

Abstract
Infection with Helicobacter pylori (H. pylori) is necessary but not sufficient for the development of gastric cancer, the third leading cause of cancer death globally. H. pylori infection affects over half of people globally; however, it does not affect populations uniformly. H. pylori infection rates are declining in Western industrialized countries but are plateauing in developing and newly industrialized countries where gastric cancer is most prevalent. Despite H. pylori infection being the primary causative agent for gastric cancer, H. pylori infection can also cause other effects, detrimental or beneficial, throughout an individual’s life, with the beneficial effects often being seen in childhood and the deleterious effects in adulthood. H. pylori is an ancient bacterium and its likelihood of affecting disease or health is dependent on both human and bacterial genetics that have co-evolved over millennia. In this review, we focus on the impact of infection and its genetic bases in different populations and diseases throughout an individual’s lifespan, highlighting the benefits of individualized treatment and argue that universal eradication of H. pylori in its host may cause more harm than good for those infected with H. pylori.
Keywords: H. pylori, Human, Genetics, Gastric cancer, Precision medicine
INTRODUCTION
Gastric cancer is the fifth most frequently diagnosed cancer and the third leading cause of cancer death globally, accounting for 1 000 000 new cases and approximately 783 000 deaths in 2018 (1). It is most prevalent in low-income countries, but incidence of gastric cancer has been increasing in high-income countries (1, 2). Helicobacter pylori (H. pylori) is the principal cause and strongest known risk factor for gastric cancer. It infects over half of the world’s population, more than 4.4 billion individuals, and in developing countries, up to 80% of middle-aged adults may be infected (3–5). H. pylori infection rates vary widely between regions with prevalence being highest in Africa (79.1%), Latin America and the Caribbean (63.4%), and Asia (54.7%) and lowest in Northern America (37.1%) and Oceania (24.4%) (Figure 1). However, the infection rate does not necessarily correlate with gastric cancer prevalence. For example, despite H. pylori infection declining in highly industrialized countries of the West, prevalence has been increasing, and in many low- and middle-income countries, disease is rare despite high infection rates countries (5, 6). Therefore, not only is there variability in prevalence of H. pylori infection, but there can also be a disconnect between infection rate and disease.
Figure 1: Global distribution of H. pylori and Gastric Cancer.
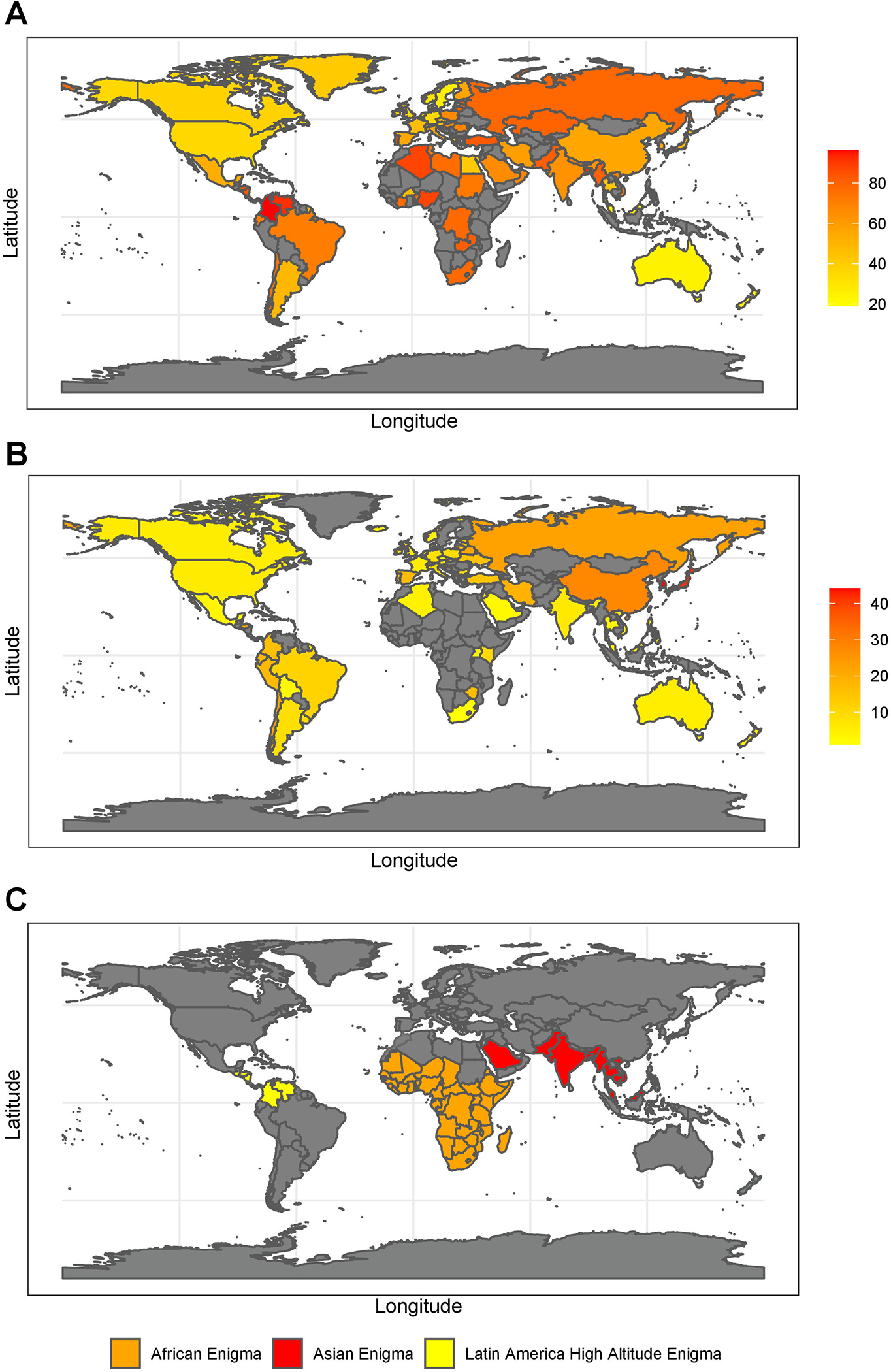
The prevalence of H. pylori infection as percent of total adult population (A), gastric cancer incidence presented as number per 100,000 (B) mapped with the most recent H. pylori prevalence data(5) and gastric cancer incidence rates from the WHO except where WHO did not have data, in which case data was supplemented from recent publications or presented as no data available (in gray) (101–103). As gastric cancer incidence is generally presented for male and females separately we took average of the two for the figures. The average for each country is presented. The locales of the three gastric cancer enigmas, the African Enigma, the Asian Enigma, and the Latin America High Altitude Enigma (C), highlighting the contrast of H. pylori infection incidence and gastric cancer prevalence.
H. pylori infection is most often thought to associate with gastric cancer and ulcers. However, H. pylori infection is not sufficient to cause either disease, as only a small percentage of H. pylori-infected develop ulcers or progress on the gastric disease cascade to gastric cancer. Therefore, dissecting both genetic and environmental factors that drive or prevent these diseases, is important for developing preventative strategies (4, 7). There is a disparity in gastric cancer in the US and globally (8). In the US, nonwhites, including Latinxs, have twice the incidence rate of whites (9). The disconnect between infection rates and disease has led to the designation of three paradoxes: the “African enigma”, the “Asian enigma”, and the “Altitude enigma” that represent inconsistencies between infection rate and gastric cancer prevalence (Figure 1). Studies have indicated that factors beyond geographical location, such as the compatibility of the genomes of the host and pathogen, can be determinants of gastric disease prevalence and severity, thereby redefining the possible causes of these enigmas (8).
H. pylori is a Gram negative spiral-shaped bacterium and one of the most diverse bacterial species (10, 11). To initiate colonization of the stomach, H. pylori neutralizes the acidic conditions, moves towards the host gastric epithelium, and binds to the host cell receptors (Figure 2) (12). The H. pylori genome is highly plastic, and a study of 1 531 genes showed that as many as 25% of the genes were absent from at least one of the 56 studied strains (13). It is thought that the plasticity and high rates of intraspecific recombination improves H. pylori’s ability to colonize the stomach (10, 11, 13).
Figure 2: H. pylori colonization.
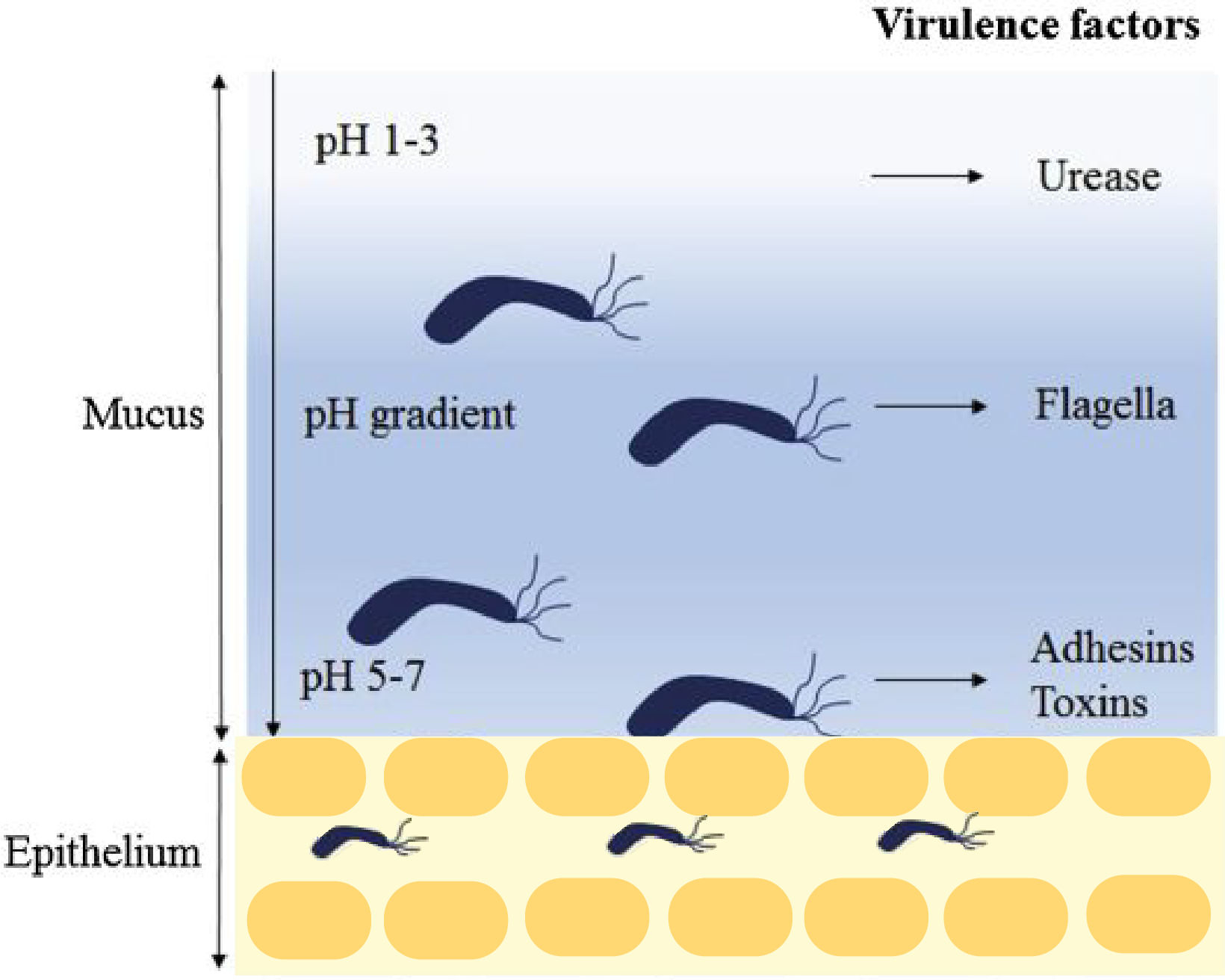
H. pylori enters the stomach, and the urease activity reduces the acidity of the local environment. The H. pylori flagella-mediated motility facilitate its movement towards the lower mucosa, and it enters the epithelium with assistance from several adhesins including babA and sabA. Once the epithelium is successfully colonized, H. pylori release toxins including cagA and vacA. The figure modified from Kao et al (12).
H. pylori is usually acquired in childhood via fecal-oral or oral-oral transmission (14). There is considerable evidence that H. pylori is an ancient bacterium as it colonized humans at least 100 000 years ago (15, 16). Therefore, it can be used to trace human migration as a commensal bacterium and, similar to infectious agents such as Mycobacterium tuberculosis and human papillomavirus, H. pylori has presumably co-evolved with humans (17–21). H. pylori is considered a dominant member of the human gastric microbiota (22, 23). Further, H. pylori is an amphibiont bacterium, as some cases of infection promote pathological conditions and others protect from pathology. These different effects of H. pylori infection may vary by life stage with protection against diseases acquired in early life but promotion of disease later in life (Table 1) (24, 25). In this review, we will describe the deleterious and protective effects of H. pylori infection and argue that treatment of H. pylori infection should vary, using a precision-medicine based approach, that incorporates a number of factors, including patient age.
Table 1:
Deleterious and Protective effects of H. pylori Infection
| Deleterious | Protective |
|---|---|
| Gastric Adenocarcinoma and Gastritis(3, 4) | Esophageal Diseases(65–67) |
| Peptic ulcers(5, 6) | Asthma and Allergy(67, 70–76) |
| MALT lymphoma(58–61) | Inflammatory Bowel Disease(23, 84–86) |
| Chronic Obstructive Pulmonary Disease (COPD) (104) | Diarrheal disease(13, 88, 89) |
| Atherosclerosis/coronary artery disease(105, 106) | Tuberculosis(13, 81) |
| Iron deficiency anemia(63, 64) | Metabolism and Obesity(13, 81) |
| Preeclampsia (PE)/Small for Gestational Age (SGA)/Spontaneous Preterm Birth (SPB) (107) | Food Allergy(23) |
| Metabolic Syndrome (MetS) in pregnancy(108) | Celiac Disease(109) |
| Idiopathic thrombocytic purpura(64) | Graves’s Disease(110) |
| Impaired Glucose Intolerance(111) |
GENE VARIANTS AND EPIGENETIC EFFECTS DRIVE GASTRIC CANCER
H. pylori Genetic Variation
Once H. pylori binds to host cell receptors, it releases effector proteins and toxins including cytotoxin-associated gene A (cagA) and vacuolating cytotoxin A (vacA) (Figure 2) (12). The cagA gene is prevalent in high gastric cancer incidence areas and presence of cagA associates with increased gastric cancer risk.(26, 27) The CagA protein sequence is variable and can be divided into Western-type CagA and East Asian-type CagA, using the repeated Glu-Pro-Ile-Tyr-Ala (EPIYA) motifs at the N-terminus of CagA. Once the translocated CagA protein is injected into the host cell cytoplasm, it can alter host cell signaling in both phosphorylation-dependent and phosphorylation-independent manners (12). VacA can disrupt the balance of cell proliferation and death by affecting cell cycle genes, can induce acute inflammatory responses through the host cell release of IL-8, and can induce the release of cytochrome C, ER stress, and apoptosis. All H. pylori strains carry the vacA gene, but the gene varies in its signal sequence (s1a, s1b, s1c, and s2), mid-region (m1, m1T, and m2), and the intermediate region (i1, i2, and i3). Thus, there is a variable genotype dependent response as vacA s1 and m1 strains are associated with high levels of inflammation in the gastric mucosa and increased risk of carcinoma in comparison to less virulent vacA s2 and m2 strains (12). Both cagA and gastric cancer associated vacA genotypes are not present in all strains of H. pylori, but they are important cancer risk factors, and are independent of host genotype (26, 27). With the general global decline of H. pylori infection, the variation of gastric cancer incidence may be tied not only to infection prevalence but to strain characteristics. For example, prevalence of high-risk CagA positive H. pylori strains may more accurately describe a population’s gastric cancer risk (28). Nonetheless, they alone are not sufficient to predict who will develop gastric diseases (26, 27).
With the availability of more H. pylori genomes and new bioinformatics methods, there is a greater understanding of H. pylori genetic diversity and evolution, virulence, and putative new targets for treatments. Recent studies have described genes associated with functions in the adaptability and pathogenicity of H. pylori to the human host including cag pathogenicity island (cagPAI), babA, babB, and sabB.(29–32) The cagPAI consists of approximately 30 genes that can deliver CagA and a bacterial cell wall component into host cells (33). Blood group antigen binding adhesin (BabA) binds to Lewis b (Leb) blood group antigen or sialic acid-binding adhesion (SabA) as the primary mode of H. pylori adhesion to the human gastric epithelium. BabA expression is regulated by phase variation and recombination between babA and babB or babC (30, 31). Thus, there is a wide variety of H. pylori genes that affect gastric disease risk and severity.
Genetic diversity of H. pylori can be substantial within a host population and can associate with differential gastric disease risk. This has been demonstrated in African, Middle East, and American populations, using multilocus sequence typing (MLST) lineages determined from sequences of seven housekeeping genes (17, 29). In one or more MLST lineages, pan-genome analyses have identified over-expressed or under-expressed genes among MLST lineages or associated with the cag pathogenicity island (34). Thus, H. pylori MLST genome sequence can be used to detect varying genetic diversity between populations and possibly disease risk.
A study using MLST genome sequence of H. pylori in North, Central, and South America found evidence of admixture between ancestries that created new H. pylori subpopulations that spread and adapted rapidly during times of demographic flux. Admixture was seen in high prevalence regions, e.g., Colombia and Nicaragua, where bottlenecks and rampant genetic exchange between H. pylori isolates has led to chimeric gene pools unique to the local population and correlated with national boundaries. This indicates that adaption of bacteria genetics to particular human ethnic groups may be indicative of interaction with the host immune system (35–37).
Host Genetic Variation
Polymorphisms in the host that modulate levels of cytokines IL-10, IL-1β, and TNF-α have been associated with elevated risk of non-cardia gastric cancer.(38, 39) Genome-wide associations studies (GWAS) in Asian populations have identified other variant associations with gastric cancer including variants in PSCA and MUC1 (40–42), variants near PRKAA1 and PTGER4(40, 43, 44), a nonsynonymous SNP located in PLCE1(41), and variants in CUX2 and ABO (45). A recent gastric cancer GWAS in European populations also showed association with MUC1. Haplotype analysis of two previously reported MUC1 variants further verifies the association of MUC1 variants with gastric cancer and is indicative of a pathogenic role for a tandem repeat in exon 2 of MUC1 that causes alternative splicing (46). Additionally, associations of SNPs in PRKAA1 and PSCA with gastric cancer have been replicated (46). Variation in PRKAA1 associated with gastric cancer in both Asian and European populations, but the specific SNPs differed between the populations. Gastric cancer in the Han Chinese populations associated with rs13361707[C] and 33 proxy variants and in the European population association with rs10036575 was more significant when adjusting for rs13361707. In a study of another European sample, two independent loss-of-function mutations in ATM, a gene with a key role in the DNA damage response, were identified (46). Therefore, associations between gastric cancer and multiple human gene variants have been found with some replicating across populations while others have not to date.
In a Colombian study population, GATA-5 genotype and promoter methylation as well as the interaction between these two factors associated with the development of gastric disease (47). Recent in vitro and in vivo studies indicate that the upregulation of GATA-5 and TFF1 correlate with a protective effect of the gastric mucosa in response to H. pylori infection (48). GATA-5 and TFF1 were upregulated in H. pylori infected human cells 48 hours post infection, and in mice with long-term (6 and 12 months) H. pylori infections. Additionally, biopsies from infected pediatric, chronic gastritis, and gastric cancer patients had epigenetic inactivation of GATA-5, indicating that methylation occurs as an early event following H. pylori infection (48). Furthermore, the GATA-5 methylation signature was suggested to be a valuable marker for past exposure to H. pylori and to assess gastric cancer risk (48). Epigenetic inactivation of GATA-5 is further seen in human gastric mucosa samples where methylation of CpG islands correlates with H. pylori infection and occurs frequently in early gastric carcinogenesis (49). Therefore, multiple lines of evidence support the role of GATA-5 in gastric disease risk.
Variation in host gene expression has also been associated with H. pylori infection in model organisms. In mice infected with H. pylori for 6 months, gastric and pulmonary tissues had increased expression of multiple immune response genes, including those for T cell activation and pro-inflammatory molecules. The expression of multiple immune response genes was increased, conserved across all mice and over time in the stomach and lungs (50). Therefore, H. pylori infection affects local histologic, physiologic, immune, and microbiologic features in both the stomach and distal organs (50). Indeed, H. pylori infection influences the microbiota and host immune responses that drive gastric disease progression in the stomach as well as distal sites, and therefore H. pylori’s widespread effects should be considered in understanding not the development of future treatments but their total impacts.
OTHER EFFECTS OF H. PYLORI INFECTION
Although H. pylori is generally thought of as the causative agent for gastric disease, it has the potential to cause other deleterious effects as well as some beneficial effects in those who are infected (Table 1) (51). The other diseases associated with H. pylori infection with the most supporting data are described below. The association with other diseases is less clear and require further studies (i.e., malaria incidence (52) and lung cancer(53)).
OTHER DELETERIOUS EFFECTS OF H. PYLORI INFECTION
PEPTIC ULCERS
H. pylori was first discovered in patients with ulcers and is the primary etiological agent for peptic ulcer disease (5, 54). CagA-positive H. pylori strains are known to associate with higher risk of several diseases including peptic ulcer disease, atrophic gastritis, and gastric cancer (6). With eradication methods for H. pylori in Western Europe, the United States, and Japan, peptic ulcer incidence has decreased (5). The decrease of H. pylori prevalence and, as a result, peptic ulcer incidence in more developed countries further demonstrates the deleterious impact of reduced sanitation, decreased access to clean water, and lower socioeconomic status in less developed countries on peptic ulcer incidence (5). An 18-year cohort study in Taiwan revealed a significant decrease in H. pylori infection, duodenal ulcer prevalence, and gastric ulcer prevalence, following reductions in infection prevalence. Interestingly, over the 18 years of the study, despite the decrease in duodenal ulcer prevalence, gastric ulcer prevalence remained the same. This may be related to human genetic variation. Genetic analysis of the urease gene of H. pylori positive individuals showed that gastric ulcer patients were more likely to have an MboI-restriction fragment length polymorphism-defined genotype 3 for the gene encoding Urease subunit alpha, UreC (55). The UreC genotype was significantly correlated with only gastric ulcers but not duodenal ulcers. Therefore, this genotype may be a target for a precision based method for predicting and treating gastric ulcers, particularly in endemic areas (55).
Peptic ulcers are more common in children, and children with peptic ulcers are at a higher risk for developing celiac disease, an immune-related, gluten sensitivity of the small intestine that induces both gastrointestinal and non-gastrointestinal symptoms (56). However, even though H. pylori prevalence is associated with peptic ulcers, it does not associate with an increase in cases of celiac disease. Therefore, the mechanism of peptic ulcer disease and H. pylori and how it may interact with celiac disease needs to be further studied (57).
MALT LYMPHOMA
Inflammation caused by chronic H. pylori infection is also associated with a rare form of lymphoma, gastric lymphoma of mucosa-associated lymphoid tissue (MALT). In a study of 110 patients with gastric MALT lymphoma, 92% were infected by H. pylori.(58, 59) H. pylori eradication caused regression of low-grade B-cell gastric MALT lymphoma with tumor size decreasing in approximately 75% of patients. Therefore, eradication treatment has been indicated for this lymphoma (59, 60). As regression of MALT lymphoma by H. pylori eradication is most successful in the early stages of disease, early treatment is recommended (61). Additionally, patients with MALT lymphomas are more likely to have the HLA-DQA1*0103, HLA-DQB1*0601 alleles and R702W mutation in the NOD2/CARD15 gene. Therefore, individuals with H. pylori infection can be genetically screened for early eradication treatment in a precision medicine approach for treating MALT lymphoma.
CORONARY ARTERY DISEASE
Coronary Artery Disease (CAD) is a leading cause of death world-wide and the most prevalent cause of myocardial infarction. Current research has shown an involvement of microbes, including H. pylori, with the development of vascular and atherosclerotic disease, heart abnormalities, and the development and progression of CAD (62). In several studies, how other CAD risk factors associate with H. pylori to affect risk are unclear but new molecular studies should elucidate the relationship (62).
IRON DEFICIENCY ANEMIA
Colonization by H. pylori leads to iron deficiency anemia (IDA), especially in children and adolescents (63, 64). Several studies have shown that H. pylori eradication therapy can improve IDA. In a meta-analysis of observational epidemiological studies and randomized controlled trials, individuals had increased hemoglobin levels and serum ferritin concentrations after eradication (64). The soluble transferrin receptor (sTFR), a significantly elevated receptor in H. pylori-infected children, may be a means for assessing iron status (64). In addition, a study examining IDA in children and adolescents examined patterns of H. pylori gene expression and showed increased expression of SabA in those with IDA (63). However, the mechanism leading to IDA via this gene is unclear, although there was some indication that there is a synergistic relationship with VacA, which is also upregulated in IDA (63).
PROTECTIVE EFFECTS OF H. PYLORI INFECTION
ESOPHAGEAL DISEASES
The absence of H. pylori, predominantly in individuals after eradication, has been linked to some esophageal diseases including Gastro-esophageal Reflux Disease (GERD), Barrett’s Esophagus, and adenocarcinoma of the esophagus and the adjacent gastroesophageal junction (65–67). Similarly, epidemiological studies have shown that over the past 50 years, the incidence of the cardia-subtype of gastric cancer has increased several fold, especially in the developed world, while non-cardia gastric cancer has declined. The timing of decreased infection and changes in disease frequency of cancer do not necessarily follow the same trend (68). This link between absence of H. pylori and esophageal disease may be due to H. pylori colonization diminishing gastric acidity and protecting against damage to the esophageal epithelium during reflux episodes. It has also been proposed that H. pylori modifies the expression of gastric hormones that effect the esophageal tissue (13).
A generalized link between the absence of H. pylori infection and esophageal disease may be overly broad. A protective H. pylori effect has been proposed for Eosinophilic esophagitis, a relatively new, allergen/immune-mediated disease in the esophagus, but this conclusion is controversial (69). Overall, the mechanism(s) by which H. pylori protects against esophageal diseases is incomplete and further mechanistic studies are required to confirm potential associations, particularly for eosinophilic esophagitis (69).
ASTHMA AND ALLERGY
A potential inverse relationship between H. pylori and asthma and allergy has been widely reported but also remains controversial (67, 70–76). The presence of H. pylori has been linked in multiple large, blinded epidemiological studies with decreased risk of childhood-onset asthma, hay fever, and cutaneous allergies. These studies support the hypothesis that the rise in asthma is in part due to lower prevalence of H. pylori and several accompanying protective immunological functions, including TLR2/NLRP3/CASP1/IL-18 axis (13, 23, 77). As part of this axis, H. pylori activates CASP1 and induces IL-1β and IL-18 secretion by dendritic cells in the Toll-like receptor 2 (TLR2), a caspase recruitment domain (ASC), and the NLR family pyrin domain containing 3 (NLRP3) (77). These proteins are critical to the H. pylori specific immune response in humans.
Interestingly, the effect of H. pylori factors vary due to ethnicity as reported in an multi-ethnic study of children where 6 year old European children with CagA-negative H. pylori were associated with an increased prevalence of asthma, but children of a non-European background were not (78). Additionally, only H. pylori positive children with a H. pylori negative mother had an increased risk of asthma; thus, the mother’s infection may protect against asthma in H. pylori positive children (78).
H. pylori infection and obesity may interact to affect risk of asthma and allergy, but the causal pathway linking these is unclear (66, 79, 80). In a case-control study using the second Nord-Trøndelag Health Study (HUNT), abdominal obesity and H. pylori infection were associated with reduced risk of asthma and allergy (70). Additionally, H. pylori eradication has been shown to change BMI. This has been hypothesized as being due to the regulation of appetite and energy expenditure by leptin and ghrelin, energy related hormones produced in the gastric mucosa (81). This reveals a likely causal pathway that increases risk of asthma and allergy from reduced H. pylori infections through obesity as well as a possible protective effect of H. pylori infection in the development of asthma and allergy.
Studies in model organisms also support an inverse relationship between H. pylori and allergy. Studies in mice reported that H. pylori protects against allergic asthma by regulating factors seen in human studies, including effector T-cell and T regulatory cells and inhibiting dendritic cells and HSP70 (23, 82). In a study of mice sensitized and challenged with house dust mite (HDM), extract of H. pylori was an effective treatment to reduce mucus production and various features of inflammation in mice rechallenged with HDMs after 1 to 3.5 months of rest (83). Thus, these in vivo studies support the protective role of H. pylori infection on asthma and allergy.
INFLAMMATORY BOWEL DISEASE
Inflammatory bowel disease (IBD) is a chronic relapsing and remitting inflammation of the gastrointestinal tract that results from genetic, environmental, and microbial factors. IBD includes the subtypes Crohn’s disease (CD) and ulcerative colitis (UC) (84, 85). Ecological studies show that IBD is more prevalent in areas with lower rates of H. pylori infection and multiple meta-analyses have shown a significant negative association between H. pylori infection and IBD. This supports a possible protective effect of H. pylori infection against the development of IBD (23, 84–86). In a meta-analysis of 32 studies, a significant negative association was identified between H. pylori infection and IBD that varies by IBD subtype (CD and UC) and by geographic region. H. pylori infection provides more protection against UC than CD, and the protection is more apparent in East Asian populations than Mediterranean ones. However, there is no evidence for an interaction between IBD subtype and region in risk (85). A systemic review of electronic databases including 63 clinical studies also proposed that Helicobacter suppresses proinflammatory products and pathways or prevent or mitigate underlying dysbiosis (84).
An inverse relationship between H. pylori and IBD may be due to the co-evolution of H. pylori strain-specific constituents, such as CagA (84). Nonetheless, heterogeneity among studies and the possibility of publication bias limits the veracity of concluding a negative association. Functional studies are warranted to investigate the effect of H. pylori eradication on the development of IBD with mechanistic studies in H. pylori mouse models defining the mechanism of the negative association, if it exists (87).
OTHER PUTATITIVE PROTECTIONS
H. pylori colonization has also been proposed to provide protection against various other infectious diseases. For example, H. pylori infection may protect against gastrointestinal infections and exogenous intestinal pathogens that cause diarrhea, but this relationship has not been consistently observed and may be due to recent changes in other factors (13, 88, 89). Specifically, as H. pylori prevalence has decreased with industrialization, other factors such as the introduction of clean water, improved sanitation, and less crowding also occurred and both therefore associate with the incidence of lethal diarrheal disease decreasing. Reduced H. pylori transmission is expected as a result of these environmental changes, making it impossible to infer a causal protective relationship between H. pylori infection and diarrheal disease (13).
Individuals infected by H. pylori also show a negative association with tuberculosis. In endemic areas of West Africa, individuals infected by H. pylori are less likely to reactivate latent tubercular infections (13). It has been suggested that H. pylori infection may induce bystander effects with continuous inflammation and T-cell signaling to enhance the host’s innate response and modify the risk of active tuberculosis in humans and non-human primates (13, 90). Ergo, functional studies are needed to elucidate the mechanism to improve protection against tuberculosis.
In summary, while the association of the absence of H. pylori with increased prevalence of chronic diseases have been observed several times as noted above, the statistical association may be due to confounding of other factors that are causative. Further studies will be needed for all disease associations described above to conclude causation.
THE CO-EVOLUTION HYPOTHESIS
H. pylori infection, human and H. pylori genetics, and the environment alone do not predict gastric cancer incidence. Therefore, the interaction of human and H. pylori genetics may play a role in disease severity. Recent work indicated that humans and H. pylori coevolved to reciprocally impact each other and that gastric cancer risk is higher in host-pathogen genomic pairs that did not co-evolve together (91). There is strong evidence for human and H. pylori co-evolution as the migration of humans and their cohabiting H. pylori dates to the original human exodus from Africa, allowing for the coordinated evolution of the two species (92). This relationship has been observed in Latin America’s “Altitude enigma” where human populations living in the mountains have gastric cancer incidence rates up to 25 times higher than the coast with nearly identical and universal H. pylori prevalence (91). When comparing two Colombian sites, gastric disease severity was less severe in H. pylori-human pairs that had similar ancestries; patients with a high proportion of African H. pylori ancestry with primarily Amerindian host ancestry had more severe disease. Contrastingly, those with primarily African H. pylori ancestry and African human ancestry had less severe disease (91, 93). In the discussion of Latin America coevolution, it is important to note that that ancestral Amerindian H. pylori strains have been mostly displaced by ones of European origin (93, 94); this may provide additional disruption. Although the role of co-evolution is difficult to definitively prove, multiple studies have shown patterns of parallel host-pathogen genetic variation that have correlated with functional and molecular changes that increase severity of disease (8).
Another hallmark of co-evolution is the adaptive microevolution in humans seen in the adhesion protein encoding H. pylori gene variant, babA2, that exhibits host-specific effects. H. pylori blood group antigen binding adhesin (BabA) is involved in the binding with receptors and can be modulated at the molecular and functional level to adapt to stress in the gastro-intestinal tract. Amerindians, who almost all carry the O blood group, harbor BabA variant strains that have up to 1500-fold greater O blood group binding affinity (93, 95). Therefore, a human and H. pylori pair with disrupted coevolution may be in part responsible for triggering more severe gastric disease.
H. PYLORI TREATMENTS AND EVOLVING DRUG RESISTANCE
In addition to the potential beneficial effects of H. pylori infection, there is also another reason to limit eradication as a strategy of dealing with it. H. pylori eradication treatments rely on aggressive and regular antibiotic treatment. This has resulted in widespread resistance against commonly used antibiotics (5). In Nigeria, an epidemiological study detected high frequency of bacterial resistance for metronidazole (99.1%) followed by commonly used treatments amoxicillin (33.3%), clarithromycin (14.4%) and tetracycline (4.5%) (96). In contrast, North-East Indian strains are highly resistant to levofloxacin, but highly sensitive to clarithromycin (97). Thus, antibiotic sensitivity varies regionally due to the predominant local antibiotic treatment options but may also be in part due to the underlying genetics of the different populations. In patients in southern Mexico with chronic gastritis, the prevalence of clarithromycin resistance is within internationally accepted ranges (17.8%), but patients with clarithromycin resistant strains (vacA s1m1/cagA+ and vacA s1m1/cagA+/babA2+) are likely at an increased risk of progression to more severe gastric disease due to failure of eradication treatment (98). Therefore, treatment methods will need to vary by region, as well as human and H. pylori genetics.
While there are varying infection rates and gastric cancer incidences, only 1.87% of males and 0.79% of females will develop gastric cancer in their lifetime (68). The majority of people who are infected with H. pylori will never progress to severe gastric disease and may benefit from a precision medicine-based treatment approach. Central America is a prime location for the implementation of precision medicine-based treatments as the infection rate is high and the gastric cancer incidence varies regionally. In a large population-based study in the high gastric cancer incidence region of Central America, using 94 hypothesis-driven variants in 54 genes, an association with gastric cancer models including covariates age, sex, and the H. pylori virulence genotype cagA was detected for three genes, one being ornithine decarboxylase (ODC) (99). ODC has been associated with colorectal cancer and is a possible target for chemoprevention with the ODC inhibitor, eflornithine (100). Therefore, the association of the ODC1 SNP (rs230615) with gastric cancer supports chemoprevention trials using the available agents, such as difluoromethylornithine (DFMO) as seen in colorectal cancer, that interact with the ODC-related polyamine pathway. The ODC1 SNP association with gastric cancer was also significant when stratified by one of the other detected SNPs, toll-like receptor-4 (TLR4), rs1927914 genotype TT. Thus, DMFO treatment may be most effective in persons harboring the TT TLR4 genotype and ODC1 CC genotype at rs230615 (99).
CONCLUSION AND FUTURE DIRECTIONS
Gastric cancer is a complex disease driven by H. pylori infection. Yet, the overwhelming majority of people infected with H. pylori suffer no consequences related to their infection. Prevalence and severity of gastric cancer varies by population and by distinct genetic factors in both host and H. pylori that may have resulted from a complex evolutionary interaction between the two species that involves both deleterious disease and beneficial health effects. Additionally, H. pylori infection’s multiple effects, both protective and deleterious ones, vary throughout an individual’s lifetime, and according to host and H. pylori genetic factors. Therefore, universal eradication for early stage gastric disease that is unlikely to proceed to gastric cancer and other deleterious effects may cause more harm than good. Treatment should vary by the individual and should target host and H. pylori genetic factors as a safer, more realistic method to reduce deleterious effects.
Acknowledgments:
A.M. was supported by T32HL007567 from the National Heart, Lung, and Blood Institute. The research content is solely the responsibility of the authors and does not necessarily represent the official views of the National Heart, Lung, and Blood Institute or the National Institutes of Health.
Footnotes
Competing Interests
The authors declare that they have no competing interests.
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. [DOI] [PubMed] [Google Scholar]
- 2.Allemani C, Matsuda T, Di Carlo V, Harewood R, Matz M, Niksic M, et al. Global surveillance of trends in cancer survival 2000–14 (CONCORD-3): analysis of individual records for 37 513 025 patients diagnosed with one of 18 cancers from 322 population-based registries in 71 countries. Lancet. 2018;391(10125):1023–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Correa P, Piazuelo MB. Helicobacter pylori Infection and Gastric Adenocarcinoma. US Gastroenterol Hepatol Rev. 2011;7(1):59–64. [PMC free article] [PubMed] [Google Scholar]
- 4.Graham DY. Helicobacter pylori update: gastric cancer, reliable therapy, and possible benefits. Gastroenterology. 2015;148(4):719–31 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hooi JKY, Lai WY, Ng WK, Suen MMY, Underwood FE, Tanyingoh D, et al. Global Prevalence of Helicobacter pylori Infection: Systematic Review and Meta-Analysis. Gastroenterology. 2017;153(2):420–9. [DOI] [PubMed] [Google Scholar]
- 6.den Hollander WJ, Holster IL, den Hoed CM, van Deurzen F, van Vuuren AJ, Jaddoe VW, et al. Ethnicity is a strong predictor for Helicobacter pylori infection in young women in a multi-ethnic European city. J Gastroenterol Hepatol. 2013;28(11):1705–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lunet N, Barros H. Helicobacter pylori infection and gastric cancer: facing the enigmas. Int J Cancer. 2003;106(6):953–60. [DOI] [PubMed] [Google Scholar]
- 8.Tavera G, Morgan DR, Williams SM. Tipping the Scale Toward Gastric Disease: A Host-Pathogen Genomic Mismatch? Curr Genet Med Rep. 2018;6(4):199–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karimi P, Islami F, Anandasabapathy S, Freedman ND, Kamangar F. Gastric cancer: descriptive epidemiology, risk factors, screening, and prevention. Cancer Epidemiol Biomarkers Prev. 2014;23(5):700–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Correa P Helicobacter pylori and gastric cancer: state of the art. Cancer Epidemiol Biomarkers Prev. 1996;5(6):477–81. [PubMed] [Google Scholar]
- 11.Suerbaum S, Smith JM, Bapumia K, Morelli G, Smith NH, Kunstmann E, et al. Free recombination within Helicobacter pylori. Proc Natl Acad Sci U S A. 1998;95(21):12619–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kao CY, Sheu BS, Wu JJ. Helicobacter pylori infection: An overview of bacterial virulence factors and pathogenesis. Biomed J. 2016;39(1):14–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cover TL, Blaser MJ. Helicobacter pylori in health and disease. Gastroenterology. 2009;136(6):1863–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wroblewski LE, Peek RM Jr., Wilson KT. Helicobacter pylori and gastric cancer: factors that modulate disease risk. Clin Microbiol Rev. 2010;23(4):713–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maixner F, Krause-Kyora B, Turaev D, Herbig A, Hoopmann MR, Hallows JL, et al. The 5300-year-old Helicobacter pylori genome of the Iceman. Science. 2016;351(6269):162–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moodley Y, Linz B, Bond RP, Nieuwoudt M, Soodyall H, Schlebusch CM, et al. Age of the association between Helicobacter pylori and man. PLoS Pathog. 2012;8(5):e1002693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Falush D, Wirth T, Linz B, Pritchard JK, Stephens M, Kidd M, et al. Traces of human migrations in Helicobacter pylori populations. Science. 2003;299(5612):1582–5. [DOI] [PubMed] [Google Scholar]
- 18.Vale FF, Vadivelu J, Oleastro M, Breurec S, Engstrand L, Perets TT, et al. Dormant phages of Helicobacter pylori reveal distinct populations in Europe. Sci Rep. 2015;5:14333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burkitt MD, Duckworth CA, Williams JM, Pritchard DM. Helicobacter pylori-induced gastric pathology: insights from in vivo and ex vivo models. Dis Model Mech. 2017;10(2):89–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ciesielski TH, Pendergrass SA, White MJ, Kodaman N, Sobota RS, Huang M, et al. Diverse convergent evidence in the genetic analysis of complex disease: coordinating omic, informatic, and experimental evidence to better identify and validate risk factors. BioData Min. 2014;7:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maixner F, Thorell K, Granehall L, Linz B, Moodley Y, Rattei T, et al. Helicobacter pylori in ancient human remains. World J Gastroenterol. 2019;25(42):6289–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bik EM, Eckburg PB, Gill SR, Nelson KE, Purdom EA, Francois F, et al. Molecular analysis of the bacterial microbiota in the human stomach. Proc Natl Acad Sci U S A. 2006;103(3):732–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Borbet TC, Zhang X, Muller A, Blaser MJ. The role of the changing human microbiome in the asthma pandemic. J Allergy Clin Immunol. 2019;144(6):1457–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blaser MJ, Chen Y, Reibman J. Does Helicobacter pylori protect against asthma and allergy? Gut. 2008;57(5):561–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blaser MJ. Equilibria of humans and our indigenous microbiota affecting asthma. Proc Am Thorac Soc. 2012;9(2):69–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tummuru MK, Cover TL, Blaser MJ. Cloning and expression of a high-molecular-mass major antigen of Helicobacter pylori: evidence of linkage to cytotoxin production. Infect Immun. 1993;61(5):1799–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blaser MJ, Perez-Perez GI, Kleanthous H, Cover TL, Peek RM, Chyou PH, et al. Infection with Helicobacter pylori strains possessing cagA is associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Res. 1995;55(10):2111–5. [PubMed] [Google Scholar]
- 28.Park JY, Forman D, Waskito LA, Yamaoka Y, Crabtree JE. Epidemiology of Helicobacter pylori and CagA-Positive Infections and Global Variations in Gastric Cancer. Toxins (Basel). 2018;10(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thorell K, Lehours P, Vale FF. Genomics of Helicobacter pylori. Helicobacter. 2017;22 Suppl 1. [DOI] [PubMed] [Google Scholar]
- 30.Thorell K, Hosseini S, Palacios Gonzales RV, Chaotham C, Graham DY, Paszat L, et al. Identification of a Latin American-specific BabA adhesin variant through whole genome sequencing of Helicobacter pylori patient isolates from Nicaragua. BMC Evol Biol. 2016;16:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berthenet E, Yahara K, Thorell K, Pascoe B, Meric G, Mikhail JM, et al. A GWAS on Helicobacter pylori strains points to genetic variants associated with gastric cancer risk. BMC Biol. 2018;16(1):84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Quintana-Hayashi MP, Rocha R, Padra M, Thorell A, Jin C, Karlsson NG, et al. BabA-mediated adherence of pediatric ulcerogenic H. pylori strains to gastric mucins at neutral and acidic pH. Virulence. 2018;9(1):1699–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boonyanugomol W, Chomvarin C, Hahnvajanawong C, Sripa B, Kaparakis-Liaskos M, Ferrero RL. Helicobacter pylori cag pathogenicity island (cagPAI) involved in bacterial internalization and IL-8 induced responses via NOD1- and MyD88-dependent mechanisms in human biliary epithelial cells. PLoS One. 2013;8(10):e77358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van Vliet AH. Use of pan-genome analysis for the identification of lineage-specific genes of Helicobacter pylori. FEMS Microbiol Lett. 2017;364(2). [DOI] [PubMed] [Google Scholar]
- 35.Thorell K, Yahara K, Berthenet E, Lawson DJ, Mikhail J, Kato I, et al. Rapid evolution of distinct Helicobacter pylori subpopulations in the Americas. PLoS Genet. 2017;13(2):e1006546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Munoz-Ramirez ZY, Pascoe B, Mendez-Tenorio A, Mourkas E, Sandoval-Motta S, Perez-Perez G, et al. A 500-year tale of co-evolution, adaptation, and virulence: Helicobacter pylori in the Americas. ISME J. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mane SP, Dominguez-Bello MG, Blaser MJ, Sobral BW, Hontecillas R, Skoneczka J, et al. Host-interactive genes in Amerindian Helicobacter pylori diverge from their Old World homologs and mediate inflammatory responses. J Bacteriol. 2010;192(12):3078–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.El-Omar EM, Carrington M, Chow WH, McColl KE, Bream JH, Young HA, et al. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature. 2000;404(6776):398–402. [DOI] [PubMed] [Google Scholar]
- 39.El-Omar EM, Rabkin CS, Gammon MD, Vaughan TL, Risch HA, Schoenberg JB, et al. Increased risk of noncardia gastric cancer associated with proinflammatory cytokine gene polymorphisms. Gastroenterology. 2003;124(5):1193–201. [DOI] [PubMed] [Google Scholar]
- 40.Shi Y, Hu Z, Wu C, Dai J, Li H, Dong J, et al. A genome-wide association study identifies new susceptibility loci for non-cardia gastric cancer at 3q13.31 and 5p13.1. Nature genetics. 2011;43(12):1215–8. [DOI] [PubMed] [Google Scholar]
- 41.Abnet CC, Freedman ND, Hu N, Wang Z, Yu K, Shu XO, et al. A shared susceptibility locus in PLCE1 at 10q23 for gastric adenocarcinoma and esophageal squamous cell carcinoma. Nature genetics. 2010;42(9):764–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Study Group of Millennium Genome Project for C, Sakamoto H, Yoshimura K, Saeki N, Katai H, Shimoda T, et al. Genetic variation in PSCA is associated with susceptibility to diffuse-type gastric cancer. Nature genetics. 2008;40(6):730–40. [DOI] [PubMed] [Google Scholar]
- 43.Song HR, Kim HN, Kweon SS, Choi JS, Shim HJ, Cho SH, et al. Genetic variations in the PRKAA1 and ZBTB20 genes and gastric cancer susceptibility in a Korean population. Mol Carcinog. 2013;52 Suppl 1:E155–60. [DOI] [PubMed] [Google Scholar]
- 44.Yan C, Zhu M, Ding Y, Yang M, Wang M, Li G, et al. Meta-analysis of genome-wide association studies and functional assays decipher susceptibility genes for gastric cancer in Chinese populations. Gut. 2020;69(4):641–51. [DOI] [PubMed] [Google Scholar]
- 45.Tanikawa C, Kamatani Y, Toyoshima O, Sakamoto H, Ito H, Takahashi A, et al. Genome-wide association study identifies gastric cancer susceptibility loci at 12q24.11–12 and 20q11.21. Cancer Sci. 2018;109(12):4015–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Helgason H, Rafnar T, Olafsdottir HS, Jonasson JG, Sigurdsson A, Stacey SN, et al. Loss-of-function variants in ATM confer risk of gastric cancer. Nat Genet. 2015;47(8):906–10. [DOI] [PubMed] [Google Scholar]
- 47.Sobota RS, Kodaman N, Mera R, Piazuelo MB, Bravo LE, Pazos A, et al. Epigenetic and genetic variation in GATA5 is associated with gastric disease risk. Hum Genet. 2016;135(8):895–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alvarez MC, Fernandes J, Michel V, Touati E, Ribeiro ML. Effect of Helicobacter pylori Infection on GATA-5 and TFF1 Regulation, Comparison Between Pediatric and Adult Patients. Dig Dis Sci. 2018;63(11):2889–97. [DOI] [PubMed] [Google Scholar]
- 49.Wen XZ, Akiyama Y, Pan KF, Liu ZJ, Lu ZM, Zhou J, et al. Methylation of GATA-4 and GATA-5 and development of sporadic gastric carcinomas. World J Gastroenterol. 2010;16(10):1201–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kienesberger S, Cox LM, Livanos A, Zhang XS, Chung J, Perez-Perez GI, et al. Gastric Helicobacter pylori Infection Affects Local and Distant Microbial Populations and Host Responses. Cell Rep. 2016;14(6):1395–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen Y, Segers S, Blaser MJ. Association between Helicobacter pylori and mortality in the NHANES III study. Gut. 2013;62(9):1262–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gupta V, Perez-Perez GI, Dorsey G, Rosenthal PJ, Blaser MJ. The seroprevalence of Helicobacter pylori and its relationship to malaria in Ugandan children. Trans R Soc Trop Med Hyg. 2012;106(1):35–42. [DOI] [PubMed] [Google Scholar]
- 53.Koshiol J, Flores R, Lam TK, Taylor PR, Weinstein SJ, Virtamo J, et al. Helicobacter pylori seropositivity and risk of lung cancer. PLoS One. 2012;7(2):e32106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marshall BJ, Warren JR. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet. 1984;1(8390):1311–5. [DOI] [PubMed] [Google Scholar]
- 55.Chen TH, Cheng HT, Yeh CT. Epidemiology changes in peptic ulcer diseases 18 years apart explored from the genetic aspects of Helicobacter pylori. Transl Res. 2020. [DOI] [PubMed] [Google Scholar]
- 56.Tumgor G, Agin M, Doran F, Cetiner S. Frequency of Celiac Disease in Children with Peptic Ulcers. Dig Dis Sci. 2018;63(10):2681–6. [DOI] [PubMed] [Google Scholar]
- 57.Agin M, Batun I, Ozdemir S, Doran F, Tumgor G. Prevalence of Helicobacter pylori in Turkish children with celiac disease and its effect on clinical, histopathological, and laboratory parameters. Arch Med Sci. 2019;15(6):1475–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wotherspoon AC, Ortiz-Hidalgo C, Falzon MR, Isaacson PG. Helicobacter pylori-associated gastritis and primary B-cell gastric lymphoma. Lancet. 1991;338(8776):1175–6. [DOI] [PubMed] [Google Scholar]
- 59.Craig VJ, Arnold I, Gerke C, Huynh MQ, Wundisch T, Neubauer A, et al. Gastric MALT lymphoma B cells express polyreactive, somatically mutated immunoglobulins. Blood. 2010;115(3):581–91. [DOI] [PubMed] [Google Scholar]
- 60.Wotherspoon AC, Doglioni C, Diss TC, Pan L, Moschini A, de Boni M, et al. Regression of primary low-grade B-cell gastric lymphoma of mucosa-associated lymphoid tissue type after eradication of Helicobacter pylori. Lancet. 1993;342(8871):575–7. [DOI] [PubMed] [Google Scholar]
- 61.Violeta Filip P, Cuciureanu D, Sorina Diaconu L, Maria Vladareanu A, Silvia Pop C. MALT lymphoma: epidemiology, clinical diagnosis and treatment. J Med Life. 2018;11(3):187–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dadashi M, Hajikhani B, Ghazi M, Yazdani S, Goudarzi M, Nasiri MJ, et al. The global prevalence of Chlamydia pneumoniae, Helicobacter pylori, Cytomegalovirus and Herpes simplex virus in patients with coronary artery disease: A systematic review and meta-analysis. Microb Pathog. 2020:104572. [DOI] [PubMed] [Google Scholar]
- 63.Kato S, Osaki T, Kamiya S, Zhang XS, Blaser MJ. Helicobacter pylori sabA gene is associated with iron deficiency anemia in childhood and adolescence. PLoS One. 2017;12(8):e0184046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Malfertheiner P, Selgrad M. Helicobacter pylori infection and current clinical areas of contention. Curr Opin Gastroenterol. 2010;26(6):618–23. [DOI] [PubMed] [Google Scholar]
- 65.Nomura A, Stemmermann GN, Chyou PH, Perez-Perez GI, Blaser MJ. Helicobacter pylori infection and the risk for duodenal and gastric ulceration. Ann Intern Med. 1994;120(12):977–81. [DOI] [PubMed] [Google Scholar]
- 66.Yap TW, Leow AH, Azmi AN, Callahan DL, Perez-Perez GI, Loke MF, et al. Global Fecal and Plasma Metabolic Dynamics Related to Helicobacter pylori Eradication. Front Microbiol. 2017;8:536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Malnick SD, Melzer E, Attali M, Duek G, Yahav J. Helicobacter pylori: friend or foe? World J Gastroenterol. 2014;20(27):8979–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rawla P, Barsouk A. Epidemiology of gastric cancer: global trends, risk factors and prevention. Prz Gastroenterol. 2019;14(1):26–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Doulberis M, Kountouras J, Rogler G. Reconsidering the “protective” hypothesis of Helicobacter pylori infection in eosinophilic esophagitis. Ann N Y Acad Sci. 2020. [DOI] [PubMed] [Google Scholar]
- 70.Ness-Jensen E, Langhammer A, Hveem K, Lu Y. Helicobacter pylori in relation to asthma and allergy modified by abdominal obesity: The HUNT study in Norway. World Allergy Organ J. 2019;12(5):100035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen Y, Blaser MJ. Helicobacter pylori colonization is inversely associated with childhood asthma. J Infect Dis. 2008;198(4):553–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen Y, Blaser MJ. Inverse associations of Helicobacter pylori with asthma and allergy. Arch Intern Med. 2007;167(8):821–7. [DOI] [PubMed] [Google Scholar]
- 73.Fouda EM, Kamel TB, Nabih ES, Abdelazem AA. Helicobacter pylori seropositivity protects against childhood asthma and inversely correlates to its clinical and functional severity. Allergol Immunopathol (Madr). 2018;46(1):76–81. [DOI] [PubMed] [Google Scholar]
- 74.Khamechian T, Movahedian AH, Ebrahimi Eskandari G, Heidarzadeh Arani M, Mohammadi A. Evaluation of the Correlation Between Childhood Asthma and Helicobacter pylori in Kashan. Jundishapur J Microbiol. 2015;8(6):e17842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Reibman J, Marmor M, Filner J, Fernandez-Beros ME, Rogers L, Perez-Perez GI, et al. Asthma is inversely associated with Helicobacter pylori status in an urban population. PLoS One. 2008;3(12):e4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Annagur A, Kendirli SG, Yilmaz M, Altintas DU, Inal A. Is there any relationship between asthma and asthma attack in children and atypical bacterial infections; Chlamydia pneumoniae, Mycoplasma pneumoniae and Helicobacter pylori. J Trop Pediatr. 2007;53(5):313–8. [DOI] [PubMed] [Google Scholar]
- 77.Koch KN, Hartung ML, Urban S, Kyburz A, Bahlmann AS, Lind J, et al. Helicobacter urease-induced activation of the TLR2/NLRP3/IL-18 axis protects against asthma. The Journal of clinical investigation. 2015;125(8):3297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.den Hollander WJ, Sonnenschein-van der Voort AM, Holster IL, de Jongste JC, Jaddoe VW, Hofman A, et al. Helicobacter pylori in children with asthmatic conditions at school age, and their mothers. Aliment Pharmacol Ther. 2016;43(8):933–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Elias N, Nasrallah E, Khoury C, Mansour B, Abu Zuher L, Asato V, et al. Associations of Helicobacter pylori seropositivity and gastric inflammation with pediatric asthma. Pediatr Pulmonol. 2020;55(9):2236–45. [DOI] [PubMed] [Google Scholar]
- 80.Tsigalou C, Konstantinidis TG, Cassimos D, Karvelas A, Grapsa A, Tsalkidis A, et al. Inverse association between Helicobacter pylori infection and childhood asthma in Greece: a case-control study. Germs. 2019;9(4):182–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Francois F, Roper J, Joseph N, Pei Z, Chhada A, Shak JR, et al. The effect of H. pylori eradication on meal-associated changes in plasma ghrelin and leptin. BMC Gastroenterol. 2011;11:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zuo ZT, Ma Y, Sun Y, Bai CQ, Ling CH, Yuan FL. The Protective Effects of Helicobacter pylori Infection on Allergic Asthma. Int Arch Allergy Immunol. 2020:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.van Wijck Y, John-Schuster G, van Schadewijk A, van den Oever RL, Obieglo K, Hiemstra PS, et al. Extract of Helicobacter pylori Ameliorates Parameters of Airway Inflammation and Goblet Cell Hyperplasia following Repeated Allergen Exposure. Int Arch Allergy Immunol. 2019;180(1):1–9. [DOI] [PubMed] [Google Scholar]
- 84.Axelrad JE, Cadwell KH, Colombel JF, Shah SC. Systematic review: gastrointestinal infection and incident inflammatory bowel disease. Aliment Pharmacol Ther. 2020;51(12):1222–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Imawana RA, Smith DR, Goodson ML. The relationship between inflammatory bowel disease and Helicobacter pylori across East Asian, European and Mediterranean countries: a meta-analysis. Ann Gastroenterol. 2020;33(5):485–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rokkas T, Gisbert JP, Niv Y, O’Morain C. The association between Helicobacter pylori infection and inflammatory bowel disease based on meta-analysis. United European Gastroenterol J. 2015;3(6):539–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Luther J, Dave M, Higgins PD, Kao JY. Association between Helicobacter pylori infection and inflammatory bowel disease: a meta-analysis and systematic review of the literature. Inflamm Bowel Dis. 2010;16(6):1077–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rothenbacher D, Blaser MJ, Bode G, Brenner H. Inverse relationship between gastric colonization of Helicobacter pylori and diarrheal illnesses in children: results of a population-based cross-sectional study. J Infect Dis. 2000;182(5):1446–9. [DOI] [PubMed] [Google Scholar]
- 89.Bode G, Rothenbacher D, Brenner H. Helicobacter pylori colonization and diarrhoeal illness: results of a population-based cross-sectional study in adults. Eur J Epidemiol. 2001;17(9):823–7. [DOI] [PubMed] [Google Scholar]
- 90.Perry S, de Jong BC, Solnick JV, de la Luz Sanchez M, Yang S, Lin PL, et al. Infection with Helicobacter pylori is associated with protection against tuberculosis. PLoS One. 2010;5(1):e8804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kodaman N, Pazos A, Schneider BG, Piazuelo MB, Mera R, Sobota RS, et al. Human and Helicobacter pylori coevolution shapes the risk of gastric disease. Proc Natl Acad Sci U S A. 2014;111(4):1455–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Linz B, Balloux F, Moodley Y, Manica A, Liu H, Roumagnac P, et al. An African origin for the intimate association between humans and Helicobacter pylori. Nature. 2007;445(7130):915–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kodaman N, Sobota RS, Mera R, Schneider BG, Williams SM. Disrupted human-pathogen co-evolution: a model for disease. Front Genet. 2014;5:290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gutierrez-Escobar AJ, Velapatino B, Borda V, Rabkin CS, Tarazona-Santos E, Cabrera L, et al. Identification of New Helicobacter pylori Subpopulations in Native Americans and Mestizos From Peru. Front Microbiol. 2020;11:601839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ansari S, Yamaoka Y. Helicobacter pylori BabA in adaptation for gastric colonization. World J Gastroenterol. 2017;23(23):4158–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Harrison U, Fowora MA, Seriki AT, Loell E, Mueller S, Ugo-Ijeh M, et al. Helicobacter pylori strains from a Nigerian cohort show divergent antibiotic resistance rates and a uniform pathogenicity profile. PLoS One. 2017;12(5):e0176454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mahant S, Sharma AK, Gehlot V, Mukhopadhyay AK, Chhawchharia A, Dutta S, et al. Geographically distinct North-East Indian Helicobacter pylori strains are highly sensitive to clarithromycin but are levofloxacin resistant. Indian J Med Microbiol. 2019;37(3):337–44. [DOI] [PubMed] [Google Scholar]
- 98.Alarcon-Millan J, Fernandez-Tilapa G, Cortes-Malagon EM, Castanon-Sanchez CA, De Sampedro-Reyes J, Cruz-Del Carmen I, et al. Clarithromycin resistance and prevalence of Helicobacter pylori virulent genotypes in patients from Southern Mexico with chronic gastritis. Infect Genet Evol. 2016;44:190–8. [DOI] [PubMed] [Google Scholar]
- 99.Miller AK, Tavera G, Dominguez R, Camargo MC, Waterboer T, Wilson KT, et al. Ornithine Decarboxylase (ODC1) gene variant (rs2302615) is associated with gastric cancer independently of Helicobacter pylori CagA serostatus. medRxiv. 2021:2021.04.13.21254467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zell JA, McLaren CE, Chen WP, Thompson PA, Gerner EW, Meyskens FL. Ornithine decarboxylase-1 polymorphism, chemoprevention with eflornithine and sulindac, and outcomes among colorectal adenoma patients. J Natl Cancer Inst. 2010;102(19):1513–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sierra MS, Cueva P, Bravo LE, Forman D. Stomach cancer burden in Central and South America. Cancer Epidemiol. 2016;44 Suppl 1:S62–S73. [DOI] [PubMed] [Google Scholar]
- 102.Corral JE, Delgado Hurtado JJ, Dominguez RL, Valdez de Cuellar M, Balmore Cruz C, Morgan DR. The descriptive epidemiology of gastric cancer in Central America and comparison with United States Hispanic populations. J Gastrointest Cancer. 2015;46(1):21–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bray F, Colombet M, Mery L, Piñeros M, Znaor A, Zanetti R and Ferlay J, editors. Cancer Incidence in Five Continents. Vol XI (electronic version) Lyon: International Agency for Research on Cancer; Available from: https://ci5iarcfr, accessed 01/15/2021. 2017. [Google Scholar]
- 104.Roussos A, Philippou N, Mantzaris GJ, Gourgoulianis KI. Respiratory diseases and Helicobacter pylori infection: is there a link? Respiration. 2006;73(5):708–14. [DOI] [PubMed] [Google Scholar]
- 105.Rozankovic PB, Huzjan AL, Cupic H, Bencic IJ, Basic S, Demarin V. Influence of CagA-positive Helicobacter pylori strains on atherosclerotic carotid disease. J Neurol. 2011;258(5):753–61. [DOI] [PubMed] [Google Scholar]
- 106.Longo-Mbenza B, Nsenga JN, Mokondjimobe E, Gombet T, Assori IN, Ibara JR, et al. Helicobacter pylori infection is identified as a cardiovascular risk factor in Central Africans. Vasc Health Risk Manag. 2012;6:455–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.den Hollander WJ, Schalekamp-Timmermans S, Holster IL, Jaddoe VW, Hofman A, Moll HA, et al. Helicobacter pylori colonization and pregnancies complicated by preeclampsia, spontaneous prematurity, and small for gestational age birth. Helicobacter. 2017;22(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Xia B, Wang W, Lu Y, Chen C. Helicobacter pylori infection increases the risk of metabolic syndrome in pregnancy: a cohort study. Ann Transl Med. 2020;8(14):875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lebwohl B, Blaser MJ, Ludvigsson JF, Green PH, Rundle A, Sonnenberg A, et al. Decreased risk of celiac disease in patients with Helicobacter pylori colonization. Am J Epidemiol. 2013;178(12):1721–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bassi V, Marino G, Iengo A, Fattoruso O, Santinelli C. Autoimmune thyroid diseases and Helicobacter pylori: the correlation is present only in Graves’s disease. World J Gastroenterol. 2012;18(10):1093–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chen Y, Blaser MJ. Association between gastric Helicobacter pylori colonization and glycated hemoglobin levels. J Infect Dis. 2012;205(8):1195–202. [DOI] [PMC free article] [PubMed] [Google Scholar]