140
Later, the SWATH-MS approach was implemented to address the dynamics of protein abundance and modification during the brewing process. Results of this research revealed that both wort and beer proteomes showed an interplay of partial proteolysis and temperature-dependent protein unfolding as well as protein modification; different types of modifications were identified including oxidation, glycation, and proteolytic cleavage in proteins of wort. Interestingly, these modifications can alter the thermal stability of proteins and their ability to survive the boil intact. Additional results indicated an increase of most proteins during the maltose rest (63 °C) and sugar rest 1 (73 °C), and then a substantial decrease of those proteins during sugar rest 2 (78 °C) and into the boil (102 °C).141 Although this study helped to uncover post-translational modifications of wort and beer, it lacks information regarding modifications of the tseed germination stage.
Quantitative proteomics by SWATH-MS has been used recently to investigate protein abundance changes in Tibetan barley responses to osmotic stress. The study provided a glimpse into the application of multidimensional proteomic data obtained by the SWATH-MS workflow with the aim of improvement of osmotic/drought stress tolerance in hull-less barley. Functional characterization analysis of differentially abundant proteins revealed that cytokinin synthesis degradation, UDP glucosyl and glucoronyl transferases, and wax-related proteins were likely to be an exclusive response in the drought-sensitive cultivar, while GDSL-motif lipase, DUF26 kinase, and plasma membrane intrinsic protein (PIP) were the three main functional terms in the drought-tolerant cultivar. In a closer examination, it was found that MAPK and PR10 showed a higher abundance in the treatment group over all time points of drought-tolerant and drought-sensitive cultivars, highlighting that these genes might play essential roles in plant defense responses to osmotic stress.148
In a recent study on hordeins, SWATH-MS acquisition was used to measure proteome-wide abundance differences between wild-type and single hordein-null (B-, C-, and D-null) barley lines to explore the effect of hordein deletion on the proteome level. Comparative analysis between single-null lines and wild type (WT) showed a significant difference between the C-null line and WT and B- and D-null lines. As a result, 1122 proteins were identified with significantly different protein abundance patterns in experimented samples. Additional GO enrichment analysis of these differentially expressed proteins showed that the top three biological processes associated with the differences were metabolic, single organism, and cellular processes. It was concluded that there is a linkage between downregulation of different storage protein families and upregulation of proteins related to primary metabolism, transcription, and enzymatic biosynthesis processes and enzyme inhibitors with the absence of B-, C-, and/or D-hordeins. Furthermore, results revealed an increase in globulins, lipid transfer proteins, and proteins rich in essential amino acids in the null lines. This increase in nongluten storage proteins occurred as the consequence of a specific reduction in hordein proteins. Implementing SWATH-MS, this study highlights one of the main applications of this advanced method in proteome-level alterations studies for investigating modified crops.149
The application of MS-based proteomic approaches for investigating cereals including barley in different steps of the food production chain has increased in the past decade. The following sections aim to shed light on the application of proteomics in the three critical sectors of barley studies including (1) breeding; (2) manufacturing and processing; and (3) final product quality and food safety.
MS Application in Barley Breeding
Barley represents a major cereal crop grown in temperate climate areas worldwide. The development of crop resilience to environmental changes is a crucial breeding goal of agricultural programs. Development of methods and techniques to address food security with an increasing population and global climate change is significant.
MS-based proteomics offers the potential to inform breeding programs and improve existing barley varieties or develop new ones that are high yielding and stress-resistant. Proteins play an important role in cellular mechanisms and are involved in various biological processes. In an organism, proteins are more directly related to the phenotypic changes when compared to gene expression profile changes, and investigating the differential abundance patterns of protein profiles can complement information obtained from genomics and transcriptomics analyses.23,166,185
Different proteomics methods including discovery (shotgun) proteomics and absolute and relative quantitative approaches have been used to broaden the knowledge of barley proteome changes under specific abiotic and biotic stress conditions (Table 3) and to explore impact of post-translational modification (PTMs) and protein interactions.29,36,152,157,168,187,188
Table 3. Proteomic Studies on Abiotic and Biotic Stress Response in Barley169a.
| plant material | tissue | methods | quantitation | ref | |
|---|---|---|---|---|---|
| Drought | |||||
| barley cv. Basrah (T), Golden Promise (S) | leaf, root | 2-DE- MALDI-TOF | yes | (170) | |
| Two Egyptian accessions; 15141 (T), 15163 (S) | leaf | 2-DE- MALDI-TOF | (171) | ||
| barley cv. Golden Promise | leaf | 2-DE-MS | (172) | ||
| spring malting barley cv. Amulet | leaf, crowns | 2-DE, MALDI-TOF/TOF | yes | (173) | |
| spring barley cv. Maresi (T), Syrian breeding line – Cam/B1/CI (T) | leaf, root | 2-DE, MALDI-TOF/TOF | yes | (174) | |
| spring barley cv. Maresi x Syrian breeding line – Cam/B1/CI (T), 100 RILs | leaf. root | MALDI-TOF/TOF | yes | (167) | |
| Low Temperature (Cold/Frost Tolerance) | |||||
| winter barley cv. Luxor (T) | crown, leaf | 2-DE-MALDI-TOF | yes | (175) | |
| spring barley cv. Aths (S) | leaf, root | 2-DE, LC-MS/MS | (176) | ||
| 10 DH lines: DH534 (T), DH602 (T), DH561 (t), DH61 (t), DH435 (t), DH584 (t), DH363 (S), DH575 (S), DH158 (S), DH65 (S) | leaf | 2-DE-MALDI-TOF/TOF | (177) | ||
| Osmotic Stress (Polyethylene Glycol PEG-6000) | |||||
| Tibetan hull-less barley (Hordeum vulgare L. var. nudum) inbred lines, drought-resistant XL (T), drought-sensitive DQ (S) | leaf | LC-MS/MS | yes | (148) | |
| Salinity | |||||
| barley cv. OUK305 (T), OUI743 (S) | root | 2-DE, LC-MS/MS | yes | (178) | |
| barley cv. Morex (T), Steptoe (S) | root | 2-DE, LC- MS/MS | yes | (179) | |
| barley cv. Afzal (T), L-527 (S) | leaf | 2-DE, MALDI-TOF/TOF | yes | (180) | |
| barley cv. Afzal(T), L-527 (S) | leaf | 2-DE, MALDI-TOF/TOF | yes | (181) | |
| barley cv. Morex (T), Steptoe (S) | leaf, root | 2-DE, MALDI-TOF; LC-qTOF MS/MS | yes | (182) | |
| barley cv. DH187 (T), DH14 (S) | seed | 2-DE, MALDI-TOF/TOF | yes | (183) | |
| barley DH population cross TX9425 and NasoNijo two pairs of NILs (N33 (S) and T46 (T), N53 (S), and T66 (T) | leaf, root | 2-DE, MALDI-TOF/TOF | yes | (184) | |
| Combined Stress | |||||
| drought and osmotic stress | barley genotypes 004223 (T), 004186 (S) | leaf | 2-DE MALDI-TOF; LC-MS/MS | yes | (185) |
| drought and heat stress | Arta (T), Keel (T) | leaf | 2-DE, MALDI-TOF/TOF | yes | (186) |
| drought and Piriformospora. indica | barley cv. Golden Promise | leaf | 2-DE, LC-MS/MS | yes | (187) |
| Pathogenes | |||||
| Fungal Pathogen - Fusarium Head Blight (FHB) | |||||
| barley cv. Scarlett (S) | spikelet | 2-DE, LC-MS/MS | yes | (188) | |
| barley cv. Scarlett (S) | seed | 2-DE, MALDI-TOF, MS/MS | yes | (189) | |
| naked barley (Hordeum vulgare ssp. nudum) | seed | 2-DE MALDI-TOF-MS, LC-MS/MS | yes | (190) | |
| Net Blotch −Pyrenophora teres | |||||
| barley cv. La Trobe, Charger, Oxford, Commander, Fairview, Compass | seed | LC-MS/MS, SWATH-MS | yes | (143) | |
| barley cv. Oxford, Commander, Compass, Scope, Shepherd, Flagship | seed | LC-MS/MS, SWATH-MS | yes | (143) | |
| barley cv. Baudin | leaf | 2-DE, LC-MS/MS, SWATH-MS | yes | (191) | |
| Leaf Rust −Puccinia hordei | |||||
| barley cv. LaTrobe, Commander, Compass, Scope, Shepherd, Fathom | seed | LC-MS/MS, SWATH-MS | yes | (143) | |
| Powdery Mildew– Blumeria graminis | |||||
| barley cv. Golden Promise | leaf | LC-MS/MS | (192) | ||
| barley cv. Golden Promise | leaf | LC-MS/MS | yes | (193) | |
Abbreviations: 2-DE - two-dimensional electrophoresis; DH - double haploid (line); LC - liquid chromatography; MALDI-TOF/TOF - matrix-assisted laser desorption ionization time-of-flight/time-of-flight (spectrometry); MS, mass spectrometry; qTOF - quadrupole time-of-flight; S - sensitive (genotype); SWATH - sequential window acquisition of all theoretical spectra; T - tolerant (genotype); t - genotype less tolerant than T.
The agricultural production of barley is limited by a wide range of biotic and abiotic stress factors. Biotic stress factors including pathogens and insects, and abiotic stress factors including drought, salinity, cold, and frost severely limit plant growth and development as well as the final yield in barley crops. Recent reviews on plant protein reactions to pathogens194 and abiotic stresses195−197 provide a critical overview of plant responses to stresses on a proteome level. These stresses pose a drastic threat to barley production and affect proteins and consequently plant phenotypes. Therefore, comprehensive knowledge about plant responses to different stresses is a prerequisite for the progress of breeding programs to deliver stress-tolerant crops. MS-based proteomics along with advances in sequencing technology and bioinformatics have greatly assisted in achieving this goal.197 One valuable method that is used to link plant proteome changes to genetic variations is proteomic quantitative trait locus (pQTL) analysis, which can be defined as a way to correlate protein abundance patterns with genetic polymorphism or QTL that controls variation in protein profiles.187,198,215,218 Using this approach, differences in protein abundance are considered as a molecular phenotype.199
Because of the essential roles of proteins in plant-stress interaction, they can be potential candidates for proteomics investigation by using pQTL approaches for producing resistant crops. This technique has been used for the identification of drought-responsive proteins in barley, and valuable reviews have been published upon this subject.187,215 Large-scale barley leaf and root proteomic analysis performed by 2-DE and MS revealed 48 pQTLs for roots and 31 pQTLs for leaves, and a genetic linkage map was established for the studied recombinant inbred lines relative to the proteomic data.167
Using the same method, a comprehensive study was undertaken on double-haploid introgression lines including wild-type line (Hs213, Hordeum vulgare subsp. spontaneum) within a modern cultivar background (H. vulgare cv. Brenda). As the primary stage of searching for QTLs, Li and his colleagues (2005) identified QTLs for malting quality and yield components, and protein content was one of the measured trait in this study.200 As a result of this research, three malting quality traits were evaluated in two years, and two QTLs for increased protein content were detected, which accounted for 11.8% and 14.6% of the phenotypic variance. Furthermore, three QTLs were identified for seed friability. The limitation of the study was that it was based on the measured protein content as a phenotypic trait. In later years, the difference in protein abundance of barley grain, considered as a molecular phenotypic trait, was used to map a QTL that regulates protein expression. Further QTL analysis was performed on the proteome level on the same population.201 Grains from 45 barley lines were analyzed, and their 2-DE proteome patterns were used for QTL analysis. MS identification for 49 segregating spots was achieved, and functional protein annotation of proteins revealed that most were involved in defense mechanisms and metabolism processes. The DE spots include α-amylase inhibitor BDAI, protein disulfide isomerase, adenosine kinase, NADP malic enzyme, peroxidase BP1 and disease-related processes proteins. Proteins showing altered expression were mapped to the same chromosomal locations as the coding genes.201
With the aim of establishing a linkage between proteins and malting characteristics, the referred QTL approach was applied to a subset of near-isogenic wild barley introgression lines. In the mentioned study, 2-DE and MS were applied to identify related proteins by using two QTLs specifically linked to malting quality, and the results identified 14 candidate proteins that affected this trait.70
The genotypic differences at a proteomic level can be exploited for the identification of candidate proteins that can be further analyzed for improving stress-specific tolerance in barley and development of biomarkers.185 The application of MS combined with the pQTL approach can be considered a practical tool to identify desirable genes for barley breeding programs that aim to deliver barley lines that are resistant to major stresses or containing desirable characteristics for the malting purposes. Further research aiming to investigate the relationship of the barley genome and proteome will allow the investigation of complex stress tolerance traits, the analysis of the molecular basis of these traits, and the development of the next-generation of barley crops adapted to the changing climate that will be critically important for food security.
MS Application in Food Manufacturing and Processing in Barley
MS proteomic applications in food sciences have increased dramatically in the past decade, largely due to significant advances in sample separation techniques and MS instrumentation. In the past, food proteomic approaches relied heavily on electrophoretic methods. The application of MS technologies allows protein profiling, protein characterization, and analysis of proteins in complex matrices, such as food.202
Food proteomics is complicated by several different factors that need consideration: (1) being complex matrices, most foods have no comprehensive proteome data available; (2) complex modifications occur in food proteins during processing, for example, the Maillard reactions that is in general not measured in classical proteomics applications; (3) unknown dynamics in the proteome during storage and processing can cause high protein degradation or modification; and (4) interference or interactions of food proteins with carbohydrates or lipids can significantly impact the food matrices and complicate proteome analysis.74,203
Food losses contribute considerably to food security, food quality, and safety, both on an economic and environmental level. According to a study, one-third of food produced globally is wasted with developed country losses as high as in developing countries at 40%.204 A reduction in postharvest losses is considered to be the most efficient and cost-effective method for ensuring food security, as postharvest savings not only increase food production, but the increases are sustainability achieved without additional land and water usage.204 Postharvest management systems and smart food processing technologies are equally necessary to guarantee the availability of safe, high-quality food in the coming years.206
After harvest, barley crops are exposed to a series of stresses. Exposure can be either abiotic stresses during, or before they reach the final consumer, for example: (1) physical stress due to handling; (2) variation in temperature and vibration caused by transportation; (3) alternation in atmospheric conditions or temperature reduction with regards to storage; or (4) a combination of biotic and abiotic stresses, such as cereals containing fungal spores that will proliferate during storage.205 During transportation and storage, barley will undergo a series of changes trying to adjust to or acclimate to imposed stress that will change the composition of the proteome. Since proteins are involved in stress responses, for example (1) as enzymes catalyzing changes in the regulation of protein levels, (2) as structural proteins of the cytoskeleton; or (3) as protein-bound receptors, proteomic studies designed to understand the physiological processes involved in stress tolerance of barley crops are of significant importance. These varying states can allow for comparative proteomic investigations related to changes in protein concentration and physiological parameters associated with stress during storage and shelf life related to susceptibility,207 for instance, the first requirement to reduce losses postharvest temperature control combined with postharvest technologies to delay product degradation.23
While the majority of studies address the genomic and transcriptomic data, there is growing interest to understand proteomic involvement in physiological variations during postharvest processing.205 Applications of such knowledge would allow for biomarker identification of postharvest disease. Additionally, this information would support selection and breeding programs, drive harvesting strategies, optimize processing, and storage conditions with the goal of reducing product losses.23
The discovery of novel biomarkers is an important and exciting part of food safety, but to impact human health biomarkers must ensure efficacy and safety. Unfortunately, verification can be time-consuming and expensive, and can limit the number of biomarkers. A typical verification process involves either developing novel antibodies or validating existing antibodies against the new biomarkers so that these new biomarkers can be tested in a broader research program using an immunoassay.208 However, both the development and validation of antibodies take time and money, particularly when the discovery program has identified a panel of biomarkers to move into verification.
In comparison to ELISA methodology, LC-MS/MS methods for biomarker verification are more efficient because they do not rely on the development of new antibodies to measure biomarkers. In addition, LC-MS/MS can accurately and precisely measure multiple biomarkers in a single sample. The advantage of this technique is that it can regulate the bioanalysis of the instrument and can quantify various biomarkers in various states, such as grain, flour, raw, and processed foods. The major disadvantage of this technique is the sensitivity of large molecules compared to the ligand binding assay.162,209
MS Application in Final-Product Quality and Food Safety in Barley
The global scale of food supply chains is a challenge for ensuring food safety. Most countries have implemented regulations and laws in an effort to guarantee food safety.162 More importantly, increased consumers’ attention to the biochemical composition, processing, and functional ingredients have led to making better-informed decisions.210 Cost-effective, efficient, robust analytical methods with greater sensitivity are important to ensure the traceability, quality, and safety of foods. Over recent years, proteomic analysis has emerged as a tool for quality control assessment in food processing and production as well as in food safety.28
Barley proteins contribute significantly to quality parameters, such as flavor, texture, color, with changes resulting from the response to stress.211 Knowledge of the proteins present, their biological role, structures, and functions in raw food materials as well as in final food products is critical for process optimization. The protein content is an essential parameter for food barley and malt barley selection. The hordein content can also be considered, given that hordeins constitute the largest portion of the barley grain and impact the endosperm texture but also play a role in influencing modification during malting and hordein peptides persistent in beer.212 Hordeins are degraded by endoproteases into smaller peptides during malting, but some of the proteins survive the heat of kilning and mashing constituting approximately one-third of the proteins present in the final beer.34,106,111 Flodrová et al.130 compared two proteomic approaches (2-DE and HPLC) to monitor hordein profiles during malting. Their results suggested that LC was more efficient due to its capacity to detect more hordein proteins in a single experiment, facilitating the creation of a barley hordein map that can be be applied to brewing optimization.111−113 Measuring hordein content, as well as protein content, can be beneficial from a food safety perspective as food intolerance is linked to hordeins.
It has been reported that during processing, increased pressure or temperatures can modify gluten proteins, resulting in changes to the protein structure through alteration of the peptide sequence.213,214 Therefore, the gluten content of products is difficult to calculate and can result in underestimation, which poses a health risk for people with CD. The detection of allergenic ingredients is often accomplished using enzyme-linked immunosorbent assays (ELISA). Although this approach has some disadvantages, ELISAs are sensitive but can be challenged by limited reproducibility in food matrices and cross-reactivity, and it is not possible to simultaneously detect multiple allergens.215 Mass spectrometry-based proteomics approaches overcome some of these disadvantages, allowing high-throughput multiplexed analyses.216
Tanner et al.(2015), compared the main characteristics between ELISA and MS-based proteomics and found that hordein (gluten) measurement in beer via ELISA analysis was problematic, where measured levels of hordein varied by four orders of magnitude.217 It was concluded that ELISA detects only the antigenic peptides that may be absent in hydrolyzed gluten present in fermented products.218 MS quantitation is carried out using peptides that are unique and specific, enabling the quantitation of individual hordein isoforms, therefore making it a more reliable approach for detection and speciation of gluten.217 Furthermore, Lock219 confirmed that in comparison to ELISA, LC-MS/MS, demonstrating that LC-MS/MS can be used to detect gluten in processed complex food matrices and food ingredients. Therefore, biomarkers can be developed for gluten and more specifically for barley hordeins.
A common proteomic workflow for the analysis of gluten in barley involves the identification of specific hordein peptide markers followed by quantitative analysis of the protein in food using multiple reaction monitoring (MRM) methods.220 This LC-MS/MS methodology has been broadly applied to gluten detection in barley, malt, and beer. However, further developmental work and standardization of methods are still required. This not only applies to the analytical approach but also food sample processing and database development.221 A further example of the application of proteomics in food science was the development of ultralow gluten barley, for food and beverage production.222 Starting with reduced hordein barley lines, Tanner et al. (2016) used a conventional breeding approach to produce an ultralow gluten barley variety Kebari.222 Proteomics was used to confirm the reduced gluten content (∼5 mg/kg) a ∼19 000-fold reduction of hordein content when compared to conventional barley, below the level recommendation for classification as gluten-free by the World Health Organization (WHO). Further proteomic analysis of this line uncovered that while the levels of α-amylase remained at levels similar to the control, the β-amylase levels were reduced remarkably by approximately 50-fold. Additionally, the overall protein content in this line was comparable to control barley, but there was a 10–15-fold increase in some free amino acid levels.222
Food and beverage proteomes are complex wherein the matrices can interfere with detection of proteins (qualitative and quantitative).206 LC-MS/MS-based proteomics methods are now available for the detection of various foodborne proteins and can be used as a complementary approach in food testing applications due to its accuracy, precision, sensitivity, and robust quantitative ability.
Conclusions
Advanced MS-based proteomics tools coupled with the progress in genomics and transcriptomics provide a platform to investigate barley, its expressed proteins, and their biological roles. Gel-based techniques such as 2-DE while time and labor intensive have been effectively utilized for protein separation and visualization. More recently, liquid chromatography has become more common to improve the speed and precision of protein separation. The application of mass spectrometry in barley proteomics has provided a robust platform for the identification and quantitation of proteins. In parallel, developments in sample preparation, protein identification algorithms, and the used background databases have enabled greater depth of coverage of the barley proteome as well as application to raw ingredients, i.e., barley, or its products: malt and beer. Proteomics can be used in molecular breeding to reveal more information about target proteins which are linked to desired malting characteristics, understanding and optimizing health-related proteins, investigating modifications of those proteins during malting, mashing, and fermentation, as well as measuring the expression level of each protein. Furthermore, proteomics coupled with other omics technologies provides a multidisciplinary system biology platform.
We are now in a position to not just explore the proteins present in barley, but we can do so from a truly quantitative standpoint. We can investigate the changes that occur as a result of environmental/experimental manipulations and that will lead to changes in barley cultivation practices, food processing, and can be used for food safety assessment. Applying global quantitative proteomics to barley will change the way that barley is bred, grown, and processed and ensure food safety.
Acknowledgments
This work was supported by an RTP scholarship from Edith Cowan University (M.B.) and an industry Ph.D. scholarship sponsored by Edith Cowan University, Intergrain, and CSIRO, Industry Ph.D. ID: G1004654 (C.E.O.).
The authors declare no competing financial interest.
References
- Lister D. L.; Jones H.; Oliveira H. R.; Petrie C. A.; Liu X.; Cockram J.; Kneale C. J.; Kovaleva O.; Jones M. K.. Barley heads east: Genetic analyses reveal routes of spread through diverse Eurasian landscapes. Sun G, ed. PLoS One 2018, 13, e0196652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FAOSTAT (Food and Agriculture Organization of the United Nations) . Crops [Online], 2019. http://www.fao.org/faostat/en/#data/QC (accessed: December 25, 2020).
- Wang Y.; Harding S. V.; Thandapilly S. J.; Tosh S. M.; Jones P. J. H.; Ames N. P. Barley β -glucan reduces blood cholesterol levels via interrupting bile acid metabolism. Br. J. Nutr. 2017, 118, 822–829. 10.1017/S0007114517002835. [DOI] [PubMed] [Google Scholar]
- AbuMweis S.; Thandapilly S. J.; Storsley J.; Ames N. Effect of barley β-glucan on postprandial glycaemic response in the healthy human population: A meta-analysis of randomized controlled trials. J. Funct. Foods 2016, 27, 329–342. 10.1016/j.jff.2016.08.057. [DOI] [Google Scholar]
- Gong L.; Cao W.; Chi H.; Wang J.; Zhang H.; Liu J.; Sun B. Whole cereal grains and potential health effects: Involvement of the gut microbiota. Food Res. Int. 2018, 103, 84–102. 10.1016/j.foodres.2017.10.025. [DOI] [PubMed] [Google Scholar]
- Ghaffarzadegan T.; Zhong Y.; Fåk Hållenius F.; Nyman M. Effects of barley variety, dietary fiber and β-glucan content on bile acid composition in cecum of rats fed low- and high-fat diets. J. Nutr. Biochem. 2018, 53, 104–110. 10.1016/j.jnutbio.2017.10.008. [DOI] [PubMed] [Google Scholar]
- Idehen E.; Tang Y.; Sang S. Bioactive phytochemicals in barley. J. Food Drug Anal. 2017, 25, 148–161. 10.1016/j.jfda.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrovska-Avramenko N.; Karklina D.; Gedrovica I. In Water soluble vitamins B1, B2 and B3 in triticale and hull-less barley grains. 11th Baltic Conference on Food Science and Technology “Food Science and Technology in a Changing World” 2017, 207–209. 10.22616/foodbalt.2017.039 [DOI] [Google Scholar]
- Kellner R. Proteomics. Concepts and perspectives. Fresenius' J. Anal. Chem. 2000, 366, 517–524. 10.1007/s002160051547. [DOI] [PubMed] [Google Scholar]
- TechNavio . Global barley market 2018–2022. http://www.technavio.com, 2018.
- Lee K. H.; Chung S. H.; Song J. H.; Yoon J. S.; Lee J. H.; Jung M. J.; Kim J. H.. Cosmetic compositions for skin-tightening and method of skin-tightening using the same. U.S. Patent 8580741, November 12, 2013.
- Qureshi N.; Saha B. C.; Dien B.; Hector R. E.; Cotta M. A. Production of butanol (a biofuel) from agricultural residues: Part I - Use of barley straw hydrolysate. Biomass Bioenergy 2010, 34, 559–565. 10.1016/j.biombioe.2009.12.024. [DOI] [Google Scholar]
- Tricase C.; Amicarelli V.; Emilia L.; Roberto Leonardo R.. Economic Analysis of the Barley Market and Related Uses. In: Grasses as Food and Feed; Tadele Z., Eds.; IntechOpen: London, UK, 2018; 10.5772/intechopen.78967. [DOI] [Google Scholar]
- Eckert E.; Wismer W.; Waduthanthri K.; Babii O.; Yang J.; Chen L. Application of Barley- and Lentil-Protein Concentrates in the Production of Protein-Enriched Doughnuts. J. Am. Oil Chem. Soc. 2018, 95, 1027–1040. 10.1002/aocs.12103. [DOI] [Google Scholar]
- Newman C. W.; Newman R. K. A brief history of barley foods. Cereal Foods World 2006, 51, 4–7. 10.1094/CFW-51-0004. [DOI] [Google Scholar]
- Baik B. K.; Ullrich S. E. Barley for food: Characteristics, improvement, and renewed interest. J. Cereal Sci. 2008, 48, 233–242. 10.1016/j.jcs.2008.02.002. [DOI] [Google Scholar]
- Lösch S.; Moghaddam N.; Grossschmidt K.; Risser D. U.; Kanz F. Stable isotope and trace element studies on gladiators and contemporary Romans from Ephesus (Turkey, 2nd and 3rd Ct. AD)-implications for differences in diet. PLoS One 2014, 9, e110489 10.1371/journal.pone.0110489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.; Tian Z.; Chen L.; Temelli F.; Liu H.; Wang Y. Functionality of barley proteins extracted and fractionated by alkaline and alcohol methods. Cereal Chem. 2010, 87, 597–606. 10.1094/CCHEM-06-10-0097. [DOI] [Google Scholar]
- Fox G. P.; Watson-Fox L.. Barley: Current Understanding of ‘Omics Data on Quality. In Comprehensive Foodomics; Cifuentes A., Eds.; Elsevier: Amsterdam, Netherlands, 2021; pp 513–527. 10.1016/B978-0-08-100596-5.22740-6. [DOI] [Google Scholar]
- Wilkins M. R.; Pasquali C.; Appel R. D.; Ou K.; Golaz O.; Sanchez J.-C.; Yan J. X.; Gooley A. A.; Hughes G.; Humphery-Smith I.; Williams K. L.; Hochstrasser D. F. From proteins to proteomes: Large scale protein identification by two-dimensional electrophoresis and amino acid analysis. Nat. Biotechnol. 1996, 14, 61–65. 10.1038/nbt0196-61. [DOI] [PubMed] [Google Scholar]
- Anderson N. L.; Matheson A. D.; Steiner S. Proteomics: Applications in basic and applied biology. Curr. Opin. Biotechnol. 2000, 11, 408–412. 10.1016/S0958-1669(00)00118-X. [DOI] [PubMed] [Google Scholar]
- Domon B.; Gallien S. Recent advances in targeted proteomics for clinical applications. Proteomics: Clin. Appl. 2015, 9, 423–431. 10.1002/prca.201400136. [DOI] [PubMed] [Google Scholar]
- Mora L.; Gallego M.; Toldrá F. New approaches based on comparative proteomics for the assessment of food quality. Curr. Opin Food Sci. 2018, 22, 22–27. 10.1016/j.cofs.2018.01.005. [DOI] [Google Scholar]
- Mock H. P.; Finnie C.; Witzel K.; Svensson B.. Barley Proteomics. In The Barley Genome. Compendium of Plant Genomes; Stein N., Muehlbauer G., Eds.; Springer: Cham, New York, 2018; pp 345–361. 10.1007/978-3-319-92528-8_19. [DOI] [Google Scholar]
- Ortea I.; O’Connor G.; Maquet A. Review on proteomics for food authentication. J. Proteomics 2016, 147, 212–225. 10.1016/j.jprot.2016.06.033. [DOI] [PubMed] [Google Scholar]
- Ramalingam A.; Kudapa H.; Pazhamala L. T.; Weckwerth W.; Varshney R. K. Proteomics and metabolomics: Two emerging areas for legume improvement. Front. Plant Sci. 2015, 6, 1–21. 10.3389/fpls.2015.01116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawade A.; Van Ham J.; Blomquist H.; Bagge O.; Alexandersson E.; Ortiz R. High-throughput field-phenotyping tools for plant breeding and precision agriculture. Agronomy 2019, 9, 258. 10.3390/agronomy9050258. [DOI] [Google Scholar]
- Andjelković U.; Josić D. Mass spectrometry based proteomics as foodomics tool in research and assurance of food quality and safety. Trends Food Sci. Technol. 2018, 77, 100–119. 10.1016/j.tifs.2018.04.008. [DOI] [Google Scholar]
- Willows R. D.; Worden P.; Mirzaei M.. Barley Grain Proteomics. In: Proteomics in Food Science; Colgrave M. L., Eds.; Elsevier: Amsterdam, Netherlands, 2017; Chapter 5, pp 75–88. 10.1016/B978-0-12-804007-2.00005-9. [DOI] [Google Scholar]
- Diaz-Mendoza M.; Diaz I.; Martinez M. Insights on the Proteases Involved in Barley and Wheat Grain Germination. Int. J. Mol. Sci. 2019, 20, 2087. 10.3390/ijms20092087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnie C.; Andersen B.; Shahpiri A.; Svensson B. Proteomes of the barley aleurone layer: A model system for plant signalling and protein secretion. Proteomics 2011, 11, 1595–1605. 10.1002/pmic.201000656. [DOI] [PubMed] [Google Scholar]
- Labuschagne M. T. A review of cereal grain proteomics and its potential for sorghum improvement. J. Cereal Sci. 2018, 84, 151–158. 10.1016/j.jcs.2018.10.010. [DOI] [Google Scholar]
- Petersen J.; Rogowska-Wrzesinska A.; Jensen O. N. Functional proteomics of barley and barley chloroplasts - strategies, methods and perspectives. Front. Plant Sci. 2013, 4, 1–10. 10.3389/fpls.2013.00052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanislava G. A review: The role of barley seed pathogenesis-related proteins (PRs) in beer production. J. Inst. Brew. 2010, 116, 111–124. 10.1002/j.2050-0416.2010.tb00407.x. [DOI] [Google Scholar]
- Wang W. Q.; Liu S. J.; Song S. Q.; Møller I. M. Proteomics of seed development, desiccation tolerance, germination and vigor. Plant Physiol. Biochem. 2015, 86, 1–15. 10.1016/j.plaphy.2014.11.003. [DOI] [PubMed] [Google Scholar]
- Ribeiro M.; Nunes-Miranda J. D.; Branlard G.; Carrillo J. M.; Rodriguez-Quijano M.; Igrejas G. One Hundred Years of Grain Omics: Identifying the Glutens That Feed the World. J. Proteome Res. 2013, 12, 4702–4716. 10.1021/pr400663t. [DOI] [PubMed] [Google Scholar]
- Chakraborty S.; Salekdeh G. H.; Yang P.; Woo S. H.; Chin C. F.; Gehring C.; Haynes P. A.; Mirzaei M.; Komatsu S. Proteomics of Important Food Crops in the Asia Oceania Region: Current Status and Future Perspectives. J. Proteome Res. 2015, 14, 2723–2744. 10.1021/acs.jproteome.5b00211. [DOI] [PubMed] [Google Scholar]
- Steinwand M. A.; Ronald P. C. Crop biotechnology and the future of food. Nat. Food. 2020, 1, 273–283. 10.1038/s43016-020-0072-3. [DOI] [Google Scholar]
- Gubatz S.; Shewry P. R.. The Development, Structure, and Composition of the Barley Grain. In Barley: Production, Improvement, and Uses; Ullrich S. E., Eds.; Wiley & Sons: New York, 2010; Chapter 13, pp 391–448. 10.1002/9780470958636.ch13. [DOI] [Google Scholar]
- Osborne T. B. The proteins of barley. J. Am. Chem. Soc. 1895, 17, 539–567. 10.1021/ja02162a008. [DOI] [Google Scholar]
- Shewry P. R.; Halford N. G. Cereal seed storage proteins: Structures, properties and role in grain utilization. J. Exp. Bot. 2002, 53, 947–958. 10.1093/jexbot/53.370.947. [DOI] [PubMed] [Google Scholar]
- Juhasz A.; Belova T.; Florides C. G.; Maulis C.; Fischer I.; Gell G.; Birinyi Z.; Ong J.; Keeble-Gagnere G.; Maharajan A.; Ma W.; Gibson P.; Jia J.; Lang D.; Mayer K. F. X.; Spannagl M.; Tye-Din J. A.; Appels R.; Olsen O.-A. Genome mapping of seed-borne allergens and immunoresponsive proteins in wheat. Sci. Adv. 2018, 4, eaar8602 10.1126/sciadv.aar8602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.Wheat Grain Avenin-like Protein Dynamics in Relation to Genotypes and Environments. Ph.D. Thesis, Murdoch University, August 2018. [Google Scholar]
- Schalk K.; Lexhaller B.; Koehler P.; Scherf K. A. Isolation and characterization of gluten protein types from wheat, rye, barley, and oats for use as reference materials. PLoS One 2017, 12, e0172819 10.1371/journal.pone.0172819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lexhaller B.; Colgrave M. L.; Scherf K. A. Characterization and relative quantitation of wheat, rye, and barley gluten protein types by liquid chromatography-tandem mass spectrometry. Front. Plant Sci. 2019, 10, 1530. 10.3389/fpls.2019.01530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrieri N.; Cavaletto M.. Cereals proteins. In Proteins in Food Processing; Yada R. Y., Eds.; Elsevier: Amsterdam, Netherlands, 2018; pp 223–244. 10.1016/B978-0-08-100722-8.00009-7. [DOI] [Google Scholar]
- Baxter E. D. Hordein in Barley and Malt—a Review. J. Inst. Brew. 1981, 87, 173–176. 10.1002/j.2050-0416.1981.tb04011.x. [DOI] [Google Scholar]
- Houde M.; Khodaei N.; Benkerroum N.; Karboune S. Barley protein concentrates: Extraction, structural and functional properties. Food Chem. 2018, 254, 367–376. 10.1016/j.foodchem.2018.01.156. [DOI] [PubMed] [Google Scholar]
- Tanner G. J.; Colgrave M. L.; Blundell M. J.; Howitt C. A.; Bacic A. Hordein Accumulation in Developing Barley Grains. Front. Plant Sci. 2019, 10, 1–10. 10.3389/fpls.2019.00649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahalingam R. Shotgun proteomics of the barley seed proteome. BMC Genomics 2017, 18, 1–11. 10.1186/s12864-016-3408-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shewry P. R.; Napier J. A.; Tatham A. S. Seed storage proteins: Structures and biosynthesis. Plant Cell 1995, 7, 945–956. 10.2307/3870049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izydorczyk M. S.; Edney M.. Barley: Grain-Quality Characteristics and Management of Quality Requirements. In Cereal Grains; Elsevier: Amsterdam, Netherlands, 2017; pp 195–234. 10.1016/B978-0-08-100719-8.00009-7. [DOI] [Google Scholar]
- Henson C. A.; Vinje M. A.; Duke S. H. Maltose Effects on Barley Malt β-Amylase Activity and Thermostability at Low Isothermal Mashing Temperatures. J. Am. Soc. Brew. Chem. 2020, 78, 207–218. 10.1080/03610470.2020.1738811. [DOI] [Google Scholar]
- Swanston J. S.; Molina-Cano J. L. Beta-amylase activity and thermostability in two mutants derived from the malting barley cv. Triumph. J. Cereal Sci. 2001, 33, 155–161. 10.1006/jcrs.2000.0364. [DOI] [Google Scholar]
- Rasmussen C. B.; Henriksen A.; Abelskov A. K.; Jensen R. B.; Rasmussen S. K.; Hejgaard J.; Welinder K. G. Purification, characterization and stability of barley grain peroxidase BP 1, a new type of plant peroxidase. Physiol. Plant. 1997, 100, 102–110. 10.1111/j.1399-3054.1997.tb03459.x. [DOI] [Google Scholar]
- Feussner I.; Wasternack C. The Lipoxygenase Pathway. Annu. Rev. Plant Biol. 2002, 53, 275–297. 10.1146/annurev.arplant.53.100301.135248. [DOI] [PubMed] [Google Scholar]
- Holtman W. L.; Van Duijn G.; Sedee N. J. A.; Douma A. C. Differential expression of lipoxygenase isoenzymes in embryos of germinating barley. Plant Physiol. 1996, 111, 569–576. 10.1104/pp.111.2.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeats T. H.; Rose J. K. C. The biochemistry and biology of extracellular plant lipid-transfer proteins (LTPs). Protein Sci. 2008, 17, 191–198. 10.1110/ps.073300108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikołajczak K.; Ogrodowicz P.; Surma M.; Adamski T.; Kuczyńska A. Introgression of LTP2 gene through marker assisted backcross in barley (Hordeum vulgare L.). Electron. J. Biotechnol. 2016, 24, 9–11. 10.1016/j.ejbt.2016.09.003. [DOI] [Google Scholar]
- Shewry P. R.; Tatham A. S. The prolamin storage proteins of cereal seeds: structure and evolution. Biochem. J. 1990, 267, 1–12. 10.1042/bj2670001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones B. L. Endoproteases of barley and malt. J. Cereal Sci. 2005, 42, 139–156. 10.1016/j.jcs.2005.03.007. [DOI] [Google Scholar]
- Shewry P. R.; Lucas J. A. Plant Proteins that Confer Resistance to Pests and Pathogens. Adv. Bot. Res. 1997, 26, 135–192. 10.1016/S0065-2296(08)60120-2. [DOI] [Google Scholar]
- Barber D.; Sanchez-Monge R.; Mendez E.; Lazaro A.; Garcia-Olmedo F.; Salcedo G. New α-amylase and trypsin inhibitors among the CM-proteins of barley (Hordeum vulgare). Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol. 1986, 869, 115–118. 10.1016/0167-4838(86)90318-3. [DOI] [PubMed] [Google Scholar]
- Bose U.; Juhász A.; Broadbent J. A.; Byrne K.; Howitt C. A.; Colgrave M. L. Identification and Quantitation of Amylase Trypsin Inhibitors Across Cultivars Representing the Diversity of Bread Wheat. J. Proteome Res. 2020, 19, 2136–2148. 10.1021/acs.jproteome.0c00059. [DOI] [PubMed] [Google Scholar]
- Nielsen P. K.; Bønsager B. C.; Fukuda K.; Svensson B. Barley α-amylase/subtilisin inhibitor: Structure, biophysics and protein engineering. Biochim. Biophys. Acta, Proteins Proteomics 2004, 1696, 157–164. 10.1016/j.bbapap.2003.09.019. [DOI] [PubMed] [Google Scholar]
- Svendsen I. B.; Boisen S.; Hejgaard J. Amino acid sequence of serine protease inhibitor CI-1 from barley. Homology with barley inhibitor CI-2, potato inhibitor I, and leech eglin. Carlsberg Res. Commun. 1982, 47, 45–53. 10.1007/BF02907796. [DOI] [Google Scholar]
- Darlington H. F.; Tecsi L.; Harris N.; Griggs D. L.; Cantrell I. C.; Shewry P. R. Starch granule associated proteins in barley and wheat. J. Cereal Sci. 2000, 32, 21–29. 10.1006/jcrs.2000.0312. [DOI] [Google Scholar]
- Osborn R. W.; Broekaert W. F. Antifungal Proteins. Seed Proteins. 1999, 727–751. 10.1007/978-94-011-4431-5_31. [DOI] [Google Scholar]
- Colgrave M. L.; Goswami H.; Howitt C. A.; Tanner G. J. Proteomics as a tool to understand the complexity of beer. Food Res. Int. 2013, 54, 1001–1012. 10.1016/j.foodres.2012.09.043. [DOI] [Google Scholar]
- March T. J.; Richter D.; Colby T.; Harzen A.; Schmidt J.; Pillen K. Identification of proteins associated with malting quality in a subset of wild barley introgression lines. Proteomics 2012, 12, 2843–2851. 10.1002/pmic.201200117. [DOI] [PubMed] [Google Scholar]
- Niu C.; Han Y.; Wang J.; Zheng F.; Liu C.; Li Y.; Li Q. Malt derived proteins: Effect of protein Z on beer foam stability. Food Biosci. 2018, 25, 21–27. 10.1016/j.fbio.2018.07.003. [DOI] [Google Scholar]
- Schulte F.; Flaschel E.; Niehaus K. Proteome-Based Analysis of Colloidal Instability Enables the Detection of Haze-Active Proteins in Beer. J. Agric. Food Chem. 2016, 64, 6752–6761. 10.1021/acs.jafc.6b02467. [DOI] [PubMed] [Google Scholar]
- Schulz B. L.; Phung T. K.; Bruschi M.; Janusz A.; Stewart J.; Meehan J.; Healy P.; Nouwens A. S.; Fox G. P.; Vickers C. E. Process Proteomics of Beer Reveals a Dynamic Proteome with Extensive Modifications. J. Proteome Res. 2018, 17, 1647–1653. 10.1021/acs.jproteome.7b00907. [DOI] [PubMed] [Google Scholar]
- Gupta M.; Abu-Ghannam N.; Gallaghar E. Barley for brewing: Characteristic changes during malting, brewing and applications of its by-products. Compr. Rev. Food Sci. Food Saf. 2010, 9, 318–328. 10.1111/j.1541-4337.2010.00112.x. [DOI] [PubMed] [Google Scholar]
- Fox G. P.; Panozzo J. F.; Li C. D.; Lance R. C. M.; Inkerman P. A.; Henry R. J. Molecular basis of barley quality. Aust. J. Agric. Res. 2003, 54, 1081–1101. 10.1071/AR02237. [DOI] [Google Scholar]
- Bamforth C. W. Current perspectives on the role of enzymes in brewing. J. Cereal Sci. 2009, 50, 353–357. 10.1016/j.jcs.2009.03.001. [DOI] [Google Scholar]
- Gous P. W.; Fox G. P. Amylopectin synthesis and hydrolysis-Understanding isoamylase and limit dextrinase and their impact on starch structure on barley (Hordeum vulgare) quality. Trends Food Sci. Technol. 2017, 62, 23–32. 10.1016/j.tifs.2016.11.013. [DOI] [Google Scholar]
- Huang Y.; Cai S.; Zhang G. The relationship of limit dextrinase, limit dextrinase inhibitor and malt quality parameters in barley and their genetic analysis. J. Cereal Sci. 2016, 70, 140–145. 10.1016/j.jcs.2016.05.027. [DOI] [Google Scholar]
- McCafferty C. A.; Jenkinson H. R.; Brosnan J. M.; Bryce J. H. Limit dextrinase - Does its malt activity relate to its activity during brewing?. J. Inst. Brew. 2004, 110, 284–296. 10.1002/j.2050-0416.2004.tb00623.x. [DOI] [Google Scholar]
- Taylor J. P.; Zannini E.; Jacob F.; Arendt E. K. A study on malt modification, used as a tool to reduce levels of beer hordeins. J. Inst. Brew. 2018, 124, 143–147. 10.1002/jib.482. [DOI] [Google Scholar]
- Sun H.; Song W.; Zhang L.; Yang X.; Zhu Z.; Ma R.; Wang D. Structural characterization and inhibition on α-glucosidase of a novel oligosaccharide from barley malt. J. Cereal Sci. 2018, 82, 82–93. 10.1016/j.jcs.2018.05.009. [DOI] [Google Scholar]
- Wenwen Y.; Tao K.; Gidley M. J.; Fox G. P.; Gilbert R. G. Molecular brewing: Molecular structural effects involved in barley malting and mashing. Carbohydr. Polym. 2019, 206, 583–592. 10.1016/j.carbpol.2018.11.018. [DOI] [PubMed] [Google Scholar]
- Bamforth C. W.; Martin H. L. the Degradation of B-Glucan During Malting and Mashing: the Role of B-Glucanase. J. Inst. Brew. 1983, 89, 303–307. 10.1002/j.2050-0416.1983.tb04190.x. [DOI] [Google Scholar]
- Rimsten L.; Haraldsson A.-K.; Andersson R.; Alminger M.; Sandberg A.-S.; Aman P. Effects of malting on β-glucanase and phytase activity in barley grain. J. Sci. Food Agric. 2002, 82, 904–912. 10.1002/jsfa.1135. [DOI] [Google Scholar]
- Jones B. L.; Marinac L. A.; Fontanini D. Quantitative study of the formation of endoproteolytic activities during malting and their stabilities to kilning. J. Agric. Food Chem. 2000, 48, 3898–3905. 10.1021/jf000458k. [DOI] [PubMed] [Google Scholar]
- Osman A. M.; Coverdale S. M.; Cole N.; Hamilton S. E.; de Jersey J.; Inkerman P. A. Characterisation and Assessment of the Role of Barley Malt Endoproteases During Malting and Mashing1. J. Inst. Brew. 2002, 108, 62–67. 10.1002/j.2050-0416.2002.tb00125.x. [DOI] [Google Scholar]
- Rossi S.; Sileoni V.; Perretti G.; Marconi O. Characterization of the volatile profiles of beer using headspace solid-phase microextraction and gas chromatography-mass spectrometry. J. Sci. Food Agric. 2014, 94, 919–928. 10.1002/jsfa.6336. [DOI] [PubMed] [Google Scholar]
- Sørensen S. B.; Ottesen M. Fractionation and characterization of beer proteins. Carlsberg Res. Commun. 1978, 43, 133–144. 10.1007/BF02914237. [DOI] [Google Scholar]
- Evans D. E.; Sheehan M. C.; Stewart D. C. The Impact of Malt Derived Proteins on Beer Foam Quality. Part II: The Influence of Malt Foam-positive Proteins and Non-starch Polysaccharides on Beer Foam Quality. J. Inst. Brew. 1999, 105, 171–178. 10.1002/j.2050-0416.1999.tb00016.x. [DOI] [Google Scholar]
- Carvalho A. d. O.; Gomes V. M. Role of plant lipid transfer proteins in plant cell physiology-A concise review. Peptides 2007, 28, 1144–1153. 10.1016/j.peptides.2007.03.004. [DOI] [PubMed] [Google Scholar]
- Salminen T. A.; Blomqvist K.; Edqvist J. Lipid transfer proteins: classification, nomenclature, structure, and function. Planta 2016, 244, 971–997. 10.1007/s00425-016-2585-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanislava G. Barley grain non-specific lipid-transfer proteins (ns-LTPs) in beer production and quality. J. Inst. Brew. 2007, 113, 310–324. 10.1002/j.2050-0416.2007.tb00291.x. [DOI] [Google Scholar]
- Picariello G.; Mamone G.; Nitride C.; Ferranti P.. Proteomic Analysis of Beer. In Proteomic Analysis of Beer; Colgrave M. L., Eds.; Elsevier: Amsterdam, Netherlands, 2017; Chapter 23, pp 383–403. 10.1016/B978-0-12-804007-2.00023-0. [DOI] [Google Scholar]
- Jegou S.; Douliez J. P.; Molle D.; Boivin P.; Marion D. Purification and structural characterization of LTP1 polypeptides from beer. J. Agric. Food Chem. 2000, 48, 5023–5029. 10.1021/jf000075m. [DOI] [PubMed] [Google Scholar]
- Lindorff-Larsen K.; Winther J. R. Surprisingly high stability of barley lipid transfer protein, LTP1, towards denaturant, heat and proteases. FEBS Lett. 2001, 488, 145–148. 10.1016/S0014-5793(00)02424-8. [DOI] [PubMed] [Google Scholar]
- Perrocheau L.; Bakan B.; Boivin P.; Marion D. Stability of barley and malt lipid transfer protein 1 (LTP1) toward heating and reducing agents: Relationships with the brewing process. J. Agric. Food Chem. 2006, 54, 3108–3113. 10.1021/jf052910b. [DOI] [PubMed] [Google Scholar]
- Gordon R.; Power A.; Chapman J.; Chandra S.; Cozzolino D. A review on the source of lipids and their interactions during beer fermentation that affect beer quality. Fermentation. 2018, 4, 89. 10.3390/fermentation4040089. [DOI] [Google Scholar]
- Hirota N.; Kuroda H.; Takoi K.; Kaneko T.; Kaneda H.; Yoshida I.; Takashio M.; Ito K.; Takeda K. Brewing performance of malted lipoxygenase-1 null barley and effect on the flavor stability of beer. Cereal Chem. 2006, 83, 250–254. 10.1094/CC-83-0250. [DOI] [Google Scholar]
- Porta H.; Rocha-Sosa M. Plant lipoxygenases. Physiological and molecular features. Plant Physiol. 2002, 130, 15–21. 10.1104/pp.010787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt N. F.; Van Mechelen J. R. Expression of lipoxygenase isoenzymes in developing barley grains. Plant Sci. 1997, 128, 141–150. 10.1016/S0168-9452(97)00152-0. [DOI] [Google Scholar]
- Kuroda H.; Kobayashi N.; Kaneda H.; Watari J.; Takashio M. Characterization of factors that transform linoleic acid into di- and trihydroxyoctadecenoic acids in mash. J. Biosci Bioeng. 2002, 93, 73–77. 10.1016/S1389-1723(02)80057-3. [DOI] [PubMed] [Google Scholar]
- Cohen M.; Davydov O.; Fluhr R. Plant serpin protease inhibitors: Specificity and duality of function. J. Exp. Bot. 2019, 70, 2077–2085. 10.1093/jxb/ery460. [DOI] [PubMed] [Google Scholar]
- Roberts T. H.; Marttila S.; Rasmussen S. K.; Hejgaard J. Differential gene expression for suicide-substrate serine proteinase inhibitors (serpins) in vegetative and grain tissues of barley. J. Exp Bot. 2003, 54, 2251–2263. 10.1093/jxb/erg248. [DOI] [PubMed] [Google Scholar]
- Steiner E.; Gastl M.; Becker T. Protein changes during malting and brewing with focus on haze and foam formation: A review. Eur. Food Res. Technol. 2011, 232, 191–204. 10.1007/s00217-010-1412-6. [DOI] [Google Scholar]
- Bobálová J.; Petry-Podgórska I.; Laštovičková M.; Chmelík J. Monitoring of malting process by characterization of glycation of barley protein Z. Eur. Food Res. Technol. 2010, 230, 665–673. 10.1007/s00217-009-1205-y. [DOI] [Google Scholar]
- Iimure T.; Sato K. Beer proteomics analysis for beer quality control and malting barley breeding. Food Res. Int. 2013, 54, 1013–1020. 10.1016/j.foodres.2012.11.028. [DOI] [Google Scholar]
- Mastanjević K.; Krstanović V.; Lukinac J.; Jukić M.; Vulin Z.; Mastanjević K. Beer-the importance of colloidal stability (non-biological haze). Fermentation. 2018, 4, 91. 10.3390/fermentation4040091. [DOI] [Google Scholar]
- Siebert K. J.; Lynn P. Y. On the mechanisms of adsorbent interactions with haze-active proteins and polyphenols. J. Am. Soc. Brew. Chem. 2008, 66, 48–54. 10.1094/ASBCJ-2007-1116-02. [DOI] [Google Scholar]
- Asano K.; Shinagawa K.; Hashimoto N. Characterization of Haze-Forming Proteins of Beer and Their Roles in Chill Haze Formation. J. Am. Soc. Brew. Chem. 1982, 40, 147–154. 10.1094/ASBCJ-40-0147. [DOI] [Google Scholar]
- Bamforth C. W.; Kanauchi M. Interactions between polypeptides derived from barley and other beer components in model foam systems. J. Sci. Food Agric. 2003, 83, 1045–1050. 10.1002/jsfa.1503. [DOI] [Google Scholar]
- Colgrave M. L.; Goswami H.; Howitt C. A.; Tanner G. J. What is in a beer? Proteomic characterization and relative quantification of hordein (gluten) in beer. J. Proteome Res. 2012, 11, 386–396. 10.1021/pr2008434. [DOI] [PubMed] [Google Scholar]
- Tanner G. J.; Colgrave M. L.; Howitt C. A. Gluten, celiac disease, and gluten intolerance and the impact of gluten minimization treatments with prolylendopeptidase on the measurement of gluten in beer. J. Am. Soc. Brew. Chem. 2014, 72, 36–50. 10.1094/ASBCJ-2014-0129-01. [DOI] [Google Scholar]
- Tye-Din J. A.; Stewart J. A.; Dromey J. A.; et al. Comprehensive, quantitative mapping of T cell epitopes in gluten in celiac disease. Sci. Transl. Med. 2010, 2, 41ra51–41ra51. 10.1126/scitranslmed.3001012. [DOI] [PubMed] [Google Scholar]
- Klose J. Protein mapping by combined isoelectric focusing and electrophoresis of mouse tissues - A novel approach to testing for induced point mutations in mammals. Hum. Genet. 1975, 26, 231–243. [DOI] [PubMed] [Google Scholar]
- O’Farrell P. H. High Resolution of Proteins Electrophoresis of Proteins. J. Biol. Chem. 1975, 250, 4007–4021. 10.1016/S0021-9258(19)41496-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorrin-Novo J. V.; Komatsu S.; Sanchez-Lucas R.; Rodríguez de Francisco L. E. Gel electrophoresis-based plant proteomics: Past, present, and future. Happy 10th anniversary Journal of Proteomics!. J. Proteomics 2019, 198, 1–10. 10.1016/j.jprot.2018.08.016. [DOI] [PubMed] [Google Scholar]
- Görg A.; Postel W.; Domscheit A.; Günther S. Two-dimensional electrophoresis with immobilized pH gradients of leaf proteins from barley (Hordeum vulgare): Method, reproducibility and genetic aspects. Electrophoresis 1988, 9, 681–692. 10.1002/elps.1150091103. [DOI] [PubMed] [Google Scholar]
- Ahsan N.; Rao R. S. P.; Gruppuso P. A.; Ramratnam B.; Salomon A. R. Targeted proteomics: Current status and future perspectives for quantification of food allergens. J. Proteomics 2016, 143, 15–23. 10.1016/j.jprot.2016.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manes N. P.; Nita-Lazar A. Application of targeted mass spectrometry in bottom-up proteomics for systems biology research. J. Proteomics 2018, 189, 75–90. 10.1016/j.jprot.2018.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teo G.; Kim S.; Tsou C. C.; Collins B.; Gingras A. C.; Nesvizhskii A. I.; Choi H. MapDIA: Preprocessing and statistical analysis of quantitative proteomics data from data independent acquisition mass spectrometry. J. Proteomics 2015, 129, 108–120. 10.1016/j.jprot.2015.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Østergaard O.; Melchior S.; Roepstorff P.; Svensson B. Initial proteome analysis of mature barley seeds and malt. Proteomics 2002, 2, 733–739. . [DOI] [PubMed] [Google Scholar]
- Finnie C.; Melchior S.; Roepstorff P.; Svensson B. Proteome analysis of grain filling and seed maturation in barley. Plant Physiol. 2002, 129, 1308–1319. 10.1104/pp.003681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bak-Jensen K. S.; Laugesen S.; Roepstorff P.; Svensson B. Two-dimensional gel electrophoresis pattern (pH 6–11) and identification of water-soluble barley seed and malt proteins by mass spectrometry. Proteomics 2004, 4, 728–742. 10.1002/pmic.200300615. [DOI] [PubMed] [Google Scholar]
- Finnie C.; Maeda K.; Østergaard O.; Bak-Jensen K. S. S.; Larsen J.; Svensson B. Proteomics of Plant Proteins Aspects of the barley seed proteome during development and germination. Biochem. Soc. Trans. 2004, 32, 517–519. 10.1042/bst0320517. [DOI] [PubMed] [Google Scholar]
- Perrocheau L.; Rogniaux H.; Boivin P.; Marion D. Probing heat-stable water-soluble proteins from barley to malt and beer. Proteomics 2005, 5, 2849–2858. 10.1002/pmic.200401153. [DOI] [PubMed] [Google Scholar]
- Bønsager B. C.; Finnie C.; Roepstorff P.; Svensson B. Spatio-temporal changes in germination and radical elongation of barley seeds tracked by proteome analysis of dissected embryo, aleurone layer, and endosperm tissues. Proteomics 2007, 7, 4528–4540. 10.1002/pmic.200700766. [DOI] [PubMed] [Google Scholar]
- Finnie C.; Svensson B. Barley seed proteomics from spots to structures. J. Proteomics 2009, 72, 315–324. 10.1016/j.jprot.2008.12.001. [DOI] [PubMed] [Google Scholar]
- Iimure T.; Nankaku N.; Hirota N.; Tiansu Z.; Hoki T.; Kihara M.; Hayashi K.; Ito K.; Sato K. Construction of a novel beer proteome map and its use in beer quality control. Food Chem. 2010, 118, 566–574. 10.1016/j.foodchem.2009.05.022. [DOI] [Google Scholar]
- Iimure T.; Kimura T.; Araki S.; Kihara M.; Sato M.; Yamada S.; Shigyou T.; Sato K. Mutation analysis of barley malt protein Z4 and protein Z7 on beer foam stability. J. Agric. Food Chem. 2012, 60, 1548–1554. 10.1021/jf2044718. [DOI] [PubMed] [Google Scholar]
- Flodrová D.; Šalplachta J.; Benkovská D.; Bobálová J. Application of proteomics to hordein screening in the malting process. Eur. J. Mass Spectrom. 2012, 18, 323–332. 10.1255/ejms.1184. [DOI] [PubMed] [Google Scholar]
- Picariello G.; Mamone G.; Nitride C.; Addeo F.; Camarca A.; Vocca I.; Gianfrani C.; Ferranti P. Shotgun proteome analysis of beer and the immunogenic potential of beer polypeptides. J. Proteomics 2012, 75, 5872–5882. 10.1016/j.jprot.2012.07.038. [DOI] [PubMed] [Google Scholar]
- Flodrová D.; Benkovská D.; Laštovičková M.; Bobálová J. HPLC Bottom-Up MS-Based Proteomics for Mapping of Specific Proteins in Several European Spring Barley Varieties. J. Am. Soc. Brew. Chem. 2015, 73, 71–77. 10.1094/ASBCJ-2015-0107-01. [DOI] [Google Scholar]
- Colgrave M. L.; Byrne K.; Blundell M.; Howitt C. A. Identification of barley-specific peptide markers that persist in processed foods and are capable of detecting barley contamination by LC-MS/MS. J. Proteomics 2016, 147, 169–176. 10.1016/j.jprot.2016.03.045. [DOI] [PubMed] [Google Scholar]
- Guo B.; Luan H.; Lin S.; Lv C.; Zhang X.; Xu R. Comparative proteomic analysis of two barley cultivars (Hordeum vulgare L.) with contrasting grain protein content. Front. Plant Sci. 2016, 7, 1–11. 10.3389/fpls.2016.00542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason K. E.; Hilmer J. K.; Maaty W. S.; Reeves B. D.; Grieco P. A.; Bothner B.; Fischer A. M. Proteomic comparison of near-isogenic barley (Hordeum vulgare L.) germplasm differing in the allelic state of a major senescence QTL identifies numerous proteins involved in plant pathogen defense. Plant Physiol. Biochem. 2016, 109, 114–127. 10.1016/j.plaphy.2016.09.008. [DOI] [PubMed] [Google Scholar]
- Schmidt D.; Gaziola S. A.; Boaretto L. F.; Azevedo R. A. Proteomic analysis of mature barley grains from C-hordein antisense lines. Phytochemistry 2016, 125, 14–26. 10.1016/j.phytochem.2016.03.001. [DOI] [PubMed] [Google Scholar]
- Lee S. H.; Gupta R.; Kim Y. J.; Min C. W.; Kim S. W.; Seo W. D.; Kim S. T. Proteomic analysis indicates activation of reactive oxygen species signaling during seed germination and seedlings growth in Hordeum vulgare (barley). J. Proteins Proteomics 2016, 7, 269–277. [Google Scholar]
- Huang X.; Sontag-Strohm T.; Stoddard F. L.; Kato Y. Oxidation of proline decreases immunoreactivity and alters structure of barley prolamin. Food Chem. 2017, 214, 597–605. 10.1016/j.foodchem.2016.07.108. [DOI] [PubMed] [Google Scholar]
- Mahalingam R. Temporal Analyses of Barley Malting Stages Using Shotgun Proteomics. Proteomics 2018, 18, 1800025. 10.1002/pmic.201800025. [DOI] [PubMed] [Google Scholar]
- Kerr E. D.; Phung T. K.; Caboche C. H.; Fox G. P.; Platz G. J.; Schulz B. L. The intrinsic and regulated proteomes of barley seeds in response to fungal infection. Anal. Biochem. 2019, 580, 30–35. 10.1016/j.ab.2019.06.004. [DOI] [PubMed] [Google Scholar]
- Bi X.; Ye L.; Lau A.; Kok Y. J.; Zheng L.; Ng D.; Tan K.; Ow D.; Ananta E.; Vafiadi C.; Muller J. Proteomic profiling of barley spent grains guides enzymatic solubilization of the remaining proteins. Appl. Microbiol. Biotechnol. 2018, 102, 4159–4170. 10.1007/s00253-018-8886-8. [DOI] [PubMed] [Google Scholar]
- Wang W.-Q.; Jensen O. N.; Møller I. M.; Hebelstrup K. H.; Rogowska-Wrzesinska A. Evaluation of sample preparation methods for mass spectrometry-based proteomic analysis of barley leaves. Plant Methods 2018, 14, 72. 10.1186/s13007-018-0341-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr E. D.; Caboche C. H.; Schulz B. L. Posttranslational Modifications Drive Protein Stability to Control the Dynamic Beer Brewing Proteome. Mol. Cell Proteomics. 2019, 18, 1721–1731. 10.1074/mcp.RA119.001526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose U.; Broadbent J. A.; Byrne K.; Hasan S.; Howitt C. A.; Colgrave M. L. Optimisation of protein extraction for in-depth profiling of the cereal grain proteome. J. Proteomics 2019, 197, 23–33. 10.1016/j.jprot.2019.02.009. [DOI] [PubMed] [Google Scholar]
- Bose U.; Byrne K.; Howitt C. A.; Colgrave M. L. Targeted proteomics to monitor the extraction efficiency and levels of barley α-amylase trypsin inhibitors that are implicated in non-coeliac gluten sensitivity. J. Chromatogr A 2019, 1600, 55–64. 10.1016/j.chroma.2019.04.043. [DOI] [PubMed] [Google Scholar]
- Mahalingam R. Analysis of the Barley Malt Rootlet Proteome. Int. J. Mol. Sci. 2020, 21, 179. 10.3390/ijms21010179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo H.; Zhang Q.; Tucek M.; Zhang X.-Q.; Li C. Phenotypic and allelic variation for wort protein Z in Australian and Canadian barleys. J. Cereal Sci. 2020, 93, 102935. 10.1016/j.jcs.2020.102935. [DOI] [Google Scholar]
- Wang Y.; Sang Z.; Xu S.; Xu Q.; Zeng X.; Jabu D.; Yuan H. Comparative proteomics analysis of Tibetan hull-less barley under osmotic stress via data-independent acquisition mass spectrometry. GigaScience 2020, 9, 1–12. 10.1093/gigascience/giaa019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose U.; Broadbent J. A.; Byrne K.; Blundell M. J.; Howitt C. A.; Colgrave M. L. Proteome Analysis of Hordein-Null Barley Lines Reveals Storage Protein Synthesis and Compensation Mechanisms. J. Agric. Food Chem. 2020, 68, 5763–5775. 10.1021/acs.jafc.0c01410. [DOI] [PubMed] [Google Scholar]
- Bateman N. W.; Goulding S. P.; Shulman N. J.; Gadok A. K.; Szumlinski K. K.; MacCoss M. J.; Wu C. C. Maximizing peptide identification events in proteomic workflows using data-dependent acquisition (DDA). Mol. Cell Proteomics. 2014, 13, 329–338. 10.1074/mcp.M112.026500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting Y. S.; Egertson J. D.; Payne S. H.; Kim S.; MacLean B.; Käll L.; Aebersold R.; Smith R. D.; Noble W. S.; MacCoss M. J. Peptide-centric proteome analysis: An alternative strategy for the analysis of tandem mass spectrometry data. Mol. Cell Proteomics. 2015, 14, 2301–2307. 10.1074/mcp.O114.047035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnie C.; Svensson B. Feasibility study of a tissue-specific approach to barley proteome analysis: Aleurone layer, endosperm, embryo and single seeds. J. Cereal Sci. 2003, 38, 217–227. 10.1016/S0733-5210(03)00033-X. [DOI] [Google Scholar]
- Finnie C.; Steenholdt T.; Noguera O. R.; Knudsen S.; Larsen J.; Brinch-Pedersen H.; Holm P. B.; Olsen O.; Svensson B. Environmental and transgene expression effects on the barley seed proteome. Phytochemistry 2004, 65, 1619–1627. 10.1016/j.phytochem.2004.04.018. [DOI] [PubMed] [Google Scholar]
- Finnie C.; Bagge M.; Steenholdt T.; Østergaard O.; Bak-Jensen K. S.; Backes G.; Jensen A.; Giese H.; Larsen J.; Roepstorff P.; Svensson B. Integration of the barley genetic and seed proteome maps for chromosome 1H, 2H, 3H, 5H and 7H. Funct. Integr. Genomics 2009, 9, 135–143. 10.1007/s10142-008-0101-z. [DOI] [PubMed] [Google Scholar]
- Mayer K. F. X.; Waugh R.; Langridge P.; Close T. J.; Wise R. P.; Graner A.; Matsumoto T.; Sato K.; et al. A physical, genetic and functional sequence assembly of the barley genome. Nature 2012, 491, 711–716. 10.1038/nature11543. [DOI] [PubMed] [Google Scholar]
- Ankney J. A.; Muneer A.; Chen X. Relative and Absolute Quantitation in Mass Spectrometry-Based Proteomics. Annu. Rev. Anal. Chem. 2018, 11, 49–77. 10.1146/annurev-anchem-061516-045357. [DOI] [PubMed] [Google Scholar]
- Anand S.; Samuel M.; Ang C. S.; Keerthikumar S.; Mathivanan S.. Label-Based and Label-Free Strategies for Protein Quantitation. In Methods Mol. Biol.; Keerthikumar S.; Mathivanan S. V., Eds.; Humana Press: New York, 2017; pp 31–43. 10.1007/978-1-4939-6740-7_4. [DOI] [PubMed] [Google Scholar]
- Lyutvinskiy Y.; Yang H.; Rutishauser D.; Zubarev R. A. In silico instrumental response correction improves precision of label-free proteomics and accuracy of proteomics-based predictive models. Mol. Cell Proteomics. 2013, 12, 2324–2331. 10.1074/mcp.O112.023804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesur A.; Domon B. Advances in high-resolution accurate mass spectrometry application to targeted proteomics. Proteomics 2015, 15, 880–890. 10.1002/pmic.201400450. [DOI] [PubMed] [Google Scholar]
- Picotti P.; Aebersold R. Selected reaction monitoring-based proteomics: Workflows, potential, pitfalls and future directions. Nat. Methods 2012, 9, 555–566. 10.1038/nmeth.2015. [DOI] [PubMed] [Google Scholar]
- Schmidlin T.; Garrigues L.; Lane C. S.; Mulder T. C.; van Doorn S.; Post H.; de Graaf E. L.; Lemeer S.; Heck A. J.; Altelaar A. M. Assessment of SRM, MRM3, and DIA for the targeted analysis of phosphorylation dynamics in non-small cell lung cancer. Proteomics 2016, 16, 2193–2205. 10.1002/pmic.201500453. [DOI] [PubMed] [Google Scholar]
- Li H.; Han J.; Pan J.; Liu T.; Parker C. E.; Borchers C. H. Current trends in quantitative proteomics - an update. J. Mass Spectrom. 2017, 52, 319–341. 10.1002/jms.3932. [DOI] [PubMed] [Google Scholar]
- Gillet L. C.; Navarro P.; Tate S.; Röst H.; Selevsek N.; Reiter L.; Bonner R.; Aebersold R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: A new concept for consistent and accurate proteome analysis. Mol. Cell Proteomics 2012, 11, O111.016717. 10.1074/mcp.O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Q.; Yang L.; Luo J.; Guo L.; Wang Z.; Yang X.; Jin W.; Fang Y.; Ye J.; Shan B.; Zhang Y. SWATH enables precise label-free quantification on proteome scale. Proteomics 2015, 15, 1215–1223. 10.1002/pmic.201400270. [DOI] [PubMed] [Google Scholar]
- Ludwig C.; Gillet L.; Rosenberger G.; Amon S.; Collins B. C.; Aebersold R. Data-independent acquisition-based SWATH - MS for quantitative proteomics: a tutorial. Mol. Syst. Biol. 2018, 14, 1–23. 10.15252/msb.20178126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosová K.; Vítámvás P.; Prášil I. T. Proteomics of stress responses in wheat and barley-search for potential protein markers of stress tolerance. Front. Plant Sci. 2014, 5, 711. 10.3389/fpls.2014.00711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodziewicz P.; Chmielewska K.; Sawikowska A.; Marczak Ł.; Łuczak M.; Bednarek P.; Mikołajczak K.; Ogrodowicz P.; Kuczyńska A.; Krajewski P.; Stobiecki M. Identification of drought responsive proteins and related proteomic QTLs in barley. J. Exp. Bot. 2019, 70, 2823–2837. 10.1093/jxb/erz075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wójcik-Jagła M.; Rapacz M.; Dubas E.; Krzewska M.; Kopeć P.; Nowicka A.; Ostrowska A.; Malaga S.; Żur I. Candidate Genes for Freezing and Drought Tolerance Selected on the Basis of Proteome Analysis in Doubled Haploid Lines of Barley. Int. J. Mol. Sci. 2020, 21, 2062. 10.3390/ijms21062062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosová K.; Vítámvás P.; Prášil I. T. Wheat and barley dehydrins under cold, drought, and salinityi“ what can LEA-II proteins tell us about plant stress response?. Front. Plant Sci. 2014, 5, 343. 10.3389/fpls.2014.00343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendelboe-Nelson C.; Morris P. C. Proteins linked to drought tolerance revealed by DIGE analysis of drought resistant and susceptible barley varieties. Proteomics 2012, 12, 3374–3385. 10.1002/pmic.201200154. [DOI] [PubMed] [Google Scholar]
- Ashoub A.; Beckhaus T.; Berberich T.; Karas M.; Brüggemann W. Comparative analysis of barley leaf proteome as affected by drought stress. Planta 2013, 237, 771–781. 10.1007/s00425-012-1798-4. [DOI] [PubMed] [Google Scholar]
- Ghabooli M.; Khatabi B.; Ahmadi F. S.; Sepehri M.; Mirzaei M.; Amirkhani A.; Jorrin-Novo J. V.; Salekdeh G. H. Proteomics study reveals the molecular mechanisms underlying water stress tolerance induced by Piriformospora indica in barley. J. Proteomics 2013, 94, 289–301. 10.1016/j.jprot.2013.09.017. [DOI] [PubMed] [Google Scholar]
- Vítámvás P.; Urban M. O.; Škodáček Z.; Kosová K.; Pitelková I.; Vítámvás J.; Renaut J.; Prášil I. T. Quantitative analysis of proteome extracted from barley crowns grown under different drought conditions. Front. Plant Sci. 2015, 6, 479. 10.3389/fpls.2015.00479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chmielewska K.; Rodziewicz P.ł; Swarcewicz B.; Sawikowska A.; Krajewski P.ł; Marczak Łu.; Ciesiołka D.; Kuczynska A.; Mikołajczak K.; Ogrodowicz P.; Krystkowiak K.; Surma M.; Adamski T.; Bednarek P.ł; Stobiecki M. Analysis of Drought-Induced Proteomic and Metabolomic Changes in Barley (Hordeum vulgare L.) Leaves and Roots Unravels Some Aspects of Biochemical Mechanisms Involved in Drought Tolerance. Front. Plant Sci. 2016, 7, 1108. 10.3389/fpls.2016.01108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hlaváčková I.; Vítámvás P.; Šantrůček J.; Kosová K.; Zelenková S.; Prášil I. T.; Ovesná J.; Hynek R.; Kodíček M. Proteins Involved in Distinct Phases of Cold Hardening Process in Frost Resistant Winter Barley (Hordeum vulgare L.) cv Luxor. Int. J. Mol. Sci. 2013, 14, 8000–8024. 10.3390/ijms14048000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo V.; Valizadeh R.; Michaletti A.; Toorchi M. Proteomic and Physiological Response of Spring Barley Leaves to Cold Stress. Int. J. Plant Biol. Res. 2017, 5, 1061. [Google Scholar]
- Gołebiowska-Pikania G.; Kopec P.ła.; Surowka E.; Krzewska M.; Dubas E.; Nowicka A.; Rapacz M.; Wojcik-Jagła M.; Malaga S.; Zur I. Changes in protein abundance and activity involved in freezing tolerance acquisition in winter barley (Hordeum vulgare L.). J. Proteomics 2017, 169, 58–72. 10.1016/j.jprot.2017.08.019. [DOI] [PubMed] [Google Scholar]
- Sugimoto M.; Takeda K. Proteomic Analysis of Specific Proteins in the Root of Salt-Tolerant Barley. Biosci., Biotechnol., Biochem. 2009, 73, 2762–2765. 10.1271/bbb.90456. [DOI] [PubMed] [Google Scholar]
- Witzel K.; Weidner A.; Surabhi G.-K.; Börner A.; Mock H.-P. Salt stress-induced alterations in the root proteome of barley genotypes with contrasting response towards salinity. J. Exp. Bot. 2009, 60, 3545–3557. 10.1093/jxb/erp198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasoulnia A.; Bihamta M. R.; Peyghambari S. A.; Alizadeh H.; Rahnama A. Proteomic response of barley leaves to salinity. Mol. Biol. Rep. 2011, 38, 5055–5063. 10.1007/s11033-010-0651-8. [DOI] [PubMed] [Google Scholar]
- Fatehi F.; Hosseinzadeh A.; Alizadeh H.; Brimavandi T.; Struik P. C. The proteome response of salt-resistant and salt-sensitive barley genotypes to long-term salinity stress. Mol. Biol. Rep. 2012, 39, 6387–6397. 10.1007/s11033-012-1460-z. [DOI] [PubMed] [Google Scholar]
- Witzel K.; Matros A.; Strickert M.; Kaspar S.; Peukert M.; Muhling K. H.; Borner A.; Mock H.-P. Salinity Stress in Roots of Contrasting Barley Genotypes Reveals Time-Distinct and Genotype-Specific Patterns for Defined Proteins. Mol. Plant 2014, 7, 336–355. 10.1093/mp/sst063. [DOI] [PubMed] [Google Scholar]
- Mostek A.; Börner A.; Badowiec A.; Weidner S. Alterations in root proteome of salt-sensitive and tolerant barley lines under salt stress conditions. J. Plant Physiol. 2015, 174, 166–176. 10.1016/j.jplph.2014.08.020. [DOI] [PubMed] [Google Scholar]
- Zhu J.; Fan Y.; Li C.; Shabala S.; Zhao C.; Hong Y.; Lv C.; Guo B.; Xu R.; Zhou M. Candidate genes for salinity tolerance in barley revealed by RNA-seq analysis of near-isogenic lines. Plant Growth Regul. 2020, 92, 571–582. 10.1007/s10725-020-00662-9. [DOI] [Google Scholar]
- Kausar R.; Arshad M.; Shahzad A.; Komatsu S. Proteomics analysis of sensitive and tolerant barley genotypes under drought stress. Amino Acids 2013, 44, 345–359. 10.1007/s00726-012-1338-3. [DOI] [PubMed] [Google Scholar]
- Rollins J. A.; Habte E.; Templer S. E.; Colby T.; Schmidt J.; Von Korff M. Leaf proteome alterations in the context of physiological and morphological responses to drought and heat stress in barley (Hordeum vulgare L.). J. Exp. Bot. 2013, 64, 3201–3212. 10.1093/jxb/ert158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaffari M. R.; Mirzaei M.; Ghabooli M.; Khatabi B.; Wu Y.; Zabet-Moghaddam M.; Mohammadi-Nejad G.; Haynes P. A.; Hajirezaei M. R.; Sepehri M.; Salekdeh G. H. Root endophytic fungus Piriformospora indica improves drought stress adaptation in barley by metabolic and proteomic reprogramming. Environ. Exp. Bot. 2019, 157, 197–210. 10.1016/j.envexpbot.2018.10.002. [DOI] [Google Scholar]
- Yang F.; Jensen J. D.; Spliid N. H.; Svensson B.; Jacobsen S.; Jørgensen L. N.; Jørgensen H. J.; Collinge D. B.; Finnie C. Investigation of the effect of nitrogen on severity of Fusarium Head Blight in barley. J. Proteomics 2010, 73, 743–752. 10.1016/j.jprot.2009.10.010. [DOI] [PubMed] [Google Scholar]
- Yang F.; Jensen J. D.; Svensson B.; Jørgensen H. J. L.; Collinge D. B.; Finnie C. Analysis of early events in the interaction between Fusarium graminearum and the susceptible barley (Hordeum vulgare) cultivar Scarlett. Proteomics 2010, 10, 3748–3755. 10.1002/pmic.201000243. [DOI] [PubMed] [Google Scholar]
- Eggert K.; Pawelzik E. Proteome analysis of Fusarium head blight in grains of naked barley (Hordeum vulgare subsp. nudum). Proteomics 2011, 11, 972–985. 10.1002/pmic.201000322. [DOI] [PubMed] [Google Scholar]
- Hassett K.; Ellwood S. R.; Zulak K. G.; Muria-Gonzalez M. J. Analysis of apoplastic proteins expressed during net form net blotch of barley. J. Plant Dis. Prot. 2020, 127, 683–694. 10.1007/s41348-020-00318-w. [DOI] [Google Scholar]
- Godfrey D.; Zhang Z.; Saalbach G.; Thordal-Christensen H. A proteomics study of barley powdery mildew haustoria. Proteomics 2009, 9, 3222–3232. 10.1002/pmic.200800645. [DOI] [PubMed] [Google Scholar]
- Lambertucci S.; Orman K. M.; Das Gupta S.; Fisher J. P.; Gazal S.; Williamson R. J.; Cramer R.; Bindschedler L. V. Analysis of Barley Leaf Epidermis and Extrahaustorial Proteomes During Powdery Mildew Infection Reveals That the PR5 Thaumatin-Like Protein TLP5 Is Required for Susceptibility Towards Blumeria graminis f. sp. hordei. Front. Plant Sci. 2019, 10, 1138. 10.3389/fpls.2019.01138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Fernandez R.; Jorrin-Novo J. V. Contribution of proteomics to the study of plant pathogenic fungi. J. Proteome Res. 2012, 11, 3–16. 10.1021/pr200873p. [DOI] [PubMed] [Google Scholar]
- Kosová K.; Vítámvás P.; Klíma M.; Prášil I. T. Breeding drought-resistant crops: G × E interactions, proteomics and pQTLS. J. Exp. Bot. 2019, 70, 2605–2608. 10.1093/jxb/erz116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma J. K.; Sihmar M.; Santal A. R.; Singh N. P. Impact assessment of major abiotic stresses on the proteome profiling of some important crop plants: a current update. Biotechnol. Genet. Eng. Rev. 2019, 35, 126–160. 10.1080/02648725.2019.1657682. [DOI] [PubMed] [Google Scholar]
- Gupta P. K.; Kulwal P. L.; Mir R. R.. QTL Mapping: Methodology and Applications in Cereal Breeding. In Cereal Genomics II.; Gupta P., Varshney R., Eds.; Springer: Dordrecht, Netherlands, 2013; pp 5–318. 10.1007/978-94-007-6401-9_11. [DOI] [Google Scholar]
- Raorane M. L.; Pabuayon I. M.; Miro B.; Kalladan R.; Reza-Hajirezai M.; Oane R. H.; Kumar A.; Sreenivasulu N.; Henry A.; Kohli A. Variation in primary metabolites in parental and near-isogenic lines of the QTL qDTY 12.1: altered roots and flag leaves but similar spikelets of rice under drought. Mol. Breed. 2015, 35, 138. 10.1007/s11032-015-0322-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salekdeh G. H.; Komatsu S. Crop proteomics: Aim at sustainable agriculture of tomorrow. Proteomics 2007, 7, 2976–2996. 10.1002/pmic.200700181. [DOI] [PubMed] [Google Scholar]
- Li J. Z.; Huang X. Q.; Heinrichs F.; Ganal M. W.; Röder M. S. Analysis of QTLs for yield, yield components, and malting quality in a BC3-DH population of spring barley. Theor. Appl. Genet. 2005, 110, 356–363. 10.1007/s00122-004-1847-x. [DOI] [PubMed] [Google Scholar]
- Witzel K.; Pietsch C.; Strickert M.; Matros A.; Röder M. S.; Weschke W.; Wobus U.; Mock H. P. Mapping of quantitative trait loci associated with protein expression variation in barley grains. Mol. Breed. 2011, 27, 301–314. 10.1007/s11032-010-9432-2. [DOI] [Google Scholar]
- Korte R.; Brockmeyer J. Novel mass spectrometry approaches in food proteomics. TrAC, Trends Anal. Chem. 2017, 96, 99–106. 10.1016/j.trac.2017.07.010. [DOI] [Google Scholar]
- Bogdan P.; Kordialik-Bogacka E. Alternatives to malt in brewing. Trends Food Sci. Technol. 2017, 65, 1–9. 10.1016/j.tifs.2017.05.001. [DOI] [Google Scholar]
- Gustavsson J.; Cederberg C.; Sonesson U.. Global Food Losses and Food Waste. In Global Food Losses and Food Waste; Save Food Congress: Düsseldorf, 2011. [Google Scholar]
- Pedreschi R.; Lurie S.; Hertog M.; Nicolaï B.; Mes J.; Woltering E. Post-harvest proteomics and food security. Proteomics 2013, 13, 1772–1783. 10.1002/pmic.201200387. [DOI] [PubMed] [Google Scholar]
- Samperi R.; Capriotti A. L.; Cavaliere C.; Colapicchioni V.; Chiozzi R. Z.; Laganà A.. Food Proteins and Peptides. In: Comprehensive Analytical Chemistry; Picó Y., Eds.; Elsevier: Amsterdam, Netherlands, 2015; Chapter 6, pp 309–357. 10.1007/978-94-007-6401-9_11. [DOI] [Google Scholar]
- Kosová K.; Vítámvás P.; Prášil I. T.; Renaut J. Plant proteome changes under abiotic stress — Contribution of proteomics studies to understanding plant stress response. J. Proteomics 2011, 74, 1301–1322. 10.1016/j.jprot.2011.02.006. [DOI] [PubMed] [Google Scholar]
- Sang S. Biomarkers of Whole Grain Intake. J. Agric. Food Chem. 2018, 66, 10347–10352. 10.1021/acs.jafc.8b04110. [DOI] [PubMed] [Google Scholar]
- Agrawal G. K.; Sarkar A.; Righetti P. G.; Pedreschi R.; Carpentier S.; Wang T.; Barkla B. J.; Kohli A.; Ndimba B. K.; Bykova N. V.; Rampitsch C.; Zolla L.; Rafudeen M. S.; Cramer R.; Bindschedler L. V.; Tsakirpaloglou N.; Ndimba R. J.; Farrant J. M.; Renaut J.; Job D.; Kikuchi S.; Rakwal R. A decade of plant proteomics and mass spectrometry: Translation of technical advancements to food security and safety issues. Mass Spectrom. Rev. 2013, 32, 335–365. 10.1002/mas.21365. [DOI] [PubMed] [Google Scholar]
- Stewart D.; Shepherd L. V. T.; Hall R. D.; Fraser P. D. Crops and Tasty, Nutritious Food - How Can Metabolomics Help?. Annu. plant rev online. 2018, 181–217. 10.1002/9781119312994.apr0467. [DOI] [Google Scholar]
- Mora L.; Toldrá F.. Proteomics and Peptidomics for Food Safety. In Comprehensive Foodomics; Cifuentes A., Eds.; Elsevier: Amsterdam, Netherlands, 2021; pp 149–156. 10.1016/B978-0-08-100596-5.22843-6. [DOI] [Google Scholar]
- Fox G. The Brewing Industry and the Opportunities for Real-Time Quality Analysis Using Infrared Spectroscopy. Appl. Sci. 2020, 10, 616. 10.3390/app10020616. [DOI] [Google Scholar]
- Juhász A.; Colgrave M. L.; Howitt C. A. Developing gluten-free cereals and the role of proteomics in product safety. J. Cereal Sci. 2020, 93, 102932. 10.1016/j.jcs.2020.102932. [DOI] [Google Scholar]
- Popping B. Why allergen detection by ELISA is insufficient to protect allergic consumers. Clin. Transl. Allergy 2013, 3, O9. 10.1186/2045-7022-3-S3-O9. [DOI] [Google Scholar]
- Stelk T.; Niemann L.; Lambrecht D. M.; Baumert J. L.; Taylor S. L. False Positive Detection of Peanut Residue in Liquid Caramel Coloring Using Commercial ELISA Kits. J. Food Sci. 2013, 78, T1091–T1093. 10.1111/1750-3841.12146. [DOI] [PubMed] [Google Scholar]
- Marzano V.; Tilocca B.; Fiocchi A. G.; Vernocchi P.; Mortera S. L.; Urbani A.; Roncada P.; Putignani L. Perusal of food allergens analysis by mass spectrometry-based proteomics. J. Proteomics 2020, 215, 103636. 10.1016/j.jprot.2020.103636. [DOI] [PubMed] [Google Scholar]
- Tanner G. J.; Colgrave M. L.; Blundell M. J.; Goswami H. P.; Howitt C. A. Measuring Hordein (Gluten) in Beer - A Comparison of ELISA and Mass Spectrometry. PLoS One 2013, 8, e56452. 10.1371/journal.pone.0056452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colgrave M. L.; Goswami H.; Blundell M.; Howitt C. A.; Tanner G. J. Using mass spectrometry to detect hydrolysed gluten in beer that is responsible for false negatives by ELISA. J. Chromatogr A 2014, 1370, 105–114. 10.1016/j.chroma.2014.10.033. [DOI] [PubMed] [Google Scholar]
- Lock S. Gluten Detection and Speciation by Liquid Chromatography Mass Spectrometry (LC-MS/MS). Foods 2014, 3, 13–29. 10.3390/foods3010013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colgrave M. L.; Byrne K.; Blundell M.; Heidelberger S.; Lane C. S.; Tanner G. J.; Howitt C. A. Comparing Multiple Reaction Monitoring and Sequential Window Acquisition of All Theoretical Mass Spectra for the Relative Quantification of Barley Gluten in Selectively Bred Barley Lines. Anal. Chem. 2016, 88, 9127–9135. 10.1021/acs.analchem.6b02108. [DOI] [PubMed] [Google Scholar]
- Fischer M.; Creydt M. Food Profiling—Analytical Strategies for Food Authentication. J. Agric. Food Chem. 2020, 68, 14321–14322. 10.1021/acs.jafc.0c03285. [DOI] [PubMed] [Google Scholar]
- Tanner G. J.; Blundell M. J.; Colgrave M. L.; Howitt C. A. Creation of the first ultra-low gluten barley (Hordeum vulgare L.) for coeliac and gluten-intolerant populations. Plant Biotechnol. J. 2016, 14, 1139–1150. 10.1111/pbi.12482. [DOI] [PMC free article] [PubMed] [Google Scholar]