

Abstract

Despite a large number of synthesis procedures for pyrazoles known today, those directly employing primary amines as substrates are rare. Herein, we report an original method for the preparation of N-alkyl and N-aryl pyrazoles from primary aliphatic or aromatic amines as a limiting reagent of the reaction. The protocol utilizes no inorganic reagents and requires a short reaction time, mild conditions, and the use of structurally simple and commercially available starting reagents. During this study, pyrazoles containing a wide variety of N-substituents were obtained using the same procedure for both aliphatic and aromatic amines.
Introduction
Pyrazoles can be obtained by numerous synthetic methods,1−5 but their versatile applicability as pharmaceuticals,6,7 crop protection chemicals,8,9 building blocks for organic and inorganic chemistry10,11 still propels research to develop new synthetic methodologies. Over the past decade, a new specific application has appeared for N-substituted pyrazoles, i.e., as a metal-coordinating directing group for transition-metal-catalyzed reactions.12−18 A large number of such chemical transformations significantly increase interest in pyrazole-functionalized molecules. Preparation of N-substituted pyrazoles from primary amines as a limiting reagent and source of N-substituent gives a wide variety of potential products that might be further functionalized.15
In contrast to N-alkyl indazoles for which synthetic methodology starting from primary aliphatic amines is well-known and broadly used,19−21N-alkyl pyrazoles are usually synthesized from difficult to handle hydrazines or hydrazine derivatives.2−4,22−24 With a great variety of synthetic methods for N-alkyl pyrazoles, there are just two reports describing primary aliphatic amine as a limiting reagent that introduces an N-linked substituent into a product’s structure (Scheme 1).25,26 The known methods are limited because of multistep functionalizations of diketone (A) or amination reagent preparation (B). The drawback of the methodologically elegant reaction B is, moreover, a complicated and time-consuming procedure (Scheme 1).
Scheme 1. Pyrazoles from Aliphatic Amines as a Limiting Reagent.

Electrophilic amination of primary aliphatic amines is a well-known strategy for the preparation of hydrazines.27−29 However, in all cases, a large excess of the amine is required due to hydrazine’s enhanced nucleophilicity relative to the corresponding origin amine.30 This problem might be solved by utilizing N-protected electrophilic amination reagents that form hydrazines that are stable under reaction conditions, but this requires a further deprotection step.25,31,32 Transformation with commercially available amination reagents that form unprotected hydrazine has practical advantages. Here we report a fast and straightforward method for the preparation of pyrazoles from primary aliphatic and aromatic amines as limiting reagents using bench-stable, commercially available amination reagent.
Results and Discussion
Initially, we tested reagents R1, R2, and R4, and we found that only R1 gives the desired product under initial conditions. Solvent, temperature, time, and proportion of reagents optimization with R1 allowed obtaining the desired N-alkyl pyrazole 1a with 44% isolated yield (Table 1, for the full table, see Table S1). Adding all three reagents simultaneously at 0 °C followed by their heating is critical to obtain the desired product with a reproducible yield. Minor changes in the reaction temperature or equivalents of reagents R1 and 1 slightly reduce the yield (Table 1, entries 3–7). The addition of a stoichiometric amount of water, acetic acid, or weak basic salts has a minor effect on yield, showing that the reaction is not promoted by this species, and it is not sensitive to them (Table 1, entries 8–12). The presence of diisopropylethylamine (DIPEA) decreases the yield of 1a, which is probably an effect of an R1 consumption for a side formation of 1,1-diisopropyl-1-ethylhydrazonium salt (entry 12). Alternative amination reagents R2–R6 were tried under optimized conditions (Table 1, entries 13–17). The product was obtained in 23%, 41%, and 53% yields for R2, R3, and R6, respectively, and was not observed (1H NMR and GC–MS) for reactions conducted with R4 and R5.
Table 1. Reaction Conditions and Amination Reagent Optimizationa.

| entry | R1–6 [equiv] | additive [1.0 equiv] | temp [°C] | yieldb [%] |
|---|---|---|---|---|
| 1 | R1 [1.5] | 0–85 | 54, 53,e 44f | |
| 2c | R1 [1.5] | 0–85 | 17 | |
| 3 | R1 [1.5] | 0–110 | 49 | |
| 4 | R1 [1.5] | 0–50 | 41 | |
| 5 | R1 [1.1] | 0–85 | 47 | |
| 6 | R1 [1.7] | 0–85 | 48 | |
| 7d | R1 [1.5] | 0–85 | 48 | |
| 8 | R1 [1.5] | H2O | 0–85 | 51 |
| 9 | R1 [1.5] | AcOH | 0–85 | 51 |
| 10 | R1 [1.5] | AcONa | 0–85 | 53 |
| 11 | R1 [1.5] | K2CO3 | 0–85 | 56 |
| 12 | R1 [1.5] | DIPEA | 0–85 | 46 |
| 13 | R2 [1.5] | 0–85 | 23e | |
| 14 | R3 [1.5] | 0–85 | 41e | |
| 15 | R4 [1.5] | 0–85 | 0e | |
| 16 | R5 [1.5] | 0–85 | 0e | |
| 17 | R6g [1.5] | 0–85 | 53e |
Reactions were carried out using a (0.2 mmol, 1.0 equiv), 1 (0.22 mmol, 1.1 equiv), R1–6 (1.1–1.7 equiv), and DMF (1.0 mL, 0.2 M) at a given temperature under air.
GC yield.
1 was added after the mixture was heated to 80 °C.
0.3 mmol (1.5 equiv) of 1 was added.
NMR yield.
Isolated yield.
Wet R6 was used.
A series of primary aliphatic and aromatic amines were reacted under optimal conditions yielding corresponding pyrazoles (Scheme 2). Reactions were typically carried out on a 1 mmol scale. It was found that aliphatic amines with quaternary carbon or sterically hindered tertiary carbon adjacent to the amine group gave around 40% yields (1a, 1b, 1d, 1h, and 1i), whereas amines with neighboring secondary or less hindered tertiary carbon gave yields close to 30% (1c, 1e, 1g, 1j, and 1k). Aromatic amines reacted under the same condition and gave pyrazoles 1l–p generally with higher yields (47–70%) than aliphatic amines. Compounds with electron-donating and withdrawing substituent at phenyl ring (1m and 1n) and substituent in ortho position to an amine group (1p) were successfully obtained. Besides ester, methoxy, and haloarene functionalities, the reaction tolerates unprotected N–H of indole and aliphatic O–H group. However, the target product was not observed in the presence of unprotected phenol (1q), probably due to competitive O-amination. Syntheses of 1a and 1g were conducted on a 3.0 and 5.0 mmol scale, respectively, to underline the method’s synthetic applicability. The structure of 1o was confirmed by X-ray single-crystal analysis (Scheme 4).
Scheme 2. Scope of Amines.

The yields refer to the isolated products. Reactions were carried out using amines a–q (1.0 mmol), 1 (1.1 mmol), R1 (1.5 mmol), and DMF (5.0 mL) at 0–85 °C in 1.5–2.0 h under air unless otherwise given.
The experiment was conducted on a 3.0 mmol scale to give 0.243 g (1.35 mmol) of the 1a.
The experiment was conducted on a 5.0 mmol scale to give 383 mg (1.71 mmol) of the 1g.
The experiment was conducted on a 0.5 mmol scale.
The reaction was carried out for 16 h at 80 °C.
Scheme 4. Additional Experiments and X-ray Structures.
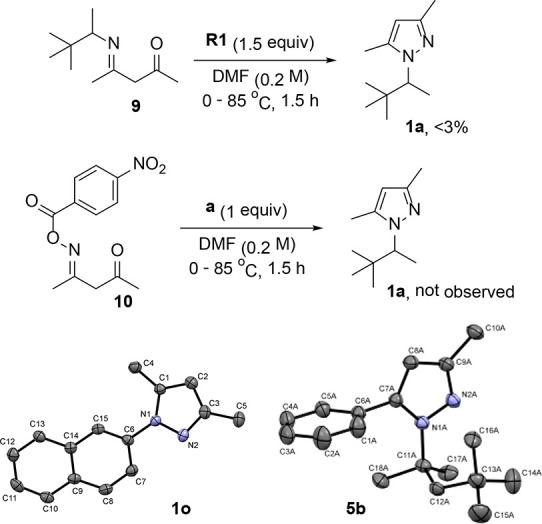
The reactions were carried out using 9 (0.1 mmol, 1.0 equiv), R1 (1.1 equiv), and DMF (0.5 mL); 10 (0.12 mmol, 1.2 equiv), a (1.0 equiv), and DMF (0.5 mL), at a given temperature under air.
ORTEP view of crystal structures of 1o (CCDC 2019272) and 5b (CCDC 2019718). Thermal ellipsoids are drawn to encompass 50% probability level.
Reactivity of a series of diketones was tested in reaction with tert-octylamine, and it was found that it strongly depends on the electronic and steric properties of diketone’s substituents (Scheme 3). 3,5-Dialkyl and 3,4,5-trialkyl pyrazoles (2b, 3b, and 6b) were obtained in 37–43% yields, whereas 4b in only 24% even with 5.0 equiv of b. Reaction conducted with diketone containing aryl substituent 5 under standard conditions gave a 20% yield. However, the use of 5.0 equiv of 5 instead of standard 1.1 allowed for 5b with a 46% yield. Sterically hindered 7 and electron-deficient 8 did not react under the standard conditions and when they were used in 5.0 equiv excess (7b and 8b). Interestingly, for reactions with diketones 5 and 6, only one isomer of pyrazoles 5b or 6b was obtained. The other was observed in the crude reaction mixture by GC–MS in a very small amount, but they were not isolated in both cases. The structure of the obtained isomer 5b was confirmed by X-ray single-crystal analysis (Scheme 4).
Scheme 3. Scope of Diketones.

The yields refer to the isolated products. Reactions were carried out using amine b (1.0 mmol), diketones 2–8 (1.1 mmol), R1 (1.5 mmol), and DMF (5.0 mL) at 0–85 °C for 1.5 h under air unless otherwise given.
The experiment was conducted on a 0.5 mmol scale for 4b and a 0.2 mmol for 5b and 7–8b with 5.0 equiv of diketones.
Additional experiments conducted for 9 and 10 showed that both compounds are not intermediates in the reaction (Scheme 4). This and selectivity of formed products from unsymmetrical diketones (Scheme 3, 5b and 6b) that indicate a nucleophilic attack of prior formed hydrazine on less hindered carbonyl gave reason to believe that the reaction starts from a nucleophilic attack of amine on II (Scheme 5). Delivered during this step, hydrazine III is trapped by diketone V before the next competitive attack on II. It is also consistent with the fact that the addition of diketone 1 with a few minutes delay caused a significantly lower yield (17%, Table 1, entry 2). Relatively strong p-nitrobenzoic acid that forms in the first step from R1 catalyzes the formation of hydrazone followed by heterocyclization to give pyrazole in Knorr condensation reaction. The observed moderate yield of the reaction explains the formation of side products VI, VII, and IX (Scheme 5). GC–MS of a crude mixture of 1a indicated corresponding imine VI, and the product of hydrazine decomposition VII was observed by the same method running reaction without diketone (see Supporting Information). The product of amination of pyrazole IX was not detected in reaction mixtures. However, deprotonated pyrazole efficiently reacts with R1,33 and the addition of the next portion of amination reagent II decreases the yield of VIII, pointing to the formation of IX (see Table S1, entries 5 and 9).
Scheme 5. Proposed Reaction Pathway and Side Product Formation.

In contrast to the multistep method of pyrazole synthesis from amines, which in showed examples gave comparable overall yields (Scheme 1, B),25 the presented method is characterized by a short time and a simple procedure that delivers products with less effort. In this context, the availability of R1 amination reagent from commercial sources offers an extra advantage over the requiring five-step synthesis of oxaziridine (15% overall yield). Alternatively, a known two-step large-scale synthesis allows to obtain R1 from basic reagents (81% overall yield).33
Conclusions
In conclusion, we have developed a new method for the preparation of N-alkyl and N-aryl substituted pyrazoles directly from primary aliphatic or aromatic amines and diketones, applying readily accessible from commercial sources electrophilic amination reagent R1. Despite the modest yields in some cases, the use of an amine as the limiting reagent, the absence of metals, short reaction times, and a simple procedure makes this method practical for functionalizing amines, giving pyrazoles containing a wide variety of N-substituents.
Experimental Section
General Information
All reactions were carried out and purified using standard, commercially available glassware. Chemicals for reactions, workup, and chromatography were reagent grade or ACS grade and were used as received. Silica gel 60 Å 0.04–0.06 mm (Macherey Nagel), Al2O3, basic/neutral alumina, Brockmann grade I, 60 mesh (Alfa Aesar), was used for product purifications. 1H NMR and 13C NMR spectra were recorded using a 500 MHz Bruker Avance spectrometer with an inverse broad-band probe. For all 1H NMR spectra, the chemical shifts are given in ppm relative to the solvent residual peaks (CDCl3, 1H = 7.26 ppm, 13C = 77.16 ppm). Coupling constants are given in hertz (Hz). 13C NMR spectra were measured with proton decoupling. HRMS spectra were recorded using Bruker Apex ultra FT-ICR (ESI) or Shimadzu q-TOF LCMS 9030 with an ESI ion source. GC–MS (EI) data were recorded using an Agilent GCMSD 7820A/5977B system. IR spectra were recorded using a Thermo Scientific Nicolet iS10 FTIR (ATR, diamond).
General Procedure
Amine (1.00 mmol) was dissolved in DMF (5.0 mL) in an 8–10 mL screw cap (silicon/PTFE septum) vial equipped with a small (10 mm × 6 mm) stir bar. The mixture was cooled in an ice–NaCl cooling bath. Then, prepared samples of O-(4-nitrobenzoyl)hydroxylamine (274 mg, 1.50 mmol) and diketone (1.10 mmol) were added one by one. The vial was immediately closed, shaken, and placed into a reaction pie block preheated on a stirrer to 85 °C for the reaction time given for a compound. In workup A, the crude mixture was poured into 1 M NaOH (100 mL) and extracted with DCM (3 × 30 mL). The organic phase was washed with brine (2 × 50 mL), dried with anhydrous MgSO4, and filtered, and the solvent was evaporated. The product was purified using column chromatography under conditions given for a compound. In workup B, the crude mixture was treated with triethylamine (0.5 mL), and DMF was partially evaporated under a nitrogen flow. The residue was adsorbed on silica gel and purified using column chromatography under conditions given for a compound.
1-(3,3-Dimethylbutan-2-yl)-3,5-dimethyl-1H-pyrazole (1a)
3,3-Dimethylbutan-2-amine a (134 μL, 1.00 mmol), 2,4-pentanedione 1 (114 μL, 1.10 mmol), O-(4-nitrobenzoyl)hydroxylamine (273 mg, 1.50 mmol), and DMF (5.0 mL) were used. The reaction was run at 85 °C (reaction block) for 1.5 h. Workup A and chromatography (basic alumina grade I, pentane–Et2O 0–60%) were applied to obtain 80 mg (0.44 mmol, 44%) of 1a as a colorless volatile liquid. For a large-scale experiment, dimethylbutan-2-amine a (410 μL, 3.00 mmol), 2,4-pentanedione 1 (342 μL, 3.30 mmol), O-(4-nitrobenzoyl)hydroxylamine (820 mg, 4.50 mmol), and DMF (20 mL) were used to obtain 243 mg (1.35 mmol, 45%) of 1a. 1H NMR (500 MHz, CDCl3): δ 5.72 (s, 1H), 3.85 (q, J = 6.9 Hz, 1H), 2.22 (s, 3H), 2.21 (s, 3H), 1.44 (d, J = 6.9 Hz, 3H), 0.93 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3): δ 146.5, 138.9, 104.0, 61.0, 36.4, 27.2, 15.9, 13.9, 12.0. IR (ATR, diamond, cm–1): 2955, 2870, 1553, 1456, 1419, 1373, 1365, 1255, 1075, 974, 773. HRMS (ESI) m/z: [M + H]+ calcd for C11H21N2, 181.1700; found, 181.1706.
3,5-Dimethyl-1-(2,4,4-trimethylpentan-2-yl)-1H-pyrazole (1b)
Compound 1b was synthesized according to the general procedure using 2,4,4-trimethylpentan-2-amine b (161 μL, 1.00 mmol), 2,4-pentanedione 1 (114 μL 1.10 mmol), O-(4-nitrobenzoyl)hydroxylamine (273 mg, 1.50 mmol), and DMF (5.0 mL). The reaction was run at 85 °C (reaction block) for 1.5 h. Workup A and chromatography (silica gel, hexane–EA 0–30%) were applied to obtain 79 mg (0.38 mmol, 38%) of 1b as a yellowish oil. 1H NMR (500 MHz, CDCl3): δ 5.70 (s, 1H), 2.32 (s, 3H), 2.12 (s, 3H), 1.76 (s, 2H), 1.62 (s, 6H), 0.71 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3): δ 144.6, 138.7, 108.1, 62.5, 53.3, 31.5, 31.0, 30.7, 15.0, 13.5. HRMS (ESI) m/z: [M + Na]+ calcd for C13H24N2Na, 231.1832; found, 231.1851. IR (ATR, diamond, cm–1): 2949, 1550, 1447, 1417, 1357, 1234, 1094, 1024, 776, 603.
3-(2-(3,5-Dimethyl-1H-pyrazol-1-yl)ethyl)-1H-indole (1c)
Tryptamine c (160 mg, 1.00 mmol), 2,4-pentanedione 1 (114 μL, 1.10 mmol), O-(4-nitrobenzoyl)hydroxylamine (273 mg, 1.50 mmol), and DMF (5.0 mL) were used. The reaction was run at 85 °C (reaction block) for 1.5 h. Workup A and chromatography (basic alumina grade I, hexane–THF 5–100%) were applied to obtain 72 mg (0.30 mmol, 30%) of 1c as a yellowish solid. 1H NMR (500 MHz, CDCl3): δ 8.34 (s, 1H), 7.55 (d, J = 8.0 Hz, 1H), 7.35 (d, J = 8.0 Hz, 1H), 7.23–7.16 (m, 1H), 7.15–7.08 (m, 1H), 6.84 (d, J = 2.3 Hz, 1H), 5.74 (s, 1H), 4.23 (t, J = 7.4 Hz, 2H), 3.25 (t, J = 7.4 Hz, 2H), 2.28 (s, 3H), 1.95 (s, 3H). 13C{1H} NMR (126 MHz, CDCl3): δ 147.5, 139.2, 136.3, 127.4, 122.5, 122.1, 119.5, 118.6, 112.6, 111.3, 104.8, 49.4, 26.7, 13.7, 10.9. IR (ATR, diamond, cm–1): 3215, 3184, 2931, 2856, 1551, 1454, 1352, 1232, 1107, 786, 765, 736. HRMS (ESI) m/z: [M + H]+ calcd for C15H18N3, 240.1496; found, 240.1493.
3,5-Dimethyl-1-(tert-pentyl)-1H-pyrazole (1d)
2-Methylbutan-2-amine d (116 μL, 1.00 mmol), 2,4-pentanedione 1 (114 μL 1.10 mmol), O-(4-nitrobenzoyl)hydroxylamine (273 mg, 1.50 mmol), and DMF (5.0 mL) were used. The reaction was run at 85 °C (reaction block) for 1.5 h. Workup A and chromatography (silica gel, hexane–EA 30%) were applied to obtain 60 mg (0.361 mmol, 36%) of 1d as a yellowish volatile liquid. 1H NMR (500 MHz, CDCl3): δ 5.75 (s, 1H), 2.33 (s, 1H), 2.16 (s, 1H), 1.85 (q, J = 7.4 Hz, 1H), 1.56 (s, 2H), 0.71 (t, J = 7.4 Hz, 1H). 13C{1H} NMR (126 MHz, CDCl3): δ 145.0, 138.7, 107.7, 62.1, 34.8, 28.0, 14.5, 13.5, 8.4. HRMS (ESI) m/z: [M + Na]+ calcd for C10H18N2Na, 189.1363; found, 189.1359. IR (ATR, diamond, cm–1): 2970, 1550, 1416, 1356, 1304, 1251, 1210, 1107, 1025, 776.
1-Dodecyl-3,5-dimethyl-1H-pyrazole (1e)
Dodecylamine e (185 mg, 1.00 mmol), 2,4-pentanedione 1 (114 μL 1.10 mmol), O-(4-nitrobenzoyl)hydroxylamine (273 mg, 1.50 mmol), and DMF (5.0 mL) were used. The reaction was run at 85 °C (reaction block) for 1.5 h. Workup A and chromatography (silica gel, hexane–EA 0–30%) were applied to obtain 87 mg (0.33 mmol, 33%) of 1e as a yellowish oil. 1H NMR (500 MHz, CDCl3): δ 5.74 (s, 1H), 3.92–3.88 (m, 2H), 2.19 (s, 6H), 1.78–1.70 (m, 2H), 1.23 (s, 18H), 0.86 (t, J = 7.0 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3): δ 147.0, 138.3, 104.6, 48.7, 31.9, 30.5, 29.6, 29.5, 29.5, 29.3, 29.2, 26.7, 22.6, 14.1, 13.4, 11.0. HRMS (ESI) m/z: [M + H]+ calcd for C17H33N2, 265.2639; found, 265.2636. IR (ATR, diamond, cm–1): 2928, 2854, 1670, 1611, 1553, 1466, 1378, 1310, 1023,774.
1-Cyclohexyl-3,5-dimethyl-1H-pyrazole (1f).34
Cyclohexanamine f (115 μL 1.00 mmol), 2,4-pentanedione 1 (114 μL 1.10 mmol), O-(4-nitrobenzoyl)hydroxylamine (273 mg, 1.50 mmol), and DMF (5.0 mL) were used. The reaction was run at 85 °C (reaction block) for 1.5 h. Workup A and flash chromatography (silica gel, hexane–THF 0–100%) were applied to obtain 63 mg (0.35 mmol, 35%) of 1f as a yellowish oil. 1H NMR (500 MHz, CDCl3): δ 5.74 (s, 1H), 3.92–3.88 (m, 2H), 2.19 (s, 6H), 1.78–1.70 (m, 2H), 1.23 (s, 18H), 0.86 (t, J = 7.0 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3): δ 147.0, 138.3, 104.6, 48.7, 31.9, 30.5, 29.6, 29.5, 29.5, 29.3, 29.2, 26.7, 22.6, 14.1, 13.4, 11.0.
6-(3,5-Dimethyl-1H-pyrazol-1-yl)-2-methylheptan-2-ol (1g)
6-Amino-2-methylheptan-2-ol g (741 mg, 5.00 mmol), 2,4-pentanedione 1 (568 μL, 5.48 mmol), O-(4-nitrobenzoyl)hydroxylamine (1.37 mg, 7.52 mmol), and DMF (50 mL) were used. The reaction was run at 80 °C (reaction block) for 2 h. Workup A and chromatography (basic alumina grade I, hexane–THF 0–80%) were applied to obtain 383 mg (1.71 mmol, 34%) of 1g as a yellowish oil. 1H NMR (500 MHz, CDCl3): δ 5.71 (s, 1H), 4.15–4.03 (m, 1H), 2.20 (s, 3H), 2.19 (s, 3H), 2.05–1.93 (m, 1H), 1.74–1.63 (m, 1H), 1.51 (s, 1H), 1.40 (m, 2H overlap with d, J = 6.7 Hz, 3H), 1.31–1.24 (m, 1H), 1.19–1.14 (m, 1H), 1.13 (s, 6H). 13C{1H} NMR (126 MHz, CDCl3): δ 147.2, 138.2, 104.5, 70.9, 53.7, 43.4, 37.1, 29.4, 29.3, 21.3, 21.3, 13.8, 11.2. IR (ATR, diamond, cm–1): 3392 (OH), 2969, 2932, 2868, 1552, 1454, 1423, 1375, 1158, 773. HRMS (ESI) m/z: [M + H]+ calcd for C13H25N2O, 225.1962; found, 225.1959.
1-Adamantan-1-yl-3,5-dimethyl-1H-pyrazole (1h).15
1-Adamantylamine h (151,25 mg, 1.00 mmol), 2,4-pentanedione 1 (114 μL 1.10 mmol), O-(4-nitrobenzoyl)hydroxylamine (273 mg, 1.50 mmol), and DMF (5.0 mL) were used. The reaction was run at 85 °C (reaction block) for 1.5 h. Workup A and chromatography (silica gel, hexane–EA 30%) were applied to obtain 97 mg (0.42 mmol, 42%) of 1h as a yellowish oil. 1H NMR (500 MHz, CDCl3): δ 5.78 (s, 1H), 2.43 (s, 3H), 2.27 (d, J = 3.1 Hz, 6H), 2.20 (s, 6H), 1.74 (s, 6H). 13C{1H} NMR (126 MHz, CDCl3): δ 145.3, 138.6, 108.0, 60.4, 42.3, 36.3, 30.0, 15.1, 13.6.
3,5-Dimethyl-1-(1-phenylethyl)-1H-pyrazole (1i).15
1-Phenylethan-1-amine i (127 μL, 1.00 mmol), 2,4-pentanedione 1 (114 μL 1.10 mmol), O-(4-nitrobenzoyl)hydroxylamine (273 mg, 1.50 mmol), and DMF (5.0 mL) were used. The reaction was run at 80 °C (reaction block) for 1.5 h. Workup A and chromatography (silicagel hexane–EA 30%) were applied to obtain 76 mg (0.38 mmol, 38%) of 1i as a yellowish oil. 1H NMR (500 MHz, CDCl3): δ 7.28–7.12 (m, 1H), 7.05–7.00 (m, 1H), 5.76 (s, 1H), 5.27 (q, J = 7.1 Hz, 1H), 2.21 (s, 1H), 2.02 (s, 1H), 1.84 (d, J = 7.1 Hz, 1H). 13C{1H} NMR (126 MHz, CDCl3): δ 146.9, 143.0, 138.9, 128.5, 127.1, 125.9, 105.5, 57.3, 21.7, 13.7, 11.1.
Ethyl 2-(3,5-Dimethyl-1H-pyrazol-1-yl)-3-phenylpropanoate (1j)
Ethyl phenylalaninate35j (193 mg, 1.00 mmol), 2,4-pentanedione 1 (114 μL, 1.10 mmol), O-(4-nitrobenzoyl)hydroxylamine (273 mg, 1.50 mmol), and DMF (5.0 mL) were used. The reaction was run at 85 °C (reaction block) for 1.5 h. Workup B and chromatography (neutral alumina grade I, hexane–THF 0–40%) were applied to obtain 73 mg (0.27 mmol, 27%) of 1j as a colorless oil. 1H NMR (500 MHz, CDCl3): δ 7.22–7.15 (m, 3H), 7.01–6.96 (m, 2H), 5.69 (s, 1H), 4.76 (dd, J = 9.8, 5.2 Hz, 1H), 4.20 (qd, J = 7.1, 2.2 Hz, 2H), 3.58–3.47 (m, 2H), 2.25 (s, 3H), 1.83 (s, 3H), 1.21 (t, J = 7.1 Hz, 3H). 13C{1H} NMR (75 MHz, CDCl3): δ 169.4, 148.4, 140.3, 137.6, 129.2, 128.5, 126.8, 105.1, 62.0, 61.8, 37.4, 14.2, 13.9, 10.7. IR (ATR, diamond, cm–1): 2980, 2925, 1745 (CO), 1557, 1455, 1262, 1210, 1173, 1028, 752, 701. HRMS (ESI) m/z: [M + H]+ calcd for C16H21N2O2, 273.1598; found, 273.1597.
1-(Bicyclo[2.2.1]heptan-2-yl)-3,5-dimethyl-1H-pyrazole (1k)
Bicyclo[2.2.1]heptan-2-amine k (60 μL, 0.50 mmol), pentane-2,4-dione 1 (56 μL 0.55 mmol), O-(4-nitrobenzoyl)hydroxylamine (136 mg, 0.75 mmol), and DMF (2.5 mL) were used. The reaction was run at 85 °C for 1.5 h. Workup A and chromatography (silica gel, hexane–THF 0–30%) were applied to obtain 33 mg (0.18 mmol, 35%) of 1k as a yellowish oil. 1H NMR (500 MHz, CDCl3): δ 5.79 (s, 1H), 3.97–3.96 (m, 1H), 2.42 (s, 1H), 2.37–2.29 (m, 1H), 2.23 (s, 1H), 2.21 (s, 1H), 2.08–2.02 (m, 1H), 1.73–1.70 (m, 1H), 1.63–1.50 (m, 1H), 1.25–1.11 (m, 2H). 13C{1H} NMR (126 MHz, CDCl3): δ 146.2, 138.4, 105.2, 60.6, 43.5, 37.7, 36.0, 35.8, 28.8, 27.7, 13.9, 11.5. IR (ATR, diamond, cm–1): 2951, 2923, 1553, 1452, 1378, 1290, 1258, 1023, 802, 773. HRMS (ESI) m/z: [M + H]+ calcd for C12H19N2, 191.1543; found, 191.1549.
3,5-Dimethyl-1-phenyl-1H-pyrazole (1l).18
Aniline l (94 μL, 1.0 mmol), 2,4-pentanedione 1 (114 μL 1.10 mmol), O-(4-nitrobenzoyl)hydroxylamine (273 mg, 1.50 mmol), and DMF (5.0 mL) were used. The reaction was run at 85 °C (reaction block) for 1.5 h. Workup A and chromatography (silica gel, hexane–EA 30%) were applied to obtain 68 mg (0.47 mmol, 47%) of 1l as a yellowish oil. 1H NMR (500 MHz, CDCl3): δ 7.34 (d, J = 4.4 Hz, 4H), 7.24 (dq, J = 8.7, 4.4 Hz, 1H), 5.91 (s, 1H), 2.22 (d, J = 10.9 Hz, 6H). 13C{1H} NMR (126 MHz, CDCl3): δ 148.9, 139.9, 139.3, 128.9, 127.2, 124.7, 106.9, 13.5, 12.3.
1-(4-Methoxyphenyl)-3,5-dimethyl-1H-pyrazole (1m).18
4-Methoxyaniline m (123 mg, 1.00 mmol), 2,4-pentanedione 1 (114 μL 1.10 mmol), O-(4-nitrobenzoyl)hydroxylamine (273 mg, 1.50 mmol), and DMF (5.0 mL) were used. The reaction was run at 85 °C (reaction block) for 1.5 h. Workup A and chromatography (silica gel, hexane–EA 30%) were applied to obtain 112 mg (0.55 mmol, 55%) of 1m as a brown oil. 1H NMR (500 MHz, CDCl3): δ 7.33–7.27 (m, 2H), 6.96–6.90 (m, 2H), 5.94 (s, 3H), 2.27 (s, 3H), 2.22 (s, 3H). 13C{1H} NMR (126 MHz, CDCl3): δ 158.7, 148.5, 139.4, 133.1, 126.4, 114.1, 106.2, 55.5, 13.5, 12.1.
1-(4-Fluorophenyl)-3,5-dimethyl-1H-pyrazole (1n).36
4-Fluoroaniline n (95 μL, 1.0 mmol), 2,4-pentanedione 1 (114 μL 1.10 mmol), O-(4-nitrobenzoyl)hydroxylamine (273 mg, 1.50 mmol), and DMF (5.0 mL) were used. The reaction was run at 85 °C (reaction block) for 1.5 h. Workup A and chromatography (silica gel, hexane–EA 30%) were applied to obtain 115 mg (0.60 mmol, 60%) of 1n as a yellowish oil. 1H NMR (500 MHz, CDCl3): δ 7.39–7.33 (m, 2H), 7.15–7.06 (m, 2H), 5.96 (s, 1H), 2.25 (d, J = 10.3 Hz, 6H). 13C{1H} NMR (126 MHz, CDCl3): δ 161.6 (d, J = 247.0 Hz), 149.1, 139.5, 136.1 (d, J = 3.0 Hz), 126.6 (d, J = 8.6 Hz), 115.9 (d, J = 22.8 Hz), 106.9, 13.5, 12.2.
3,5-Dimethyl-1-(naphthalen-2-yl)-1H-pyrazole (1o)
Naphthalen-2-amine o (143 mg, 1.00 mmol), 2,4-pentanedione 1 (114 μL 1.10 mmol), O-(4-nitrobenzoyl)hydroxylamine (273 mg, 1.50 mmol), and DMF (5.0 mL) were used. The reaction was run at 85 °C (reaction block) for 1.5 h. Workup A and chromatography (silica gel, hexane–EA 0–30%) were applied to obtain 155 mg (0.70 mmol, 70%) of 1o as a red oil. 1H NMR (500 MHz, CDCl3): δ 7.90 (d, J = 8.7 Hz, 1H), 7.88–7.82 (m, 3H), 7.59 (dd, J = 8.7, 2.1 Hz, 1H), 7.54–7.43 (m, 2H), 6.02 (s, 1H), 2.35 (s, 3H), 2.32 (s, 3H). 13C{1H} NMR (126 MHz, CDCl3): δ 148.5, 133.1, 132.7, 129.5, 128.2, 127.9, 127.1, 126.9, 123.6, 123.1, 107.5, 12.9, 12.4. HRMS (ESI) m/z: [M + Na]+ calcd for C15H14N2Na 245.1050; found, 245.1045. IR (ATR, diamond, cm–1): 3053, 2920, 2852, 1633, 1508, 1379, 1266, 857, 815, 783, 472.
1-(5-Bromo-2-methylphenyl)-3,5-dimethyl-1H-pyrazole (1p)
5-Bromo-2-methylaniline p (191 mg, 1.00 mmol), 2,4-pentanedione 1 (114 μL, 1.10 mmol), O-(4-nitrobenzoyl)hydroxylamine (273 mg, 1.50 mmol), and DMF (5.0 mL) were used. The reaction was run at 80 °C (reaction block) for 16 h. Workup A and chromatography (basic alumina grade I, hexane–THF 0–30%) were applied to obtain 149 mg (0.56 mmol, 56%) of 1p as a yellowish oil. 1H NMR (500 MHz, CDCl3): δ 7.45 (dd, J = 8.2, 2.1 Hz, 1H), 7.39 (d, J = 2.1 Hz, 1H), 7.17 (d, J = 8.2 Hz, 1H), 5.96 (s, 1H), 2.27 (s, 3H), 2.06 (s, 3H), 2.01 (s, 3H). 13C{1H} NMR (126 MHz, CDCl3): δ 149.2, 140.4, 140.0, 135.6, 132.2, 132.1, 131.1, 119.2, 105.5, 17.0, 13.7, 11.4. IR (ATR, diamond, cm–1): 2954, 2924, 2857, 1594, 1556, 1496, 1424, 1360, 1036, 823, 782. HRMS (ESI) m/z: [M + H]+ calcd for C12H14N2Br, 265.0335; found, 265.0335.
3,4,5-Ttrimethyl-1-(2,4,4-trimethylpentan-2-yl)-1H-pyrazole (2b)
2,4,4-Trimethylpentan-2-amine b (161 μL, 1.00 mmol), 3-methylpentane-2,4-dione 2 (128 μL 1.10 mmol), O-(4-nitrobenzoyl)hydroxylamine (273 mg, 1.50 mmol), and DMF (5 mL) were used. The reaction was run at 85 °C (reaction block) for 1.5 h. Workup A and chromatography (silica gel, hexane–EA 0–30%) were applied to obtain 95 mg (0.43 mmol, 43%) of 2b as a yellowish oil. 1H NMR (500 MHz, CDCl3): δ 2.30 (s, 1H), 2.21–2.08 (m, 1H), 1.86 (s, 1H), 1.82 (s, 1H), 1.68 (s, 2H), 0.77 (s, 3H). 13C{1H} NMR (126 MHz, CDCl3): δ 143.3, 135.2, 113.1, 62.3, 53.2, 31.5, 30.7, 29.5, 13.1, 11.8, 8.0. IR (ATR, diamond, cm–1): 2954, 2252, 2669, 1669, 1568, 1540, 1415, 1050, 905, 729 HRMS (ESI) m/z: [M + Na]+ calcd for C14H26N2Na, 245.1989; found, 245.1987.
3,5-Diethyl-1-(2,4,4-trimethylpentan-2-yl)-1H-pyrazole (3b)
2,4,4-Trimethylpentan-2-amine b (161 μL, 1.00 mmol), heptane-3,5-dione 3 (150 μL 1.1 mmol), O-(4-nitrobenzoyl)hydroxylamine (273 mg, 1.50 mmol), and DMF (5 mL) were used. The reaction was run at 85 °C (reaction block) for 1.5 h. Workup A and chromatography (silica gel, hexane–EA 0–30%) were applied to obtain 88 mg (0.37 mmol, 37%) of 3b as a yellowish oil. 1H NMR (500 MHz, CDCl3): δ 5.87 (s, 1H), 2.77 (q, J = 7.4 Hz, 2H), 2.56 (q, J = 7.6 Hz, 2H), 1.80 (s, 2H), 1.68 (s, 7H), 1.26 (t, J = 7.4 Hz, 3H), 1.18 (t, J = 7.6 Hz, 3H), 0.73 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3): δ 150.8, 145.5, 103.6, 62.6, 53.5, 31.6, 31.3, 30.7, 21.9, 21.6, 14.4, 13.6. HRMS (ESI) m/z: [M + H]+ calcd for C15H29N2 237.2326; found, 237.2323. IR (ATR, diamond, cm–1): 3001, 2929, 1545, 1465, 1365, 1253 1227, 1089, 956, 773.
4-Ethyl-3,5-dimethyl-1-(2,4,4-trimethylpentan-2-yl)-1H-pyrazole (4b)
2,4,4-Trimethylpentan-2-amine b (67 mg, 0.50 mmol), 3-ethylpentane-2,4-dione 4 (336 μL, 2.50 mmol), O-(4-nitrobenzoyl)hydroxylamine (136 mg, 0.75 mmol), and DMF (2.5 mL) were used. The reaction was run at 85 °C (reaction block) for 1.5 h. Workup A and chromatography (basic alumina grade I, hexane-(DCM-MeOH 10%) 0–20%) were applied to obtain 28 mg (0.118 mmol, 26%) of 4b as a colorless oil. 1H NMR (500 MHz, CDCl3): δ 2.33–2.28 (m, 5H), 2.16 (s, 3H), 1.82 (s, 2H), 1.69 (s, 6H), 1.00 (t, J = 7.6 Hz, 3H), 0.75 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3): δ 142.9, 135.1, 120.4, 62.4, 53.4, 31.7, 31.2, 30.9, 17.0, 15.6, 13.0, 12.0. IR (ATR, diamond, cm–1): 2956, 2925, 2855, 1463, 1366, 1355, 1272, 1256, 1235. HRMS (ESI) m/z: [M + H]+ calcd for C15H29N2, 237.2326; found, 237.2326.
3-Methyl-5-phenyl-1-(2,4,4-trimethylpentan-2-yl)-1H-pyrazole (5b)
2,4,4-Trimethylpentan-2-amine b (26 mg, 0.20 mmol), 1-phenylbutane-1,3-dione 5 (162 mg, 1.0 mmol), O-(4-nitrobenzoyl)hydroxylamine (55 mg, 0.30 mmol), and DMF (1 mL) were used. The reaction was run at 85 °C (reaction block) for 1.5 h. Workup A and chromatography (silica gel, hexane–EA 0–30%) were applied to obtain 25 mg (0.092 mmol, 46%) of 5b as a white solid. 1H NMR (500 MHz, CDCl3): δ 7.67–7.26 (m, 5H), 5.88 (s, 1H), 2.26 (s, 3H), 1.78 (s, 2H), 1.46 (s, 6H), 0.79 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3): δ 144.6, 143.3, 135.0, 130.5, 128.1, 127.6, 109.3, 64.1, 54.8, 31.8, 31.5, 30.7, 13.5, 13.5. IR (ATR, diamond, cm–1): 2955, 1545, 1498, 1442, 1395, 1365, 1253, 1196, 793, 704. HRMS (ESI) m/z: [M + Na]+ calcd for C18H26N2Na, 293.1989; found, 293.1989.
5-Isobutyl-3-methyl-1-(2,4,4-trimethylpentan-2-yl)-1H-pyrazole (6b)
2,4,4-Trimethylpentan-2-amine b (161 μL, 1.00 mmol), 6-methylheptane-2,4-dione 6 (173 μL, 1.10 mmol), O-(4-nitrobenzoyl)hydroxylamine (273 mg, 1.50 mmol), and DMF (5.0 mL) were used. The reaction was run at 85 °C (reaction block) for 2 h. Workup A and chromatography (basic alumina grade I, hexane–THF 0–20%) were applied to obtain 97 mg (0.37 mmol, 37%) of 6b as a yellowish oil. 1H NMR (500 MHz, CDCl3): δ 5.84 (s, 1H), 2.61 (d, J = 7.1 Hz, 2H), 2.20 (s, 3H), 2.03–1.94 (m, 1H), 1.83 (s, 2H), 1.69 (s, 6H), 1.01 (d, J = 6.6 Hz, 6H), 0.77 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3): δ 144.6, 143.3, 106.6, 62.8, 53.8, 37.9, 31.7, 31.5, 30.9, 28.5, 23.1, 13.8. IR (ATR, diamond, cm–1): 2954, 2870, 1546, 1468, 1425, 1387, 1363, 1241, 1021, 777, 600. HRMS (ESI) m/z: [M + H]+ calcd for C16H31N2, 251.2482; found, 251.2481.
Acknowledgments
The authors gratefully acknowledge the National Science Centre Poland (NCN 2017/26/D/ST5/00112 and NCN 2018/31/M/ST5/00450) for financial support of this research.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.1c00606.
Copies of 1H and 13C spectra of products, X-ray data, additional experiments details, and full optimization table (PDF)
Accession Codes
CCDC 2019272 (1n), and 2019718 (4b) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: + 44 1223 336033.
The authors declare no competing financial interest.
Supplementary Material
References
- Mykhailiuk P. K. Fluorinated Pyrazoles: From Synthesis to Applications. Chem. Rev. 2021, 121 (3), 1670–1715. 10.1021/acs.chemrev.0c01015. [DOI] [PubMed] [Google Scholar]
- Neto J. S. S.; Zeni G. Alkynes and Nitrogen Compounds: Useful Substrates for the Synthesis of Pyrazoles. Chem. - Eur. J. 2020, 26 (37), 8175–8189. 10.1002/chem.201905276. [DOI] [PubMed] [Google Scholar]
- Fustero S.; Sánchez-Roselló M.; Barrio P.; Simón-Fuentes A. From 2000 to Mid-2010: A Fruitful Decade for the Synthesis of Pyrazoles. Chem. Rev. 2011, 111 (11), 6984–7034. 10.1021/cr2000459. [DOI] [PubMed] [Google Scholar]
- Fustero S.; Simón-Fuentes A.; Sanz-Cervera J. F. Recent Advances in the Synthesis of Pyrazoles. A Review. Org. Prep. Proced. Int. 2009, 41 (4), 253–290. 10.1080/00304940903077832. [DOI] [Google Scholar]
- Dadiboyena S.; Nefzi A. Synthesis of Functionalized Tetrasubstituted Pyrazolyl Heterocycles – A Review. Eur. J. Med. Chem. 2011, 46 (11), 5258–5275. 10.1016/j.ejmech.2011.09.016. [DOI] [PubMed] [Google Scholar]
- Bennani F. E.; Doudach L.; Cherrah Y.; Ramli Y.; Karrouchi K.; Ansar M.; Faouzi M. E. A. Overview of Recent Developments of Pyrazole Derivatives as an Anticancer Agent in Different Cell Line. Bioorg. Chem. 2020, 97, 103470. 10.1016/j.bioorg.2019.103470. [DOI] [PubMed] [Google Scholar]
- Küçükgüzel Ş. G.; Şenkardeş S. Recent Advances in Bioactive Pyrazoles. Eur. J. Med. Chem. 2015, 97, 786–815. 10.1016/j.ejmech.2014.11.059. [DOI] [PubMed] [Google Scholar]
- Ma Y.; Yang L.; Liu X.; Yang J.; Sun X. Development of Celecoxib-Derived Antifungals for Crop Protection. Bioorg. Chem. 2020, 97, 103670. 10.1016/j.bioorg.2020.103670. [DOI] [PubMed] [Google Scholar]
- Lamberth C. Pyrazole Chemistry in Crop Protection. Heterocycles 2007, 71 (7), 1467–1502. 10.3987/REV-07-613. [DOI] [Google Scholar]
- Pettinari C.; Tăbăcaru A.; Galli S. Coordination Polymers and Metal–Organic Frameworks Based on Poly(Pyrazole)-Containing Ligands. Coord. Chem. Rev. 2016, 307, 1–31. 10.1016/j.ccr.2015.08.005. [DOI] [Google Scholar]
- Halcrow M. A. Pyrazoles and Pyrazolides—Flexible Synthons in Self-Assembly. Dalton Trans. 2009, (12), 2059–2073. 10.1039/b815577a. [DOI] [PubMed] [Google Scholar]
- Zhang C.; Chen S.; Ye C.-X.; Harms K.; Zhang L.; Houk K. N.; Meggers E. Asymmetric Photocatalysis by Intramolecular Hydrogen-Atom Transfer in Photoexcited Catalyst–Substrate Complex. Angew. Chem., Int. Ed. 2019, 58 (41), 14462–14466. 10.1002/anie.201905647. [DOI] [PubMed] [Google Scholar]
- Lutter F. H.; Grokenberger L.; Perego L. A.; Broggini D.; Lemaire S.; Wagschal S.; Knochel P. Regioselective Functionalization of Aryl Azoles as Powerful Tool for the Synthesis of Pharmaceutically Relevant Targets. Nat. Commun. 2020, 11 (1), 4443. 10.1038/s41467-020-18188-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onodera S.; Ishikawa S.; Kochi T.; Kakiuchi F. Direct Alkenylation of Allylbenzenes via Chelation-Assisted C–C Bond Cleavage. J. Am. Chem. Soc. 2018, 140 (31), 9788–9792. 10.1021/jacs.8b03718. [DOI] [PubMed] [Google Scholar]
- Gulia N.; Daugulis O. Palladium-Catalyzed Pyrazole-Directed sp3 C–H Bond Arylation for the Synthesis of β-Phenethylamines. Angew. Chem., Int. Ed. 2017, 56 (13), 3630–3634. 10.1002/anie.201611407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moselage M.; Li J.; Kramm F.; Ackermann L. Ruthenium(II)-Catalyzed C–C Arylations and Alkylations: Decarbamoylative C–C Functionalizations. Angew. Chem., Int. Ed. 2017, 56 (19), 5341–5344. 10.1002/anie.201701231. [DOI] [PubMed] [Google Scholar]
- Boerth J. A.; Hummel J. R.; Ellman J. A. Highly Stereoselective Cobalt(III)-Catalyzed Three-Component C–H Bond Addition Cascade. Angew. Chem., Int. Ed. 2016, 55 (41), 12650–12654. 10.1002/anie.201603831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak S. H.; Gulia N.; Daugulis O. Synthesis of Unsymmetrical 2,6-Diarylanilines by Palladium-Catalyzed C–H Bond Functionalization Methodology. J. Org. Chem. 2018, 83 (10), 5844–5850. 10.1021/acs.joc.8b00659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar M. R.; Park A.; Park N.; Lee S. Consecutive Condensation, C–N and N–N Bond Formations: A Copper- Catalyzed One-Pot Three-Component Synthesis of 2H-Indazole. Org. Lett. 2011, 13 (13), 3542–3545. 10.1021/ol201409j. [DOI] [PubMed] [Google Scholar]
- Genung N. E.; Wei L.; Aspnes G. E. Regioselective Synthesis of 2H-Indazoles Using a Mild, One-Pot Condensation–Cadogan Reductive Cyclization. Org. Lett. 2014, 16 (11), 3114–3117. 10.1021/ol5012423. [DOI] [PubMed] [Google Scholar]
- Zhu J. S.; Li C. J.; Tsui K. Y.; Kraemer N.; Son J.-H.; Haddadin M. J.; Tantillo D. J.; Kurth M. J. Accessing Multiple Classes of 2H-Indazoles: Mechanistic Implications for the Cadogan and Davis–Beirut Reactions. J. Am. Chem. Soc. 2019, 141 (15), 6247–6253. 10.1021/jacs.8b13481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niesobski P.; Klukas F.; Berens H.; Makhloufi G.; Janiak C.; Müller T. J. J. De Novo Ring-Forming Consecutive Four-Component Syntheses of 4-Pyrazolyl-1,2,3-triazoles from (Triisopropylsilyl)butadiyne as a C4 Building Block. J. Org. Chem. 2018, 83 (8), 4851–4858. 10.1021/acs.joc.8b00430. [DOI] [PubMed] [Google Scholar]
- Zhu C.; Zeng H.; Liu C.; Cai Y.; Fang X.; Jiang H. Regioselective Synthesis of 3-Trifluoromethylpyrazole by Coupling of Aldehydes, Sulfonyl Hydrazides, and 2-Bromo-3,3,3-trifluoropropene. Org. Lett. 2020, 22 (3), 809–813. 10.1021/acs.orglett.9b04228. [DOI] [PubMed] [Google Scholar]
- Wang H.; Ning Y.; Sun Y.; Sivaguru P.; Bi X. Cycloaddition of Trifluoroacetaldehyde N-Triftosylhydrazone (TFHZ-Tfs) with Alkynes for Synthesizing 3-Trifluoromethylpyrazoles. Org. Lett. 2020, 22 (5), 2012–2016. 10.1021/acs.orglett.0c00395. [DOI] [PubMed] [Google Scholar]
- Armstrong A.; Jones L. H.; Knight J. D.; Kelsey R. D. Oxaziridine-Mediated Amination of Primary Amines: Scope and Application to a One-Pot Pyrazole Synthesis. Org. Lett. 2005, 7 (4), 713–716. 10.1021/ol0474507. [DOI] [PubMed] [Google Scholar]
- Wang S.; Li Y.; Bi X.; Liu Q. Synthesis of 1,3,4-Trisubstituted Pyrazoles from α-(1,3-Dithian-2-yl) Enamine Ketones via [4 + 1] Cyclization. Synlett 2015, 26 (13), 1895–1899. 10.1055/s-0034-1378858. [DOI] [Google Scholar]
- Ragnarsson U. Synthetic Methodology for Alkyl Substituted Hydrazines. Chem. Soc. Rev. 2001, 30 (4), 205–213. 10.1039/b010091a. [DOI] [Google Scholar]
- Gever G.; Hayes K. Alkylhydrazines. J. Org. Chem. 1949, 14 (5), 813–818. 10.1021/jo01157a014. [DOI] [Google Scholar]
- Audrieth L. F.; Diamond L. H. Preparation of N-Substituted Hydrazines by Modification of the Raschig Synthesis. J. Am. Chem. Soc. 1954, 76 (19), 4869–4871. 10.1021/ja01648a034. [DOI] [Google Scholar]
- Nigst T. A.; Antipova A.; Mayr H. Nucleophilic Reactivities of Hydrazines and Amines: The Futile Search for the α-Effect in Hydrazine Reactivities. J. Org. Chem. 2012, 77 (18), 8142–8155. 10.1021/jo301497g. [DOI] [PubMed] [Google Scholar]
- Polat D. E.; Brzezinski D. D.; Beauchemin A. M. Formation of Complex Hydrazine Derivatives via Aza-Lossen Rearrangement. Org. Lett. 2019, 21 (12), 4849–4852. 10.1021/acs.orglett.9b01742. [DOI] [PubMed] [Google Scholar]
- Sarnowski M. P.; Kang C. W.; Elbatrawi Y. M.; Wojtas L.; Del Valle J. R. Peptide N-Amination Supports β-Sheet Conformations. Angew. Chem., Int. Ed. 2017, 56 (8), 2083–2086. 10.1002/anie.201609395. [DOI] [PubMed] [Google Scholar]
- Parlanti L.; Discordia R. P.; Hynes J.; Miller M. M.; O'Grady H. R.; Shi Z. Amination of Heterocyclic Compounds with O-Benzoylhydroxylamine Derivatives. Org. Lett. 2007, 9 (19), 3821–3824. 10.1021/ol701730r. [DOI] [PubMed] [Google Scholar]
- Klein J. F.; Pommelet J. C.; Chuche J.; Elguero J.; Goya P.; Martinez A. SO2 Extrusion in 1,2,6-Thiadiazine 1,1-Dioxides: A Novel Synthesis of Pyrazoles. Can. J. Chem. 1993, 71 (3), 410–412. 10.1139/v93-060. [DOI] [Google Scholar]
- Lin H.; Wang C.; Bannister T. D.; Kamenecka T. M. Site-Selective γ-C(sp3)–H and γ-C(sp2)–H Arylation of Free Amino Esters Promoted by a Catalytic Transient Directing Group. Chem. - Eur. J. 2018, 24 (38), 9535–9541. 10.1002/chem.201802465. [DOI] [PubMed] [Google Scholar]
- Yang C.-H.; Li S.-W.; Chi Y.; Cheng Y.-M.; Yeh Y.-S.; Chou P.-T.; Lee G.-H.; Wang C.-H.; Shu C.-F. Heteroleptic Cyclometalated Iridium(III) Complexes Displaying Blue Phosphorescence in Solution and Solid State at Room Temperature. Inorg. Chem. 2005, 44 (22), 7770–7780. 10.1021/ic050311g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.