

Abstract
A fundamental role of pancreatic β-cells to maintain proper blood glucose level is controlled by the Ras superfamily of small GTPases that undergo post-translational modifications, including prenylation. This covalent attachment with either a farnesyl or a geranylgeranyl group controls their localization, activity, and protein–protein interactions. Small GTPases are critical in maintaining glucose homeostasis acting in the pancreas and metabolically active tissues such as skeletal muscles, liver, or adipocytes. Hyperglycemia-induced upregulation of small GTPases suggests that inhibition of these pathways deserves to be considered as a potential therapeutic approach in treating T2D. This Perspective presents how inhibition of various points in the mevalonate pathway might affect protein prenylation and functioning of diabetes-affected tissues and contribute to chronic inflammation involved in diabetes mellitus (T2D) development. We also demonstrate the currently available molecular tools to decipher the mechanisms linking the mevalonate pathway’s enzymes and GTPases with diabetes.
1. Introduction
The incidence of diabetes has increased tremendously over the last 50 years, affecting approximately 463 million adults. By 2045, there will be 700 million patients with diabetes.1 This epidemic is predominantly caused by a rise in the prevalence of type 2 diabetes (T2D), a complex disorder that is characterized by pancreatic β-cell failure with up to 50% cell loss at diagnosis coupled with impaired insulin sensitivity of target tissues, termed insulin resistance (IR). Initially, insulin resistance causes β-cells to secrete more insulin as a way to compensate for the deficiency. Increased metabolic activity of β-cells leads to the formation of reactive oxygen species (ROS) and induction of endoplasmic reticulum (ER) stress that promote inflammation. Initially, a low-grade local inflammation exerts favorable effects, inducing β-cell proliferation and insulin secretion. However, prolonged secretion of inflammatory mediators by β-cells results in proliferation of resident macrophages and recruitment of immune cells from the circulation. Immune cells further contribute to the inflammation that impairs β-cells function and leads to exhaustion.2
Enhanced insulin production results in hyperinsulinemia that promotes de novo lipogenesis, hyperlipidemia, and adipose tissue expansion. Expanded adipose tissue supports local and systemic inflammation by enhancing pro-inflammatory mediators secretion, including cytokines, chemokines, and adipokines. Both increased systemic fat and inflammation contribute to the development of IR in the liver and skeletal muscles. Insulin resistance can be observed decades before T2D onset and, together with low-grade chronic inflammation, represents one of the earliest pathogenic events in diabetes-related complications, including cardiovascular disease, diabetic retinopathy, and diabetic kidney disease (DKD) as well as nonalcoholic fatty liver disease (NAFLD). Moreover, insulin resistance, hyperinsulinemia, hyperglycemia, and chronic inflammation are the mechanisms of T2D-associated cancer occurrence and progression.3 Despite the large panel of treatment options for T2D, including insulin analogues, biguanides, meglitinides, sodium-glucose cotransporter-2 inhibitors, incretin-based therapies, dipeptidyl peptidase 4, α-glucosidase inhibitors, thiazolidinediones, and sulfonylureas, currently available therapies cause side effects and none of them have shown promise in halting the underlying causes of T2D, namely, insulin resistance.4
The factors associated with IR, T2D and related comorbidities are complex. However, altered activity and prenylation of small GTPases appears to constitute the link with the pathogenesis. Protein prenylation by isoprenoid groups is a crucial eukaryotic post-translational modification (PTM) of lipids predicted to affect hundreds of proteins in the human proteome.5 This ubiquitous covalent attachment of farnesyl or geranylgeranyl modulates localization and function of the plethora of signaling proteins. Most prenylated proteins belong to the Ras-related G proteins, particularly Ras, Rab, and Rho that control cell growth, differentiation, proliferation, biomolecule synthesis, and membrane trafficking.6 Of interest in this regard, hyperinsulinemia was shown to upregulate prenyltransferases,7 and selective inhibitors of prenylation markedly increased insulin sensitivity.8,9 Moreover, sustained inflammation-induced prenylation of Rho GTPase mediated inhibition of insulin-promoted glucose uptake, causing fasting hyperglycemia.10
The isoprenoids used for prenylation are produced by the mevalonate pathway, which is also responsible for cholesterol generation and can be blocked by statins, inhibitors of 3-hydroxymethyl-3-methylglutaryl coenzyme A (HMG-CoA) reductase. Moreover, statins hamper the production of downstream intermediates, such as FPP (farnesyl pyrophosphate) and GGPP (GRG, geranylgeranyl pyrophosphate, geranylgeranyl diphosphate). However, although statins were reported to improve insulin resistance and reduce systemic inflammation, some studies have shown that statins might have increased the incidence of diabetes.11 Farnesyl diphosphate synthase (FPPS) and geranylgeranyl diphosphate synthase (GGPPS), downstream of HMG-CoA reductase, catalyze the production of FPP and GGPP, respectively. Bisphosphonates (BPs), the inhibitors of FPPS, constitute one of the main classes of drugs used to treat bone-associated diseases. In retrospective cohort studies, the exposure to BPs (alendronate, risedronate) was associated with reduced T2D incidence.12 Moreover, the administration of BPs was shown to positively affect diabetes-related indices, insulin, fasting plasma glucose (FPG), and hemoglobin A1c (HbA1c).13 On the other hand, overexpression of muscle,14 adipose,15 and liver16 GGPPS may contribute to insulin resistance pathogenesis. Therefore, inhibition of FPPS and GGPPS may be considered a strategy for insulin resistance treatment. However, additional large-scale trials are needed to verify these relationships.
The mechanisms by which statins and bisphosphonate treatments induce or bypass T2D are not fully understood. It is accepted that their pleiotropic effects might result from changes occurring downstream from these enzymes and that small GTPases are implicated here. Small GTPases are regulated by several protein–protein interactions (PPIs) and PTMs. One of the most studied PTMs is protein prenylation, which is crucial for glucose-stimulated insulin secretion (GSIS) by pancreatic β-cells.17 However, several proteins within the mevalonate pathway may be implicated in T2D development. Here, we discuss the mechanisms of small GTPase prenylation and how inhibition of various points in the mevalonate pathway might affect protein prenylation and functioning of pancreas and liver, skeletal muscle, kidneys, adipose tissue, and contribute to chronic inflammation involved in T2D development.
2. Overview of Superfamily of Small GTPases and Enzymes within the Mevalonate Pathway
The human Ras superfamily of small GTPases, including over 150 proteins, comprises five major subfamilies: Ras, Rab, Rho, Ran, and Arf. Six major subgroups (Ras, Ral, Rap, Rad, Rheb, and Rit) have been identified within the Ras subfamily, which includes 36 human members. The Ras branch regulates cell proliferation, differentiation, and survival.18 With over 60 members in humans, Rab proteins (Ras-related in the brain) form the largest subgroup of the small GTPase superfamily with the principal function of coordinating the transport of proteins and membranes between organelles. Twenty-two genes in humans encode 20 Rho GTPases (Ras homologue) distributed into eight subfamilies (Rac, Cdc42, Rho, RhoD/RhoF, RhoH, RhoU/RhoV, RhoBTB, and Rnd). The Rho family members are essential coordinators of the actin filament network, synchronizing cell shape and movement with intercellular communication, propagation, and differentiation.19 The single Ran (Ras-related nuclear protein) is one-of-a-kind among other GTPases due to its acidic tail at the C-terminus and the lack of the CAAX motif that precludes attachment to lipid membranes. Ran regulates the transport of molecules between the nucleus and cytoplasm and controls cell cycle progression. The adenosine diphosphate-ribosylation factor (Arf) family comprises 29 members in humans and includes Arf isoforms, Arf-like proteins (Arl), and Sar1 proteins. Arf family lacks the C-terminal prenylation signal. Many of Arf family members are myristoylated at the N-terminus for membrane targeting and control vesicular trafficking, motility, division, apoptosis, and transcriptional regulation.18
Small GTPases are guanine nucleotide-dependent molecular switches, active when in complex with GTP and inactive when in complex with GDP. Active small G proteins recruit effectors to the membranes and trigger signal cascades. It requires a tight regulation and small GTPases have three types of controllers, the GTPase-activating proteins (GAPs), the guanine nucleotide exchange factors (GEFs), and the guanine nucleotide dissociation inhibitors (GDIs). GEFs are positive regulators by promoting GDP dissociation, while GAPs are negative regulators by binding to the GTPase and enhancing hydrolysis of GTP. In the case of Rho and Rab, GDIs perturb GAP and GEF regulation and mask the prenyl moiety, thus preventing the association with target membranes (Figure 1A).18 Abnormal activity of some regulatory proteins is linked to diabetic conditions, e.g., dysregulated production of GDI2 contributes to IR.20
Figure 1.
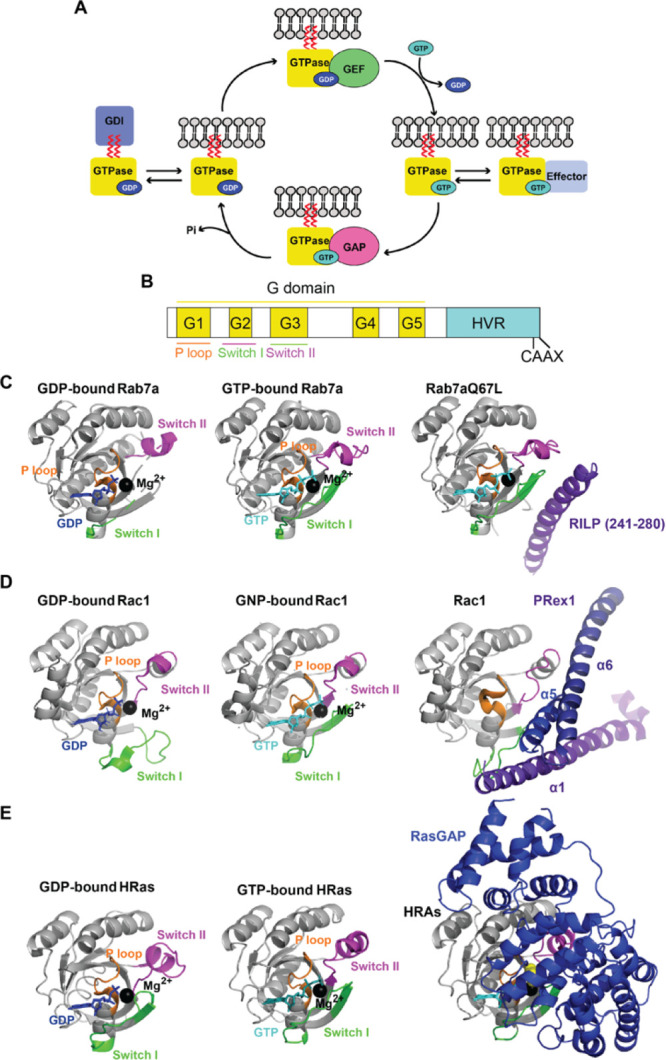
Small GTPase cycle: (A) Interaction with GEF mediates the exchange of GDP for GTP, allows activation, interaction with effectors, and initiation of the signal cascade. Interaction with GAP increases GTP hydrolysis, leading to G protein deactivation. Interaction with GDI keeps small GTPase in an off-state and prevents membrane localization. (B) The conserved architecture of the G domain present in small GTPases (for sequence alignment of Rab, Rho and Ras GTPases implicated in diabetes, see Supplementary Figure S1). (C) Crystal structures of Rab7a: left, inactivated (GDP-bound, PDB; 1VG1); middle, activated (GTP-bound, PDB: 1VG8); right, with its effector RILP (PDB: 1YHN, only part of RILP interacting with Rab7a is shown). (D) Crystal structures of Rac1: left, inactivated (GDP-bound, PDB: 6AGP), middle, activated (GNP-bound, PDB: 3TH5); right, with its effector PRex1 (PDB: 4YON, only domains of PREx1 interacting with Rac1(a1, a5, and a6) are shown). (E) Crystal structures of HRas: left, inactivated (GDP-bound, PDB: 4Q21); middle, activated (GTP-bound; PDB: 1QRA); right, with RasGAP (PDB: 1WQ1). The P loop is represented in orange, switch I in green, switch II in magenta, coordinated magnesium ion in black, GDP in dark blue, and GTP or GTP analogues in cyan. GNP: phosphoaminophosphonic acid-guanylate ester nonhydrolyzable GTP analogue. The corresponding Supplementary Table 1 contains the list of PDB codes for mammalian small GTPases implicated in diabetes, in GDP and GTP-bound form, with effector/GEF/GAP, when available.
Members of the small GTPases share a conserved G domain composed of five loops (G1–G5) that are capable of GTP binding and hydrolysis (Figure 1B, in yellow). The G1 motif (P-loop, Figure 1B, in orange) binds the phosphate groups of GTP and GDP, the G2 motif (switch I, Figure 1B, in green) involved in coordinating of Mg2+ ion with the β- and γ-phosphate is a site for effector and GAP attachment (Figure 1E: HRas-RasGAP; Supplementary Table 1), the G3 motif (switch II, Figure 1B, in magenta) activates a catalytic water molecule for GTP to GDP hydrolysis, the G4 motif provides hydrogen bonds with guanine rings, and the G5 region interacts with guanine via water-mediated hydrogen bonds. Upon exchange of GDP to GTP, effector binding is governed by switch I and switch II, very flexible regions, for which the dynamics differ depending on whether GTP or GDP is attached (Figure 1C–E; Supplementary Table 1). The additional C-terminal hypervariable region (HVR), which accommodates a polybasic region (PBR) and cysteines, regulates GTPase association with target membranes (Figure 1B, Supplementary Figure S1).18
Small G proteins regulate various effectors (Table 1). GTP binding energy is used to stabilize the switch I and II regions, required for effector recognition (Figure 1C: Rab7a-RILP, 1D: Rac1-PRex1). GTP hydrolysis induces conformational change and a flexibility in the region interacting with the effector. The binding of some effectors slows down GTP hydrolysis, while interaction with GAPs speeds it up.18
Table 1. Small GTPases Involved in Insulin Release from Pancreatic β-Cells under Physiological Conditions.
| GTPase | localization | interacting protein | function | refs |
|---|---|---|---|---|
| Rab GTPases | ||||
| Rab1a | ER-Golgi membranes | Golgin-84 | conversion of proinsulin to insulin; maintaining Golgi stability | Liu et al.32 |
| Rab2a | ERGIC | GAPDH | vesicular transport of proinsulin from ERGIC to the Golgi; a switch protein that facilitates ER-associated degradation or secretion of (pro)insulin | Sugawara et al.33 |
| Noc2 | ternary Rab2a-Noc2-Rab27a complex mediates processing proinsulin to insulin | Matsunaga et al.34 | ||
| Rab3 | ISG | RIMs | Rim2α–Rab3a interaction is required for the docking of insulin granule | Yasuda et al.35 |
| granuphilin | granuphilin-Rab3a augments insulin granule exocytosis | Coppola et al.36 | ||
| Noc2 | Noc2-Rab3 positively regulates insulin secretion required for maintenance of RRP | Matsumoto et al.37 | ||
| all Rab3, except for Rab3c, are required for Ca2+-dependent insulun secretion | Cazares et al.38 | |||
| Rab7 | late endosomes, lysosomes | RILP | insulin secretion is inhibited by RILP, which controls lysosomal degradation of proinsulin by interacting with lysosome-located Rab7 | Zhou et al.39 |
| Rab8a | PM, ISG | regulation of Kir6.2 membrane trafficking | Uchida et al.40 | |
| Rab11b | ISG | Rip11 | cAMP (but not glucose)-induced insulin release by modulating the recycling of the proteins associated with the exocytotic back to immature granules | Sugawara et al.41 |
| Rab26 | ISG | RILP | insulin secretion is inhibited by RILP, which controls lysosomal degradation of proinsulin | Zhou et al.39 |
| Rab27a | ISG | defines the total quantity of RP and RRP | Cazares et al.38 | |
| granuphilin | granuphilin forms a regulated Rab27a complex with Munc18-1 and Syntaxin1a, regulates docking of insulin granules, and inhibits subsequent fusion of docked granules | Yi et al.42 | ||
| Torii et al.43 | ||||
| exophilin-7 | movement of the granule along the actin filament | Wang et al.44 | ||
| exophilin-8 | tripartite complex of exophilin-8, Rab27a, and myosin Va mediates the fusion of undocked granules with the cell surface phospholipids | Mizuno et al.45 | ||
| Noc2 | Noc2–Rab27a complex on peripheral mature granules mediates vesicle priming and insulin exocytosis | Matsunaga et al.34 | ||
| coronin 3 | Rab27a–GDP–coronin 3, in complex with IQGAP1, is crucial for endocytosis of insulin granules | Kimura et al.46 | ||
| Rab37 | ISG | final steps of insulin exocytosis | Ljubicic et al.47 | |
| Rho GTPases | ||||
| RhoA | PM | ROCK | actin cytoskeleton stabilization and GSIS inhibition | Hammar et al.48 |
| Cdc42 | cytosol, ISG, PM | N-WASP | N-WASP binds Cdc42 to actin via the Arp2/3 complex necessary for GSIS | Uenishi et al.49 |
| PAK-1 | F-actin remodeling and granule recruitment to the plasma membrane during the first phase of insulin release | Wang et al.,50 Kalwat et al.51 | ||
| syntaxin 1, syntaxin 4, VAMP2 | Cdc42 and VAMP2 form heterotrimeric complexes with syntaxin 1 and 4 | Nevins et al.,52 Daniel et al.53 | ||
| caveolin-1 | caveolin-1 binds to Cdc42 present on ISG. The complex translocates to the plasma membrane and dissociates | Nevins et al.54 | ||
| coronin 3, IQGAP1 | endocytosis of the insulin secretory membrane requires a complex containing IQGAP1, GDP-bound Rab27a, and coronin 3. | Kimura et al.55 | ||
| Rac1 | cytosol, PM | insulin secretion via depolymerization of F-actin | Asahara et al.56 | |
| PAK1 | glucose-induced Rac1-mediated F-actin remodeling and insulin secretion | Kalwat et al.51 | ||
| Tiam1 (GEF) | modulation of Tiam1/Rac1-dependent signaling step in GSIS | Veluthakal et al.17 | ||
| Vav2 | Vav2-Rac1 required for glucose-induced actin depolymerization and GSIS | Veluthakal et al.57 | ||
| P-Rex1 (GEF) | initiates the cascade of events leading to GSIS | Thamilselvan et al.58 | ||
| Trio (GEF) | rearrangement of Rac1 to the cell surface required for GSIS | Dufurrena et al. | ||
| Kalirin (GEF) | rearrangement of Rac1 to the cell surface required for GSIS | Dufurrena et al. | ||
| Ras GTPases | ||||
| Rap1 | PM | Epac2 (GEF) | Epac2, a cAMP binding protein, regulates insulin exocytosis | Shibasaki et al.59 |
| RalA | PM, ISG | RalGDS | modulates the dynamics of the actin cytoskeleton | Ljubicic et al.60 |
| Sec6 | tethers secretory granules through its regulated association with the exocyst (Sec6) complex | Lopez et al.61 | ||
| Cavα2δ-1 subunit of VDCC | RalA binds α2δ-1 on insulin granules to tether granules to plasma membrane Ca2+channels (a step to prepare for the assembly of excitosome and exocyst complexes required for biphasic insulin secretion) | Xie et al.62 | ||
Besides GDP/GTP binding, small GTPases usually carry a post-translationally attached prenyl tail at cysteine residues present in or located close to the CAAX motif. For that purpose, the farnesyl and geranylgeranyl chains are added to GTPases, and the substrates, FPP and GGPP, are synthesized via the mevalonate pathway (Figure 2). The mevalonate pathway is an essential biosynthetic step that produces components for the cholesterol biosynthesis or FPP and GGPP, and it starts from the condensation of the monomers, isopentenyl diphosphate (IPP) with its isomer, dimethylallyl pyrophosphate (DMAPP).21
Figure 2.

Schematic representation of mevalonate pathway. HMG-CoA reductase catalyzes the formation of mevalonate from HMG-CoA. FPPS mediates further conversion to GPP and FPP. FTase catalyzes attachment of FPP to Ras, Rho, and Rheb proteins (in the process called farnesylation). GGPPS catalyzes the conversion of FPP to GGPP that can be post-translationally added to RhoA, RAc1, Cd42, Ral, and Rap by GGTase-I, Rab proteins by GGTase-II, and Ykt6 and FBXL2 by GGTase-III.
HMG-CoA reductase produces mevalonate in the rate-limiting step in the pathway. Mammalian HMG-CoA reductase functions as a homotetramer (Figure 3A; Supplementary Table 2). Each monomer consists of the cytosolic C-terminal catalytic domain, the L domain responsible for substrate binding, the S domain binding NADPH, and the N-terminal segment for anchoring to the ER membrane. Statins bind stronger to the L domain than HMG-CoA, e.g., with the inhibitory concentration values of 3.8–6.2 nM for atorvastatin.22
Figure 3.

Structural overview of enzymes within the mevalonate pathway and prenyltransferases. (A) HMG-CoA reductase (PDB: 1DQ9) is a homotetramer. Each subunit comprises an N domain (in green), large L domains (in magenta), and an S domain (in light blue). (B) FPPS (PDB: 5JA0) PO4 in red. (C) GGPPS (PDB: 2Q80) is a hexameter composed of three dimers: chain A–B (in pink), chain C–D (in green), and chain E–F (in blue). Mg2+ is represented in black, and GRG in dark blue. (D) Comparison of structures of prenyltransferases: FTase (PDB: 1FPP), GGTase-I (PDB: 1N4P), GGTase-II (PDB: 3DST), and GGTase-III (PDB: 6J6X). The α and β subunits are color-coded, and the shared domains have the same color. Zn2+ is presented in black. The corresponding Supplementary Table S2 contains the list of PDB codes for mammalian enzymes within the mevalonate pathway and prenyltransferases implicated in diabetes, in GDP and GTP-bound form, with substrate/product/inhibitor, when available.
FPPS catalyzes the synthesis of 10-carbon geranyl pyrophosphate (GPP) and the 15-carbon FPP, whereas GGPPS synthesizes the 20-carbon GGPP. Even though free GPP has been detected in cultured human cells,23 as far as we know, the geranylated entities have not been detected in human cells yet. The majority of the studies on protein prenylation concentrate on farnesylated and geranylgeranylated proteins and developing the suitable tools.24
Although human FPPS exists as a homodimer (Figure 3B; Supplementary Table 2), human GGPPS is a hexamer assembled from three dimers (Figure 3C; Supplementary Table 2). Despite low sequence identity, both isoprenoid synthases adopt a similar all α-helical structure. At least three small-molecule binding sites are present in the structure of FFPS, namely, allosteric pocket, allylic substrate (DMAPP and GPP) binding site, and homoallylic substrate (IPP) binding site, with the latter two having high similarity to those found in FPPS. The product inhibitor pocket has been identified in GGPPS as well.21
FPP and GGPP moieties are utilized by four distinct prenyltransferases, namely, farnesyltransferase (FTase), geranylgeranyltransferase I (GGTase-I), Rab geranylgeranyl transferase (GGTase-II/RGGT), and geranylgeranyltransferase III (GGTase-III). All enzymes catalyze the formation of the thioether linkage with the Cys residue located in the prenylation recognition sequence at the C terminus of selected proteins. FTase and GGTase-I transfer a respective prenyl group to protein substrates containing carboxyl-terminal CAAX motifs where C is cysteine, A is aliphatic, and X is any residue. Usually, FTase prefers Cys, Ser, Met, Ala, or Gln while GGTase-I selects Leu, Ile, or Phe at the X position.25 Ras, RhoB, and Rheb have been identified as substrates of FTase while GTPases geranylgeranylated by GGTase-I include Rho, Ral, and Rap. There are examples when a protein is either farnesylated or geranylgeranylated, for instance, RhoB. On the other hand, in the case of K-Ras, inhibition of FTase was linked to a compensatory GGTase-I upregulation that can be a reason for the insufficient clinical efficacy of anticancer FTase inhibitors. Therefore, dual FTase/GGTase-I inhibitors may prove a more effective therapeutic approach.26
GGTase-II (Rab geranylgeranyl transferase; RGGT) exclusively geranylgeranylates C-terminally localized CXC and CC motifs in Rab family members. Unlike FTase and GGTase-I, prenylation of Rab proteins by RGGT must be associated with REP1/2 chaperone proteins (Rab escort protein 1/2). Most Rab proteins are doubly geranylgeranylated in a sequential fashion without dissociation of the monoprenyl intermediate.25
The fourth type of protein prenyltransferase, GGTase-III, has been discovered very recently. This enzyme catalyzes the double prenylation of the FBXL2 ubiquitin ligase and Golgi SNARE protein Ykt6 in collaboration with FTase. Chaperone SKP1 protein is required for geranylgeranylation by GGTase-III.27,28 According to the authors’ knowledge, no inhibitors of this enzyme have been reported yet.
Each prenyltransferase exists as a heterodimer with the active site formed at these proteins’ interface and made up of α- and β-subunits (Figure 3D; Supplementary Table 2). FTase and GGTase-I have different catalytic β-subunits (FNTB/FTβ and GGT1β, respectively) and share a common α-subunit (FNTA/FTα). In turn, RGGT and GGTase-III share identical β subunit (RABGGTβ) but contain distinct α subunits (RABGGTα and PTAR1, respectively). The RABGGTβ subunit of RGGT and GGTase-III is probably necessary for double prenylation due to its hydrophobic tunnel structure.28
All protein prenyltransferases are metalloenzymes. A Zn2+ ion (a thiolate) is bound by the catalytic domain of the β subunit of GGTases. Additionally, FTase requires Mg2+ that stabilizes PPi leaving group of FPP.
3. Small GTPases as Regulators of the Insulin Trafficking and Exocytosis in Pancreatic β-Cells
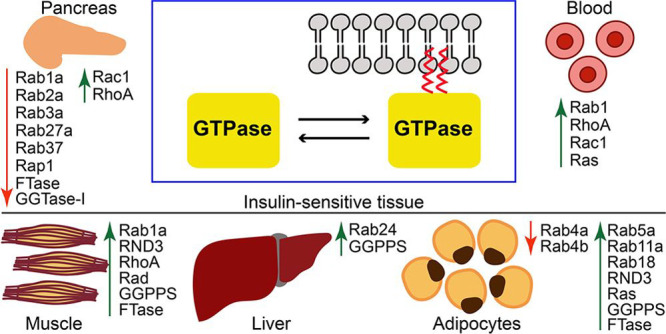
Small GTPases are critical in maintaining whole-body glucose homeostasis acting predominantly in metabolically active tissues, including the pancreas, skeletal muscles, liver and adipocytes. The pancreas plays a key role in this network by secreting the blood-glucose-lowering hormone insulin, produced by β-cells located within islets of Langerhans. Preproinsulin is synthesized on the cytoplasmic side of the ER and translocated to the ER, where the signal peptide is cleaved. The resulting proinsulin is transported to the cis-face of the Golgi apparatus and starts to be packaged after reaching Trans-Golgi Network (TGN). Proteolytic cleavage of proinsulin results in the formation of insulin. Insulin crystallizes with zinc and calcium in the form of dense-core granules during the granule maturation process. The readily releasable pools (RRP) and the reserved pool are two intracellular pools of dense-core insulin granules. When blood glucose level is low, the actin cytoskeleton prevents insulin secretory granules (ISGs) from reaching their release sites.29
When plasma glucose levels are high in humans, glucose enters the β-cells, primarily through the cell membrane glucose transporters GLUT1 and GLUT3, although GLUT2 expression was also demonstrated by several groups.30 Upon uptake, glucose is metabolized and a high ATP-to-ADP ratio triggers membrane depolarization by closing ATP-dependent potassium channels (KATP). Consequently, voltage-gated calcium channels (VGCC) open and that results in calcium influx, which induces docking and fusion with the plasma membrane (exocytosis of insulin granule). The docking and fusion of insulin granules are orchestrated by the soluble N-ethylmaleimide sensitive factor attachment receptor (SNARE) complex. The target-localized (t-SNARE) proteins in the cell surface (SNAP25 and Syntaxin) interact with VAMP (vesicle-associated membrane protein, v-SNARE) on the insulin granules (Figure 4). Under high glucose, the actin cytoskeleton is reorganized, allowing them to move to the plasma membrane. Such glucose-mediated exocytosis of different functional granule pools occurs in response to elevated glucose concentration in a biphasic manner. The rapid first phase (usually the first 10 min) results from fusion and secretion of a subset of plasma membrane-docked granules that are primed with a fully assembled exocytosis machinery (RRP). F-actin filaments are important for the short-range movement of RRP. The second step entails the recruitment of granules from the inside of the cell and microtubule transport.29
Figure 4.

Schematic representation of insulin synthesis and trafficking and exocytosis of insulin containing granules (created in BioRender.com). Proinsulin processing occurs in the lumen of ER and insulin is stored as a hexamer in complex with Zn2+. Glucose enters the cells and via mitochondrial ATP synthesis raises the ATP-to-ADP ratio, causing the ATP-sensitive K+ (KATP) channels to close. Following cellular depolarization, VGGC is activated, causing extracellular Ca2+ influx and insulin granule fusion with the plasma membrane. Specific sets of Rab GTPases regulate insulin secretory granule transport, endocytosis, and the three main stages of insulin granule exocytosis (docking, priming, and fusion). For the sake of simplicity, we have not included all the specific Rabs involved that have been described in Table 1.
The trafficking of the insulin granules is controlled by several Ras family GTPases and their effectors. Various Rab proteins are associated with the secretory granules and regulate the transport, priming, docking, and fusion of ISGs at the plasma membrane (Figure 3 and Table 1). For example, Rab3 allows ISG docking and tethering at the correct target membrane by interacting with RIM2α and the clustering of the SNARE Syntaxin1 and its binding partner munc18-1. In turn, the Rho family, including Cdc42, Rac, and RhoA, is instrumental in insulin secretion via F-actin remodeling and vesicle fusion regulation. Cdc42 was also shown to be crucial for endocytosis of insulin vesicles. Rap1 and RalA, although less studied, also elicit regulatory effects in insulin release.19,29 The detailed information on specific functions of small G proteins in insulin secretion by pancreatic β-cells is summarized in Table 1.
Most small GTPases involved in insulin trafficking and secretion are required to be prenylated to function for their biological role and interaction with their respective effectors. FTase, GGTase-I, and GGTase-II are expressed in β-cell lines and pancreatic islets. Studies utilizing inhibitors of HMG-CoA reductase (atorvastatin, lovastatin, simvastatin), GGPPS (digeranyl bisphosphonate), FTase (FTI-277, FTI-2628, allyl- or vinyl-farnesols, limonene, manumycin, perillic acid), and GGTase-I (GGTI-298, GGTI-2133, GGTI-2147; GGTI-2368, allyl- or vinyl- geraniols) as well as siRNA-mediated silencing of Rggta and Rggtb revealed that prenylation of small GTPases is essential for β-cell function and insulin secretion.31
4. Small GTPases as Regulators of GLUT4 Trafficking
Insulin-stimulated glucose uptake into skeletal muscle cells and adipocytes assumes a central role in glucose homeostasis in the body. Most (80–90%) of the infused glucose is absorbed by skeletal muscles that store glucose as glycogen and utilize it in glycolysis; however, adipocytes also exert a critical control in the regulation of blood glucose levels. Insulin promotes the exocytosis of intracellular vesicles containing GLUT4 glucose transporters, the most abundant glucose transporter in muscle and fat cells. In the basal state, GLUT4 locates intracellularly in endosomes, TGN, specialized perinuclear glucose transporter storage vesicles (GSVs), and more peripheral insulin-responsive vesicles (IRVs).63
The insulin binding to the tyrosine kinase receptor activates its autophosphorylation and initiates a signaling cascade starting from phosphorylation of insulin receptor substrates (IRS1 and IRS2). IRS, in turn, phosphorylates phosphatidyl inositol-3-kinase (PI3K) and promotes downstream signaling. PI3K constitutes a branch point in insulin signaling activating Akt and Rac1, which in parallel promote GLUT4 transport to the plasma membrane, permitting glucose intake.64 Akt phosphorylates various GAPs (e.g., TBC1D1, TBC1D4), reducing the inactivation of their cognate GTPases (Figure 5). Several Rab GTPases, including Rab4, Rab5, Rab7, Rab8a, Rab10, Rab11, Rab13, Rab14, Rab28, and Rab35, with effector proteins were demonstrated to confer directionality to GLUT4 vesicle traffic. Insulin also activates Rho and Ras GTPases mainly affecting actin remodeling (Table 2). Glucose uptake by GLUT4 also occurs upon muscle contraction; however, muscle contraction and insulin target separate GLUT4 pools. During muscle contraction, the AMP/ATP ratio increases, leading to activation of AMP-activated protein kinase (AMPK), the cellular energy sensor. AMPK, in turn, phosphorylates TBC1D1 and TBC1D4 activating target Rabs.65 Rac1 acts as another contributor to contraction-stimulated glucose transport mediating the stretch-sensitive component.66
Figure 5.

Scheme of the insulin-regulated transport of GLUT4 vesicles translocation and exocytosis (created in BioRender.com). Insulin binds the insulin receptor that induces the translocation of GLUT4 storage vesicles by activating the PI3K signaling cascade. PI3K catalyzes the formation of phosphatidylinositol (3,4,5) trisphosphate leading to the action of PDK1, which in turn stimulates Akt. Activated Akt phosphorylates and inactivates GAPs (e.g., TBC1D1, TBC1D4, RGC1/2). GAPs inhibition shifts small GTPases from the GDP- to a more active GTP-loaded state. Rac1 facilitates GLUT4 plasma membrane association via actin filament remodeling. GTP-loaded Rabs and other Ras superfamily members permit GLUT4 storage vesicle translocation to the cell surface for fusion. In addition to the main PI3K pathway, the Rho family GTPases (e.g., RhoA, Cdc42, TC10) mediate insulin signaling in regulating GLUT4 translocation. For the sake of clarity, we have not included all the specific Rabs involved that have been described in Table 2.
Table 2. Small GTPases Involved in Insulin-Induced GLUT4 Translocation.
| GTPase | localization | interacting protein | function | refs |
|---|---|---|---|---|
| adipocytes | ||||
| Rab GTPases | ||||
| Rab4a, Rab4b | IRV | syntaxin 4 | involvement in GSV sorting and fusion | Li et al.67 |
| recycling of GLUT4 via endosomes | Chen et al.68 | |||
| Rab5a | early endosomes | dynein | insulin signaling deactivates Rab5 and impedes dynein microtubule interaction, slowing GLUT4 inward movement | Tessneer et al.69 |
| Rab8a | endosomes, TGN, GSV | TBC1D4 (GAP) | GLUT4 translocation; cell surface endosome cycling of GLUT4 | Mîinea et al.,70 Chen et al.68 |
| MyoVa | insulin-mediated signaling augments Rab8a–MyoVa interaction to drive GLUT4-containing vesicles to the cell surface. | Sun et al.71 | ||
| Rab10 | perinuclear endosome/TGN, GSV | TBC1D4 (GAP) | accumulation of GLUT4-containing vesicles at the cell surface | Mîinea et al.,70 Sadacca et al.72 |
| MyoVa | Rab10–MyoVa interaction facilitates the transport of GSVs and docking at the cell surface. | Chen et al.68 | ||
| SEC16A | SEC16A–Rab10 interaction promotes GLUT4 mobilization from the intracellular compartments to the cell to accelerate formation of the GSV | Bruno et al.73 | ||
| Exoc6/6b | Rab10-Exoc6/6b promotes the fusion of GLUT4-containing vesicles with the cell surface | Sano et al.74 | ||
| Exoc7 | Exoc7 exerts a critical function in insulin-stimulated GLUT4 exocytosis | Wang et al.75 | ||
| Rlf (GEF) | Rab10 promotes RalA activation by recruiting Rlf. | Karunanithi et al.76 | ||
| RABIF (GEF) | RABIF enhances Rab10 stability and GLUT4 exocytosis | Gulbranson et al.77 | ||
| Dennd4C (GEF) | primary GEF required for GLUT4 translocation | Sano et al.78 | ||
| Rab11 | Golgi, endosomes | Rip11 | Rip11 is a scaffolding protein in the coupling of GLUT4-containing vesicles with the cell surface | Welsh et al.79 |
| GLUT4 transport from the endosomal compartments to GSV | Zeigerer et al.80 | |||
| Rab14 | TGN, endosomes, GSV | GLUT4 transport to the plasma membrane via transferrin receptor-positive endosomal structures. | Chen et al.68 | |
| early endosomes-to-TGN transport of GLUT4 | Reed et al.81 | |||
| Rab14 is a controller of GLUT4 sorting into vesicles (upstream of Rab10) | Sadacca et al.72 | |||
| TBC1D4 (GAP) | GLUT4 sorting into GSV | Mîinea et al.70 | ||
| Rab28 | TBC1D1 (GAP), TBC1D4 (GAP) | GLUT4 trafficking | Zhou et al.82 | |
| Rab35 | PM | TBC1D13 (GAP) | GLUT4 translocation (a trafficking pathway from early endosomes) | Davey et al.83 |
| Rho GTPases | ||||
| TC10 | lipid rafts in PM | CIP4/2 | GLUT4 trafficking, docking, and fusion with the cell surface | Chang et al.84 |
| N-WASP | N-WASP-Arp2/3 is required to mobilize cortical F-actin and GLUT4 translocation | Jiang et al.85 | ||
| RhoA | PM | RhoA regulates glucose transport via remodeling of actin cytoskeleton | Duong and Chun86 | |
| RhoA modulates the activity of IRS-1 | Takaguri et al.87 | |||
| ROCK1 | GLUT4 translocation and actin cytoskeleton remodeling | Chun et al.88 | ||
| Cdc42 | perinuclear cytosol, PM | GLUT4 translocation and glucose transport | Usui et al.89 | |
| Rac1 | cytosol, PM | P-Rex1 (GEF) | P-Rex1-facilitated GLUT4 plasma membrane association via regulation of the actin cytoskeleton at physiological insulin concentrations | Balamatsias et al.90 |
| Ras GTPases | ||||
| RalA | vesicles derived from endosomes, GSV | RGC1/2 (GAP) | mobilization of the exocyst complex to facilitate trafficking of GLUT4 vesicles | Chen et al.91 |
| Myo1c | trafficking of GLUT4 vesicles to the cell surface; Myo1c-RalA interaction is modulated by calmodulin | Chen et al.92 | ||
| Sec5 and Exo84 | Sec5 and Exo84 (in the exocyst complex) play a role in vesicle tethering to the cell surface | Chen et al.93 | ||
| RalGAP | GLUT4 cycling | Skorobogatko et al.94 | ||
| muscle cells | ||||
| Rab GTPase | ||||
| Rab7 | TBC1D15 (GAP) | TBC1D15 is a master regulator of GLUT4 translocation through late endosomal pathway | Wu et al.95 | |
| Rab8a | vesicles in perinuclear region | TBC1D1 (GAP), TBC1D4 (GAP), MyoVb | TBC1D4 in myoblasts and TBC1D1 in myotubes are involved in intracellular retention of GLUT4; Rab8A interacts with MyoVb to translocate GLUT4 | Ishikura and Klip96 |
| MyoVa | Rab8A-MyoVa mobilizes GLUT4 vesicles toward the plasma membrane | Sun et al.71 | ||
| Rab13 | peripheral vesicles | TBC1D4 (GAP) | Rab13 acts at a peripheral step in GLUT4 translocation | Sun et al.97 |
| MICAL-L2 | MICAL-L2 links to GLUT4 through filamentous cortical α-actinin-4 enabling their fusion with the membrane | Sun et al.98 | ||
| Rab14 | vesicles in perinuclear region | TBC1D1 (GAP), TBC1D4 (GAP) | sorting of GLUT4 from the recycling endosome to the insulin-sensitive compartments | Ishikura et al.99 |
| Rab28 | TBC1D1 (GAP), TBC1D4 (GAP) | GLUT4 trafficking | Zhou et al.82 | |
| Rho GTPases | ||||
| Rac1 | cytosol, ruffling area of the dorsal cell membrane | Rac1 stimulates actin cytoskeleton reorganization and activates PAK | JeBailey et al.100 | |
| insulin-stimulated glucose uptake is regulated by Rac1 and Akt in parallel pathways; Rac1 involves the actin cytoskeleton reorganization | Sylow et al.101 | |||
| Elmo2 | Elmo2 regulates Akt membrane compartmentalization and Rac1 activation, resulting in enhanced insulin-stimulated GLUT4 translocation | Sun et al.102 | ||
| Tiam1 (GEF) | AMPK-Tiam1-Rac1 axis mediates contraction stimulated glucose uptake | Yue et al.103 | ||
| FLJ00068 (GEF) | FLJ00068-mediated Rac1 activation in membrane ruffles mobilizes GLUT4 vesicles | Ueda et al.104 | ||
| FLJ00068 is a pivotal controller of Akt2-mediated Rac1 activation | Takenaka et al.105 | |||
| RhoGDIα | RhoGDIα acts as a negative regulator of Rac1 activity and GLUT4 surface transport | M?ller et al.19 | ||
| PAK1 | insulin-promoted GLUT4 translocation | Wang et al.106 | ||
| PAK1/2 | PAK2 is needed, while PAK1 is dispensable for insulin-stimulated glucose absorption in glycotic muscle | Møller et al.107 | ||
| Arp2/3 | Arp2/3 and cofilin coordinate actin cortex remodeling essential for insulin-mediated GLUT4 translocation | Chiu et al.108 | ||
| RhoA | RhoA regulates glucose transport via remodeling of actin cytoskeleton remodeling | Duong and Chun86 | ||
| ROCK1 | GLUT4 translocation and actin cytoskeleton remodeling | Chun et al.88 | ||
| Ras GTPases | ||||
| RalA | RalA, regulated downstream of Rac1, exerts a crucial function in GLUT4 surface transport | Nozaki et al.109 | ||
5. Small GTPases and Enzymes of the Mevalonate Pathway in Pathological States of Diabetes and Its Complications
Small GTPases are pivotal in maintaining glucose homeostasis, and aberrant function and regulation of this class of proteins are implicated in the pathological cellular machinery triggered by hyperglycemia. Some reports clearly show glucose-induced upregulation of small GTPases, suggesting that inhibition of such pathways deserves to be considered as a potential therapeutic target in the treatment of T2D and its complications. While expression or activity of Rab members tends to be downregulated under conditions that favor the development of diabetes, overactivated RhoA and Rac1 are involved in many of the pathologies observed in T2D individuals (Table 3). Rac1 is the cytosolic regulatory subunit of the NADPH oxidase (NOX) multicomponent system responsible for ROS generation. Rac1 signaling pathway is implicated in diabetes pathogenesis, mainly by the generation of oxidative stress and islet dysfunction. Hyperactivation of GTP-bound Rac1 is detected in islets derived from T2D patients and animal models.110 Importantly, prenylation of Rac1 might be essential for membrane localization and subsequent activation of NOX.111 Rac1 activation is also linked to abnormal retinal neovascularization and ROS production, leading to diabetic retinopathy and vascular dysfunction.112,113 In the pancreas, hyperglycemic conditions increase RhoA/ROCK activity that contributes to the diminished GSIS114 and insulin resistance in muscles.115 The progression of diabetic kidney disease116 and vascular complications such as diabetic retinopathy or atherosclerosis117 have also been connected with elevated levels of RhoA. Taken together, Rac1 and RhoA/ROCK are candidates as new promising targets for pharmacological prevention of islet dysfunction in T2D and T2D-related comorbidities.
Table 3. Diabetes-Related Alterations in Ras GTPases and Associated Enzymes of Mevalonate Pathway.
| GTPase | abnormality | refs |
|---|---|---|
| β-cells | ||
| Ras GTPases | ||
| Rab1a | Rab1a expression is down-regulated in islets of Goto-Kakizaki rats with T2D | Liu et al.32 |
| Rab2a | under chronic high glucose, Rab2A effector GAPDH undergoes poly(ADP-ribosyl)ation and dissociation that impairs Rab2A activity | Sugawara et al.33 |
| Rab3a | Decreased Rab3a expression under exposure to conditions that promote the development of T2D (proinflammatory cytokines, fatty acids, or oxidized low-density lipoproteins) | Ljubicic et al.47 |
| Rab7 | Rab7-dependent upregulated RILP expression in diabetic rats or mice causes a reduction of ISGs and promotes proinsulin degradation | Zhou et al.39 |
| Rab27a | decreased Rab27a expression upon exposure to conditions mimicking T2D | Abderrahmani et al.129 |
| Rab37 | decreased Rab37 expression under exposure to conditions that promote the development of T2D (proinflammatory cytokines, fatty acids, or oxidized low-density lipoproteins) | Ljubicic et al.47 |
| RhoA | hyperglycemic conditions increase RhoA/ROCK activity that enhances the growth of stress fibers and diminishes GSIS | Kong et al.114 |
| RhoA | RhoA mRNA levels are higher under lipotoxic conditions in INS cells | Malmgren et al.130 |
| Rac1 | glucotoxicity results in a sustained hyperactivation of Rac1 targeted to nuclear fraction and induces Rac1-mediated expression of CD36, p53, p38MAPK, and JNK1/2 activation (apoptotic signals, activation of NOX2); Tiam1 and Vav2 contribute to sustained Rac1 activation; prenylation is not essential for nuclear association of active Rac1 | Baidwan et al.110 |
| Rac1 prenylation is indispensable for glucose-stimulated NOX2 activation and ROS production | Syed et al.131 | |
| Rac1 is translocated to the membrane under hyperglycemia, hyperlipoidemia and increased ROS production | Zhou et al.132 | |
| Tiam1 and prenylation-dependent Rac1 activation is pivotal for cytokine-stimulated NOX2 activation and ROS production | Veluthakal et al.133 | |
| hyperglycemic conditions increase association between β-PIX (GEF) and Rac1 | Damacharla et al.134 | |
| Tiam1-Rac1-NOX2 signaling mediates impaired mitochondrial function in the β-cell in response to increased glucose, lipids, or pro-inflammatory cytokines; prenylation of Rac1 is crucial for its membrane translocation and activation of NOX2 | Subasinghe et al.111 | |
| Syed et al.135 | ||
| boosts PP2A-Rac1-mediated signaling in metabolic stress-caused β-cell dysfunction | Kowluru136 | |
| Rac1- NOX2 signaling pathway induces CD36 trafficking to the cell surface and amplifies influx of free fatty acids resulting in the dysfunction of β-cells | Elumalai et al.137 | |
| enzymes of the mevalonate pathway | ||
| FTase/GGTase-I | high glucose stimulates the expression of the common α-subunit of FTase/GGTase-I without affecting β-subunits and increases the activities of FTase and GGTase-I | Goalstone et al.126 |
| gluco- and lipotoxic ER stress conditions activate caspase-3-mediated cleavage of the α-subunit of FTase and GGTase-I, leading to their inactivation | Veluthakal et al.127 | |
| adipocytes | ||
| Ras GTPases | ||
| Rab4a, Rab4b | Rab4a and Rab4b mRNA and protein levels are reduced in epididymal fat in obese diabetic db/db mice; Rab4b mRNA expression is decreased in subcutaneous fat in pathologically obese patients with diabetes | Kaddai et al.138 |
| Rab5a | Rab5a mRNA expression is increased in subcutaneous fat in pathologically obese diabetic patients | Kaddai et al.138 |
| Rab11a | Rab11a mRNA expression is increased in subcutaneous fat in pathologically obese diabetic patients | Kaddai et al.138 |
| Rab18 | the presence of Rab18 in human adipose tissue is correlated to obesity; Rab18 overexpression participates in hydrolysis of triacylglycerols | Pulido et al.139 |
| dysregulated production of lumican and GDI2 contributes to IR in obese individuals through modification of collagen I organization and alters lipid storage by inhibiting binding of Rab18 to lipid droplets | Guzmán-Ruiz et al.20 | |
| RND3 | RND3 mRNA is elevated in obesity and associates positively with insulin resistance; RND3-mediated stimulation of lipolysis leads to insulin resistance; RND3 is farnesylated but it has no intrinsic GTPase activity (insensitive to GAPs) | Dankel et al.140 |
| Ras | GGPPS-induced Ras prenylation leads to chronic Erk1/2 signaling in hyperinsulinemia | Shen et al.15 |
| enzymes of the mevalonate pathway | ||
| GGPPS | Elevated GGPPS expression in insulin-resistant adipose tissues of ob/ob mice | Vicent et al.14 |
| hyperinsulinemia stimulates GGPPS and K-Ras by increasing geranylgeranylation; Ras/MAPK/Erk1/2 signaling leads to IRS-1 phosphorylation and insulin resistance; knock-down of Ggpps in insulin-resistant adipocytes restores insulin sensitivity | Shen et al.15 | |
| FTase | hyperinsulinemia promotes the phosphorylation of the α-subunit of FTase and potentiates activation of p21Ras by growth factors | Goalstone et al.141 |
| Goalstone et al.142 | ||
| skeletal muscle | ||
| Ras GTPases | ||
| Rab1A | Rab1a is upregulated in skeletal muscles of HFD-fed mice and in mitochondria of skeletal muscle from T2D patients | Chae et al.143 |
| RND3 | defective ROCK1 activity due to increased RND3 expression is connected with insulin resistance in skeletal muscles of obese T2D humans; in mice, ROCK1 deficiency causes whole-body IR as well as defects in insulin signaling in skeletal muscle | Chun et al.144 |
| RhoA | RhoA/ROCK signaling under obese and insulin-resistant conditions strains insulin pathway via phosphorylation of IRS-1 | Kanda et al.115 |
| RhoA | upregulation of mitochondrial RhoA in T2D patients | Chae et al.143 |
| Rad | Rad mRNA is increased in muscles of T2D individuals; Rad lacks typical prenylation motifs resulting in a primary cytosolic location | Reynet and Kahn145 |
| Rad is increased following insulin stimulation in nonexercised subjects which may be involved in developing insulin resistance in T2D | Coletta et al.146 | |
| Rad overexpression inhibits glucose transport in muscle cells | Moyers et al.147 | |
| interaction between increased expression of Rad and high-fat diet creates insulin resistance and alters lipid metabolism in T2D | Ilany et al.148 | |
| enzymes of the mevalonate pathway | ||
| GGPPS | GGPPS fosters lipid-induced IR in muscle by activating of the RhoA/ROCK signaling; GGPPS is overexpressed in skeletal muscles of ob/ob mice | Vicent et al.14 |
| GGPPS-controlled prenylation mediates lipid-induced insulin resistance by augmenting RhoA/ROCK signaling. ROCK2, but not ROCK1, mediates the GGPPS-regulated PI3K/Akt pathway and glucose transport | Tao et al.124 | |
| FTase | Reduced insulin-stimulated glucose uptake in muscle is related with augmented FTase expression and more farnesylated proteins | Nakazawa et al.128 |
| liver and nonalcoholic fatty liver disease (NAFLD) | ||
| Rab24 | Rab24 is upregulated in the livers of obese NAFLD patients and positively correlates with increased body fat content. Rab24 inhibition in the liver improves autophagic flux and mitochondrial connectivity, resulting in a reduction in hepatic steatosis | Seitz et al.149 |
| GGPPS | GGPPS is highly abundant in mice with obesity and IR | Vicent et al.14 |
| GGPPS is highly expressed in the livers of NAFLD patients; mice with liver-specific GGPPS knockout are protected from HFD-inflicted hepatic steatosis | Liu et al.150 | |
| GGPPS deficiency alters the FPP/GGPP ratio; accumulated FPP inhibits de novo lipogenesis by activating farnesoid X receptor | Xu et al.151 | |
| GGPPS expression is enhanced by lipid overload and regulates hepatocyte-derived extracellular vesicles secretion through Rab27A geranylgeranylation; mice with liver-specific Ggpps knockout have a lower fat deposition | Zhao et al.16 | |
| diabetic kidney disease (DKD) | ||
| RhoA | RhoA level is increased in human mesangial cells induced by hyperglycemia and subsequently Rho/ROCK signaling | Chen et al.152 |
| RhoA/ROCK signaling plays a role in the pathogenesis of diabetic kidney disease through glomerular sclerosis signaling pathways and extracellular matrix deposition | Wu et al.116 | |
| RhoA translocation to cell membrane is increased in diabetic renal cortex | Massey et al.153 | |
| diabetic retinopathy | ||
| Rac1 | activation of Tiam1-Rac1-NOX2 axis in the diabetic retina results in oxidative stress, mitochondrial damage, and cell death. | Kowluru and co-workers154,155 |
| Vav2-Rac1-NOX2 axis is activated in diabetic retinopathy. GDI is decreased in diabetic retinopathy | Mohammad et al.156 | |
| Sos1-Rac1-NOX2 axis increases ROS and leads to the pathogenesis of diabetic retinopathy | Mishra et al.112 | |
| Rac1 activation is related to impaired retinal neovascularization | Li et al.157 | |
| Rac1 activates p38 MAPK and contributes to disruption in the tight junctions, increased vascular permeability and activation of matrix metalloproteinases | Sahajpal et al.158 | |
| H-Ras and its effector, Raf-1, are increased in diabetic retinopathy; prenylation of Ras is essential for glucose-mediated effects in the retina in diabetes | Kowluru et al.159 | |
| FTase | higher FTase levels in retinal microvasculature from humans with diabetic retinopathy; FNTA knock-down inhibits glucose-stimulated Rac1-Nox2 signaling | Mohammad et al.156 |
| diabetes-accelerated macrovascular complications | ||
| RhoA | high glucose increases the growth of VSMCs (vascular smooth muscle cells) and c-fos gene expression through RhoA/ROCK | Ishiko et al.117 |
| Rac1 | high glucose results in membrane translocation of Rac1 leading to NOX activation and ROS generation that promotes proliferation of VSMCs and vascular impairment | Zhu et al.113 |
| Ras | high glucose stimulates VSMC proliferation through Ras-Raf-ERK1/2 pathway responsible for atherosclerosis progression | Chen et al.160 |
| hyperglycemic conditions result in Rac1 and endothelial dysfunction with abnormal platelet function. | Schiattarella et al.161 | |
| HMG-CoA reductase | high glucose induces HMG-CoA reductase overexpression in aortas from diabetics and cultured VSMCs | Chen et al.5 |
| FPPS | high glucose induces FPPS overexpression in aortas from diabetics and cultured VSMCs | Chen et al.7 |
| GGPPS | high glucose induces GGPPS overexpression in aortas from diabetics and cultured VSMCs | Chen et al.7 |
| FTase | high glucose induces FTase overexpression in aortas from diabetics and cultured VSMCs | Chen et al.7 |
| induction of FTase by hyperinsulinemia may account for the proliferative and atherogenic effects of insulin | Draznin162 | |
| GGTase-I | high glucose induces GGTase-I overexpression in aortas from diabetics and cultured VSMCs | Chen et al.7 |
GTPase can be targeted directly, through their regulatory proteins or prenylating enzymes. This strategy seems to represent a reasonable approach because increased activity of enzymes within the mevalonate pathway was observed in pathological states of insulin resistance, diabetes, and several T2D-related complications (Table 3).
FPPS expression was elevated in cardiomyocytes and aorta cells from diabetic mice with diabetic cardiomyopathy118 and atherosclerosis,119 respectively. FPPS inhibition by alendronate improved fasting plasma glucose, HbA1c, and insulin resistance,13 lowered the high glucose-stimulated proliferation of VSMCs,7 and reduced glucose uptake and formation of advanced glycation end products by retinal cells.120 Notably, in several clinical trials, treatment with bisphosphonates was correlated with a lower risk of T2D (Table 3). In the context of NAFLD, zoledronic acid attenuated hepatic lipid accumulation and improved liver injury by suppressing RhoA activation via decreasing FPP and GGPP farnesyl diphosphate levels.121
GGPPS inhibition may be another therapeutic strategy in T2D settings characterized by GGPPS overexpression. Although GGPPS was reported to decrease in the islets of T2D patients,122 this enzyme shows a high expression in the liver, fat and muscles of mice with obesity, IR, and hyperinsulinemia. GGPPS is a crucial mediator linking protein prenylation and metabolic reprogramming, causing NAFLD and subsequent fibrosis development. GGPPS expression was elevated in the livers of mice with obesity-induced hepatic steatosis and NAFLD patients and reduced in hepatocellular carcinoma patients.123 In adipocytes, chronic exposure to hyperinsulinism makes GGPPS constantly activated. GGPPS further increased prenylation of K-Ras and induced Erk1/2 activation, IRS phosphorylation, contributing to insulin resistance. Knock-down of Ggpps in insulin-resistant adipocytes restored IRS1 phosphorylation and increased insulin sensitivity.15 Similarly, in mice fed standard chow and high fat diets, knocking out Ggpps in the skeletal muscle increased systemic insulin sensitivity and glucose homeostasis and ameliorated palmitate-induced IR. GGPPS promoted lipid-inflicted IR in skeletal muscles by inducing IRS1 phosphorylation through the geranylgeranylated RhoA/ROCK pathway. Additionally, it was found that ROCK2, and not ROCK1, is involved in the GGPPS-regulated glucose transport in muscle cells, and Rock2 deficiency increases IRS-1/PI3K/Akt signaling in skeletal muscle and insulin sensitivity in the body. Importantly, any changes in muscle properties in the muscle-specific Ggpps knockout mice were not observed, suggesting that a deficit of GGPP alone probably does not affect muscle morphology and performance.124 Therefore, GGPPS in skeletal muscle and adipose tissue may be a potential pharmacological target for the prophylaxis of insulin resistance and T2D treatment. This method seems to be more selective for GGTase than FPPS targets, as the second approach decreases cellular FPP, which is used in both prenylation and cholesterol synthesis. As a consequence, a GGPPS targeting drug should have a less off-target effect.125
Interestingly, short-term exposure of INS 832/13 β-cells and normal rat islets to an insulinotropic concentration of glucose (20 mM) was shown to stimulate the activities of both FTase and GGTase-I along with increased expression of the α-subunit shared between FTase and GGTase-I.126 Successively, exposure of INS-1 832/13 cells and normal rodent and human islets to diabetogenic conditions, including long-term exposure to high glucose (30 mM), resulted in a caspase-3-dependent decline in FTase/GGTase-I α-subunit and accumulation of unprenylated Rap1 proteins.127 These data provide novel mechanistic insights into regulation of FTase and GGTase activities in the β-cells under normal and glucotoxic conditions. Further studies are required to identify factors regulating the expression and activity of pancreatic prenyltransferases under physiological and diabetic conditions. Especially in insulin-sensitive cells (e.g., muscle, liver, and adipose tissue), significant alterations in FTase and GGTases are connected with insulin resistance (Table 3). For example, in skeletal muscles, increased FTase expression and more farnesylated proteins were linked to decreased insulin-stimulated glucose uptake and metabolic changes. FTase inhibitors induce anti-inflammatory effect preventing inducible nitric oxide synthase (iNOS) expression under pathophysiological conditions.128
6. Strategies toward Regulation of Activity of Small GTPases via Their Direct Targeting or Inhibition of Mevalonate Pathway Enzymes
The involvement of small GTPases and their prenylation in regulating glucose and lipid homeostasis make this class of proteins important in metabolic disorders.163 Here, we summarize the approaches used to regulate GTPase activity that were reported to be associated with T2D. We concentrate on small molecule modulators that have already been used in diabetes-related studies. Simultaneously, we indicate more recent achievements in the field. The stimulus for widening the range of molecular tools comes from the common use of insufficiently potent inhibitors with not fully validated target(s) and selectivity, which might lead to erroneous results.164 Therefore, here we highlight the recently introduced compounds of high potency and known selectivity. In many cases, the proposed new molecular tools were applied for cancer-related studies, as small GTPases are commonly dysregulated in malignancies, including pancreatic cancer. We believe that their applicability can be extended to other pathological states.
One of the most typical starting points for studies on the mevalonate pathway and GTPases begins with the observation of the effect of statins on diverse cellular processes. Statins target HMG-CoA reductase, the enzyme at the top of the mevalonate pathway. The question arises as to how the observed effect depends on the more downstream elements of the signaling pathway. It can be further investigated by supplying the system with the missing (due to upstream enzyme inhibition) molecules, geranylgeraniol (GGOH) or farnesol (FOH), or their pyrophosphate analogues GGPP and FPP, respectively. If prenyl alcohols are used, they are converted to the corresponding pyrophosphates in cells and can rescue the effect of the inhibitor. The other solution is to use the inhibitors of more downstream enzymes or compounds interrupting protein–protein interactions to define the genuine target responsible for a particular cellular effect;165−167 however, this approach is still under-represented in the literature.
Several strategies can be proposed for the control of small GTPases. First, inhibition of the mevalonate pathway’s enzymes, responsible for supplying the farnesyl or geranylgeranyl pyrophosphates, leads to downregulation of small GTPases. Second, a similar result can be expected from the inhibition of enzymes, which use up these pyrophosphates for prenylation of small GTPases. The third approach involves the interruption of regulatory proteins, such as GEFs, GAPs, and GDIs.168,169 Fourth, direct targeting of GTPase, e.g., by modulating oncogenic mutant, K-RasG12C, already resulted in the compound investigated in clinical trials.170 Here, we discuss the above strategies and present selected molecular tools that already have been or can be in the future used in studies which aim at deciphering the diabetes–prenylation mutual dependence.
6.1. Inhibition of HMG-CoA: Statins
The prenylation of small GTPases requires farnesyl and geranylgeranyl pyrophosphates serving as lipid-donating substrates. These are synthesized via the mevalonate pathway. This route is currently targeted by two classes of drugs, statins, inhibitors of HMG-CoA reductase, and bisphosphonates, inhibitors of FPPS. Their pleiotropic effects are the subject of many studies, aimed at determining the extent to which indirect inhibition of downstream enzymes is responsible for these effects.165−167
Statins are the most prescribed drug regimen for treating cardiovascular disease. Their mechanism of action is based on inhibition of HMG-CoA reductase. However, their structural features differentiate them in terms of potency, solubility, and capability to cross the blood–brain barrier.166 Various studies have been devoted to the role of statins in several diseases, besides their original target, cardiovascular disorders. Their effect was observed in cancer, viral diseases, or parasite infections171,172 to name just a few. American Diabetes Association 2019 guidelines recommend the use of statins to T2D patients.173 Statins have been considered to be anti-inflammatory by inducing the production of anti-inflammatory cytokines which seems to be beneficial for alleviating the systemic inflammation present in diabetic patients. Hyperglycemia promotes inflammation in diabetes by increasing circulating cytokines, activating immune cells, and enhancing their migratory and adhesive capacity. Statin therapy resulted in lower circulating levels of proinflammatory mediators, including C-reactive protein (CRP), IL-1β, IL-6, tumor necrosis factor α (TNF-α), resistin, leptin, visfatin, monocyte chemoattractant protein-1 (MCP-1), intracellular adhesion molecule 1 (ICAM-1), and vascular cell adhesion molecule 1 (VCAM-1), and increased concentration of anti-inflammatory adipokine adopinectin174−182 (Figure 6, Table 4). A human pro-monocytic cell line cultured in high glucose and stimulated with LPS showed reduced release of TNF-α, IL-1β, IL-6, and MMP1 after statin treatment.183 Inhibition of MMP1 expression by statins was achieved through targeting protein prenylation-mediated ERK activation and could be partially rescued by GGPP. The effect was due to Ras and Rac prenylation as the addition of GGTase-I inhibitor exerted a similar effect to statins.184 Moreover, statins lowered resistin expression in 3T3-L1 adipocytes, human preadipocytes and monocytes/macrophages.175 Immune cells from diabetic patients who underwent statin therapy showed lower expression of activation markers, lymphocyte function-associated antigen-1 (LFA-1), very late activation antigen-4 (VLA-4), and CD18, and reduced activation potential.185,186 Pravastatin and fluvastatin decreased the adherence of neutrophils and monocytes to human endothelial cells under high glucose conditions by reducing the surface expression of endothelial adhesion molecules (intercellular adhesion molecule-1 (ICAM-1), P-selectin, and E-selectin).187,188 Furthermore, statin treatment inhibited NF-κBp65 and MAPK proinflammatory signaling pathways in monocytes from T1D patients, muscle cells from streptozotocin (STZ)-treated rats, and aortic endothelial cells cultured under high glucose.174,189,190 The effect was H-Ras-mediated, as dominant-negative H-RAs (S17N) exerted an effect similar to that with statin treatment.190 Atorvastatin and rosuvastatin improved antigen-specific immunity and cytotoxic activity of T cells in diabetic mice.191
Figure 6.
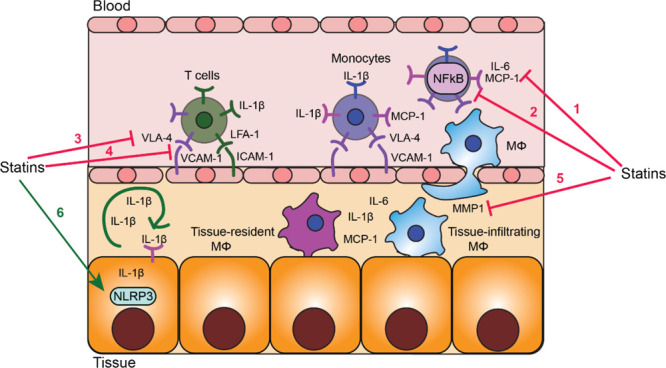
Dual effect of statins on inflammation in diabetes. Statins exert anti-inflammatory effects via (1) reducing chemoattractant levels in the circulation; (2) reducing proinflammatory signaling pathways in blood leukocytes; (3) reducing VLA-4 and FLA-1 integrin levels on blood monocytes and lymphocytes; (4) reducing VCAM-1 and ICAM-1 levels on endothelial cells; (5) reducing MMP1 production by macrophages. These effects result in the inhibition of leukocyte recruitment from the blood into the tissue. Statins exert proinflammatory effects via (6) activation of the NLRP3 inflammasome in insulin-sensitive tissue that leads to enhanced production of IL-1β. IL-1β autostimulation amplifies inflammation and attracts immune cells
Table 4. Selected Statins and Their Application as Tools to Study Diabetes and Inflammation202−205a.


Proinflammatory cytokines: IL-1β, IL-2, IL-6, TNF-α. Proinflammatory chemokines: IL-8, MCP-1. Proinflammatory adipokines: leptin, resistin, visfatin. Anti-inflammatory adipokines: adiponectin. Adhesion molecules: ICAM-1, VCAM-1, E-selectin, P-selectin. Proteases: MMP-1. Signaling pathways: ERK, NF-κB.
However, statins were also demonstrated to contribute to the proinflammatory environments in diabetes. Statins can activate the NLRP3 inflammasome in adipose tissue via p38 and mTOR.192 Activation of NLRP3 inflammasome regulates IL-1β, promotes adipose tissue inflammation and leads to IR. The effect of statins was via inhibition of prenylation and not by lowering cholesterol metabolites. The authors studied LPS-primed adipose explants in the presence of either cholesterol derivatives (LDL-cholesterol, free cholesterol or 25-hydroxycholesterol) or GGPP or FOH. They observed rescue in atorvastatin-induced suppression of the insulin signal in fat tissue in the presence of GGPP but not with FOH.193
The above studies did not report which of the small GTPases contributed to inflammasome activation and were affected by inhibition of the prenylation. The possible candidates are Rac1, Rap1A, and Rabs. In either statin-treated or GGTase-I-deficient macrophages stimulated with LPS, nonprenylated Rac1 showed increased interaction with its effector proteins, was hyperactivated, and triggered inflammasomes. Preincubating the macrophages with GGPP mostly abrogated the statin effect on cytokine production.194 In a statin-treated THP-1 monocytic cell line stimulated with LPS, prenylation of Rabs and Rap1A was inhibited and IL-1β production was induced. The addition of geranylgeraniol (GGOH) restored normal protein prenylation and abolished inflammasome formation and IL-1β and IL-18 release.195 In LPS-treated bone marrow-derived macrophages, overexpression of Rab1 increased NLRP3 inflammasomes and IL-1β and IL-18 cytokines, while knockdown of Rab1 or overexpression of its dominant-negative form (Rab1 N124I) had the opposite effect. Whether the effect of Rab1 on inflammasome activation was dependent on its prenylation remains to be assessed.196
Overall, treatment of β-cells with statins contributed to a substantial decrease in insulin release. High concentrations of statins induced β-cell apoptosis and further reduced insulin secretion. In addition, by suppressing GLUT4, statins reduce glucose uptake in human skeletal muscle cells and adipocytes.87,197 Also, treatment with statins, which results in an increase of cholesterol uptake in the β-cell, leads to reduced protein expression of GLUT2, hence limiting glucose uptake.197,198 Inhibition of prenylation using either statins or inhibitors of FTase induced a caspase-3-mediated decline in the levels of prenylated proteins, such as nuclear lamins, leading to β-cell dysregulation and death.199 High-dose statin treatment slowed the progression of coronary atherosclerosis, resulting in disease regression in both diabetic and nondiabetic patients.200
Although several questions remain unanswered, statins increase T2D risk, with some statins showing a stronger association (e.g., simvastatin, rosuvastatin, and atorvastatin) than others (e.g., pravastatin).11 Additionally, as the generation of mevalonate derivatives is blocked by statins and the former regulates the expression of HMG-CoA reductase via multiple feedback mechanisms, there is an observed remarkable increase in HMG-CoA levels. This restricts the effectiveness of the drug and instigates more intensive treatments that may lead to side effects.201 Thus, treatment of insulin resistance, T2D, and T2D-related complications with HMG-CoA reductase inhibitors may be a viable option.
6.2. Inhibition of FPPS: Bisphosphonates and Nonphosphorus Analogues
The most potent inhibitors of FPPS and GGPPS belong to the bisphosphonates, chemically stable analogues of pyrophosphates, the natural substrates of these enzymes. Bisphosphonate inhibitors of FPPS constitute a known drug class. They bind to hydroxyapatite in bone tissue because of the Ca2+ chelating properties of the α,α-bisphosphonic acid motif. They show high selectivity for osteoclasts deposited in bone minerals, and therefore, they are used to restrain osteoclast-mediated bone resorption. Bisphosphonates are also used in patients with cancers causing osteolysis, and some studies show their antitumor activity. However, the charged nature of this group makes them challenging to employ for other therapeutic applications, due to high bone affinity and low serum levels in nonbone applications, low cell membrane permeability, and high clearance by the kidneys. Still, a number of reports have shown that administration of bisphosphonates could be associated with a reduction in the risk of incident T2D,12 reduced glucose uptake, formation of glycation end products, insulin resistance,120 and hepatic lipid accumulation.121 These effects were observed in various tissues affected by diabetes, including the retina and liver (Table 5).
Table 5. Selected Inhibitors of FPPS211−217.

Nitrogen-containing bisphosphonates (N-BP), such as zoledronic acid, risedronic acid, alendronic acid, pamidronic acid, and minodronic acid, belong to the clinically validated inhibitors of FPPS (Table 5 and 6). They compete for binding in the allylic site of FPPS with the natural substrates, DMAPP and GPP. The search for inhibitors of human FPPS binding at the active site did not bring nanomolar potency inhibitors without bisphosphonic moiety. Therefore, attempts were directed at identifying inhibitors targeting the allosteric site near the C-terminus of the enzyme.207 Several such nonbisphosphonate classes of inhibitors were proposed,207−210e.g., 1–4, although not all of them bind inside the FPPS allosteric pocket.210 Although these compounds were designed to have superior “druglike” properties in comparison to the bisphosphonates, none of them showed notable antitumor activity in cell-based tests. To the best of our knowledge, their potential in diabetes-related studies has not been investigated yet. That is why here we show only selected examples, limiting cases to those tested for human FPPS and showing nanomolar potency (Table 6).
Table 6. Bisphosphonate and Non-bisphosphonate Inhibitors of FPPS with Potential to Be Used in Diabetes-Related Studies218−220.

6.3. Inhibition of GGPPS: Lipophilic Bisphosphonates
The enzyme responsible for the synthesis of geranylgeranyl pyrophosphate is GGPPS, and it is now intensively studied as a potential drug target.221
The elevated expression of GGPPS was induced by high glucose levels.7 Its high abundance was observed in a number of tissues of obese and/or diabetic patients, promoting, for example, lipid-induced muscle insulin resistance.14 However, up to now, the GGPPS inhibitors were not used in diabetes-related studies. Instead, inhibitors of upstream enzymes in the mevalonate pathway were applied or the experiments were run on cells with GGPPS knock-down. Therefore, here we show that direct inhibitors of GGPPS do exist and we present the selective and the most potent among them as available chemical tools to study diabetes-related processes.
The number of selective GGPPS inhibitors is limited, partially due to the previously held conviction that dual FPPS and GGPPS inhibitors are more efficient as antitumor agents. Despite the low sequence identity between human FFPS and GGPPS (17%), their tertiary (but not quaternary) structures are surprisingly similar and their catalytic mechanisms are probably similar.207 Therefore, many attempts at obtaining GGPPS inhibitors led to the development of dual FPPS and GGPPS inhibitors, such as compound 8 (Figure 7), which is about 100 times more potent than zoledronic acid in obstructing tumor growth,222 or compound 7, which represents another chemotype of GGPPS bisphosphonate inhibitors and shows ∼15× higher activity toward GGPPS, compared with FPPS.223
Figure 7.

Structures of the selected GGPPS inhibitors not used in diabetes studies.
The FPPS inhibitors are usually smaller molecules, having a shorter alkyl chain and a positive-charge feature. The GGPPS bisphosphonate inhibitors contain one or two large hydrophobic groups, they lack hydroxyl group in C-α, and there is no positive charge required. Therefore, they are more lipophilic, which makes them more prone to targeting nonbone tissues.207
The broadest class of GGPPS inhibitors contains a bisphosphonic acid moiety, which is a substitute of the unstable pyrophosphate residue. It turned out that digeranylated bisphosphonic acid 5, representing the so-called V-shaped molecules, shows 0.2 μM activity against GGPPS and no inhibition of farnesylation.221,224 At least one geranyl or longer isoprenoid chain is required for inhibition of GGPPS; these prenyl chains occupy the substrate and product binding sites, FPP and GGPP, respectively.225 Several such V-shaped compounds,224,226 including those that contain an ether bond, 6,226 and the so-called U-shaped analogues were prepared.227
Recent works show the anticancer therapeutic potential of several hydrophobic bisphosphonates. However, the most interesting group is constituted by triazoles228 that carry an isoprenoid chain (Figure 7). The homogeranyl and homoneryl triazole analogues, 9, turned out to be the most potent GGPPS inhibitors reported, demonstrating high selectivity in inhibiting GGPPS vs FPPS. They can slow pancreatic tumor growth in vivo.229 The preliminary studies on metabolic stability and pharmacokinetics indicate that they are metabolically stable in human liver microsomes.230 Most analogues showed a higher potency of the Z isomer. An interesting property was observed for 9, as studies demonstrated that the two isomers interact synergistically, making the mixture more potent than a single isomer. It is tentatively explained as resulting from synergistic binding in both the substrate, FPP, and product, GGPP, inhibitory channels.221 In the case of analogues bearing a methyl group at C-α, compound 10, the activity against GGPPS was similar for both isomers, 0.086 mM for (Z)-10 and 0.125 mM for (E)-10.231 Additionally, such a design, with the locked C-α, enables the prodrug form preparation to overcome the bioavailability hurdles of bisphosphonic drugs.231
6.4. Inhibition of Prenylating Enzyme, FTase, and Direct Targeting of Ras Proteins
Ras proteins regulate cell proliferation, differentiation and survival. The most known members of the Ras subfamily are Harvey-Ras (H-Ras), neuroblastoma-Ras (N-Ras), and Kirsten-Ras (K-Ras). K-Ras is the most commonly mutated protein in many cancers, accounting for almost 85% of all Ras mutations.232 The K-RasG12D mutation is the most prevalent in pancreatic and colorectal cancers. G12 is located at the protein active site, interacting with a phosphate-binding loop (P-loop) and two switch regions, which control binding to effector and regulatory proteins. The oncogenic K-Ras mutation inhibits GTP hydrolysis (by weakening its GTPase activity or hampering the GAP-stimulated GTP hydrolysis), making such mutants constantly active and activating downstream effectors.233
In the early efforts to control the activity of Ras, the inhibition of FTase was the most widely developed approach. FTase is responsible for PTMs of Ras, enabling their proper localization in the membrane, often after additional modifications, such as palmitoylation. While several FTIs (FTase inhibitors) were developed, they failed in clinical trials due to alternative prenylation with GGTase-I, which restored their membrane association. There is renewed interest in FTase inhibitors, as their efficacy against the regulation of H-Ras activity has been verified. Out of a few dozen trials, one FTI small molecule drug, lonafarnib (commercially available from Sigma-Aldrich), has been recently approved by the U.S. Food and Drugh Administration [FDA; https://www.fda.gov/drugs/drug-approvals-and-databases/drug-trials-snapshots-zokinvy] for the therapy of Hutchinson-Gilford Progeria Syndrome and certain progeroid laminopathies. Several other drug candidates are at various stages of preclinical or clinical trials to prevent or treat cancer, such as manumycin-A, FTI-277, tipifarnib, L778123, and BMS-214662.170
Several other strategies directly targeting Ras proteins have been developed. Besides the use of biologics, such as monoclonal antibodies, mimetics of antibody variable fragments, and antisense oligonucleotides,234 efforts have been undertaken to interrupt the association between Ras and regulatory or effector proteins, such as phosphodiesterase-δ, Sos, Raf, or Tiam1. A breakthrough strategy has been developed for selective targeting of a mutant variant of K-RasG12C and small molecules, such as AMG510, MRTX849, ARS3248, and LY3499446 covalently modifying the mutant cysteine, that has progressed to clinical trials (e.g., NCT04380753, NCT04667234).235 Recently, Crews and collaborators have shown the potential of a PROTAC molecule, LC-2, developed from the covalent K-RasG12C inhibitor (MRTX849) linked with the VHL (von Hippel-Lindau ligase) ligand, which turned out to be an efficient K-Ras degrader.236 Several reviews have been recently published covering these topics [see refs (232) and (235)].
Few studies were devoted to selective targeting of another mutant K-RasG12D, the most prevalent in pancreatic cancer. Sakamoto et al. introduced K-RasG12D KS-58, derived from KRpep-2d (Ac-RRRRCPLYISYDPVCRRRR-NH2), which inhibited interactions with two proteins, RasGDP-Sos1 (GDP-GTP exchange) and RasGDP-BRaf. It inhibits both GDP- and GTP-bound K-RasG12D. Despite its molecular weight (1333.6 g/mol) and negatively charged polar residue, it showed anticancer activity in vivo, making it a potential lead compound.234
To the best of our knowledge, Ras proteins have not been directly associated with diabetes yet, as their misregulation is more connected with cancer. However, several reports indicate that hyperglycemia and/or hyperinsulinemia stimulate the expression and/or activation of FTase (Table 3). Therefore, we listed some FTase inhibitors (Table 7), concentrating on those that have been already used in diabetes-related studies or are at various stages in clinical trials. Most of them are commercially available, which makes them accessible for many laboratories. On the other hand, the repurposing strategy for already studied (potential) therapeutics has many advantages. Such agents have already undergone thorough examinations in terms of their toxicity, bioavailability, and other aspects, which need consideration in drug development. For more information on the plethora of FTase inhibitors, please refer to recent reviews [see refs (232) and (235)].
Table 7. Selected Inhibitors of FTase, GGTase-I, and Ras Proteins237−240,243.

6.5. Inhibition of Prenylating Enzymes: GGTase-I
GGTase-I inhibitors have received less attention than inhibitors of FTase. GGT-I inhibitors often serve in combination with FTIs in order to inhibit prenylation and function of oncogenesis drivers, K-Ras and N-Ras proteins. Blocking only FTase activity led to alternative prenylation of FTase substrates by GGTase-I. Therefore, several dual inhibitors of these two prenyl transferases were also developed.244
Interestingly, this research area also evolved in a different direction: the development of agents directly targeting the GGTase-I substrates, Rho GTPases. This gives an alternative pathway for the selective regulation of particular GTPases. This topic is covered in the following paragraph.
Although GGTase-I is an attractive target for cancer-related studies, its inhibitors are rarely used in diabetes research. GGTase-I might be overexpressed under high glucose concentrations (Table 3), while its knock-down blocked diabetes-accelerated atherosclerosis,251 which might be related to interfering with Rac1 geranylgeranylation, finally inhibiting ROS production, and ERK1/2 and JNK signaling.
Peptidomimetics of the CAAX motif in protein substrate and dihydropyrrole or tetrahydropyridine-based analogues constitute two main classes of GGTase-I inhibitors. Here, we listed inhibitors of GGTase-I, giving priority to molecules that have already been used in diabetes-related studies. Among them, we find selective a GGTase-I inhibitor, GGTI-2147, and FGTI-2734, which show dual inhibition of FTase and GGTase-I.244 The representative of dihydropyrrole analogues, P61-A6,242 was applied in the design of targeted delivery of P61-A6 to pancreatic cancer cells.241 For that purpose, the GGTase-I inhibitor (or in combination with FTase inhibitor) was encapsulated into liposomes, which upon exposure to the lower pH of cancerous cells was released.
There are some representatives of GGT-I inhibitors, which have potential in future studies as they are of nanomolar potency, are commercially available and commonly applied in biological studies, or show different degrees of selectivity against FTase vs GGTase-I. We also include GGTI-2418 as the only GGTase-I inhibitor currently in clinical trials. Selected examples of such compounds are listed in the Tables 7 and 8.
Table 8. Selective and Dual Inhibitors of FTase and GGTase-I and Direct Inhibitor of K-Ras that Have Potential to Be Used in Diabetes-Related Studies245−250.

6.6. Direct Targeting of Rho GTPases
The strategy based on inhibition of GGTase-I alone or in combination with FTase is limited by its nonselectivity in terms of affecting many GTPases. The efforts to directly and selectively target Rho GTPase ended with success. The most studied representatives of Rho GTPases are Rac1, RhoA, and Cdc42, which are often overexpressed in malignancies, as they are regulators of cancer cell migration and invasion. The subfamilies of Rho GTPases interact with each other and are controlled by regulatory proteins and effectors.252 Their hyperactivation can result from their mutations, downregulation of GAPs, or upregulation of GEFs. The latter interaction is the most commonly targeted. As the topic of regulation of Rho GTPases has been widely summarized recently,253,252 here we concentrate on selected inhibitors, directly targeting Rac1 and RhoA, as the connections of these with diabetes-related malfunctions are the most broadly reported (Table 9).
Table 9. Compounds Interrupting the Protein–Protein Interactions of Rho GTPases Applied in Diabetes-Related Research (Part A) and Those That Have Potential to Be Used in Future Diabetes-Related Studies (Part B)259−276.


As has been already mentioned, one of the most popular strategies to inhibit Rac1 activation is the interruption of its binding with GEFs. There are several Rac1-Tiam1 (GEF) (T-cell lymphoma invasion and metastasis 1) inhibitors.254−257 The structural studies identified the specific amino acid residues.253 In addition to small molecule inhibitors, there were attempts to develop peptide-derived Rac1-Tiam1 inhibitors.258
In the case of RhoA regulation, it was found that GGPPS promotes lipid-induced insulin resistance in muscle by enhancing RhoA/ROCK signaling.124 It could be prevented by inhibition of GGPPS or RhoA/ROCK interaction. Several ROCK kinase inhibitors have been developed and used as tools in diabetes-related studies (Table 9). However, one needs to remember that the ROCK pathway is essential for many cellular processes and Rac and Cdc42 are crucial regulators of a plethora of cell signaling receptors.253 Therefore, more selective approached are needed.
In Table 9, we present inhibitors that can potentially be used as probes, as they interrupt protein–protein interactions that are important in diabetes. Among them, we can distinguish inhibitors of Rac1 interaction with GEFs such as P-Rex1, Vav2, or Trio. Another mechanism works for compound 12 and 13 that by blocking interaction with nucleotide disrupts binding between Rac1 and PAK1.
6.7. Inhibition of Prenylating Enzymes: GGTase-II
The abnormal activities of GGTase-II and some Rab proteins have been identified in several diseases, including cancer, such as pancreas, breast, skin, colon, lung, ovarian, and prostate, to name just a few.277 GGTase-II alone was not reported to be up- or downregulated in diabetes, but some Rab GTPases can be associated with various aspects of T2D (Table 3). Up to now, in most identified cases, the pathological effect of dysregulation of Rab GTPases was associated with their impaired activity. However, in a few cases, Rab GTPase was upregulated, e.g., Rab24 in the livers of obese NAFLD patients correlated with body fat content.149 Since the current state of knowledge implies that, in diabetes, the upregulation of Rabs is required to reverse the pathological state, new strategies need to be developed. Here, we discuss the approaches that have been studied to date to present the currently available tools.
Several attempts have been made to control GTPases; however, these approaches are not very diversified. One of the most studied strategies is based on the development of inhibitors of GGTase-II. This enzyme was proven to be a druggable target. Several classes of small molecule inhibitors have been developed (compounds representing these classes (15–24) are presented in Figure 8),242,278−281 differing in their mode of action (e.g., inhibitors of first or second geranylgeranylation), selectivity (versus other prenyltransferases), and potency. GGTase-II inhibition is limited by the lack of substrate selectivity, as it affects all or most Rab GTPases. The most active analogues contain a tetrahydrobenzodiazepine motif (compound 15).279 Only in the case of α-phosphonocarboxylates (19–23), the selectivity toward different Rabs was reported. This class of inhibitors prohibits the introduction only of the second geranylgeranyl group to Rabs, leaving the monogeranylated Rabs unaffected. Among the currently known phosphonocarboxylates, the most active ones contain imidazo[1,2-a]282,283,281 or the imidazole ring.284,285
Figure 8.

Structures of the selected GGTase-II (RGGT) inhibitors not used in diabetes studies. LED: lowest effective dose toward inhibition of Rab11 prenylation.
Another strategy is based on the direct targeting of Rab GTPases. Only few such attempts have been reported in the literature. These studies involved analysis of the protein–protein interaction surfaces in order to design molecules mimicking them. These studies resulted in the development of stapled peptides, StRIP16, which targets Rab8a, mimicking its interaction with RIP,286 and RFP14, blocking Rab25:FIP complex formation, in which FIP is the effector protein.287 Although these studies were also dedicated to optimizing the stability and bioavailability of these inhibitors, they need further refinement.
7. Recent Strategies for Selective Targeting of Inhibitors to Diabetes-Affected Organs
The small GTPases and their regulatory proteins are omnipresent in all kinds of cells. Therefore, when planning to use the inhibitors in diabetes-related studies, specific delivery to certain tissues needs to be considered to increase their efficiency and bioavailability while reducing toxicity and dosing frequency. A number of reviews exist that describe organ-specific delivery systems288 and prodrug strategies, including those that show a possible masking of ionic phosphonic groups, with the latter being so popular among the compounds described in this Perspective.289 Here we selected several approaches targeting tissues related with diabetes.
The development of various types of antidiabetic drugs has been accompanied by the constant progress in the field of their delivery, especially in terms of the effective and convenient transport of insulin, a protein, which due to its unstable nature cannot be delivered orally. Peptide-derived therapeutics have limited oral bioavailability due to their destruction by gastric acid and proteolytic enzymes and the limited absorption from the intestine. However, medicinal chemistry has developed several strategies to overcome these hurdles, based on various structural modifications (e.g., PEGylation, attachment of cell-penetrating peptides) or coapplication of enzyme inhibitors. That topic has been broadly described in many medicinal chemistry textbooks. In the case of peptides and other classes of therapeutics, the transportation and targeting can be improved by the use of nanocarrier delivery systems, which include liposomes, niosomes, polymeric nanoparticles or micelles, and dendrimers.290 When the drug is encapsulated within a nanostructure, such a nanomaterial presents both opportunities, such as the possibility of surface modification with a tissue-targeting moiety as well as safety concerns, variable efficiency, outcome of biomaterial degradation, and possible side effects. The field of nanodelivery is under constant development, and one needs to be aware that such studies require additional caution, but the potential of nanocarriers cannot be denied. Here, we present examples of the recently reported strategies or reviews for selectively targeting drugs to β-cells, liver cells, adipocytes, and muscle cells.
The interesting feature of β-cells is an exceptionally high concentration of zinc ions (up to ∼30 mM) while the zinc concentration in the cytosol in most cells is ∼400 pM.291 Zn(II) can catalyze hydrolytic reactions, which can be used to ignite the activity of the released cargo. Because of the above features, many attempts were reported to design a system for imaging β-cells.292
That feature was used for attaching a zinc-chelating residue onto a β-cell replication-inducing compound.293 Another study involved designing a prodrug consisting of an inactivated drug linked with a Zn(II)-binding ligand. Such an approach was applied for the targeted release of fluorochromes and β-cell mitogenic compounds in human β-cells.292 In both cases, the hybrid compounds preferentially accumulated within β-cells. Upon reaching the Zn(II)-abundant environment, the bond between the cargo and the Zn(II)-binding scaffold was cleaved, releasing the active cargo.
In the last 20 years, diverse strategies have been developed for noninvasive imaging of β-cells for diagnostics. For that purpose, a number of β-cell-surface-specific proteins, often overexpressed, were used, such as vesicular monoamine transporter 2 (VMAT2), sulphonylurea receptor (SUR-1), glucagon-like peptide 1 (GLP-1), free fatty acid receptor 1 (FFAR1), and β-cell-specific antigens. Some of the markers used for β-cell imaging can be used to design targeting molecules, such as monoclonal antibodies, to selectively deliver a drug, which will be cleaved upon reaching the target.294 To recognize the surface-specific protein, antibody–drug conjugates could be used, which recently have gained importance as an attractive approach for cell-specific targeting. Although challenging, GPCR-specific monoclonal antibodies are also being developed, and the first ones, erenumab and mogamulizumab, were recently approved by the FDA.295
These strategies were developed for certain tissues affected by nondiabetes-related pathological states, such as cancer, liver fibrosis, and muscle aging. Analogous strategies can be applied for the targeted delivery of drugs to the tissues affected by diabetes. Still, careful evaluation needs to be conducted to determine to what extent the developed methods can be applied for diabetes-stricken organs.
For selective targeting to the liver, several delivery methods, including the ones that use surface markers, were developed for liver cancer cells296 and proposed for liver fibrosis.297 In the case of muscle cells and adipocytes, selective targeting is challenging because of their high representation in the body. However, for skeletal muscle, surface recognition elements were identified and used for selective uptake. In addition to small molecules like carnitine (a drug linked with carnitine shows improved muscle uptake via OCTN2 transport), monoclonal antibodies, or viral vectors,298 aptamers have also been proposed as a muscle-specific delivery vehicle.299
8. Future Perspective
The involvement of small GTPases and their prenylation in regulating glucose and lipid homeostasis makes this class of proteins important in metabolic disorders. Inhibitors of protein prenylation have been investigated as potential therapeutics to treat multiple diseases. Statins, used primarily as cholesterol-lowering drugs, were also found to reduce systemic inflammatory responses independently of cholesterol. Various clinical trials demonstrated that treatment with statins decreased soluble proinflammatory mediators and lowered the activation capacity of monocytes and lymphocytes.176,177,179,182,206In vitro studies identified statin targets as being small GTPases (Ras, Rac and Rho).174,184,190 On the other hand, accumulating evidence suggests that statins enhance the inflammatory responses and elevate the risk of diabetes.11 The evidence for statin-mediated effects points toward the NLRP3 inflammasome/caspase-1 complex, and this could be a new target in the treatment of inflammation in diabetes.192,193 However, there may be more still-unexplored prenylation targets that contribute to increased inflammation upon exposure to statins. Thus, decreasing the activity of enzymes that are downstream from HMG-CoA reductase in the mevalonate pathway may be a promising strategy for treating insulin resistance and diabetes. Pro- and anti-inflammatory effects of statins could be explained by the opposite outcomes of the mevalonate pathway’s inhibition, depending on the tissue, euglycemia versus hyperglycemia, and target type. Enhancing prenylation may localize specific GTPase and thus enhance its function. It may also sequester it away from its effectors and reduce the effect. Further studies should be conducted to assess how prenylation controls inflammation and insulin sensitivity in muscle, liver, and adipose tissue, and insulin production and secretion by pancreatic islets. Statins, inhibitors of other enzymes in the mevalonate pathway, as well as GTPase activation inhibitors should be employed to identify the specific factors that enhance or reduce inflammation and contribute to insulin resistant β-cell dysfunction. It will further our knowledge about the function of prenylation in diabetes and allow the development of more context-specific treatments.
Defective or upregulated prenylation can contribute to the decrease of metabolic cell viability and dysfunction in pancreatic β-cells.127 Several enzymes are decreased in the islets of T2D patients while they are upregulated in the liver, adipose tissue, and muscles in individuals with obesity, insulin resistance, and hyperinsulinemia (Table 3). Therefore, further studies are required to identify factors regulating the expression and activity of pancreatic prenyltransferases under physiological and diabetic conditions. More work needs to be done to show which signaling pathway is essential for desired efficacy. Moreover, a better understanding of how the beneficial effect from preclinical T2D models can be effectively translated to T2D patients is needed.
After a broad search for the interconnections between small GTPases and different proteins and processes in T2D, we summarized the approaches that can be used to regulate GTPases activity in pathological cellular machinery triggered by hyperglycemia. We concentrated on small molecules. It is crucial to be cautious when using inhibitors, both those newly reported as well as such that are known for some time. The proper molecular probe should be potent and selective toward the validated molecular target. Otherwise, such studies might repeatedly generate uncertain or even erroneous results.164 Therefore, here, besides showing the previously used chemical probes, sometimes not of the highest quality,164 we highlight the recently introduced compounds of high potency and known selectivity.
We described the most common strategies used to control small GTPases, via inhibition of the mevalonate pathway and prenylating enzymes, or the interactions between GTPases and their regulatory proteins, such as GEFs. In the case of most GTPases, there has been significant progress in developing chemical tools—potent and selective inhibitors—allowing further studies. However, most approaches studied involve the downregulation of GTPases, while expression or activity of Rab GTPases tends to be downregulated under conditions that favor the development of diabetes. In addition to targeting the gene expression, no other strategy to achieve Rab upregulation has been applied yet. Here, the opportunity might be spotted at targeting the interactions with regulatory proteins, such as GAP and GDI, which bind Rabs and inactivates them under normal circumstances. Also, downstream effectors, or other post-translational modifications, such as phosphorylation/dephosphorylation, ubiquitination, palmitoylation, and serotonylation, can be targeted.253,300
In diabetes-related studies, the apparent targets among GAPs constitute TBC1D1 and TBC1D4, which are Akt targets in insulin-stimulated GLUT4 traffic. Mutations in TBC1D1 and TBC1D4 are linked with obesity and insulin resistance in humans. Phosphorylation of TBC1D1 and TBC1D4 is thought to shut down their GAP function, leading to increased levels of active Rab GTPases, which triggers GLUT4 translocation.301
However, these different approaches are not straightforward. Individual functions of the different Rab proteins that undergo various post-translational modifications, such as phosphorylation, serotonylation, AMPylation, phosphocholination, palmitoylation, and ubiquitination, often occur at localization, which affects the interaction with diverse proteins GAPs, GDIs, and effectors. Only a few such interactions have been already identified, and only in a few cases it was determined when the interaction with the effector is taking place, after or before particular post-translational modification. Phosphorylation of Rabs is still poorly recognized in terms of its role, mechanistic implications, and regulation via kinase-phosphatase-mediated modifications. The different sites might be phosphorylated by different kinases, leading to diverse effects and distinct distribution of Rabs, altering the activity of GAPs, GEFs, effectors, and others. Also, phosphorylation of Rab GTPases may be reversible through the action of protein phosphatases, which may reverse the signaling cascade. The four locations of phosphorylation were recently distinguished. For example, the phosphorylation at switch II may interfere with Rab–GAP interaction, simultaneously increasing or decreasing the interaction with the effector protein. On the other hand, phosphorylation within the α3/β5 loop antagonizes the catalytic activity of another kinase, LRRK2.302
It is the future task to comprehend how small GTPases are linked to diabetes and related disorders. In addition to the application of existing small molecular tools, continuously developing technologies, such as (phospho)proteome- and genome-wide screening, could be used as a measure to identify the various partners of small GTPases, including their mutual dependencies.
Glossary
Abbreviations
- Akt
protein kinase B
- Arp2/3
actin-related protein 2/3 complex
- BP
bisphosphonate
- CD
cluster of differentiation
- CRP
C-reactive protein
- DKD
diabetic kidney disease
- DMAPP
dimethylallyl pyrophosphate
- ER
endoplasmic reticulum
- ERGIC
ER-Golgi intermediate compartment
- ERK
extracellular-signal-regulated kinase
- FOH
farnesol
- FPP
farnesyl pyrophosphate
- FPPS
farnesyl pyrophosphate synthase
- FTase
farnesyltransferase
- GPP
geranyl pyrophosphate
- FTI
FTase inhibitor
- GGOH
geranylgeraniol
- GGPP (or GRG)
geranylgeranyl pyrophosphate, geranylgeranyl diphosphate
- GGPPS
geranylgeranyl pyrophosphate synthase
- GGTase-I
geranylgeranyltransferase type I
- GGTase-II
Rab geranylgeranyl transferase
- GGTase-III
geranylgeranyltransferase III
- GLUT
glucose transporter
- GSIS
glucose-stimulated insulin secretion
- GSV
GLUT4 storage vesicles
- HbA1c
hemoglobin A1c
- HMG-CoA
3-hydroxymethyl-3-methylglutaryl coenzyme A
- ICAM-1
intracellular adhesion molecule 1
- IgG
immunoglobulin G
- IL
interleukin
- IR
insulin resistance
- ISG
insulin secretory granule
- IPP
isopentenyl diphosphate
- IR
insulin, resistance
- IRS
insulin receptor substrate
- IRV
insulin-responsive vesicles
- Kir2
inwardly rectifying potassium channel 2
- LFA-1
lymphocyte function-associated antigen
- LPS
lipopolysaccharides
- MCP-1
monocyte chemoattractant protein-1
- MMP-1
matrix metalloproteinase-1
- NAFLD
nonalcoholic fatty liver disease
- NF-κB
nuclear factor kappa-light-chain-enhancer of activated B cells
- NOX
NADPH oxidase
- p65
LNR3NOD-like receptor family pyrin domain containing 3 inflammasome
- PAK1
P21-activated kinase 1
- PDK
phosphoinositide-dependent kinase
- PHA
phytohemagglutinin
- PM
plasma membrane
- RGGT
Rab geranylgeranyl transferase
- ROS
reactive oxygen species
- RRP
readily releasable pool
- SNARE
soluble N-ethylmaleimide sensitive factor attachment receptor
- STZ
streptozotocin
- SUR1
sulfonylurea receptor-1, a regulatory subunit of ATP-sensitive potassium channel
- TCA cycle
tricarboxylic acid cycle
- T2D
type 2 diabetes
- TGN
Trans-Golgi Network
- TNF-α
tumor necrosis factor α
- VCAM-1
vascular cell adhesion molecule 1
- VGCC
voltage-gated calcium channel
- VSMC
vascular smooth muscle cell
Biographies
Edyta Gendaszewska-Darmach is a Professor at at Lodz University of Technology. She is also a member of the University Senate. Prof. Gendaszewska-Darmach graduated from the Lodz University with a diploma in molecular biology. She received her Ph.D. in chemical sciences from the Centre of Molecular and Macromolecular Studies, Polish Academy of Sciences, and completed habilitation in biotechnology from the Lodz University of Technology. Her ongoing research has resulted in over 40 publications. She has collaborated extensively with scientific groups to search for compounds with antidiabetic activity in the group of GPCR ligands, inhibitors of small GTPases prenylation, and is studying the molecular mechanism of the prohealth action of phytochemicals (fatty acids and their derivatives). She has served as a Principal Investigator or Investigator on several research grants.
Katarzyna Błażewska is a Professor at the Lodz University of Technology. She graduated from the Lodz University of Technology. She was a postdoctoral fellow at the University of Southern California and Fulbright scholar at Boston College. Her main research interests concentrate on Rab GTPases prenylation, using as tools differentially modified inhibitors of GGTase-II. She shares her chemistry expertise with Prof. Gendaszewska-Darmach, using it to design and synthesise lipid-derived probes. She was a Principal Investigator on a number of grants.
Malgorzata A. Garstka is a professor at the Second Affiliated Hospital of Xi’an Jiaotong University, China. She was awarded her Ph.D. in Biochemistry at Jacob University, Bremen, Germany, and received postdoctoral training at the Leiden University Medical Center and The Netherlands Cancer Institute, The Netherlands. Her research team is investigating the adaptive immune system in health and diabetes and develops diagnostic tools. Prof. Garstka published her work in the Journal of Experimental Medicine, EMBO Journal, and PNAS, among others, and has been a Principal Investigator on several grants.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.1c00410.
List of crystal structures of small GTPases playing a role in diabetes, corresponding to Figure 1; list of crystal structures of the enzymes of mevalonate pathway playing a role in diabetes mellitus, corresponding to Figure 3; amino acid sequence alignment of human GTPases involved in diabetes and insulin resistance (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work was financially supported by the National Science Centre, Poland grants: Opus (2018/31/B/NZ9/02433 to E.G.-D.) and Sonata Bis (2014/14/E/ST5/00491 to K.M.B.), Start up Budget from the Second Affiliated Hospital of Xi’an Jiaotong University (number 82668428 to M.A.G.), Personal Fellowship from the Second Affiliated Hospital of Xi’an Jiaotong University (number 87679215 to M.A.G.) and Free Exploration Project grant from the Second Affiliated Hospital of Xi’an Jiaotong University (82668330 to M.A.G.).
The authors declare no competing financial interest.
Supplementary Material
References
- Saeedi P.; Petersohn I.; Salpea P.; Malanda B.; Karuranga S.; Unwin N.; Colagiuri S.; Guariguata L.; Motala A. A.; Ogurtsova K.; Shaw J. E.; Bright D.; Williams R. Global and Regional Diabetes Prevalence Estimates for 2019 and Projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9th Edition. Diabetes Res. Clin. Pract. 2019, 157, 107843. 10.1016/j.diabres.2019.107843. [DOI] [PubMed] [Google Scholar]
- Donath M. Y. Targeting Inflammation in the Treatment of Type 2 Diabetes: Time to Start. Nat. Rev. Drug Discovery 2014, 13, 465–476. 10.1038/nrd4275. [DOI] [PubMed] [Google Scholar]
- Roden M.; Shulman G. I. The Integrative Biology of Type 2 Diabetes. Nature 2019, 576, 51–60. 10.1038/s41586-019-1797-8. [DOI] [PubMed] [Google Scholar]
- Gendaszewska-Darmach E.; Drzazga A.; Koziołkiewicz M. Targeting GPCRs Activated by Fatty Acid-Derived Lipids in Type 2 Diabetes. Trends Mol. Med. 2019, 25, 915–929. 10.1016/j.molmed.2019.07.003. [DOI] [PubMed] [Google Scholar]
- Nguyen U. T. T.; Wu Y.; Goodall A.; Alexandrov K.. Analysis of Protein Prenylation In Vitro and In Vivo Using Functionalized Phosphoisoprenoids. In Current Protocols in Protein Science; Blackwell Publishing Inc., 2010; Chapter 14. 10.1002/0471140864.ps1403s62 [DOI] [PubMed] [Google Scholar]
- Brioschi M.; Martinez Fernandez A.; Banfi C. Exploring the Biochemistry of the Prenylome and Its Role in Disease through Proteomics: Progress and Potential. Expert Rev. Proteomics 2017, 14, 515–528. 10.1080/14789450.2017.1332998. [DOI] [PubMed] [Google Scholar]
- Chen G. P.; Zhang X. Q.; Wu T.; Li L.; Han J.; Du C. Q. Alteration of Mevalonate Pathway in Proliferated Vascular Smooth Muscle from Diabetic Mice: Possible Role in High-Glucose-Induced Atherogenic Process. J. Diabetes Res. 2015, 2015, 379287. 10.1155/2015/379287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goalstone M. L.; Wall K.; Leitner J. W.; Kurowski T.; Ruderman N.; Pan S. J.; Ivy J. L.; Moller D. E.; Draznin B. Increased Amounts of Farnesylated P21Ras in Tissues of Hyperinsulinaemic Animals. Diabetologia 1999, 42, 310–316. 10.1007/s001250051156. [DOI] [PubMed] [Google Scholar]
- Draznin B.; Miles P.; Kruszynska Y.; Olefsky J.; Friedman J.; Golovchenko I.; Stjernholm R.; Wall K.; Reitman M.; Accili D.; Cooksey R.; McClain D.; Goalstone M. Effects of Insulin on Prenylation as a Mechanism of Potentially Detrimental Influence of Hyperinsulinemia. Endocrinology 2000, 141, 1310–1316. 10.1210/endo.141.4.7411. [DOI] [PubMed] [Google Scholar]
- Okin D.; Medzhitov R. The Effect of Sustained Inflammation on Hepatic Mevalonate Pathway Results in Hyperglycemia. Cell 2016, 165, 343–356. 10.1016/j.cell.2016.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galicia-Garcia U.; Jebari S.; Larrea-Sebal A.; Uribe K. B.; Siddiqi H.; Ostolaza H.; Benito-Vicente A.; Martín C. Statin Treatment-Induced Development of Type 2 Diabetes: From Clinical Evidence to Mechanistic Insights. Int. J. Mol. Sci. 2020, 21, 4725. 10.3390/ijms21134725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toulis K. A.; Nirantharakumar K.; Ryan R.; Marshall T.; Hemming K. Bisphosphonates and Glucose Homeostasis: A Population-Based, Retrospective Cohort Study. J. Clin. Endocrinol. Metab. 2015, 100, 1933–1940. 10.1210/jc.2014-3481. [DOI] [PubMed] [Google Scholar]
- Karimi Fard M.; Aminorroaya A.; Kachuei A.; Salamat M. R.; Hadi Alijanvand M.; Aminorroaya Yamini S.; Karimifar M.; Feizi A.; Amini M. Alendronate Improves Fasting Plasma Glucose and Insulin Sensitivity, and Decreases Insulin Resistance in Prediabetic Osteopenic Postmenopausal Women: A Randomized Triple-Blind Clinical Trial. J. Diabetes Invest. 2019, 10, 731–737. 10.1111/jdi.12944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicent D.; Maratos-Flier E.; Kahn C. R. The Branch Point Enzyme of the Mevalonate Pathway for Protein Prenylation Is Overexpressed in Theob/Ob Mouse and Induced by Adipogenesis. Mol. Cell. Biol. 2000, 20, 2158–2166. 10.1128/MCB.20.6.2158-2166.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen N.; Yu X.; Pan F. Y.; Gao X.; Xue B.; Li C. J. An Early Response Transcription Factor, Egr-1, Enhances Insulin Resistance in Type 2 Diabetes with Chronic Hyperinsulinism. J. Biol. Chem. 2011, 286, 14508–14515. 10.1074/jbc.M110.190165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Zhao M. F.; Jiang S.; Wu J.; Liu J.; Yuan X. W.; Shen D.; Zhang J. Z.; Zhou N.; He J.; Fang L.; Sun X. T.; Xue B.; Li C. J. Liver Governs Adipose Remodelling via Extracellular Vesicles in Response to Lipid Overload. Nat. Commun. 2020, 11, 1–17. 10.1038/s41467-020-14450-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veluthakal R.; Madathilparambil S. V.; McDonald P.; Olson L. K.; Kowluru A. Regulatory Roles for Tiam1, a Guanine Nucleotide Exchange Factor for Rac1, in Glucose-Stimulated Insulin Secretion in Pancreatic β-Cells. Biochem. Pharmacol. 2009, 77, 101–113. 10.1016/j.bcp.2008.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu L.; Pan C.; He S. M.; Lang B.; Gao G. D.; Wang X. L.; Wang Y. The Ras Superfamily of Small Gtpases in Non-Neoplastic Cerebral Diseases. Front. Mol. Neurosci. 2019, 12, 121. 10.3389/fnmol.2019.00121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Møller L. L. V.; Klip A.; Sylow L. Rho GTPases—Emerging Regulators of Glucose Homeostasis and Metabolic Health. Cells 2019, 8, 434. 10.3390/cells8050434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzmán-Ruiz R.; Tercero-Alcázar C.; Rabanal-Ruiz Y.; Díaz-Ruiz A.; El Bekay R.; Rangel-Zuñiga O. A.; Navarro-Ruiz M. C.; Molero L.; Membrives A.; Ruiz-Rabelo J. F.; Pandit A.; López-Miranda J.; Tinahones F. J.; Malagón M. M. Adipose Tissue Depot-specific Intracellular and Extracellular Cues Contributing to Insulin Resistance in Obese Individuals. FASEB J. 2020, 34, 7520–7539. 10.1096/fj.201902703R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J.; Matralis A. N.; Berghuis A. M.; Tsantrizos Y. S. Human Isoprenoid Synthase Enzymes as Therapeutic Targets. Front. Chem. 2014, 2, 50. 10.3389/fchem.2014.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Costa R. F.; Freire V. N.; Bezerra E. M.; Cavada B. S.; Caetano E. W. S.; De Lima Filho J. L.; Albuquerque E. L. Explaining Statin Inhibition Effectiveness of HMG-CoA Reductase by Quantum Biochemistry Computations. Phys. Chem. Chem. Phys. 2012, 14, 1389–1398. 10.1039/C1CP22824B. [DOI] [PubMed] [Google Scholar]
- Holstein S. A.; Tong H.; Kuder C. H.; Hohl R. J. Quantitative Determination of Geranyl Diphosphate Levels in Cultured Human Cells. Lipids 2009, 44, 1055–1062. 10.1007/s11745-009-3355-x. [DOI] [PubMed] [Google Scholar]
- Suazo K. F.; Park K.-Y.; Distefano M. D. A Not-So-Ancient Grease History: Click Chemistry and Protein Lipid Modifications. Chem. Rev. 2021, 10.1021/acs.chemrev.0c01108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palsuledesai C. C.; Distefano M. D. Protein Prenylation: Enzymes, Therapeutics, and Biotechnology Applications. ACS Chem. Biol. 2015, 10, 51–62. 10.1021/cb500791f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zverina E. A.; Lamphear C. L.; Wright E. N.; Fierke C. A. Recent Advances in Protein Prenyltransferases: Substrate Identification, Regulation, and Disease Interventions. Curr. Opin. Chem. Biol. 2012, 16, 544–552. 10.1016/j.cbpa.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchay S.; Wang H.; Marzio A.; Jain K.; Homer H.; Fehrenbacher N.; Philips M. R.; Zheng N.; Pagano M. GGTase3 Is a Newly Identified Geranylgeranyltransferase Targeting a Ubiquitin Ligase. Nat. Struct. Mol. Biol. 2019, 26, 628–636. 10.1038/s41594-019-0249-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirakawa R.; Goto-Ito S.; Goto K.; Wakayama S.; Kubo H.; Sakata N.; Trinh D. A.; Yamagata A.; Sato Y.; Masumoto H.; Cheng J.; Fujimoto T.; Fukai S.; Horiuchi H. A SNARE Geranylgeranyltransferase Essential for the Organization of the Golgi Apparatus. EMBO J. 2020, 39, e104120. 10.15252/embj.2019104120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omar-Hmeadi M.; Idevall-Hagren O. Insulin Granule Biogenesis and Exocytosis. Cell. Mol. Life Sci. 2021, 78, 1957–1970. 10.1007/s00018-020-03688-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger C.; Zdzieblo D. Glucose Transporters in Pancreatic Islets. Pfluegers Arch. 2020, 472, 1249–1272. 10.1007/s00424-020-02383-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowluru A.; Kowluru R. A. Protein Prenylation in Islet β-Cell Function in Health and Diabetes: Putting the Pieces of the Puzzle Together. Biochem. Pharmacol. 2015, 98, 363–370. 10.1016/j.bcp.2015.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.; Wang Z.; Yang Y.; Li Q.; Zeng R.; Kang J.; Wu J. Rab1A Mediates Proinsulin to Insulin Conversion in β-Cells by Maintaining Golgi Stability through Interactions with Golgin-84. Protein Cell 2016, 7, 692–696. 10.1007/s13238-016-0298-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara T.; Kano F.; Murata M. Rab2A Is a Pivotal Switch Protein That Promotes Either Secretion or ER-Associated Degradation of (pro)Insulin in Insulin-Secreting Cells. Sci. Rep. 2015, 4, 1–14. 10.1038/srep06952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunaga K.; Taoka M.; Isobe T.; Izumi T. Rab2a and Rab27a Cooperatively Regulate the Transition from Granule Maturation to Exocytosis through the Dual Effector Noc2. J. Cell Sci. 2016, 130, 541–550. 10.1242/jcs.195479. [DOI] [PubMed] [Google Scholar]
- Yasuda T.; Shibasaki T.; Minami K.; Takahashi H.; Mizoguchi A.; Uriu Y.; Numata T.; Mori Y.; Miyazaki J. I.; Miki T.; Seino S. Rim2α Determines Docking and Priming States in Insulin Granule Exocytosis. Cell Metab. 2010, 12, 117–129. 10.1016/j.cmet.2010.05.017. [DOI] [PubMed] [Google Scholar]
- Coppola T.; Frantz C.; Perret-Menoud V.; Gattesco S.; Hirling H.; Regazzi R. Pancreatic β-Cell Protein Granuphilin Binds Rab3 and Munc-18 and Controls Exocytosis. Mol. Biol. Cell 2002, 13, 1906–1915. 10.1091/mbc.02-02-0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto M.; Miki T.; Shibasaki T.; Kawaguchi M.; Shinozaki H.; Nio J.; Saraya A.; Koseki H.; Miyazaki M.; Iwanaga T.; Seino S. Noc2 Is Essential in Normal Regulation of Exocytosis in Endocrine and Exocrine Cells. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 8313–8318. 10.1073/pnas.0306709101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazares V. A.; Subramani A.; Saldate J. J.; Hoerauf W.; Stuenkel E. L. Distinct Actions of Rab3 and Rab27 Gtpases on Late Stages of Exocytosis of Insulin. Traffic 2014, 15, 997–1015. 10.1111/tra.12182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y.; Liu Z.; Zhang S.; Zhuang R.; Liu H.; Liu X.; Qiu X.; Zhang M.; Zheng Y.; Li L.; Hong W.; Wang T. RILP Restricts Insulin Secretion through Mediating Lysosomal Degradation of Proinsulin. Diabetes 2020, 69, 67–82. 10.2337/db19-0086. [DOI] [PubMed] [Google Scholar]
- Uchida K.; Nomura M.; Yamamoto T.; Ogawa Y.; Teramoto N. Rab8a Is Involved in Membrane Trafficking of Kir6.2 in the MIN6 Insulinoma Cell Line. Pfluegers Arch. 2019, 471, 877–887. 10.1007/s00424-018-02252-1. [DOI] [PubMed] [Google Scholar]
- Sugawara K.; Shibasaki T.; Mizoguchi A.; Saito T.; Seino S. Rab11 and Its Effector Rip11 Participate in Regulation of Insulin Granule Exocytosis. Genes Cells 2009, 14, 445–456. 10.1111/j.1365-2443.2009.01285.x. [DOI] [PubMed] [Google Scholar]
- Yi Z.; Yokota H.; Torii S.; Aoki T.; Hosaka M.; Zhao S.; Takata K.; Takeuchi T.; Izumi T. The Rab27a/Granuphilin Complex Regulates the Exocytosis of Insulin-Containing Dense-Core Granules. Mol. Cell. Biol. 2002, 22, 1858–1867. 10.1128/MCB.22.6.1858-1867.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torii S.; Zhao S.; Yi Z.; Takeuchi T.; Izumi T. Granuphilin Modulates the Exocytosis of Secretory Granules through Interaction with Syntaxin 1a. Mol. Cell. Biol. 2002, 22, 5518–5526. 10.1128/MCB.22.15.5518-5526.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Ishizaki R.; Xu J.; Kasai K.; Kobayashi E.; Gomi H.; Izumi T. The Rab27a Effector Exophilin7 Promotes Fusion of Secretory Granules That Have Not Been Docked to the Plasma Membrane. Mol. Biol. Cell 2013, 24, 319–330. 10.1091/mbc.e12-04-0265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno K.; Ramalho J. S.; Izumi T. Exophilin8 Transiently Clusters Insulin Granules at the Actin-Rich Cell Cortex Prior to Exocytosis. Mol. Biol. Cell 2011, 22, 1716–1726. 10.1091/mbc.e10-05-0404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura T.; Taniguchi S.; Niki I. Actin Assembly Controlled by GDP-Rab27a Is Essential for Endocytosis of the Insulin Secretory Membrane. Arch. Biochem. Biophys. 2010, 496, 33–37. 10.1016/j.abb.2010.01.017. [DOI] [PubMed] [Google Scholar]
- Ljubicic S.; Bezzi P.; Brajkovic S.; Nesca V.; Guay C.; Ohbayashi N.; Fukuda M.; Abderrhamani A.; Regazzi R. The GTPase Rab37 Participates in the Control of Insulin Exocytosis. PLoS One 2013, 8, e68255. 10.1371/journal.pone.0068255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammar E.; Tomas A.; Bosco D.; Halban P. A. Role of the Rho-ROCK (Rho-Associated Kinase) Signaling Pathway in the Regulation of Pancreatic β-Cell Function. Endocrinology 2009, 150, 2072–2079. 10.1210/en.2008-1135. [DOI] [PubMed] [Google Scholar]
- Uenishi E.; Shibasaki T.; Takahashi H.; Seki C.; Hamaguchi H.; Yasuda T.; Tatebe M.; Oiso Y.; Takenawa T.; Seino S. Actin Dynamics Regulated by the Balance of Neuronal Wiskott-Aldrich Syndrome Protein (N-WASP) and Cofilin Activities Determines the Biphasic Response of Glucose-Induced Insulin Secretion. J. Biol. Chem. 2013, 288, 25851–25864. 10.1074/jbc.M113.464420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Oh E.; Thurmond D. C. Glucose-Stimulated Cdc42 Signaling Is Essential for the Second Phase of Insulin Secretion. J. Biol. Chem. 2007, 282, 9536–9546. 10.1074/jbc.M610553200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalwat M. A.; Yoder S. M.; Wang Z.; Thurmond D. C. A P21-Activated Kinase (PAK1) Signaling Cascade Coordinately Regulates F-Actin Remodeling and Insulin Granule Exocytosis in Pancreatic β Cells. Biochem. Pharmacol. 2013, 85, 808–816. 10.1016/j.bcp.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevins A. K.; Thurmond D. C. A Direct Interaction between Cdc42 and Vesicle-Associated Membrane Protein 2 Regulates SNARE-Dependent Insulin Exocytosis. J. Biol. Chem. 2005, 280, 1944–1952. 10.1074/jbc.M409528200. [DOI] [PubMed] [Google Scholar]
- Daniel S.; Noda M.; Cerione R. A.; Sharp G. W. G. A Link between Cdc42 and Syntaxin Is Involved in Mastoparan-Stimulated Insulin Release. Biochemistry 2002, 41, 9663–9671. 10.1021/bi025604p. [DOI] [PubMed] [Google Scholar]
- Nevins A. K.; Thurmond D. C. Caveolin-1 Functions as a Novel Cdc42 Guanine Nucleotide Dissociation Inhibitor in Pancreatic β-Cells. J. Biol. Chem. 2006, 281, 18961–18972. 10.1074/jbc.M603604200. [DOI] [PubMed] [Google Scholar]
- Kimura T.; Yamaoka M.; Taniguchi S.; Okamoto M.; Takei M.; Ando T.; Iwamatsu A.; Watanabe T.; Kaibuchi K.; Ishizaki T.; Niki I. Activated Cdc42-Bound IQGAP1 Determines the Cellular Endocytic Site. Mol. Cell. Biol. 2013, 33, 4834–4843. 10.1128/MCB.00895-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asahara S.; Shibutani Y.; Teruyama K.; Inoue H. Y.; Kawada Y.; Etoh H.; Matsuda T.; Kimura-Koyanagi M.; Hashimoto N.; Sakahara M.; Fujimoto W.; Takahashi H.; Ueda S.; Hosooka T.; Satoh T.; Inoue H.; Matsumoto M.; Aiba A.; Kasuga M.; Kido Y. Ras-Related C3 Botulinum Toxin Substrate 1 (RAC1) Regulates Glucose-Stimulated Insulin Secretion via Modulation of F-Actin. Diabetologia 2013, 56, 1088–1097. 10.1007/s00125-013-2849-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veluthakal R.; Tunduguru R.; Arora D. K.; Sidarala V.; Syeda K.; Vlaar C. P.; Thurmond D. C.; Kowluru A. VAV2, a Guanine Nucleotide Exchange Factor for Rac1, Regulates Glucose-Stimulated Insulin Secretion in Pancreatic Beta Cells. Diabetologia 2015, 58, 2573–2581. 10.1007/s00125-015-3707-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thamilselvan V.; Gamage S.; Harajli A.; Chundru S. A.; Kowluru A. P-Rex1 Mediates Glucose-Stimulated Rac1 Activation and Insulin Secretion in Pancreatic β-Cells. Cell. Physiol. Biochem. 2021, 54, 1218–1230. 10.33594/000000310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibasaki T.; Takahashi H.; Miki T.; Sunaga Y.; Matsumura K.; Yamanaka M.; Zhang C.; Tamamoto A.; Satoh T.; Miyazaki J. I.; Seino S. Essential Role of Epac2/Rap1 Signaling in Regulation of Insulin Granule Dynamics by CAMP. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 19333–19338. 10.1073/pnas.0707054104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ljubicic S.; Bezzi P.; Vitale N.; Regazzi R. The GTPase RalA Regulates Different Steps of the Secretory Process in Pancreatic β-Cells. PLoS One 2009, 4, e7770. 10.1371/journal.pone.0007770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez J. A.; Kwan E. P.; Xie L.; He Y.; James D. E.; Gaisano H. Y. The RalA GTPase Is a Central Regulator of Insulin Exocytosis from Pancreatic Islet Beta Cells. J. Biol. Chem. 2008, 283, 17939–17945. 10.1074/jbc.M800321200. [DOI] [PubMed] [Google Scholar]
- Xie L.; Kang Y.; Liang T.; Dolai S.; Xie H.; Parsaud L.; Lopez J. A.; He Y.; Chidambaram S.; Lam P. P.; James D. E.; Sugita S.; Gaisano H. Y. RalA GTPase Tethers Insulin Granules to L- and R-Type Calcium Channels Through Binding A2δ-1 Subunit. Traffic 2013, 14, 428–439. 10.1111/tra.12047. [DOI] [PubMed] [Google Scholar]
- Jaldin-Fincati J. R.; Pavarotti M.; Frendo-Cumbo S.; Bilan P. J.; Klip A. Update on GLUT4 Vesicle Traffic: A Cornerstone of Insulin Action. Trends Endocrinol. Metab. 2017, 28, 597–611. 10.1016/j.tem.2017.05.002. [DOI] [PubMed] [Google Scholar]
- Tsuchiya A.; Kanno T.; Shimizu T.; Tanaka A.; Nishizaki T. Rac1 and ROCK Are Implicated in the Cell Surface Delivery of GLUT4 under the Control of the Insulin Signal Mimetic DiDCP-LA-PE. J. Pharmacol. Sci. 2015, 128, 179–184. 10.1016/j.jphs.2015.07.001. [DOI] [PubMed] [Google Scholar]
- Richter E. A.; Hargreaves M. Exercise, GLUT4, and Skeletal Muscle Glucose Uptake. Physiol. Rev. 2013, 93, 993–1017. 10.1152/physrev.00038.2012. [DOI] [PubMed] [Google Scholar]
- Sylow L.; Nielsen I. L.; Kleinert M.; Møller L. L. V.; Ploug T.; Schjerling P.; Bilan P. J.; Klip A.; Jensen T. E.; Richter E. A. Rac1 Governs Exercise-Stimulated Glucose Uptake in Skeletal Muscle through Regulation of GLUT4 Translocation in Mice. J. Physiol. 2016, 594, 4997–5008. 10.1113/JP272039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L.; Omata W.; Kojima I.; Shibata H. Direct Interaction of Rab4 with Syntaxin 4. J. Biol. Chem. 2001, 276, 5265–5273. 10.1074/jbc.M003883200. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Wang Y.; Zhang J.; Deng Y.; Jiang L.; Song E.; Wu X. S.; Hammer J. A.; Xu T.; Lippincott-Schwartz J. Rab10 and Myosin-va Mediate Insulin-Stimulated GLUT4 Storage Vesicle Translocation in Adipocytes. J. Cell Biol. 2012, 198, 545–560. 10.1083/jcb.201111091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessneer K. L.; Jackson R. M.; Griesel B. A.; Olson A. L. Rab5 Activity Regulates GLUT4 Sorting into Insulin-Responsive and Non-Insulin-Responsive Endosomal Compartments: A Potential Mechanism for Development of Insulin Resistance. Endocrinology 2014, 155, 3315–3328. 10.1210/en.2013-2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mîinea C. P.; Sano H.; Kane S.; Sano E.; Fukuda M.; Peränen J.; Lane W. S.; Lienhard G. E. AS160, the Akt Substrate Regulating GLUT4 Translocation, Has a Functional Rab GTPase-Activating Protein Domain. Biochem. J. 2005, 391, 87–93. 10.1042/BJ20050887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y.; Chiu T. T.; Foley K. P.; Bilan P. J.; Klip A. Myosin Va Mediates Rab8A-Regulated GLUT4 Vesicle Exocytosis in Insulin-Stimulated Muscle Cells. Mol. Biol. Cell 2014, 25, 1159–1170. 10.1091/mbc.e13-08-0493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadacca L. A.; Bruno J.; Wen J.; Xiong W.; McGraw T. E. Specialized Sorting of GLUT4 and Its Recruitment to the Cell Surface Are Independently Regulated by Distinct Rabs. Mol. Biol. Cell 2013, 24, 2544–2557. 10.1091/mbc.e13-02-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno J.; Brumfield A.; Chaudhary N.; Iaea D.; McGraw T. E. SEC16A Is a RAB10 Effector Required for Insulinstimulated GLUT4 Trafficking in Adipocytes. J. Cell Biol. 2016, 214, 61–76. 10.1083/jcb.201509052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano H.; Peck G. R.; Blachon S.; Lienhard G. E. A Potential Link between Insulin Signaling and GLUT4 Translocation: Association of Rab10-GTP with the Exocyst Subunit Exoc6/6b. Biochem. Biophys. Res. Commun. 2015, 465, 601–605. 10.1016/j.bbrc.2015.08.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S.; Crisman L.; Miller J.; Datta I.; Gulbranson D. R.; Tian Y.; Yin Q.; Yu H.; Shen J. Inducible Exoc7/Exo70 Knockout Reveals a Critical Role of the Exocyst in Insulin-Regulated GLUT4 Exocytosis. J. Biol. Chem. 2019, 294, 19988–19996. 10.1074/jbc.RA119.010821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karunanithi S.; Xiong T.; Uhm M.; Leto D.; Sun J.; Chen X. W.; Saltiel A. R. A Rab10:RalA G Protein Cascade Regulates Insulin-Stimulated Glucose Uptake in Adipocytes. Mol. Biol. Cell 2014, 25, 3059–3069. 10.1091/mbc.e14-06-1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulbranson D. R.; Davis E. M.; Demmitt B. A.; Ouyang Y.; Ye Y.; Yu H.; Shen J. RABIF/MSS4 Is a Rab-Stabilizing Holdase Chaperone Required for GLUT4 Exocytosis. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, E8224–E8233. 10.1073/pnas.1712176114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano H.; Peck G. R.; Kettenbach A. N.; Gerber S. A.; Lienhard G. E. Insulin-Stimulated GLUT4 Protein Translocation in Adipocytes Requires the Rab10 Guanine Nucleotide Exchange Factor Dennd4C. J. Biol. Chem. 2011, 286, 16541–16545. 10.1074/jbc.C111.228908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh G. I.; Leney S. E.; Lloyd-Lewis B.; Wherlock M.; Lindsay A. J.; McCaffrey M. W.; Tavaré J. M. Rip11 Is a Rab11- and AS160-RabGAP-Binding Protein Required for Insulin-Stimulated Glucose Uptake in Adipocytes. J. Cell Sci. 2007, 120, 4197–4208. 10.1242/jcs.007310. [DOI] [PubMed] [Google Scholar]
- Zeigerer A.; Lampson M. A.; Karylowski O.; Sabatini D. D.; Adesnik M.; Ren M.; McGraw T. E. GLUT4 Retention in Adipocytes Requires Two Intracellular Insulin-Regulated Transport Steps. Mol. Biol. Cell 2002, 13, 2421–2435. 10.1091/mbc.e02-02-0071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed S. E.; Hodgson L. R.; Song S.; May M. T.; Mastick C. C.; Verkade P.; Tavare J. M. A Role for Rab14 in the Endocytic Trafficking of GLUT4 in 3T3-L1 Adipocytes. J. Cell Sci. 2013, 126, 1931–1941. 10.1242/jcs.104307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z.; Menzel F.; Benninghoff T.; Chadt A.; Du C.; Holman G. D.; Al-Hasani H. Rab28 Is a TBC1D1/TBC1D4 Substrate Involved in GLUT4 Trafficking. FEBS Lett. 2017, 591, 88–96. 10.1002/1873-3468.12509. [DOI] [PubMed] [Google Scholar]
- Davey J. R.; Humphrey S. J.; Junutula J. R.; Mishra A. K.; Lambright D. G.; James D. E.; Stöckli J. TBC1D13 Is a RAB35 Specific GAP That Plays an Important Role in GLUT4. Traffic 2012, 13, 1429–1441. 10.1111/j.1600-0854.2012.01397.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L.; Adams R. D.; Saltiel A. R. The TC10-Interacting Protein CIP4/2 Is Required for Insulin-Stimulated Glut4 Translocation in 3T3L1 Adipocytes. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 12835–12840. 10.1073/pnas.202495599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Z. Y.; Chawla A.; Bose A.; Way M.; Czech M. P. A Phosphatidylinositol 3-Kinase-Independent Insulin Signaling Pathway to N-WASP/Arp2/3/F-Actin Required for GLUT4 Glucose Transporter Recycling. J. Biol. Chem. 2002, 277, 509–515. 10.1074/jbc.M108280200. [DOI] [PubMed] [Google Scholar]
- Duong K. H. M.; Chun K. H. Regulation of Glucose Transport by RhoA in 3T3-L1 Adipocytes and L6Myoblasts. Biochem. Biophys. Res. Commun. 2019, 519, 880–886. 10.1016/j.bbrc.2019.09.083. [DOI] [PubMed] [Google Scholar]
- Takaguri A.; Satoh K.; Itagaki M.; Tokumitsu Y.; Ichihara K. Effects of Atorvastatin and Pravastatin on Signal Transduction Related to Glucose Uptake in 3T3L1 Adipocytes. J. Pharmacol. Sci. 2008, 107, 80–89. 10.1254/jphs.FP0072403. [DOI] [PubMed] [Google Scholar]
- Chun K. H.; Araki K.; Jee Y.; Lee D. H.; Oh B. C.; Huang H.; Park K. S.; Lee S. W.; Zabolotny J. M.; Kim Y. B. Regulation of Glucose Transport by ROCK1 Differs from That of ROCK2 and Is Controlled by Actin Polymerization. Endocrinology 2012, 153, 1649–1662. 10.1210/en.2011-1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usui I.; Imamura T.; Huang J.; Satoh H.; Olefsky J. M. Cdc42 Is a Rho TGPase Family Member That Can Mediate Insulin Signaling to Glucose Transport in 3T3-L1 Adipocytes. J. Biol. Chem. 2003, 278, 13765–13774. 10.1074/jbc.M208904200. [DOI] [PubMed] [Google Scholar]
- Balamatsias D.; Kong A. M.; Waters J. E.; Sriratana A.; Gurung R.; Bailey C. G.; Rasko J. E. J.; Tiganis T.; Macaulay S. L.; Mitchell C. A. Identification of P-Rex1 as a Novel Rac1-Guanine Nucleotide Exchange Factor (GEF) That Promotes Actin Remodeling and GLUT4 Protein Trafficking in Adipocytes. J. Biol. Chem. 2011, 286, 43229–44340. 10.1074/jbc.M111.306621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X. W.; Leto D.; Xiong T.; Yu G.; Cheng A.; Decker S.; Saltiel A. R. A Ral GAP Complex Links PI 3-Kinase/Akt Signaling to RalA Activation in Insulin Action. Mol. Biol. Cell 2011, 22, 141–152. 10.1091/mbc.e10-08-0665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X. W.; Leto D.; Chiang S. H.; Wang Q.; Saltiel A. R. Activation of RalA Is Required for Insulin-Stimulated Glut4 Trafficking to the Plasma Membrane via the Exocyst and the Motor Protein Myo1c. Dev. Cell 2007, 13, 391–404. 10.1016/j.devcel.2007.07.007. [DOI] [PubMed] [Google Scholar]
- Chen X. W.; Leto D.; Xiao J.; Goss J.; Wang Q.; Shavit J. A.; Xiong T.; Yu G.; Ginsburg D.; Toomre D.; Xu Z.; Saltiel A. R. Exocyst Function Regulated by Effector Phosphorylation. Nat. Cell Biol. 2011, 13, 580–588. 10.1038/ncb2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skorobogatko Y.; Dragan M.; Cordon C.; Reilly S. M.; Hung C. W.; Xia W.; Zhao P.; Wallace M.; Lackey D. E.; Chen X. W.; Osborn O.; Bogner-Strauss J. G.; Theodorescu D.; Metallo C. M.; Olefsky J. M.; Saltiel A. R. RalA Controls Glucose Homeostasis by Regulating Glucose Uptake in Brown Fat. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 7819–7824. 10.1073/pnas.1801050115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J.; Cheng D.; Liu L.; Lv Z.; Liu K. TBC1D15 Affects Glucose Uptake by Regulating GLUT4 Translocation. Gene 2019, 683, 210–215. 10.1016/j.gene.2018.10.025. [DOI] [PubMed] [Google Scholar]
- Ishikura S.; Klip A. Muscle Cells Engage Rab8A and Myosin Vb in Insulin-Dependent GLUT4 Translocation. American Journal of Physiology - Cell Physiology 2008, 295, C1016–C1025. 10.1152/ajpcell.00277.2008. [DOI] [PubMed] [Google Scholar]
- Sun Y.; Bilan P. J.; Liu Z.; Klip A. Rab8A and Rab13 Are Activated by Insulin and Regulate GLUT4 Translocation in Muscle Cells. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 19909–19914. 10.1073/pnas.1009523107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y.; Jaldin-Fincati J.; Liu Z.; Bilan P. J.; Klip A. A Complex of Rab13 with MICAL-L2 and α-Actinin-4 Is Essential for Insulin-Dependent GLUT4 Exocytosis. Mol. Biol. Cell 2016, 27, 75–89. 10.1091/mbc.E15-05-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikura S.; Bilan P. J.; Klip A. Rabs 8A and 14 Are Targets of the Insulin-Regulated Rab-GAP AS160 Regulating GLUT4 Traffic in Muscle Cells. Biochem. Biophys. Res. Commun. 2007, 353, 1074–1079. 10.1016/j.bbrc.2006.12.140. [DOI] [PubMed] [Google Scholar]
- JeBailey L.; Wanono O.; Niu W.; Roessler J.; Rudich A.; Klip A. Ceramide- and Oxidant-Induced Insulin Resistance Involve Loss of Insulin-Dependent Rac-Activation and Actin Remodeling in Muscle Cells. Diabetes 2007, 56, 394–403. 10.2337/db06-0823. [DOI] [PubMed] [Google Scholar]
- Sylow L.; Kleinert M.; Pehmøller C.; Prats C.; Chiu T. T.; Klip A.; Richter E. A.; Jensen T. E. Akt and Rac1 Signaling Are Jointly Required for Insulin-Stimulated Glucose Uptake in Skeletal Muscle and Downregulated in Insulin Resistance. Cell. Signalling 2014, 26, 323–331. 10.1016/j.cellsig.2013.11.007. [DOI] [PubMed] [Google Scholar]
- Sun Y.; Côté J. F.; Du K. Elmo2 Is a Regulator of Insulin-Dependent Glut4Membrane Translocation. J. Biol. Chem. 2016, 291, 16150–16161. 10.1074/jbc.M116.731521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Y.; Zhang C.; Zhao X.; Liu S.; Lv X.; Zhang S.; Yang J.; Chen L.; Duan H.; Zhang Y.; Yao Z.; Niu W. Tiam1Mediates Rac1 Activation and Contraction-induced Glucose Uptake in Skeletal Muscle Cells. FASEB J. 2021, 35, e21210. 10.1096/fj.202001312R. [DOI] [PubMed] [Google Scholar]
- Ueda S.; Kataoka T.; Satoh T. Activation of the Small GTPase Rac1 by a Specific Guanine-Nucleotide-Exchange Factor Suffices to Induce Glucose Uptake into Skeletal-Muscle Cells. Biology of the Cell 2008, 100, 645–661. 10.1042/BC20070160. [DOI] [PubMed] [Google Scholar]
- Takenaka N.; Nihata Y.; Satoh T. Rac1 Activation Caused by Membrane Translocation of a Guanine Nucleotide Exchange Factor in Akt2-Mediated Insulin Signaling in Mouse Skeletal Muscle. PLoS One 2016, 11, e0155292. 10.1371/journal.pone.0155292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Oh E.; Clapp D. W.; Chernoff J.; Thurmond D. C. Inhibition or Ablation of P21-Activated Kinase (PAK1) Disrupts Glucose Homeostatic Mechanisms in Vivo. J. Biol. Chem. 2011, 286, 41359–41367. 10.1074/jbc.M111.291500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Møller L. L. V.; Jaurji M.; Kjøbsted R.; Joseph G. A.; Madsen A. B.; Knudsen J. R.; Lundsgaard A. M.; Andersen N. R.; Schjerling P.; Jensen T. E.; Krauss R. S.; Richter E. A.; Sylow L. Insulin-Stimulated Glucose Uptake Partly Relies on P21-Activated Kinase (PAK)2, but Not PAK1, in Mouse Skeletal Muscle. J. Physiol. 2020, 598, 5351–5377. 10.1113/JP280294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu T. T.; Patel N.; Shaw A. E.; Bamburg J. R.; Klip A. Arp2/3- and Cofilin-Coordinated Actin Dynamics Is Required for Insulin-Mediated GLUT4 Translocation to the Surface of Muscle Cells. Mol. Biol. Cell 2010, 21, 3529–3539. 10.1091/mbc.e10-04-0316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozaki S.; Ueda S.; Takenaka N.; Kataoka T.; Satoh T. Role of RalA Downstream of Rac1 in Insulin-Dependent Glucose Uptake in Muscle Cells. Cell. Signalling 2012, 24, 2111–2117. 10.1016/j.cellsig.2012.07.013. [DOI] [PubMed] [Google Scholar]
- Baidwan S.; Chekuri A.; Hynds D. A. L.; Kowluru A. Glucotoxicity Promotes Aberrant Activation and Mislocalization of Ras-Related C3 Botulinum Toxin Substrate 1 [Rac1] and Metabolic Dysfunction in Pancreatic Islet β-Cells: Reversal of Such Metabolic Defects by Metformin. Apoptosis 2017, 22, 1380–1393. 10.1007/s10495-017-1409-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subasinghe W.; Syed I.; Kowluru A. Phagocyte-like NADPH Oxidase Promotes Cytokine-Induced Mitochondrial Dysfunction in Pancreatic β-Cells: Evidence for Regulation by Rac1. American Journal of Physiology - Regulatory Integrative and Comparative Physiology 2011, 300, R12–R20. 10.1152/ajpregu.00421.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra M.; Duraisamy A. J.; Bhattacharjee S.; Kowluru R. A. Adaptor Protein P66Shc: A Link between Cytosolic and Mitochondrial Dysfunction in the Development of Diabetic Retinopathy. Antioxid. Redox Signaling 2019, 30, 1621–1634. 10.1089/ars.2018.7542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L.-H.; Wang L.; Wang D.; Jiang H.; Tang Q.-Z.; Yan L.; Bian Z.-Y.; Wang X.-A.; Li H. Puerarin Attenuates High-Glucose-and Diabetes-Induced Vascular Smooth Muscle Cell Proliferation by Blocking PKCβ2/Rac1-Dependent Signaling. Free Radical Biol. Med. 2010, 48, 471–482. 10.1016/j.freeradbiomed.2009.10.040. [DOI] [PubMed] [Google Scholar]
- Kong X.; Yan D.; Sun J.; Wu X.; Mulder H.; Hua X.; Ma X. Glucagon-like Peptide 1 Stimulates Insulin Secretion via Inhibiting RhoA/ROCK Signaling and Disassembling Glucotoxicity-Induced Stress Fibers. Endocrinology 2014, 155, 4676–4685. 10.1210/en.2014-1314. [DOI] [PubMed] [Google Scholar]
- Kanda T.; Wakino S.; Homma K.; Yoshioka K.; Tatematsu S.; Hasegawa K.; Takamatsu I.; Sugano N.; Hayashi K.; Saruta T. Rho-kinase as a Molecular Target for Insulin Resistance and Hypertension. FASEB J. 2006, 20, 169–171. 10.1096/fj.05-4197fje. [DOI] [PubMed] [Google Scholar]
- Wu S.-Z.; Peng F.-F.; Li J.-L.; Ye F.; Lei S.-Q.; Zhang B.-F. Akt and RhoA Activation in Response to High Glucose Require Caveolin-1 Phosphorylation in Mesangial Cells. American Journal of Physiology-Renal Physiology 2014, 306, F1308–F1317. 10.1152/ajprenal.00447.2013. [DOI] [PubMed] [Google Scholar]
- Ishiko K.; Sakoda T.; Akagami T.; Naka T.; Doi T.; Tsujino T.; Masuyama T.; Ohyanagi M. Hyperglycemia Induced Cell Growth and Gene Expression via the Serum Response Element through Rhoa and Rho-Kinase in Vascular Smooth Muscle Cells. Prep. Biochem. Biotechnol. 2010, 40, 139–151. 10.1080/10826060903558927. [DOI] [PubMed] [Google Scholar]
- Li Z.; Zhang J.; Wang M.; Qiu F.; Jin C.; Fu G. Expression of Farnesyl Pyrophosphate Synthase Is Increased in Diabetic Cardiomyopathy. Cell Biol. Int. 2021, 10.1002/cbin.11573. [DOI] [PubMed] [Google Scholar]
- Chen G. P.; Zhang X. Q.; Wu T.; Han J.; Ye D. Inhibition of Farnesyl Pyrophosphate Synthase Attenuates High Glucose-Induced Vascular Smooth Muscle Cells Proliferation. Mol. Med. Rep. 2017, 15, 3153–3160. 10.3892/mmr.2017.6360. [DOI] [PubMed] [Google Scholar]
- Panagiotakou A.; Yavropoulou M.; Nasiri-Ansari N.; Makras P.; Basdra E. K.; Papavassiliou A. G.; Kassi E. N. Extra-Skeletal Effects of Bisphosphonates. Metab., Clin. Exp. 2020, 110, 154264. 10.1016/j.metabol.2020.154264. [DOI] [PubMed] [Google Scholar]
- Tang Q.; Jiang S.; Jia W.; Shen D.; Qiu Y.; Zhao Y.; Xue B.; Li C. Zoledronic Acid, an FPPS Inhibitor, Ameliorates Liver Steatosis through Inhibiting Hepatic de Novo Lipogenesis. Eur. J. Pharmacol. 2017, 814, 169–177. 10.1016/j.ejphar.2017.08.010. [DOI] [PubMed] [Google Scholar]
- Jiang S.; Shen D.; Jia W. J.; Han X.; Shen N.; Tao W.; Gao X.; Xue B.; Li C. J. GGPPS-Mediated Rab27A Geranylgeranylation Regulates β Cell Dysfunction during Type 2 Diabetes Development by Affecting Insulin Granule Docked Pool Formation. Journal of Pathology 2016, 238, 109–119. 10.1002/path.4652. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Wu T. Y.; Zhao M. F.; Li C. J. The Balance of Protein Farnesylation and Geranylgeranylation during the Progression of Nonalcoholic Fatty Liver Disease. J. Biol. Chem. 2020, 295, 5152–5162. 10.1074/jbc.REV119.008897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao W.; Wu J.; Xie B. X.; Zhao Y. Y.; Shen N.; Jiang S.; Wang X. X.; Xu N.; Jiang C.; Chen S.; Gao X.; Xue B.; Li C. J. Lipid-Induced Muscle Insulin Resistance Is Mediated by GGPPS via Modulation of the RhoA/Rho Kinase Signaling Pathway. J. Biol. Chem. 2015, 290, 20086–20097. 10.1074/jbc.M115.657742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manaswiyoungkul P.; de Araujo E. D.; Gunning P. T. Targeting Prenylation Inhibition through the Mevalonate Pathway. RSC Medicinal Chemistry 2020, 11, 51–71. 10.1039/C9MD00442D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goalstone M.; Kamath V.; Kowluru A. Glucose Activates Prenyltransferases in Pancreatic Islet β-Cells. Biochem. Biophys. Res. Commun. 2010, 391, 895–898. 10.1016/j.bbrc.2009.11.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veluthakal R.; Arora D. K.; Goalstone M. L.; Kowluru R. A.; Kowluru A. Metabolic Stress Induces Caspase-3 Mediated Degradation and Inactivation of Farnesyl and Geranylgeranyl Transferase Activities in Pancreatic β-Cells. Cell. Physiol. Biochem. 2016, 39, 2110–2120. 10.1159/000447907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa H.; Yamada M.; Tanaka T.; Kramer J.; Yu Y. M.; Fischman A. J.; Martyn J. A. J.; Tompkins R. G.; Kaneki M. Role of Protein Farnesylation in Burn-Induced Metabolic Derangements and Insulin Resistance in Mouse Skeletal Muscle. PLoS One 2015, 10, e0116633. 10.1371/journal.pone.0116633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abderrahmani A.; Cheviet S.; Ferdaoussi M.; Coppola T.; Waeber G.; Regazzi R. ICER Induced by Hyperglycemia Represses the Expression of Genes Essential for Insulin Exocytosis. EMBO J. 2006, 25, 977–986. 10.1038/sj.emboj.7601008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malmgren S.; Spégel P.; Danielsson A. P. H.; Nagorny C. L.; Andersson L. E.; Nitert M. D.; Ridderstrale M.; Mulder H.; Ling C. Coordinate Changes in Histone Modifications, MRNA Levels, and Metabolite Profiles in Clonal INS-1 832/13 β-Cells Accompany Functional Adaptations to Lipotoxicity. J. Biol. Chem. 2013, 288, 11973–11987. 10.1074/jbc.M112.422527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syed I.; Kyathanahalli C. N.; Kowluru A. Phagocyte-like NADPH Oxidase Generates ROS in INS 832/13 Cells and Rat Islets: Role of Protein Prenylation. American Journal of Physiology - Regulatory Integrative and Comparative Physiology 2011, 300, R756–R762. 10.1152/ajpregu.00786.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S.; Yu D.; Ning S.; Zhang H.; Jiang L.; He L.; Li M.; Sun M. Augmented Rac1 Expression and Activity Are Associated with Oxidative Stress and Decline of β Cell Function in Obesity. Cell. Physiol. Biochem. 2015, 35, 2135–2148. 10.1159/000374019. [DOI] [PubMed] [Google Scholar]
- Veluthakal R.; Sidarala V.; Kowluru A. NSC23766, a Known Inhibitor of Tiam1-Rac1 Signaling Module, Prevents the Onset of Type 1 Diabetes in the NOD Mouse Model. Cell. Physiol. Biochem. 2016, 39, 760–767. 10.1159/000445666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damacharla D.; Thamilselvan V.; Zhang X.; Mestareehi A.; Yi Z.; Kowluru A. Quantitative Proteomics Reveals Novel Interaction Partners of Rac1 in Pancreatic β-Cells: Evidence for Increased Interaction with Rac1 under Hyperglycemic Conditions. Mol. Cell. Endocrinol. 2019, 494, 110489. 10.1016/j.mce.2019.110489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syed I.; Jayaram B.; Subasinghe W.; Kowluru A. Tiam1/Rac1 Signaling Pathway Mediates Palmitate-Induced, Ceramide-Sensitive Generation of Superoxides and Lipid Peroxides and the Loss of Mitochondrial Membrane Potential in Pancreatic β-Cells. Biochem. Pharmacol. 2010, 80, 874–883. 10.1016/j.bcp.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowluru A. Potential Roles of PP2A-Rac1 Signaling Axis in Pancreatic β-Cell Dysfunction under Metabolic Stress: Progress and Promise. Biochem. Pharmacol. 2020, 180, 114138. 10.1016/j.bcp.2020.114138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elumalai S.; Karunakaran U.; Lee I. K.; Moon J. S.; Won K. C. Rac1-NADPH Oxidase Signaling Promotes CD36 Activation under Glucotoxic Conditions in Pancreatic Beta Cells. Redox Biol. 2017, 11, 126–134. 10.1016/j.redox.2016.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaddai V.; Gonzalez T.; Keslair F.; Grémeaux T.; Bonnafous S.; Gugenheim J.; Tran A.; Gual P.; Le Marchand-Brustel Y.; Cormont M. Rab4b Is a Small GTPase Involved in the Control of the Glucose Transporter GLUT4 Localization in Adipocyte. PLoS One 2009, 4, e5257. 10.1371/journal.pone.0005257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulido M. R.; Diaz-Ruiz A.; Jiménez-Gómez Y.; Garcia-Navarro S.; Gracia-Navarro F.; Tinahones F.; López-Miranda J.; Frühbeck G.; Vázquez-Martínez R.; Malagón M. M. Rab18 Dynamics in Adipocytes in Relation to Lipogenesis, Lipolysis and Obesity. PLoS One 2011, 6, e22931. 10.1371/journal.pone.0022931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dankel S. N.; Røst T. H.; Kulyté A.; Fandalyuk Z.; Skurk T.; Hauner H.; Sagen J. V.; Rydén M.; Arner P.; Mellgren G. The Rho GTPase RND3 Regulates Adipocyte Lipolysis. Metab., Clin. Exp. 2019, 101, 153999. 10.1016/j.metabol.2019.153999. [DOI] [PubMed] [Google Scholar]
- Goalstone M. L.; Draznin B. Effect of Insulin on Farnesyltransferase Activity in 3T3-L1 Adipocytes. J. Biol. Chem. 1996, 271 (44), 27585–27589. 10.1074/jbc.271.44.27585. [DOI] [PubMed] [Google Scholar]
- Goalstone M.; Carel K.; Leitner J. W.; Draznin B. Insulin Stimulates the Phosphorylation and Activity of Farnesyltransferase via the Ras-Mitogen-Activated Protein Kinase Pathway. Endocrinology 1997, 138, 5119–5124. 10.1210/endo.138.12.5621. [DOI] [PubMed] [Google Scholar]
- Chae S.; Kim S. J.; Do Koo Y.; Lee J. H.; Kim H.; Ahn B. Y.; Ha Y. C.; Kim Y. H.; Jang M. G.; Koo K. H.; Choi S. H.; Lim S.; Park Y. J.; Jang H. C.; Hwang D.; Lee S. W.; Park K. S. A Mitochondrial Proteome Profile Indicative of Type 2 Diabetes Mellitus in Skeletal Muscles. Exp. Mol. Med. 2018, 50, 1–14. 10.1038/s12276-018-0154-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun K. H.; Choi K. D.; Lee D. H.; Jung Y. S.; Henry R. R.; Ciaraldi T. P.; Kim Y. B. In Vivo Activation of ROCK1 by Insulin Is Impaired in Skeletal Muscle of Humans with Type 2 Diabetes. American Journal of Physiology - Endocrinology and Metabolism 2011, 300, E536–E542. 10.1152/ajpendo.00538.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynet C.; Kahn C. R. Rad: A Member of the Ras Family Overexpressed in Muscle of Type II Diabetic Humans. Science 1993, 262, 1441–1444. 10.1126/science.8248782. [DOI] [PubMed] [Google Scholar]
- Coletta D. K.; Balas B.; Chavez A. O.; Baig M.; Abdul-Ghani M.; Kashyap S. R.; Folli F.; Tripathy D.; Mandarino L. J.; Cornell J. E.; DeFronzo R. A.; Jenkinson C. P. Effect of Acute Physiological Hyperinsulinemia on Gene Expression in Human Skeletal Muscle in Vivo. American Journal of Physiology - Endocrinology and Metabolism 2008, 294, E910–E917. 10.1152/ajpendo.00607.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyers J. S.; Bilan P. J.; Reynet C.; Kahn C. R. Overexpression of Rad Inhibits Glucose Uptake in Cultured Muscle and Fat Cells. J. Biol. Chem. 1996, 271, 23111–23116. 10.1074/jbc.271.38.23111. [DOI] [PubMed] [Google Scholar]
- Ilany J.; Bilan P. J.; Kapur S.; Caldwell J. S.; Patti M. E.; Marette A.; Kahn C. R. Overexpression of Rad in Muscle Worsens Diet-Induced Insulin Resistance and Glucose Intolerance and Lowers Plasma Triglyceride Level. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 4481–4486. 10.1073/pnas.0511246103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seitz S.; Kwon Y.; Hartleben G.; Jülg J.; Sekar R.; Krahmer N.; Najafi B.; Loft A.; Gancheva S.; Stemmer K.; Feuchtinger A.; Hrabe de Angelis M.; Müller T. D.; Mann M.; Blüher M.; Roden M.; Berriel Diaz M.; Behrends C.; Gilleron J.; Herzig S.; Zeigerer A. Hepatic Rab24 Controls Blood Glucose Homeostasis via Improving Mitochondrial Plasticity. Nature Metabolism 2019, 1, 1009–1026. 10.1038/s42255-019-0124-x. [DOI] [PubMed] [Google Scholar]
- Liu J.; Jiang S.; Zhao Y.; Sun Q.; Zhang J.; Shen D.; Wu J.; Shen N.; Fu X.; Sun X.; Yu D.; Chen J.; He J.; Shi T.; Ding Y.; Fang L.; Xue B.; Li C. Geranylgeranyl Diphosphate Synthase (GGPPS) Regulates Non-Alcoholic Fatty Liver Disease (NAFLD)–fibrosis Progression by Determining Hepatic Glucose/Fatty Acid Preference under High-Fat Diet Conditions. Journal of Pathology 2018, 246, 277–288. 10.1002/path.5131. [DOI] [PubMed] [Google Scholar]
- Xu N.; Shen N.; Wang X. X.; Jiang S.; Xue B.; Li C. J. Protein Prenylation and Human Diseases: A Balance of Protein Farnesylation and Geranylgeranylation. Sci. China: Life Sci. 2015, 58, 328–335. 10.1007/s11427-015-4836-1. [DOI] [PubMed] [Google Scholar]
- Chen F.; Zhang N.; Ma D.; Ma X.; Huang T.; Wang Q. Effect of the Rho Kinase Inhibitor Y-27632 and Fasudil on Inflammation and Fibrosis in Human Mesangial Cells (HMCs) under High Glucose via the Rho/ROCK Signaling Pathway. Int. J. Clin. Exp. Med. 2017, 10, 13224–13234. [Google Scholar]
- Massey A. R.; Miao L.; Smith B. N.; Liu J.; Kusaka I.; Zhang J. H.; Tang J. Increased RhoA Translocation in Renal Cortex of Diabetic Rats. Life Sci. 2003, 72, 2943–2952. 10.1016/S0024-3205(03)00228-5. [DOI] [PubMed] [Google Scholar]
- Kowluru R. A.; Kowluru A.; Veluthakal R.; Mohammad G.; Syed I.; Santos J. M.; Mishra M. TIAM1-RAC1 Signalling Axis-Mediated Activation of NADPH Oxidase-2 Initiates Mitochondrial Damage in the Development of Diabetic Retinopathy. Diabetologia 2014, 57, 1047–1056. 10.1007/s00125-014-3194-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowluru R. A.; Mishra M.; Kumar B. Diabetic Retinopathy and Transcriptional Regulation of a Small Molecular Weight G-Protein, Rac1. Exp. Eye Res. 2016, 147, 72–77. 10.1016/j.exer.2016.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammad G.; Duraisamy A. J.; Kowluru A.; Kowluru R. A. Functional Regulation of an Oxidative Stress Mediator, Rac1, in Diabetic Retinopathy. Mol. Neurobiol. 2019, 56, 8643–8655. 10.1007/s12035-019-01696-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Li Y.; Zhang M.; Hu Z. Silencing of Rac1 Expression via RNA Interference Inhibits Retinal Neovascularization in Rats. Mol. Vision 2012, 18, 1354–1360. [PMC free article] [PubMed] [Google Scholar]
- Sahajpal N.; Kowluru A.; Kowluru R. A. The Regulatory Role of Rac1, a Small Molecular Weight GTPase, in the Development of Diabetic Retinopathy. J. Clin. Med. 2019, 8, 965. 10.3390/jcm8070965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowluru R. A.; Kowluru A.; Chakrabarti S.; Khan Z. Potential Contributory Role of H-Ras, A Small G-Protein, in the Development of Retinopathy in Diabetic Rats. Diabetes 2004, 53, 775–783. 10.2337/diabetes.53.3.775. [DOI] [PubMed] [Google Scholar]
- Chen J.; Dai M.; Wang Y. Paeonol Inhibits Proliferation of Vascular Smooth Muscle Cells Stimulated by High Glucose via Ras-Raf-ERK1/2 Signaling Pathway in Coculture Model. J. Evidence-Based Complementary Altern. Med. 2014, 2014, 484269. 10.1155/2014/484269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiattarella G. G.; Carrizzo A.; Ilardi F.; Damato A.; Ambrosio M.; Madonna M.; Trimarco V.; Marino M.; Angelis E.; De; Settembrini S.; Perrino C.; Trimarco B.; Esposito G.; Vecchione C. Rac 1 Modulates Endothelial Function and Platelet Aggregation in Diabetes Mellitus. J. Am. Heart Assoc. 2018, 7, e007322. 10.1161/JAHA.117.007322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draznin B. Mitogenic Action of Insulin: Friend, Foe or “Frenemy”?. Diabetologia 2010, 53, 229–233. 10.1007/s00125-009-1558-6. [DOI] [PubMed] [Google Scholar]
- Gilleron J.; Gerdes J. M.; Zeigerer A. Metabolic Regulation through the Endosomal System. Traffic 2019, 20, 552–570. 10.1111/tra.12670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrowsmith C. H.; Audia J. E.; Austin C.; Baell J.; Bennett J.; Blagg J.; Bountra C.; Brennan P. E.; Brown P. J.; Bunnage M. E.; Buser-Doepner C.; Campbell R. M.; Carter A. J.; Cohen P.; Copeland R. A.; Cravatt B.; Dahlin J. L.; Dhanak D.; Edwards A. M.; Frye S. V.; Gray N.; Grimshaw C. E.; Hepworth D.; Howe T.; Huber K. V. M.; Jin J.; Knapp S.; Kotz J. D.; Kruger R. G.; Lowe D.; Mader M. M.; Marsden B.; Mueller-Fahrnow A.; Müller S.; O’Hagan R. C.; Overington J. P.; Owen D. R.; Rosenberg S. H.; Roth B.; Ross R.; Schapira M.; Schreiber S. L.; Shoichet B.; Sundström M.; Superti-Furga G.; Taunton J.; Toledo-Sherman L.; Walpole C.; Walters M. A.; Willson T. M.; Workman P.; Young R. N.; Zuercher W. J. The Promise and Peril of Chemical Probes. Nat. Chem. Biol. 2015, 11, 536–541. 10.1038/nchembio.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y.; Xie Y.; Yu Z.; Xiao H.; Jiang G.; Zhou X.; Yang Y.; Li X.; Zhao M.; Li L.; Zheng M.; Han S.; Zong Z.; Meng X.; Deng H.; Ye H.; Fa Y.; Wu H.; Oldfield E.; Hu X.; Liu W.; Shi Y.; Zhang Y. The Mevalonate Pathway Is a Druggable Target for Vaccine Adjuvant Discovery. Cell 2018, 175, 1059–1073.e21. 10.1016/j.cell.2018.08.070. [DOI] [PubMed] [Google Scholar]
- Jeong A.; Suazo K. F.; Wood W. G.; Distefano M. D.; Li L. Isoprenoids and Protein Prenylation: Implications in the Pathogenesis and Therapeutic Intervention of Alzheimer’s Disease. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 279–310. 10.1080/10409238.2018.1458070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers M. J.; Mönkkönen J.; Munoz M. A. Molecular Mechanisms of Action of Bisphosphonates and New Insights into Their Effects Outside the Skeleton. Bone 2020, 139, 115493. 10.1016/j.bone.2020.115493. [DOI] [PubMed] [Google Scholar]
- Müller M. P.; Goody R. S. Molecular Control of Rab Activity by GEFs, GAPs and GDI. Small GTPases 2018, 9, 5–21. 10.1080/21541248.2016.1276999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray J. L.; von Delft F.; Brennan P. E. Targeting the Small GTPase Superfamily through Their Regulatory Proteins. Angew. Chem., Int. Ed. 2020, 59, 6342–6366. 10.1002/anie.201900585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klochkov S. G.; Neganova M. E.; Yarla N. S.; Parvathaneni M.; Sharma B.; Tarasov V. V.; Barreto G.; Bachurin S. O.; Ashraf G. M.; Aliev G. Implications of Farnesyltransferase and Its Inhibitors as a Promising Strategy for Cancer Therapy. Semin. Cancer Biol. 2019, 56, 128–134. 10.1016/j.semcancer.2017.10.010. [DOI] [PubMed] [Google Scholar]
- Ahmadi M.; Amiri S.; Pecic S.; Machaj F.; Rosik J.; Łos M. J.; Alizadeh J.; Mahdian R.; da Silva Rosa S. C.; Schaafsma D.; Shojaei S.; Madrakian T.; Zeki A. A.; Ghavami S. Pleiotropic Effects of Statins: A Focus on Cancer. Biochim. Biophys. Acta, Mol. Basis Dis. 2020, 1866, 165968. 10.1016/j.bbadis.2020.165968. [DOI] [PubMed] [Google Scholar]
- Kumar G. A.; Roy S.; Jafurulla M.; Mandal C.; Chattopadhyay A. Statin-Induced Chronic Cholesterol Depletion Inhibits Leishmania Donovani Infection: Relevance of Optimum Host Membrane Cholesterol. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 2088–2096. 10.1016/j.bbamem.2016.06.010. [DOI] [PubMed] [Google Scholar]
- Cardiovascular Disease and Risk Management: Standards of Medical Care in Diabetes—2019. Diabetes Care 2019, 42, S103–S123. 10.2337/dc19-s010. [DOI] [PubMed] [Google Scholar]
- Jialal I.; Miguelino E.; Griffen S. C.; Devaraj S. Concomitant Reduction of Low-Density Lipoprotein-Cholesterol and Biomarkers of Inflammation with Low-Dose Simvastatin Therapy in Patients with Type 1 Diabetes. J. Clin. Endocrinol. Metab. 2007, 92, 3136–3140. 10.1210/jc.2007-0453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichida Y.; Hasegawa G.; Fukui M.; Obayashi H.; Ohta M.; Fujinami A.; Ohta K.; Nakano K.; Yoshikawa T.; Nakamura N. Effect of Atorvastatin on in Vitro Expression of Resistin in Adipocytes and Monocytes/Macrophages and Effect of Atorvastatin Treatment on Serum Resistin Levels in Patients with Type 2 Diabetes. Pharmacology 2005, 76, 34–39. 10.1159/000088948. [DOI] [PubMed] [Google Scholar]
- Bahammam M. A.; Attia M. S. Effects of Systemic Simvastatin on the Concentrations of Visfatin, Tumor Necrosis Factor-α, and Interleukin-6 in Gingival Crevicular Fluid in Patients with Type 2 Diabetes and Chronic Periodontitis. J. Immunol. Res. 2018, 2018, 8481735. 10.1155/2018/8481735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dworacka M.; Krzyzagórska E.; Wesołowska A.; Zharmakhanova G.; Iskakova S.; Dworacki G. Circulating Monocyte Chemotactic Protein 1 (MCP-1), Vascular Cell Adhesion Molecule 1 (VCAM-1) and Angiogenin in Type 2 Diabetic Patients Treated with Statins in Low Doses. Eur. J. Pharmacol. 2014, 740, 474–479. 10.1016/j.ejphar.2014.06.041. [DOI] [PubMed] [Google Scholar]
- Usharani P.; Mateen A. A.; Naidu M. U. R.; Raju Y. S. N.; Chandra N. Effect of NCB-02, Atorvastatin and Placebo on Endothelial Function, Oxidative Stress and Inflammatory Markers in Patients with Type 2 Diabetes Mellitus: A Randomized, Parallel-Group, Placebo-Controlled, 8-Week Study. Drugs R&D 2008, 9, 243–250. 10.2165/00126839-200809040-00004. [DOI] [PubMed] [Google Scholar]
- Von Eynatten M.; Schneider J. G.; Hadziselimovic S.; Hamann A.; Bierhaus A.; Nawroth P. P.; Dugi K. A. Adipocytokines as a Novel Target for the Anti-Inflammatory Effect of Atorvastatin in Patients with Type 2 Diabetes. Diabetes Care 2005, 28, 754–755. 10.2337/diacare.28.3.754. [DOI] [PubMed] [Google Scholar]
- Soran H.; Liu Y.; Adam S.; Siahmansur T.; Ho J. H.; Schofield J. D.; Kwok S.; Gittins M.; France M.; Younis N.; Gibson J. M.; Durrington P. N.; Rutter M. K. A Comparison of the Effects of Low- and High-Dose Atorvastatin on Lipoprotein Metabolism and Inflammatory Cytokines in Type 2 Diabetes: Results from the Protection Against Nephropathy in Diabetes with Atorvastatin (PANDA) Randomized Trial. J. Clin. Lipidol. 2018, 12, 44–55. 10.1016/j.jacl.2017.10.011. [DOI] [PubMed] [Google Scholar]
- Lin Y.; Ye S.; Chen Y.; Li X.; Yang G. w.; Fan A.; Wang Y. The Effect of Simvastatin on the Serum Monocyte Chemoattractant Protein-1 and Intracellular Adhesion Molecule-1 Levels in Diabetic Rats. J. Diabetes Complications 2009, 23, 214–218. 10.1016/j.jdiacomp.2007.09.003. [DOI] [PubMed] [Google Scholar]
- Milajerdi A.; Sadeghi A.; Mousavi S. M.; Larijani B.; Esmaillzadeh A. Influence of Statins on Circulating Inflammatory Cytokines in Patients With Abnormal Glucose Homeostasis: A Meta-Analysis of Data From Randomized Controlled Trials. Clin. Ther. 2020, 42, e13–e31. 10.1016/j.clinthera.2019.12.009. [DOI] [PubMed] [Google Scholar]
- Nareika A.; Maldonado A.; He L.; Game B. A.; Slate E. H.; Sanders J. J.; London S. D.; Lopes-Virella M. F.; Huang Y. High Glucose-Boosted Inflammatory Responses to Lipopolysaccharide Are Suppressed by Statin. J. Periodontal Res. 2007, 42, 31–38. 10.1111/j.1600-0765.2006.00911.x. [DOI] [PubMed] [Google Scholar]
- Sundararaj K. P.; Samuvel D. J.; Li Y.; Nareika A.; Slate E. H.; Sanders J. J.; Lopes-Virella M. F.; Huang Y. Simvastatin Suppresses LPS-Induced MMP-1 Expression in U937 Mononuclear Cells by Inhibiting Protein Isoprenylation-Mediated ERK Activation. J. Leukocyte Biol. 2008, 84, 1120–1129. 10.1189/jlb.0108064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krysiak R.; Gdula-Dymek A.; Okopien B. Effect of Simvastatin and Fenofibrate on Cytokine Release and Systemic Inflammation in Type 2 Diabetes Mellitus with Mixed Dyslipidemia. Am. J. Cardiol. 2011, 107, 1010–1018.e1. 10.1016/j.amjcard.2010.11.023. [DOI] [PubMed] [Google Scholar]
- Štulc T.; Češka R.; Marinov I.; Škrha J. The Effect of Simvastatin and Fenofibrate on the Expression of Leukocyte Adhesion Molecules and Lipopolysaccharide Receptor CD14 in Type 2 Diabetes Mellitus. Neuro Endocrinol. Lett. 2012, 33, 73–77. [PubMed] [Google Scholar]
- Omi H.; Okayama N.; Shimizu M.; Fukutomi T.; Imaeda K.; Okouchi M.; Itoh M. Statins Inhibit High Glucose-Mediated Neutrophil–endothelial Cell Adhesion through Decreasing Surface Expression of Endothelial Adhesion Molecules by Stimulating Production of Endothelial Nitric Oxide. Microvasc. Res. 2003, 65, 118–124. 10.1016/S0026-2862(02)00033-X. [DOI] [PubMed] [Google Scholar]
- Park J.; Hwang I.; Kim S. J.; Youn S. W.; Hur J.; Kim H. S. Atorvastatin Prevents Endothelial Dysfunction in High Glucose Condition through Skp2-Mediated Degradation of FOXO1 and ICAM-1. Biochem. Biophys. Res. Commun. 2018, 495, 2050–2057. 10.1016/j.bbrc.2017.08.023. [DOI] [PubMed] [Google Scholar]
- Riad A.; Du J.; Stiehl S.; Westermann D.; Mohr Z.; Sobirey M.; Doehner W.; Adams V.; Pauschinger M.; Schultheiss H. P.; Tschöpe C. Low-Dose Treatment with Atorvastatin Leads to Anti-Oxidative and Anti-Inflammatory Effects in Diabetes Mellitus. Eur. J. Pharmacol. 2007, 569, 204–211. 10.1016/j.ejphar.2007.04.065. [DOI] [PubMed] [Google Scholar]
- Takata M.; Urakaze M.; Temaru R.; Yamazaki K.; Nakamura N.; Nobata Y.; Kishida M.; Sato A.; Kobayashi M. Pravastatin Suppresses the Interleukin-8 Production Induced by Thrombin in Human Aortic Endothelial Cells Cultured with High Glucose by Inhibiting the P44/42 Mitogen Activated Protein Kinase. Br. J. Pharmacol. 2001, 134, 753–762. 10.1038/sj.bjp.0704305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.; Lee Y.; Kim J.; An J.; Kim K.; Lee H.; Kong H.; Song Y.; Kim K. Atorvastatin and Rosuvastatin Improve Physiological Parameters and Alleviate Immune Dysfunction in Metabolic Disorders. Biochem. Biophys. Res. Commun. 2016, 478, 1242–1247. 10.1016/j.bbrc.2016.08.101. [DOI] [PubMed] [Google Scholar]
- Henriksbo B. D.; Tamrakar A. K.; Phulka J. S.; Barra N. G.; Schertzer J. D. Statins Activate the NLRP3 Inflammasome and Impair Insulin Signaling via P38 and MTOR. American Journal of Physiology - Endocrinology and Metabolism 2020, 319, E110–E116. 10.1152/ajpendo.00125.2020. [DOI] [PubMed] [Google Scholar]
- Henriksbo B. D.; Tamrakar A. K.; Xu J.; Duggan B. M.; Cavallari J. F.; Phulka J.; Stampfli M. R.; Ashkar A. A.; Schertzer J. D. Statins Promote Interleukin-1β-Dependent Adipocyte Insulin Resistance through Lower Prenylation, Not Cholesterol. Diabetes 2019, 68, 1441–1448. 10.2337/db18-0999. [DOI] [PubMed] [Google Scholar]
- Akula M. K.; Shi M.; Jiang Z.; Foster C. E.; Miao D.; Li A. S.; Zhang X.; Gavin R. M.; Forde S. D.; Germain G.; Carpenter S.; Rosadini C. V.; Gritsman K.; Chae J. J.; Hampton R.; Silverman N.; Gravallese E. M.; Kagan J. C.; Fitzgerald K. A.; Kastner D. L.; Golenbock D. T.; Bergo M. O.; Wang D. Control of the Innate Immune Response by the Mevalonate Pathway. Nat. Immunol. 2016, 17, 922–929. 10.1038/ni.3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner O. P.; Jurczyluk J.; Baker P. J.; Masters S. L.; Rios Wilks A. G.; Clearwater M. S.; Robertson A. A. B.; Schroder K.; Mehr S.; Munoz M. A.; Rogers M. J. Lack of Protein Prenylation Promotes NLRP3 Inflammasome Assembly in Human Monocytes. J. Allergy Clin. Immunol. 2019, 143, 2315–2317.e3. 10.1016/j.jaci.2019.02.013. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Wang L.; Lv Y.; Jiang C.; Wu G.; Dull R. O.; Minshall R. D.; Malik A. B.; Hu G. The GTPase Rab1 Is Required for NLRP3 Inflammasome Activation and Inflammatory Lung Injury. J. Immunol. 2019, 202, 194–206. 10.4049/jimmunol.1800777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W.; Zhao S. P. Different Effects of Statins on Induction of Diabetes Mellitus: An Experimental Study. Drug Des., Dev. Ther. 2015, 9, 6211–6223. 10.2147/DDDT.S87979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruit J. K.; Brunham L. R.; Verchere C. B.; Hayden M. R. HDL and LDL Cholesterol Significantly Influence β-Cell Function in Type 2 Diabetes Mellitus. Curr. Opin. Lipidol. 2010, 21, 178–185. 10.1097/MOL.0b013e328339387b. [DOI] [PubMed] [Google Scholar]
- Syeda K. G.; Kowluru A. Inhibition of Prenylation Promotes Caspase 3 Activation, Lamin B Degradation and Loss in Metabolic Cell Viability in Pancreatic β-Cells. Cell. Physiol. Biochem. 2017, 43, 1052–1063. 10.1159/000481702. [DOI] [PubMed] [Google Scholar]
- Stegman B.; Puri R.; Cho L.; Shao M.; Ballantyne C. M.; Barter P. J.; Chapman M. J.; Erbel R.; Libby P.; Raichlen J. S.; Uno K.; Kataoka Y.; Nissen S. E.; Nicholls S. J. High-Intensity Statin Therapy Alters the Natural History of Diabetic Coronary Atherosclerosis: Insights from SATURN. Diabetes Care 2014, 37, 3114–3120. 10.2337/dc14-1121. [DOI] [PubMed] [Google Scholar]
- Jiang S. Y.; Li H.; Tang J. J.; Wang J.; Luo J.; Liu B.; Wang J. K.; Shi X. J.; Cui H. W.; Tang J.; Yang F.; Qi W.; Qiu W. W.; Song B. L. Discovery of a Potent HMG-CoA Reductase Degrader That Eliminates Statin-Induced Reductase Accumulation and Lowers Cholesterol. Nat. Commun. 2018, 9, 5138. 10.1038/s41467-018-07590-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho C.; Hsu Y. C.; Tseng C. C.; Wang F. S.; Lin C. L.; Wang J. Y. Simvastatin Alleviates Diabetes-Induced VEGF-Mediated Nephropathy via the Modulation of Ras Signaling Pathway. Renal Failure 2008, 30, 557–565. 10.1080/08860220802064457. [DOI] [PubMed] [Google Scholar]
- Zhou J.; Li W.; Xie Q.; Hou Y.; Zhan S.; Yang X.; Xu X.; Cai J.; Huang Z. Effects of Simvastatin on Glucose Metabolism in Mouse MIN6 Cells. J. Diabetes Res. 2014, 2014, 376570. 10.1155/2014/376570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohsawa M.; Aasato M.; Hayashi S. S.; Kamei J. RhoA/Rho Kinase Pathway Contributes to the Pathogenesis of Thermal Hyperalgesia in Diabetic Mice. Pain 2011, 152, 114–122. 10.1016/j.pain.2010.10.005. [DOI] [PubMed] [Google Scholar]
- Sun H.; Li Y.; Sun B.; Hou N.; Yang J.; Zheng M.; Xu J.; Wang J.; Zhang Y.; Zeng X.; Shan C.; Chang B.; Chen L.; Chang B. Atorvastatin Inhibits Insulin Synthesis by Inhibiting the Ras/Raf/ERK/CREB Pathway in INS-1 Cells. Medicine 2016, 95, e4906. 10.1097/MD.0000000000004906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadoglou N. P. E.; Sailer N.; Kapelouzou A.; Lampropoulos S.; Vitta I.; Kostakis A.; Liapis C. D. Effects of Atorvastatin on Apelin, Visfatin (Nampt), Ghrelin and Early Carotid Atherosclerosis in Patients with Type 2 Diabetes. Acta Diabetol. 2012, 49, 269–276. 10.1007/s00592-011-0310-0. [DOI] [PubMed] [Google Scholar]
- Waller D. D.; Park J.; Tsantrizos Y. S. Inhibition of Farnesyl Pyrophosphate (FPP) and/or Geranylgeranyl Pyrophosphate (GGPP) Biosynthesis and Its Implication in the Treatment of Cancers. Crit. Rev. Biochem. Mol. Biol. 2019, 54, 41–60. 10.1080/10409238.2019.1568964. [DOI] [PubMed] [Google Scholar]
- Jahnke W.; Rondeau J. M.; Cotesta S.; Marzinzik A.; Pellé X.; Geiser M.; Strauss A.; Götte M.; Bitsch F.; Hemmig R.; Henry C.; Lehmann S.; Glickman J. F.; Roddy T. P.; Stout S. J.; Green J. R. Allosteric Non-Bisphosphonate FPPS Inhibitors Identified by Fragment-Based Discovery. Nat. Chem. Biol. 2010, 6, 660–666. 10.1038/nchembio.421. [DOI] [PubMed] [Google Scholar]
- Liu J.; Liu W.; Ge H.; Gao J.; He Q.; Su L.; Xu J.; Gu L. Q.; Huang Z. S.; Li D. Syntheses and Characterization of Non-Bisphosphonate Quinoline Derivatives as New FPPS Inhibitors. Biochim. Biophys. Acta, Gen. Subj. 2014, 1840, 1051–1062. 10.1016/j.bbagen.2013.11.006. [DOI] [PubMed] [Google Scholar]
- Liu Y. L.; Cao R.; Wang Y.; Oldfield E. Farnesyl Diphosphate Synthase Inhibitors with Unique Ligand-Binding Geometries. ACS Med. Chem. Lett. 2015, 6, 349–354. 10.1021/ml500528x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergstrom J. D.; Bostedor R. G.; Masarachia P. J.; Reszka A. A.; Rodan G. Alendronate Is a Specific, Nanomolar Inhibitor of Farnesyl Diphosphate Synthase. Arch. Biochem. Biophys. 2000, 373, 231–241. 10.1006/abbi.1999.1502. [DOI] [PubMed] [Google Scholar]
- Chan D.-C.; Yang R.-S.; Ho C.-H.; Tsai Y.-S.; Wang J.-J.; Tsai K.-T. The Use of Alendronate Is Associated with a Decreased Incidence of Type 2 Diabetes Mellitus—A Population-Based Cohort Study in Taiwan. PLoS One 2015, 10, e0123279. 10.1371/journal.pone.0123279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maugeri D.; Panebianco P.; Rosso D.; Calanna A.; Speciale S.; Santangelo A.; Rizza I.; Motta M.; Lentini A.; Malaguarnera M. Alendronate Reduces the Daily Consumption of Insulin (DCI) in Patients with Senile Type I Diabetes and Osteoporosis. Arch. Gerontol. Geriatr. 2002, 34, 117–122. 10.1016/S0167-4943(01)00202-3. [DOI] [PubMed] [Google Scholar]
- Lee N. Y.; Park H. J.; Kang Y. S. Effects of Bisphosphonates on Glucose Transport in a Conditionally Immortalized Rat Retinal Capillary Endothelial Cell Line (TR-IBRB Cells). Biomol. Ther. 2016, 24, 94–98. 10.4062/biomolther.2015.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durgia H.; Sahoo J.; Kamalanathan S.; Palui R.; Sridharan K.; Raj H. Role of Bisphosphonates in the Management of Acute Charcot Foot. World Journal of Diabetes 2018, 9, 115–126. 10.4239/wjd.v9.i7.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavanagh K. L.; Guo K.; Dunford J. E.; Wu X.; Knapp S.; Ebetino F. H.; Rogers M. J.; Russell R. G. G.; Oppermann U. The Molecular Mechanism of Nitrogen-Containing Bisphosphonates as Antiosteoporosis Drugs. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 7829–7834. 10.1073/pnas.0601643103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L.; Zhu L.; Shi W. H.; Zhang J.; Ma D.; Yu B. Zoledronate Inhibits the Proliferation, Adhesion and Migration of Vascular Smooth Muscle Cells. Eur. J. Pharmacol. 2009, 602, 124–131. 10.1016/j.ejphar.2008.10.043. [DOI] [PubMed] [Google Scholar]
- Dunford J. E.; Thompson K.; Coxon F. P.; Luckman S. P.; Hahn F. M.; Poulter C. D.; Ebetino F. H.; Rogers M. J. Structure-Activity Relationships for Inhibition of Farnesyl Diphosphate Synthase in Vitro and Inhibition of Bone Resorption in Vivo by Nitrogen-Containing Bisphosphonates. J. Pharmacol. Exp. Ther. 2001, 296, 235–242. [PubMed] [Google Scholar]
- Marzinzik A. L.; Amstutz R.; Bold G.; Bourgier E.; Cotesta S.; Glickman J. F.; Götte M.; Henry C.; Lehmann S.; Hartwieg J. C. D.; Ofner S.; Pellé X.; Roddy T. P.; Rondeau J. M.; Stauffer F.; Stout S. J.; Widmer A.; Zimmermann J.; Zoller T.; Jahnke W. Discovery of Novel Allosteric Non-Bisphosphonate Inhibitors of Farnesyl Pyrophosphate Synthase by Integrated Lead Finding. ChemMedChem 2015, 10, 1884–1891. 10.1002/cmdc.201500338. [DOI] [PubMed] [Google Scholar]
- Park J.; Leung C. Y.; Matralis A. N.; Lacbay C. M.; Tsakos M.; Fernandez De Troconiz G.; Berghuis A. M.; Tsantrizos Y. S. Pharmacophore Mapping of Thienopyrimidine-Based Monophosphonate (ThP-MP) Inhibitors of the Human Farnesyl Pyrophosphate Synthase. J. Med. Chem. 2017, 60, 2119–2134. 10.1021/acs.jmedchem.6b01888. [DOI] [PubMed] [Google Scholar]
- Haney S. L.; Wills V. S.; Wiemer D. F.; Holstein S. A. Recent Advances in the Development of Mammalian Geranylgeranyl Diphosphate Synthase Inhibitors. Molecules 2017, 22, 886. 10.3390/molecules22060886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Cao R.; Yin F.; Hudock M. P.; Guo R. T.; Krysiak K.; Mukherjee S.; Gao Y. G.; Robinson H.; Song Y.; No J. H.; Bergan K.; Leon A.; Cass L.; Goddard A.; Chang T. K.; Lin F. Y.; Beek E.; Van; Papapoulos S.; Wang A. H. J.; Kubo T.; Ochi M.; Mukkamala D.; Oldfield E. Lipophilic Bisphosphonates as Dual Farnesyl/Geranylgeranyl Diphosphate Synthase Inhibitors: An X-Ray and Nmr Investigation. J. Am. Chem. Soc. 2009, 131, 5153–5162. 10.1021/ja808285e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacbay C. M.; Waller D. D.; Park J.; Palou M. G.; Vincent F.; Huang X. F.; Ta V.; Berghuis A. M.; Sebag M.; Tsantrizos Y. S. Unraveling the Prenylation-Cancer Paradox in Multiple Myeloma with Novel Geranylgeranyl Pyrophosphate Synthase (GGPPS) Inhibitors. J. Med. Chem. 2018, 61, 6904–6917. 10.1021/acs.jmedchem.8b00886. [DOI] [PubMed] [Google Scholar]
- Wiemer A. J.; Tong H.; Swanson K. M.; Hohl R. J. Digeranyl Bisphosphonate Inhibits Geranylgeranyl Pyrophosphate Synthase. Biochem. Biophys. Res. Commun. 2007, 353, 921–925. 10.1016/j.bbrc.2006.12.094. [DOI] [PubMed] [Google Scholar]
- Chen C. K. M.; Hudock M. P.; Zhang Y.; Guo R. T.; Cao R.; Joo H. N.; Liang P. H.; Ko T. P.; Chang T. H.; Chang S. C.; Song Y.; Axelson J.; Kumar A.; Wang A. H. J.; Oldfield E. Inhibition of Geranylgeranyl Diphosphate Synthase by Bisphosphonates: A Crystallographic and Computational Investigation. J. Med. Chem. 2008, 51, 5594–5607. 10.1021/jm800325y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X.; Reilly J. E.; Loerch K. A.; Hohl R. J.; Wiemer D. F. Synthesis of Isoprenoid Bisphosphonate Ethers through C-P Bond Formations: Potential Inhibitors of Geranylgeranyl Diphosphate Synthase. Beilstein J. Org. Chem. 2014, 10, 1645–1650. 10.3762/bjoc.10.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foust B. J.; Allen C.; Holstein S. A.; Wiemer D. F. A New Motif for Inhibitors of Geranylgeranyl Diphosphate Synthase. Bioorg. Med. Chem. 2016, 24, 3734–3741. 10.1016/j.bmc.2016.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthiesen R. A.; Wills V. S.; Metzger J. I.; Holstein S. A.; Wiemer D. F. Stereoselective Synthesis of Homoneryl and Homogeranyl Triazole Bisphosphonates. J. Org. Chem. 2016, 81, 9438–9442. 10.1021/acs.joc.6b01693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haney S. L.; Varney M. L.; Chhonker Y. S.; Shin S.; Mehla K.; Crawford A. J.; Smith H. J.; Smith L. M.; Murry D. J.; Hollingsworth M. A.; Holstein S. A. Inhibition of Geranylgeranyl Diphosphate Synthase Is a Novel Therapeutic Strategy for Pancreatic Ductal Adenocarcinoma. Oncogene 2019, 38, 5308–5320. 10.1038/s41388-019-0794-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haney S. L.; Chhonker Y. S.; Varney M. L.; Talmon G.; Murry D. J.; Holstein S. A. Preclinical Investigation of a Potent Geranylgeranyl Diphosphate Synthase Inhibitor. Invest. New Drugs 2018, 36, 810–818. 10.1007/s10637-018-0571-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthiesen R. A.; Varney M. L.; Xu P. C.; Rier A. S.; Wiemer D. F.; Holstein S. A. α-Methylation Enhances the Potency of Isoprenoid Triazole Bisphosphonates as Geranylgeranyl Diphosphate Synthase Inhibitors. Bioorg. Med. Chem. 2018, 26, 376–385. 10.1016/j.bmc.2017.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer-Smith R.; O’Bryan J. P. Direct Inhibition of RAS: Quest for the Holy Grail?. Semin. Cancer Biol. 2019, 54, 138–148. 10.1016/j.semcancer.2017.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey P.; Chang D. K.; Nones K.; Johns A. L.; Patch A. M.; Gingras M. C.; Miller D. K.; Christ A. N.; Bruxner T. J. C.; Quinn M. C.; Nourse C.; Murtaugh L. C.; Harliwong I.; Idrisoglu S.; Manning S.; Nourbakhsh E.; Wani S.; Fink L.; Holmes O.; Chin V.; Anderson M. J.; Kazakoff S.; Leonard C.; Newell F.; Waddell N.; Wood S.; Xu Q.; Wilson P. J.; Cloonan N.; Kassahn K. S.; Taylor D.; Quek K.; Robertson A.; Pantano L.; Mincarelli L.; Sanchez L. N.; Evers L.; Wu J.; Pinese M.; Cowley M. J.; Jones M. D.; Colvin E. K.; Nagrial A. M.; Humphrey E. S.; Chantrill L. A.; Mawson A.; Humphris J.; Chou A.; Pajic M.; Scarlett C. J.; Pinho A. V.; Giry-Laterriere M.; Rooman I.; Samra J. S.; Kench J. G.; Lovell J. A.; Merrett N. D.; Toon C. W.; Epari K.; Nguyen N. Q.; Barbour A.; Zeps N.; Moran-Jones K.; Jamieson N. B.; Graham J. S.; Duthie F.; Oien K.; Hair J.; Grützmann R.; Maitra A.; Iacobuzio-Donahue C. A.; Wolfgang C. L.; Morgan R. A.; Lawlor R. T.; Corbo V.; Bassi C.; Rusev B.; Capelli P.; Salvia R.; Tortora G.; Mukhopadhyay D.; Petersen G. M.; Munzy D. M.; Fisher W. E.; Karim S. A.; Eshleman J. R.; Hruban R. H.; Pilarsky C.; Morton J. P.; Sansom O. J.; Scarpa A.; Musgrove E. A.; Bailey U. M. H.; Hofmann O.; Sutherland R. L.; Wheeler D. A.; Gill A. J.; Gibbs R. A.; Pearson J. V.; Waddell N.; Biankin A. V.; Grimmond S. M. Genomic Analyses Identify Molecular Subtypes of Pancreatic Cancer. Nature 2016, 531, 47–52. 10.1038/nature16965. [DOI] [PubMed] [Google Scholar]
- Sakamoto K.; Masutani T.; Hirokawa T. Generation of KS-58 as the First K-Ras(G12D)-Inhibitory Peptide Presenting Anti-Cancer Activity in Vivo. Sci. Rep. 2020, 10, 21671. 10.1038/s41598-020-78712-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore A. R.; Rosenberg S. C.; McCormick F.; Malek S. RAS-Targeted Therapies: Is the Undruggable Drugged?. Nat. Rev. Drug Discovery 2020, 19, 533–552. 10.1038/s41573-020-0068-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond M. J.; Chu L.; Nalawansha D. A.; Li K.; Crews C. M. Targeted Degradation of Oncogenic KRASG12Cby VHL-Recruiting PROTACs. ACS Cent. Sci. 2020, 6, 1367–1375. 10.1021/acscentsci.0c00411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt A.; Qian Y.; McGuire T. F.; Hamilton A. D.; Sebti S. M. Protein Geranylgeranylation, Not Farnesylation, Is Required for the G1 to S Phase Transition in Mouse Fibroblasts. Oncogene 1996, 13, 1991–1999. [PubMed] [Google Scholar]
- Cohen L. H.; Pieterman E.; Van Leeuwen R. E. W.; Du J.; Negre-Aminou P.; Valentijn A. R. P. M.; Overhand M.; Van Der Marel G. A.; Van Boom J. H. Inhibition of Human Smooth Muscle Cell Proliferation in Culture by Farnesyl Pyrophosphate Analogues, Inhibitors of in Vitro Protein:Farnesyl Transferase. Biochem. Pharmacol. 1999, 57, 365–373. 10.1016/S0006-2952(98)00322-0. [DOI] [PubMed] [Google Scholar]
- Sugita M.; Sugita H.; Kaneki M. Farnesyltransferase Inhibitor, Manumycin A, Prevents Atherosclerosis Development and Reduces Oxidative Stress in Apolipoprotein E-Deficient Mice. Arterioscler., Thromb., Vasc. Biol. 2007, 27, 1390–1395. 10.1161/ATVBAHA.107.140673. [DOI] [PubMed] [Google Scholar]
- Sun J.; Blaskovich M. A.; Knowles D.; Qian Y.; Ohkanda J.; Bailey R. D.; Hamilton A. D.; Sebti S. M. Antitumor Efficacy of a Novel Class of Non-Thiol-Containing Peptidomimetic Inhibitors of Farnesyltransferase and Geranylgeranyltransferase I: Combination Therapy with the Cytotoxic Agents Cisplatin, Taxol, and Gemcitabine. Cancer Res. 1999, 59, 4919–4926. [PubMed] [Google Scholar]
- Lu J.; Yoshimura K.; Goto K.; Lee C.; Hamura K.; Kwon O.; Tamanoi F. Nanoformulation of Geranylgeranyltransferase-I Inhibitors for Cancer Therapy: Liposomal Encapsulation and PH-Dependent Delivery to Cancer Cells. PLoS One 2015, 10, e0137595. 10.1371/journal.pone.0137595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M.; Fiji H. D. G.; Guo L.; Chan L.; Kinderman S. S.; Slamon D. J.; Kwon O.; Tamanoi F. Inhibitors of Protein Geranylgeranyltransferase I and Rab Geranylgeranyltransferase Identified from a Library of Allenoate-Derived Compounds. J. Biol. Chem. 2008, 283, 9571–9579. 10.1074/jbc.M706229200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan L. N.; Fiji H. D. G.; Watanabe M.; Kwon O.; Tamanoi F. Identification and Characterization of Mechanism of Action of P61-E7, a Novel Phosphine Catalysis-Based Inhibitor of Geranylgeranyltransferase-I. PLoS One 2011, 6, e26135. 10.1371/journal.pone.0026135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazi A.; Xiang S.; Yang H.; Chen L.; Kennedy P.; Ayaz M.; Fletcher S.; Cummings C.; Lawrence H. R.; Beato F.; Kang Y.; Kim M. P.; Delitto A.; Underwood P. W.; Fleming J. B.; Trevino J. G.; Hamilton A. D.; Sebti S. M. Dual Farnesyl and Geranylgeranyl Transferase Inhibitor Thwarts Mutant KRAS-Driven Patient-Derived Pancreatic Tumors. Clin. Cancer Res. 2019, 25, 5984–5996. 10.1158/1078-0432.CCR-18-3399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt J. T.; Ding C. Z.; Batorsky R.; Bednarz M.; Bhide R.; Cho Y.; Chong S.; Chao S.; Gullo-Brown J.; Guo P.; Soong Hoon Kim; Lee F. Y. F.; Leftheris K.; Miller A.; Mitt T.; Patel M.; Penhallow B. A.; Ricca C.; Rose W. C.; Schmidt R.; Slusarchyk W. A.; Vite G.; Manne V. Discovery of (R)-7-Cyano-2,3,4,5-Tetrahydro-1-(1H-Imidazol-4-Ylmethyl)-3-(Phenylmethyl)-4- (2-Thienysulfonyl)-1H-1,4-Benzodiazepine (BMS-214662), a Farnesyltransferase Inhibitor with Potent Preclinical Antitumor Activity. J. Med. Chem. 2000, 43, 3587–3595. 10.1021/jm000248z. [DOI] [PubMed] [Google Scholar]
- Liu M.; Bryant M. S.; Chen J.; Lee S.; Yaremko B.; Lipari P.; Malkowski M.; Ferrari E.; Nielsen L.; Prioli N.; Dell J.; Sinha D.; Syed J.; Korfmacher W. A.; Nomeir A. A.; Lin C.-C.; Wang L.; Taveras A. G.; Doll R. J.; Njoroge F. G.; Mallams A. K.; Remiszewski S.; Catino J. J.; Girijavallabhan V. M.; Kirschmeier P.; Bishop W. R. Antitumor Activity of SCH 66336, an Orally Bioavailable Tricyclic Inhibitor of Farnesyl Protein Transferase, in Human Tumor Xenograft Models and Wap-Ras Transgenic Mice. Cancer Res. 1998, 58, 4947–4956. [PubMed] [Google Scholar]
- Puntambekar D. S.; Giridhar R.; Yadav M. R. Inhibition of Farnesyltransferase: A Rational Approach to Treat Cancer?. J. Enzyme Inhib. Med. Chem. 2007, 22, 127–140. 10.1080/14756360601072841. [DOI] [PubMed] [Google Scholar]
- Lobell R. B.; Liu D.; Buser C.; Davide J.; Depuy E.; Hamilton K.; Koblan K.; Lee Y.; Mosser S.; Motzel S.; Abbruzzese J.; Fuchs C.; Rowinsky E.; Rubin E.; Sharma S.; Deutsch P.; Mazina K.; Morrison B. W.; Wildonger L.; Yao S.-L.; Kohl N. Preclinical and Clinical Pharmacodynamic Assessment of L-778,123, a Dual Inhibitor of Farnesyl:Protein Transferase and Geranylgeranyl:Protein Transferase Type-I. Mol. Cancer Ther. 2002, 1, 747–758. [PubMed] [Google Scholar]
- Tsubamoto M.; Le T. K.; Li M.; Watanabe T.; Matsumi C.; Parvatkar P.; Fujii H.; Kato N.; Sun J.; Ohkanda J. A Guanidyl-Based Bivalent Peptidomimetic Inhibits K-Ras Prenylation and Association with C-Raf. Chem. - Eur. J. 2019, 25, 13531–13536. 10.1002/chem.201903129. [DOI] [PubMed] [Google Scholar]
- Peterson Y. K.; Kelly P.; Weinbaum C. A.; Casey P. J. A Novel Protein Geranylgeranyltransferase-I Inhibitor with High Potency, Selectivity, and Cellular Activity. J. Biol. Chem. 2006, 281, 12445–12450. 10.1074/jbc.M600168200. [DOI] [PubMed] [Google Scholar]
- Chen G. P.; Yang J.; Qian G. F.; Xu W. W.; Zhang X. Q. Geranylgeranyl Transferase-I Knockout Inhibits Oxidative Injury of Vascular Smooth Muscle Cells and Attenuates Diabetes-Accelerated Atherosclerosis. J. Diabetes Res. 2020, 2020, 7574245. 10.1155/2020/7574245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng C. W.; Zeng R. J.; Xu L. Y.; Li E. M. Rho GTPases: Promising Candidates for Overcoming Chemotherapeutic Resistance. Cancer Lett. 2020, 475, 65–78. 10.1016/j.canlet.2020.01.018. [DOI] [PubMed] [Google Scholar]
- Del Mar Maldonado M.; Dharmawardhane S. Targeting Rac and Cdc42 GTPases in Cancer. Cancer Res. 2018, 78, 3101–3111. 10.1158/0008-5472.CAN-18-0619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferri N.; Bernini S. K.; Corsini A.; Clerici F.; Erba E.; Stragliotto S.; Contini A. 3-Aryl-N-Aminoylsulfonylphenyl-1H-Pyrazole-5-Carboxamides: A New Class of Selective Rac Inhibitors. MedChemComm 2013, 4, 537–541. 10.1039/c2md20328f. [DOI] [Google Scholar]
- Ferri N.; Corsini A.; Bottino P.; Clerici F.; Contini A. Virtual Screening Approach for the Identification of New Rac1 Inhibitors. J. Med. Chem. 2009, 52, 4087–4090. 10.1021/jm8015987. [DOI] [PubMed] [Google Scholar]
- Gao Y.; Dickerson J. B.; Guo F.; Zheng J.; Zheng Y. Rational Design and Characterization of a Rac GTPase-Specific Small Molecule Inhibitor. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 7618–7623. 10.1073/pnas.0307512101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruffoni A.; Ferri N.; Pinto A.; Pellegrino S.; Contini A.; Clerici F. Identification of the First Enantiopure Rac1-Tiam1 Protein-Protein Interaction Inhibitor and Its Optimized Synthesis: Via Phosphine Free Remote Group Directed Hydroarylation. MedChemComm 2019, 10, 310–314. 10.1039/C8MD00477C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contini A.; Ferri N.; Bucci R.; Lupo M. G.; Erba E.; Gelmi M. L.; Pellegrino S. Peptide Modulators of Rac1/Tiam1 Protein-protein Interaction: An Alternative Approach for Cardiovascular Diseases. Peptide Science 2018, 110, e23089. 10.1002/bip.23089. [DOI] [PubMed] [Google Scholar]
- Sidarala V.; Veluthakal R.; Syeda K.; Vlaar C.; Newsholme P.; Kowluru A. Phagocyte-like NADPH Oxidase (Nox2) Promotes Activation of P38MAPK in Pancreatic β-Cells under Glucotoxic Conditions: Evidence for a Requisite Role of Ras-Related C3 Botulinum Toxin Substrate 1 (Rac1). Biochem. Pharmacol. 2015, 95, 301–310. 10.1016/j.bcp.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheshachalam A.; Baier A.; Eitzen G. The Effect of Rho Drugs on Mast Cell Activation and Degranulation. J. Leukocyte Biol. 2017, 102, 71–81. 10.1189/jlb.2A0616-279RRR. [DOI] [PubMed] [Google Scholar]
- Shang X.; Marchioni F.; Sipes N.; Evelyn C. R.; Jerabek-Willemsen M.; Duhr S.; Seibel W.; Wortman M.; Zheng Y. Rational Design of Small Molecule Inhibitors Targeting Rhoa Subfamily Rho GTPases. Chem. Biol. 2012, 19, 699–710. 10.1016/j.chembiol.2012.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M.; Liu A.; Ouyang Y.; Huang Y.; Chao X.; Pi R. Fasudil and Its Analogs: A New Powerful Weapon in the Long War against Central Nervous System Disorders?. Expert Opin. Invest. Drugs 2013, 22, 537–550. 10.1517/13543784.2013.778242. [DOI] [PubMed] [Google Scholar]
- Kikuchi Y.; Yamada M.; Imakiire T.; Kushiyama T.; Higashi K.; Hyodo N.; Yamamoto K.; Oda T.; Suzuki S.; Miura S. A Rho-Kinase Inhibitor, Fasudil, Prevents Development of Diabetes and Nephropathy in Insulin-Resistant Diabetic Rats. J. Endocrinol. 2007, 192, 595–603. 10.1677/JOE-06-0045. [DOI] [PubMed] [Google Scholar]
- Hofni A.; Shehata Messiha B. A.; Mangoura S. A. Fasudil Ameliorates Endothelial Dysfunction in Streptozotocin-Induced Diabetic Rats: A Possible Role of Rho Kinase. Naunyn-Schmiedeberg's Arch. Pharmacol. 2017, 390, 801–811. 10.1007/s00210-017-1379-y. [DOI] [PubMed] [Google Scholar]
- Gu L.; Gao Q.; Ni L.; Wang M.; Shen F. Fasudil Inhibits Epithelial-Myofibroblast Transdifferentiation of Human Renal Tubular Epithelial HK-2 Cells Induced by High Glucose. Chem. Pharm. Bull. 2013, 61, 688–694. 10.1248/cpb.c13-00066. [DOI] [PubMed] [Google Scholar]
- Li H.; Peng W.; Jian W.; Li Y.; Li Q.; Li W.; Xu Y. ROCK Inhibitor Fasudil Attenuated High Glucose-Induced MCP-1 and VCAM-1 Expression and Monocyte-Endothelial Cell Adhesion. Cardiovasc. Diabetol. 2012, 11, 65. 10.1186/1475-2840-11-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimi E.; Kumakura F.; Hatori C.; Hamachi E.; Iwashita A.; Ishii N.; Terasawa T.; Shimizu Y.; Takeshita N. Antinociceptive Effects of AS1892802, a Novel Rho Kinase Inhibitor, in Rat Models of Inflammatory and Noninflammatory Arthritis. J. Pharmacol. Exp. Ther. 2010, 334, 955–963. 10.1124/jpet.110.167924. [DOI] [PubMed] [Google Scholar]
- Ishizaki T.; Uehata M.; Tamechika I.; Keel J.; Nonomura K.; Maekawa M.; Narumiya S. Pharmacological Properties of Y-27632, a Specific Inhibitor of Rho- Associated Kinases. Mol. Pharmacol. 2000, 57, 976–983. [PubMed] [Google Scholar]
- Petersen N.; Frimurer T. M.; Terndrup Pedersen M.; Egerod K. L.; Wewer Albrechtsen N. J.; Holst J. J.; Grapin-Botton A.; Jensen K. B.; Schwartz T. W. Inhibiting RHOA Signaling in Mice Increases Glucose Tolerance and Numbers of Enteroendocrine and Other Secretory Cells in the Intestine. Gastroenterology 2018, 155, 1164–1176.e2. 10.1053/j.gastro.2018.06.039. [DOI] [PubMed] [Google Scholar]
- Tamura M.; Nakao H.; Yoshizaki H.; Shiratsuchi M.; Shigyo H.; Yamada H.; Ozawa T.; Totsuka J.; Hidaka H. Development of Specific Rho-Kinase Inhibitors and Their Clinical Application. Biochim. Biophys. Acta, Proteins Proteomics 2005, 1754, 245–252. 10.1016/j.bbapap.2005.06.015. [DOI] [PubMed] [Google Scholar]
- Ghazizadeh Z.; Kao D. I.; Amin S.; Cook B.; Rao S.; Zhou T.; Zhang T.; Xiang Z.; Kenyon R.; Kaymakcalan O.; Liu C.; Evans T.; Chen S. ROCKII Inhibition Promotes the Maturation of Human Pancreatic Beta-like Cells. Nat. Commun. 2017, 8, 1–12. 10.1038/s41467-017-00129-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardama G.; Comin M.; Hornos L.; Gonzalez N.; Defelipe L.; Turjanski A.; Alonso D.; Gomez D.; Menna P. Preclinical Development of Novel Rac1-GEF Signaling Inhibitors Using a Rational Design Approach in Highly Aggressive Breast Cancer Cell Lines. Anti-Cancer Agents Med. Chem. 2014, 14, 840–851. 10.2174/18715206113136660334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphries-Bickley T.; Castillo-Pichardo L.; Hernandez-O’Farrill E.; Borrero-Garcia L. D.; Forestier-Roman I.; Gerena Y.; Blanco M.; Rivera-Robles M. J.; Rodriguez-Medina J. R.; Cubano L. A.; Vlaar C. P.; Dharmawardhane S. Characterization of a Dual Rac/Cdc42 Inhibitor MBQ-167 in Metastatic Cancer. Mol. Cancer Ther. 2017, 16, 805–818. 10.1158/1535-7163.MCT-16-0442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnst J. L.; Hein A. L.; Taylor M. A.; Palermo N. Y.; Contreras J. I.; Sonawane Y. A.; Wahl A. O.; Ouellette M. M.; Natarajan A.; Yan Y. Discovery and Characterization of Small Molecule Rac1 Inhibitors. Oncotarget 2017, 8, 34586–34600. 10.18632/oncotarget.16656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oprea T. I.; Sklar L. A.; Agola J. O.; Guo Y.; Silberberg M.; Roxby J.; Vestling A.; Romero E.; Surviladze Z.; Murray-Krezan C.; Waller A.; Ursu O.; Hudson L. G.; Wandinger-Ness A. Novel Activities of Select NSAID Renantiomers against Rac1 and Cdc42 GTPases. PLoS One 2015, 10, e0142182. 10.1371/journal.pone.0142182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z.; Zhang H.; Zhang Y.; Liao L.; Zhou W.; Zhang F.; Lian F.; Huang J.; Xu P.; Zhang R.; Lu W.; Zhu M.; Tao H.; Yang F.; Ding H.; Chen S.; Yue L.; Zhou B.; Zhang N.; Tan M.; Jiang H.; Chen K.; Liu B.; Liu C.; Dang Y.; Luo C. Covalent Inhibitors Allosterically Block the Activation of Rho Family Proteins and Suppress Cancer Cell Invasion. Advanced Science 2020, 7, 2000098. 10.1002/advs.202000098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhuin T.; Roy J. K. Rab11 in Disease Progression. Int. J. Mol. Cell. Med. 2015, 4, 1–8. [PMC free article] [PubMed] [Google Scholar]
- Tan K. T.; Guiu-Rozas E.; Bon R. S.; Guo Z.; Delon C.; Wetzel S.; Arndt S.; Alexandrov K.; Waldmann H.; Goody R. S.; Wu Y. W.; Blankenfeldt W. Design, Synthesis, and Characterization of Peptide-Based Rab Geranylgeranyl Transferase Inhibitors. J. Med. Chem. 2009, 52, 8025–8037. 10.1021/jm901117d. [DOI] [PubMed] [Google Scholar]
- Stigter E. A.; Guo Z.; Bon R. S.; Wu Y. W.; Choidas A.; Wolf A.; Menninger S.; Waldmann H.; Blankenfeldt W.; Goody R. S. Development of Selective, Potent RabGGTase Inhibitors. J. Med. Chem. 2012, 55, 8330–8340. 10.1021/jm300624s. [DOI] [PubMed] [Google Scholar]
- Deraeve C.; Guo Z.; Bon R. S.; Blankenfeldt W.; DiLucrezia R.; Wolf A.; Menninger S.; Stigter E. A.; Wetzel S.; Choidas A.; Alexandrov K.; Waldmann H.; Goody R. S.; Wu Y. W. Psoromic Acid Is a Selective and Covalent Rab-Prenylation Inhibitor Targeting Autoinhibited Rabggtase. J. Am. Chem. Soc. 2012, 134, 7384–7391. 10.1021/ja211305j. [DOI] [PubMed] [Google Scholar]
- McKenna C. E.; Kashemirov B. A.; Błazewska K. M.; Mallard-Favier I.; Stewart C. A.; Rojas J.; Lundy M. W.; Ebetino F. H.; Baron R. A.; Dunford J. E.; Kirsten M. L.; Seabra M. C.; Bala J. L.; Marma M. S.; Rogers M. J.; Coxon F. P. Synthesis, Chiral High Performance Liquid Chromatographic Resolution and Enantiospecific Activity of a Potent New Geranylgeranyl Transferase Inhibitor, 2-Hydroxy-3-Imidazo[1,2-a]Pyridin-3-Yl-2-Phosphonopropionic Acid. J. Med. Chem. 2010, 53, 3454–3464. 10.1021/jm900232u. [DOI] [PubMed] [Google Scholar]
- Kaźmierczak A.; Kusy D.; Niinivehmas S. P.; Gmach J.; Joachimiak Ł.; Pentikäinen O. T.; Gendaszewska-Darmach E.; Błazewska K. M. Identification of the Privileged Position in the Imidazo[1,2-a]Pyridine Ring of Phosphonocarboxylates for Development of Rab Geranylgeranyl Transferase (RGGT) Inhibitors. J. Med. Chem. 2017, 60, 8781–8800. 10.1021/acs.jmedchem.7b00811. [DOI] [PubMed] [Google Scholar]
- Kusy D.; Marchwicka A.; Małolepsza J.; Justyna K.; Gendaszewska-Darmach E.; Błażewska K. M. Synthesis of the 6-Substituted Imidazo[1,2-a]Pyridine-3-Yl-2- Phosphonopropionic Acids as Potential Inhibitors of Rab Geranylgeranyl Transferase. Front. Chem. 2021, 8, 596162. 10.3389/fchem.2020.596162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coxon F.; Joachimiak Ł.; Najumudeen A. K.; Breen G.; Gmach J.; Oetken-Lindholm C.; Way R.; Dunford J.; Abankwa D.; Błazewska K. M. Synthesis and Characterization of Novel Phosphonocarboxylate Inhibitors of RGGT. Eur. J. Med. Chem. 2014, 84, 77–89. 10.1016/j.ejmech.2014.06.062. [DOI] [PubMed] [Google Scholar]
- Joachimiak L.; Marchwicka A.; Gendaszewska-Darmach E.; Błazewska K. M. Synthesis and Biological Evaluation of Imidazole-Bearing α-Phosphonocarboxylates as Inhibitors of Rab Geranylgeranyl Transferase (RGGT). ChemMedChem 2018, 13, 842–851. 10.1002/cmdc.201700791. [DOI] [PubMed] [Google Scholar]
- Cromm P. M.; Spiegel J.; Küchler P.; Dietrich L.; Kriegesmann J.; Wendt M.; Goody R. S.; Waldmann H.; Grossmann T. N. Protease-Resistant and Cell-Permeable Double-Stapled Peptides Targeting the Rab8a GTPase. ACS Chem. Biol. 2016, 11, 2375–2382. 10.1021/acschembio.6b00386. [DOI] [PubMed] [Google Scholar]
- Mitra S.; Montgomery J. E.; Kolar M. J.; Li G.; Jeong K. J.; Peng B.; Verdine G. L.; Mills G. B.; Moellering R. E. Stapled Peptide Inhibitors of RAB25 Target Context-Specific Phenotypes in Cancer. Nat. Commun. 2017, 8, 660. 10.1038/s41467-017-00888-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsaggar M.; Liu D. Organ-Based Drug Delivery. J. Drug Targeting 2018, 26, 385–397. 10.1080/1061186X.2018.1437919. [DOI] [PubMed] [Google Scholar]
- Rautio J.; Meanwell N. A.; Di L.; Hageman M. J. The Expanding Role of Prodrugs in Contemporary Drug Design and Development. Nat. Rev. Drug Discovery 2018, 559–587. 10.1038/nrd.2018.46. [DOI] [PubMed] [Google Scholar]
- Hu C.; Jia W. Therapeutic Medications against Diabetes: What We Have and What We Expect. Adv. Drug Delivery Rev. 2019, 139, 3–15. 10.1016/j.addr.2018.11.008. [DOI] [PubMed] [Google Scholar]
- Carpenter M. C.; Lo M. N.; Palmer A. E. Techniques for Measuring Cellular Zinc. Arch. Biochem. Biophys. 2016, 611, 20–29. 10.1016/j.abb.2016.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M.; Maji B.; Manna D.; Kahraman S.; Elgamal R. M.; Small J.; Kokkonda P.; Vetere A.; Goldberg J. M.; Lippard S. J.; Kulkarni R. N.; Wagner B. K.; Choudhary A. Native Zinc Catalyzes Selective and Traceless Release of Small Molecules in β-Cells. J. Am. Chem. Soc. 2020, 142, 6477–6482. 10.1021/jacs.0c00099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton T. M.; Allegretti P. A.; Lee S.; Moeller H. P.; Smith M.; Annes J. P. Zinc-Chelating Small Molecules Preferentially Accumulate and Function within Pancreatic β Cells. Cell Chem. Biol. 2019, 26, 213–222.e6. 10.1016/j.chembiol.2018.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W.; Ehlerding E. B.; Lan X.; Luo Q. Y.; Cai W. Molecular Imaging of β-Cells: Diabetes and Beyond. Adv. Drug Delivery Rev. 2019, 139, 16–31. 10.1016/j.addr.2018.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y.; Ding Y.; Song X.; Ma X.; Li X.; Zhang N.; Song Y.; Sun Y.; Shen Y.; Zhong W.; Hu L. A.; Ma Y.; Zhang M. Y. Structure-Guided Discovery of a Single-Domain Antibody Agonist against Human Apelin Receptor. Science Advances 2020, 6, eaax7379. 10.1126/sciadv.aax7379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shilpi S. Drug Targeting Strategies for Liver Cancer and Other Liver Diseases. MOJ. Drug Design Development & Therapy 2018, 2, 171–177. 10.15406/mojddt.2018.02.00044. [DOI] [Google Scholar]
- Mahdinloo S.; Kiaie S. H.; Amiri A.; Hemmati S.; Valizadeh H.; Zakeri-Milani P. Efficient Drug and Gene Delivery to Liver Fibrosis: Rationale, Recent Advances, and Perspectives. Acta Pharm. Sin. B 2020, 10, 1279–1293. 10.1016/j.apsb.2020.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebner D.; Bialek P.; El-Kattan A.; Ambler C.; Tu M. Strategies for Skeletal Muscle Targeting in Drug Discovery. Curr. Pharm. Des. 2015, 21, 1327–1336. 10.2174/1381612820666140929095755. [DOI] [PubMed] [Google Scholar]
- Philippou S.; Mastroyiannopoulos N. P.; Makrides N.; Lederer C. W.; Kleanthous M.; Phylactou L. A. Selection and Identification of Skeletal-Muscle-Targeted RNA Aptamers. Mol. Ther.--Nucleic Acids 2018, 10, 199–214. 10.1016/j.omtn.2017.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homma Y.; Hiragi S.; Fukuda M. Rab Family of Small GTPases: An Updated View on Their Regulation and Functions. FEBS J. 2021, 288, 36–55. 10.1111/febs.15453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mafakheri S.; Chadt A.; Al-Hasani H. Regulation of RabGAPs Involved in Insulin Action. Biochem. Soc. Trans. 2018, 46, 683–690. 10.1042/BST20170479. [DOI] [PubMed] [Google Scholar]
- Waschbüsch D.; Khan A. R. Phosphorylation of Rab GTPases in the Regulation of Membrane Trafficking. Traffic 2020, 21, 712–719. 10.1111/tra.12765. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.