

Abstract
Idiopathic pulmonary fibrosis (IPF) is a disease of progressive scarring caused by excessive extracellular matrix (ECM) deposition and activation of α-SMA-expressing myofibroblasts. Human antigen R (HuR) is an RNA binding protein that promotes protein translation. Upon translocation from the nucleus to the cytoplasm, HuR functions to stabilize mRNA to increase protein levels. However, the role of HuR in promoting ECM production, myofibroblast differentiation and lung fibrosis is unknown. Human lung fibroblasts (HLFs) treated with TGFβ1 showed a significant increase in translocation of HuR from the nucleus to the cytoplasm. TGF-β-treated HLFs that were transfected with HuR siRNA had a significant reduction in α-SMA protein as well as the ECM proteins COL1A1, COL3A and FN1. HuR was also bound to mRNA for ACTA2, COL1A1, COL3A1 and FN. HuR knock-down affected the mRNA stability of ACTA2 but not that of the ECM genes COL1A1, COL3A1 or FN. In mouse models of pulmonary fibrosis, there was higher cytoplasmic HuR in lung structural cells compared to control mice. In human IPF lungs, there was also more cytoplasmic HuR. This study is the first to show that HuR in lung fibroblast controls their differentiation to myofibroblasts and consequent ECM production. Further research on HuR could assist in establishing the basis for the development of new target therapy for fibrotic diseases such as IPF.
Keywords: Idiopathic pulmonary fibrosis, human antigen R, bleomycin, radiation, ELAVL1, α-smooth muscle actin, collagen
1. INTRODUCTION
Interstitial lung disease (ILD) is a heterogeneous collection of more than 200 lung disorders characterized by varying degrees of fibrosis and inflammation of the lung parenchyma. Of these, idiopathic pulmonary fibrosis (IPF) is the most common and deadly and although the etiology of IPF is unknown, risk factors include environmental agents such as cigarette smoke (Martin, Chung, & Kanne, 2016) silica and wood dust (Spagnolo et al., 2015); gastroesophageal reflux disease (GERD) (Antoniou et al., 2014; Spagnolo et al., 2015); viral infections (Naik & Moore, 2010) and presence of autoimmune conditions (Hoyne, Elliott, Mutsaers, & Prele, 2017). IPF is a progressive and irreversible disease where there is permanent destruction of the lung architecture due to scar formation that stiffens the lung and disrupts oxygen exchange. Incidence of IPF ranges from 4.6 and 7.4 cases per 100,000 in Europe and USA (Martin et al., 2016; Raghu, Nyberg, & Morgan, 2004) and 18 cases per 100,000 in Canada (Hopkins, Burke, Fell, Dion, & Kolb, 2016). This rose to 93.7 per 100,000 person years over the age of 65 (Raghu et al., 2014). Treatment options for IPF are limited and have been shown to slow, but not reverse, disease progression. The current standard-of-care is typically centered on supplemental oxygen, pulmonary rehabilitation and prescription of anti-fibrotic medication- either pirfenidone (Esbriet®) or nintedanib (Ofev®). These two drugs may slow fibrosis progression but unfortunately do not improve survival (Raghu et al., 2015). The only definitive cure for IPF is lung transplantation, which is only available to a minority of patients. A major impediment to the development of effective therapies for IPF is our limited understanding of the molecular and genetic pathways that contribute to the disease process.
Despite extensive research on IPF, the exact pathogenesis is not clear. The current paradigm is that recurrent damage to the alveolar epithelium in a susceptible individual drives an abnormal wound-healing response that results in fibrosis rather than repair. Mechanistically, it is thought that alveolar epithelial type II cell damage by microinjuries disrupts the continuity of the basal lamina within the alveoli (Loomis-King, Flaherty, & Moore, 2013). Such damage to the epithelium drives the accumulation of myofibroblasts, largely differentiated from resident lung fibroblasts (Wuyts et al., 2013). Myofibroblasts are α-smooth muscle actin (α-SMA)-expressing cells that are the key effector cells in pulmonary fibrosis, forming characteristic “fibroblastic foci’ within the lung. Myofibroblasts produce copious amounts of extracellular matrix (ECM) proteins such as collagens (COL) and fibronectin (FN) that contribute to fibrosis. The differentiation of fibroblasts into ECM-producing myofibroblasts occurs under the direction of cytokines and growth factors, particularly transforming growth factor-β1 (TGF-β1). TGF-β levels are significantly increased in the lungs of IPF patients (Khalil et al., 1991) and over-expression of TGF-β in animal models potently induces pulmonary fibrosis (Sime, Xing, Graham, Csaky, & Gauldie, 1997), supporting that TGF-β is a key driver in IPF development.
Studies have shown that myofibroblasts from patients with IPF have increased protein translation, especially from mRNA that encode ECM (Larsson et al., 2008). Although the importance of post-transcriptional events in the context of pulmonary fibrosis remains largely unknown, several RNA binding proteins including human antigen R (HuR), have bene shown to play an important role in the etiology of several human pathologies (Giaginis et al., 2017). HuR is a member of the Hu/embryonic lethal, abnormal vision (ELAV) family of RNA binding proteins that is encoded by the ELAVL1 gene. In the lung, HuR is found in many cell types including epithelial cells and fibroblasts (J. Fan et al., 2011; Zago et al., 2013). Members of the ELAV family control the fate of mRNA by modulating their turnover (stability or decay), localization and translation (Giaginis et al., 2017; Mukherjee et al., 2011; W. Wang et al., 2002). HuR is best-known to stabilize target mRNA by binding to adenylate-and uridylate-rich elements (AREs) in the 3’ untranslated regions (3’UTRs) (Myer, Fan, & Steitz, 1997). In a resting cell, HuR is predominantly localized in the nucleus. Upon activation by stress conditions such as ultraviolet (UV) radiation, proliferation, nutrient depletion or immune activation, HuR binds to and transports target mRNA to the cytoplasm where it regulates their stability and/or translation (Singh, Martinez, Govindaraju, & Lee, 2013; Xu, Di Marco, Gallouzi, Rola-Pleszczynski, & Radzioch, 2005). Therefore, we hypothesized that HuR contributes to pulmonary fibrosis by post-transcriptionally regulating the expression of mRNAs encoding proteins that are implicated in multiple facets of disease pathogenesis. Herein we report that HuR is essential for the differentiation of lung fibroblasts into myofibroblasts and consequent ECM production in response to TGF-β. This was due to increased translocation of HuR from the nucleus to the cytoplasm, which was also observed in animal models of lung fibrosis and lung samples taken from individuals with IPF. HuR also bound to fibrotic mRNA. Thus, HuR may be involved in the pathogenesis of pulmonary fibrosis by facilitating the differentiation of myofibroblasts and increasing ECM production in a susceptible individual.
2. MATERIALS AND METHODS
2.1. Chemicals
All chemicals were purchased from Sigma-Aldrich (St Louis, MO, USA) unless otherwise indicated.
2.2. Derivation and culture of primary lung fibroblasts
Human lung fibroblasts (HLFs) used in this study were derived from lung tissue obtained from subjects undergoing lung resection surgery at McMaster University as we have previously described (Sheridan et al., 2015). All cells used in this study were obtained from a subject with no smoking history or relevant risk factors (e.g. radiation or medication) for lung fibrosis. This study was approved by the Research Ethics Board of St, Joseph’s Healthcare Hamilton and an informed written consent was obtained from each patient. Derivation of mouse lung fibroblasts (MLFs) is also as previously described (Baglole et al., 2008; Hecht et al., 2014). Lung fibroblasts were cultured in Gibco™ Minimum Essential Media (MEM) (Thermo Fisher Scientific, USA) containing 10% fetal bovine serum (FBS; Hyclone Laboratories, Logan, UT) supplemented with gentamycin (WISENT Inc, Canada), Antibiotic-Antimycotic (WISENT Inc, Canada) and glutamax (Thermo Fisher Scientific, USA). Cell at passages between 4 and 11 were used for all experiments.
2.3. Western blot
Fibroblasts were cultured with serum-free MEM for 18 hours before treatment with TGF-β1 from 12-72 hrs. HLFs were then rinsed with PBS and lysed by RIPA buffer (Thermo Scientific, Rockford) containing protease inhibitors (PIC, Roche, US). Protein concentrations were determined using the bicinchoninic acid (BCA) protein assay kit (Thermo Fisher Scientific, USA). Cell lysates were mixed with loading buffer and boiled for 10 min. Protein samples (20 μg per lane) were electrophoresed on 7.5% SDS PAGE and resolved to PVDF membrane (Bio-Rad Laboratories, Hercules, CA). After blocking for 1 h with 5% non-fat dry milk, the membranes were incubated at 4 °C overnight with an anti-HuR antibody (1:2000; Santa Cruz, CA). The next day, the membranes were probed with the horseradish peroxidase (HRP)-linked anti-mouse IgG (1:10000, Cell Signaling Technologies, CA). Membranes were then visualized with ClarityTM western ECL substrate (Bio-Rad Laboratories, Mississauga, ON) or AmershamTM western ECL substrate (GE Healthcare, Italy). Protein bands were visualized using a ChemiDoc™ MP Imaging System (Bio-Rad, CA). Densitometric analysis was performed using Image Lab™ Software Version 5 (Bio-Rad, CA). Tubulin (1:50000; Sigma, CA) was used as the loading control. Protein expression was normalized to tubulin. Additional antibodies included anti-α-SMA (1:5000; Sigma, CA), anti-COL1A1 (1:200; Santa Cruz, CA), anti-COL3A1 (1:200; Santa Cruz, CA) and anti-FN (1:200; Santa Cruz, CA); images of uncropped blots are in the online supplement.
2.4. Mouse models of pulmonary fibrosis
Thoracic radiation and bleomycin were administered to mice age 8-10 weeks to induce pulmonary fibrosis as previously described (Lemay & Haston, 2018). After euthanasia, the lungs were removed, and the single left lobe of each mouse was perfused with 10% neutral buffered formalin and submitted for histological processing for H& E staining and Masson’s trichrome staining as described (Bergeron, Stefanov, & Haston, 2018). Samples were also processed for immunofluorescence (IF) for HuR as indicated below. All animal procedures were approved by the McGill University Animal Care Committee.
2.5. Quantitative RT-PCR
Total RNA was isolated with Trizol and the concentration and quality of the RNA confirmed using NanoDrop 1000 spectrophotometer (Infinite M200 pro, TECAN, CA). Reverse transcription of 25 ng of RNA to cDNA was performed using iScript™ Reverse Transcription Supermix (Bio-Rad Laboratories, Mississauga, ON). Quantitative PCR (qPCR) was done by addition of 1 μl of cDNA and 0.5 μM primers with SsoFast™ EvaGreen® (BioRad Laboratories, Mississauga, ON). PCR amplification was performed using a CFX96 Real-Time PCR Detection System (Bio-Rad, CA). Thermal cycling was initiated at 95°C for 3 minutes and followed by 39 cycles of denaturation at 95°C for 10 seconds and annealing at 59°C for 5 seconds. Gene expression was analyzed using the -ΔΔCt method, and results are presented as fold-change normalized to housekeeping gene GAPDH. The primers were designed and purchased from Integrated DNA Technologies (Marlton, NJ). Primer sequences are in Table 1.
Table 1.
Primer Sequences
| Gene | Forward primer sequence | Reverse primer sequence |
|---|---|---|
| ELAVL1 | AACGCCTCCTCCGGCTGGTGC | GCGGTAGCCGTTCAGGCT GGC |
| ACTA2 | GACCGAATGCAGAAGGAGAT | CACCGATCCAGACAGAGTATTT |
| COL1A1 | CAGACTGGCAACCTCAAGAA | CAGTGACGCTGTAGGTGAAG |
| COL3A1 | GCTCTGCTTCATCCCACTATTA | CTGGCTTCCAGACATCTCTATC |
| FN1 | CTGAGACCACCATCACCATTAG | GATGGTTCTCTGGATTGGAGTC |
| GAPDH | GTCTCCTCTGACTTCAACAGC | ACCACCCTGTTGCTGTAGCCA |
2.6. Immunofluorescence (IF)
Cells were treated with TGF-β1 for 6, 24 and 48 hours. In HLFs whereby HuR was knocked-down by siRNA, cells were treated with TGF-β1 for 72 hours. The cells were then fixed with 4% paraformaldehyde (PFA) for 15 min and permeabilized for 30 min in PBS containing 0.5 % Triton. After incubation with blocking buffer (Dako) for 1 hour at room temperature, cells were incubated in a 1:50 or 1:300 dilution of anti-HuR antibody in blocking buffer (Dako) for 2 h at room temperature. Cells were washed with 1x PBS, incubated for 1 h with the secondary antibody (Alexa fluor 488, 1:1000). For α-SMA, cells were incubated with the primary antibody at a dilution of 1:1000. Cells were washed with PBS and nucleus was stained with DAPI or Hoechst for 15 min (1:1000). Cell images were acquired with a Zeiss LSM 780 confocal microscope (Zeiss, Oberkochen, Baden-Württemberg, German). ImageJ (National Institutes of Health, USA) (Schneider, Rasband, & Eliceiri, 2012) was used to process and analyze the images for quantification HuR expression.
2.7. Cytoplasmic and nuclear protein fractionation
Cytoplasmic and nuclear protein fractions were obtained using a nuclear extraction kit as per manufacturer instructions (Active Motif, Carlsbad, CA). For these experiments, HLFs were untreated or treated with TGF-β1 (5 ng/ml) for various times. In addition, cells were also treated with actinomycin D (ActD; 0.5 µg/ml) for 6 h. Protein concentrations were determined by the BCA protein assay kit. Western blot and antibodies used were described in Section 2.3. Lamin A/C (1:1000; Cell Signaling Technologies, CA) was used as a marker for the nuclear fraction.
2.8. HuR knock-down
Approximately 100,000 HLFs were seeded into 6-well plates containing 2 ml of 10% FBS/MEM without antibiotics and allowed to grow overnight for 24 hours. Transfections were performed with either 60 nM of HuR small interfering RNA (siHuR) or control (scrambled) siRNA (siCtrl; Santa Cruz, CA). siRNA-transfected cells were incubated for an additional 24 hours, followed by serum starvation for 18 h. Cells were harvested 4, 8, 24 and 48 h after 5 ng/ml TGF-β1 (protein) or at 3, 6, 24 and 48 hours for RNA. HuR, α-SMA and ECM expression was assessed by western blot and qRT-PCR, respectively.
2.9. RNA Immunoprecipitation- qPCR (RIP-qPCR):
HLFs were grown to approximately 70-80% confluence and cultured with serum-free MEM for 18 hours before the treatment. Then, cells were treated with 5 ng/ml TGF-β1 for 48h. After treatment, cells were rinsed with PBS and then collected by cell scraper in PBS. Cells were centrifuged at 1500 rpm, 4°C for 5 minutes, then the PBS were discarded. The cell pellets were harvested in the lysis buffer (50mM Tris PH 8; 0.5% Triton X100; 450mM NaCl; protease inhibitor cocktail; phosphatase inhibitor (Roche, US)), incubated for 15 min on ice and then centrifuged at 10,000 rpm, 4°C for 15 min. The cell extracts were transferred into a new tube and another buffer was added (50mM Tris PH 8; 0.5% Triton X100; 10% glycerol; protease inhibitor cocktail; phosphatase inhibitor). The protein concentration was measured by BCA Protein Assay Kit. Thirty-five μl of protein G Sepharose™ 4 fast glow beads (GE Healthcare) were pre-coated with 2 μg of IgG (Cell Signaling Technologies, CA) or 2 μg of anti-HuR (Santa Cruz Biotechnology) antibodies overnight on a rotator at 4°C. Beads were washed three times with buffer (50mM Tris PH 8; 0.5% Triton X100; 150mM NaCl) and incubated with cell extracts for 2 hours at 4°C. Beads were washed three times to remove unbound material. RNAs were then extracted, reverse transcribed and analyzed by qPCR (qRT-PCR) as described above. RNA expression was normalized to GAPDH mRNA bound in a non-specific manner to IgG (Keene, Komisarow, & Friedersdorf, 2006; Mubaid et al., 2019).
2.10. Actinomycin D pulse-chase experiments
HLFs transfected with HuR siRNA oligos were treated with or without TGF-β1 for 24 hours. The cells were subsequently treated with ActinomycinD (1μg/ml) for 1, 3 or 6 hours (Zago et al., 2013). RNA was extracted from the cells and quantified by qRT-PCR. mRNA decay was calculated as the percentage of mRNA remaining over time compared with the amount before the addition of actinomycin D.
2.11. Human lung tissue acquisition and IF
Lung tissue from patients with IPF were obtained from the University of Michigan Interstitial Lung Disease Biorepository. Non-fibrotic control lung tissue was also obtained from the University of Michigan Lung Biorepository through donor lungs provided by Gift of Life, Michigan. All IPF lung tissues were collected at the time of lung transplantation, and all cases of IPF were diagnosed by multidisciplinary consensus conference at the University of Michigan prior to transplantation. Cases of IPF were further confirmed diagnostically after transplantation to show a histologic pattern of usual interstitial pneumonia. All tissues were acquired using research protocols and informed consent that were approved by the Michigan Medicine Institutional Review Board (HUM00105694). All materials were de-identified to the research team. Formalin-fixed paraffin-embedded lung tissue from IPF and control subjects were sectioned and stained for HuR using the Discovery Ultra Ventana (Roche, CA). Briefly, slides were de-parafinized and rehydrated. Antigen retrieval was done using CC1 buffer for 32 mins. Slides were incubated with primary vimentin antibody (Cell Signaling #5741 clone D21H3) at 37°C in dilution 1:50 for 24 min After washing, anti-rabbit HRP was added for 20 mins at RT followed by a wash. FITC was used to detect the signal (green). Slides were then incubated with HuR antibody (Santa Cruz, sc-5261) at 37 °C in dilution 1:100 for 24 min followed by anti- mouse HRP for 20 mins at RT; detection was via rhodamine (red). Finally, slides were washed and stained with DAPI at a dilution of 1:8000 and mounted with aqueous mounting media (Permafluor; Thermo Scientific. Cat TA-030-FM).
2.12. Statistical analysis
All values are expressed as mean ± SEM. Statistical analysis was performed using analysis of variance (ANOVA) (for multiple comparisons) or an unpaired two tailed t test to analyze the differences between two groups. A p value < 0.05 was considered statistically significant. All statistical analyses were performed using GraphPad Prism 6 (GraphPad Software Inc. USA). For mRNA stability, we calculated the half-lives of mRNAs on a one-phase exponential decay model. The semi-logarithmic curves were also analyzed by GraphPad Prism software.
3. RESULTS
3.1. TGF-β1-induced differentiation of lung fibroblasts into myofibroblasts is accompanied by cytoplasmic translocation of HuR
Myofibroblasts play a dominant role in pulmonary fibrosis by increasing the production of ECM proteins. TGF-β1 is a pro-fibrotic cytokine that potently induces myofibroblast differentiation characterized by upregulation of α-SMA, COL1A1, COL3A1 and FN1. This was observed in our system with there being an increase in α-SMA expression at 48 and 72 hrs (Figure 1A) and those of COL1A1 at the 72 hr timepoint (Figure 1B). In addition, protein levels of COL3A1 increased significantly at 24, 48 and 72 hours (Figure 1C) while those of FN began to increase at 12 hrs (Figure 1D). Therefore, we used TGF-β at a concentration of 5 ng/ml for the remainder of the experimental conditions to evaluate the contribution of HuR to myofibroblast differentiation and ECM production.
FIGURE 1.
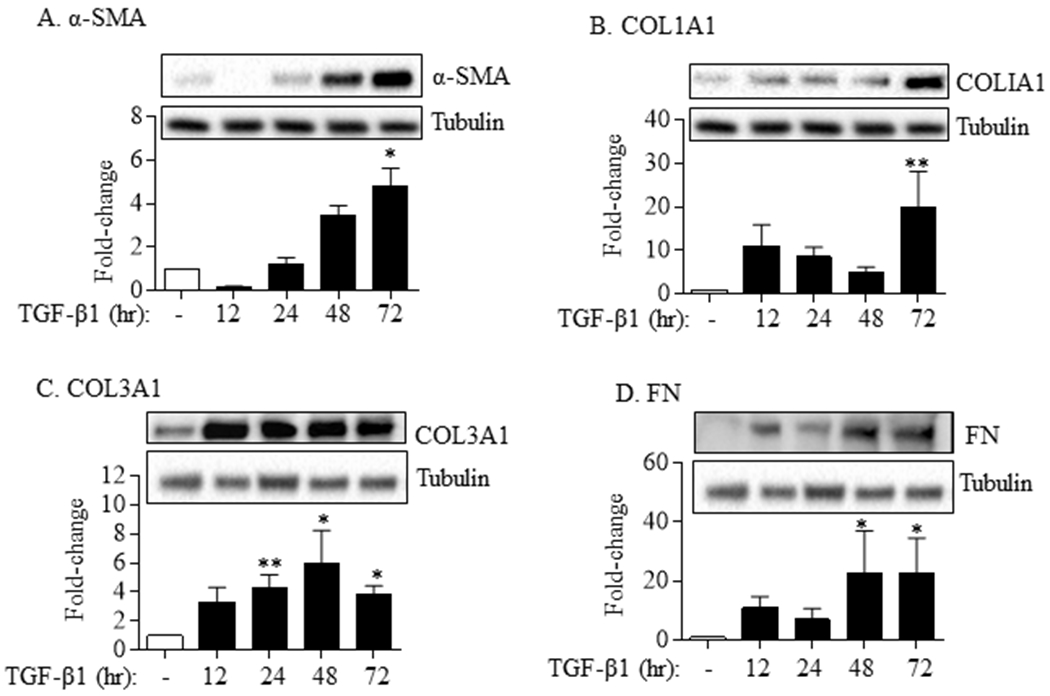
Lung fibroblast differentiation and ECM production in response to TGF-β1. TGF-β1 (5 ng/ml) significantly increased the protein levels of α-SMA (A), COLI (B), COLIII (C) and FN (D) in HLFs in a time-dependent fashion. Representative western blots are shown. Note that the same tubulin is presented between (A) and (B) and between (C) and (D) as these tubulin were from the same western blot/experiment used for α-SMA (A) and COL1A1 (B) and the same western blot/experiment for COL3A1 (C) and FN (D). Data are expressed as the mean ± SEM (n = 6 independent experiments). *p< 0.05 and **p< 0.01, as compared to control.
Although HuR is ubiquitous, its expression in lung fibroblasts in response to pro-fibrotic stimulation is not known. Therefore, we treated HLFs with TGF-β1 from 12–72 hrs and evaluated HuR (ELAVL1) mRNA and total cellular protein levels. TGF-β1 did not significantly alter total mRNA or protein expression of HuR over this time period (Figure 2A–B). We next evaluated the cellular localization of HuR in response to TGF-β because in order to post-transcriptionally regulate its target mRNA, HuR must translocate from the nucleus (where it normally resides) to the cytoplasm (Atasoy, Watson, Patel, & Keene, 1998). First, we treated HLFs with TGF-β1 for 6 or 24 hrs and separated total cell lysates into cytosolic and nuclear fractions for western blotting. There was an increase in cytosolic levels of HuR by approximately six-fold after 6hrs exposure to TGF-β1 (Figure 2C). As a complementary technique, we also assessed nuclear versus cytoplasmic localization of HuR using IF. In quiescent HLFs, HuR is predominantly in the nucleus (Figure 2D- left panel). After TGF-β1 treatment for 6 hours, there was accumulation of HuR in the cytoplasm (Figure 2D- middle panel). Actinomycin D, used as a positive control for HuR translocation to the cytoplasm (X. C. Fan & Steitz, 1998; Sengupta et al., 2003), elicited dramatic increase in cytosolic HuR localization (Figure 2D- right panel). Quantification of cytoplasmic HuR revealed that there was a significant increase in HuR in the cytoplasm of cells treated with TGF-β at all the times points evaluated (Figure 2E). Finally, we performed these experiments in MLFs, where there was also robust translocation of HuR to the cytoplasm upon exposure to TGF-β (Figure 2F). Thus, the pro-fibrotic cytokine TGF-β increases the translocation of HuR to the cytoplasm in conjunction with lung fibroblast differentiation and ECM production.
FIGURE 2.
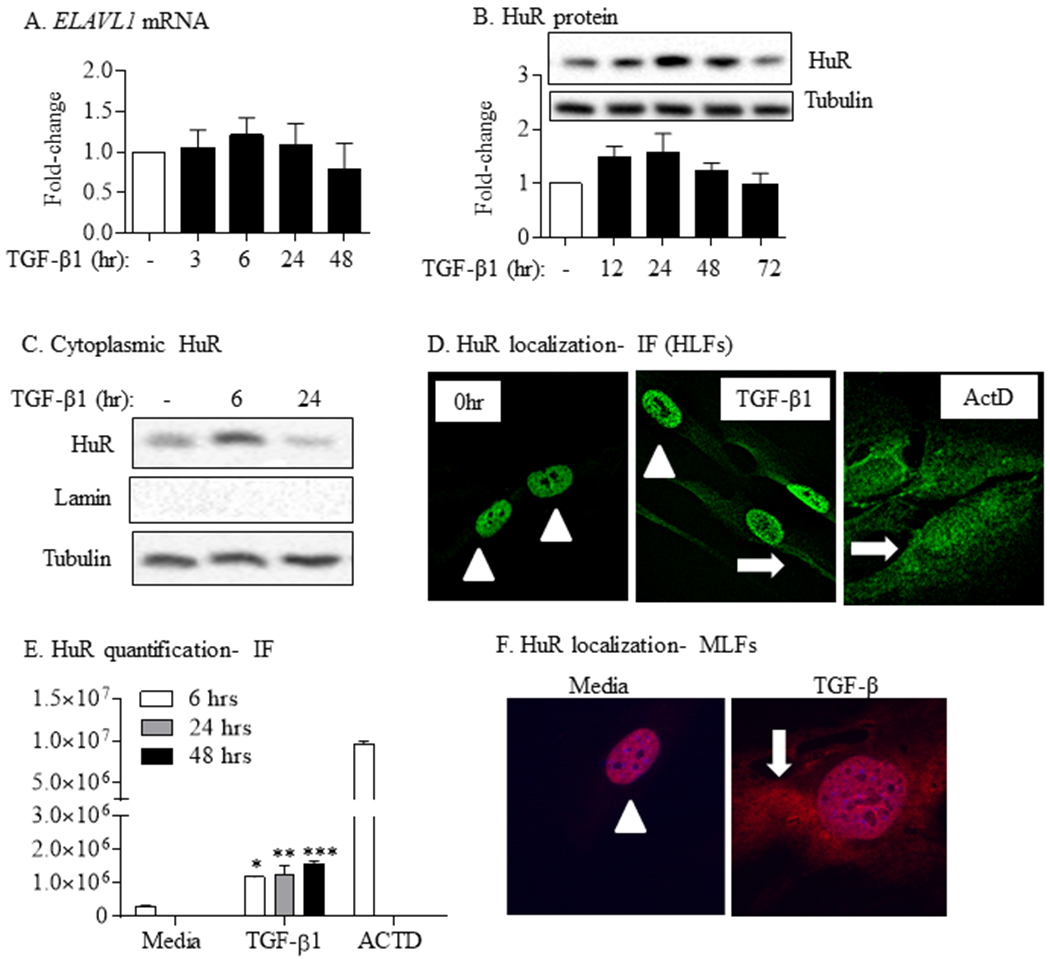
TGF-β1 increases cytoplasmic HuR localization in primary lung fibroblasts. TGF-β1 did not alter total HuR mRNA (ELAVL1) (A) or protein (B) expression. Representative western blot is shown. Note the same tubulin is also presented in Figure 1A and 1B as the HuR western blot selected was from the same experiment/western blot as that selected for α-SMA and COL1A1. Values are means ± SEM (n = 6). (C) Cytoplasmic HuR was increases in response to TGF-β compared to media-only cells. Tubulin was used as a housekeeping and Lamin for cytoplasmic purity. (D) HuR localization- IF (HLFs): HuR is predominantly nuclear (arrowheads) in media-only cells (0 hr). In response to TGF-β1, there is robust translocation of HuR to the cytoplasm (arrow). ActD was used as a positive control; note the re-distribution to the cytoplasm in response to ActD (arrows). (E) HuR quantification-IF: there was a significant increase in HuR cytoplasmic levels in response to TGF-β at all time-points. (F) HuR localization- MLFs: there was also translocation of HuR to the cytoplasm (arrow) in primary MLFs treated with TGF-β. Representative images are shown.
3.2. HuR is required for fibroblast differentiation into myofibroblasts and ECM production in response to TGF-β1
To show whether HuR is necessary for lung fibroblast differentiation and ECM production, we used siRNA to knock-down HuR in primary HLFs and assessed the effect of TGF-β1 on the expression of α-SMA and ECM markers. Transfection of HLFs with HuR-specific siRNA oligos triggered more than a 50% decrease in ELAVL1 mRNA (Figure 3A). Reducing HuR expression resulted in significant attenuation of TGFβ1-induced increase in ACTA2 (gene encoding for α-SMA) mRNA (Figure 3B) but had no significant effect on mRNA expression of COL1A1 (Figure 3C), COL3A1 (Figure 3D) or FN (Figure 3E). In contrast, analysis of protein levels by immunoblot in siHuR-transfected cells demonstrated decreased HuR expression as well as TGF-β-induced α-SMA protein (Figure 4A–4B) as well as COL1A1 (Figure 4C), COL3A1 (Figure 4D) and FN (Figure 4E). In addition, there was a robust change in the morphology of HLFs upon TGF-β1 treatment for 72 hours (siCtrl) as visualized by α-SMA staining (Figure 5); however, when HuR was reduced (siHuR), this change in cellular morphology was abrogated. Note the relative decrease in red fluorescence intensity (HuR) in the siHuR cells, indicative of HuR knockdown in these experiments (Figure 5- right panel). Thus, these data indicate that knock-down of HuR significantly reduces TGFβ1-induced fibroblast differentiation and consequent ECM production.
FIGURE 3.
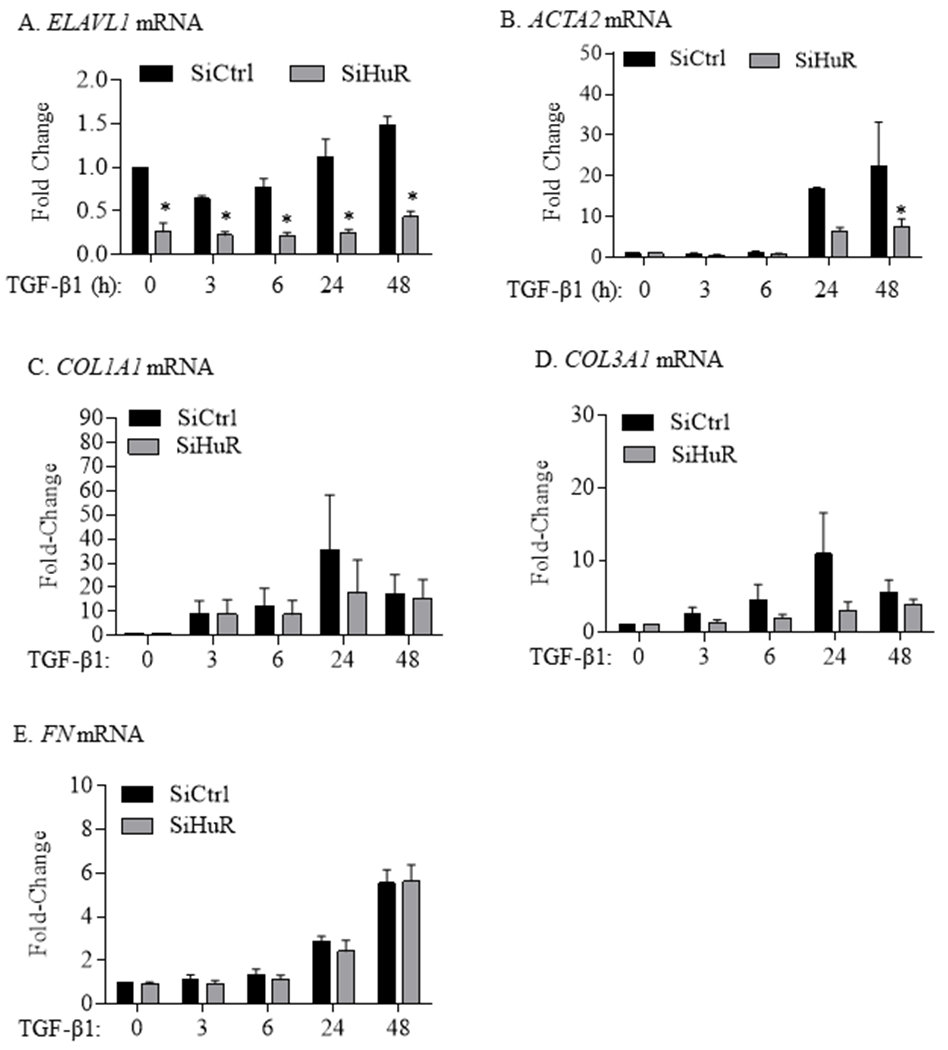
HuR knockdown does not affect ECM gene induction by TGF-β. (A) ELAVL1 mRNA: ELAVL1 mRNA levels were significantly reduced in HLFs transfected with HuR siRNA oligos and treated with TGF-β1 as well as those untreated. ELAVL1 mRNA values (means ± SEM) are expressed as fold change from values measured in cells transfected with control siRNA and untreated with TGF-β1 (*p< 0.05). (B) ACTA2 mRNA: There was significant attenuation of TGF-β1-induced increase ACTA2 mRNA expression (*p< 0.05 compared to control siRNA oligos) only at the 48-hour timepoint. There was no significant difference in the expression levels of COL1A1 (C), COL3A1 (D) or FN1 (E) mRNA between the SiCtrl and the SiHuR-transfected HLFs.
FIGURE 4.
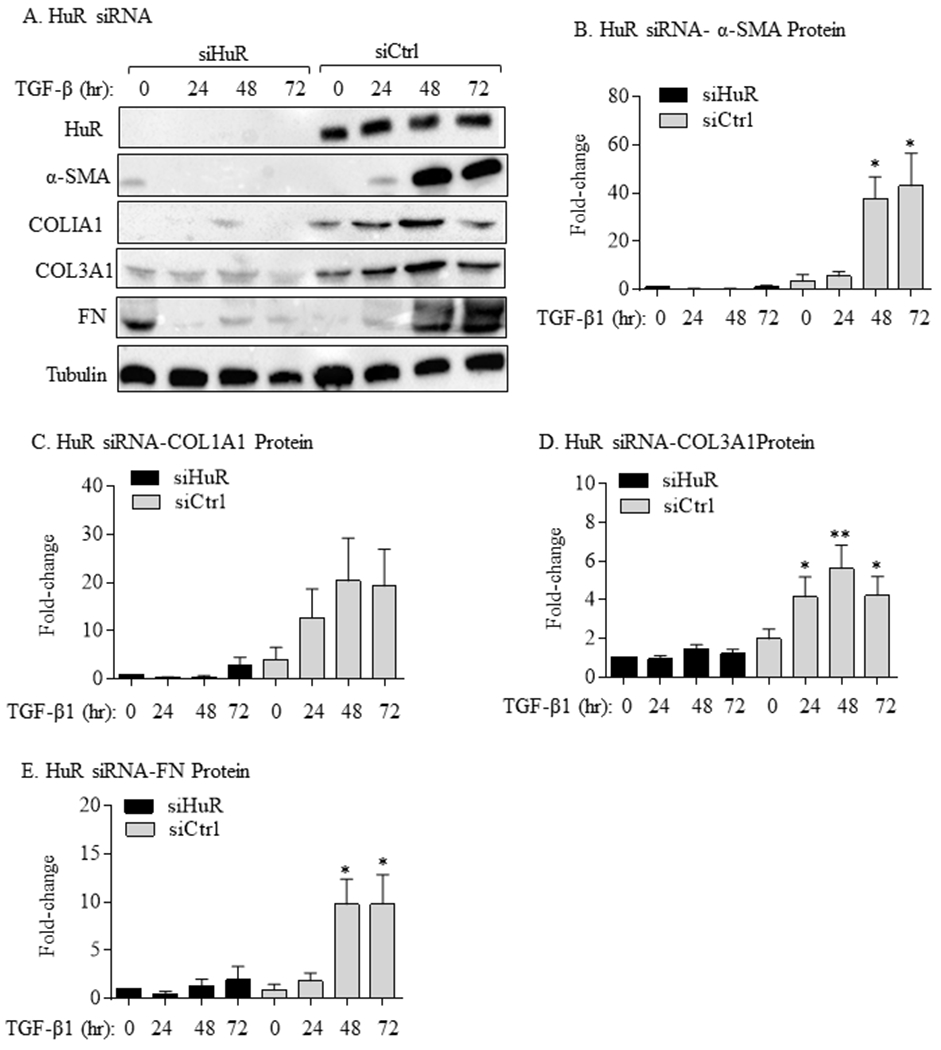
HuR knockdown decreases TGFβ1-induced myofibroblast differentiation and ECM protein production. (A) HuR siRNA: representative western blots showing reductions in the expression of HuR, α-SMA, COL1A1, COL3A1 and FN proteins. Equal loading was confirmed with tubulin. Quantification revealed that there was reduction in α-SMA (B), COL1A1 (C), COL3A1 (D) and FN (E) protein in TGFβ1-treated HLFs transfected with HuR siRNA (SiHuR) oligos mRNA compared to control siRNA (SiCtrl)-transfected cells. Results are expressed as the mean ± SEM; n = 3 independent experiments.
FIGURE 5.
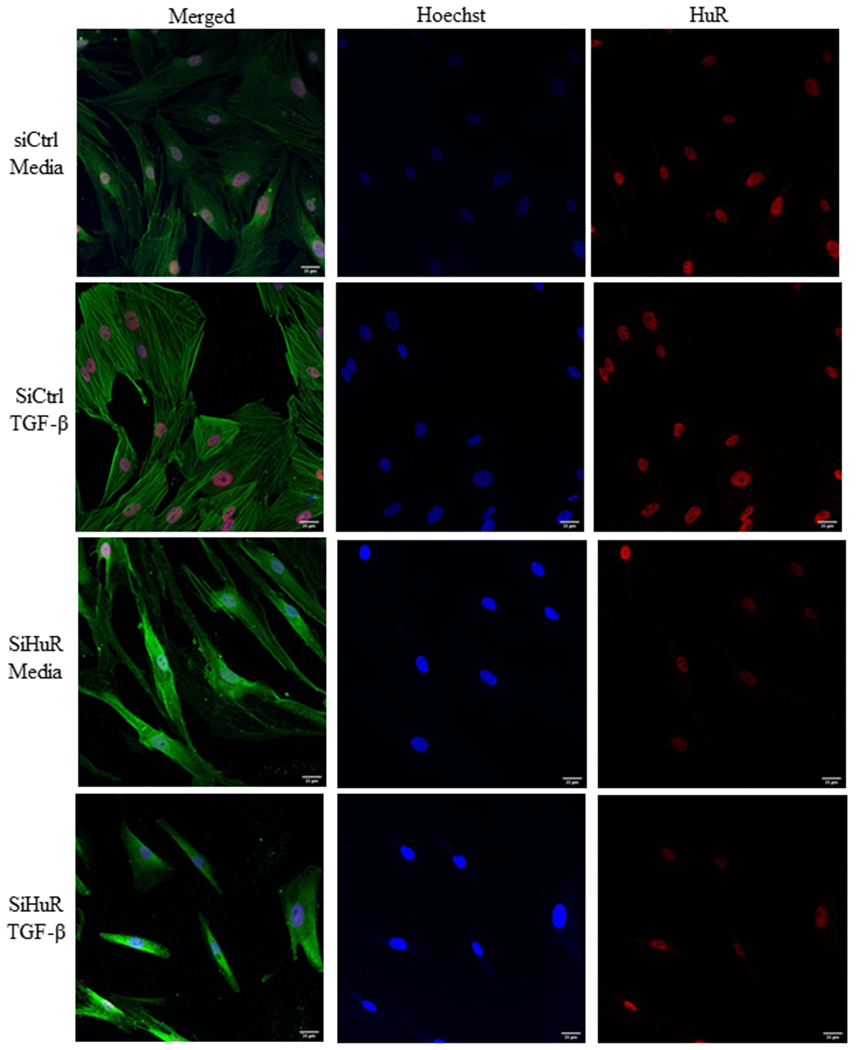
HuR controls myofibroblast differentiation in response to TGF-β. Using siRNA-mediated knockdown, HLFs were subsequently treated with TGF-β1 for 72 hours. Cells were then stained using antibodies against HuR (red) or α-SMA (green); nuclei were visualized with Hoechst (blue). Note that in cells whereby HuR was present (siCtrl) and treated with TGF-β, there was a dramatic increase in a-SMA as well as a change in morphology indicative of myofibroblasts. In HLFs where HuR was reduced, this change in cellular morphology was abrogated.
3.3. There is enrichment of ACTA2 and ECM mRNA bound to HuR in response to TGF-β1
An important step in the overall function of HuR is HuR binding target mRNA. Therefore, we next assessed HuR:mRNA interaction using RIP-qPCR (Keene et al., 2006; Mubaid et al., 2019; von Roretz et al., 2013), an antibody-based technique used to map RNA-protein interactions. We used RIP to determine whether HuR binds directly to fibrotic mRNA in HLFs treated with or without TGF-β1 for 48 hrs. IPs underwent western blotting (verification of HuR-IP) (Figure 6A) or total RNA extraction for the detection of gene enrichment using qPCR. We first verified that β-Actin mRNA (a known mRNA target of HuR (Calaluce et al., 2010)) significantly bound in HuR-IPs compared to (control) IgG-IP (Figure 6B). There is also binding of HuR to mRNA for ACTA2 (Figure 6C) and COL1A1 (Figure 6D). COL3A1 was also enriched but there was no further increase with TGF-β (Figure 6E). Finally, there was significant enrichment for FN1 after treatment with TGF-β (Figure 6F). Thus, HuR directly binds to fibrotic mRNA in primary HLFs under myofibroblast-differentiating conditions.
FIGURE 6.
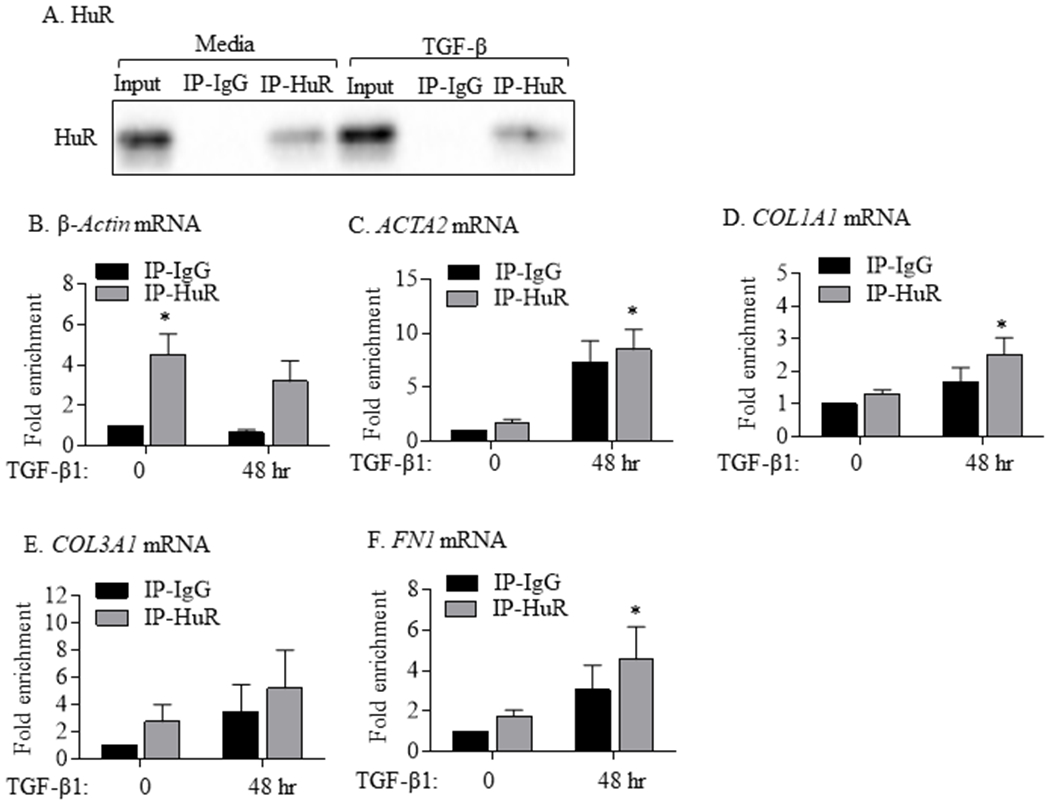
Binding of HuR to mRNAs of ACTA2 and ECM genes in HLFs treated with TGF-β1. (A) HuR: Representative western blot of HuR IP in HLFs treated with TGF-β1 for 48 hrs. Input refers to crude cell lysates. IP-IgG refers to immunoprecipitation (IP) with control IgG antibody while IP-HuR refers to the IP with anti-HuR IgG antibody. Note the presence of HuR protein in IP-HuR but not in IP-IgG. Detection of mRNA for β-Actin (B; positive control), ACTA2 (C), COL1A1 (D), COL3A1 (E), and FN1 (F) in IP-IgG and IP-HuR was done using qPCR. Values are expressed as fold change to values measured in IP-IgG in HLFs untreated with TGF-β1 (0 hr). Note the enrichment of β-Actin, ACTA2, COL1A1, and FN in IP-HuR of cells treated with TGF-β1. Results are presented as the mean ± SEM (n = 3 independent experiments) (p< 0.05 compared to IP-IgG).
3.4. HuR silencing destabilizes ACTA2 mRNA in TGF-β treated lung fibroblasts
As one of the mechanisms through which HuR controls protein expression is via mRNA stability, we next evaluated whether HuR stabilizes ACTA2, COL1A1, COL3A1 and FN mRNA in HLFs exposed to TGF-β1. For these experiments, HLFs were initially transfected 24 hrs with control or HuR siRNA oligos before treatment with TGF-β1 for 24 hrs. Cells were subsequently exposed to ActD (2.5 μg/ml) to inhibit new transcription and mRNA levels were then quantified by qPCR 1, 3 and 6 hrs after ActD treatment (Figure 7A) (Zago et al., 2013). ELAVL1 mRNA remained significantly lower in the knockdown cells for the duration of the experiments (Figure 7B). These experiments demonstrate that HuR knockdown had no significant effect on the decay of ELAVL1 (Figure 7B) but destabilized ACTA2 mRNA by 6 hours post-ActD treatment (Figure 7C). There was also a significantly less ACTA2 mRNA remaining 6 hours post-ActD in the siHuR cells compared to Time 0. However, silencing HuR had no impact on the stability of the ECM genes COL1A1 (Figure 7D), COL3A1 (Figure 7E) or FN (Figure 7F). Although there was a trend towards a decrease in mRNA levels, these did not achieve statistical significance. These data suggest that HuR differentially affects mRNA stability of ACTA2 but not of ECM markers in HLFs treated with TGF-β1.
FIGURE 7.
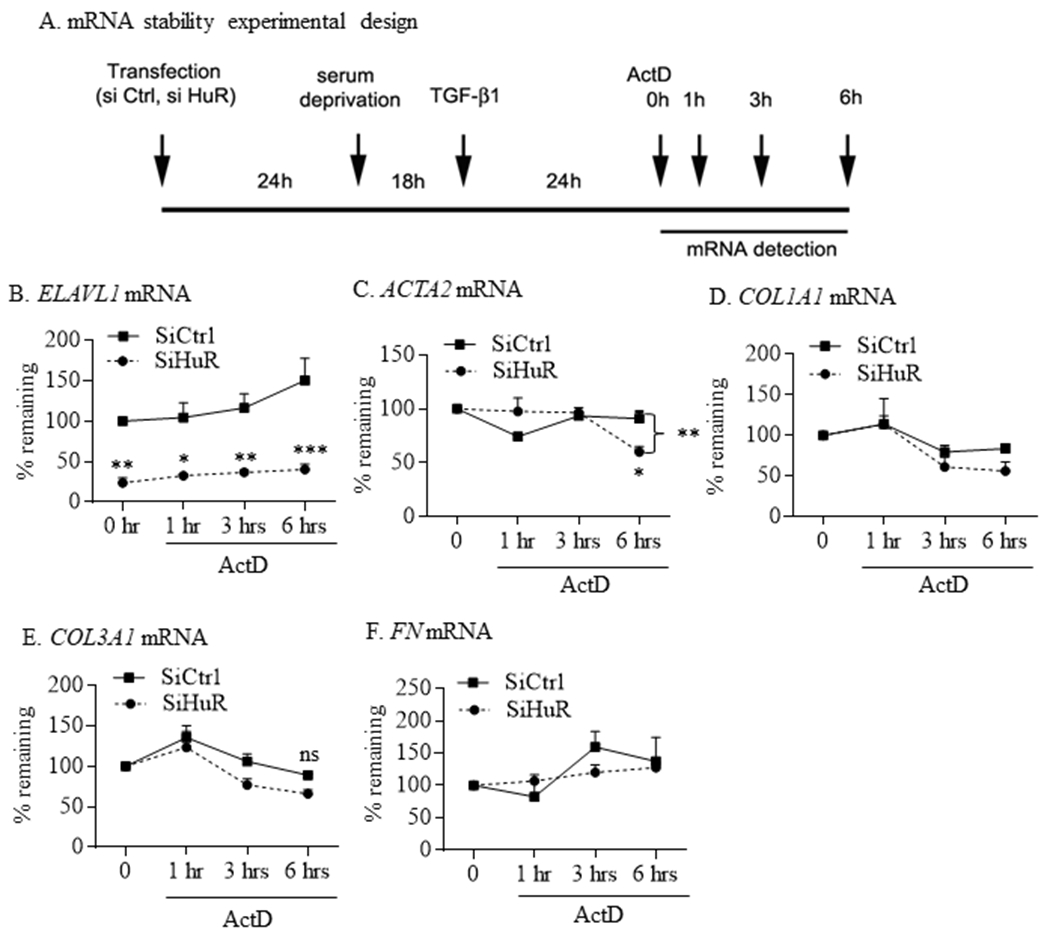
HuR differentially influence mRNA stability of ACTA2 but not the ECM genes COL1A1, COL3A1 or FN. (A) mRNA stability experimental design: experimental protocol for measuring the role of HuR in mRNA stability. Note that 0 hr refers to the time point immediately after the addition of ActD. HLFs were transfected 24 hrs earlier with control or HuR siRNA oligos were treated for 24 hrs with 5 ng/ml of TGF-β1. Cells were then treated with ActD and mRNA levels of ELVAL1 (B), ACTA2 (C), COL1A1 (D), COL3A1 (E) and FN (F) were measured after 1, 3 and 6 hrs using qPCR. Values are means ± SEM and are expressed as percent of values measured at time 0. All values were set to 100% except for analysis of ELAVL1 mRNA to reflect the level of reduction following siHuR (*p<0.05; **p<0.01 and ***p<0.001). There was a significant difference in mRNA stability for ACTA2 (C) between siCtrl and SiHuR cells at 6 hours post-ActD (**p<0.01) and between siHuR at time 0 and 6 hours post-ActD (*p<0.01). Results are presented as the mean ± SEM (n = 4 independent experiments).
3.5. Cytoplasmic HuR localization in pulmonary cells in response to in vivo exposure to bleomycin and thoracic radiation
To test whether there is an increase in cytoplasmic HuR that corresponds to the development of pulmonary fibrosis in vivo, we exposed mice to either bleomycin, the most commonly-used agent to induce lung fibrosis in animals (Barkauskas & Noble, 2014; Della Latta, Cecchettini, Del Ry, & Morales, 2015; Kulkarni et al., 2013; Lemay & Haston, 2005; Stefanov, Fox, Depault, & Haston, 2013) or thoracic radiation, a clinically-relevant injury that results in fibrosis (B et al., 2013; Ding, Li, & Sun, 2013; Rube et al., 2000). Induction of fibrosis was measured by Masson’s Trichrome staining. We further performed IF experiments to evaluate cellular HuR localization under these conditions. These data reveal that both bleomycin and radiation caused fibrosis in the lungs as evidenced by H&E staining (Figure 8A) as well as Masson’s trichrome (Figure 8B). Fibrosis in the lungs of these treated mice was marked by the extensive accumulation of HuR in the cytoplasm of lung cells, compared with nuclear localization seen in the lungs of unexposed mice (Figure 8C). Altogether, our data demonstrate that pro-fibrotic stimuli both in vitro and in vivo (e.g. TGF-β, bleomycin, radiation) promote the nuclear-to-cytoplasmic shuttling of HuR, a key feature in its activation.
FIGURE 8.
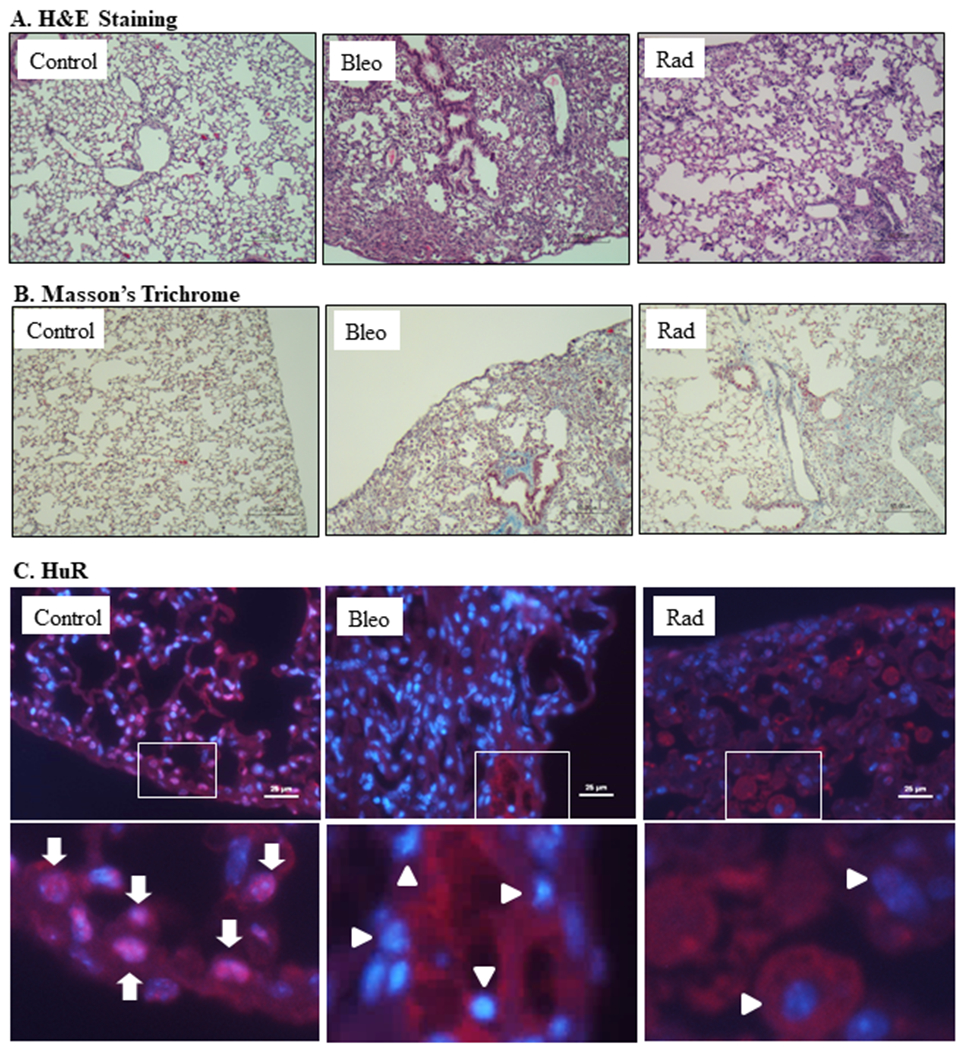
Induction of pulmonary fibrosis is associated with increased cytoplasmic HuR in the mouse lung. (A) H&E staining of lung tissue from unexposed mice as well as mice exposed to bleomycin (Bleo) and radiation (Rad) indicate lung tissue damage. (B) Masson’s Trichrome-confirmation of induction of fibrosis in the bleomycin and radiation-treated mice (blue color). (C) HuR- Lung tissue (serial sections used for Panels A and B) were used to visualize cellular HuR localization in lung tissue (red); nuclei are indicated by blue (DAPI). Only the merged images are shown and co-localization of HuR within the nucleus is a pink color (arrows). Original magnification (top panel is 20x) and bottom is a digital enlargement to show co-localization (arrows). Note that HuR is localized to both the nucleus (arrows) as well as cytoplasm (arrowheads) in the control lung whereas in the mice exposed to bleomycin or radiation, HuR was predominantly cytoplasmic (arrowheads) with almost no HuR in the nucleus (predominant blue color).
3.6. Increased cytoplasmic HuR in vimentin-positive cells in the lungs of IPF subjects
Finally, we evaluated whether there was an increase in cytoplasmic HuR in the lungs of subjects with IPF. Lung sections from individuals with and without IPF were immuno-stained for HuR and its expression evaluated in vimentin-positive cells (green colour) (Figure 9). Note that in the control lung, HuR is predominantly nuclear in the fibroblasts whereas in the IPF lung cells, there is extensive cytoplasmic localization of HuR in vimentin-positive cells (Figure 9). Collectively, these data support the hypothesis that HuR controls fibroblast differentiation into myofibroblasts and a consequent increase in ECM production. Thus, HuR may be involved in the development of fibrotic lung disease.
FIGURE 9.
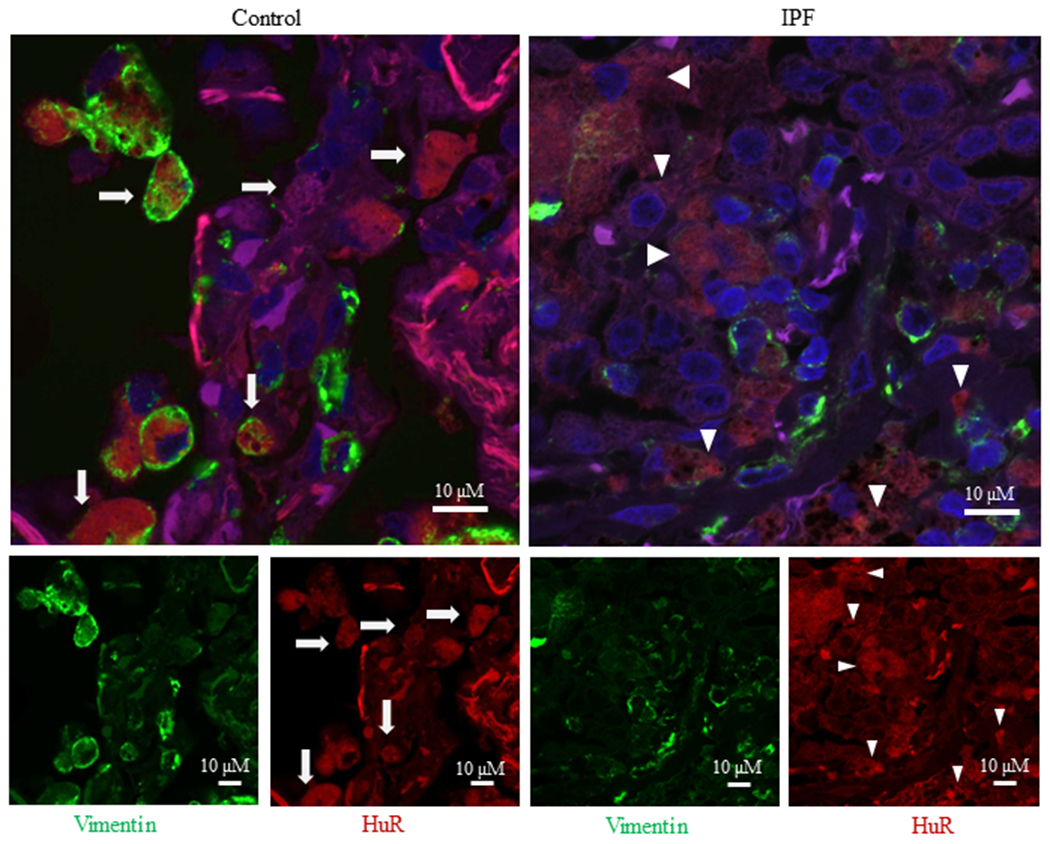
Cytoplasmic HuR in IPF lung. There is extensive cytoplasmic localization of HuR in IPF lung. A representative slide is shown of control (left panel) and IPF (right panel) lung that was stained for vimentin (green), HuR (red) or DAPI (blue); the larger merge image is shown above. Note the extensive cytoplasmic localization of HuR to fibroblasts in the IPF lung (arrowhead) compared to the largely nuclear HuR (arrows) in the control (non-fibrotic) lung.
4. DISCUSSION
IPF is a devastating, progressive, and typically fatal lung disease with a median survival of 3-5 years from diagnosis (Liu, Nepali, & Liou, 2017). IPF is characterized by up-regulation of the pro-fibrotic cytokine TGF-β1 which is implicated in the recruitment, proliferation and differentiation of lung fibroblasts to myofibroblast and the enhanced production of ECM proteins including collagens (mainly type I and III), proteoglygans and glycoproteins such as FN. Despite its known roles in many pathologies, the functional importance of HuR in IPF pathogenesis- including fibroblast differentiation- is unknown. Recent studies highlight the roles of RNA binding proteins in several disease processes such as cancer, cardiovascular disease and autoimmunity, with HuR receiving significant attention. The best-known function of HuR is to regulate mRNA stability. Although the function of HuR differs according to cell type and stimuli (Cammas et al., 2014), HuR is well-known to promote cell proliferation, differentiation and angiogenesis, and recent studies have focused on HuR in cardiac and liver fibrosis. These studies found a significant increase in HuR levels in hepatic stellate cells (HSC) and cardiac fibroblasts. This rise in HuR levels correlated with the degree of liver and cardiac fibrosis (Bai et al., 2012; Woodhoo et al., 2012). However, despite this recent progress in elucidating a potential role for HuR in fibrotic disease, its involvement in lung fibrosis and specifically IPF remain unexplored. Our study is the first to explore the relation between HuR and lung fibroblast differentiation and ECM deposition (α-SMA, COL1A1, COL3A1 and FN). Herein, we show the importance of HuR in the differentiation of fibroblasts into myofibroblasts as evidenced by α-SMA expression and ECM levels.
Mobilization of HuR from the nucleus to the cytosol is critical for HuR activity, as its presence in the cytoplasm can stabilize and prevent degradation of specific mRNA. Cytoplasmic HuR expression levels are known to correlate with poor disease outcome in several cancers, inflammatory conditions and fibrotic diseases (Kotta-Loizou, Giaginis, & Theocharis, 2014; Woodhoo et al., 2012). Indeed, histological evaluation shows that in non-malignant (normal) tissue, HuR is predominantly found in the nucleus, whereas in cancerous tumors, HuR is cytoplasmic (Stoppoloni et al., 2008; J. Wang et al., 2009). This increase in cytoplasmic HuR also strongly correlates with higher tumor grade and poorer patient outcomes (Giaginis et al., 2017), including worse prognosis (Stoppoloni et al., 2008; J. Wang et al., 2009) Our data show that there is a notable increase in cytoplasmic HuR in vimentin-positive cells from IPF lung compared to the largely nuclear HuR in control (non-fibrotic) lung. In our HLF model, we further evaluated the mechanistic aspects of HuR by studying the effects of TGF-β1 on total cellular levels and subcellular localization of HuR. Although total levels of HuR mRNA and protein were not affected by TGF-β1, this exposure significantly increased cytoplasmic levels of HuR protein. To our knowledge, we are the first to describe increased HuR cytoplasmic localization by TGF-β1 in lung fibroblasts. Our observations are in agreement with those of Bai et al. who reported that TGF-β increased HuR shuttling to the cytoplasm in cardiac fibroblasts within 6 hours (Bai et al., 2012). Although the exact mechanism through which TGF-β1 induces cytoplasmic translocation of HuR in lung fibroblasts is unknown, it may involve the activation of the p38 MAPK pathway. This hypothesis is based on several reports indicating that TGF-β1 activates several MAPK pathways including p38 (Ferrari et al., 2012; Khalil, Xu, O’Connor, & Duronio, 2005; Tsukada, Westwick, Ikejima, Sato, & Rippe, 2005). In a study conducted on vascular endothelial cells, TGF-β1 activated p38 MAPKs (Ferrari et al., 2012). In primary interstitial lung fibroblasts, TGF-β1 induced cell proliferation through phosphorylation of p38 and JNK (c-Jun amino-terminal kinases), but not the ERK1/2 (extracellular signal-regulated kinases) pathways (Khalil et al., 2005). It should also be noted that HuR phosphorylation by p38 MAPK increases HuR cytoplasmic translocation through HuR nucleocytoplasmic shuttling domain (HNS) as shown in colon cancer and human bone osteosarcoma cell line (Lafarga et al., 2009).
An important hypothesis for our study was that HuR plays a role in TGF-β1-driven control over ECM protein expression and markers for fibroblast differentiation. We found that HuR silencing significantly decreased TGFβ1-induced αSMA mRNA and protein. Our data show that under myofibroblast-differentiating conditions, HuR binds to ACTA2 mRNA and promotes stability of the transcript to indirectly increase its protein expression (e.g. α-SMA) It is well-established that HuR regulates the mRNA stability of its target genes by protecting them from degradation machinery via HuR-mRNA interaction. For example, binding of HuR to Cyclin D1 mRNA in mesangial cells treated with Angiotensin II triggers a significant increase in fibrogenic processes inside the kidney (Che et al., 2014). However, our data suggest that the mechanism of action for HuR does not involve mRNA stability for all genes, as silencing HuR expression affected the decay of ACTA2 but not that of ECM marker mRNAs. Yet there was binding of HuR to mRNA encoding for collagen and fibronectin. While all of the biological functions of Hu proteins are believed to be the result of their binding to target mRNA (Hinman & Lou, 2008), and altering mRNA stability is the best-studied function of HuR, our data suggest that HuR may have additional functions in promoting collagen and fibronectin proteins expression. In line with this is the recent finding that HuR promotes the expression of STAT3 without affecting stability of the STAT3 transcript by a mechanism that involved competitive interplay with the microRNA-330 (miR-330) (Mubaid et al., 2019). HuR can also cooperate with other RNA binding proteins to promote protein translation. For example, the RNA-binding protein Hzf (hematopoietic zinc finger) works with HuR to promote p53 expression (Nakamura et al., 2011). Thus, it is possible that interaction with other RNA binding proteins and miRNAs may play a role in regulating the stability of ECM marker mRNAs or that HuR is controlling translation. Further support for these possibilities comes from a study in cardiac fibroblasts, where miR-33a increases the expression of COL1A1 and COL3A1 (in vivo and in vitro) and that knockdown of miR-33a is sufficient to decrease their expression (Chen et al., 2018). This effect of miR-33a on COL1A1 and COL3A1 expression is mediated through the p38 MAPK pathway but not through the TGFβ1/SMAD pathway (Chen et al., 2018) In human dermal fibroblasts, TGFβ1 upregulates the expression of COL1A1 and FN through activation of the RNA-binding protein PTB (polypyrimidine-tract-binding protein), and that knockdown of PTB was associated with a significant decrease in COL3A1 and FN expression (Jiao et al., 2016). Taken together, our findings support the notion that HuR plays a differential role in TGF-β1-induced fibroblast differentiation, and ongoing research is currently aimed at assessing how HuR controls lung fibroblast differentiation and ECM production.
In summary, we report for the first time how TGF-β1 affects cellular levels and subcellular localization of HuR in human lung fibroblasts. We found that TGF-β1 has no effect on total HuR levels in these cells, but it significantly increases cytoplasmic translocation of HuR. We also demonstrate that HuR is required for the expression of TGF-β1-induced myofibroblast markers and that there is increased cytoplasmic HuR in the lungs of mice exposure to fibrotic agents as well as in the lungs of IPF subjects. Our findings support the notion that HuR may be responsible for promoting the development of lung fibrosis and that targeting HuR may prove to be beneficial in preventing the progression of fibrotic diseases such as IPF.
ACKNOWLEDGEMENTS
Histology services were by the Histopathology Platform of the RI-MUHC. This work was supported by the Canada Foundation for Innovation (CFI), the Canadian Institutes for Health Research (CIHR; Project Grant PJT-168836) and a Boehringer-Ingelheim Grant for Understanding Interstitial Lung Disease (BUILD). CJB. was supported by a salary award from the Fonds de recherche du Quebec-Sante (FRQ-S). NA was supported by a scholarship from Taibah University, Saudi Arabia. SKH is supported by a grant from the NHLBI (HL127203). This work was also supported by a CIHR Project Grant (PJT-159618) to IEG.
Footnotes
CONFICTS OF INTEREST
The authors declare there are no conflicts of interest.
DATA SHARING
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- Antoniou KM, Margaritopoulos GA, Tomassetti S, Bonella F, Costabel U, & Poletti V (2014). Interstitial lung disease. Eur Respir Rev, 23(131), 40–54. doi: 10.1183/09059180.00009113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atasoy U, Watson J, Patel D, & Keene JD (1998). ELAV protein HuA (HuR) can redistribute between nucleus and cytoplasm and is upregulated during serum stimulation and T cell activation. J Cell Sci, 111 (Pt 21), 3145–3156. [DOI] [PubMed] [Google Scholar]
- B BM, Lawson WE, Oury TD, Sisson TH, Raghavendran K, & Hogaboam CM (2013). Animal models of fibrotic lung disease. Am J Respir Cell Mol Biol, 49(2), 167–179. doi:rcmb.2013-0094TR 10.1165/rcmb.2013-0094TR [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baglole CJ, Maggirwar SB, Gasiewicz TA, Thatcher TH, Phipps RP, & Sime PJ (2008). The aryl hydrocarbon receptor attenuates tobacco smoke-induced cyclooxygenase-2 and prostaglandin production in lung fibroblasts through regulation of the NF-kappaB family member RelB. J Biol Chem, 283(43), 28944–28957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai D, Gao Q, Li C, Ge L, Gao Y, & Wang H (2012). A conserved TGFbeta1/HuR feedback circuit regulates the fibrogenic response in fibroblasts. Cell Signal, 24(7), 1426–1432. doi: 10.1016/j.cellsig.2012.03.003 [DOI] [PubMed] [Google Scholar]
- Barkauskas CE, & Noble PW (2014). Cellular mechanisms of tissue fibrosis. 7. New insights into the cellular mechanisms of pulmonary fibrosis. Am J Physiol Cell Physiol, 306(11), C987–996. doi:ajpcell.00321.2013 10.1152/ajpcell.00321.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergeron ME, Stefanov A, & Haston CK (2018). Fine mapping of the major bleomycin-induced pulmonary fibrosis susceptibility locus in mice. Mamm Genome, 29(9-10), 670–679. doi: 10.1007/s00335-018-9774-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calaluce R, Gubin MM, Davis JW, Magee JD, Chen J, Kuwano Y,… Atasoy U (2010). The RNA binding protein HuR differentially regulates unique subsets of mRNAs in estrogen receptor negative and estrogen receptor positive breast cancer. BMC Cancer, 10, 126. doi: 10.1186/1471-2407-10-126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cammas A, Sanchez BJ, Lian XJ, Dormoy-Raclet V, van der Giessen K, Lopez de Silanes I,… Gallouzi IE (2014). Destabilization of nucleophosmin mRNA by the HuR/KSRP complex is required for muscle fibre formation. Nat Commun, 5, 4190. doi: 10.1038/ncomms5190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Che Y, Yi L, Akhtar J, Bing C, Ruiyu Z, Qiang W, & Rong W (2014). AngiotensinII induces HuR shuttling by post-transcriptional regulated CyclinD1 in human mesangial cells. Mol Biol Rep, 41(2), 1141–1150. doi: 10.1007/s11033-013-2960-1 [DOI] [PubMed] [Google Scholar]
- Chen Z, Ding HS, Guo X, Shen JJ, Fan D, Huang Y, & Huang CX (2018). MiR-33 promotes myocardial fibrosis by inhibiting MMP16 and stimulating p38 MAPK signaling. Oncotarget, 9(31), 22047–22057. doi: 10.18632/oncotarget.25173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Della Latta V, Cecchettini A, Del Ry S, & Morales MA (2015). Bleomycin in the setting of lung fibrosis induction: From biological mechanisms to counteractions. Pharmacol Res, 97, 122–130. doi:S1043-6618(15)00080-8 10.1016/j.phrs.2015.04.012 [DOI] [PubMed] [Google Scholar]
- Ding NH, Li JJ, & Sun LQ (2013). Molecular mechanisms and treatment of radiation-induced lung fibrosis. Curr Drug Targets, 14(11), 1347–1356. doi:CDT-EPUB-04-54762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Ishmael FT, Fang X, Myers A, Cheadle C, Huang SK,… Stellato C (2011). Chemokine transcripts as targets of the RNA-binding protein HuR in human airway epithelium. J Immunol, 186(4), 2482–2494. doi: jimmunol.0903634 10.4049/jimmunol.0903634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan XC, & Steitz JA (1998). HNS, a nuclear-cytoplasmic shuttling sequence in HuR. Proc Natl Acad Sci U S A, 95(26), 15293–15298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari G, Terushkin V, Wolff MJ, Zhang X, Valacca C, Poggio P,… Mignatti P (2012). TGF-beta1 induces endothelial cell apoptosis by shifting VEGF activation of p38(MAPK) from the prosurvival p38beta to proapoptotic p38alpha. Mol Cancer Res, 10(5), 605–614. doi: 10.1158/1541-7786.MCR-11-0507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaginis C, Sampani A, Kotta-Loizou I, Giannopoulou I, Danas E, Politi E,… Theocharis S (2017). Elevated Hu-Antigen Receptor (HuR) Expression is Associated with Tumor Aggressiveness and Poor Prognosis but not with COX-2 Expression in Invasive Breast Carcinoma Patients. Pathol Oncol Res. doi: 10.1007/s12253-017-0288-1 [DOI] [PubMed] [Google Scholar]
- Hecht E, Zago M, Sarill M, Rico de Souza A, Gomez A, Matthews J,… Baglole CJ (2014). Aryl hydrocarbon receptor-dependent regulation of miR-196a expression controls lung fibroblast apoptosis but not proliferation. Toxicol Appl Pharmacol. doi:S0041-008X(14)00315-9[pii] 10.1016/j.taap.2014.08.023 [DOI] [PubMed] [Google Scholar]
- Hinman MN, & Lou H (2008). Diverse molecular functions of Hu proteins. Cell Mol Life Sci, 65(20), 3168–3181. doi: 10.1007/s00018-008-8252-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins RB, Burke N, Fell C, Dion G, & Kolb M (2016). Epidemiology and survival of idiopathic pulmonary fibrosis from national data in Canada. Eur Respir J, 48(1), 187–195. doi: 10.1183/13993003.01504-2015 [DOI] [PubMed] [Google Scholar]
- Hoyne GF, Elliott H, Mutsaers SE, & Prele CM (2017). Idiopathic pulmonary fibrosis and a role for autoimmunity. Immunol Cell Biol, 95(7), 577–583. doi: 10.1038/icb.2017.22 [DOI] [PubMed] [Google Scholar]
- Jiao H, Dong P, Yan L, Yang Z, Lv X, Li Q,… Xiao R (2016). TGF-beta1 Induces Polypyrimidine Tract-Binding Protein to Alter Fibroblasts Proliferation and Fibronectin Deposition in Keloid. Sci Rep, 6, 38033. doi: 10.1038/srep38033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keene JD, Komisarow JM, & Friedersdorf MB (2006). RIP-Chip: the isolation and identification of mRNAs, microRNAs and protein components of ribonucleoprotein complexes from cell extracts. Nat Protoc, 1(1), 302–307. doi:nprot.2006.47[pii] 10.1038/nprot.2006.47 [DOI] [PubMed] [Google Scholar]
- Khalil N, O’Connor RN, Unruh HW, Warren PW, Flanders KC, Kemp A,… Greenberg AH (1991). Increased production and immunohistochemical localization of transforming growth factor-beta in idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol, 5(2), 155–162. [DOI] [PubMed] [Google Scholar]
- Khalil N, Xu YD, O’Connor R, & Duronio V (2005). Proliferation of pulmonary interstitial fibroblasts is mediated by transforming growth factor-beta1-induced release of extracellular fibroblast growth factor-2 and phosphorylation of p38 MAPK and JNK. J Biol Chem, 280(52), 43000–43009. doi: 10.1074/jbc.M510441200 [DOI] [PubMed] [Google Scholar]
- Kotta-Loizou I, Giaginis C, & Theocharis S (2014). Clinical significance of HuR expression in human malignancy. Med Oncol, 31(9), 161. doi: 10.1007/s12032-014-0161-y [DOI] [PubMed] [Google Scholar]
- Kulkarni AA, Thatcher TH, Hsiao HM, Olsen KC, Kottmann RM, Morrissette J,… Sime PJ (2013). The triterpenoid CDDO-Me inhibits bleomycin-induced lung inflammation and fibrosis. PLoS One, 8(5), e63798. doi: 10.1371/journal.pone.0063798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafarga V, Cuadrado A, Lopez de Silanes I, Bengoechea R, Fernandez-Capetillo O, & Nebreda AR (2009). p38 Mitogen-activated protein kinase- and HuR-dependent stabilization of p21(Cip1) mRNA mediates the G(1)/S checkpoint. Mol Cell Biol, 29(16), 4341–4351. doi:MCB.00210-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson O, Diebold D, Fan D, Peterson M, Nho RS, Bitterman PB, & Henke CA (2008). Fibrotic myofibroblasts manifest genome-wide derangements of translational control. PLoS One, 3(9), e3220. doi: 10.1371/journal.pone.0003220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemay AM, & Haston CK (2005). Bleomycin-induced pulmonary fibrosis susceptibility genes in AcB/BcA recombinant congenic mice. Physiol Genomics, 23(1), 54–61. doi:23/1/54 [DOI] [PubMed] [Google Scholar]
- Lemay AM, & Haston CK (2018). A Chromosome 6, not Natural Killer Cell, Contribution to Radiation- and Bleomycin-Induced Lung Disease in Mice. Radiat Res, 190(6), 605–611. doi: 10.1667/RR15144.1 [DOI] [PubMed] [Google Scholar]
- Liu YM, Nepali K, & Liou JP (2017). Idiopathic Pulmonary Fibrosis: Current Status, Recent Progress, and Emerging Targets. J Med Chem, 60(2), 527–553. doi: 10.1021/acs.jmedchem.6b00935 [DOI] [PubMed] [Google Scholar]
- Loomis-King H, Flaherty KR, & Moore BB (2013). Pathogenesis, current treatments and future directions for idiopathic pulmonary fibrosis. Curr Opin Pharmacol, 13(3), 377–385. doi: 10.1016/j.coph.2013.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin MD, Chung JH, & Kanne JP (2016). Idiopathic Pulmonary Fibrosis. J Thorac Imaging, 31(3), 127–139. doi: 10.1097/RTI.0000000000000204 [DOI] [PubMed] [Google Scholar]
- Mubaid S, Ma JF, Omer A, Ashour K, Lian XJ, Sanchez BJ,… Gallouzi IE (2019). HuR counteracts miR-330 to promote STAT3 translation during inflammation-induced muscle wasting. Proc Natl Acad Sci U S A, 116(35), 17261–17270. doi: 10.1073/pnas.1905172116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee N, Corcoran DL, Nusbaum JD, Reid DW, Georgiev S, Hafner M,… Keene JD (2011). Integrative regulatory mapping indicates that the RNA-binding protein HuR couples pre-mRNA processing and mRNA stability. Mol Cell, 43(3), 327–339. doi: 10.1016/j.molcel.2011.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myer VE, Fan XC, & Steitz JA (1997). Identification of HuR as a protein implicated in AUUUA-mediated mRNA decay. EMBO J, 16(8), 2130–2139. doi: 10.1093/emboj/16.8.2130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik PK, & Moore BB (2010). Viral infection and aging as cofactors for the development of pulmonary fibrosis. Expert Rev Respir Med, 4(6), 759–771. doi: 10.1586/ers.10.73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura H, Kawagishi H, Watanabe A, Sugimoto K, Maruyama M, & Sugimoto M (2011). Cooperative role of the RNA-binding proteins Hzf and HuR in p53 activation. Mol Cell Biol, 31(10), 1997–2009. doi: 10.1128/MCB.01424-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghu G, Chen SY, Yeh WS, Maroni B, Li Q, Lee YC, & Collard HR (2014). Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: incidence, prevalence, and survival, 2001-11. Lancet Respir Med, 2(7), 566–572. doi: 10.1016/S2213-2600(14)70101-8 [DOI] [PubMed] [Google Scholar]
- Raghu G, Nyberg F, & Morgan G (2004). The epidemiology of interstitial lung disease and its association with lung cancer. Br J Cancer, 91 Suppl 2, S3–10. doi: 10.1038/sj.bjc.6602061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghu G, Rochwerg B, Zhang Y, Garcia CA, Azuma A, Behr J,… Latin American Thoracic, A. (2015). An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline: Treatment of Idiopathic Pulmonary Fibrosis. An Update of the 2011 Clinical Practice Guideline. Am J Respir Crit Care Med, 192(2), e3–19. doi: 10.1164/rccm.201506-1063ST [DOI] [PubMed] [Google Scholar]
- Rube CE, Uthe D, Schmid KW, Richter KD, Wessel J, Schuck A,… Rube C (2000). Dose-dependent induction of transforming growth factor beta (TGF-beta) in the lung tissue of fibrosis-prone mice after thoracic irradiation. Int J Radiat Oncol Biol Phys, 47(4), 1033–1042. doi:S0360-3016(00)00482-X [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, & Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nature Methods, 9(7), 671–675. doi: 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta S, Jang BC, Wu MT, Paik JH, Furneaux H, & Hla T (2003). The RNA-binding protein HuR regulates the expression of cyclooxygenase-2. J Biol Chem, 278(27), 25227–25233. doi: 10.1074/jbc.M301813200 [DOI] [PubMed] [Google Scholar]
- Sheridan JA, Zago M, Nair P, Li PZ, Bourbeau J, Tan WC,… Baglole CJ (2015). Decreased expression of the NF-kappaB family member RelB in lung fibroblasts from Smokers with and without COPD potentiates cigarette smoke-induced COX-2 expression. Respir Res, 16, 54. doi: 10.1186/s12931-015-0214-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sime PJ, Xing Z, Graham FL, Csaky KG, & Gauldie J (1997). Adenovector-mediated gene transfer of active transforming growth factor-beta1 induces prolonged severe fibrosis in rat lung. J Clin Invest, 100(4), 768–776. doi: 10.1172/JCI119590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M, Martinez AR, Govindaraju S, & Lee BS (2013). HuR inhibits apoptosis by amplifying Akt signaling through a positive feedback loop. J Cell Physiol, 228(1), 182–189. doi: 10.1002/jcp.24120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spagnolo P, Sverzellati N, Rossi G, Cavazza A, Tzouvelekis A, Crestani B, & Vancheri C (2015). Idiopathic pulmonary fibrosis: an update. Ann Med, 47(1), 15–27. doi: 10.3109/07853890.2014.982165 [DOI] [PubMed] [Google Scholar]
- Stefanov AN, Fox J, Depault F, & Haston CK (2013). Positional cloning reveals strain-dependent expression of Trim16 to alter susceptibility to bleomycin-induced pulmonary fibrosis in mice. PLoS Genet, 9(1), e1003203. doi: 10.1371/journal.pgen.1003203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoppoloni D, Cardillo I, Verdina A, Vincenzi B, Menegozzo S, Santini M,… Galati R (2008). Expression of the embryonic lethal abnormal vision-like protein HuR in human mesothelioma: association with cyclooxygenase-2 and prognosis. Cancer, 113(10), 2761–2769. doi: 10.1002/cncr.23904 [DOI] [PubMed] [Google Scholar]
- Tsukada S, Westwick JK, Ikejima K, Sato N, & Rippe RA (2005). SMAD and p38 MAPK signaling pathways independently regulate alpha1(I) collagen gene expression in unstimulated and transforming growth factor-beta-stimulated hepatic stellate cells. J Biol Chem, 280(11), 10055–10064. doi: 10.1074/jbc.M409381200 [DOI] [PubMed] [Google Scholar]
- von Roretz C, Lian XJ, Macri AM, Punjani N, Clair E, Drouin O,… Gallouzi IE (2013). Apoptotic-induced cleavage shifts HuR from being a promoter of survival to an activator of caspase-mediated apoptosis. Cell Death Differ, 20(1), 154–168. doi: 10.1038/cdd.2012.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Zhao W, Guo Y, Zhang B, Xie Q, Xiang D,… Chen Z (2009). The expression of RNA-binding protein HuR in non-small cell lung cancer correlates with vascular endothelial growth factor-C expression and lymph node metastasis. Oncology, 76(6), 420–429. doi:000216837 [DOI] [PubMed] [Google Scholar]
- Wang W, Fan J, Yang X, Furer-Galban S, Lopez de Silanes I, von Kobbe C,… Gorospe M (2002). AMP-activated kinase regulates cytoplasmic HuR. Mol Cell Biol, 22(10), 3425–3436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhoo A, Iruarrizaga-Lejarreta M, Beraza N, Garcia-Rodriguez JL, Embade N, Fernandez-Ramos D,… Martinez-Chantar ML (2012). Human antigen R contributes to hepatic stellate cell activation and liver fibrosis. Hepatology, 56(5), 1870–1882. doi: 10.1002/hep.25828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuyts WA, Agostini C, Antoniou KM, Bouros D, Chambers RC, Cottin V,… Verleden GM (2013). The pathogenesis of pulmonary fibrosis: a moving target. Eur Respir J, 41(5), 1207–1218. doi:09031936.00073012 [DOI] [PubMed] [Google Scholar]
- Xu YZ, Di Marco S, Gallouzi I, Rola-Pleszczynski M, & Radzioch D (2005). RNA-binding protein HuR is required for stabilization of SLC11A1 mRNA and SLC11A1 protein expression. Mol Cell Biol, 25(18), 8139–8149. doi: 10.1128/MCB.25.18.8139-8149.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zago M, Sheridan JA, Nair P, Rico de Souza A, Gallouzi IE, Rousseau S,… Baglole CJ (2013). Aryl Hydrocarbon Receptor-Dependent Retention of Nuclear HuR Suppresses Cigarette Smoke-Induced Cyclooxygenase-2 Expression Independent of DNA-Binding. PLoS One, 8(9), e74953. doi: 10.1371/journal.pone.0074953 [DOI] [PMC free article] [PubMed] [Google Scholar]