

Abstract
The development of stable, efficient chemoselective self-immolative systems, for use in applications such as sensors, requires the optimization of the reactivity and degradation characteristics of the self-immolative unit. In this paper, we describe the effect that the structure of the reporter group has upon the self-immolative efficacy of a prototype system designed for the disclosure of electrophilic alkylating agents. The amine of the reporter group (a nitroaniline unit) was a constituent part of a carbamate that functioned as the self-immolative unit. The number and position of substituents on the nitroaniline unit were found to play a key role in the rate of self-immolative degradation and release of the reporter group. The position of the nitro substituent (meta- vs para-) and the methyl groups in the ortho-position relative to the carbamate exhibited an influence on the rate of elimination and stability of the self-immolative system. The ortho-methyl substituents imparted a twist on the N–C (aromatic) bond leading to increased resonance of the amine nitrogen’s lone pair into the carbonyl moiety and a decrease of the leaving character of the carbamate group; concomitantly, this may also make it a less electron-withdrawing group and lead to less acidification of the eliminated β-hydrogen.
Introduction
Stimuli-responsive compounds,1−3 such as self-immolative molecules and polymers,4−7 have become significant targets in organic-based material development as systems of this type offer enormous potential in a diverse range of applications that span drug delivery,8−10 biological and chemical sensors,11,12 diagnostics,13 and degradable polymers or degrade-on-demand adhesives.14−19 The seminal report by Katzenellenbogen and co-workers,20 based upon the “prodrug” concept discussed by Albert,21 set in place the key molecular design for self-immolative materials that have been explored and refined by several groups in recent years, one in which a substrate-specific trigger is coupled via a degradable linker group to a reporter moiety. Activation or deprotection of the trigger moiety, by either nucleophiles4 or electrophiles,22−24 renders the immolative linker unit labile via generation of an electron-rich center that then initiates a cascade of electrons and culminates in the release of the reporter group from the linker group. When designing a self-immolative system for a specific application, the delicate balance of reactivity and stability of each of the key components (i.e., trigger, linker, reporter) must be assessed and optimized. The system must feature a selective trigger unit to react with the desired substrate; equally, the linker to the reporter group must be sufficiently stable to withstand unwanted side reactions (such as environment-induced degradation) that can lead to “false-positive” results. The effect of the structure of the linker group upon self-immolative pathway and degradation rates has been shown to be pivotal in this design rationale.25,26 In a detailed study, Phillips and co-workers have elegantly shown27 the effect that the structure of aromatic self-immolative linkers has on a self-immolative process and were able to tune the controlled release of phenols under neutral conditions.
In a recent communication,23 we reported the first examples of selective solution-phase self-immolative systems triggered by a nonacidic electrophilic species, such as methyl, allyl, and benzylic halides, to afford a facile colorimetric visual disclosure of toxic electrophilic alkylating agents (Scheme 1).
Scheme 1. Self-Immolative System Triggered by Nonacidic, Electrophilic Alkylating Agents in the Presence of a Hindered Base.

These systems, based upon the rarely utilized carbamate-based 2-(methylthio)ethoxycarbonyl (Mteoc) protecting group developed28 by Kunz and the phosphorus analogue advanced by Verducci et al.,29 afforded a visual colorimetric response to such electrophiles following exposure to mild, basic conditions. A diphenylphosphine nucleophile was used as it offered the best balance between reactivity toward electrophiles while also offering reasonable stability toward aerobic oxidation. Recently, a related approach for detection of alkylating agents has been reported using a Förster resonance energy-transfer (FRET)-based profluorescent probe.24 In this system, alkylation of the probe results in the formation of a quaternary ammonium salt, which increases its leaving group capacity allowing the probe to facilitate the 1,6-elimination of the self-immolative process. When the elimination proceeds, the fluorophore is no longer quenched and can emit a fluorescence signal.
To the best of our knowledge, a systematic study on the effect of the structure of the reporter group (red moiety in Scheme 1) on self-immolative processes, embracing conformational and basicity variations, has not been reported in the literature. Indeed, in Katzenellenbogen’s seminal paper,20 and in a later paper by Shabat,4b the nature of the reporter group was considered to have only a limited impact on the rate of self-immolative elimination of their carbamate systems. A related observation was made in respect of carbonate linkers highlighted by Schmidt and Jullien; interestingly, consideration was also given to conformational effects in the leaving group.4c Hay has varied the substituent in the para-position of an aniline reporter group that fragmented from a 4-hydroxylaminobenzyl alcohol linker, observing that the extent of release was not affected by its leaving group ability, but rather by its subsequent reactions.4d In this paper, we describe a systematic study that reveals valuable information on the role of the reporter group in relation to how this key component influences the self-immolative molecule’s stability and degradation rate.
Results and Discussion
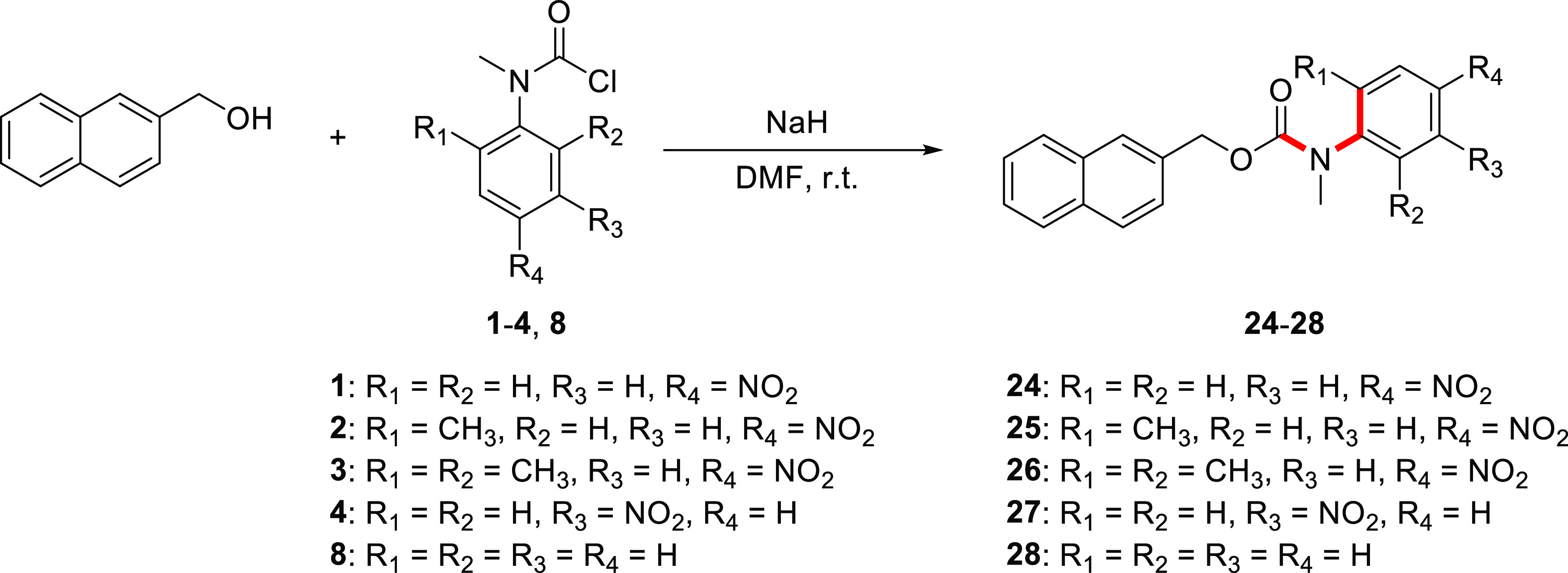
In view of employing self-immolative systems in real-world environments for the specific disclosure of toxic alkylating agents, it is important that these materials possess suitable shelf-lives and are resistant toward fluctuations in ambient conditions (such as moisture and/or temperature) while stored. We have recently reported a novel self-immolative system23 for the selective disclosure of reactive electrophilic alkylating agents that is stable in solution (>72 h in CD3CN) and when exposed to water (10% D2O in CD3CN). However, disappointingly, release of the reporter group was observed when stored in the bulk. To study the influence of the reporter group structure on the stability and reactivity of the self-immolative system, a variety of analogue structures were prepared; these analogues have been designed to probe both electronic and steric effects. To this end, carbamoyl chloride precursors 1–9 were first synthesized (see Figure 1).
Figure 1.

Structure of carbamoyl chlorides 1–9.
Carbamoyl chloride 3, substituted by a nitro group in the para-position relative to the carbamoyl group, was synthesized according to the route displayed in Scheme 2.30,31 Protection of the amine group using 4-toluenesulfonyl chloride (TsCl) was carried out prior to nitration to promote regioselective reaction para- to the N-tosylate group. The carbamoyl chloride 3 was then obtained via a two-pot, three-step process; that involved initial N-methylation of the protected aniline followed by deprotection of the tosylate group under acidic conditions to yield 10, which was then converted to the corresponding carbamoyl chlorides using triphosgene (see Figures S1–S4 in the Supporting Information (SI) for analytical data for 10 and 3).
Scheme 2. Synthesis of Carbamoyl Chloride 3.
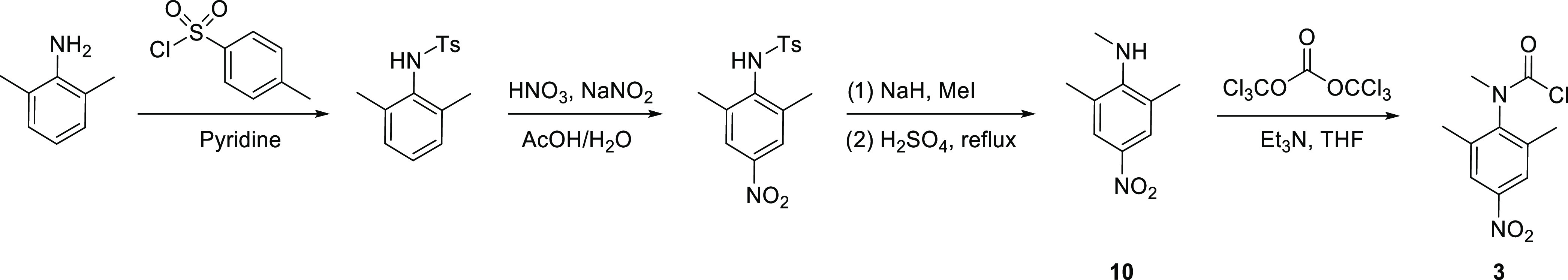
The carbamoyl chlorides 2 and 5–7 were synthesized utilizing an alternative synthetic pathway (Scheme 3). Regioselective nitration of the anilines leading to 5–7 afforded the corresponding meta-derivatives (2-methyl-4-nitroaniline was purchased), and then selective monomethylation was carried out according to the procedure reported by Lebleu et al.,32 using methyl triflate as an alkylating agent in hexafluoroisopropanol (HFIP), affording the desired N-methylated aniline products (11–14) in yields ranging from 68 to 83%. As in the case of 3, the carbamoyl chlorides 2 and 5–7 were then obtained by reaction of the N-methylated aniline derivatives with triphosgene (see Figures S5–S20 in the SI for analytical data for 11–14 and 2, 5–7). The carbamoyl chlorides 1, 4, 8, and 9 were obtained using procedures reported previously (see Figures S21–S28 in the SI for analytical data for 1, 4, 8, and 9).31,33,34
Scheme 3. Synthesis of Carbamoyl Chlorides 2 and 5–7.

The target self-immolative systems were obtained by conjugation of 2-diphenylphosphinoethanol to the N-methyl carbamoyl chloride derivatives 1–9 using 4-dimethylaminopyridine (DMAP) in a catalytic amount and heating under reflux in tetrahydrofuran (THF): this procedure afforded the self-immolative systems 15–23 (Scheme 4; see Figures S29–S60 in the SI for analytical data for 15–23).23
Scheme 4. Synthesis of the Self-Immolative Systems 15–23.
NMR spectroscopic analysis of the different self-immolative systems containing one or two methyl groups ortho- to the carbamate revealed the presence of two rotamers as a result of restricted rotation about their carbamate C–N bonds (see Scheme 5), and it is evident that the rotamer ratios for these disclosure systems were perturbed by changing the solvent (e.g., compound 19; see Figure 2).a In addition, the rotameric ratio increased with the number of electron-donating methyl groups present on the aromatic ring (see Table 1: 19 vs 20 vs 21).35
Scheme 5. Syn–anti Rotamer Equilibrium Caused by the Restricted Rotation of the Carbamate C–N Bond.

Figure 2.
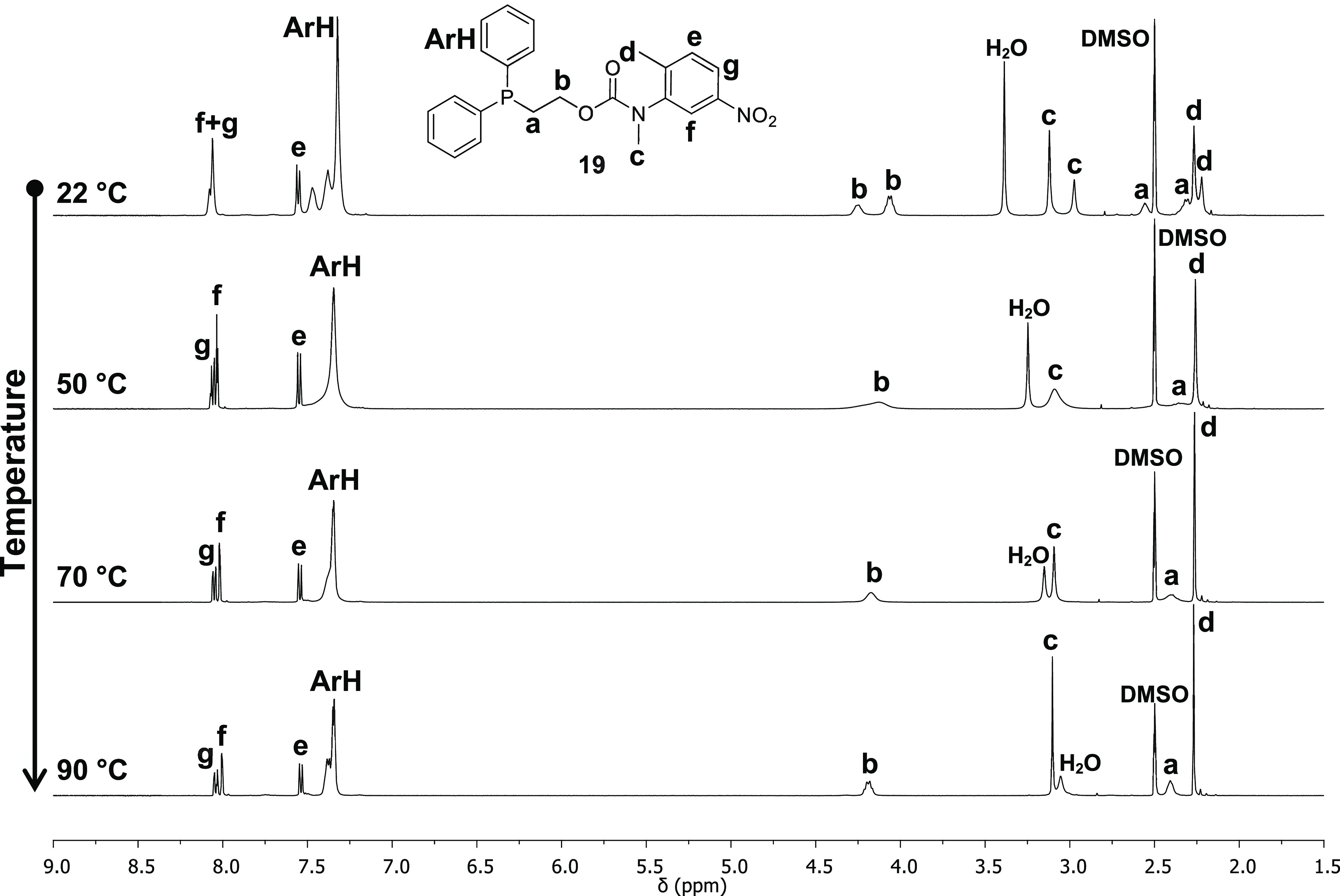
VT 1H NMR spectroscopic analysis of self-immolative system 19: rotamer ratio 62/38 recorded in DMSO-d6 (500 MHz).
Table 1. Rotamer Ratios and Stability Information in Neat/Solution Form for the Self-Immolative Systems 15–23, Determined Using 1H NMR Spectroscopic Analysis (Errors Associated with the Integral Values Were ±5%).

Rotameric ratio is not determined as the coalescence temperature is below 22 °C.
The observed degradation occurs within the first 24 h, after which the physical state of the neat liquid changes to a paste-like consistency.
Since the energy barrier to free rotation of the C–N bonds of the carbamate group can be overcome at elevated temperatures, variable-temperature (VT) 1H and 31P NMR spectroscopic studies were conducted on 16, 19, and 20 to specifically facilitate two pairwise comparisons, 16/19 and 19/20 (see Figures S61–S65). The pair 16/19 was selected to evaluate the relative effect of a para- and meta-positioned nitro group on the double-bond character of the nominally C–N single bond of the carbamate, whereas 19/20 permitted the effect of one vs two ortho-methyl groups to be assessed. These results could then be correlated to the observed stability/reactivity of the systems. Thus, 16 and 19 were analyzed in DMSO-d6 in increments between 25 and 90 °C: Figure 2 shows the 1H NMR spectra obtained for the self-immolative system 19 at four selected temperatures. The resonances at 2.98–3.12 ppm (c, CH3–N), 4.06–4.25 ppm (b, CH2–O), and 8.06 ppm (f and g, CHAr) began to broaden as the temperature was increased and eventually coalesced by ca. 50 °C into two single broad resonances. Meanwhile, the resonances corresponding to the aromatic protons CHArf and g sharpened from a broad signal to a doublet for proton f and a doublet of doublets for proton g. Upon heating to 90 °C, the resonances corresponding to the protons CH3–N and CH2–O sharpened into a singlet and an apparent quartet, respectively. Compound 16 showed similar spectroscopic changes but with coalescence being achieved at ca. 40 °C (see Figures S62 and S63). The determination of the coalescence temperature (Tc), in addition to the value of the rotamer population difference (ΔP) and the limiting chemical shift difference (Δν), obtained from spectra at temperatures below coalescence for both 16 and 19 allowed the calculation of the free energy of activation (ΔG‡) for the rotational equilibrium of 16 and 19 by treating the data as an exchange between two unequally populated sites (see eqs S1–S6 in the SI).36 Lower values of the energy barriers were found for 16 (ΔGC,rotamer A‡ = 65.2 kJ·mol–1 and ΔGC,rotamer B = 64.0 kJ·mol–1, respectively) when compared to 19 (ΔGC,rotamer A‡ = 67.8 kJ·mol–1 and ΔGC,rotamer B = 66.5 kJ·mol–1, respectively), showing the influence of the nitro group position (para- vs meta-) on the carbamate C–N rotation equilibrium. As anticipated, the ability of the nitrogen lone pair to resonate into the carbamate increases when the nitro group was moved from the para- to the meta-position, consistent with the change in coalescence temperature.
In contrast to 19, 20 revealed only broadening of the key resonances (see Figures S64 and S65), without coalescence at 100 °C, indicating a greater resonance of the nitrogen lone pair into the carbamate carbonyl and a larger steric barrier to rotation (ΔGC,rotamer A‡ > 79.3 kJ·mol–1 and ΔGC,rotamer B > 77.3 kJ·mol–1, respectively) when compared to 19. A similar trend was observed by comparing the VT 1H and 31P NMR spectroscopic analysis obtained for 16 and 17 (with the nitro group in the para-position) (see Figures S62–S63 and S66–S67). The reduced resonance into the aromatic ring arises from a substantial torsion angle between the ring and the carbamate group (Figure 3).
Figure 3.
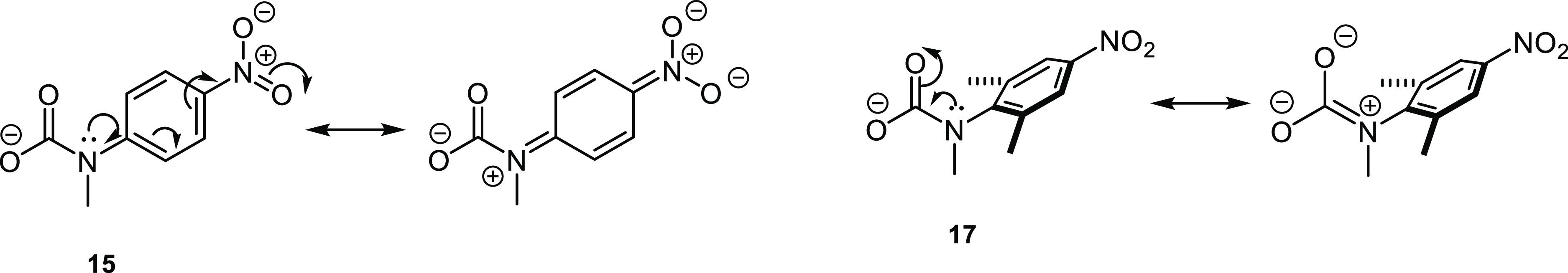
Resonance structures contributing to the stability of the leaving groups of 15 and 17.
To investigate the thermal stability of these self-immolative systems, the solutions were then cooled down from 100 to 25 °C and the sharp, well-resolved resonances observed at an elevated temperature for 16, 17, 19, and 20 returned to their original shape, indicating their thermal stability across this temperature range (see Figures S68–S75). In addition, the different self-immolative systems 15–23 were found to be stable at both 0.1 and 0.025 M (reaction conditions for the self-immolative study) solutions in CD3CN over >72 h at 22 °C (see Figures S76–S84). However, a small amount of oxidation to the corresponding phosphine oxide was noted.
The stability of the different self-immolative systems as neat liquids was then investigated (see Table 1). The stability of 15, bearing a nitro group in the para-position relative to the carbamate group, was first assessed. When left to stand at room temperature as a neat liquid, it proved to be unstable and release of the reporter group (N-methyl-para-nitroaniline) was observed, with 32% degradation evident after 24 h (see Figure S85). Lowering the storage temperature to −20 °C was ineffective in preventing degradation. The instability of 15 was attributed to the structure of the N-methyl-para-nitroaniline reporter unit; in particular, the resonance of the nitrogen lone pair into the aromatic ring and nitro group rendered the carbamate less electron-rich and more electrophilic. In contrast, the self-immolative system 18, with the nitro group in the meta-position (wherein its influences are limited to inductive and field effects), proved more stable. However, while the stability of 18 improved relative to 15, the observed release of 4% of the reporter group (N-methyl-meta-nitroaniline) over 72 h would still risk a false-positive response if used in real-world applications (see Figure S86). Removal of the nitro group afforded phenyl carbamate 22 that proved to be stable under identical conditions (see Figure S87). These observations indicated the influence of the presence of the nitro group on the carbamate unit stability. However, as a colorimetric response is required for visual disclosure, the presence of the nitro group was essential in the reporter moiety. Therefore, an alternative route to enhance the stability of the self-immolative system was investigated that involved the introduction of methyl groups in the ortho-position of the reporter unit (relative to the carbamate linkage). Introduction of the methyl groups in the ortho-position of the reporter group should impart a twist on the N–C (aromatic) bond, reducing the ability of the lone pair of the carbamate nitrogen to resonate into the aromatic ring (Figure 3). Indeed, resonance into a carbamate anion was noted by Johnson as significant in determining the pKa of its conjugate acid.37
When added to the increase in steric size of the reporter group, this should have the effect of reducing the carbamate’s electrophilicity. Regarding the self-immolative system 16, featuring a single ortho-methyl group and the nitro group in the para-position, relative to the carbamate group, a decrease of degradation rate was observed when compared to 15. However, after 72 h, a similar amount of reporter group release was observed for 16 and 15 (see Figures S85 and S88). In contrast, regarding the self-immolative system 19, with the nitro group located in the meta-position and a single methyl group in the ortho-position, relative to the carbamate group, degradation was not observed after 72 h (see Figure S89). In the cases of the self-immolative systems 17, 20, 21, and 23, substituted by two ortho-methyl groups, degradation was not observed at room temperature after 72 h (see Figures S90–S93). These results highlight the strong influence of the presence of the methyl groups in the ortho-position, relative to the carbamate group, on the stability of the self-immolative system.
These effects will also bear upon the ability of the carbamate to function as a leaving group in the manner identified in Schemes 1 and 8 and Figure 5. However, it is worth noting that such effects will be attenuated as compared to the leaving group ability of the aniline itself. Johnson has noted that while the pKaH+ of the amine in a carbamate can vary by as much as 8 pKa units, the corresponding carbamic acids vary by around 1 pKa unit.37 However, though the differences are attenuated, they do vary in the same direction as the amine pKaH+.
Scheme 8. Degradation of Alkylated Self-Immolative Systems 15a–23a using 2 molar equiv of DIPEA as a Base in CD3CN.

Figure 5.

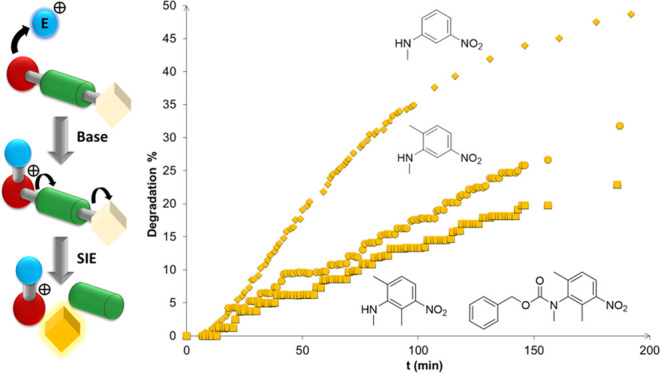
Top: reaction schematic of the self-immolative degradation of disclosure systems 18–20. Bottom: reaction kinetics calculated from the 1H NMR spectra (20 °C) obtained following the addition of 10 molar equiv of benzyl bromide to 18–20 and N,N-diisopropylethylamine (DIPEA) in a one-pot approach (18–20, blue diamonds; 18a–20a, red squares; reporter groups, yellow triangles). Errors associated with the integral values for these 1H NMR studies were ±5%.
To quantify the torsion angle imparted on the CCNC moiety, as highlighted in red (Scheme 6), the model compounds 24–28 that possess similar structures to the self-immolative systems 15, 16, 17, 18, and 22 were synthesized. 2-Naphthalenemethanol was selected as a result of its crystalline nature. Crystalline carbamates were obtained via the conjugation of 2-naphthalenemethanol to N-methyl-carbamoyl chloride derivatives 1–4 and 8 using sodium hydride (NaH) in dimethylformamide (DMF) (see Figures S94–S103).
Scheme 6. Synthesis of the Crystalline Carbamate Model Compounds 24–28.
The solid-state structures of 22–28 were obtained using X-ray crystallographic analysis. Partial structures are shown in Figure 4 to emphasize the CCNC torsion angle (for full structures, see Figures S104–S110 and Tables S2–S8).b
Figure 4.

Partial solid-state structures of model compounds 22–28.
The analysis showed no significant changes in geometry when the nitro group was located in either the para- or the meta-position, with torsion angles of 41(1) and 44(1)° for 24 and 27, respectively. However, when the nitro group was not present (e.g., 28), a higher torsion angle of 57(1)° was observed. In the cases of 24 and 27, the torsion angles reflect the ability of the CCNC linkage to adopt a more planar geometry as a consequence of electronic effects when the electron-withdrawing nitro group is present. These results are in good agreement with the stability profiles observed for the self-immolative systems 15, 18, and 22 in their neat liquid form. In contrast to 24 and 27, the solid-state structures of 25 and 26 that possess one and two ortho-methyl groups, respectively, revealed an increase in the torsion angle to 72(1)° for 25 and 81(1) or 88(1)° for 26 (two conformers were evident in the unit cell). Assuming that the solid-state data of the model carbamates translates to the self-immolative system in their liquid form, the additional twist imparted on the N–C (aromatic) bond by the presence of ortho-methyl groups should lead to a reduction in the electrophilicity of the carbamate linker. These effects could serve to explain the improved stability, in their neat liquid forms, of the self-immolative systems 17 when compared to that of 15 as well as the improved stability of 19 and 20 when compared to that of 18.
The solid-state structures of the self-immolative systems 22 and 23 were also obtained. The CCNC torsion angle for 23 (that features two ortho-methyl groups but no nitro residue) was determined to be 73(1) or 78(1)° (two conformers were evident in the unit cell), comparable in value relative to that of the model carbamate 26 (81(1) or 88(1)°). However, the CCNC torsion angle of the phenyl carbamate 22 (78(1)°) was found to be higher than expected when compared to that of the model compound 28; the difference between the torsion angles values could be attributed to crystal packing forces involving the planar aromatic units.
To assess the ability of the different self-immolative systems to disclose the presence of reactive electrophilic alkylating agents via a two-step process involving alkylation followed by elimination/decarboxylation (Scheme 1), the reactivity of 15–23 toward alkylation was first investigated, using benzyl bromide as the alkylating agent (Scheme 7).38,39
Scheme 7. Alkylation of Self-Immolative Systems 15–23 Using BnBr as an Alkylating Agent in CD3CN.

Cognizant that these are type II SN2 reactions,38,39 alkylation of self-immolative systems 15–23 was conducted by dissolution of the self-immolative system in the polar aprotic solvent CD3CN, followed by the addition of 10 molar equiv of benzyl bromide and recording the 1H NMR spectra at regular time intervals at 20 °C (see Figures S111–S119). Following analysis of the reactions under these pseudo-first-order conditions, it was found that the introduction of different substituents on the aromatic ring of the reporter moiety had no dramatic influence on the alkylation rate, with half-life times (t1/2) measured between 10.4 and 28.6 min (see Table 2 and Figure S120). Given that there is a developing positive charge at the phosphorus center, during alkylation, it is understandable that self-immolative systems with otherwise equivalent reporter groups, but with the nitro group meta- to the carbamate alkylate faster than those with it in the para-position, e.g., 18 vs 15. Incorporation of ortho-methyl groups relative to the carbamate slowed the rate of alkylation, e.g., 18 vs 19, vs 20. Also evident was the slower alkylation of the nitro-substituted aromatic derivatives in comparison to those systems without nitro substituents, e.g., 22 vs 18. Whether this arises purely by a charge–dipole interaction between the developing phosphonium center and the carbamate, or a more specific alignment of the developing σ*(C–P) orbital is not clear.
Table 2. Alkylation Rate Data Obtained Following the Addition of Benzyl Bromide to the Self-Immolative Systems 15–23 and β-Elimination Rate Data Obtained Following the Addition of 2 molar equiv of DIPEA to Alkylated Self-Immolative Systems 15a–23a Calculated Using 1H NMR Spectroscopic Data at 20 °C (Errors Associated with the Integral Values were ±5%).

Percentage of β-elimination after 24 h.
The β-elimination of the alkylated systems 15a–23a was then performed using 2 molar equiv of N,N-diisopropylethylamine (DIPEA) (see Figures S121–S131). All compounds underwent the expected elimination to produce the reporter unit and CO2 (Scheme 8). When the elimination of 21 was conducted in D2O/CD3CN, at partial conversion, no incorporation of deuterium was observed (1H NMR) in the PCH2CH2 resonance of residual 21 or the vinyl group of phosphonium salt 29. However, both species were deuterated at the benzylic methylene. This limits the mechanism to either E2, E1cB (anion), or E1cB (irreversible).
In contrast to the alkylation profiles, the rate of elimination dramatically changed with respect to the structure of reporter groups (see Table 2). The highest elimination rates were observed for the reporter groups most able to withdraw electron density from the carbamate nitrogen, thus affording both the best leaving group and, concomitantly, acidifying the β-hydrogen removed during the elimination reaction. Therefore, for equivalently substituted reporter groups bearing the same substituents but located in different positions on the aromatic ring, a para-nitro substituent is more effective than a meta-substituent, e.g., 15a vs 18a, whereas additional methyl groups slowed the elimination, e.g., 18a vs 19a vs 20a. In addition, these methyl groups are modestly electron donating and would be expected to restrict solvation of the leaving group. Other solvents were investigated to enhance the sensitivity of the alkylation and β-elimination pathways using 19 (see Figures S132–S136). The rate of alkylation varied with the solvent, with the observed order being DMSO-d6 > 10% MeOD/CD3CN > CD3CN > DMF; however, β-elimination was observed at a respectable rate in CD3CN in comparison to DMSO-d6 and DMF-d7. The variation in the rate of elimination in these three solvents may relate to changes in the pKaH+ for iPr2EtNH+ in them. For example, pKaH+ Et3NH+ (CH3CN) > pKaH+ Et3NH+ (DMSO).40
The rate of elimination was enhanced using 10% MeOD as a cosolvent with CD3CN in comparison to neat CD3CN;27 however, for consistency within these studies, alkylation and elimination experiments were conducted in CD3CN.
In addition, under the pseudo-first-order conditions used, depending on the reporter group structure, it was observed that post degradation of the self-immolative system, alternative alkylation pathways could occur involving the excess benzyl bromide present. Therefore, three distinct reporter group categories have been determined: (i) partial alkylation of the carbamate leaving group was observed for the reporter groups featuring two ortho-methyl groups leading to the formation of Cbz-protected amines (observed for 17, 20, 21, and 23; see Figures S123, S126–S127, S129, and S130), (ii) full N-alkylation of the reporter group was observed for 22 after the release of N-methylaniline (see Figures S128 and S131), and (iii) in the case of reporter groups possessing either one or no ortho-methyl groups and a nitro substituent, alkylation was not observed (15, 16, 18, and 19; see Figures S121–S122 and S124–S125). As a result of increasing the steric bulk of the reporter groups (case (i): 17, 20, 21, and 23), the lifetime of the carbamate anion generated upon initial degradation was sufficient to permit its alkylation. In the case of (ii), the increased nucleophilicity of N-methylaniline released from 22a led to rapid N-alkylation with the excess benzyl bromide present. The optimum degradation pathway was obtained in the case of (iii), whereby the reporter units featured a nitro substituent and either a hydrogen or a single methyl group in the ortho-position. Since slow decarboxylation and subsequent alkylation lead to loss of the color of the reporter group, this is an important design point for this self-immolative system.
In the light of the stability of 19 in both the neat and solution forms, taken together with its reactivity profile, this self-immolative system was selected for degradation studies, whereby the disclosure system and base were present and then exposed to the alkylating agent. As a result, a one-pot alkylation–elimination sequence was carried out by mixing system 19 with 2 molar equiv of DIPEA before the addition of benzyl bromide. From the plot shown in Figure 5, it can be observed that after addition of 10 molar equiv of benzyl bromide the reporter group N-methyl-2-methyl-5-nitroaniline was released, demonstrating the viability of this one-pot alkylation–elimination process (see Figure S137 and Table S10). Furthermore, an intense yellow coloration was evident within 15 min following the addition of the alkylating agent. In addition, identical degradation studies conducted on self-immolative systems 18 and 20 confirmed the viability of the one-pot alkylation–elimination process as well as the relative reactivities of these compounds relative to 19 (e.g., 18 > 19 > 20; see Figures S138–S139 and Tables S9 and S11). In addition to these degradation studies, self-immolative system 19 was further studied via UV–visible spectroscopy, utilizing a solution of alkylated 19 and in relation to an external calibration plot. In an analogous one-pot study, the concentration of both 19 and benzyl bromide was decreased 10-fold and the release of the reporter group was monitored with respect to time (see Figures S140–S143).
Conclusions
In summary, a range of selective self-immolative systems, triggered by a nonacidic electrophilic alkylating agent to afford a facile colorimetric visual disclosure, have been synthesized. The effect of the structure of the reporter group upon the rate of degradation and stability of the compounds has been determined. A significant influence on both the stability and the rate of elimination was determined in relation to the position of the nitro substituent (meta- vs para-) on the aniline reporter group. In addition, both the stability and reactivity of the self-immolative system could be tuned by the introduction of methyl groups in the ortho-position relative to the carbamate moiety of the reporter group. The ortho-methyl groups were found to have imparted an increased twist on the N–C (aromatic) bond, which led to a reduction of the ability of the carbamate nitrogen’s lone pair to resonate within the aromatic ring, resulting in a decrease of the leaving character of the carbamate group. Additionally, this should render the carbamate less electron withdrawing and so less able to acidify the β-hydrogen undergoing elimination. This study revealed that the structural modifications to the reporter group had a modest influence on the alkylation rate, with similar half-lives being observed for the benzylation of self-immolative systems 15–23. In contrast, the β-elimination rates were affected by both the position of the nitro group on the aromatic ring (pKa) and the introduction of ortho-methyl groups relative to the carbamate group (conformation). The position and nature of the substituents on the reporter group have led to these being divided into three groups, depending on their fate post release. Additionally, the rate of the present 1,2-elimination appears to be more substantially affected by the nature of the reporter group than the original 1,6-elimination reported by Katzenellenbogen, and by Hay and 1,4- and 1,6- examples reported by Shabat.4b,20,4d From the series of self-immolative disclosure systems synthesized, 19 was selected as the candidate for the disclosure of reactive alkylating agents, as it exhibited the optimum balance between stability and reactivity. Owing to the potential of these self-immolative systems to offer a practical route for the selective disclosure of reactive electrophilic species, such as chemical warfare agents, outside of the laboratory environment and without need for instrumentation, further studies are currently underway to produce clear and robust disclosure events for a broader range of electrophiles.
Experimental Section
General Information
All chemical reagents were purchased from Sigma-Aldrich and used as received, without purification. 2-Diphenylphosphinoethanol41,42 and N-(2,6-dimethyl-4-nitrophenyl)-4-methylbenzenesulfonamide30 were synthesized by modifications of literature procedures. Solvents were purchased from Fisher Scientific except for ethyl acetate and hexane, which were purchased from Sigma-Aldrich. All solvents were used as supplied except for THF that was distilled under argon from sodium and benzophenone prior to use. Fisher Scientific Silica 60A (particle size 35–70 μm) was used to perform column chromatography. Thin-layer chromatography (TLC) was performed on aluminum sheets coated with Merck 5735 Kieselgel 60 F254. Developed plates were air-dried and stained using a potassium permanganate solution. 1H (400 MHz), 31P{1H} (162 MHz), and 13C{1H} (100 MHz) NMR spectra were recorded at 20 °C on a Bruker Nanobay 400 MHz (9.39 T) or a Bruker DPX 400 (9.39 T) instrument. 1H (500 MHz), 31P{1H} (203 MHz), and 13C{1H} (125 MHz) variable-temperature NMR (VT-NMR) were recorded on a Bruker Avance III 500 MHz instrument (11.74 T). This instrument was calibrated at installation with ethylene glycol with an associated error of ±0.1 °C. 1H NMR spectra recorded in CDCl3 were referenced to tetramethylsilane (TMS) as the internal standard, whereas those recorded in CD3CN, DMF-d7, or DMSO-d6 were referenced to residual solvent. Chemical shifts (δ) are reported in parts per million (ppm) from low to high field. Coupling constants (J) are reported in hertz (Hz). Standard abbreviations indicating multiplicity are used as follows: br. = broad, s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, and app. = apparent. Fourier transform infrared (FTIR) spectra were recorded on a PerkinElmer Spectrum FTIR directly using a diamond ATR sampling accessory. Mass spectrometry was conducted using a ThermoFisher Scientific Orbitrap XL LCMS. The sample was introduced by liquid chromatography, and sample ionization was achieved by electrospray ionization (ESI). Elemental microanalyses of 2, 3, 6, and 7 were performed by MEDAC Ltd. Melting points were recorded using a Stuart MP10 melting point. UV–visible spectra were measured with a Varian Cary 300 spectrophotometer using a 10 mm inner width Quartz cuvette in the wavelength range of 250–800 nm.
Crystals of 22, 23, 24, 25, 26, 27, and 28 were mounted under paratone-N oil and flash-cooled to either 100 K (22, 23, 26, 27, 28) or 150 K (24, 25) under nitrogen in an Oxford Cryosystems Cryostream. Single-crystal X-ray intensity data were collected using either an Agilent Gemini S Ultra diffractometer (Mo Kα radiation (λ = 0.71073 Å)) (24, 25) or a Rigaku XtaLAB Synergy diffractometer (Cu Kα radiation (λ = 1.54184 Å)) (22, 23, 26, 27, 28). The data were reduced within CrysAlisPro software.43 The structures were solved using the program Superflip,44 and all nonhydrogen atoms were located. Least-squares refinements on F were carried out using the CRYSTALS suite of programs.45 The nonhydrogen atoms were refined anisotropically. All of the hydrogen atoms were located in difference Fourier maps and then placed geometrically with a C–H distance of 0.95 Å and a Uiso of 1.2 times the value of Ueq of the parent C atom. The hydrogen atoms attached to C were then refined with riding constraints. The crystallographic details for compounds 22–28 are presented in the Supporting Information.
Synthesis of N,2,6-Trimethyl-4-nitroaniline (10)
N-(2,6-Dimethyl-4-nitrophenyl)-4-methylbenzenesulfonamide30 (4.0 g, 12.49 mmol) was dissolved in THF (20 mL) and cooled to room temperature. Upon cooling, 60% sodium hydride dispersion in mineral oil (0.55 g, 13.73 mmol) was added followed by iodomethane (1.17 mL, 18.73 mmol) and left to stir overnight. The volatiles were removed in vacuo to yield a white crystalline solid and used without further purification. N-(2,6-Dimethyl-4-nitrophenyl)-N-4-dimethylbenzenesulfonamide was dissolved in H2SO4 (15 mL) and water (1 mL) and warmed at 40 °C (using an aluminum heating block) for 16 h. The reaction mixture was poured slowly into an ice/water/NaOH mixture. This was extracted with ethyl acetate, dried over MgSO4, filtered, and the solvent was removed in vacuo to yield yellow crystals (1.89 g, 84%). Mp 90–92 °C. 1H NMR (CDCl3, 400 MHz) δ 7.86 (s, 2H), 3.88 (br. s, 1H), 3.06 (s, 3H), 2.33 (s, 6H). 13C{1H} NMR (CDCl3, 100 MHz) δ 153.8, 139.4, 125.4, 124.9, 24.9, 19.6. FTIR (ATR/νmax) 3425, 2920, 2383, 1592, 1525, 1421, 1352, 1254, 1235, 1153, 1107, 1029 cm–1. HRMS (ESI) m/z: [M + H]+ calcd for C9H13O2N2 181.0972; found 181.0972.
General Procedure 1: Synthesis of N-Methylated Anilines (11–14)
To a solution of HFIP (hexafluoroisopropanol) (10 equiv) and aniline (1 equiv) was added methyl trifluoromethanesulfonate (MeOTf) (1.5 equiv). The mixture was stirred for 1 h at room temperature and then quenched by a solution of 2 N HCl. Volatiles were evaporated in vacuo. The resulting mixture was neutralized with a saturated aqueous solution of NaHCO3 and extracted with CH2Cl2 (3 × 30 mL). The organic phases were dried over MgSO4, filtered, and the solvent was removed in vacuo. The crude product was further purified by column chromatography.
Synthesis of N,2-Dimethyl-4-nitroaniline (11)
Following general procedure 1, 2-methyl 4-nitroaniline (2.0 g, 13.14 mmol) was treated with HFIP (13.8 mL, 131.45 mmol) and MeOTf (2.16 mL, 19.72 mmol) to provide 11 (1.58 g, 72%) as a yellow solid after silica-gel column chromatography (THF/hexane 10/90). Mp 138–140 °C. 1H NMR (CDCl3, 400 MHz) δ 8.09 (dd, 3J = 9.0 Hz, 4J = 2.5 Hz, 1H), 7.99–7.96 (d, 4J = 2.5 Hz, 1H), 6.53 (d, 3J = 9.0 Hz, 1H), 4.37 (br. s, 1H), 3.00 (d, 3J = 5.0 Hz, 3H), 2.17 (s, 3H). 13C{1H} NMR (CDCl3, 100 MHz) δ 152.6, 137.6, 126.0, 124.9, 121.0, 107.4, 30.5, 17.3. FTIR (ATR/νmax) 3378, 2908, 1608, 1587, 1541, 1486, 1463, 1407, 1386, 1290, 1261, 1189, 1118, 1094, 1036, 1001 cm–1. HRMS (ESI) m/z: [M + H]+ calcd for C8H11O2N2 167.0815; found 167.0810.
Synthesis of N,2-Dimethyl-5-nitroaniline (12)
Following general procedure 1, 2-methyl-5-nitroaniline (2.0 g, 13.14 mmol) was treated with HFIP (13.8 mL, 131.45 mmol), and MeOTf (2.16 mL, 19.72 mmol) to provide 12 (1.49 g, 68%) as a yellow solid after silica-gel column chromatography (THF/hexane 10/90). Mp 110–112 °C. 1H NMR (CDCl3, 400 MHz) δ 8.31 (dd, 3J = 8.0 Hz, 4J = 2.5 Hz, 1H), 7.37 (d, 4J = 2.5 Hz, 1H), 7.13 (d, 3J = 8.0 Hz, 1H), 3.85 (br. s, 1H), 2.96 (d, 3J = 4.0 Hz, 3H), 2.19 (s, 3H). 13C{1H} NMR (CDCl3, 100 MHz) δ 148.0, 147.9, 130.0, 129.3, 111.9, 103.1, 30.7, 17.7. FTIR (ATR/νmax) 3429, 2923, 2801, 1621, 1524, 1498, 1336, 1284, 1162, 1093, 1067 cm–1. HRMS (ESI) m/z: [M + H]+ calcd for C8H11O2N2 167.0815; found 167.0816.
Synthesis of N,2,6-Trimethyl-3-nitroaniline (13)
Following general procedure 1, 2,6-dimethyl-3-nitroaniline (2.0 g, 12.04 mmol) was treated with HFIP (12.64 mL, 120.35 mmol) and MeOTf (1.97 mL, 18.05 mmol) to provide 13 (1.67 g, 77%) as a yellow oil after silica-gel column chromatography (THF/hexane 10/90). 1H NMR (CDCl3, 400 MHz) δ 7.39 (d, 3J = 8.5 Hz, 1H), 7.07 (d, 3J = 8.5 Hz, 1H), 3.23 (br. s, 1H), 2.80 (s, 3H), 2.42 (s, 3H), 2.33 (s, 3H). 13C{1H} NMR (CDCl3, 100 MHz) δ 150.2, 149.5, 134.4, 128.3, 124.3, 117.6, 35.5, 18.9, 14.9. FTIR (ATR/νmax) 3407, 2953, 1599, 1465, 1376, 1293, 1260, 1229, 1144, 1158, 1013 cm–1. HRMS (ESI) m/z: [M + H]+ calcd for C9H13O2N2 181.0972; found 181.0970.
Synthesis of N,2,4,6-Tetramethyl-3-nitroaniline (14)
Following general procedure 1, 2,4,6-trimethyl-3-nitroaniline (2.0 g, 11.10 mmol) was treated with HFIP (11.66 mL, 110.98 mmol) and MeOTf (1.82 mL, 16.65 mmol) to provide 14 (1.79 g, 83%) as a yellow solid after silica-gel column chromatography (THF/hexane 10/90). Mp 59–60 °C. 1H NMR (CDCl3, 400 MHz) δ 6.89 (app. s, 1H), 2.98 (br. s, 1H), 2.75 (s, 3H), 2.27 (s, 3H), 2.20 (s, 6H). 13C{1H} NMR (CDCl3, 100 MHz) δ 151.6, 146.5, 131.8, 130.7, 122.3, 121.2, 35.6, 18.4, 16.9, 13.3. FTIR (ATR/νmax) 3407, 2930, 1582, 1507, 1476, 1432, 1377, 1356, 1309, 1281, 1249, 1216, 1151, 1063, 1038, 1013 cm–1. HRMS (ESI) m/z: [M + H]+ calcd for C10H15O2N2 195.1128; found 195.1123.
General Procedure 2: Synthesis of Carbamoyl Chlorides (1–9)
To a solution of triphosgene (2 equiv) in dry hexane (10 mL), aliquat 336 (0.5 equiv) was added and left to stir at room temperature for 16 h. A solution of the N-methylated aniline derivative (1 equiv) and triethylamine (2 equiv) in THF (20 mL) was added dropwise to the solution at 0 °C. After stirring the solution at room temperature for 2 h, the precipitate was filtered and the solvent was removed in vacuo. The crude product was further purified by column chromatography.
Synthesis of Methyl(4-nitrophenyl)carbamic Chloride (1)31
Following the general procedure 2, N-methyl-4-nitroaniline (0.77 g, 5.05 mmol) was treated with triphosgene (3.00 g, 10.11 mmol), aliquat 336 (1.00 g, 2.53 mmol), and triethylamine (1.41 mL, 10.11 mmol) to provide 1 (0.91 g, 84% yield) as a white solid after silica-gel column chromatography (10 → 20% EtOAc/hexane) followed by a recrystallization from EtOAc/hexane (9/1 v/v). Mp 101–103 °C. 1H NMR (CDCl3, 400 MHz) δ 8.31 (app. d, 2H), 7.48 (app. d, 2H), 3.48 (s, 3H). 13C{1H} NMR (CDCl3, 100 MHz) δ 143.3, 142.9, 141.6 (br.), 122.5, 119.7, 35.3 (br.). FTIR (ATR/νmax) 3111–3081, 1726, 1594, 1512, 1495, 1418, 1342, 1255 cm–1.
Synthesis of Methyl(2-methyl-4-nitrophenyl)carbamic Chloride (2)
Following the general procedure 2, 11 (0.84 g, 5.05 mmol) was treated with triphosgene (3.00 g, 10.11 mmol), aliquat 336 (1.00 g, 2.53 mmol), and triethylamine (1.86 mL, 13.30 mmol) to provide 2 (0.89 g, 76% yield) as a white solid with an 83:17 mixture of rotamers A/B after silica-gel column chromatography (10 → 20% EtOAc/hexane). Mp 85–87 °C. 1H NMR (CDCl3, 400 MHz) δ 8.23–8.17 (s, 1H, rotamer A + rotamer B), 8.17–8.10 (m, 1H, rotamer A + rotamer B), 7.37 (app. d, 1H, rotamer A + rotamer B), 3.46 (s, 3H, rotamer B), 3.32 (s, 3H, rotamer A), 2.42 (s, 3H rotamer A), 2.38 (s, 3H rotamer B). 13C{1H} NMR (CDCl3, 100 MHz) δ 149.1, 148.5, 147.7, 147.4, 147.1, 147.0, 138.1, 137.3, 129.4, 127.9, 126.6, 122.9, 122.7, 41.3, 39.0, 17.9, 17.8. FTIR (ATR/νmax) 3081–2932, 1724, 1586, 1529, 1513, 1491, 1344, 1304, 1252, 1113, 1085 cm–1. Anal. calcd for C9H9ClN2O3: C, 47.28; H, 3.97; N, 12.25. Found: C, 46.97; H, 3.75; N, 11.97.
Synthesis of (2,6-Dimethyl-4-nitrophenyl)(methyl)carbamic Chloride (3)
Following the general procedure 2, 10 (0.91 g, 5.05 mmol) was treated with triphosgene (3.00 g, 10.11 mmol), aliquat 336 (1.00 g, 2.53 mmol), and triethylamine (1.86 mL, 13.30 mmol) to provide 3 (0.97 g, 72% yield) as a white solid with an 88:12 mixture of rotamers A/B after silica-gel column chromatography (10 → 20% EtOAc/hexane). Mp 124–126 °C. 1H NMR (CDCl3, 400 MHz) δ 8.02 (s, 1H, rotamer A), 8.00 (s, 1H, rotamer B), 3.37 (s, 3H, rotamer B), 3.26 (s, 3H, rotamer A), 2.39 (s, 6H, rotamer A), 2.36 (s, 6H, rotamer B). 13C{1H} NMR (CDCl3, 100 MHz) δ 148.8, 147.4, 146.0, 138.1, 137.2, 124.0, 39.7, 37.3, 18.1 (×2). FTIR (ATR/νmax) 3086–2966, 1744, 1587, 1516, 1473, 1348, 1299, 1238, 1118, 1090 cm–1. Anal. calcd for C10H11ClN2O3: C, 49.50; H, 4.57; N, 11.54. Found: C, 49.13; H, 4.60; N, 11.18.
Synthesis of Methyl(3-nitrophenyl)carbamic Chloride (4)33
Following the general procedure 2, N-methyl-3-nitroaniline (0.77 g, 5.05 mmol) was treated with triphosgene (3.00 g, 10.11 mmol), aliquat 336 (1.00 g, 2.53 mmol), and triethylamine (1.41 mL, 10.11 mmol) to provide 4 (0.75 g, 70% yield) as a white solid after silica-gel column chromatography (10 → 20% EtOAc/hexane). Mp 92–94 °C. 1H NMR (CDCl3, 400 MHz) δ 8.26 (m, 1H), 8.18 (s, 1H), 7.76–7.58 (m, 2H) 3.47 (br. s, 3H). 13C{1H} NMR (CDCl3, 100 MHz) δ 148.8, 143.9, 133.8 (br.), 130.6, 123.3 (br.), 40.4. FTIR (ATR/νmax) 3073, 1727, 1532, 1480, 1418, 1352, 1252 cm–1.
Synthesis of Methyl(2-methyl-5-nitrophenyl)carbamic Chloride (5)34
Following the general procedure 2, 12 (0.84 g, 5.05 mmol) was treated with triphosgene (3.00 g, 10.11 mmol), aliquat 336 (1.00 g, 2.53 mmol), and triethylamine (1.86 mL, 13.30 mmol) to provide 5 (1.01 g, 87% yield) as a colorless oil with an 83:17 mixture of rotamers A/B after silica-gel column chromatography (10 → 20% EtOAc/hexane). 1H NMR (CDCl3, 400 MHz) δ 8.16 (dd, 3J = 8.5 Hz, 4J = 2.5 Hz, 1H rotamer A), 8.11 (dd, 3J = 8.5 Hz, 4J = 2.5 Hz, 1H, rotamer B), 8.09–8.05 (m, 1H, rotamer A + rotamer B), 7.49 (d, 3J = 8.5 Hz, 1H, rotamer A), 7.46 (d, 3J = 8.5 Hz, 1H, rotamer B), 3.48 (s, 3H, rotamer B), 3.33 (s, 3H, rotamer A), 2.40 (s, 3H, rotamer A), 2.36 (s, 3H, rotamer B).13C{1H} NMR (CDCl3, 100 MHz) δ 149.4 (rotamer B), 148.8 (rotamer A), 147.1 (rotamer A), 147.0 (rotamer B), 144.0 (rotamer A), 143.3 (rotamer B), 142.5 (rotamer A), 142.1 (rotamer B), 132.3 (rotamer A), 132.2 (rotamer B), 123.9 (rotamer A), 123.5 (rotamer A), 123.4 (rotamer B), 122.3 (rotamer B), 41.4 (rotamer B), 39.1 (rotamer A), 17.9 (rotamer B), 17.8 (rotamer A). FTIR (ATR/νmax) 2923–2852, 1733, 1520, 1480, 1347, 1325, 1270, 1253 cm–1.
Synthesis of (2,6-Dimethyl-3-nitrophenyl)(methyl)carbamic Chloride (6)
Following the general procedure 2, 13 (1.20 g, 6.67 mmol) was treated with triphosgene (3.95 g, 13.30 mmol), aliquat 336 (1.32 g, 3.33 mmol), and triethylamine (1.86 mL, 13.30 mmol) to provide 6 (1.20 g, 74% yield) as a white solid with an 87:13 mixture of rotamers A/B after silica-gel column chromatography (10% EtOAc/hexane). Mp 64–66 °C. 1H NMR (CDCl3, 400 MHz) δ 7.88 (d, 3J = 8.5 Hz, 1H, rotamer A), 7.84 (d, 3J = 8.5 Hz, 1H rotamer B), 7.30 (d, 3J = 8.5 Hz, 1H, rotamer A + rotamer B), 3.38 (s, 3H, rotamer B), 3.26 (s, 3H, rotamer A), 2.45 (s, 3H, rotamer A), 2.44 (s, 3H, rotamer B), 2.36 (s, 3H, rotamer A), 2.34 (s, 3H, rotamer B). 13C{1H} NMR (CDCl3, 100 MHz) δ 151.5, 146.4, 131.7, 130.5, 122.2, 121.1, 40.0, 37.5, 18.3, 14.2. Anal. calcd for C10H11ClN2O3: C, 49.50; H, 4.57; N, 11.54. found: C, 49.38; H, 4.40; N, 11.38.
Synthesis of Methyl(2,4,6-trimethyl-3-nitrophenyl)carbamic Chloride (7)
Following the general procedure 2, 14 (0.55 g, 2.84 mmol) was treated with triphosgene (1.68 g, 5.67 mmol), aliquat 336 (0.56 g, 1.42 mmol), and triethylamine (0.79 mL, 5.67 mmol) to provide 7 (0.97 g, 72% yield) as a colorless oil with an 88:12 mixture of rotamers A/B after silica-gel column chromatography (10% EtOAc/hexane). 1H NMR (CDCl3, 400 MHz) δ 7.09 (s, 1H, rotamer A), 7.07 (s, 1H, rotamer B), 3.35 (s, 3H, rotamer B), 3.23 (s, 3H, rotamer A), 2.30 (s, 3H, rotamer A), 2.28 (s, 3H, rotamer A), 2.27 (s, 3H, rotamer B), 2.26 (s, 3H, rotamer B), 2.19 (s, 3H, rotamer A), 2.17 (s, 3H, rotamer B). 13C{1H} NMR (CDCl3, 100 MHz) δ 150.9, 149.4, 139.3, 138.4, 137.5, 131.3, 130.2, 129.9, 128.0, 40.1, 37.6, 17.7, 17.4, 12.9. FTIR (ATR/νmax) 2931, 1734, 1524, 1477, 1415, 1366, 1350, 1330, 1304, 1219, 1119, 1058, 1015 cm–1. Anal. calcd for C11H13ClN2O3: C, 51.47; H, 5.10; N, 10.91. Found: C, 51.48; H, 5.11; N, 10.78.
Synthesis of Methyl(phenyl)carbamic Chloride (8)31
Following the general procedure 2, N-methylaniline (0.77 g, 7.19 mmol) was treated with triphosgene (4.27 g, 14.40 mmol), aliquat 336 (1.00 g, 2.53 mmol), and triethylamine (2 mL, 14.4 mmol) to provide 8 (0.91 g, 84% yield) as a white solid after silica-gel column chromatography (10% EtOAc/hexane) followed by a recrystallization from hexane. Mp 88–90 °C. 1H NMR (CDCl3, 400 MHz) δ 7.50–7.32 (m, 3H), 7.32–7.18 (m, 2H), 3.35 (br. s, 3H). 13C{1H} NMR (CDCl3, 100 MHz) δ 149.2, 143.3, 129.6, 128.5, 127.4, 40.4. FTIR (ATR/νmax) 2942, 1727, 1598, 2587, 1497, 1454, 1418, 1360, 1329, 1263, 1160, 1113, 1071, 1034, 1018 cm–1.
Synthesis of Mesityl(methyl)carbamic Chloride (9)33
Following the general procedure 2, N-methyl-2,4,6-trimethylaniline (1.0 g, 6.70 mmol) was treated with triphosgene (3.98 g, 13.40 mmol), aliquat 336 (1.65 g, 3.34 mmol), and triethylamine (1.9 mL, 13.62 mmol) to provide 9 (1.10 g, 50% yield) as a white solid with a 90:10 mixture of rotamers A/B after silica-gel column chromatography (10% EtOAc/hexane) followed by a recrystallization from hexane. Mp 73–75 °C. 1H NMR (CDCl3, 400 MHz) δ 6.94 (m, 2H rotamer A), 6.92 (m, 2H, rotamer B), 3.34 (s, 3H, rotamer B), 3.23 (s, 3H, rotamer A), 2.30 (s, 3H, rotamer A), 2.27 (s, 3H rotamer B), 2.22 (s, 6H, rotamer A + rotamer B). 13C{1H} NMR (CDCl3, 100 MHz) δ 150.0, 138.6, 138.4, 135.2, 129.5, 37.6, 21.0, 17.5. FTIR (ATR/νmax) 2920, 1727, 1483, 1409, 1379, 1354, 1333, 1302, 1248, 1169, 1100, 1035, 1011 cm–1.
General Procedure 3: Synthesis of Self-Immolative Systems (15–23)
Carbamoyl chloride (1 equiv), 2-(diphenylphosphino)ethanol (1 equiv), 4-dimethylaminopyridine (0.1 equiv), and triethylamine (2 equiv) were dissolved in THF (20 mL), and the solution was heated (using an aluminum heating block) under reflux overnight. The solvent was removed in vacuo, and the crude product was purified via column chromatography.
Synthesis of 2-(Diphenylphosphanyl)ethyl Methyl(4-nitrophenyl)carbamate (15)23
Following the general procedure 3, 1 (0.30 g, 1.40 mmol) was treated with 2-(diphenylphosphino)ethanol (0.32 g, 1.40 mmol), 4-dimethylaminopyridine (0.018 g, 0.14 mmol), and triethylamine (0.38 mL, 2.80 mmol) to provide 15 (0.40 g, 73%) as a pale yellow oil after silica-gel column chromatography (hexane/EtOAc 75/25). 1H NMR (CDCl3, 400 MHz) δ 8.18 (2H, AA′XX′), 7.48–7.38 (m, 6H), 7.38–7.28 (m, 6H), 4.35 (dt, J = 9.5, 7.0 Hz, 2H), 3.25 (s, 3H), 2.48 (t, J = 7.0 Hz, 2H). 13C{1H} NMR (CDCl3, 100 MHz) δ 154.5, 148.8, 144.3, 137.6 (d, 1JCP = 12.0 Hz), 132.6 (d, 2JCP = 19.0 Hz), 128.9, 128.6 (d, 3JCP = 7.0 Hz), 124.4, 124.2, 64.2 (d, 2JCP = 23.3 Hz), 36.8, 28.1 (d, 1JCP = 14.4 Hz). 31P{1H} NMR (CDCl3, 162 MHz) −22.27. FTIR (ATR/νmax) 3051–2954, 1705, 1593, 1513, 1498, 1433, 1325, 1254, 1151, 1103 cm–1. HRMS (ESI) m/z: [M + H]+ calcd for C22H22N2O4P 409.1312; found 409.1304.
Synthesis of 2-(Diphenylphosphanyl)ethyl Methyl(2-methyl-4-nitrophenyl)carbamate (16)
Following the general procedure 3, 2 (0.30 g, 1.31 mmol) was treated with 2-(diphenylphosphino)ethanol (0.30 g, 1.31 mmol), 4-dimethylaminopyridine (0.016 g, 0.13 mmol), and triethylamine (0.37 mL, 2.62 mmol) to provide 16 (0.38 g, 69%) as a colorless oil after silica-gel column chromatography (hexane/EtOAc 75/25). 1H NMR (CDCl3, 400 MHz) δ 8.18–7.95 (m, 2H rotamer A + rotamer B), 7.55–7.15 (m, 11H, rotamer A + rotamer B), 4.44–4.29 (m, 2H, rotamer B), 4.29–4.11 (m, 2H, rotamer A), 3.20 (s, 3H, rotamer A), 3.06 (s, 3H, rotamer B), 2.59–2.18 (m, 5H, rotamer A + rotamer B). 13C{1H} NMR (CDCl3, 100 MHz) δ 154.9, 147.5, 146.7, 137.7, 132.6 (d, 1Jcp =18.5 Hz), 129.1–128.1 (m, 7C), 126.2, 122.2, 63.9 (d, 2J = 25.0 Hz), 37.2, 28.2 (d, 1Jcp = 15.0 Hz), 18.0 (br.). 31P{1H} NMR (CDCl3, 162 MHz) δ −22.08 (rotamer B), −22.80 (rotamer A). FTIR (ATR/νmax) 3070–2925, 1704, 1586, 1520, 1492, 1433, 1345, 1154, 1086 cm–1. HRMS (ESI) m/z: [M + H]+ calcd for C23H24O4N2P 423.14468; found 423.1459.
Synthesis of 2-(Diphenylphosphanyl)ethyl (2,6-Dimethyl-4-nitrophenyl)(methyl)carbamate (17)
Following the general procedure 3, 3 (0.30 g, 1.24 mmol) was treated with 2-(diphenylphosphino)ethanol (0.29 g, 1.24 mmol), 4-dimethylaminopyridine (0.015 g, 0.12 mmol), and triethylamine (0.35 mL, 2.48 mmol) to provide 17 (0.37 g, 68%) as a colorless oil after silica-gel column chromatography (hexane/CH2Cl2 1/1 → hexane/EtOAc 75/25). 1H NMR (CDCl3, 400 MHz) δ 7.95 (s, 2H, rotamer B), 7.91 (s, 2H rotamer A), 7.52–7.27 (m, 10H, rotamer A + rotamer B), 4.36 (dt, J = 9.5, 7.5 Hz, 2H, rotamer B), 4.18 (m, 2H, rotamer A), 3.13 (s, 3H, rotamer A), 2.99 (s, 3H, rotamer B), 2.53 (t, 2J = 7.5 Hz, 2H, rotamer B), 2.33–2.24 (m, 8H rotamer A + rotamer B). 13C{1H} NMR (CDCl3, 100 MHz) δ 155.1 (rotamer A), 154.5 (rotamer B), 146.7 (rotamer B), 146.6 (rotamer A), 146.5 (rotamer B), 146.0 (rotamer A), 138.2 (rotamer B), 137.9 (rotamer A), 137.8 (d, 1Jcp = 12.0 Hz, rotamer B), 137.7 (d, 1Jcp = 12.5 Hz, rotamer A), 132.8 (d, 2Jcp = 19.0 Hz, rotamer B), 132.6 (d, 2Jcp = 19.0 Hz, rotamer A), 129.0 (rotamer B), 128.9 (rotamer A), 128.7 (d, 3Jcp = 7.0 Hz, rotamer B), 128.6 (d, 3Jcp = 7.0 Hz, rotamer A), 123.7 (rotamer B), 123.6 (rotamer A), 63.9 (d, 2Jcp = 25.5 Hz, rotamer A), 63.8 (d, 2Jcp = 24.0 Hz, rotamer B), 35.6 (rotamer B), 35.4 (rotamer A), 28.5 (d, 1Jcp = 14.5 Hz, rotamer B), 28.3 (d, 1Jcp = 15.0 Hz, rotamer A), 18.2 (rotamer A and rotamer B). 31P{1H} NMR (CDCl3, 162 MHz) δ −22.17 (rotamer B), −22.99 (rotamer A). FTIR (ATR/νmax) 2923–2852, 2361, 1702, 1591, 1520, 1478, 1433, 1389, 1345, 1298, 1153, 1090 cm–1. HRMS (ESI) m/z: [M + H]+ calcd for C24H26O4N2P 437.162; found 437.1621.
Synthesis of 2-(Diphenylphosphanyl)ethyl Methyl(3-nitrophenyl)carbamate (18)
Following the general procedure 3, 4 (0.30 g, 1.40 mmol) was treated with 2-(diphenylphosphino)ethanol (0.32 g, 1.40 mmol), 4-dimethylaminopyridine (0.018 g, 0.14 mmol), and triethylamine (0.38 mL, 2.80 mmol) to provide 18 (0.40 g, 73%) as a colorless oil after silica-gel column chromatography (hexane/CH2Cl2 1/1 → hexane/EtOAc 75/25). 1H NMR (CDCl3, 400 MHz) δ 8.10 (s, 1H), 8.03 (d, J = 8.0 Hz, 1H), 7.61 (d, J = 8.0 Hz, 1H), 7.48 (t, J = 8.0 Hz, 1H), 7.42 (m, 4H), 7.33 (m, 6H), 4.34 (q, 3J = 7.0 Hz, 2H), 3.24 (s, 3H), 2.46 (t, 2J = 7.0 Hz, 2H). 13C{1H} NMR (CDCl3, 100 MHz) δ 154.9, 148.5, 144.3, 137.7 (d, 1JCP = 12.1 Hz), 132.8 (d, 2JCP = 19.0 Hz), 131.3, 129.5, 129.0, 128.7 (d, 3JCP = 6.9 Hz), 120.4, 120.0, 64.2 (d, 2JCP = 23.4 Hz), 37.2, 28.2 (d, 1JCP = 14.3 Hz). 31P{1H} NMR (CDCl3, 162 MHz) δ −22.3. FTIR (ATR/νmax) 3071–2954, 1702, 1526, 1481, 1433, 1386, 1342, 1261, 1154, 1126 cm–1. HRMS (ESI) m/z: [M + H]+ calcd for C22H22N2O4P 409.1312; found 409.1312.
Synthesis of 2-(Diphenylphosphanyl)ethyl Methyl(2-methyl-5-nitrophenyl)carbamate (19)
Following the general procedure 3, 5 (0.32 g, 1.40 mmol) was treated with 2-(diphenylphosphino)ethanol (0.32 g, 1.40 mmol), 4-dimethylaminopyridine (0.018 g, 0.14 mmol), and triethylamine (0.38 mL, 2.80 mmol) to provide 19 (0.18 g, 30%) as a colorless oil with a 65:35 mixture of rotamers A/B after silica-gel column chromatography (hexane/acetone 95/5). 1H NMR (CDCl3, 400 MHz) δ 8.04 (app. d, 3J = 8.5 Hz, 1H, rotamer A + rotamer B), 7.99 (s, 1H, rotamer B), 7.94 (s, 1H, rotamer A), 7.59–7.27 (m, 10H, rotamer A + rotamer B), 4.38 (br. s, 2H, rotamer B), 4.28–4.10 (m, 2H, rotamer A), 3.21 (s, 3H, rotamer A), 3.06 (s, 3H, rotamer B), 2.54 (br. s, 2H, rotamer B), 2.30 (br. s, 2H, rotamer A), 2.30 (s, 3H, rotamer A + rotamer B). 13C{1H} NMR (CDCl3, 100 MHz) δ 155.2 (rotamer A), 155.0 (rotamer B), 146.9 (rotamer A + rotamer B), 144.3 (rotamer B), 143.1 (rotamer A), 143.0 (rotamer B), 142.5 (rotamer A), 138.0–137.5 (m, rotamer A + rotamer B), 133.1–132.4 (m, rotamer A + rotamer B), 131.8 (rotamer B), 131.7 (rotamer A), 128.8 (d, 2Jcp = 22.5 Hz, rotamer A + rotamer B), 128.6 (rotamer A + rotamer B), 123.1 (rotamer A + rotamer B), 122.5 (br. s, rotamer A + rotamer B), 64.0 (d, 2Jcp = 25.5 Hz, rotamer A + rotamer B), 37.4 (rotamer A), 37.3 (rotamer B), 28.4 (d, 1Jcp = 15.0 Hz, rotamer B), 28.3 (d, 1Jcp = 14.5 Hz, rotamer A), 18.1 (rotamer A + rotamer B). 31P{1H} NMR (CDCl3, 162 MHz) δ −21.8 (rotamer B), −23.1 (rotamer A). FTIR(ATR/νmax) 3071–2954, 1702, 1519, 1433, 1340, 1153, 1123 cm–1. HRMS (ESI) m/z: [M + H]+ calcd for C23H24N2O4P 423.1468; found 423.1463.
Synthesis of 2-(Diphenylphosphanyl)ethyl (2,6-Dimethyl-3-nitrophenyl)(methyl)carbamate (20)
Following the general procedure 3, 6 (0.34 g, 1.40 mmol) was treated with 2-(diphenylphosphino)ethanol (0.32 g, 1.40 mmol), 4-dimethylaminopyridine (0.018 g, 0.14 mmol), and triethylamine (0.38 mL, 2.80 mmol) to provide 20 (0.10 g, 33%) as a colorless oil with a 72:28 mixture of rotamers A/B after silica-gel column chromatography (hexane/acetone 95/5). 1H NMR (CDCl3, 400 MHz) δ 7.77 (app. d, J = 8.5 Hz, 1H, rotamer A + rotamer B), 7.54–7.27 (m, 10H, rotamer A + rotamer B), 7.21 (app. d, J = 8.5 Hz, 1H rotamer B), 7.20 (app. d, 3J = 8.5 Hz, 1H, rotamer A), 4.38 (dt, 3JHP = 8.5 Hz, 3J = 7.5 Hz, 2H, rotamer B), 4.23–4.13 (m, 2H, rotamer A), 3.14 (s, 3H rotamer A), 3.00 (s, 3H, rotamer B), 2.55 (t, J = 7.5 Hz, 2H, rotamer B), 2.34–2.29 (m, 2H, rotamer A), 2.28 (s, 3H, rotamer A), 2.27 (s, 3H, rotamer B). 13C{1H} NMR (CDCl3, 100 MHz) δ 155.3 (rotamer A), 154.7 (rotamer B), 149.1 (rotamer B), 149.0 (rotamer A), 142.6 (rotamer B), 142.4 (rotamer B), 142.3 (rotamer A), 141.9 (rotamer A), 141.3 (rotamer A), 137.85 (d, 1Jcp = 12.5 Hz, rotamer B, Ph′ or Ph″), 137.84 (d, 1Jcp = 12.5 Hz, rotamer B, Ph′ or Ph″), 137.66 (d, 1Jcp = 12.5 Hz, rotamer A, Ph″), 137.64 (d, 1Jcp = 12.5 Hz, rotamer A, Ph′), 132.74 (d, 2Jcp = 18.0 Hz, rotamer B, Ph′ or Ph″), 132.72 (d, 2Jcp = 18.5 Hz, rotamer B, Ph′ or Ph″), 132.60 (d, 2Jcp = 18.5 Hz, rotamer A, Ph″), 132.56 (d, 2Jcp = 18.5 Hz, rotamer A, Ph′), 132.0 (rotamer B), 131.7 (rotamer A), 128.9 (rotamer B), 128.8 (rotamer A), 128.74 (d, 3Jcp = 6.0 Hz, rotamer B), 128.70 (d, 3Jcp = 7.5 Hz, rotamer A), 128.5 (rotamer A + rotamer B), 123.8 (rotamer B), 123.7 (rotamer A), 63.7 (d, 3Jcp = 26.0 Hz, rotamer A), 63.6 (d, 3Jcp = 23.0 Hz, rotamer B), 35.8 (rotamer B), 35.7 (rotamer A), 28.4 (d, 3Jcp = 14.5 Hz, rotamer B), 28.3 (d, 3Jcp = 15.0 Hz, rotamer A), 18.43 (rotamer A), 18.40 (rotamer B), 14.3 (rotamer A + rotamer B). 31P{1H} NMR (CDCl3, 162 MHz) δ −22.15 (rotamer B), −23.37 (rotamer A). FTIR (ATR/νmax) 3081–2932, 1724, 1586, 1529, 1513, 1491, 1422, 1344, 1304, 1252, 1113, 1085 cm–1. HRMS (ESI) m/z: [M + H]+ calcd for C24H26N2O4P 437.1625; found 437.1624. As an example of signal NMR assignment for these self-immolative systems, further NMR analyses were carried out for 20 (see Figures S44–S51).
Synthesis of 2-(Diphenylphosphanyl)ethyl Methyl(2,4,6-trimethyl-3-nitrophenyl)carbamate (21)
Following the general procedure 3, 7 (0.36 g, 0.82 mmol) was treated with 2-(diphenylphosphino)ethanol (0.32 g, 1.40 mmol), 4-dimethylaminopyridine (0.018 g, 0.14 mmol), and triethylamine (0.38 mL, 2.80 mmol) to provide 21 (0.11 g, 30%) as a colorless oil with a 74:26 mixture of rotamers A/B after silica-gel column chromatography (hexane/acetone 95/5). 1H NMR (CDCl3, 400 MHz) δ 7.50–7.27 (m, 10H, rotamer A + rotamer B), 7.01 (s, 1H, rotamer B), 6.99 (s, 1H, rotamer A), 4.35 (dt, 2H, 3JHP = 9.5 Hz, 3J = 7.5 Hz rotamer B), 4.16 (AA′BB′, 2H, rotamer A), 3.11 (s, 3H, rotamer A), 2.97 (s, 3H, rotamer B), 2.52 (app. t, 3J = 8.0 Hz, 3H rotamer B), 2.31 (app. t, 3J = 8.0 Hz, 2H, rotamer A), 2.27 (s, 3H, rotamer A), 2.26 (s, 3H, rotamer B), 2.19 (s, 3H, rotamer A + rotamer B), 2.12 (s, 3H, rotamer A), 2.11 (s, 3H, rotamer B). 13C{1H} NMR (CDCl3, 100 MHz) δ 155.5 (rotamer A), 154.9 (rotamer B), 151.0 (rotamer B), 150.9 (rotamer A), 139.6 (rotamer B), 139.1 (rotamer A), 138.8 (rotamer B), 138.6 (rotamer A), 137.90 (d, 1Jcp = 12.0 Hz, rotamer B, Ph′ or Ph″), 137.87 (d, 1Jcp = 12.0 Hz, rotamer B, Ph′ or Ph″), 137.72 (d, 1Jcp = 12.0 Hz, rotamer A, Ph′ or Ph″), 137.68 (d, 1Jcp = 12.0 Hz, rotamer A, Ph′ or Ph″), 132.85 (d, 2JCP = 18.0 Hz, rotamer B, Ph′ or Ph″), 132.82 (d, 2JCP = 19.0 Hz, rotamer B, Ph′ or Ph″), 132.77 (d, 2Jcp = 18.0 Hz, rotamer A, Ph″), 132.69 (d, 2Jcp = 18.5 Hz, rotamer A, Ph′), 131.0 (rotamer B), 130.9 (rotamer A), 129.0–128.5 (m, 7C), 128.3 (rotamer B), 128.0 (rotamer A), 63.74 (d, 2Jcp = 25.5 Hz, rotamer A), 63.68 (d, 2Jcp = 24.0 Hz, rotamer B), 36.0 (rotamer B), 35.9 (rotamer A), 29.2 (d, 1Jcp = 14.5 Hz, rotamer B), 28.4 (d, 1Jcp = 15.0 Hz, rotamer A), 18.0 (rotamer A), 17.9 (rotamer B), 17.5 (rotamer A), 17.4 (rotamer B), 13.1 (rotamer A + rotamer B). 31P{1H} NMR (CDCl3, 162 MHz) δ −22.1 (rotamer B), −23.3 (rotamer A). FTIR (ATR/νmax) 3054–2932, 1702, 1524, 1480, 1454, 1433, 1388, 1345, 1311, 1284, 1220, 1152, 1124, 1095, 1027 cm–1. HRMS (ESI) m/z: [M + H]+ calcd for C25H28N2O4P 451.1781; found 451.1766.
Synthesis of 2-(Diphenylphosphanyl)ethyl Methyl(phenyl)carbamate (22)
Following the general procedure 3, 8 (0.24 g, 1.40 mmol) was treated with 2-(diphenylphosphino)ethanol (0.32 g, 1.40 mmol), 4-dimethylaminopyridine (0.018 g, 0.14 mmol), and triethylamine (0.38 mL, 2.80 mmol) to provide 22 (0.30 g, 59%) as a white solid after silica-gel column chromatography (hexane/EtOAc 95/5 → 90/10) followed by crystallization via vapor diffusion using a mixture of n-hexane and ethyl acetate to obtain colorless single crystals. Mp 53–55 °C. 1H NMR (CDCl3, 400 MHz) δ 7.47–7.37 (m, 4H), 7.37–7.29 (m, 8H), 7.25–7.17 (m, 3H), 4.27 (q, J = 7.5 Hz, 2H), 3.22 (s, 3H), 2.44 (t, J = 7.5 Hz, 2H). 13C{1H} NMR (CDCl3, 100 MHz) δ 155.5, 143.3, 137.9 (d, 1JCP = 12.2 Hz), 132.8 (d, 2JCP = 18.9 Hz), 128.9 (×2), 128.7 (d, 3JCP = 6.7 Hz), 126.1, 125.8, 63.5 (d, 2JCP = 25.0 Hz), 37.7, 28.4 (d, 1JCP = 14.0 Hz). 31P{1H} NMR (CDCl3, 162 MHz) δ −22.5. FTIR (ATR/νmax) 3066–2961, 1688, 1595, 1493, 1451, 1433, 1393, 1351, 1307, 1295, 1283, 1151, 1040 cm–1. HRMS (ESI) m/z: [M + H]+ calcd for C22H23NO2P 364.1461; found 364.1458.
Synthesis of 2-(Diphenylphosphanyl)ethyl Mesityl(methyl)carbamate (23)
Following the general procedure 3, 9 (0.26 g, 1.40 mmol) was treated with 2-(diphenylphosphino)ethanol (0.32 g, 1.40 mmol), 4-dimethylaminopyridine (0.018 g, 0.14 mmol), and triethylamine (0.38 mL, 2.80 mmol) to provide 23 (0.20 g, 35%) as a white solid after silica-gel column chromatography (hexane/acetone 95/5) followed by crystallization via vapor diffusion using a mixture of n-hexane and ethyl acetate to obtain colorless single crystals. Mp 93–95 °C. 1H NMR (CDCl3, 400 MHz) δ 7.53–7.23 (m, 10H, rotamer A + rotamer B), 6.88 (s, 2H, rotamer A + rotamer B), 4.33 (AA′BB′, 2H, rotamer B), 4.15 (AA′BB′, 2H, rotamer A), 3.11 (s, 3H, rotamer A), 2.98 (s, 3H, rotamer B), 2.52 (AA′BB′, 2H, rotamer B), 2.33 (AA′BB′, 2H, rotamer A), 2.28 (s, 3H, rotamer A), 2.26 (s, 3H, rotamer B), 2.14 (s, 6H, rotamer A + rotamer B). 13C{1H} NMR (CDCl3, 100 MHz) δ 156.0 (rotamer A) 155.0 (rotamer A), 138.2 (rotamer A + rotamer B), 137.9 (d, 1Jcp = 12.5 Hz, rotamer A), 137.7 (d, 1Jcp = 12.5 Hz, rotamer B), 137.7 (rotamer A), 137.1 (br. s, rotamer A + rotamer B), 135.4 (rotamer B), 135.3 (rotamer A), 132.8 (d, 2Jcp = 19.0 Hz, rotamer B), 132.7 (d, 2Jcp = 19.0 Hz, rotamer A), 129.3 (rotamer B), 129.1 (rotamer A), 128.8 (rotamer B), 128.7 (rotamer A), 128.6 (d, 3Jcp = 7.0 Hz, rotamer B), 128.5 (d, 3Jcp = 6.5 Hz, rotamer A), 63.3 (d, 2Jcp = 28.0 Hz, rotamer A), 63.2 (d, 2Jcp = 23.0 Hz, rotamer B), 35.99 (rotamer B), 35.97 (rotamer A), 28.5 (d, 1Jcp = 14.5 Hz, rotamer B), 28.3 (d, 1Jcp = 14.5 Hz, rotamer A), 21.11 (rotamer A), 21.06 (rotamer B), 17.75 (rotamer A), 17.72 (rotamer B). 31P{1H} NMR (CDCl3, 162 MHz) δ −22.0 (rotamer B), −23.1 (rotamer A). FTIR (ATR/νmax) 3049–2915, 1707, 1655, 1584, 1483, 1454, 1432, 1390, 1348, 1308, 1277, 1217, 1172, 1148, 1097, 1070, 1027 cm–1. HRMS (ESI) m/z: [M + H]+ calcd for C25H29NO2P 406.1924; found 406.1930.
General Procedure 4 for the Synthesis of Carbamate Model Compounds (24–28)
Sodium hydride (NaH, 60% dispersion in mineral oil, 1.1 equiv) was added to a solution of 2-naphthalenemethanol (1 equiv) in anhydrous DMF (10 mL) at 0 °C and left to stir for 15 min. Carbamoyl chloride (1 equiv) was added, and the mixture was left to stir overnight. The salts formed were filtered off, and volatiles were removed in vacuo. The crude product was further purified by column chromatography, followed by recrystallization via vapor diffusion using a mixture of n-hexane and ethyl acetate to obtain colorless single crystals.
Synthesis of Naphthalen-2-ylmethyl Methyl(4-nitrophenyl)carbamate (24)
Following the general procedure 4, 2-naphthalenemethanol (0.122 g, 0.77 mmol) was treated with NaH (0.034 g, 0.85 mmol) and 1 (0.166, 0.77 mmol) to provide 24 (0.200 g, 77%) as a white solid after silica-gel column chromatography (hexane/EtOAc 75/25). Mp 109–111 °C. 1H NMR (CDCl3, 400 MHz) δ 8.21 (d, J = 9.0 Hz, 2H), 7.88–7.80 (m, 4H), 7.67–7.34 (m, 5H), 5.38 (s, 2H), 3.42 (s, 3H). 13C{1H} NMR (CDCl3, 100 MHz) δ 154.8, 149.0, 144.6, 133.31, 133.28, 128.6, 128.1, 127.9, 127.7, 126.6, 125.9, 124.7, 124.4, 68.5, 37.2. FTIR (ATR/νmax) 3057–2963, 1709, 1602, 1590, 1512, 1442, 1329, 1257, 1160, 1104 cm–1. HRMS (ESI) m/z: [M + Na]+ calcd for C19H16O4N2Na 359.1002; found 359.0999.
Synthesis of Naphthalen-2-ylmethyl Methyl(2-methyl-4-nitrophenyl)carbamate (25)
Following the general procedure 4, 2-naphthalenemethanol (0.122 g, 0.77 mmol) was treated with NaH (0.034 g, 0.85 mmol) and 2 (0.176 g, 0.77 mmol) to provide 25 (0.197 g, 73%) as an off-white solid after silica-gel column chromatography (hexane/EtOAc 75/25). Mp 99–101 °C. 1H NMR (CDCl3, 400 MHz) δ 8.08 (br. s, 1H, rotamer A + rotamer B), 8.09–8.04 (m, 1H, rotamer A + rotamer B), 7.90–7.71 (m, 3H, rotamer A + rotamer B), 7.63 (br. s, 1H, rotamer A + rotamer B), 7.55–7.44 (m, 2H, rotamer A + rotamer B), 7.38–7.27 (m, 2H, rotamer A + rotamer B), 5.43–5.18 (m, 2H, rotamer A + rotamer B), 3.24 (br. s, 3H, rotamer A + rotamer B), 2.32 (s, 3H, rotamer B), 2.24 (s, 3H, rotamer A). 13C{1H} NMR (CDCl3, 100 MHz) δ 154.8, 147.4, 146.6, 137.7, 133.5, 133.0, 128.4, 128.3, 127.9, 127.7, 127.4, 127.1, 126.3, 126.3, 126.0, 125.8, 125.5, 122.2, 67.9, 37.3, 17.9. FTIR (ATR/νmax) 3070–2956, 1698, 1582, 1514, 1431, 1304, 1155, 1088 cm–1. HRMS (ESI) m/z: [M + Na]+ calcd for C20H18O4N2Na 373.1159; found 373.1155.
Synthesis of Naphthalen-2-ylmethyl (2,6-Dimethyl-4-nitrophenyl)(methyl)carbamate (26)
Following the general procedure 4, 2-naphthalenemethanol (0.100 g, 0.63 mmol) was treated with NaH (0.028 g, 0.70 mmol) and 3 (0.153 g, 0.63 mmol) to provide 26 (0.178 g, 77%) as an off-white solid after silica-gel column chromatography (hexane/EtOAc 75/25). Mp 87–89 °C. 1H NMR (CDCl3, 400 MHz) δ 7.97 (s, 2H, rotamer B), 7.95 (s, 2H, rotamer A), 7.91–7.84 (m, 1H, rotamer A + rotamer B), 7.82–7.79 (m, 1H, rotamer A + rotamer B), 7.77 (app. d, J = 8.0 Hz, 1H, rotamer A + rotamer B), 7.74–7.72 (m, 1H, rotamer A + rotamer B), 7.62 (s, 1H, rotamer A), 7.55 (s, 1H, rotamer B), 7.54–7.44 (m, 2H, rotamer A + rotamer B), 7.28–7.23 (m, 1H, rotamer A + rotamer B), 5.40 (s, 2H, rotamer B), 5.25 (s, 2H, rotamer A), 3.18 (s, 3H, rotamer B), 3.15 (s, 3H, rotamer A), 2.30 (s, 6H, rotamer B), 2.22 (s, 6H, rotamer A). 13C{1H} NMR (CDCl3, 100 MHz) δ 155.0, 154.6, 146.7, 146.1, 138.2, 138.0, 133.7, 133.3, 133.2, 133.2, 128.6, 128.4, 128.1, 128.1, 127.9, 127.8, 127.4, 127.2, 126.5, 126.5, 126.4, 126.4, 125.9, 125.7, 123.7, 123.6, 68.0, 67.8, 35.8, 35.4, 18.1. FTIR (ATR/νmax) 3052–2928, 1693, 1602, 1518, 1479, 1435, 1303, 1168, 1088 cm–1. HRMS (ESI) m/z: [M + Na]+ calcd for C21H20O4N2Na 387.1315; found 387.1316.
Synthesis of Naphthalen-2-ylmethyl Methyl(3-nitrophenyl)carbamate (27)
Following the general procedure 4, 2-naphthalenemethanol (0.122 g, 0.77 mmol) was treated with NaH (0.034 g, 0.85 mmol) and 4 (0.166 g, 0.77 mmol) to provide 27 (0.160 g, 62%) as a white solid after silica-gel column chromatography (hexane/EtOAc 75/25). Mp 85–87 °C. 1H NMR (CDCl3, 400 MHz) δ 8.19 (app. t, J = 2.0 Hz, 1H), 8.06 (dd, J = 8.0, 2.0 Hz, 1H), 7.87–7.78 (m, 4H), 7.65 (d, J = 8.0 Hz, 1H), 7.54–7.41 (m, 4H), 5.36 (s, 2H), 3.41 (s, 3H). 13C{1H} NMR (CDCl3, 100 MHz) δ 155.1, 148.6, 144.4, 133.4, 133.3, 131.3, 129.6, 128.6, 128.2, 127.9, 127.5, 126.5, 126.5, 125.9, 120.6, 120.3, 68.3, 37.5. FTIR (ATR/νmax) 3090–2925, 1703, 1615, 1579, 1528, 1486, 1443, 1348, 1336, 1251, 1169, 1122, 1026 cm–1. HRMS (ESI) m/z: [M + Na]+ calcd for C19H16O4N2Na 359.1002; found 359.0998.
Synthesis of Naphthalen-2-ylmethyl Methyl(phenyl)carbamate (28)
Following the general procedure 4, 2-naphthalenemethanol (0.122 g, 0.77 mmol) was treated with NaH (0.034 g, 0.85 mmol) and 8 (0.131 g, 0.77 mmol) to provide 28 (0.180 g, 80%) as a white solid after silica-gel column chromatography (hexane/EtOAc 75/25). Mp 93–95 °C. 1H NMR (CDCl3, 400 MHz) δ 7.84–7.70 (m, 4H), 7.50–7.45 (m, 2H), 7.44–7.39 (m, 1H), 7.39–7.32 (m, 2H), 7.25–7.19 (m, 3H), 5.32 (s, 2H), 3.34 (s, 3H). 13C{1H} NMR (CDCl3, 100 MHz) δ 155.6, 143.4, 134.2, 133.3, 133.1, 129.0, 128.3, 128.1, 127.8, 126.9 (br. s), 126.3 (br. s), 126.2, 126.0 (br. s), 125.7, 67.5, 38.0. FTIR (ATR/νmax) 3060–2946, 1697, 1597, 1438, 1397, 1362, 1337, 1261, 1180, 1119 cm–1. HRMS (ESI) m/z: [M + H]+ calcd for C19H18O2N 292.1332; found 292.1332.
General Procedure for the Alkylation Reactions of Self-Immolative Systems (15–23)
1H NMR spectroscopic studies were conducted by dissolution of the various disclosure systems (15–23) in MeCN-d3 (0.5 mL, [disclosure system]0 = 0.025 mol·L–1), followed by the addition of 10 molar equiv of benzyl bromide (BnBr) directly to the NMR tube. The 1H NMR spectra were recorded at regular time intervals. The area of the singlet resonance CH3N of 15–23 was standardized to the area of the MeCN solvent resonance at 1.94 ppm and was used to calculate the percentage of disclosure system that remained nonalkylated over time with respect to the initial area of the methyl resonance (i.e., before the addition of the alkylating agent, time point = 0). The same procedure was employed for the solvent screening of self-immolative system 19.
General Procedure for the β-Elimination Reactions of Self-Immolative Systems 15a–23a
1H NMR spectroscopic studies were conducted by addition of 2 molar equiv of N,N-diisopropylethylamine (DIPEA) to the corresponding alkylated disclosure system (15a–23a) directly to the NMR tube. The percentage of alkylated disclosure system 15a–23a remaining over time was calculated by comparison of the integrations of the CH3N or aromatic ortho- aromatic proton of the reporter moiety prior to β-elimination and standardized to the area of the MeCN solvent resonance at 1.94 ppm. The same procedure was employed for the solvent screening of self-immolative system 19a.
General Procedure for the “One-Pot” Reactions of Self-Immolative Systems 18–20
1H NMR spectroscopic studies were conducted by addition of 10 molar equiv of benzyl bromide to a solution of the corresponding self-immolative disclosure system 18–20 (V = 0.5 mL, [disclosure system] = 0.025 mol·L–1) and 2 molar equiv of DIPEA directly to the NMR tube. The 1H NMR spectra were recorded at regular time intervals. The area of the singlet CH3N for the self-immolative disclosure systems 18 and 20, the area of the singlet CHAr for the self-immolative disclosure system 19, the area of the multiplet resonance CH2P+ for the self-immolative alkylated disclosure system 18a–20a as well as the area for the vinyl proton corresponding to the phosphonium salt 29 were used to calculate the percentage of the different species remaining over time (i.e., before the addition of BnBr, time point = 0).
Acknowledgments
This study was supported by Dstl (contracts DSTLX1000116213R and DSTLX1000114599) for A.G.G. and F.L. The authors thank the University of Reading for access to resources in the Chemical Analysis Facility and Nick Spencer (University of Reading) for his help in collecting the single-crystal X-ray diffraction data.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.1c00996.
1H NMR, 13C NMR, and 31P spectra; calculations of rotational barriers; and X-ray crystallographic data of CCDC 1980913 (22), 1962710 (23), 1908144 (24), 1908143 (25), 1908145 (26), 1908146 (27), and 1980598 (28) (PDF)
Accession Codes
CCDC 1908143–1908146, 1962710, 1980598, and 1980913 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no competing financial interest.
Footnotes
When the reporter group is unsymmetrically substituted, the molecule is chiral and, as a result, the phenyl groups attached to the phosphine are diastereotopic.
With respect to the syn/anti conformation of the carbamate unit, in agreement with Smith et al., we observe in solution that the methylene protons in the α-position to the carbamate group of the major rotamer are always at the higher field.35
Supplementary Material
References
- Herbert K. M.; Schrettl S.; Rowan S. J.; Weder C. 50th Anniversary Perspective: Solid-State Multistimuli, Multiresponsive Polymeric Materials. Macromolecules 2017, 50, 8845–8870. 10.1021/acs.macromol.7b01607. [DOI] [Google Scholar]
- Hart L. R.; Harries J. L.; Greenland B. W.; Colquhoun H. M.; Hayes W. Healable Supramolecular Polymers. Polym. Chem. 2013, 4, 4860–4870. 10.1039/c3py00081h. [DOI] [Google Scholar]
- Gillies E. R. Reflections on the Evolution of Smart Polymers. Isr. J. Chem. 2019, 75–85. 10.1002/ijch.201900075. [DOI] [Google Scholar]
- a Roth M. E.; Green O.; Gnaim S.; Shabat D. Dendritic, Oligomeric, and Polymeric Self-Immolative Molecular Amplification. Chem. Rev. 2016, 116, 1309–1352. 10.1021/acs.chemrev.5b00372. [DOI] [PubMed] [Google Scholar]; b Erez R.; Shabat D. The azaquinone-methide elimination: comparison study of 1,6- and 1,4-eliminations under physiological conditions. Org. Biomol. Chem. 2008, 6, 2669–2672. 10.1039/b808198k. [DOI] [PubMed] [Google Scholar]; c Alouane A.; Labruère R.; Le Saux T.; Schmidt F.; Jullien L. Self-Immolative Spacers: Kinetic Aspects, Structure–Property Relationships, and Applications. Angew. Chem., Int. Ed. 2015, 54, 7492–7509. 10.1002/anie.201500088. [DOI] [PubMed] [Google Scholar]; d Sykes B. M.; Hay M. P.; Bohinc-Herceg D.; Helsby N. A.; O’Connor C. J.; Denny W. A. Leaving group effects in reductively triggered fragmentation of 4-nitrobenzyl carbamates. J. Chem. Soc., Perkin Trans. 1 2000, 1601–1608. 10.1039/b000135j. [DOI] [Google Scholar]
- Peterson G. I.; Larsen M. B.; Boydston A. J. Controlled Depolymerization: Stimuli-Responsive Self-Immolative Polymer. Macromolecules 2012, 45, 7317–7328. 10.1021/ma300817v. [DOI] [Google Scholar]
- a Blencowe C. A.; Russell A. T.; Greco F.; Hayes W.; Thornthwaite D. W. Self-Immolative Linkers in Polymeric Delivery Systems. Polym. Chem. 2011, 2, 773–790. 10.1039/C0PY00324G. [DOI] [Google Scholar]; b Blencowe C. A.; Thornthwaite D. W.; Hayes W.; Russell A. T. Self-immolative base-mediated conjugate release from triazolylmethylcarbamates. Org. Biomol. Chem. 2015, 13, 8703–8707. 10.1039/C5OB00984G. [DOI] [PubMed] [Google Scholar]; c Roberts D. A.; Pilgrim B. S.; Dell T. N.; Stevens M. M. Dynamic pH responsivity of triazole-based self-immolative linkers. Chem. Sci., 2020, 11, 3713–3718. 10.1039/D0SC00532K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yardley R. E.; Kenaree A. R.; Gillies E. R. Triggering Depolymerization: Progress and Opportunities for Self-Immolative Polymers. Macromolecules 2019, 52, 6342–6360. 10.1021/acs.macromol.9b00965. [DOI] [Google Scholar]
- Tranoy-Opalinski I.; Fernandes A.; Thomas M.; Gesson J. P.; Papot S. Design of Self-Immolative Linkers for Tumour-Activated Prodrug Therapy. Anti-Cancer Agents Med. Chem. 2008, 8, 618–637. 10.2174/187152008785133065. [DOI] [PubMed] [Google Scholar]
- Sagi A.; Segal E.; Satchi-Fainaro R.; Shabat D. Remarkable Drug-Release Enhancement with an Elimination-Based AB3 Self-Immolative Dendritic Amplifier. Bioorg. Med. Chem. 2007, 15, 3720–3727. 10.1016/j.bmc.2007.03.054. [DOI] [PubMed] [Google Scholar]
- Kratz F.; Müller I. A.; Ryppa C.; Warnecke A. Prodrug Strategies in Anticancer Chemotherapy. ChemMedChem 2008, 3, 20–53. 10.1002/cmdc.200700159. [DOI] [PubMed] [Google Scholar]
- Ho N. H.; Weissleder R.; Tung C. H. A Self-Immolative Reporter for β-Galactosidase Sensing. ChemBioChem 2007, 8, 560–566. 10.1002/cbic.200600386. [DOI] [PubMed] [Google Scholar]
- Avital-Shmilovici M.; Shabat D. Enzymatic Activation of Hydrophobic Self-Immolative Dendrimers: The Effect of Reporters with Ionizable Functional Groups. Bioorg. Med. Chem. Lett. 2009, 19, 3959–3962. 10.1016/j.bmcl.2009.03.002. [DOI] [PubMed] [Google Scholar]
- Sella E.; Shabat D. Dendritic Chain Reaction. J. Am. Chem. Soc. 2009, 131, 9934–9936. 10.1021/ja903032t. [DOI] [PubMed] [Google Scholar]
- DeWit M. A.; Gillies E. R. A Cascade Biodegradable Polymer Based on Alternating Cyclization and Elimination Reaction. J. Am. Chem. Soc. 2009, 131, 18327–18334. 10.1021/ja905343x. [DOI] [PubMed] [Google Scholar]
- Esser-Kahn A. P.; Sottos N. R.; White S. R.; Moore J. S. Programmable Microcapsules from Self-Immolative Polymers. J. Am. Chem. Soc. 2010, 132, 10266–10268. 10.1021/ja104812p. [DOI] [PubMed] [Google Scholar]
- Seo W.; Phillips S. T. Patterned Plastics That Change Physical Structure in Response to Applied Chemical Signals. J. Am. Chem. Soc. 2010, 132, 9234–9235. 10.1021/ja104420k. [DOI] [PubMed] [Google Scholar]
- DiLauro A. M.; Phillips S. T. End-capped poly(4,5-dichlorophthalaldehyde): a stable self-immolative poly(aldehyde) for translating specific inputs into amplified outputs, both in solution and the solid state. Polym. Chem. 2015, 6, 3252–3258. 10.1039/C5PY00190K. [DOI] [Google Scholar]
- Dilauro A. M.; Lewis G. G.; Phillips S. T. Self-Immolative Poly(4,5-dichlorophthalaldehyde) and its Applications in Multi-Stimuli-Responsive Macroscopic Plastics. Angew. Chem., Int. Ed. 2015, 54, 6200–6205. 10.1002/anie.201501320. [DOI] [PubMed] [Google Scholar]
- Babra T. S.; Trivedi A.; Warriner C. N.; Bazin N.; Castiglione D.; Sivour C.; Hayes W.; Greenland B. W. Fluoride degradable and thermally debondable polyurethane based adhesive. Polym. Chem. 2017, 8, 7207–7216. 10.1039/C7PY01653K. [DOI] [Google Scholar]
- Carl P. L.; Chakravarty P. K.; Katzenellenbogen J. A. A Novel Connector Linkage Applicable in Prodrug Design. J. Med. Chem. 1981, 24, 479–480. 10.1021/jm00137a001. [DOI] [PubMed] [Google Scholar]
- Albert A. Chemical Aspects of Selective Toxicity. Nature 1958, 182, 421–423. 10.1038/182421a0. [DOI] [PubMed] [Google Scholar]
- Gnaim S.; Shabat D. Quinone-Methide Species, a Gateway to Functional Molecular Systems: From Self-Immolative Dendrimers to Long-Wave Fluorescent Dyes. Acc. Chem. Res. 2014, 47, 2970–2984. 10.1021/ar500179y. [DOI] [PubMed] [Google Scholar]
- Acton A. L.; Leroux F.; Feula A.; Melia K.; Sambrook M. R.; Hayes W.; Russell A. T. Self-Immolative Systems for the Disclosure of Reactive Electrophilic Alkylating Agents. Chem. Commun. 2019, 55, 5219–5222. 10.1039/C8CC09728C. [DOI] [PubMed] [Google Scholar]
- Tuo W.; Bouqet J.; Taran F.; Le Gall T. A FRET probe for the detection of alkylating agents. Chem. Commun. 2019, 55, 8655–8658. 10.1039/C9CC04391H. [DOI] [PubMed] [Google Scholar]
- Warnecke A.; Kratz F. 2,4-Bis(Hydroxymethyl)Aniline as a Building Block for Oligomers with Self-Eliminating and Multiple Release Properties. J. Org. Chem. 2008, 73, 1546–1552. 10.1021/jo702484z. [DOI] [PubMed] [Google Scholar]
- Perry-Feigenbaum R.; Baran P. S.; Shabat D. The Pyridinone-Methide Elimination. Org. Biomol. Chem. 2009, 7, 4825–4828. 10.1039/b915265b. [DOI] [PubMed] [Google Scholar]
- Schmid K. M.; Jensen L.; Phillips S. T. A Self-Immolative Spacer That Enables Tunable Controlled Release of Phenols under Neutral Conditions. J. Org. Chem. 2012, 77, 4363–4374. 10.1021/jo300400q. [DOI] [PubMed] [Google Scholar]
- Kunz H. Der 2-(Triphenylphosphonio)äthoxycarbonyl-Rest als Schutzgruppe für die Aminofunktion in Aminosäuren und Peptiden. Chem. Ber. 1976, 109, 2670–2683. 10.1002/cber.19761090803. [DOI] [Google Scholar]
- Chantreux D.; Gamet J. P.; Jacquier R.; Verducci J. The 2-(diphenylphosphino) ethyl group (DPPE) as a new carboxyl-protecting group in peptide chemistry. Tetrahedron 1984, 40, 3087–3094. 10.1016/S0040-4020(01)82433-9. [DOI] [Google Scholar]
- Cockroft S. L.; Perkins J.; Zonta C.; Adams H.; Spey S. E.; Low C. M. R.; Vinter J. G.; Lawson K. R.; Urch C. J.; Hunter C. A. Substituent Effects on Aromatic Stacking Interactions. Org. Biomol. Chem. 2007, 5, 1062–1080. 10.1039/b617576g. [DOI] [PubMed] [Google Scholar]
- Iwai T.; Fujihara T.; Terao J.; Tsuji Y. Iridium-Catalyzed Annulation of N-Arylcarbamoyl Chlorides with Internal Alkynes. J. Am. Chem. Soc. 2010, 132, 9602–9603. 10.1021/ja104153k. [DOI] [PubMed] [Google Scholar]
- Lebleu T.; Ma X.; Maddaluno J.; Legros J. Selective Monomethylation of Primary Amines with Simple Electrophiles. Chem. Commun. 2014, 50, 1836–1838. 10.1039/c3cc48997c. [DOI] [PubMed] [Google Scholar]
- Patel J. Z.; Nevalainen T. J.; Savinainen J. R.; Adams Y.; Laitinen T.; Runyon R. S.; Vaara M.; Ahenkorah S.; Kaczor A. A.; Navia-Paldanius D.; Gynther M.; Aaltonen N.; Joharapurkar A. A.; Jain M. R.; Haka A. S.; Maxfield F. R.; Laitinen J. T.; Parkkari T. Optimization of 1,2,5-Thiadiazole Carbamates as Potent and Selective ABHD6 Inhibitors. ChemMedChem 2015, 10, 253–265. 10.1002/cmdc.201402453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukano C.; Okuno M.; Takemoto Y. Palladium-Catalyzed Amidation by Chemoselective C(sp3)-H Activation: Concise Route to Oxindoles Using a Carbamoyl Chloride Precursor. Angew. Chem., Int. Ed. 2012, 51, 2763–2766. 10.1002/anie.201108889. [DOI] [PubMed] [Google Scholar]
- Smith B. D.; Goodenough-Lashua D. A. M.; D’Souza C. J. E.; Norton K. J.; Schmidt L. M.; Tung J. C. Substituent Effects on the Barrier to Carbamate C-N Rotation. Tetrahedron Lett. 2004, 45, 2747–2749. 10.1016/j.tetlet.2004.02.037. [DOI] [Google Scholar]
- Shanan-Atidi H.; Bar-Eli K. H. A Convenient Method for Obtaining Free Energies of Activation by the Coalescence Temperature of an Unequal Doublet. J. Phys. Chem. A. 1970, 74, 961–963. 10.1021/j100699a054. [DOI] [Google Scholar]
- Johnson S. L.; Morrison D. L. Kinetics and mechanism of decarboxylation of N-arylcarbamates. Evidence for kinetically important zwitterionic carbamic acid species of short lifetime. J. Am. Chem. Soc. 1972, 94, 1323–1334. 10.1021/ja00759a045. [DOI] [PubMed] [Google Scholar]
- Hughes E. D.; Ingold C. K.; Shapiro U. G. Mechanism of Substitution at a Saturated Carbon Atom. Part VI. Hydrolysis of isopropyl bromide. J. Chem. Soc. 1936, 225–236. 10.1039/jr9360000225. [DOI] [Google Scholar]
- Ingold C. K.Structure and Mechanism in Organic Chemistry; G. Bell and Sons Ltd.: London, 1953; p 346. [Google Scholar]
- Kaljurand I.; Kütt A.; Sooväli L.; Rodima T.; Mäemets V.; Leito I.; Koppel I. A. Extension of the Self-Consistent Spectrophotometric Basicity Scale in Acetonitrile to a Full Span of 28 pKa Units: Unification of Different Basicity Scales. J. Org. Chem. 2005, 70, 1019–1028. 10.1021/jo048252w. [DOI] [PubMed] [Google Scholar]
- Kanbayashi N.; Onitsuka K. Ruthenium-Catalyzed Regio- and Enantioselective Allylic Substitution with Water: Direct Synthesis of Chiral Allylic Alcohols. Angew. Chem., Int. Ed. 2011, 50, 5197–5199. 10.1002/anie.201101078. [DOI] [PubMed] [Google Scholar]
- Vargas S.; Rubio M.; Suárez A.; Del Río D.; Álvarez E.; Pizzano A. Iridium Complexes with Phosphine - Phosphite Ligands. Structural Aspects and Application in the Catalytic Asymmetric Hydrogenation of N-Aryl Imines. Organometallics 2006, 25, 961–973. 10.1021/om050885t. [DOI] [Google Scholar]
- Agilent CrysAlis PRO; Agilent Technologies, Ltd.: Yarnton, Oxfordshire, England, 2014.
- Palatinus L.; Chapuis G. SUPERFLIP– a computer program for the solution of crystal structures by charge flipping in arbitrary dimensions. J. Appl. Crystallogr. 2007, 40, 786–790. 10.1107/S0021889807029238. [DOI] [Google Scholar]
- Betteridge P. W.; Carruthers J. R.; Cooper R. I.; Prout K.; Watkin D. J. CRYSTALS version 12: software for guided crystal structure analysis. J. Appl. Crystallogr. 2003, 36, 1487. 10.1107/S0021889803021800. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.