

Abstract
Sirtuin 6 (SIRT6) is an NAD+-dependent protein deacylase and mono-ADP-ribosyltransferase of the sirtuin family with a wide substrate specificity. In vitro and in vivo studies have indicated that SIRT6 overexpression or activation has beneficial effects for cellular processes such as DNA repair, metabolic regulation, and aging. On the other hand, SIRT6 has contrasting roles in cancer, acting either as a tumor suppressor or promoter in a context-specific manner. Given its central role in cellular homeostasis, SIRT6 has emerged as a promising target for the development of small-molecule activators and inhibitors possessing a therapeutic potential in diseases ranging from cancer to age-related disorders. Moreover, specific modulators allow the molecular details of SIRT6 activity to be scrutinized and further validate the enzyme as a pharmacological target. In this Perspective, we summarize the current knowledge about SIRT6 pharmacology and medicinal chemistry and describe the features of the activators and inhibitors identified so far.
Introduction
The sirtuin family is a class of enzymes that employs NAD+ as cofactor.1 Although initially classified as class III HDACs, sirtuins (SIRTs) are capable of catalyzing different reactions and possess a wide range of substrates far beyond histones.2 Among them, sirtuin 6 (SIRT6) is a pivotal chromatin homeostasis modulator that deacetylates both histone and nonhistone proteins, including DNA repair factors and glucose homeostasis regulators. In addition, SIRT6 promotes the deacylation of long-chain fatty-acid groups and catalyzes the mono-ADP-ribosylation of chromatin silencing DNA repair proteins,3 including self-mono-ADP-ribosylation.4 Through its enzymatic activity, SIRT6 facilitates the removal of acyl groups from the ε-amino group of lysines and transfers ADP–ribose moieties to lysine and arginine residues of protein substrates (Figure 1).
Figure 1.

(A) Deacetylation/deacylation reaction catalyzed by SIRT6. The acetyl/acyl group is transferred to an NAD+ acceptor, coupled with removal of nicotinamide. (B) Mono-ADP-ribosylation reaction. In this case, ADP–ribose is transferred onto the ε-amino group of lysine from an NAD+ donor. Nicotinamide and ADP–ribosyl protein are the products.
Given the requirement of NAD+ for their activity, SIRTs have been regarded as pivotal proteins connecting metabolism to cellular physiology.5 SIRT6, being a nuclear member of this family, tightly regulates DNA repair and genome maintenance and has a pivotal role in glucose and lipid metabolism. These activities are tightly related to the central roles that SIRT6 has in aging, stem cell differentiation, and tumorigenesis.
Loss-of-function studies performed in mouse models indicated the crucial roles that SIRT6 plays for organism wellbeing. Indeed, SIRT6-deficient mice displayed alteration of glycolysis and genomic instability, ultimately leading to premature aging and shortened lifespan.6−8 In addition, SIRT6 deletion was associated with increased tumor aggressiveness, and later studies in human cancers identified mutations impairing SIRT6 activity.9 Conversely, recent studies also described SIRT6 as a tumor promoter, hence highlighting the context-dependent role of this enzyme in cellular homeostasis.10,11
Homozygous mutations leading to SIRT6 loss of activity in humans caused fetal loss associated with muscle and brain developmental deficiencies.12 Similarly, cynomolgus monkeys bearing a SIRT6 knockout obtained through CRISPR-Cas9 suggested a primary role of SIRT6 for primates’ fetal development.13
Conversely, SIRT6 overexpression in male mice determined an increased lifespan, and another study indicated that SIRT6 levels increase in cultured cells, mice, and rats under conditions of caloric restriction, a dietary program that protects against many aging-related changes.14
SIRT6 has been initially described as an HDAC, having histone H3 as a substrate and catalyzing the deacetylation of lysines Lys9, Lys18, and Lys56.15−18 Histone deacetylation is associated with compaction of chromatin and consequent transcriptional repression as well as DNA-damage response. Nevertheless, recent reports indicated that SIRT6 deacetylase catalytic activity is 100 to 1000 times lower compared to the most active SIRTs.19 In addition, the deacylase efficiency of SIRT6 has been shown to be higher compared to deacetylation, which can be in turn activated by small molecules, including free fatty acids (FFAs).20,21 For instance, in vitro demyristoylation activity is roughly 300 times higher than deacetylation.22 Nonetheless, the majority of studies on SIRT6 indicate deacetylation as the main reaction responsible for its cellular functions, while deacylation has only been reported in the case of TNF-α22 and R-Ras223 so far. These features, along with the ability of SIRT6 to catalyze mono-ADP-ribosylation, depict a complicated picture of SIRT6 biological functions. Many details connecting the biochemical activity of SIRT6 and the observed phenotypes in both physiological and pathological conditions are still missing; hence, the main goals for future investigations consist of uncovering new SIRT6 substrates and elucidating its molecular interactors.
An important strategy for further elucidation of SIRT6 activity is played by chemical probes that through activation or inhibition of SIRT6 enzymatic activity may help to clarify the connection between SIRT6 function and the observed phenotypes. In addition, given the central role that SIRT6 plays in processes such as DNA repair, metabolism, aging, and tumorigenesis, small-molecule modulators could represent potential weapons for SIRT6-targeted treatment of diseases such as diabetes, obesity, cancer, and neurodegeneration.
SIRT6 Structure and Catalytic Mechanism
A key role in the investigation of SIRT6 function is played by the elucidation of its structural features. A decade has passed since the first structure of SIRT6 in complex with ADP–ribose has been solved,19 followed by the structure of SIRT6 bound to both ADP–ribose and myristoylated H3K9 peptide (Figure 2A).22 SIRT6 has two globular domains: a large Rossmann fold and a small zinc-binding region. The Rossman fold consists of six β-sheets sandwiched between four α-helices on one side and two α-helices on the other side. This domain contains the NAD+ binding site as well as a hydrophobic pocket to accommodate the acyl chains of SIRT6 substrates. Differently from other SIRTs, SIRT6 has been reported to bind NAD+ in the absence of the acylated substrate.19 This feature is explained by the structural differences in the NAD+-binding region. Indeed, SIRT6 lacks the cofactor-binding loop24−26 but presents a helix (α3) that keeps its ordered structure even in the absence of the acylated peptide.19
Figure 2.

(A) Structure of SIRT6 in complex with H3K9-Myr (green) and ADP–ribose (yellow) bound (PDB ID: 3ZG6). (B) Focus on the hydrophobic pocket in the Rossman fold accommodating the myristoyl chain. (C) Catalytic mechanism of SIRT6-mediated deacylation.
The hydrophobic channel is shaped by residues belonging to different loops engaging hydrophobic interactions with the fatty acyl chain, as shown by Lin and colleagues (Figure 2B).22 In the presence of a myristoylated peptide, the N-terminus of SIRT6, which covers part of the hydrophobic pocket, becomes structured. The structural ordering induced by the myristoylated peptide may facilitate the catalytic process and explain the higher catalytic efficiency of long-fatty-chain deacylation compared to deacetylation.22 It can also account for the increased deacetylation activity in the presence of FFA and small molecules. Nonetheless, there are no SIRT6 structures bound to an acetylated substrate; hence, this hypothesis is yet to be proven.
Finally, the zinc-binding motif is only structural and does not participate in the catalysis; this feature is shared by all SIRTs and differentiates them from class I, II, and IV HDACs possessing in the active site a zinc ion essential for catalysis.27
Notably, the in vitro deacetylase activity of SIRT6 is much lower than that of other SIRTs, probably because of SIRT6 peculiar structural features. Nevertheless, several cell-based assays suggested that the deacetylation is the most prominent activity of SIRT6, and H3K9 was indicated as the SIRT6 main substrate.19 This is explained by the fact that SIRT6 preferably associates with histones when they are packaged in nucleosomes. Conversely, SIRT1 exhibits higher deacetylation activity toward unpacked histones. Thus, interaction with packaged histones may trigger a transition toward an active SIRT6 conformation. Hence, SIRT6 activity depends on histone packaging, thereby being lower when tested in vitro using free histones.28 It is therefore possible that in the case of other substrates SIRT6 deacetylase activity is affected by the presence of interactors contributing to the formation of multiprotein complexes.
As mentioned above, SIRT6 deacylase activity has been reported to be higher than deacetylation.22 However, a functional role for SIRT6-mediated deacylation has only been described in the case of TNF-α22 and R-Ras2.23 Importantly, histone deacylation has only been indicated in preliminary in vitro studies. In the same study in which TNF-α deacylation was described for the first time, Jiang et al. also showed that SIRT6 can catalyze the removal of octanoyl, myristoyl, and palmitoyl groups from H3K9 and of myristoyl from H2BK12 using synthetic histone peptides as substrates.22 A subsequent analysis was performed using a chemical biology approach, in which the SIRT6-acylated substrate was the octenoylated H3 incorporated in the nucleosome. The terminal olefin selectively could react with a tetrazine probe allowing nucleosome labeling. This study suggested that SIRT6 catalyzes the efficient deacylation of H3K9, H3K18, and H3K27 while having low activity toward H3K4 and H3K23.29 Nonetheless, the precise physiological role of histones’ acylation/deacylation equilibria need further elucidation.
The SIRT6 residue Gly60 is pivotal for deacetylation; indeed, the G60A mutant has its deacetylase activity abolished while retaining deacylase activity. Mechanistically, Gly60 is crucial for NAD+ binding, and fatty-acylated substrates, but not acetylated ones, are able to reverse the conformational change induced by its mutation.30 This is in line with the above-mentioned activation of SIRT6-mediated deacetylation in the presence of FFA.20,21
Beyond deacetylation and deacylation, SIRT6 also catalyzes mono-ADP-ribosylation. This was initially demonstrated using mouse SIRT6 (mSIRT6), which was shown to self-mono-ADP-ribosylate, and suggested that SIRT6 may self-modulate its activity through this post-translational modification (PTM).4 Further studies indicated that SIRT6 mono-ADP-ribosylates different factors, including the poly(ADP–ribose) polymerase 1 (PARP1),31 the transcription factor KAP1,32 the BAF chromatin remodeling complex subunit BAF170,33 and the histone lysine demethylase KDM2A.34
Given its peculiar structure, SIRT6 can bind NAD+ before the acylated protein, and following binding of both substrates, a slow conformational change allows the formation of the alkylimidate intermediate (Figure 2C, step I). The rate of this step is enhanced by FFA and small molecules and has been shown to be faster during demyristoylation.21 This reaction consists of a nucleophilic attack of the acyl carbonyl on C1′ of nicotinamide-bound ribose and consequent formation of a C1′-O-alkylimidate intermediate, along with release of nicotinamide. Subsequently, His133 acts as a general base on ribose C3′ thereby triggering an intramolecular nucleophilic attack of the C2′ hydroxyl toward the C1′-O-alkylimidate, thus yielding the C1′-C2′ cyclic intermediate (Figure 2C, step II). A conserved water molecule then catalyzes the hydrolysis of the cyclic intermediate, affording the tetrahedral intermediate (Figure 2C, step III). The imino group then attacks His133, which is now positively charged, thus gaining a proton and resulting in the cleavage of the C–N bond. This leads to the final products O-acyl-ADP–ribose and deacylated lysine (Figure 2C, step IV), which are then released from the enzyme (Figure 2C, step V).3,21
Biological Functions and Disease Relevance of SIRT6
Genome Maintenance
Initial observations on SIRT6-knockout mice revealed hypersensitivity to DNA-damaging agents and genomic instability, indicating aberrant functioning of DNA double-strand break (DSB) repair and base excision repair (BER) mechanisms.35 Following these early studies, a growing body of reports indicated that SIRT6 associates with damaged chromatin sites36 and coordinates the recruitment of different factors to initiate DNA-damage repair (DDR).36−39 In particular, Onn and colleagues suggested that a SIRT6 dimer is able to directly bind to open-ended DSBs, whereby each monomer interacts with one DNA strand.40
SIRT6-mediated recruitment of repair factors is triggered by the deacetylation of nucleosomes. For instance, H3K56 deacetylation facilitates the recruitment of the ISWI-chromatin remodeller SNF2H, which increases chromatin accessibility, thereby promoting the binding to damaged DNA of repair factors such as BRCA1, RPA, and 53BP1.37 Remarkably, a recent study indicated the crucial role of the SIRT6-SNF2H dimer at the neurological level. Indeed, animals lacking SIRT6 in the brain showed AD symptoms, including increased levels of hyperphosphorylated Tau protein.41
Recent studies indicated that SIRT6 is phosphorylated on Ser10 by c-Jun N-terminal kinase (JNK) under oxidative stress conditions. This PTM enables the binding of SIRT6 to DSBs and subsequent recruitment of PARP1, which mediates nonhomologous end-joining (NHEJ) and homology-directed repair (HDR). PARP1 is also mono-ADP-ribosylated by SIRT6 on Lys521,31 a modification that is required for PARP1 activity in BER. In addition, mono-ADP ribosylation of the histone lysine demethylase KDM2A has been shown to augment H3K36me2 level at DNA-damage sites, thereby promoting H3K9 trimethylation and consequent recruitment of NHEJ factors to DSBs (Figure 3).34
Figure 3.
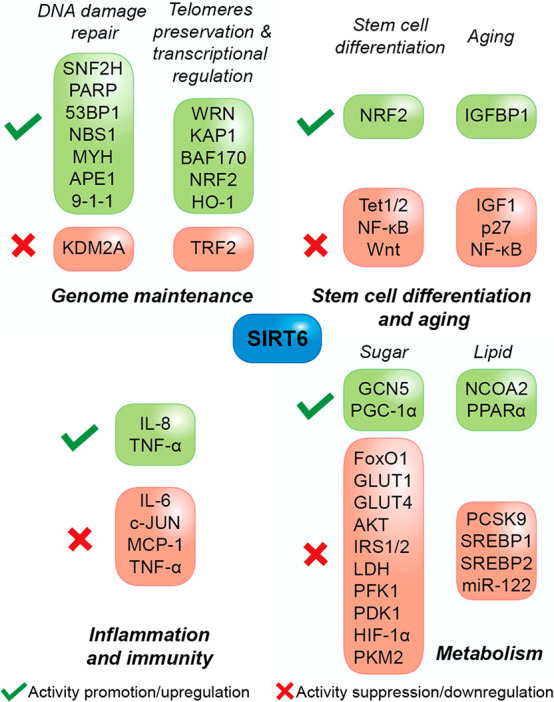
Roles of SIRT6 in cellular and organism homeostasis. The figure indicates the main proteins involved in the most important processes regulated by SIRT6, except cancer.
As anticipated above, the role of SIRT6 in DNA repair has implications in pathology and therapy, particularly in neurodegeneration as the frequency and precision of repair mechanisms declines with age. Accordingly, SIRT6 levels have been shown to decrease with cellular senescence and its overexpression is able to stimulate HDR through the PARP1 pathway.
SIRT6 mediates DNA repair also through BER as indicated by reports showing that overexpression of SIRT6 increases 2-fold the efficiency of this DNA repair mechanism.42 Moreover, under oxidative DNA damage, SIRT6 interacts with and stimulates MYH DNA glycosylase and the endonuclease APE1, two enzymes involved in BER. The process is aided by the checkpoint clamp Rad9-Rad1-Hus1 (9-1-1), which forms a multiprotein complex with MYH, APE1, and SIRT6 that is pivotal for whole genome and telomere stability in mammalian cells (Figure 3).43
SIRT6 is also responsible for telomeric preservation in mammalian cells through deacetylation of H3K9 and H3K56 in telomeric regions.15,16 SIRT6-mediated H3K9 deacetylation determines chromatin conformational changes that allow the binding of the Werner syndrome ATP-dependent helicase (WRN), the DNA-processing factor that is mutated in the Werner syndrome, a premature aging disorder.15 Furthermore, SIRT6 interacts with telomere repeat binding factor 2 (TRF2), a pivotal regulator of telomere homeostasis and DNA-damage response, and their interaction is increased during DNA-damage events, in a PARP1-dependent manner. SIRT6 catalyzes TRF2 deacetylation, triggering its ubiquitination finally leading to its proteolysis. In line with this, the levels of the two proteins were negatively correlated in a cohort of colorectal cancer (CRC) patients. These results indicate a regulation mechanism of TRF2 levels in response to DNA damage and oncogenesis, whereby SIRT6-induced degradation of TRF2 impairs DNA-damage repair leading to cancer cell death.44
Apart from its roles in DNA repair and telomeres, SIRT6 is mainly responsible for transcriptional silencing. SIRT6-mediated H3K9 and H3K56 deacetylation contributes to the repression of proteins involved in lipid metabolism, inflammation (NF-κB-dependent proteins), as well as c-Myc targets, ribosomal proteins, and early developmental genes.8,45−47 Furthermore, SIRT6 promotes the silencing of long interspersed element-1 (LINE-1) retrotransposable elements (RTEs), a class of retrotransposons linked to mutagenesis and genomic instability.48 SIRT6 facilitates heterochromatin packaging of these RTEs, hence suppressing transposition. Notably, recent findings indicate that this function is directed by SIRT6-mediated mono-ADP-ribosylation of the transcriptional corepressor KAP1 (Figure 3).32
Moreover, SIRT6 is responsible for pericentric chromatin silencing through H3K18 deacetylation, and this function is mediated by KAP1, although in a different manner compared to LINE-1 RTEs. Evidence indicates that H3K18 deacetylation is necessary for KAP1 retention at pericentric satellite repeats and consequent transcriptional repression. Conversely, SIRT6 knockout and consequent H3K18 hyperacetylation causes KAP1 detachment and transcriptional derepression.18 SIRT6-deficient cells display accumulation of pathological pericentric transcripts causing genomic instability, mitotic errors, and cellular senescence, defects associated with aging and tumorigenesis.
As previously mentioned, SIRT6 has also been indicated to catalyze the ADP-ribosylation of the BAF chromatin remodeling complex subunit BAF170 at Lys312. This modification enhances the transcription upon oxidative stress of a subset of the nuclear factor erythroid 2-related factor (NRF2) responsive genes such as HO-1.33
Stem Cell Differentiation
Embryonic stem cell (ESC) pluripotency maintenance is guaranteed by the expression of Oct4, Sox2, and Nanog genes, which are lost upon differentiation.49,50 Recent studies indicated that SIRT6 mediates ESC differentiation through H3K9 and H3K56 deacetylation, which determines the repression of ten-eleven translocation methylcytosine dioxygenase 1 and 2 (TET1 and TET2). These enzymes convert 5-methylcytosine (5-mC) into 5-hydroxymethylcytosine (5-hmC) and regulate cell lineage choice during ESC differentiation (Figure 3).51
SIRT6 has also a role in mesenchymal stem cell (MSC) and hematopoietic stem cell (HSC) homeostasis through H3K56 deacetylation. Through this activity, SIRT6 seems to coactivate transcription of NRF2 target genes and protect MSCs from oxidative stress.52 In addition, SIRT6-mediated H3K56 deacetylation was shown to suppress the NF-κB signaling pathway, thereby promoting osteogenic differentiation and new bone formation and repair in rats.53 In case of HSCs, SIRT6 interacts with LEF1 and, through H3K56 deacylation, corepresses Wnt target genes, thus blocking aberrant HSC proliferation.54
In addition, SIRT6 expression is associated with higher reprogramming efficiency of induced pluripotent stem cells (iPSCs).55 Given the increasing evidence supporting iPSC-based therapies in the context of neurodegenerative diseases, SIRT6 activation may represent a useful strategy to increase the success rate of these treatments.
Aging
Given its roles in genomic maintenance and stem cell homeostasis, SIRT6 has an indirect influence on aging, a process tightly related to DNA damage, telomere maintenance, and differentiation. In addition, SIRT6 plays a direct role in senescence and aging-related conditions through its activity on specific substrates at both the cytoplasmic and nuclear level.
SIRT6 overexpression determined a 15% increase of male mice life expectancy along with reduction of insulin-like growth factor 1 (IGF1) signaling through increased levels of IGF-binding protein 1 (IGFBP1) and altered phosphorylation of proteins involved in IGF1 downstream signaling.7 Moreover, SIRT6 deacetylates the cyclin-dependent kinase inhibitor p27, a factor involved in cellular senescence, hence promoting its proteasome-dependent degradation.56 Similarly, the transcription factor NF-κB, which induces the expression of aging-related genes, is negatively regulated by SIRT6 through a double mechanism. Indeed, at the transcriptional level, SIRT6 deacetylates H3K9 at NF-κB promoters, thereby reducing the expression of its components, while at the protein level, SIRT6 catalyzed deacetylation of the NF-κB p65 subunit (RelA) at Lys310 results in NF-κB nuclear export (Figure 3).57
Cancer
DNA damage and cell cycle dysregulation are two of the most important hallmarks of cancer; hence, it comes with no surprise that SIRT6 has been regarded as a tumor suppressor, as indicated by early studies in knockout mice showing genomic instability.6 Further investigations indicated that SIRT6 knockout in MEFs leads to tumorigenesis without activation of known oncogenes, and deletion of SIRT6 in vivo correlates with an increased number, size, and aggressiveness of tumors (Figure 4).8,11 The tumor-suppressor role of SIRT6 was associated with the suppression of glycolytic genes crucial for the Warburg effect, a metabolic shift common in cancer cells where ATP is obtained mostly through glycolysis rather than mitochondrial oxidative phosphorylation, in order to generate immediate energy to support fast proliferation and related cellular processes.58 These genes, including the glucose transporter-1 (GLUT1), lactate dehydrogenase (LDH), phosphofructokinase-1 (PFK1), and pyruvate dehydrogenase kinase-1 (PDK1), are regulated by the hypoxia-inducible factor 1α (HIF-1α), which is corepressed by SIRT6.59 SIRT6 also deacetylates pyruvate kinase M2 (PKM2), a nuclear isozyme that enhances aerobic glycolysis even under hypoxia conditions and promotes tumor growth. SIRT6-mediated deacetylation triggers PKM2 transport to the cytoplasm and repression of its functions.60 In addition, glycolytic genes are downregulated through direct deacetylation of H3K9 at their promoters.59
Figure 4.

Roles of SIRT6 in cancer. The figure indicates the main factors modulated by SIRT6 in the context of both tumor suppression and promotion. Different mechanisms are involved, including the regulation of DNA-damage response, glycolysis, apoptosis, cell migration, and inflammation.
In line with this, SIRT6 is selectively downregulated in CRC and pancreatic ductal adenocarcinoma (PDAC), which display increased expression of glycolysis-related genes.8 The following studies confirmed these findings and expanded the role of SIRT6 as a main regulator of glycolysis in prostate, bladder,61 and breast cancers.62 Interestingly, SIRT6 activity is antagonized by the Runt-related transcription factor 2 (RUNX2), which represses SIRT6 transcription in low-glucose conditions.62 In addition, E2 transcription factor 1 (E2F1) negatively regulates SIRT6 in response to hypoxia, hence facilitating the Warburg effect.61
SIRT6-mediated H3K9 deacetylation has effects on multiple oncogenes beyond glycolytic genes. These include two proteins involved in apoptosis inhibition and consequently tumor progression: the caspase activation inhibitor survivin63 and the RNA-binding oncofetal protein Lin28b.64 Liver cancer mouse models also showed that survivin activity is impaired through the inhibition of NF-κB activation and consequent binding to a survivin promoter.65 NF-κB is also involved in the activation of other antiapoptotic proteins (FLIP, c-IAP1/2, and XIAP)66 and its expression is antagonized by SIRT6 in nasopharyngeal carcinoma (NPC).67 Lin28b expression is downregulated through deacetylation of both H3K9 and H3K56. In PDAC, SIRT6 deficiency was associated with H3K9 and H3K56 hyperacetylation at the Lin28b promoter and poor patient prognosis; moreover, in a mouse model of pancreatic cancer, a SIRT6 deficit led to increased tumor aggressiveness and metastasis.68 Given the severity of PDAC and the important role played by SIRT6 in this subset of tumors, targeting this pathway through activation of SIRT6 may represent a successful approach for this type of malignancy.
Lin28b is also a target gene of the c-Myc oncogene. In PDAC, Lin28b promoter hyperacetylation is associated with c-Myc recruitment and consequent augmentation of cancer progression and metastasis.68 Notably, c-Myc activity is antagonized by SIRT6, which represses the transcription of c-Myc and its target genes and leads to cell cycle arrest and inhibition of tumor growth.69,70
SIRT6 deacylase activity also contributes to its action as a tumor suppressor. Indeed, SIRT6 deacylates the GTPase R-Ras2, a Ras-family protein that contributes to tumorigenesis and metastasis.23 SIRT6-mediated deacylation of R-Ras2 shifts its location toward intracellular vesicles rather than the plasma membrane, where it usually sits, hence blocking its signaling and cell proliferation.23
Notwithstanding the great number of reports indicating the tumor-suppressor role of SIRT6 in many forms of cancer, some evidence points toward an oncogenic role of SIRT6 under specific conditions (Figure 4). For instance, in hepatocellular carcinoma (HCC), the activity of SIRT6 in DNA-damage repair and cellular senescence prevention play in favor of cancer cell growth.71,72 In addition, SIRT6 deacetylates H3K9 at the promoter of the proapoptotic factor Bax, resulting in evasion from apoptosis.73 SIRT6 also facilitates the epithelial–mesenchymal transition (EMT) in HCC through deacetylation of the autophagy regulator Beclin-1 leading to the autophagic degradation of E-cadherin, a crucial receptor involved in cell adhesion.74 The controversial role of SIRT6 in cancer is confirmed by a separate study that suggests a protective role of SIRT6 in HCC mediated by the inhibition of ERK1/2 phosphorylation and the suppression of its downstream pathway.75
A connection between SIRT6 and MAPK signaling has also been described in multiple myeloma (MM), although at a transcriptional level. In MM, high SIRT6 levels have been associated with poor prognosis. This disease is characterized by high genome instability; hence, SIRT6 overexpression may be a compensatory response to facilitate DNA repair. While this mechanism may be favorable for cancer cell survival, Cea and colleagues demonstrated that in MM cells, SIRT6 interacts with ELK1 and deacetylates H3K9 at the promoters of MAPK signaling genes, thus stopping cell proliferation. On the other hand, SIRT6-mediated suppression of ERK2 and p90RSK signaling increases resistance to DNA-damaging therapeutics.76 Upregulation of SIRT6 has also been observed in other blood cancers, including acute myeloid leukemia,77 chronic lymphocytic leukemia,78 and diffuse large B-cell lymphoma (DLBCL).79 In DLBCL cells, knockdown of SIRT6 leads to higher sensitivity to chemotherapy, altered cell proliferation, augmented rates of apoptosis, and cell cycle arrest. These phenotypes were associated with inhibition of the PI3K/AKT/mTOR signaling pathway.79
SIRT6 catalytic activity determines an increase of the intracellular ADP–ribose concentration, which activates the Ca2+ channel TRPM2. Increased Ca2+ concentration finally leads to the activation of the Ca2+-dependent nuclear factor of activated T cells (NFAT), which upregulates the expression of TNF-α and IL-8, two proangiogenetic and proinflammatory cytokines that promote tumor growth and metastasis.80
A recent study indicated that SIRT6 is overexpressed in NSCLC cells, and its silencing determined activation of the p53/p21 pathway and consequent inhibition of cell proliferation associated with cell cycle arrest and apoptosis.81 Conversely, an earlier study indicated that SIRT6 suppresses NSCLC proliferation through inhibition of Twist1 expression, a factor that facilitates EMT and metastasis.82
These examples indicate the complicated role played by SIRT6 in tumorigenesis, suggesting a context dependency. If we take into account the involvement of SIRT6 in DNA-damage repair, depending on the stage of cancer progression, this pathway may have tumor-promoting or tumor-suppressing effects.11 Moreover, high levels of SIRT6 associated with tumors may also represent a compensating response rather than a causality. Therefore, it is vital to distinguish the potential of SIRT6 as a therapeutic target or as a biomarker in each type of tumor.
Inflammation and Immunity
In the context of immune regulation, SIRT6 has been shown to upregulate proinflammatory cytokines, as explained above in the case of TNF-α and IL-8.80 SIRT6 catalyzes the demyristoylation of TNF-α at Lys19 and Lys20, triggering its secretion22,30 during inflammatory response, while acylated TNF-α is retained and finally degraded in lysosomes. In addition, TNF-α levels are positively regulated by NAD+ concentration, and SIRT6 was identified as the mediator of the increased translation efficiency of Tnf mRNA.83
On the other hand, SIRT6 also exerts anti-inflammatory roles through negative regulation of NF-κB, and this is supported by studies in macrophages where SIRT6 deletion promotes NF-κB activation and IL-6 production.84 Studies performed on SIRT6-knockout mice indicated a chronic liver inflammation and fibrosis. Moreover, SIRT6-deficient lymphocytes and myeloid-derived cells presented aberrant activation. Mechanistically, SIRT6 repressed the transcription of genes controlled by the oncogenic transcription factor c-JUN (Figure 3).85
Sugar and Lipid Metabolism
SIRT6 is undoubtedly a multitasking protein, and beyond its involvement in DNA maintenance and cancer progression, its main function is probably the regulation of glucose and lipid metabolism. As outlined in the context of cancer, SIRT6 corepresses HIF-1α and deacetylates H3K9 at glycolytic genes promoters,59 thus channeling glucose catabolism from glycolysis toward more energy-efficient pathways (Figure 4). Indeed, SIRT6-knockout mice display increased glycolytic pathway associated with high glucose uptake, increased insulin signaling, and severe hypoglycemia as a compensatory response.6,59
SIRT6 modulates glucose homeostasis also through control of gluconeogenesis. SIRT6 has been found to deacetylate the acetyltransferase general control nonderepressible 5 (GCN5), a regulator of cell cycle progression involved in the onset of different tumors,86−88 leading to an increased enzymatic activity. In the liver, increased GCN5 activity results in acetylated PGC-1α,89 thus leading to reduced gluconeogenesis gene expression,90 which prevents hyperglycemia in diabetic/obese mice.89 Another important transcription factor for gluconeogenesis is FoxO1, which activates the transcription of the rate-limiting gluconeogenesis enzymes glucose-6-phosphatase (G6PC) and phosphoenolpyruvate carboxykinase (PCK1).91 FoxO1 is deacetylated by SIRT6, triggering its nuclear export and reduced transcription of its target genes (Figure 3).92
SIRT6 activity has also an effect on insulin signaling through downregulation of glucose transporters GLUT1 and GLUT4 and decreased phosphorylation of AKT,93 an important regulator of cellular glucose uptake.94 Mechanistically, SIRT6 is involved in the inactivation of AKT upstream proteins, including the insulin receptor substrates IRS1/2.93 Therefore, the absence of SIRT6 sensitizes the organism to insulin action, giving a complementary explanation to glycolytic gene suppression for the observed hypoglycemia in SIRT6-knockout mice.
In the case of lipid metabolism, SIRT6 reduces triglyceride synthesis and fatty-acid uptake while promoting β-oxidation, as indicated by mice-knockout studies.47 SIRT6 also contributes to keeping low the levels of LDL-C (Figure 3).95 Mechanistically, SIRT6 has been shown to decrease acetylation of the PPARα coactivator NCOA2, although it is not clear whether NCOA2 is a direct substrate of SIRT6 enzymatic activity. This determines activation of PPARα, a key transcription factor for hepatic β-oxidation genes.96 Furthermore, SIRT6 represses the expression of the proprotein convertase subtilisin/kexin type 9 (PCSK9), which controls the degradation of LDL-C in lysosomes (Figure 3).97 Through interaction with FoxO3α, SIRT6 is recruited at the PCSK9 promoter, where it deacetylates H3K9 and H3K56, hence suppressing its transcription.98 SIRT6 also deacetylates H3K9 and H3K56 at the promoters of genes regulated by the sterol regulatory element-binding protein 1 and 2 (SREBP1/2).99 These transcriptional regulators activate transcription of lipogenic genes and are also directly controlled by SIRT6 at the transcriptional level through H3K56 deacetylation at promoters. Notably, micro-RNAs miR-33a and mi33b, which are expressed from the introns of SREBP1/2, are associated with repression of SIRT6 levels, contributing to a negative feedback loop in the SIRT6-SREBP1/2 axis.100 Another micro-RNA involved in SIRT6-mediated pathways is miR-122, the most abundant hepatic miRNA, which negatively regulates SIRT6 expression and is in turn negatively regulated by SIRT6 (Figure 3). In addition, while SIRT6 positively regulates genes involved in fatty-acid β-oxidation, miR-122 performs an opposite action.101
The connection between SIRT6 activity and fatty-acid metabolism is fascinating given the evidence indicating that FFA are capable of increasing SIRT6 activity in vitro.20 Therefore, SIRT6 may act as a fatty-acid sensor that amplifies metabolic signals into epigenetic responses that affect crucial homeostatic mechanisms beyond metabolism itself; these include all the pathways analyzed in this section such as genomic maintenance, immunity, cellular differentiation, and transformation.
Pharmacological Modulation of SIRT6
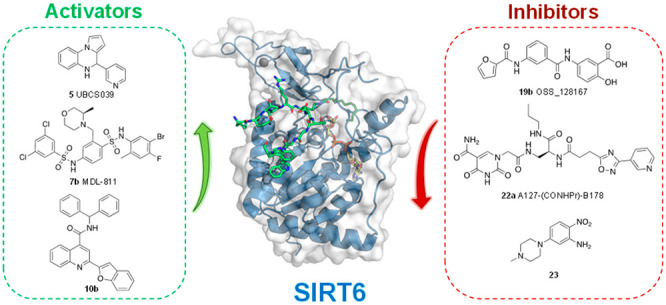
The implications of SIRT6 as a positive regulator of metabolism and aging, along with the discovery that the deacetylase activity may be enhanced by FFA, has stimulated research groups toward the development of SIRT6 activators (Table 1). On the other hand, given the dual role of SIRT6 in inflammation and cancer, inhibitors have also been developed (Table 2). The possibility of either activating or inhibiting SIRT6 in a context-dependent manner paves the way for personalized pharmacology. From a wider perspective, highly potent and selective SIRT6 modulators (both activators and inhibitors) allow the molecular details of its activity to be better scrutinized and further validate the enzyme as a pharmacological target.
Table 1. Most Relevant SIRT6 Activators.

Table 2. Most Relevant SIRT6 Inhibitors.

In the following section, we will first discuss the most relevant SIRT6 activators followed by a detailed description of SIRT6 inhibitors.
SIRT6 Activators
As already mentioned, early studies on SIRT6 activity indicated that FFA containing 14 to 18 carbons (Figure 5, upper panel) stimulated SIRT6 activity. In particular, myristic acid (1a) increased deacetylase activity up to 10.8 times, with an EC50 of 246 μM with a 35-fold increase in catalytic efficiency (kcat/Km value, i.e., the ability of SIRT6 to capture a substrate for catalysis) at 400 μM, suggesting increased affinity of SIRT6 for the acetylated substrate.20 Oleic (1b) and linoleic acid (1c) displayed EC50 values of 90 and 100 μM, yielding an increase in deacetylase activity of 5.8 and 6.8 times, respectively. In the same study, 1a was shown to act as a competitive inhibitor for demyristoylation, suggesting that the same hydrophobic pocket occupied by FFA during deacetylation is necessary to accommodate the long acyl chain of fatty-acid substrates for deacylation. These findings are the basis for the development of small-molecule SIRT6 activators.20
Figure 5.

SIRT6 modulators based on endogenous ligands (upper panel) and natural products (lower panel).
Following the studies on FFA, it has been shown that myristoylethanolamide (MEA, 2a) and oleoylethanolamide (OEA, 2b), the ethanolamine derivatives of 1a and 1b, showed a 2-fold maximum activation of SIRT6 and EC50 values of 7.5 and 3.1 μM, respectively.102 In the same study, 1b and 1c were tested, yielding SIRT6 maximum-fold activation of 4.6 and 3.7 along with EC50 values of 89 and 230 μM, respectively. Rahnasto-Rilla et al. also evaluated the influence of the flavonoids luteolin (3a) and quercetin (3b) on SIRT6 activity. The skeleton of flavonoids consists of a benzene ring (A) fused with a heterocyclic pyran ring (C) presenting a further phenyl group (ring B) in position 2. All the compounds described here present hydroxyl groups on carbons 5 and 7 in ring A (Figure 5, lower panel). 3b belongs to the subclass of flavonols and are characterized by an oxidized pyran ring, bearing a carbonyl group in position 4 and an additional hydroxyl group in position 3. Differently, 3a is a flavon and lacks the hydroxyl group in position 3. Both compounds demonstrated a dose-dependent role, whereby they exert inhibitory activity at low concentrations with IC50 values of 1.9 μM (3a) and 24 μM (3b) while increasing the deacetylase activity at higher concentrations. In particular, 3a showed a 6-fold maximum activation with an EC50 value of 270 μM, while 3b yielded 10-fold maximum activation and an EC50 of 990 μM.102 Although the EC50 values for these two flavonoids are very high and with scarce pharmacological relevance, these results suggest multiple binding sites for small molecules, which may interact with an inhibition site at low concentrations while inducing favorable conformational changes that activate the enzyme at higher concentrations.
Following these studies, further flavonoids were tested for their capability of altering SIRT6 enzymatic activity.103 The flavonol myricetin (3c) has the same structure of 3b with an extra hydroxyl group in 3′. This compound displayed an EC50 of 404 μM and 7.7-fold maximal SIRT6 activation, like observed for 3a.
Anthocyanidins are a subgroup of flavonoids which, compared to flavonols, lack the carbonyl group in position 4 of ring B. Delphinidin (3d), the anthocyanidin analogue of 3c, showed decreased activating potency, with an EC50 of 760 μM and 6.3-fold maximal SIRT6 activation. Remarkably, removal of the 3′ hydroxyl group in the case of cyanidin (3e) led to a massive increase in activation efficiency as indicated by the 55-fold maximum activation and EC50 of 460 μM.103 Notably, 3e exhibited in-cell effects when tested on colon adenocarcinoma Caco-2 cells, with a dose-dependent SIRT6 upregulation along with modulation of SIRT6-associated genes such as FoxO3a, Twist1, and GLUT1. In particular, the authors observed a dose-dependent increase in FoxO3a expression, while Twist1 and GLUT1 were decreased.
Although the activity of these molecules toward other SIRTs was not assessed in this study, another report evaluated 3b action on SIRT1–3, SIRT5, and SIRT6. In this case, 3b showed a 2-fold maximum SIRT6 activation with an EC50 of 1.2 mM, and no inhibition was observed at low concentrations. Conversely, 3b inhibited SIRT1–3 deacetylation activity and SIRT5-mediated desuccinylation in a concentration-dependent manner.104 In particular, at a 312.5 μM concentration, the enzymatic activities of SIRT1/2/3/5 were 60–70% compared to the respective controls, while SIRT6 activity was about 150%. Given their polyphenolic nature, flavonoids are known to present pleiotropic activities and have been shown to inhibit a diverse subset of enzymes. Among others, the starch digestive enzyme α-glucosidase is inhibited by compounds 3a–c and 3e(105−107) with IC50 values in the low–mid μM range, while α-amylase was shown to be inhibited by 3a–c with IC50 values of ∼300 μM.105 Moreover, compounds 3a–e have been reported to inhibit topoisomerases I and II108−112 and to affect the epigenetic regulation of transcription through inhibition of DNA methyltransferase 1 (DNMT1).113,1143b was also shown to suppress the activity of other epigenetic enzymes such as HDAC1115 and the histone acetyltransferase (HAT) p300.116 In contrast, a different study indicated that 3b administration results in increased histone H3 acetylation, associated with HAT activation.117 Moreover, compounds 3a–c have been reported to interfere with multiple bioassays, thus being classified among the pan assay interference compounds (PAINS)118,119 and suggesting caution in interpreting the results of biological studies on them.
Nonetheless, flavonoids could represent useful hit compounds for the development of SIRT6 activators thanks to the release of SIRT6–3b and SIRT6–3e cocrystal structures (PDB IDs: 6QCD and 6QCH, respectively). These structures indicated that 3b and 3e interact with SIRT6 at the distal end of the hydrophobic acyl-binding pocket, with surface contacts with the β6/α6 loop that caps this channel (Figure 6).104 In both cases, the catechol portion (ring B) is inserted in the acyl-binding pocket with the 4′-hydroxyl group forming a hydrogen bond with Pro62 backbone oxygen and with a conserved water molecule that in turn forms a hydrogen bond with the backbone oxygens of Ala53 and Ile61. Similarly, the 3′-hydroxyls of both molecules are hydrogen-bonded with another conserved water molecule that is in contact with the side-chain oxygen of Asp116. In the case of 3b, the chromen-4-one moiety (rings A and C) forms hydrophobic contacts with Phe64/82/86, Val70/115, and Met136/157 (Figure 6B). As for 3e, although the density of this portion was weaker, it was positioned in the same area as 3b, suggesting a similar binding mode.104 Compared to 3b, 3e lacks the carbonyl group pointing toward Met136/157, which may be one of the reasons for its higher potency. Indeed, this group may represent a steric hindrance, and its absence allows optimal hydrophobic contacts between ring C and Met136/157 (Figure 6C).
Figure 6.

(A) Structure of SIRT6 in complex with ADP–ribose (yellow) and quercetin (3b) (green) (PDB ID: 6QCD). (B) Focus on 3b binding site showing the presence of key water molecules (red spheres) mediating protein-compound interaction. (C) Structure of SIRT6 in complex with ADP–ribose (yellow) and cyanidin (3e) (green) with a focus on the 3e binding site (PDB ID: 6QCH). Key residues for compounds’ binding are labeled, and polar interactions are shown as dashed orange lines.
The crystal structures of SIRT6 in complex with 3b and 3e enable the identification of key features for ligand binding and, likely, could be exploited to develop new compounds containing only the hydroxyl groups essential for the interaction with the target, thus decreasing the polyphenolic character. Moreover, computational scaffold hopping120,121 approaches integrated with AI-driven drug discovery122 could allow the design of derivatives bearing a different core but retaining the moieties important for SIRT6 interaction. Overall, these strategies could enable molecules with increased specificity and potency to be obtained.
Another naturally occurring molecule showing SIRT6 activation is fucoidan (4), a heterogeneous sulfated polysaccharide present in brown algae. Its backbone consists of repeating (1 → 3) or (1 → 3) and (1 → 4) linked α-l-fucopyranose residues, in which some hydroxyl groups form sulfated esters (Figure 5, lower panel).123 The oversulfated fucoidan subtype extracted from Fucus vesiculosus displayed a 355-fold increase of SIRT6 activity at a 100 μg/mL concentration. In addition, when tested against other SIRTs (SIRT1/2/3), it did not display significant changes in activity, suggesting a specific action toward SIRT6. 4 was also able to activate SIRT6 acetylation toward H3K9 in vitro. According to the authors of the study, sulfate esters may play a central role in SIRT6–4 interaction and hence SIRT6 activation.123 However, the heterogeneity of the mixture, the polymeric nature of the compound, and the absence of kinetic data makes it difficult to compare this macromolecule to small molecules and to devise structure–activity relationships.
The first synthetic SIRT6 activator is the pyrrolo[1,2-a]quinoxaline derivative UBCS039 (5, Figure 7a, upper panel), which exhibited an EC50 of 38 μM and 3.5 maximum activation of SIRT6 in H3K9Ac peptide deacetylation assays.1245 showed specific binding on SIRT6, with no significant effects on basal SIRT1, 2, and 3 deacetylation activities. Notably, it stimulated SIRT5 desuccinylation activity (2-fold increase at 100 μM), the physiologically dominant activity of this enzyme. The 5-SIRT6 cocrystal (solved at 1.87 Å resolution, PDB ID: 5MF6) indicated a similar binding mode to 3b and 3e, with the compound occupying the exit of the acyl channel pocket and exposing the benzene moiety of the quinoxaline to solvent. The tricyclic portion of the molecule likely forms a methionine–aromatic ring interaction with Met136 along with weak hydrophobic interactions with Trp71, Phe82, Phe86, Ile185, and Met157. In addition, the pyridine nitrogen forms a hydrogen bond with the backbone carbonyl of Pro62 (Figure 7a, lower panel); this interaction represents a key anchoring point as a shift of the position of the nitrogen led to decreased SIRT6 affinity and activation. Comparison of this crystal structure with the cocrystal of SIRT6 and myristoylated peptide indicates that UBCS039 overlaps with the last seven carbons of the myristoyl chain. In addition, comparison with the SIRT6/ADP–ribose/3b cocrystal indicates that the compounds share a similar binding site. The 5 pyridine portion overlaps with 3b cathechol moiety, and both engage in the key interaction with Pro62. In addition, the pyrrolo[1,2-a]quinoxaline moiety of 5 and the chromen-4-one are involved in similar hydrophobic interactions. One difference relies on the fact that 3b possesses a carbonyl group pointing toward Met136/157, which may impair optimal hydrophobic contacts between the aromatic ring and the methionine residues. Differently, the 5 tricyclic system is positioned in a privileged location for aromatic and hydrophobic interactions with Met136/157, thus explaining its higher potency compared to 3b. Although 5 did not display significant inhibition of SIRT6-mediated demyristoylation, as the binding affinity for the myristoylated peptide is much higher, addition of myristoylated peptide decreased 5 binding by an order of magnitude, thus indicating competition for the same binding site. Compound 5 was also tested using physiological substrates, such as full-length histones extracted from calf thymus and HeLa nucleosomes. In both cases, Western blot analysis indicated H3K18 deacetylation in the presence of 5.124 Follow-up studies indicated that 5 causes SIRT6 activation in a different subset of cancer cell lines, including NSCLC, colon and epithelial cervix carcinoma, and fibrosarcoma. 5-mediated SIRT6 activation led to decreased H3K9 and H3K56 acetylation and autophagy-related cell death.125 This study represents the first evidence of in-cell small-molecule-mediated SIRT6 activation, suggesting a potential therapeutic exploitation of this activity.
Figure 7.

(A) Upper panel: Molecular structure of UBCS039 (5). Lower panel: Structure of SIRT6 in complex with ADP–ribose (yellow) and 5 (green) with a focus on the 5 binding site (PDB ID: 5MF6). (B) Upper panel: Molecular structure of fluvastatin (6). Structure of SIRT6 in complex with ADP–ribose (yellow) and 6 (green) with a focus on the 6 binding site (PDB ID: 6ZU4). Key residues for compounds’ binding are labeled, and polar interactions are shown as dashed orange lines.
A compound screening for drug repurposing recently identified the HMG-CoA reductase inhibitor fluvastatin (6, Figure 7B, upper panel), already approved for hypercholesterolemia treatment, as a SIRT6 activator.1266 showed an EC50 = 7.1 μM and decreased H3K9 and H3K56 acetylation in HepG2 cell lines. This effect was accompanied by increased nuclear translocation of SIRT6. In addition, 6 treatment increased levels of phosphorylated AMPKα, which in turn promoted SREBP1 phosphorylation at Ser372. In addition, cleaved SREBP1 was negatively regulated. These results are in line with previous reports suggesting that SIRT6 overexpression represses SREBP1/2 through the AMPK pathway.99 Interestingly, a subsequent study found a much higher EC50 (>250 μM) for 6-mediated SIRT6 activation, though it could reach 3.5-fold maximum activation at 1 mM.127 Compound 6 also displayed weak inhibition of SIRT1/2/3, while it did not affect SIRT5 activity. Nonetheless, the authors managed to cocrystallize 6 with the N-terminally truncated SIRT6 (13–308) and ADP–ribose and solved the structure at 2.46 Å (PDB ID: 6ZU4, Figure 7B, lower panel). 6 interacts with SIRT6 at the exit of its acyl channel in its acid form, rather than lactone, forming a hydrogen bond with Trp188 through its carboxyl group. In addition, the fluorophenyl and isopropyl residues point toward the channel exit, while the heptenoic acid moiety interacts with a surface formed by Lys15, Trp71, and Glu74. The indole moiety has a similar positioning of the pyridine ring of 5,124 as it is oriented toward the hydrophobic pocket formed by Phe64/82/86, Ile61, Pro62, and Met136. However, bulky substituents, such as the isopropyl and fluorophenyl groups, obstruct the entrance in the pocket, thereby impairing the key polar interactions with the backbone oxygen of Pro62 seen with 5 and other ligands. In summary, the authors of this study suggest that the initially measured low EC50126 may be a result of an assay artifact and that the reported cellular effects may be due to an indirect action of 6.127 Nevertheless, the elucidation of the 6 binding mode aids the development of modulators possessing the same core scaffold, but different substituents, in order to maximize polar interactions.
Virtual screening followed by in vitro evaluation led to the discovery of a new selective and cellularly active SIRT6 activator, the N-phenyl-4-(phenylsulfonamido)benzenesulfonamide derivative MDL-800 (7a, Figure 8A).1287a displayed an EC50 value of 10.3 μM, enhancing SIRT6 activity by more than 22 times (at 100 μM), using a synthetic acetylated peptide (RHKK-ac-AMC) as a substrate. When tested on SIRT6 using the H3K9Ac peptide (KQTARK-ac-STGGWW), 7a exhibited 18-fold maximal SIRT6 activation. In addition, 7a increased the deacetylation of H3K9 and H3K56 on HeLa-extracted nucleosome substrates in a dose-dependent manner. 7a did not display any effect on the enzymatic activities of SIRT1/3/4 and HDAC1–11 at concentrations up to 50 or 100 μM. It displayed weak inhibition of SIRT2 (IC50 = 100.4 μM) and weak activation of SIRT5 (IC50 = 104.6 μM) and SIRT7 (IC50 = 187.1 μM). Since the IC50/EC50 values are 10 times (or more) greater than SIRT6 EC50, the compound is considered selective. The analogue MDL-801 (7b), in which the methyl carboxylate ester in position 2 of the central benzenesulfonamide ring is replaced by a carboxylic group (Figure 8A), exhibited overlapping SIRT6 activation features with an EC50 = 5.7 μM. However, while 7a was highly cell permeable and accumulated in cells, 7b had poor cellular permeability and a high efflux ratio. Therefore, the only compound tested for cellular activity was 7a. This molecule caused a dose-dependent decrease of H3K9Ac and H3K56Ac in HCC cells (specifically Bel7405, PLC/PRF/5, and Bel7402 cell lines), leading to inhibition of their proliferation through SIRT6-mediated cell cycle arrest. In particular, the observed IC50 for cell growth (IC50-growth) was between 18.6 and 24 μM, depending on the cell line. These results were confirmed in mouse xenograft models, where 7a suppressed HCC tumor growth through SIRT6 activation. A recent investigation indicated that 7a inhibits the proliferation of 12 NSCLC cell lines in a dose-dependent manner and caused cell cycle arrest at the G0/G1 phase in NSCLC HCC827 and PC9 cells, consistent with studies indicating the role of SIRT6 in cell cycle regulation.16,70 Notably, it exhibited synergistic activity with epidermal growth factor receptor tyrosine kinase inhibitors (EGFR-TKIs) in osimertinib-resistant HCC827 and PC9 cells and in patient-derived primary tumor cells. Moreover, 7a suppressed tumor growth in HCC827 cell-derived xenograft nude mice and caused H3 deacetylation and downregulation of p-MEK and p-ERK in tumor tissues.129
Figure 8.

(A) Molecular structures of MDL compounds (7a–c). (B) Structure of SIRT6 in complex with ADP–ribose (yellow), H3K9-Myr (myristoyl chain in dark green), and MDL-801 (7b) (green) with a focus on the 7b binding site, as reported by Huang et al. (PDB ID: 5Y2F). (C) Structure of SIRT6 in complex with ADP–ribose (yellow) as well as 7b (green) with a focus on the 7b binding site, as reported by You and Steegborn (PDB ID: 6XVG). Key residues for compounds’ binding are labeled, and polar interactions are shown as dashed orange lines.
Huang et al. solved the cocrystal structure of the complex formed by SIRT6, ADP–ribose, H3K9 myristoylated peptide, and 7b (PDB ID: 5Y2F, Figure 8B).128 Given the structural similarities between 7b and 7a, the observed features are likely shared between the two compounds. Interestingly, 7b appeared to interact with SIRT6 in a unique pocket, distinct from the binding site of 3b, 3e, 5, and 6 located in the acyl-binding hydrophobic channel. Indeed, 7b was shown to interact with a surface-exposed distal region defined by the N-terminal residues 1–7, Val70, Glu74, Phe82, Phe86, Val153, and Met157. The 3,5-dichlorobenzene moiety of 7b is involved in weak polar interactions with Asn40, Val70, and Glu74 and engages π-stacking interactions with Phe82 (Figure 8B). The central 2-carboxybenzenesulfonamide ring is also involved in π-stacking interactions with Phe86, whose importance was confirmed by single-residue mutation experiments showing decreased potency of both 7a and 7b toward SIRT6-F86A.128 However, a recent report from You and Steegborn argued that the observed electron density could be attributed to a molecule of morpholinoethanesulfonic acid (MES), used as crystallization buffer, rather than 7b.130 Therefore, they determined new crystal structures for SIRT6 in complex with 7b. They solved the cocrystal of N-terminal truncated SIRT613–308 in complex with ADP–ribose and 7b (PDB ID: 6XV1) as well as in the absence of 7b (PDB ID: 6XUY). Similarly, they solved the structure for SIRT63–308 (comprising the N-terminus) in complex with ADP–ribose and 7b (PDB ID: 6XVG, Figure 8C), along with a reference structure without the activator (PDB ID: 6XV6).130 These structures indicate a different binding mode for 7b, which does not bind at the distal end of the acyl-binding hydrophobic channel but in the same region as the previously described activators 3b, 3e, and 5. In both SIRT613–308–7b and SIRT63–308–7b structures, the activator engages in extensive hydrophobic interactions, the central 2-carboxybenzenesulfonamide is packed between Val70, Trp71, and Met157, and the 5-bromo-4-fluoro-2-methylaniline portion interacts with Phe64, Val70, Phe82, Phe86, and Val115. In addition, bromine forms a halogen bond with the backbone amide oxygen of Pro62, which has been shown to be a key residue for small-molecule interactions with SIRT6 (Figure 8C). Notably, the interaction with Pro62 is missing in the binding mode illustrated by Huang et al.128 The 3,5-dichlorobenzene moiety is less defined and seems to be largely solvent-exposed. Hence, the structures from the two groups display rather different binding modes for 7b, whose orientations within SIRT6 are perpendicular to each other in the two studies. In response to this report, Huang et al. crystallized SIRT6 with and without 7b using the same conditions as in their original publication (PDB ID: 7CL0 for SIRT6 in complex with ADP–ribose and H3K9 myristoyl peptide; PDB ID: 7CL1 for SIRT6 in complex with ADP–ribose, H3K9 myristoyl peptide, and MDL-801).128,131 They showed that, in the absence of 7b, the buffer molecule MES does not fit properly the originally proposed ligand-binding pocket. In addition, the newly solved cocrystal in the presence of 7b, although possessing lower overall resolution (3.2 Å for 7CL1 vs 2.53 Å for 5Y2F), has better electron density for the activator and confirms their initial findings.131 Importantly, Huang et al. crystallized SIRT6 in the presence of the H3K9 myristoyl peptide, which is absent in the crystallization mixture of You and Steegborn. Overall, the observed differences in the 7b binding mode may be ascribed to the presence of the substrate, which influences the interaction between the small molecule and SIRT6. Indeed, the crystal structure represents just a conformational state of the protein, whose conformational dynamics can be altered by the presence of ligands, thereby leading to alteration of key interactions between a small molecule and their target. Hence, structures of SIRT6–7b from both groups may be equally valid and represent two different states of the protein, regulated by the presence of substrate. Nonetheless, further experiments, including the structure of 7b in the presence of acetylated substrate may help to further clarify this controversy.
The replacement of the methyl carboxylate with an N-methyl-3-methylmorpholine at the C3 position of the central benzene ring of 7a led to compound MDL-811 (7c, Figure 8AA) with improved activity (EC50 = 5.7 μM) and bioavailability in C57BL/6J mice (F%MDL-800 = 71.33% vs F%MDL-811 = 92.96%).132 The improved activity may be explained by the higher number of interactions that the N-methyl-3-methylmorpholine moiety can establish. Indeed, it presents an exposed oxygen that can act as a hydrogen bond acceptor and a methyl group potentially involved in hydrophobic interactions. The compound enhanced deacetylation of H3K9, H3K18, and H3K56 in nucleosomes extracted from HeLa cells as well as in HEK293T cells in a dose-dependent manner. 7c was shown to be specific for SIRT6, as it was not able to affect the activity of SIRT1–3, SIRT5, SIRT7, and HDAC1–11 at concentrations up to 100 μM. According to docking studies, the 3-methylmorpholine moiety may participate in hydrophobic interactions with Phe82, Thr85, and Phe86 of SIRT6, and the oxygen can form a hydrogen bond with the backbone amide of Phe86. As already mentioned, CRC is a type of cancer characterized by heavy downregulation of SIRT6.8 Notably, 7c displayed dose-dependent reduction of H3K9Ac, H3K18Ac, and H3K56Ac levels in different CRC cell lines and antiproliferative effects associated with marked G0/G1 cell cycle arrest. In line with this, 7c suppressed CRC growth in patient-derived organoids and showed antitumor efficacy in cell-line-derived and patient-derived xenograft (CDX and PDX, respectively) models as well as in a spontaneous CRC mouse model. Mechanistically, cytochrome P450 family 24 subfamily A member 1 (CYP24A1), which had been previously shown to be aberrantly overexpressed in CRC,133,134 was identified as a new target gene of SIRT6. 7c also exhibited synergistic activity with vitamin D3 in suppressing CRC proliferation. Interestingly, vitamin D3 is both a substrate and transcriptional regulator of CYP24A1 and has shown antitumor efficacy in CRC.135,136
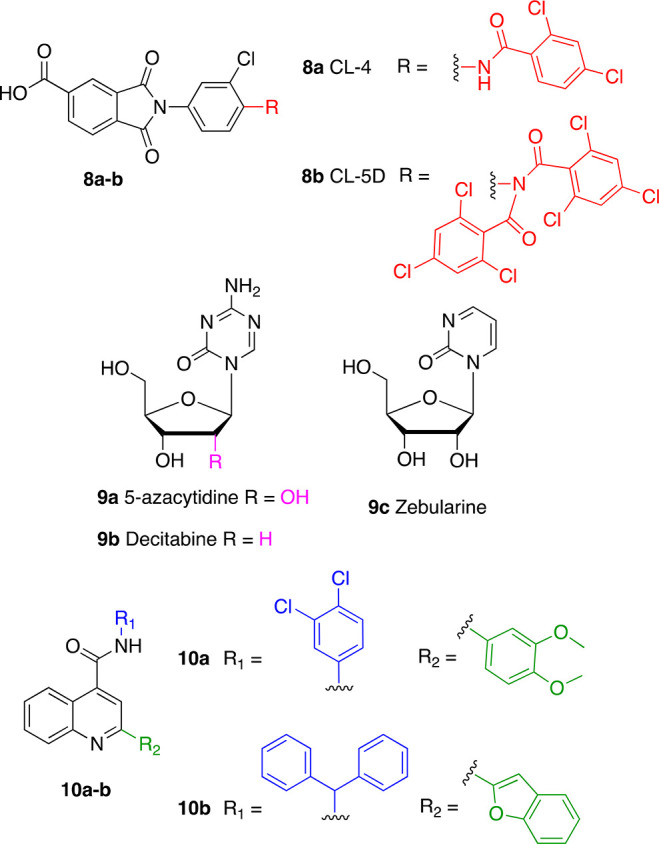
Activity-based screening of lipid-like molecules led to the discovery and optimization of CL4 (8a), a compound consisting of a 4-carboxyphthalimide conjugated to an N-(2-chlorophenyl)2,5-dichlorobenzamide. 8a displayed an EC50 = 97 μM and 17 maximum-fold activation of SIRT6 deacetylase activity. In addition, it displayed selectivity over SIRT1–3 and SIRT5.21 Therefore, 8a represented an ideal lead compound for the development of selective SIRT6 activators. Removal of chlorine atoms from either the 2,5-dichlorophenyl moiety or both benzene rings led to the suppression of SIRT6 activation, while progressive addition of chlorine restored this ability (Figure 9, upper panel). Finally, addition of a double-trichlorobenzoyl group at the aniline nitrogen led to CL5D (8b), which showed 7-fold increased potency over 8a, with an EC50 = 15.5 μM. Notably, the methyl ester of 8b did not show any activity. The data obtained from the development of 8b indicate that electron-withdrawing groups on the aromatic rings are crucial for SIRT6 activation in this series of molecules. In addition, the anionic headgroup (the carboxylic acid) is also essential for activity, and it is probably involved in hydrogen bond interactions. The maximum-fold activation of 8b was measured in terms of the kcat/Km ratio, which was ∼50 under steady-state conditions. 8b displayed competitive inhibition of demyristoylation (Ki = 13.4 μM), suggesting occupation of the acyl-binding pocket, although structural data are missing. 8b also stimulated SIRT6 deacetylase activity in a time-dependent fashion in full-length histones extracted from HEK293T cells.3
Figure 9.
Further synthetic SIRT6 activators.
A recent study that evaluated the influence of the FDA-approved DNA hypomethylating agents (DHAs) on sirtuin family members showed that the nucleoside analogues 5-azacytidine (5AC, 9a), decitabine (DAC, 9b), and zebularine (9c) increase SIRT6 enzymatic activity (Figure 9, middle panel).1379a and 9b increased SIRT6 activity after 12, 24, and 48 h of incubation at 0.25 and 0.5 μM; albeit, no dose-dependency was observed. Moreover, while the maximum activation (1.3-fold activation) for 9a was observed after 48 h, 9b exhibited 1.5-fold activation after 12 h, followed by a decrease in activation efficiency at 24 and 48 h. 9c could also activate SIRT6 deacetylase activity, although at higher concentrations (0.5 and 1 μM), with 1.4 maximum-fold activation observed after 48 h of incubation. In addition, both 9a and 9c (but not 9b) reduced the enzymatic activity of SIRT1, while the activity of SIRT2, SIRT3, and SIRT5 was not affected by any of these compounds. Although these data indicate that these compounds activate SIRT6, the lack of dose dependency suggests that they have been tested far below their EC50; hence, the maximum activation values presented here should be taken cautiously. In line with these results, U937 leukemia cells treated with 0.5 μM 9b for 24 and 48 h displayed decreased levels of H3K9Ac and H3K56Ac, according to Western blot experiments. Further ChIP-Seq analysis of bone marrow cells derived from six AML patients and treated with 9b indicated changes of H3K9 acetylation at 187 gene loci; specifically, 102 genes displayed an acetylation decrease, while 85 genes showed an acetylation increase. The authors of this study speculated that the unexpected increase in acetylation may be a consequence of differential effects of 9b on both HATs and HDACs or possible upregulation of the HAT enzymes targeting H3K9. Signaling pathway analysis showed that H3K9 acetylation changes are related to pathways like EGF/EGFR and Wnt/Hedgehog/Notch, which are associated with AML. Although the study lacks details about the connection between SIRT6 inhibition and the overall antitumor effect of 9b, it highlighted a possible second mechanism of action of nucleoside analogues, which is worth exploring. Indeed, these molecules seem to be active at relatively low concentrations; hence, a complete biochemical evaluation by SAR studies may lead to the development of selective SIRT6 inhibitors.
A virtual screening campaign performed using the SIRT6–5 (PDB ID: 5MF6)124 complex led to the identification of the initial hit 10a that was further optimized to provide the 2-(1-benzofuran-2-yl)-N-(diphenylmethyl) quinoline-4-carboxamide (10b) as a potent and selective small-molecule activator of SIRT6 (Figure 9, lower panel).138 In docking experiments, 10a appeared to bind at the distal end of the hydrophobic channel and engage in π–π interactions with Phe86 with its quinoline scaffold as well as σ–π interactions with the amide backbone of Ala7 through the 3,4-dichlorobenzene moiety. When tested in vitro, compound 10a increased SIRT6 activity by 50% (EC1.5) at ∼27 μM. From the docking model, it appeared that the space around the 3,4-dichlorobenzene group and 3,4-dimethoxybenzene group was not occupied; hence, modifications were executed to add moieties that would strengthen the interactions between the molecule and SIRT6. This led to compound 10b, where the 3,4-dichlorobenzene and the 3,4-dimethoxybenzene were replaced by a diphenylmethane group and a 2-benzofuranyl moiety, respectively. Compound 10b displayed activation toward both SIRT6 deacetylase and deacylase activities, with EC50 values of 5.35 and 8.91 μM for deacetylation and demyristoylation, respectively. 10b showed no influence on the enzymatic activity of SIRT2, SIRT3, SIRT5, and HDAC1–11. It weakly inhibited SIRT1, but the IC50 value for SIRT1-mediated deacetylation (IC50 = 171 μM) is more than 30 times higher compared to SIRT6 EC50, thereby indicating in any case an appreciable selectivity. According to docking experiments, compound 10b interacts with SIRT6 in a similar way to its parent compound and presents some extra interactions given by the different substituents. Indeed, the benzofuran forms hydrogen bonds with Met157 and Lys160 and a π–σ interaction with the amide group of Thr156; in addition, the N-benzhydryl group is inserted in the allosteric pocket and establishes hydrophobic interactions with Tyr5, Val70, Phe82, Pro62, and Pro80, and one of its benzene rings forms π–π interactions with Phe86. According to this model, although located in a similar region, compound 10b binds SIRT6 more toward the end of the hydrophobic channel compared to 5, which may justify 10b-mediated enhancement of SIRT6 demyristoylation activity. Compound 10b suppressed the proliferation and caused cell cycle arrest in the G2 phase of PANC-1 and BXPC-3 PDAC cell lines. Cellular thermal shift assay (CETSA)139 performed in intact cells confirmed that 10b (at 25 μM concentration) interacts with SIRT6 in cells. In addition, 10b exhibited antitumor activity in a human pancreatic tumor xenograft mouse model associated with a decrease of H3K9 acetylation levels. A preliminary study in male Sprague-Dawley rats also indicated a promising pharmacokinetic profile, although the bioavailability was only 4%. Although 10b with its low micromolar EC50 values is a promising lead compound, we cannot exclude that the effects observed in cells and in vivo are related to interactions with off-target proteins beyond SIRT6. Therefore, further functional and target engagement assays such as mass-spectrometry-based thermal profiling,140,141 histone deacetylase assay homogeneous (HDASH) procedures,142 fluorescence resonance energy transfer imaging (FRET) probes,142 and affinity-based protein profiling (ABPP)143 seem necessary to clarify this point. Moreover, genetic studies144,145 alone and in combination with compound treatment in both cellular and animal PDAC models would be required to confirm the causal link between the observed phenotypes and SIRT6 activation and to conclusively assess the therapeutic potential of 10b in this tumor context.
SIRT6 Inhibitors
Given the double-faced involvement of SIRT6 in cancer and inflammation, inhibition of SIRT6 in specific contexts may represent a successful strategy for cancer management. Indeed, inhibitors may target different SIRT6-mediated pathways that contribute to cancer progression such as DNA repair mechanisms, cell differentiation inhibition, and inflammatory response (Table 2).
Nicotinamide (11a, Figure 10) is one of the products of the sirtuin-mediated deacylation reaction and may act as a weak product inhibitor of SIRTs without subclass specificity.146−14811a has been validated as a SIRT6 deacylation inhibitor through two different assays using H3K9 myristoyl peptides: an HPLC assay yielded an IC50 = 153 μM; similarly in a fluorogenic assay, 11a displayed an IC50 = 184 μM.149 In a subsequent study, 11a displayed an IC50 for a demyristoylation reaction of 73 μM while showing increased inhibitory potency toward deacylation of H3K9 decanoyl peptide (IC50 = 45 μM) and lower potency using H3K9 hexanoyl peptide as a substrate (IC50 = 184 μM).39
Figure 10.

Product-based SIRT6 inhibitors.
Based on the 11a analogue pyrazinamide (PZA), Bolivar et al. developed two derivatives with improved SIRT6 inhibition activity (Figure 10): 5-MeO-PZA (11b, IC50 = 40.4 μM) and 5-Cl-PZA (11c, IC50 = 33.2 μM). Remarkably, these compounds did not show NAD+ competition, hence indicating a different mechanism of action from 11a.15011c was reported to be active toward SIRT1, but not SIRT2/3, while 11b was not evaluated against SIRT1–3. Nonetheless, selectivity against other SIRTs and HDACs need to be ascertained.
ADP–ribose (12, Figure 10) also inhibits SIRT6 activity and showed higher potency than 11a with IC50 values of 74 μM (deoctanoylation) and 89 μM (demyristoylation), compared to values of 150 and 120 μM, respectively, for 11a.151
Another class of inhibitors directly related to the SIRT6 enzymatic mechanism are Nε-thioacyl lysine peptides, which cause a stall of the catalysis after the nucleophilic attack of the (thio)carbonyl group to the C1′ of nicotinamide-bound ribose that happens in the first step of the catalytic mechanism.152 Early reports following the thioacyl peptide strategy let to Nε-thioacyl lysine pentapeptides 13a and 13b (Figure 11, upper panel) showing IC50 values toward SIRT6 deacetylase activity of 78 and 47 μM, respectively.153 These data indicate that replacement of a His residue with an Ala residue improves inhibitor activity. Both compounds inhibit SIRT1/2 with higher potency compared to SIRT6. Indeed, they both abolish SIRT1/2 almost completely at a 200 μM concentration, while the inhibition of SIRT6 was 62% (13a) and 91% (13b) at the same concentration. In the case of 13b, the IC50 values for SIRT1 and SIRT2 were 0.38 and 8.5 μM, respectively. These data are in line with the fact that both peptides are based on the sequence of p53, a known substrate of both SIRT1 and SIRT2. Nevertheless, although these molecules are not selective against SIRT6, they represent the first successful example of synthetic SIRT6 inhibitors.
Figure 11.

Peptide-based SIRT6 inhibitors.
A later study described the development of thiomyristoyl peptides designed on the basis of SIRT6 natural substrates. In particular, compounds BHJH-TM1 (14a), BHJH-TM3 (14b), and BH-TM4 (14c) (Figure 11, middle panel) displayed SIRT6 inhibition in the low micromolar range with IC50 values for demyristoylation of 2.8, 8.1, and 1.7 μM, respectively.154 These compounds were based on TNFα-K20, TNFα-K19, and H3K9 peptides, respectively. All three peptides were active against SIRT1/2/3, with IC50 values between 2.3 and 8.0 μM for all the isoforms, thus indicating a lack of selectivity and a mixed mode of action. Interestingly, they all displayed SIRT6 inhibition and increased TNFα fatty acylation in HEK293T cells with 14b being the most potent.
More recently, cyclic pentapeptides (15a–f) harboring a central Nε-dodecyl- or Nε-myristoyl-thiocarbamoyl-lysine (Figure 11, middle and lower panels) showed inhibitory activity toward SIRT6 in the nanomolar range (IC50 (15a) = 256 nM, IC50 (15b) = 282 nM, IC50 (15c) = 368 nM, IC50 (15d) = 319 nM, IC50 (15e) = 495 nM, IC50 (15f) = 319 nM). Compounds 15a–e had comparable IC50 values for SIRT1, while 15f an IC50 toward SIRT1 2.3 higher compared to SIRT6. Compounds 15e and 15f, bearing the same macrocycle bridging unit, were also tested against SIRT2 and SIRT3. Compound 15e showed moderate selectivity over SIRT2 and SIRT3 (∼2.9-fold and ∼1.5-fold, respectively), while 15f exhibited high selectivity over the two isoforms (20-fold and 11-fold, respectively). Finally, 15f was tested against SIRT5, where the results indicated that the molecule is substantially inactive towards this enzyme (IC50 > 300 μM). This analysis suggests that the only selective SIRT6 inhibitor is 15f.155 Despite that, 15f was not able to inhibit SIRT6 inside the human pancreatic cancer BxPC3 cells, likely because of poor cellular permeability given its peptide nature and high molecular weight. Nonetheless, these peptides represent valuable lead compounds for the development of peptidomimetics inhibiting SIRT6.
Recently, Sociali et al. developed a lysine-based compound targeting SIRT6 deacetylase and deacylase activities (16, Figure 11, lower panel).156 This molecule consists of a lysine residue whereby the Nα-amine group is protected with an acetyl group, while the carboxy group is coupled with a 12-carbon alkyl chain amine. This compound inhibited SIRT6 deacetylation (IC50 = 95 μM) without isoform specificity, as it inhibited also SIRT1 and SIRT2 with comparable potency (IC50s = 51 and 102 μM, respectively). Remarkably, compound 16 behaved as a deacylation activator showing 52% activation of demyristoylation (EC50 = 70 μM) and 80% activation of depalmitoylation at 100 μM while still acting as an inhibitor for SIRT1/2 deacylation (IC50 (SIRT1)= 157 μM, IC50 (SIRT2)= 177 μM). 16 displayed competitive inhibition toward acetylated peptide, but not NAD+, and increased H3K9 acetylation in the MCF-7 breast cancer cell line. Moreover, the activities of key glycolysis enzymes were increased, in line with SIRT6 involvement in downregulation of glycolytic enzymes, and TNF-α secretion was reduced, consistently with the ability of SIRT6 to trigger TNF-α secretion.22,30 The results obtained in this study are rather surprising in light of the evidence reported by Feldman et al. that FFAs determine enhancement of deacetylation activity and inhibition of deacylation.20 Based on in silico data, the authors speculate that the acetyl moiety bound to the Cα amine group may mimic the acetylated substrate, being close to NAD+, in agreement with the observed competition with acetylated substrate and not with NAD+. However, further experimental evidence is necessary to clarify the binding mode and account for the differential SIRT6 modulation profile.
Interestingly, the 3b derivatives (−)-catechin gallate (17a) and (−)-gallocatechin gallate (17b) displayed inhibition of SIRT6-mediated deacetylation in the low micromolar range (IC50 (17a) = 2.5 μM; IC50 (17b) = 5.4 μM).103 The epimers of compounds 17a and 17b, (−)-epicatechin gallate (17c) and (−)-epigallocatechin gallate (17d), displayed lower activity toward SIRT6, with ∼60 and ∼40% inhibition at 100 μM, respectively, compared to ∼85–90% inhibition of 17a–b at the same concentration. Structurally, these compounds differ from 3b in ring C, which is reduced and presents a 3,4,5-trihydroxybenzoyl substitution. The 17a–SIRT6 cocrystal (PDB ID: 6QCJ, Figure 12A) indicated that the inhibitor shares the same binding site as 3b with identical conformations of the catechol groups, while the chromen-4-one of 17a was rotated to accommodate the bulky trihydroxybenzoyl moiety. Ring C interacts with Trp71 of the acyl channel exit, and the trihydroxybenzoyl portion forms hydrophobic interactions with the other side of the channel and a hydrogen bond with the backbone of Gly155. It appears that the main difference between 3b-derived activators and inhibitors consists of the presence of the bulky substituent on ring C and consequent tilted position of the chroman, which is saturated in inhibitors 17a,b. This is supported by the fact that the orientation of the pyrrolo[1,2-a]quinoxaline of the SIRT6 activator 5 is similar to the ring C of 3b derivatives, rather than 17a. Nonetheless, these compounds were not tested against other SIRTs or HDAC isoforms, so their selectivity needs to be further investigated. In addition, given their polyphenolic structure, both compounds very likely display pleiotropic off-target effects, as previously described for compounds 3a–e. Indeed, 17a–b also inhibit α-glucosidase,157 while inhibition of topoisomerases has been widely reported for the 17b epimer 17d.158,159 This compound also exhibited dual activity toward SIRT3, acting as either an activator or inhibitor depending on the cellular context.160 In addition, 17d inhibits DNMT1113,114 and different HAT enzymes such as p300, CBP, PCAF, and Tip60.86,87 Even though 17a–b have poor specificity, the availability of the SIRT6–17a cocrystal structure could be exploited by medicinal chemists for drug design, as previously mentioned in the case of activators 3b and 3e.
Figure 12.

(A) Upper panel: Flavonole-based SIRT6 inhibitors. Lower panel: Structure of SIRT6 in complex with ADP–ribose (yellow) and catechin gallate (17a) (green) with a focus on the 17a binding site (PDB ID: 6QCJ). (B) Upper panel: TSA (18) structure. Lower panel: Structure of SIRT6 in complex with ADP–ribose (yellow) and 18 (green) with a focus on the 18 binding site (PDB ID: 6HOY). The binding mode is substantially different from 17a, although some interactions are shared, such as the key water-mediated hydrogen bond with Pro62 and the hydrophobic contacts with Trp71, Phe82, and Phe86. Key residues for compounds’ binding are labeled, key water molecules for the protein-compound interaction are represented as red spheres, and polar interactions are shown as dashed orange lines.
Trichostatin A (18, TSA), a hydroxamate derivative known for its nanomolar inhibitory activity of class I and II HDACs given its zinc-chelating properties, was recently found to inhibit SIRT6.161 Though no IC50 was calculated, the Ki values for 18-mediated SIRT6 deacetylation were 2.02 μM when using H3K9Ac peptide and 4.62 μM when using p53K382Ac peptide. No inhibitory activity was observed against SIRT1–3 and SIRT5 up to a 50 μM concentration. Kinetic analysis indicated competitive inhibition toward the acetylated peptide but not NAD+. The crystal structure of the SIRT6/ADP–ribose/18 complex (PDB ID: 6HOY, Figure 12B) indicated the binding of 18 to the acyl channel extension of SIRT6, explaining its isoform specificity.162 The hydroxamate moiety of 18 engages in polar interactions, including a water-mediated hydrogen bond between 18 nitrogen and the backbone oxygens of Ile 61 and Pro62. The carbonyl group is involved in hydrogen bonds with the backbone nitrogen of Val115 and Asp116. In addition, the 18 hydroxyl moiety acts as a hydrogen bond donor in its interaction with the side chains of Asn114 and Asp116 (Figure 12B). The 18 hydroxamate group mimics 11a interactions, as confirmed by a competition binding assay in the presence of 11a. Since no 18/NAD+ competition was observed in activity assays, the authors propose a mechanism whereby 18 interacts with the 11a binding region following the release of the 11a moiety from NAD+. They also argue that the reported acetylated peptide competition is caused indirectly, by inducing conformational changes leading to clashes with the acylated substrate.
Beyond drug repurposing, the first synthetic small-molecule compounds displaying SIRT6 inhibition were identified by Parenti et al. following an in silico screening.163 This approach led to the discovery of the derivatives 19a–c (Figure 13) possessing IC50 values of 106, 89, and 181 μM, respectively. Among them, compounds 19b (subsequently named OSS_128167) and 19c displayed selectivity over SIRT1 and SIRT2 (IC50 values 8.44 to 19.15 times higher), while 19a was mildly selective over SIRT1 (IC50 = 314 μM) but not over SIRT2 (IC50 = 114 μM). The three compounds increased H3K9 acetylation in BxPC3 cells and induced GLUT1 upregulation and consequent augmented glucose uptake in L6 rat myoblasts and BxPC3 cells. This is consistent with reports indicating the role of SIRT6 in GLUT1 downregulation.59 Furthermore, the compounds were able to reduce TNF-α secretion. This study shows that small-molecule-mediated SIRT6 inhibition mimics the effects of SIRT6 knockdown. When tested in a murine model of type 2 diabetes, 19a improved glucose tolerance and reduced plasma levels of insulin, triglycerides, and cholesterol.164 Remarkably, 19b reduced the recruitment of SIRT6 to DNA-damage locations and sensitized primary MM cells, along with melphalan- and doxorubicin-resistant MM cell lines, to DNA-damaging chemotherapeutics.165 Compound 19b also decreased the viability of DLBCL cells, usually displaying SIRT6 overexpression, and inhibited their proliferation in a time- and dose-dependent fashion, through induction of apoptosis and cell cycle arrest at G2/M phase. When tested in a mouse xenograft model with human DLBCL cells, 19b reduced tumor growth and decreased the levels of the proliferative marker Ki-67.79 Nevertheless, these reports lack of target engagement studies demonstrating that 19b does bind to SIRT6 at least in the cellular context. Hence, the observed in vivo phenotype may be a consequence of off-target effects, particularly considering the weak in vitro potency of 19b. To shed light on this, cellular and in vivo target engagement studies should be performed.139−143 Moreover, the comparison between the phenotypes induced by 19b treatment, by SIRT6 gene knockdown, and by a combination of the two should also be carried out to clarify the mechanism of action144,145 and unambiguously link the observed anticancer effects to SIRT6 inhibition.
Figure 13.

Synthetic small-molecule SIRT6 inhibitors.
Optimization of compound 19a led to the quinazolinedione derivatives 20a–c (Figure 13, left).166 Compounds 20a and 20b are characterized by different substituents on the sulphonamide residue; in addition to this, in compound 20c, the nitrogen atoms of the quinazolinedione core are methylated. These substitutions led to improved SIRT6 inhibition (IC50 (20a) = 60 μM; IC50 (20b) = 37 μM; IC50 (20c) = 49 μM).Compound 20a was slightly selective over SIRT1 and SIRT2 with IC50 values of 238 and 159 μM, respectively. Compounds 20b–c exhibited good selectivity over SIRT1 (IC50 values were 11 and 133 times higher, respectively) and low selectivity over SIRT2 (2.30-fold and 4.94-fold, respectively), although the activity against other isoforms remains to be tested. It appears that removal of oxygen in the sulphonamide side-chain and the extension of the aliphatic spacer between the aromatic groups (see 20b) improves the inhibitory efficiency of these derivatives. In addition, the simultaneous oxygen removal from the side chain and methylation of the quinazolinedione nitrogens increases isoform specificity. These derivatives increased H3K9 acetylation in BxPC3, but only compounds 20b and 20c caused increased glucose uptake in L6 rat myoblasts and BxPC3 cells. Remarkably, 20a and 20b were able to sensitize BxPC3 cells to the chemotherapeutic gemcitabine. Compound 20c was not evaluated in vivo, since it was found to be cytotoxic at a concentration close to its IC50 (30 μM). Compounds 19a and 20b were found to effectively enhance the anticancer activity of the PARP inhibitor olaparib in Capan-1 cells (a BRCA2-deficient pancreatic cancer cell line). These observations are consistent with previous findings suggesting that SIRT6 knockdown improves the efficacy of chemotherapeutics.167
The salicylate derivative 19b was further optimized yielding the highly selective SIRT6 inhibitors 21a–c (Figure 13, right). Compound 21a is an analogue of 19b, in which the furan-2-carboxamide moiety is shifted from 3′ to 4′. Compound 21b presents the furan-2-carboxamide at the same position as 21a but has the hydroxyl and carboxylic groups swapped with each other. 21a and 21b have IC50 values of 34 and 22 μM, respectively. These data indicate that the presence of the furan-2-carboxamide at para position massively increases the SIRT6 inhibitory activity, while the swap of hydroxyl and carboxylic groups leads to only a slight improvement of the inhibition. In compound 21c, a phthalimide moiety replaces the furan-2-carboxamide in 3′, while the carboxylic and hydroxyl groups are in positions 2 and 4, respectively. These modifications furnished a compound with slightly improved inhibitory efficacy, having an IC50 of 20 μM.168 All compounds displayed selectivity over SIRT1 and SIRT2 (IC50 values between 13 and 27 times higher), although the selectivity over other SIRT isoforms needs to be evaluated. Compounds 21a and 21b increased H3K9 acetylation and glucose uptake in human peripheral blood mononuclear cells (PBMCs), in line with previous studies and with the roles of SIRT6 in cell homeostasis. Conversely, compound 21c did not show any effect in cell-based assays, probably due to a lack of cell permeability. Compounds 21a and 21b also impaired TNF-α secretion and sensitized pancreatic cancer cells to gemcitabine. Compound 21b also presented antiproliferative properties in PBMCs.
Compound screening based on a DNA-encoded library designed for NAD+-binding pockets led to the identification of two SIRT6 inhibitors with a 5-aminocarbonyl-uracil core (Figure 13, lower panel): A127-(CONHPr)-B178 (22a) and A127-(CONHMe)-B178 (22b). Both molecules were evaluated in a demyristoylation assay and displayed IC50 values of 6.7 and 9.2 μM, respectively.169 Compound 22a was selective over other SIRTs, as inhibition of SIRT1–3, SIRT5, and SIRT7 was less than 10% at 10 μM and was stable in serum after 72 h. It caused an increase of DNA-damage markers and telomere-dysfunction-induced foci in primary human umbilical venous endothelial cells (HUVECs), like what was observed following SIRT6 knockdown.170 Similarly to other SIRT6 inhibitors, 22a caused a dose-dependent decrease in the TNF-α levels.
Recently, a series of 1-phenylpiperazine derivatives have been reported as a SIRT6 inhibitors. Among them, 5-(4-methylpiperazin-1-yl)-2-nitroaniline (23,Figure 13, lower panel) displayed an IC50 of 4.93 μM in a peptide deacetylation assay and showed no activity against SIRT1–3 and HDAC1–11 up to 200 μM concentration.171 When tested in BxPC-3 cells, compound 23 augmented the level of both H3K9 and H3K18 acetylation in a dose-dependent manner and increased GLUT1 expression levels. In addition, it reduced the blood glucose content in a mouse model of type 2 diabetes, thus demonstrating promising lead-like properties.
Conclusions
Mounting evidence supports the critical roles of SIRT6 in multiple processes regulating both physiological and pathological states. Although SIRT6 shares mechanistic features with other SIRTs, it differs from them, as it greatly depends on FFA activation to increase the efficiency of its enzymatic activities.20,22 Through its multiple enzymatic activities, SIRT6 finely regulates not only genome maintenance and DNA repair but also stem cell differentiation, metabolism, and aging. The involvement of SIRT6 in these key processes may explain its dual role in cancer. For instance, DNA repair promotion may help evasion from tumorigenic transformation at early phases of cancer. On the other hand, the same mechanism may facilitate cancer progression at later stages or decrease the effectiveness of cytotoxic drug chemotherapy. It is worth noticing that the upregulation of SIRT6 in certain types of cancers76−79 may be representative of a compensatory effect rather than the cause itself of tumor initiation and/or progression.76
SIRT6 also regulates crucial proteins involved in sugar homeostasis, as it promotes the expression of glycolytic genes,59,60 suppressing gluconeogenesis and increasing insulin secretion, hence having a favorable role in diabetes. The downregulation of glycolytic genes also acts as a tumor-suppressor pathway, since it suppresses the Warburg effect.58 SIRT6 also regulates fat metabolism by reducing LDL-cholesterol levels95 and triglyceride synthesis as well as promoting fatty-acid β-oxidation,47 being a key player in obesity prevention. Like the double-faced role in cancer, SIRT6 has contrasting actions in the regulation of inflammation.80,84,85
Although the growing knowledge about SIRT6 biology has been uncovering multifaceted functions in human diseases, the discovery of potent and selective SIRT6 modulators is at its infancy. The notion that FFAs increase the deacetylation efficiency of SIRT6 led to the investigation and discovery of the first SIRT6 activators. Initial hit compounds were derived from simple modifications of fatty acids, such as the ethanolamides 2a and 2b.102 Subsequent synthetic activators overcame issues directly related to the lipidic structure, such as metabolic instability, poor cellular permeability, and low water solubility. Ligand-based drug design efforts led to 5, the first synthetic activator yielding cellular effects at mid-μM concentrations.124,125 This discovery paved the way for the development of further activators, such as 7a.128 Although the binding mode of the analogue 7b raised some discussion,130,131 it is possible that both proposed models are valid in different conditions, and this controversy reminds us that ligand–protein interactions cannot be always recapitulated by a single-crystal structure. In any case, both 7a and its derivative 7c(132) inhibited tumor growth in xenograft models, showing SIRT6 activation efficacy in vivo for the first time.129,132 The recently described activator 10b,138 developed using the 5 binding mode as a model, displayed efficacy at low micromolar concentrations, being an activator of both deacetylation and demyristoylation activities. Remarkably, it also possessed antitumor activity in vivo, even though it showed poor water solubility and very low bioavailability. Notably, CETSA measurements demonstrated that 10b binds to SIRT6 in cells. Nonetheless, further studies are necessary to verify whether the observed phenotypic effects are genuinely related only to SIRT6 activation. Anyhow, 10b represents a good lead compound that still necessitates a full validation and optimization of the pharmacodynamic and pharmacokinetic properties to be considered as a therapeutic option in the PDAC context.
In the case of inhibitors, the development of substrate-based peptidomimetics led to compounds 15a–f that displayed SIRT6 inhibition in the nanomolar range, although the only compound tested in vivo (15f) did not show any effect. Nonetheless, given the high potency, these compounds represent optimal starting scaffolds for further developments, first aimed at improving the cellular permeability. Differently, compounds 19b, 20b, and 21b, developed following structure- and ligand-based drug design strategies, were cellularly active, although they inhibited SIRT6 enzymatic activity only in the micromolar range. 19b also displayed efficacy in a mouse xenograft model of DLBCL; however, additional analyses are necessary to demonstrate a causal correlation between its anticancer activity and SIRT6 inhibition. Therefore, currently, 19b can be considered only a hit molecule that needs complete validation and, in case, extensive optimization of potency and selectivity.
Innovative approaches relying on high-throughput compound testing hold great potential for drug discovery. Compound 22a has been discovered by means of DNA-encoded libraries,169 a combinatorial approach in which the structure of each molecule is encoded by a conjugated DNA identifier sequence.172,173 This method allows quick testing of millions of combinations of fragments using micrograms of protein, offering the exploration of a vast chemical space. The successful application of this approach to SIRT6 led to a low micromolar inhibitor (22a) endowed with cellular activity. The application of this technique also to SIRT6 activator discovery would be very interesting.
Finally, compound 23 represents an exciting prospect. It showed low micromolar activity in vitro and was able to cause blood glucose reduction in a mouse model of diabetes.171 Moreover, its simple structure is amenable of modifications that may lead to more potent derivatives upon a proper structural optimization.
Different challenges have been characterizing the path to the discovery of SIRT6 modulators. These include initial difficulties of properly separating activation and inhibition and the suboptimal efficacy of currently discovered modulators, as explained by the absence of nanomolar activators thus far.
The recent discoveries of in vivo active compounds (particularly 7c and 10b) bring good hopes for the development of further potent and selective SIRT6 activators. In the case of inhibitors, researchers managed to obtain nanomolar or low micromolar compounds such as 15f, 22a, and 23, plus the mid micromolar inhibitor 19b, which was active both in cell and in vivo, although its SIRT6 target engagement needs to be demonstrated. These molecules cover different chemical classes, ranging from peptidomimetics to small molecules, and some of them represent ideal hit/lead compounds for further development.
Anyway, additional efforts are necessary to improve the potency of the currently available SIRT6 modulators, since usually only nanomolar compounds have concrete chances to progress to preclinical and clinical phases.
To this end, structure-based drug design approaches might be particularly beneficial. Indeed, the availability of the crystal structures of SIRT6 in complex with both activators and inhibitors of polyphenolic nature, such as 3b, 3e, and 17a, enable the identification of key features for target recognition and activity modulation. These cocrystals could be exploited to develop new activating or inhibiting compounds whereby only the important hydroxyl groups are kept while removing the nonessential ones, thus abolishing their pleiotropic effects and increasing their specificity. Moreover, in order to increase potency and selectivity, scaffold hopping120,121 approaches could be applied to develop molecules bearing different core chemotypes but retaining the key moieties for SIRT6 interaction. The inspection of the X-ray solved crystal structures of SIRT6 in complex with different modulators that bind in the same pocket with similar binding modes such as the activators 3b (Figure 6B) and 5 (Figure 7A) could be also leveraged for the structural optimization by combining crucial chemical features. For instance, compound 5 might be modified through the addition of polar groups to the pyridine moiety to form hydrogen bonds with the conserved water molecules bridging to Val115 and Asp116. Moreover, the addition of large hydrophobic groups to the benzene ring could allow further hydrophobic interactions to be established with the pocket formed by Ile61, Phe82 and Phe86.
In conclusion, the available cocrystal structures, along with cutting-edge approaches such as artificial-intelligence-driven drug design and DNA-encoded libraries, have great potential in allowing the evaluation of a more diverse chemical space to obtain molecules possessing drug-like properties to facilitate the discovery of new SIRT6 modulators. To date, the ideal scenario for the initial evaluation of SIRT6 targeting molecules relies on the integration of structural approaches with classic biophysical assays174 and modern, label-free methods such as those based on mass spectrometry,175−177 to allow reliable assessment of protein–ligand interactions and avoid false positives and negatives that may impair the following steps of a drug discovery campaign.
Acknowledgments
This work was supported by PRIN 2016 (prot. 20152TE5PK) (A.M.), AIRC 2016 (n. 19162) (A.M.), and Progetto di Ateneo “Sapienza” 2017 n. RM11715C7CA6CE53 (D.R.).
Glossary
Abbreviations Used
- 5AC
5-azacytidine
- 5-hmC
5-hydroxymethylcytosine
- 5-mC
5-methylcytosine
- ABPP
affinity-based protein profiling
- AMPK
AMP-activated protein kinase
- Bax
Bcl-2-associated X protein
- BER
base excision repair
- CETSA
cellular thermal shift assay
- CREB
cAMP response element-binding protein
- CRC
colorectal cancer
- DAC
decitabine
- DDR
DNA-damage repair
- DLBCL
diffuse large B-cell lymphoma
- DNMT1
DNA methyltransferase 1
- DSB
double-strand break
- EMT
epithelial–mesenchymal transition
- E2F1
E2 transcription factor 1
- FFA
free fatty acid
- G6PC
glucose-6-phosphatase
- FRET
fluorescence resonance energy transfer
- GCN5
general control nonderepressible 5
- GLUT
glucose transporter
- HAT
histone acetyltransferase
- HCC
hepatocellular carcinoma
- HDASH
histone deacetylase assay homogeneous
- HIF-1α
hypoxia-inducible factor 1α
- HR
homologous recombination
- HSCs
hematopoietic stem cells
- IGF
insulin-like growth factor
- IGFBP
insulin-like growth factor-binding protein
- IL-6
interleukin-6
- IL-8
interleukin-8
- iPSC
induced pluripotent stem cell
- LDH
lactate dehydrogenase
- LINE-1
long interspersed element-1
- lncRNA
long noncoding RNA
- MES
morpholinoethanesulfonic acid
- MDM2
mouse double minute 2 homologue
- MEFs
mouse embryonic fibroblasts
- miRNA
micro-RNA
- MM
multiple myeloma
- MSCs
mesenchymal stem cells
- NHEJ
nonhomologous end-joining
- NPC
nasopharyngeal carcinoma
- NRF2
nuclear factor erythroid 2-related factor
- PAINS
pan assay interference compounds
- PARP1
poly(ADP–ribose)polymerase 1
- PBMC
peripheral blood mononuclear cell
- PCPB2
poly(C)-binding protein 2
- PCD
programmed cell death
- PCK1
phosphoenolpyruvate carboxykinase
- PCSK9
proprotein convertase subtilisin/kexin type 9
- PDAC
pancreatic ductal adenocarcinoma
- PDK1
pyruvate dehydrogenase kinase-1
- PFK1
phosphofructokinase-1
- PKM2
pyruvate kinase M2
- PTM
post-translational modification
- RTE
retrotransposable element
- RUNX2
runt-related transcription factor 2
- SIRTs
sirtuins
- SIRT6
sirtuin 6
- SUMO
small ubiquitin-related modifier
- SREBP
sterol regulatory element-binding protein
- TET
ten-11 translocation methylcytosine dioxygenase
- TRF2
telomere repeat binding factor 2
- TSA
trichostatin A
- UTR
untranslated region
- WRN
Werner syndrome ATP-dependent helicase
- XIAP
X-linked inhibitor of apoptosis protein
Biographies
Francesco Fiorentino graduated in Medicinal Chemistry at the University of Rome “La Sapienza” (Italy) in 2016. He received his Ph.D. in Biophysical Chemistry at University of Oxford (UK) in 2020 under the supervision of Prof. Dame Carol Robinson, working on the elucidation of the structure and regulation of membrane proteins using mass spectrometry. He is now a Postdoctoral Research Associate at the University of Oxford. His research activity has been directed toward the investigation of the molecular mechanisms underpinning protein function and modulation. To this end, he is applying native mass spectrometry and other biophysical techniques to investigate the protein complexes involved in bacterial membrane biogenesis and epigenetics.
Antonello Mai graduated in Pharmacy at the University of Rome “La Sapienza”, Italy, in 1984. He received his Ph.D. in 1992 in Pharmaceutical Sciences, with a thesis entitled “Researches on New Polycyclic Benzodiazepines Active on Central Nervous System”, with advisor Prof. M. Artico. In 1998, he was appointed Associate Professor of Medicinal Chemistry at the same University. In 2011, Prof. Mai was appointed Full Professor of Medicinal Chemistry at the Faculty of Pharmacy and Medicine, Sapienza University of Rome. He has published more than 250 papers in peer-reviewed high-impact factor journals. His research interests include the synthesis and biological evaluation of new bioactive small-molecule compounds, in particular modulators of epigenetic targets. In addition, he is working in the field of antibacterial/antimycobacterial, antiviral, and CNS agents.
Dante Rotili graduated in Medicinal Chemistry at the University of Rome “La Sapienza” (Italy) in 2003. He received his Ph.D. in Pharmaceutical Sciences at the same University in 2007. In 2009/2010, he was a research associate at the Department of Chemistry of the University of Oxford, where he worked in collaboration with Prof. C. Schofield in the development of chemoproteomic probes for the characterization of 2-oxoglutarate-dependent enzymes. In 2020, he was appointed as Associate Professor of Medicinal Chemistry at the University of Rome “La Sapienza”. Since 2017, he has had the Italian National Habilitation to Full Professor of Medicinal Chemistry. His research activity has been focusing mainly on the development of modulators of epigenetic enzymes with potential applications in cancer, neurodegenerative, metabolic, and infectious diseases.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
References
- Finkel T.; Deng C. X.; Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature 2009, 460, 587–591. 10.1038/nature08197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang A. R.; Ferrer C. M.; Mostoslavsky R. SIRT6, a mammalian deacylase with multitasking abilities. Physiol. Rev. 2020, 100, 145–169. 10.1152/physrev.00030.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein M. A.; Denu J. M. Biological and catalytic functions of sirtuin 6 as targets for small-molecule modulators. J. Biol. Chem. 2020, 295, 11021–11041. 10.1074/jbc.REV120.011438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liszt G.; Ford E.; Kurtev M.; Guarente L. Mouse Sir2 homolog SIRT6 is a nuclear ADP-ribosyltransferase. J. Biol. Chem. 2005, 280, 21313–21320. 10.1074/jbc.M413296200. [DOI] [PubMed] [Google Scholar]
- Imai S.-I.; Guarente L. It takes two to tango: NAD(+) and sirtuins in aging/longevity control. NPJ. Aging Mech. Dis. 2016, 2, 16017. 10.1038/npjamd.2016.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostoslavsky R.; Chua K. F.; Lombard D. B.; Pang W. W.; Fischer M. R.; Gellon L.; Liu P.; Mostoslavsky G.; Franco S.; Murphy M. M.; Mills K. D.; Patel P.; Hsu J. T.; Hong A. L.; Ford E.; Cheng H. L.; Kennedy C.; Nunez N.; Bronson R.; Frendewey D.; Auerbach W.; Valenzuela D.; Karow M.; Hottiger M. O.; Hursting S.; Barrett J. C.; Guarente L.; Mulligan R.; Demple B.; Yancopoulos G. D.; Alt F. W. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 2006, 124, 315–329. 10.1016/j.cell.2005.11.044. [DOI] [PubMed] [Google Scholar]
- Kanfi Y.; Naiman S.; Amir G.; Peshti V.; Zinman G.; Nahum L.; Bar-Joseph Z.; Cohen H. Y. The sirtuin SIRT6 regulates lifespan in male mice. Nature 2012, 483, 218–221. 10.1038/nature10815. [DOI] [PubMed] [Google Scholar]
- Sebastián C.; Zwaans B. M. M.; Silberman D. M.; Gymrek M.; Goren A.; Zhong L.; Ram O.; Truelove J.; Guimaraes A. R.; Toiber D.; Cosentino C.; Greenson J. K.; MacDonald A. I.; McGlynn L.; Maxwell F.; Edwards J.; Giacosa S.; Guccione E.; Weissleder R.; Bernstein B. E.; Regev A.; Shiels P. G.; Lombard D. B.; Mostoslavsky R. The histone deacetylase SIRT6 Is a tumor suppressor that controls cancer metabolism. Cell 2012, 151, 1185–1199. 10.1016/j.cell.2012.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kugel S.; Feldman J. L.; Klein M. A.; Silberman D. M.; Sebastián C.; Mermel C.; Dobersch S.; Clark A. R.; Getz G.; Denu J. M.; Mostoslavsky R. Identification of and Molecular Basis for SIRT6 Loss-of-Function Point Mutations in Cancer. Cell Rep. 2015, 13, 479–488. 10.1016/j.celrep.2015.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Ceu Teixeira M.; Sanchez-Lopez E.; Espina M.; Garcia M. L.; Durazzo A.; Lucarini M.; Novellino E.; Souto S. B.; Santini A.; Souto E. B. Sirtuins and SIRT6 in Carcinogenesis and in Diet. Int. J. Mol. Sci. 2019, 20, 4945. 10.3390/ijms20194945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorentino F.; Carafa V.; Favale G.; Altucci L.; Mai A.; Rotili D. The Two-Faced Role of SIRT6 in Cancer. Cancers 2021, 13, 1156. 10.3390/cancers13051156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer C. M.; Alders M.; Postma A. V.; Park S.; Klein M. A.; Cetinbas M.; Pajkrt E.; Glas A.; van Koningsbruggen S.; Christoffels V. M.; Mannens M. M. A. M.; Knegt L.; Etchegaray J. P.; Sadreyev R. I.; Denu J. M.; Mostoslavsky G.; van Maarle M. C.; Mostoslavsky R. An inactivating mutation in the histone deacetylase SIRT6 causes human perinatal lethality. Genes Dev. 2018, 32, 373–388. 10.1101/gad.307330.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W.; Wan H.; Feng G.; Qu J.; Wang J.; Jing Y.; Ren R.; Liu Z.; Zhang L.; Chen Z.; Wang S.; Zhao Y.; Wang Z.; Yuan Y.; Zhou Q.; Li W.; Liu G. H.; Hu B. SIRT6 deficiency results in developmental retardation in cynomolgus monkeys. Nature 2018, 560, 661–665. 10.1038/s41586-018-0437-z. [DOI] [PubMed] [Google Scholar]
- Kanfi Y.; Shalman R.; Peshti V.; Pilosof S. N.; Gozlan Y. M.; Pearson K. J.; Lerrer B.; Moazed D.; Marine J. C.; de Cabo R.; Cohen H. Y. Regulation of SIRT6 protein levels by nutrient availability. FEBS Lett. 2008, 582, 543–548. 10.1016/j.febslet.2008.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michishita E.; McCord R. A.; Berber E.; Kioi M.; Padilla-Nash H.; Damian M.; Cheung P.; Kusumoto R.; Kawahara T. L. A.; Barrett J. C.; Chang H. Y.; Bohr V. A.; Ried T.; Gozani O.; Chua K. F. SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature 2008, 452, 492–496. 10.1038/nature06736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michishita E.; McCord R. A.; Boxer L. D.; Barber M. F.; Hong T.; Gozani O.; Chua K. F. Cell cycle-dependent deacetylation of telomeric histone H3 lysine K56 by human SIRT6. Cell Cycle 2009, 8, 2664–2666. 10.4161/cc.8.16.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B.; Zwaans B. M. M.; Eckersdorff M.; Lombard D. B. The sirtuin SIRT6 deacetylates H3 K56Ac in vivo to promote genomic stability. Cell Cycle 2009, 8, 2662–2663. 10.4161/cc.8.16.9329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasselli L.; Xi Y.; Zheng W.; Tennen R. I.; Odrowaz Z.; Simeoni F.; Li W.; Chua K. F. SIRT6 deacetylates H3K18ac at pericentric chromatin to prevent mitotic errors and cellular senescence. Nat. Struct. Mol. Biol. 2016, 23, 434–440. 10.1038/nsmb.3202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan P. W.; Feldman J. L.; Devries M. K.; Dong A.; Edwards A. M.; Denu J. M. Structure and biochemical functions of SIRT6. J. Biol. Chem. 2011, 286, 14575–14587. 10.1074/jbc.M111.218990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman J. L.; Baeza J.; Denu J. M. Activation of the protein deacetylase SIRT6 by long-chain fatty acids and widespread deacylation by Mammalian Sirtuins. J. Biol. Chem. 2013, 288, 31350–31356. 10.1074/jbc.C113.511261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein M. A.; Liu C.; Kuznetsov V. I.; Feltenberger J. B.; Tang W.; Denu J. M. Mechanism of activation for the sirtuin 6 protein deacylase. J. Biol. Chem. 2020, 295, 1385–1399. 10.1016/S0021-9258(17)49896-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H.; Khan S.; Wang Y.; Charron G.; He B.; Sebastian C.; Du J.; Kim R.; Ge E.; Mostoslavsky R.; Hang H. C.; Hao Q.; Lin H. SIRT6 regulates TNF-α secretion through hydrolysis of long-chain fatty acyl lysine. Nature 2013, 496, 110–113. 10.1038/nature12038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Spiegelman N. A.; Nelson O. D.; Jing H.; Lin H. SIRT6 regulates Ras-related protein R-Ras2 by lysine defatty-acylation. eLife 2017, 6, e25158 10.7554/eLife.25158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnin M. S.; Donigian J. R.; Pavletich N. P. Structure of the histone deacetylase SIRT2. Nat. Struct. Biol. 2001, 8, 621–625. 10.1038/89668. [DOI] [PubMed] [Google Scholar]
- Schuetz A.; Min J.; Antoshenko T.; Wang C. L.; Allali-Hassani A.; Dong A.; Loppnau P.; Vedadi M.; Bochkarev A.; Sternglanz R.; Plotnikov A. N. Structural Basis of Inhibition of the Human NAD+-Dependent Deacetylase SIRT5 by Suramin. Structure 2007, 15, 377–389. 10.1016/j.str.2007.02.002. [DOI] [PubMed] [Google Scholar]
- Jin L.; Wei W.; Jiang Y.; Peng H.; Cai J.; Mao C.; Dai H.; Choy W.; Bemis J. E.; Jirousek M. R.; Milne J. C.; Westphal C. H.; Perni R. B. Crystal structures of human SIRT3 displaying substrate-induced conformational changes. J. Biol. Chem. 2009, 284, 24394–24405. 10.1074/jbc.M109.014928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders B. D.; Jackson B.; Marmorstein R. Structural basis for sirtuin function: What we know and what we don’t. Biochim. Biophys. Acta, Proteins Proteomics 2010, 1804, 1604–1616. 10.1016/j.bbapap.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil R.; Barth S.; Kanfi Y.; Cohen H. Y. SIRT6 exhibits nucleosome-dependent deacetylase activity. Nucleic Acids Res. 2013, 41, 8537–8545. 10.1093/nar/gkt642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W. W.; Zeng Y.; Wu B.; Deiters A.; Liu W. R. A Chemical Biology Approach to Reveal Sirt6-targeted Histone H3 Sites in Nucleosomes. ACS Chem. Biol. 2016, 11, 1973–1981. 10.1021/acschembio.6b00243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Khan S.; Jiang H.; Antonyak M. A.; Chen X.; Spiegelman N. A.; Shrimp J. H.; Cerione R. A.; Lin H. Identifying the functional contribution of the defatty-Acylase activity of SIRT6. Nat. Chem. Biol. 2016, 12, 614–620. 10.1038/nchembio.2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Meter M.; Simon M.; Tombline G.; May A.; Morello T. D.; Hubbard B. P.; Bredbenner K.; Park R.; Sinclair D. A.; Bohr V. A.; Gorbunova V.; Seluanov A. JNK Phosphorylates SIRT6 to Stimulate DNA Double-Strand Break Repair in Response to Oxidative Stress by Recruiting PARP1 to DNA Breaks. Cell Rep. 2016, 16, 2641–2650. 10.1016/j.celrep.2016.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Meter M.; Kashyap M.; Rezazadeh S.; Geneva A. J.; Morello T. D.; Seluanov A.; Gorbunova V. SIRT6 represses LINE1 retrotransposons by ribosylating KAP1 but this repression fails with stress and age. Nat. Commun. 2014, 5, 5011. 10.1038/ncomms6011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezazadeh S.; Yang D.; Tombline G.; Simon M.; Regan S. P.; Seluanov A.; Gorbunova V. SIRT6 promotes transcription of a subset of NRF2 targets by mono-ADP-ribosylating BAF170. Nucleic Acids Res. 2019, 47, 7914–7928. 10.1093/nar/gkz528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezazadeh S.; Yang D.; Biashad S. A.; Firsanov D.; Takasugi M.; Gilbert M.; Tombline G.; Bhanu N. V.; Garcia B. A.; Seluanov A.; Gorbunova V. SIRT6 mono-ADP ribosylates KDM2A to locally increase H3K36me2 at DNA damage sites to inhibit transcription and promote repair. Aging 2020, 12, 11165–11184. 10.18632/aging.103567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostoslavsky R.; Chua K. F.; Lombard D. B.; Pang W. W.; Fischer M. R.; Gellon L.; Liu P. F.; Mostoslavsky G.; Franco S.; Murphy M. M.; Mills K. D.; Patel P.; Hsu J. T.; Hong A. L.; Ford E.; Cheng H. L.; Kennedy C.; Nunez N.; Bronson R.; Frendewey D.; Auerbach W.; Valenzuela D.; Karow M.; Hottiger M. O.; Hursting S.; Barrett J. C.; Guarente L.; Mulligan R.; Demple B.; Yancopoulos G. D.; Alt F. W. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 2006, 124, 315–329. 10.1016/j.cell.2005.11.044. [DOI] [PubMed] [Google Scholar]
- McCord R. A.; Michishita E.; Hong T.; Berber E.; Boxer L. D.; Kusumoto R.; Guan S.; Shi X.; Gozani O.; Burlingame A. L.; Bohr V. A.; Chua K. F. SIRT6 stabilizes DNA-dependent protein kinase at chromatin for DNA double-strand break repair. Aging 2009, 1, 109–121. 10.18632/aging.100011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toiber D.; Erdel F.; Bouazoune K.; Silberman D.; Zhong L.; Mulligan P.; Sebastian C.; Cosentino C.; Martinez-Pastor B.; Giacosa S.; D’Urso A.; Näär A.; Kingston R.; Rippe K.; Mostoslavsky R. SIRT6 recruits SNF2H to DNA break sites, preventing genomic instability through chromatin remodeling. Mol. Cell 2013, 51, 454–468. 10.1016/j.molcel.2013.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Z.; Hine C.; Tian X.; Van Meter M.; Au M.; Vaidya A.; Seluanov A.; Gorbunova V. SIRT6 promotes DNA repair under stress by activating PARP1. Science 2011, 332, 1443–1446. 10.1126/science.1202723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman J. L.; Dittenhafer-Reed K. E.; Kudo N.; Thelen J. N.; Ito A.; Yoshida M.; Denu J. M. Kinetic and structural basis for Acyl-group selectivity and NAD+ dependence in sirtuin-catalyzed deacylation. Biochemistry 2015, 54, 3037–3050. 10.1021/acs.biochem.5b00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onn L.; Portillo M.; Ilic S.; Cleitman G.; Stein D.; Kaluski S.; Shirat I.; Slobodnik Z.; Einav M.; Erdel F.; Akabayov B.; Toiber D. SIRT6 is a DNA double-strand break sensor. eLife 2020, 9, e51636 10.7554/eLife.51636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaluski S.; Portillo M.; Besnard A.; Stein D.; Einav M.; Zhong L.; Ueberham U.; Arendt T.; Mostoslavsky R.; Sahay A.; Toiber D. Neuroprotective Functions for the Histone Deacetylase SIRT6. Cell Rep. 2017, 18, 3052–3062. 10.1016/j.celrep.2017.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z.; Zhang L.; Zhang W.; Meng D.; Zhang H.; Jiang Y.; Xu X.; Van Meter M.; Seluanov A.; Gorbunova V.; Mao Z. SIRT6 rescues the age related decline in base excision repair in a PARP1-dependent manner. Cell Cycle 2015, 14, 269–276. 10.4161/15384101.2014.980641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang B. J.; Jin J.; Gao Y.; Shi G.; Madabushi A.; Yan A.; Guan X.; Zalzman M.; Nakajima S.; Lan L.; Lu A. L. SIRT6 protein deacetylase interacts with MYH DNA glycosylase, APE1 endonuclease, and Rad9-Rad1-Hus1 checkpoint clamp. BMC Mol. Biol. 2015, 16, 12. 10.1186/s12867-015-0041-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzo A.; Iachettini S.; Salvati E.; Zizza P.; Maresca C.; D’Angelo C.; Benarroch-Popivker D.; Capolupo A.; Del Gaudio F.; Cosconati S.; Di Maro S.; Merlino F.; Novellino E.; Amoreo C. A.; Mottolese M.; Sperduti I.; Gilson E.; Biroccio A. SIRT6 interacts with TRF2 and promotes its degradation in response to DNA damage. Nucleic Acids Res. 2017, 45, 1820–1834. 10.1093/nar/gkw1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara T. L. A.; Michishita E.; Adler A. S.; Damian M.; Berber E.; Lin M.; McCord R. A.; Ongaigui K. C. L.; Boxer L. D.; Chang H. Y.; Chua K. F. SIRT6 Links Histone H3 Lysine 9 Deacetylation to NF-κB-Dependent Gene Expression and Organismal Life Span. Cell 2009, 136, 62–74. 10.1016/j.cell.2008.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etchegaray J. P.; Chavez L.; Huang Y.; Ross K. N.; Choi J.; Martinez-Pastor B.; Walsh R. M.; Sommer C. A.; Lienhard M.; Gladden A.; Kugel S.; Silberman D. M.; Ramaswamy S.; Mostoslavsky G.; Hochedlinger K.; Goren A.; Rao A.; Mostoslavsky R. The histone deacetylase SIRT6 controls embryonic stem cell fate via TET-mediated production of 5-hydroxymethylcytosine. Nat. Cell Biol. 2015, 17, 545–557. 10.1038/ncb3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. S.; Xiao C.; Wang R. H.; Lahusen T.; Xu X.; Vassilopoulos A.; Vazquez-Ortiz G.; Jeong W. I.; Park O.; Ki S. H.; Gao B.; Deng C. X. Hepatic-specific disruption of SIRT6 in mice results in fatty liver formation due to enhanced glycolysis and triglyceride synthesis. Cell Metab. 2010, 12, 224–236. 10.1016/j.cmet.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp J. R.; Longworth M. S. Crossing the LINE toward genomic instability: LINE-1 retrotransposition in cancer. Front. Chem. 2015, 3, 68. 10.3389/fchem.2015.00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Y.; Xue Y.; Song C.; Grunstein M. Acetylated histone H3K56 interacts with Oct4 to promote mouse embryonic stem cell pluripotency. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 11493–11498. 10.1073/pnas.1309914110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W.; Song C.; Young N. L.; Sperling A. S.; Xu F.; Sridharan R.; Conway A. E.; Garcia B. A.; Plath K.; Clark A. T.; Grunstein M. Histone H3 Lysine 56 Acetylation Is Linked to the Core Transcriptional Network in Human Embryonic Stem Cells. Mol. Cell 2009, 33, 417–427. 10.1016/j.molcel.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh K. P.; Yabuuchi A.; Rao S.; Huang Y.; Cunniff K.; Nardone J.; Laiho A.; Tahiliani M.; Sommer C. A.; Mostoslavsky G.; Lahesmaa R.; Orkin S. H.; Rodig S. J.; Daley G. Q.; Rao A. Tet1 and Tet2 regulate 5-hydroxymethylcytosine production and cell lineage specification in mouse embryonic stem cells. Cell Stem Cell 2011, 8, 200–213. 10.1016/j.stem.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan H.; Guan D.; Liu X.; Li J.; Wang L.; Wu J.; Zhou J.; Zhang W.; Ren R.; Zhang W.; Li Y.; Yang J.; Hao Y.; Yuan T.; Yuan G.; Wang H.; Ju Z.; Mao Z.; Li J.; Qu J.; Tang F.; Liu G. H. SIRT6 safeguards human mesenchymal stem cells from oxidative stress by coactivating NRF2. Cell Res. 2016, 26, 190–205. 10.1038/cr.2016.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H.; Wu Y.; Fu D.; Liu Y.; Huang C. SIRT6 regulates osteogenic differentiation of rat bone marrow mesenchymal stem cells partially via suppressing the nuclear factor-κB signaling pathway. Stem Cells 2014, 32, 1943–1955. 10.1002/stem.1671. [DOI] [PubMed] [Google Scholar]
- Wang H.; Diao D.; Shi Z.; Zhu X.; Gao Y.; Gao S.; Liu X.; Wu Y.; Rudolph K. L.; Liu G.; Li T.; Ju Z. SIRT6 Controls Hematopoietic Stem Cell Homeostasis through Epigenetic Regulation of Wnt Signaling. Cell Stem Cell 2016, 18, 495–507. 10.1016/j.stem.2016.03.005. [DOI] [PubMed] [Google Scholar]
- Sharma A.; Diecke S.; Zhang W. Y.; Lan F.; He C.; Mordwinkin N. M.; Chua K. F.; Wu J. C. The role of SIRT6 protein in aging and reprogramming of human induced pluripotent stem cells. J. Biol. Chem. 2013, 288, 18439–18447. 10.1074/jbc.M112.405928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao G.; Wang H.; Xu C.; Wang P.; Chen J.; Wang P.; Sun Z.; Su Y.; Wang Z.; Han L.; Tong T. SIRT6 delays cellular senescence by promoting p27Kip1 ubiquitinproteasome degradation. Aging 2016, 8, 2308–2323. 10.18632/aging.101038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N.; Li Z.; Mu W.; Li L.; Liang Y.; Lu M.; Wang Z.; Qiu Y.; Wang Z. Calorie restriction-induced SIRT6 activation delays aging by suppressing NF-κB signaling. Cell Cycle 2016, 15, 1009–1018. 10.1080/15384101.2016.1152427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O. On the origin of cancer cells. Science 1956, 123, 309–314. 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- Zhong L.; D’Urso A.; Toiber D.; Sebastian C.; Henry R. E.; Vadysirisack D. D.; Guimaraes A.; Marinelli B.; Wikstrom J. D.; Nir T.; Clish C. B.; Vaitheesvaran B.; Iliopoulos O.; Kurland I.; Dor Y.; Weissleder R.; Shirihai O. S.; Ellisen L. W.; Espinosa J. M.; Mostoslavsky R. The Histone Deacetylase Sirt6 Regulates Glucose Homeostasis via Hif1α. Cell 2010, 140, 280–293. 10.1016/j.cell.2009.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhardwaj A.; Das S. SIRT6 deacetylates PKM2 to suppress its nuclear localization and oncogenic functions. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, E538–E547. 10.1073/pnas.1520045113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M.; Seto E.; Zhang J. E2F1 enhances glycolysis through suppressing Sirt6 transcription in cancer cells. Oncotarget 2015, 6, 11252–11263. 10.18632/oncotarget.3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe M.; Brusgard J. L.; Chumsri S.; Bhandary L.; Zhao X. F.; Lu S.; Goloubeva O. G.; Polster B. M.; Fiskum G. M.; Girnun G. D.; Kim M. S.; Passaniti A. The RUNX2 Transcription Factor Negatively Regulates SIRT6 Expression to Alter Glucose Metabolism in Breast Cancer Cells. J. Cell. Biochem. 2015, 116, 2210–2226. 10.1002/jcb.25171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah N. K.; Khan Z.; Khan G. J.; Bisen P. S. Structural, functional and therapeutic biology of survivin. Cancer Lett. 2006, 244, 164–171. 10.1016/j.canlet.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Lin X.; Shen J.; Peng D.; He X.; Xu C.; Chen X.; Tanyi J. L.; Montone K.; Fan Y.; Huang Q.; Zhang L.; Zhong X. RNA-binding protein LIN28B inhibits apoptosis through regulation of the AKT2/FOXO3A/BIM axis in ovarian cancer cells. Signal Transduct. Target. Ther. 2018, 3, 23. 10.1038/s41392-018-0026-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min L.; Ji Y.; Bakiri L.; Qiu Z.; Cen J.; Chen X.; Chen L.; Scheuch H.; Zheng H.; Qin L.; Zatloukal K.; Hui L.; Wagner E. F. Liver cancer initiation is controlled by AP-1 through SIRT6-dependent inhibition of survivin. Nat. Cell Biol. 2012, 14, 1203–1211. 10.1038/ncb2590. [DOI] [PubMed] [Google Scholar]
- Xia Y.; Shen S.; Verma I. M. NF-κB, an active player in human cancers. Cancer Immunol. Res. 2014, 2, 823–830. 10.1158/2326-6066.CIR-14-0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang L.; Yi L.; Li J.; Yi S.; Li S.; Liu P.; Yang X. SIRT6 overexpression induces apoptosis of nasopharyngeal carcinoma by inhibiting NF-kappaB signaling. OncoTargets Ther. 2018, 11, 7613–7624. 10.2147/OTT.S179866. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Kugel S.; Sebastián C.; Fitamant J.; Ross K. N.; Saha S. K.; Jain E.; Gladden A.; Arora K. S.; Kato Y.; Rivera M. N.; Ramaswamy S.; Sadreyev R. I.; Goren A.; Deshpande V.; Bardeesy N.; Mostoslavsky R. SIRT6 suppresses pancreatic cancer through control of Lin28b. Cell 2016, 165, 1401–1415. 10.1016/j.cell.2016.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai J.; Zuo Y.; Wang T.; Cao Y.; Cai R.; Chen F. L.; Cheng J.; Mu J. A crucial role of SUMOylation in modulating Sirt6 deacetylation of H3 at lysine 56 and its tumor suppressive activity. Oncogene 2016, 35, 4949–4956. 10.1038/onc.2016.24. [DOI] [PubMed] [Google Scholar]
- Lin Z.; Yang H.; Tan C.; Li J.; Liu Z.; Quan Q.; Kong S.; Ye J.; Gao B.; Fang D. USP10 Antagonizes c-Myc transcriptional activation through SIRT6 stabilization to suppress tumor formation. Cell Rep. 2013, 5, 1639–1649. 10.1016/j.celrep.2013.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee N.; Ryu H. G.; Kwon J. H.; Kim D. K.; Kim S. R.; Wang H. J.; Kim K. T.; Choi K. Y. SIRT6 depletion suppresses tumor growth by promoting cellular senescence induced by DNA damage in HCC. PLoS One 2016, 11, e0165835. 10.1371/journal.pone.0165835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng X. X.; Luo J.; Liu M.; Yan W.; Zhou Z. Z.; Xia Y. J.; Tu W.; Li P. Y.; Feng Z. H.; Tian D. A. Sirtuin 6 promotes transforming growth factor-β1/H2O2/HOCl-mediated enhancement of hepatocellular carcinoma cell tumorigenicity by suppressing cellular senescence. Cancer Sci. 2015, 106, 559–566. 10.1111/cas.12632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran L. K.; Chen Y.; Zhang Z. Z.; Tao N. N.; Ren J. H.; Zhou L.; Tang H.; Chen X.; Chen K.; Li W. Y.; Huang A. L.; Chen J. SIRT6 overexpression potentiates apoptosis evasion in hepatocellular carcinoma via BCL2-associated X protein-dependent apoptotic pathway. Clin. Cancer Res. 2016, 22, 3372–3382. 10.1158/1078-0432.CCR-15-1638. [DOI] [PubMed] [Google Scholar]
- Han L. L.; Jia L.; Wu F.; Huang C. Sirtuin6 (SIRT6) Promotes the EMT of Hepatocellular Carcinoma by Stimulating Autophagic Degradation of E-Cadherin. Mol. Cancer Res. 2019, 17, 2267–2280. 10.1158/1541-7786.MCR-19-0321. [DOI] [PubMed] [Google Scholar]
- Zhang Z.-G.; Qin C.-Y. Sirt6 suppresses hepatocellular carcinoma cell growth via inhibiting the extracellular signal-regulated kinase signaling pathway. Mol. Med. Rep. 2014, 9, 882–888. 10.3892/mmr.2013.1879. [DOI] [PubMed] [Google Scholar]
- Cea M.; Cagnetta A.; Adamia S.; Acharya C.; Tai Y. T.; Fulciniti M.; Ohguchi H.; Munshi A.; Acharya P.; Bhasin M. K.; Zhong L.; Carrasco R.; Monacelli F.; Ballestrero A.; Richardson P.; Gobbi M.; Lemoli R. M.; Munshi N.; Hideshima T.; Nencioni A.; Chauhan D.; Anderson K. C. Evidence for a role of the histone deacetylase SIRT6 in DNA damage response of multiple myeloma cells. Blood 2016, 127, 1138–1150. 10.1182/blood-2015-06-649970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cagnetta A.; Soncini D.; Orecchioni S.; Talarico G.; Minetto P.; Guolo F.; Retali V.; Colombo N.; Carminati E.; Clavio M.; Miglino M.; Bergamaschi M.; Nahimana A.; Duchosal M.; Todoerti K.; Neri A.; Passalacqua M.; Bruzzone S.; Nencioni A.; Bertolini F.; Gobbi M.; Lemoli R. M.; Cea M. Depletion of SIRT6 enzymatic activity increases acute myeloid leukemia cells’ vulnerability to DNA-damaging agents. Haematologica 2018, 103, 80–90. 10.3324/haematol.2017.176248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. C.; Kafeel M. I.; Avezbakiyev B.; Chen C.; Sun Y.; Rathnasabapathy C.; Kalavar M.; He Z.; Burton J.; Lichter S. Histone Deacetylase in Chronic Lymphocytic Leukemia. Oncology 2011, 81, 325–329. 10.1159/000334577. [DOI] [PubMed] [Google Scholar]
- Yang J.; Li Y.; Zhang Y.; Fang X.; Chen N.; Zhou X.; Wang X. Sirt6 promotes tumorigenesis and drug resistance of diffuse large B-cell lymphoma by mediating PI3K/Akt signaling. J. Exp. Clin. Cancer Res. 2020, 39, 142. 10.1186/s13046-020-01623-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer I.; Grozio A.; Lasiglie D.; Basile G.; Sturla L.; Magnone M.; Sociali G.; Soncini D.; Caffa I.; Poggi A.; Zoppoli G.; Cea M.; Feldmann G.; Mostoslavsky R.; Ballestrero A.; Patrone F.; Bruzzone S.; Nencioni A. The NAD+-dependent histone deacetylase SIRT6 promotes cytokine production and migration in pancreatic cancer cells by regulating Ca 2+ responses. J. Biol. Chem. 2012, 287, 40924–40937. 10.1074/jbc.M112.405837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamoorthy V.; Vilwanathan R. Silencing Sirtuin 6 induces cell cycle arrest and apoptosis in non-small cell lung cancer cell lines. Genomics 2020, 112, 3703–3712. 10.1016/j.ygeno.2020.04.027. [DOI] [PubMed] [Google Scholar]
- Han Z.; Liu L.; Liu Y.; Li S. Sirtuin SIRT6 suppresses cell proliferation through inhibition of Twist1 expression in non-small cell lung cancer. Int. J. Clin. Exp. Pathol. 2014, 7, 4774–4781. [PMC free article] [PubMed] [Google Scholar]
- Van Gool F.; Gallí M.; Gueydan C.; Kruys V.; Prevot P. P.; Bedalov A.; Mostoslavsky R.; Alt F. W.; De Smedt T.; Leo O. Intracellular NAD levels regulate tumor necrosis factor protein synthesis in a sirtuin-dependent manner. Nat. Med. 2009, 15, 206–210. 10.1038/nm.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y.; Ka S. O.; Cha H. N.; Chae Y. N.; Kim M. K.; Park S. Y.; Bae E. J.; Park B. H. Myeloid sirtuin 6 deficiency causes insulin resistance in high-fat diet-fed mice by eliciting macrophage polarization toward an M1 phenotype. Diabetes 2017, 66, 2659–2668. 10.2337/db16-1446. [DOI] [PubMed] [Google Scholar]
- Xiao C.; Wang R. H.; Lahusen T. J.; Park O.; Bertola A.; Maruyama T.; Reynolds D.; Chen Q.; Xu X.; Young H. A.; Chen W. J.; Gao B.; Deng C. X. Progression of chronic liver inflammation and fibrosis driven by activation of c-JUN signaling in Sirt6 mutant mice. J. Biol. Chem. 2012, 287, 41903–41913. 10.1074/jbc.M112.415182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorentino F.; Mai A.; Rotili D. Lysine acetyltransferase inhibitors: structure-activity relationships and potential therapeutic implications. Future Med. Chem. 2018, 10, 1067–1091. 10.4155/fmc-2017-0244. [DOI] [PubMed] [Google Scholar]
- Fiorentino F.; Mai A.; Rotili D. Lysine Acetyltransferase Inhibitors From Natural Sources. Front. Pharmacol. 2020, 11, 1243. 10.3389/fphar.2020.01243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorentino F.; Mai A.; Rotili D. HAT inhibitors in cancer therapy. Histone Modifications in Therapy 2020, 20, 51–80. 10.1016/B978-0-12-816422-8.00003-9. [DOI] [Google Scholar]
- Dominy J. E. Jr; Lee Y.; Jedrychowski M. P.; Chim H.; Jurczak M. J.; Camporez J. P.; Ruan H. B.; Feldman J.; Pierce K.; Mostoslavsky R.; Denu J. M.; Clish C. B.; Yang X.; Shulman G. I.; Gygi S. P.; Puigserver P. The Deacetylase Sirt6 Activates the Acetyltransferase GCN5 and Suppresses Hepatic Gluconeogenesis. Mol. Cell 2012, 48, 900–913. 10.1016/j.molcel.2012.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puigserver P.; Rhee J.; Donovan J.; Walkey C. J.; Yoon J. C.; Oriente F.; Kitamura Y.; Altomonte J.; Dong H. J.; Accili D.; Spiegelman B. M. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1 alpha interaction. Nature 2003, 423, 550–555. 10.1038/nature01667. [DOI] [PubMed] [Google Scholar]
- Schilling M. M.; Oeser J. K.; Boustead J. N.; Flemming B. P.; O’Brien R. M. Gluconeogenesis: Re-evaluating the FOXO1-PGC-1α connection. Nature 2006, 443, E10–E11. 10.1038/nature05288. [DOI] [PubMed] [Google Scholar]
- Zhang P.; Tu B.; Wang H.; Cao Z.; Tang M.; Zhang C.; Gu B.; Li Z.; Wang L.; Yang Y.; Zhao Y.; Wang H.; Luo J.; Deng C. X.; Gao B.; Roeder R. G.; Zhu W. G. Tumor suppressor p53 cooperates with SIRT6 to regulate gluconeogenesis by promoting FoxO1 nuclear exclusion. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 10684–10689. 10.1073/pnas.1411026111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao C.; Kim H. S.; Lahusen T.; Wang R. H.; Xu X.; Gavrilova O.; Jou W.; Gius D.; Deng C. X. SIRT6 deficiency results in severe hypoglycemia by enhancing both basal and insulin-stimulated glucose uptake in mice. J. Biol. Chem. 2010, 285, 36776–36784. 10.1074/jbc.M110.168039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L.; Chiang S. H.; Saltiel A. R. Insulin signaling and the regulation of glucose transport. Mol. Med. 2004, 10, 65–71. 10.2119/2005-00029.Saltiel. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanfi Y.; Peshti V.; Gil R.; Naiman S.; Nahum L.; Levin E.; Kronfeld-Schor N.; Cohen H. Y. SIRT6 protects against pathological damage caused by diet-induced obesity. Aging Cell 2010, 9, 162–173. 10.1111/j.1474-9726.2009.00544.x. [DOI] [PubMed] [Google Scholar]
- Naiman S.; Huynh F. K.; Gil R.; Glick Y.; Shahar Y.; Touitou N.; Nahum L.; Avivi M. Y.; Roichman A.; Kanfi Y.; Gertler A. A.; Doniger T.; Ilkayeva O. R.; Abramovich I.; Yaron O.; Lerrer B.; Gottlieb E.; Harris R. A.; Gerber D.; Hirschey M. D.; Cohen H. Y. SIRT6 Promotes Hepatic Beta-Oxidation via Activation of PPAR alpha. Cell Rep. 2019, 29, 4127–4143. 10.1016/j.celrep.2019.11.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abifadel M.; Varret M.; Rabès J. P.; Allard D.; Ouguerram K.; Devillers M.; Cruaud C.; Benjannet S.; Wickham L.; Erlich D.; Derré A.; Villéger L.; Farnier M.; Beucler I.; Bruckert E.; Chambaz J.; Chanu B.; Lecerf J. M.; Luc G.; Moulin P.; Weissenbach J.; Prat A.; Krempf M.; Junien C.; Seidah N. G.; Boileau C. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 2003, 34, 154–156. 10.1038/ng1161. [DOI] [PubMed] [Google Scholar]
- Tao R.; Xiong X.; DePinho R. A.; Deng C. X.; Dong X. C. FoxO3 transcription factor and Sirt6 deacetylase regulate low density lipoprotein (LDL)-cholesterol homeostasis via control of the proprotein convertase subtilisin/kexin type 9 (Pcsk9) gene expression. J. Biol. Chem. 2013, 288, 29252–29259. 10.1074/jbc.M113.481473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao R.; Xiong X.; DePinho R. A.; Deng C. X.; Dong X. C. Hepatic SREBP-2 and cholesterol biosynthesis are regulated by FoxO3 and Sirt6. J. Lipid Res. 2013, 54, 2745–2753. 10.1194/jlr.M039339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elhanati S.; Kanfi Y.; Varvak A.; Roichman A.; Carmel-Gross I.; Barth S.; Gibor G.; Cohen H. Multiple regulatory layers of SREBP1/2 by SIRT6. Cell Rep. 2013, 4, 905–912. 10.1016/j.celrep.2013.08.006. [DOI] [PubMed] [Google Scholar]
- Elhanati S.; Ben-Hamo R.; Kanfi Y.; Varvak A.; Glazz R.; Lerrer B.; Efroni S.; Cohen H. Y. Reciprocal Regulation between SIRT6 and miR-122 Controls Liver Metabolism and Predicts Hepatocarcinoma Prognosis. Cell Rep. 2016, 14, 234–242. 10.1016/j.celrep.2015.12.023. [DOI] [PubMed] [Google Scholar]
- Rahnasto-Rilla M.; Kokkola T.; Jarho E.; Lahtela-Kakkonen M.; Moaddel R. N-Acylethanolamines Bind to SIRT6. ChemBioChem 2016, 17, 77–81. 10.1002/cbic.201500482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahnasto-Rilla M.; Tyni J.; Huovinen M.; Jarho E.; Kulikowicz T.; Ravichandran S.; Bohr V. A.; Ferrucci L.; Lahtela-Kakkonen M.; Moaddel R. Natural polyphenols as sirtuin 6 modulators. Sci. Rep. 2018, 8, 4163. 10.1038/s41598-018-22388-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You W.; Zheng W.; Weiss S.; Chua K. F.; Steegborn C. Structural basis for the activation and inhibition of Sirtuin 6 by quercetin and its derivatives. Sci. Rep. 2019, 9, 19176. 10.1038/s41598-019-55654-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadera K.; Minami Y.; Takamatsu K.; Matsuoka T. Inhibition of α-Glucosidase and α-Amylase by Flavonoids. J. Nutr. Sci. Vitaminol. 2006, 52, 149–153. 10.3177/jnsv.52.149. [DOI] [PubMed] [Google Scholar]
- Proenca C.; Freitas M.; Ribeiro D.; Oliveira E. F. T.; Sousa J. L. C.; Tome S. M.; Ramos M. J.; Silva A. M. S.; Fernandes P. A.; Fernandes E. alpha-Glucosidase inhibition by flavonoids: an in vitro and in silico structure-activity relationship study. J. Enzyme Inhib. Med. Chem. 2017, 32, 1216–1228. 10.1080/14756366.2017.1368503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J.; Chen C.; Zhang B.; Huang Q. The inhibitory effects of flavonoids on alpha-amylase and alpha-glucosidase. Crit. Rev. Food Sci. Nutr. 2020, 60, 695–708. 10.1080/10408398.2018.1548428. [DOI] [PubMed] [Google Scholar]
- Boege F.; Straub T.; Kehr A.; Boesenberg C.; Christiansen K.; Andersen A.; Jakob F.; Kohrle J. Selected novel flavones inhibit the DNA binding or the DNA religation step of eukaryotic topoisomerase I. J. Biol. Chem. 1996, 271, 2262–2270. 10.1074/jbc.271.4.2262. [DOI] [PubMed] [Google Scholar]
- Chowdhury A. R.; Sharma S.; Mandal S.; Goswami A.; Mukhopadhyay S.; Majumder H. K. Luteolin, an emerging anti-cancer flavonoid, poisons eukaryotic DNA topoisomerase I. Biochem. J. 2002, 366, 653–661. 10.1042/bj20020098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Lazaro M.; Willmore E.; Austin C. A. The dietary flavonoids myricetin and fisetin act as dual inhibitors of DNA topoisomerases I and II in cells. Mutat. Res., Genet. Toxicol. Environ. Mutagen. 2010, 696, 41–47. 10.1016/j.mrgentox.2009.12.010. [DOI] [PubMed] [Google Scholar]
- Cantero G.; Campanella C.; Mateos S.; Cortes F. Topoisomerase II inhibition and high yield of endoreduplication induced by the flavonoids luteolin and quercetin. Mutagenesis 2006, 21, 321–325. 10.1093/mutage/gel033. [DOI] [PubMed] [Google Scholar]
- Habermeyer M.; Fritz J.; Barthelmes H. U.; Christensen M. O.; Larsen M. K.; Boege F.; Marko D. Anthocyanidins Modulate the Activity of Human DNA Topoisomerases I and II and Affect Cellular DNA Integrity. Chem. Res. Toxicol. 2005, 18, 1395–1404. 10.1021/tx050039n. [DOI] [PubMed] [Google Scholar]
- Lee W. J.; Shim J. Y.; Zhu B. T. Mechanisms for the inhibition of DNA methyltransferases by tea catechins and bioflavonoids. Mol. Pharmacol. 2005, 68, 1018–1030. 10.1124/mol.104.008367. [DOI] [PubMed] [Google Scholar]
- Zwergel C.; Valente S.; Mai A. DNA Methyltransferases Inhibitors from Natural Sources. Curr. Top. Med. Chem. 2015, 16, 680–696. 10.2174/1568026615666150825141505. [DOI] [PubMed] [Google Scholar]
- Priyadarsini R. V.; Vinothini G.; Murugan R. S.; Manikandan P.; Nagini S. The flavonoid quercetin modulates the hallmark capabilities of hamster buccal pouch tumors. Nutr. Cancer 2011, 63, 218–226. 10.1080/01635581.2011.523503. [DOI] [PubMed] [Google Scholar]
- Xiao X.; Shi D.; Liu L.; Wang J.; Xie X.; Kang T.; Deng W. Quercetin suppresses cyclooxygenase-2 expression and angiogenesis through inactivation of P300 signaling. PLoS One 2011, 6, e22934 10.1371/journal.pone.0022934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W. J.; Chen Y. R.; Tseng T. H. Quercetin induces FasL-related apoptosis, in part, through promotion of histone H3 acetylation in human leukemia HL-60 cells. Oncol. Rep. 2011, 25, 583–591. 10.3892/or.2010.1097. [DOI] [PubMed] [Google Scholar]
- Bisson J.; McAlpine J. B.; Friesen J. B.; Chen S. N.; Graham J.; Pauli G. F. Can Invalid Bioactives Undermine Natural Product-Based Drug Discovery?. J. Med. Chem. 2016, 59, 1671–1690. 10.1021/acs.jmedchem.5b01009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baell J. B. Feeling Nature’s PAINS: Natural Products, Natural Product Drugs, and Pan Assay Interference Compounds (PAINS). J. Nat. Prod. 2016, 79, 616–628. 10.1021/acs.jnatprod.5b00947. [DOI] [PubMed] [Google Scholar]
- Bohm H. J.; Flohr A.; Stahl M. Scaffold hopping. Drug Discovery Today: Technol. 2004, 1, 217–224. 10.1016/j.ddtec.2004.10.009. [DOI] [PubMed] [Google Scholar]
- Hoffmann T.; Gastreich M. The next level in chemical space navigation: going far beyond enumerable compound libraries. Drug Discovery Today 2019, 24, 1148–1156. 10.1016/j.drudis.2019.02.013. [DOI] [PubMed] [Google Scholar]
- Paul D.; Sanap G.; Shenoy S.; Kalyane D.; Kalia K.; Tekade R. K. Artificial intelligence in drug discovery and development. Drug Discovery Today 2021, 26, 80–93. 10.1016/j.drudis.2020.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahnasto-Rilla M. K.; McLoughlin P.; Kulikowicz T.; Doyle M.; Bohr V. A.; Lahtela-Kakkonen M.; Ferrucci L.; Hayes M.; Moaddel R. The Identification of a SIRT6 Activator from Brown Algae Fucus distichus. Mar. Drugs 2017, 15, 190. 10.3390/md15060190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You W.; Rotili D.; Li T. M.; Kambach C.; Meleshin M.; Schutkowski M.; Chua K. F.; Mai A.; Steegborn C. Structural Basis of Sirtuin 6 Activation by Synthetic Small Molecules. Angew. Chem., Int. Ed. 2017, 56, 1007–1011. 10.1002/anie.201610082. [DOI] [PubMed] [Google Scholar]
- Iachettini S.; Trisciuoglio D.; Rotili D.; Lucidi A.; Salvati E.; Zizza P.; Di Leo L.; Del Bufalo D.; Ciriolo M. R.; Leonetti C.; Steegborn C.; Mai A.; Rizzo A.; Biroccio A. Pharmacological activation of SIRT6 triggers lethal autophagy in human cancer cells. Cell Death Dis. 2018, 9, 996. 10.1038/s41419-018-1065-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. H.; Lee J. M.; Kim J. H.; Kim K. R. Fluvastatin activates sirtuin 6 to regulate sterol regulatory element-binding proteins and AMP-activated protein kinase in HepG2 cells. Biochem. Biophys. Res. Commun. 2018, 503, 1415–1421. 10.1016/j.bbrc.2018.07.057. [DOI] [PubMed] [Google Scholar]
- You W.; Steegborn C. Structural Basis for Activation of Human Sirtuin 6 by Fluvastatin. ACS Med. Chem. Lett. 2020, 11, 2285–2289. 10.1021/acsmedchemlett.0c00407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z.; Zhao J.; Deng W.; Chen Y.; Shang J.; Song K.; Zhang L.; Wang C.; Lu S.; Yang X.; He B.; Min J.; Hu H.; Tan M.; Xu J.; Zhang Q.; Zhong J.; Sun X.; Mao Z.; Lin H.; Xiao M.; Chin Y. E.; Jiang H.; Xu Y.; Chen G.; Zhang J. Identification of a cellularly active SIRT6 allosteric activator. Nat. Chem. Biol. 2018, 14, 1118–1126. 10.1038/s41589-018-0150-0. [DOI] [PubMed] [Google Scholar]
- Shang J. L.; Ning S. B.; Chen Y. Y.; Chen T. X.; Zhang J. MDL-800, an allosteric activator of SIRT6, suppresses proliferation and enhances EGFR-TKIs therapy in non-small cell lung cancer. Acta Pharmacol. Sin. 2021, 42, 120–131. 10.1038/s41401-020-0442-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You W.; Steegborn C. Binding site for activator MDL-801 on SIRT6. Nat. Chem. Biol. 2021, 17, 519. 10.1038/s41589-021-00749-y. [DOI] [PubMed] [Google Scholar]
- Huang Z.; Zhao J.; Deng W.; Chen Y.; Shang J.; Song K.; Zhang L.; Wang C.; Lu S.; Yang X.; He B.; Min J.; Hu H.; Tan M.; Xu J.; Zhang Q.; Zhong J.; Sun X.; Mao Z.; Lin H.; Xiao M.; Chin Y. E.; Jiang H.; Shen H.; Xu Y.; Chen G.; Zhang J. Reply to: Binding site for MDL-801 on SIRT6. Nat. Chem. Biol. 2021, 17, 522–523. 10.1038/s41589-021-00750-5. [DOI] [PubMed] [Google Scholar]
- Shang J.; Zhu Z.; Chen Y.; Song J.; Huang Y.; Song K.; Zhong J.; Xu X.; Wei J.; Wang C.; Cui L.; Liu C. Y.; Zhang J. Small-molecule activating SIRT6 elicits therapeutic effects and synergistically promotes anti-tumor activity of vitamin D3 in colorectal cancer. Theranostics 2020, 10, 5845–5864. 10.7150/thno.44043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobaus J.; Hummel D. M.; Thiem U.; Fetahu I. S.; Aggarwal A.; Mullauer L.; Heller G.; Egger G.; Mesteri I.; Baumgartner-Parzer S.; Kallay E. Increased copy-number and not DNA hypomethylation causes overexpression of the candidate proto-oncogene CYP24A1 in colorectal cancer. Int. J. Cancer 2013, 133, 1380–1388. 10.1002/ijc.28143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H.; Wang C.; Hao M.; Sun R.; Wang Y.; Liu T.; Cong X.; Liu Y. CYP24A1 is a potential biomarker for the progression and prognosis of human colorectal cancer. Hum. Pathol. 2016, 50, 101–108. 10.1016/j.humpath.2015.11.008. [DOI] [PubMed] [Google Scholar]
- Pereira F.; Larriba M. J.; Muñoz A. Vitamin D and colon cancer. Endocr.-Relat. Cancer 2012, 19, R51–R71. 10.1530/ERC-11-0388. [DOI] [PubMed] [Google Scholar]
- Ng K.; Nimeiri H. S.; McCleary N. J.; Abrams T. A.; Yurgelun M. B.; Cleary J. M.; Rubinson D. A.; Schrag D.; Miksad R.; Bullock A. J.; Allen J.; Zuckerman D.; Chan E.; Chan J. A.; Wolpin B. M.; Constantine M.; Weckstein D. J.; Faggen M. A.; Thomas C. A.; Kournioti C.; Yuan C.; Ganser C.; Wilkinson B.; Mackintosh C.; Zheng H.; Hollis B. W.; Meyerhardt J. A.; Fuchs C. S. Effect of High-Dose vs Standard-Dose Vitamin D3 Supplementation on Progression-Free Survival Among Patients With Advanced or Metastatic Colorectal Cancer: The SUNSHINE Randomized Clinical Trial. JAMA 2019, 321, 1370–1379. 10.1001/jama.2019.2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carraway H. E.; Malkaram S. A.; Cen Y.; Shatnawi A.; Fan J.; Ali H. E. A.; Abd Elmageed Z. Y.; Buttolph T.; Denvir J.; Primerano D. A.; Fandy T. E. Activation of SIRT6 by DNA hypomethylating agents and clinical consequences on combination therapy in leukemia. Sci. Rep. 2020, 10, 10325. 10.1038/s41598-020-67170-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Sun W.; Huang S.; Zhang H.; Lin G.; Li H.; Qiao J.; Li L.; Yang S. Discovery of Potent Small-Molecule SIRT6 Activators: Structure-Activity Relationship and Anti-Pancreatic Ductal Adenocarcinoma Activity. J. Med. Chem. 2020, 63, 10474–10495. 10.1021/acs.jmedchem.0c01183. [DOI] [PubMed] [Google Scholar]
- Jafari R.; Almqvist H.; Axelsson H.; Ignatushchenko M.; Lundback T.; Nordlund P.; Martinez Molina D. The cellular thermal shift assay for evaluating drug target interactions in cells. Nat. Protoc. 2014, 9, 2100–2122. 10.1038/nprot.2014.138. [DOI] [PubMed] [Google Scholar]
- Savitski M. M.; Reinhard F. B.; Franken H.; Werner T.; Savitski M. F.; Eberhard D.; Molina D. M.; Jafari R.; Dovega R. B.; Klaeger S.; Kuster B.; Nordlund P.; Bantscheff M.; Drewes G. Tracking cancer drugs in living cells by thermal profiling of the proteome. Science 2014, 346, 1255784. 10.1126/science.1255784. [DOI] [PubMed] [Google Scholar]
- Franken H.; Mathieson T.; Childs D.; Sweetman G. M.; Werner T.; Togel I.; Doce C.; Gade S.; Bantscheff M.; Drewes G.; Reinhard F. B.; Huber W.; Savitski M. M. Thermal proteome profiling for unbiased identification of direct and indirect drug targets using multiplexed quantitative mass spectrometry. Nat. Protoc. 2015, 10, 1567–93. 10.1038/nprot.2015.101. [DOI] [PubMed] [Google Scholar]
- Hau M.; Zenk F.; Ganesan A.; Iovino N.; Jung M. Cellular analysis of the action of epigenetic drugs and probes. Epigenetics 2017, 12, 308–322. 10.1080/15592294.2016.1274472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon G. M.; Niphakis M. J.; Cravatt B. F. Determining target engagement in living systems. Nat. Chem. Biol. 2013, 9, 200–205. 10.1038/nchembio.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y.; Furuno M.; Arakawa T.; Takizawa S.; de Hoon M.; Suzuki H.; Arner E. A framework for identification of on- and off-target transcriptional responses to drug treatment. Sci. Rep. 2019, 9, 17603. 10.1038/s41598-019-54180-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacca R.; Engle S. J.; Qin W.; Stock J. L.; McNeish J. D. Genetically Engineered Mouse Models in Drug Discovery Research. Methods Mol. Biol. 2010, 602, 37–54. 10.1007/978-1-60761-058-8_3. [DOI] [PubMed] [Google Scholar]
- Bitterman K. J.; Anderson R. M.; Cohen H. Y.; Latorre-Esteves M.; Sinclair D. A. Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast Sir2 and human SIRT1. J. Biol. Chem. 2002, 277, 45099–45107. 10.1074/jbc.M205670200. [DOI] [PubMed] [Google Scholar]
- Jackson M. D.; Schmidt M. T.; Oppenheimer N. J.; Denu J. M. Mechanism of nicotinamide inhibition and transglycosidation by Sir2 histone/protein deacetylases. J. Biol. Chem. 2003, 278, 50985–50998. 10.1074/jbc.M306552200. [DOI] [PubMed] [Google Scholar]
- Avalos J. L.; Bever K. M.; Wolberger C. Mechanism of sirtuin inhibition by nicotinamide: Altering the NAD(+) cosubstrate specificity of a Sir2 enzyme. Mol. Cell 2005, 17, 855–868. 10.1016/j.molcel.2005.02.022. [DOI] [PubMed] [Google Scholar]
- Hu J.; He B.; Bhargava S.; Lin H. A fluorogenic assay for screening Sirt6 modulators. Org. Biomol. Chem. 2013, 11, 5213–5216. 10.1039/c3ob41138a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolivar B. E.; Welch J. T. Studies of the Binding of Modest Modulators of the Human Enzyme, Sirtuin 6, by STD NMR. ChemBioChem 2017, 18, 931–940. 10.1002/cbic.201600655. [DOI] [PubMed] [Google Scholar]
- Madsen A. S.; Andersen C.; Daoud M.; Anderson K. A.; Laursen J. S.; Chakladar S.; Huynh F. K.; Colaco A. R.; Backos D. S.; Fristrup P.; Hirschey M. D.; Olsen C. A. Investigating the Sensitivity of NAD(+)-dependent Sirtuin Deacylation Activities to NADH. J. Biol. Chem. 2016, 291, 7128–7141. 10.1074/jbc.M115.668699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatkins D. G.; Monnot A. D.; Zheng W. N-epsilon-thioacetyl-lysine: A multi-facet functional probe for enzymatic protein lysine N-epsilon-deacetylation. Bioorg. Med. Chem. Lett. 2006, 16, 3651–3656. 10.1016/j.bmcl.2006.04.075. [DOI] [PubMed] [Google Scholar]
- Kokkonen P.; Rahnasto-Rilla M.; Kiviranta P. H.; Huhtiniemi T.; Laitinen T.; Poso A.; Jarho E.; Lahtela-Kakkonen M. Peptides and Pseudopeptides as SIRT6 Deacetylation Inhibitors. ACS Med. Chem. Lett. 2012, 3, 969–974. 10.1021/ml300139n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B.; Hu J.; Zhang X.; Lin H. Thiomyristoyl peptides as cell-permeable Sirt6 inhibitors. Org. Biomol. Chem. 2014, 12, 7498–7502. 10.1039/C4OB00860J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Zheng W. Cyclic peptide-based potent human SIRT6 inhibitors. Org. Biomol. Chem. 2016, 14, 5928–5935. 10.1039/C5OB02339D. [DOI] [PubMed] [Google Scholar]
- Sociali G.; Liessi N.; Grozio A.; Caffa I.; Parenti M. D.; Ravera S.; Tasso B.; Benzi A.; Nencioni A.; Del Rio A.; Robina I.; Millo E.; Bruzzone S. Differential modulation of SIRT6 deacetylase and deacylase activities by lysine-based small molecules. Mol. Diversity 2020, 24, 655–671. 10.1007/s11030-019-09971-2. [DOI] [PubMed] [Google Scholar]
- Kamiyama O.; Sanae F.; Ikeda K.; Higashi Y.; Minami Y.; Asano N.; Adachi I.; Kato A. In vitro inhibition of α-glucosidases and glycogen phosphorylase by catechin gallates in green tea. Food Chem. 2010, 122, 1061–1066. 10.1016/j.foodchem.2010.03.075. [DOI] [Google Scholar]
- Berger S. J.; Gupta S.; Belfi C. A.; Gosky D. M.; Mukhtar H. Green tea constituent (−)-epigallocatechin-3-gallate inhibits topoisomerase I activity in human colon carcinoma cells. Biochem. Biophys. Res. Commun. 2001, 288, 101–105. 10.1006/bbrc.2001.5736. [DOI] [PubMed] [Google Scholar]
- Bandele O. J.; Osheroff N. (−)-Epigallocatechin Gallate, A Major Constituent of Green Tea, Poisons Human Type II Topoisomerases. Chem. Res. Toxicol. 2008, 21, 936–943. 10.1021/tx700434v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao L.; Park J. Y.; Lambert J. D. Differential prooxidative effects of the green tea polyphenol, (−)-epigallocatechin-3-gallate, in normal and oral cancer cells are related to differences in sirtuin 3 signaling. Mol. Nutr. Food Res. 2015, 59, 203–211. 10.1002/mnfr.201400485. [DOI] [PubMed] [Google Scholar]
- Wood M.; Rymarchyk S.; Zheng S.; Cen Y. Trichostatin A inhibits deacetylation of histone H3 and p53 by SIRT6. Arch. Biochem. Biophys. 2018, 638, 8–17. 10.1016/j.abb.2017.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You W.; Steegborn C. Structural Basis of Sirtuin 6 Inhibition by the Hydroxamate Trichostatin A: Implications for Protein Deacylase Drug Development. J. Med. Chem. 2018, 61, 10922–10928. 10.1021/acs.jmedchem.8b01455. [DOI] [PubMed] [Google Scholar]
- Parenti M. D.; Grozio A.; Bauer I.; Galeno L.; Damonte P.; Millo E.; Sociali G.; Franceschi C.; Ballestrero A.; Bruzzone S.; Del Rio A.; Nencioni A. Discovery of Novel and Selective SIRT6 Inhibitors. J. Med. Chem. 2014, 57, 4796–4804. 10.1021/jm500487d. [DOI] [PubMed] [Google Scholar]
- Sociali G.; Magnone M.; Ravera S.; Damonte P.; Vigliarolo T.; von Holtey M.; Vellone V. G.; Millo E.; Caffa I.; Cea M.; Parenti M. D.; Del Rio A.; Murone M.; Mostoslavsky R.; Grozio A.; Nencioni A.; Bruzzone S. Pharmacological Sirt6 inhibition improves glucose tolerance in a type 2 diabetes mouse model. FASEB J. 2017, 31, 3138–3149. 10.1096/fj.201601294R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cea M.; Cagnetta A.; Adamia S.; Acharya C.; Tai Y.-T.; Fulciniti M.; Ohguchi H.; Munshi A.; Acharya P.; Bhasin M. K.; Zhong L.; Carrasco R.; Monacelli F.; Ballestrero A.; Richardson P.; Gobbi M.; Lemoli R. M.; Munshi N.; Hideshima T.; Nencioni A.; Chauhan D.; Anderson K. C. Evidence for a role of the histone deacetylase SIRT6 in DNA damage response of multiple myeloma cells. Blood 2016, 127, 1138–1150. 10.1182/blood-2015-06-649970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sociali G.; Galeno L.; Parenti M. D.; Grozio A.; Bauer I.; Passalacqua M.; Boero S.; Donadini A.; Millo E.; Bellotti M.; Sturla L.; Damonte P.; Puddu A.; Ferroni C.; Varchi G.; Franceschi C.; Ballestrero A.; Poggi A.; Bruzzone S.; Nencioni A.; Del Rio A. Quinazolinedione SIRT6 inhibitors sensitize cancer cells to chemotherapeutics. Eur. J. Med. Chem. 2015, 102, 530–539. 10.1016/j.ejmech.2015.08.024. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Xie Q. R.; Wang B.; Shao J.; Zhang T.; Liu T.; Huang G.; Xia W. Inhibition of SIRT6 in prostate cancer reduces cell viability and increases sensitivity to chemotherapeutics. Protein Cell 2013, 4, 702–710. 10.1007/s13238-013-3054-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damonte P.; Sociali G.; Parenti M. D.; Soncini D.; Bauer I.; Boero S.; Grozio A.; von Holtey M.; Piacente F.; Becherini P.; Sanguineti R.; Salis A.; Damonte G.; Cea M.; Murone M.; Poggi A.; Nencioni A.; Del Rio A.; Bruzzone S. SIRT6 inhibitors with salicylate-like structure show immunosuppressive and chemosensitizing effects. Bioorg. Med. Chem. 2017, 25, 5849–5858. 10.1016/j.bmc.2017.09.023. [DOI] [PubMed] [Google Scholar]
- Yuen L. H.; Dana S.; Liu Y.; Bloom S. I.; Thorsell A. G.; Neri D.; Donato A. J.; Kireev D.; Schuler H.; Franzini R. M. A Focused DNA-Encoded Chemical Library for the Discovery of Inhibitors of NAD(+)-Dependent Enzymes. J. Am. Chem. Soc. 2019, 141, 5169–5181. 10.1021/jacs.8b08039. [DOI] [PubMed] [Google Scholar]
- Cardus A.; Uryga A. K.; Walters G.; Erusalimsky J. D. SIRT6 protects human endothelial cells from DNA damage, telomere dysfunction, and senescence. Cardiovasc. Res. 2013, 97, 571–579. 10.1093/cvr/cvs352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W.; Chen X.; Huang S.; Li W.; Tian C.; Yang S.; Li L. Discovery of 5-(4-methylpiperazin-1-yl)-2-nitroaniline derivatives as a new class of SIRT6 inhibitors. Bioorg. Med. Chem. Lett. 2020, 30, 127215. 10.1016/j.bmcl.2020.127215. [DOI] [PubMed] [Google Scholar]
- Shi B.; Zhou Y.; Huang Y.; Zhang J.; Li X. Recent advances on the encoding and selection methods of DNA-encoded chemical library. Bioorg. Med. Chem. Lett. 2017, 27, 361–369. 10.1016/j.bmcl.2016.12.025. [DOI] [PubMed] [Google Scholar]
- Franzini R. M.; Randolph C. Chemical Space of DNA-Encoded Libraries. J. Med. Chem. 2016, 59, 6629–6644. 10.1021/acs.jmedchem.5b01874. [DOI] [PubMed] [Google Scholar]
- Renaud J. P.; Chung C. W.; Danielson U. H.; Egner U.; Hennig M.; Hubbard R. E.; Nar H. Biophysics in drug discovery: impact, challenges and opportunities. Nat. Rev. Drug Discovery 2016, 15, 679–98. 10.1038/nrd.2016.123. [DOI] [PubMed] [Google Scholar]
- Fiorentino F.; Sauer J. B.; Qiu X.; Corey R. A.; Cassidy C. K.; Mynors-Wallis B.; Mehmood S.; Bolla J. R.; Stansfeld P. J.; Robinson C. V. Dynamics of an LPS translocon induced by substrate and an antimicrobial peptide. Nat. Chem. Biol. 2021, 17, 187–195. 10.1038/s41589-020-00694-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDowell M. A.; Heimes M.; Fiorentino F.; Mehmood S.; Farkas A.; Coy-Vergara J.; Wu D.; Bolla J. R.; Schmid V.; Heinze R.; Wild K.; Flemming D.; Pfeffer S.; Schwappach B.; Robinson C. V.; Sinning I. Structural Basis of Tail-Anchored Membrane Protein Biogenesis by the GET Insertase Complex. Mol. Cell 2020, 80, 72–86.e7. 10.1016/j.molcel.2020.08.012. [DOI] [PubMed] [Google Scholar]
- Bolla J. R.; Howes A. C.; Fiorentino F.; Robinson C. V. Assembly and regulation of the chlorhexidine-specific efflux pump AceI. Proc. Natl. Acad. Sci. U. S. A. 2020, 117, 17011–17018. 10.1073/pnas.2003271117. [DOI] [PMC free article] [PubMed] [Google Scholar]