

Abstract
The Brazilian buffy-tufted-ear marmoset (Callithrix aurita), one of the world’s most endangered primates, is threatened by anthropogenic hybridization with exotic, invasive marmoset species. As there are few genetic data available for C. aurita, we developed a PCR-free protocol with minimal technical requirements to rapidly generate genomic data with genomic skimming and portable nanopore sequencing. With this direct DNA sequencing approach, we successfully determined the complete mitogenome of a marmoset that we initially identified as C. aurita. The obtained nanopore-assembled sequence was highly concordant with a Sanger sequenced version of the same mitogenome. Phylogenetic analyses unexpectedly revealed that our specimen was a cryptic hybrid, with a C. aurita phenotype and C. penicillata mitogenome lineage. We also used publicly available mitogenome data to determine diversity estimates for C. aurita and three other marmoset species. Mitogenomics holds great potential to address deficiencies in genomic data for endangered, non-model species such as C. aurita. However, we discuss why mitogenomic approaches should be used in conjunction with other data for marmoset species identification. Finally, we discuss the utility and implications of our results and genomic skimming/nanopore approach for conservation and evolutionary studies of C. aurita and other marmosets.
Subject terms: Evolutionary genetics, Genetic hybridization
Introduction
‘Genomic skimming’ is an emerging technique involving random shotgun sequencing of a small percentage of total genomic DNA, which results in a comparatively large sampling of high-copy portions of the genome like the mitogenome and repetitive elements1. As genetic characterization of a species is fundamental for conservation and evolutionary studies, this technique is expanding the use of mitogenomics for phylogenetics, species identification, and biodiversity assessments of under-studied organisms2,3. While genomic skimming originates from short-read, next generation sequencing, recent implementations of the technique instead have utilized portable, long-read nanopore sequencing to successfully generate de novo mitogenomic assemblies of genomic-data deficient vertebrates without the need for PCR4,5. Thus, the coupling of genomic skimming with nanopore sequencing holds great potential to address genomic data deficiencies in non-model organisms to assist in species conservation efforts and evolutionary studies.
In this work, we aimed to develop a rapid, cost-effective protocol to obtain genomic data for one of the world’s most endangered primates, the small buffy-tufted-ear marmoset C. aurita (Fig. 1), for which there is relatively little genetic knowledge. This species is native to the mountainous regions of southeastern Brazil6–8, and there are about 10,000 mature C. aurita remaining in the wild9. The IUCN Red List considers this species Endangered, and C. aurita conservation threats include habitat deforestation, urbanization, and logging9. The buffy-tufted-ear marmoset also faces ecological and hybridization threats from two invasive marmoset species that have been introduced by humans into southeastern Brazil, C. jacchus and C. penicillata (natural ranges and species shown in Fig. 1)7–11. Currently, a captive breeding program is underway for C. aurita, with the aim to eventually reintroduce the species into the wild. As genetics and genomics are an integral part of the C. aurita program for species identification, breeding, and population management8,12, rapid access to genetic data is crucial for the long-term success of C. aurita conservation.
Figure 1.

The distinct natural distributions and phenotypes of C. jacchus, C. penicillata, and C. aurita. Cities of origin (São Jose dos Campos) and collection (Guarulhos) of individual BJT022 are shown as orange squares. Callithrix species distributions are based on 2012 IUCN Red List Spatial Data (http://www.iucnredlist.org/technical-documents/spatial-data) and the accompanying map was produced with the R 4.1.0 scripting language18. The photographs on the right hand side show the phenotype of individual BJT022 as matching that of C. aurita, as well as the phenotypes of C. jacchus and C. penicillata for reference.
Most Callithrix genetic work is based on phylogenetic analysis of short mitochondrial DNA (mtDNA) regions such as COI, COII, and the control region to resolve phylogenetic relationships (e.g.,10,13–15). However, due to the relatively recent divergence times between Callithrix mtDNA lineages8,16, these Callithrix phylogenies usually show polytomies or lack strong statistical support. Nonetheless, mitogenomics alleviates some of these difficulties in Callithrix phylogenetics studies7,16, and the C. aurita mitogenome was recently assembled and annotated with Sanger sequencing and short-read high-throughput sequencing7. However, one drawback to current Callithrix mitogenomics work is the use of relatively time-consuming molecular techniques and lag time for sequencing data generation from several weeks to months (pers. obs., Malukiewicz).
As a subsequent step in developing genomic resources for C. aurita, we present here a rapid, PCR-free, genomic skimming technique for mitogenomic reconstruction with the hand-held, long-read Oxford Nanopore Technologies (ONT) minION sequencer. We assessed whether the mitogenome of a single buffy-tufted-ear marmoset could reliably be reconstructed with ONT sequencing, and then be used for downstream phylogenetic and genetic diversity analyses. We checked the accuracy of the ONT mitogenome assembly against a mitogenome assembly generated from the same marmoset individual using long range PCR and Sanger sequencing7,16. There was not only very high concordance between the ONT and Sanger-derived sequences, but with this technique we also uncovered unexpected discordance between the phenotype and mitogenomic lineage of the sampled marmoset individual. Our analyses showed that our marmoset individual was actually a cryptic hybrid possessing a mitogenome lineage belonging to C. penicillata, but a C. aurita phenotype. This finding represents the first recorded case of genetic introgression of C. penicillata genetic material into C. aurita. Our approach involving ONT-based genomic skimming increased the available genomic information for Callithrix taxa and provides a robust and accessible tool for genomic studies and conservation of Callithrix, particularly C. aurita.
Results
Phenotypic taxon identification of sampled marmoset individual
We collected a small skin biopsy from a single individual (BJT022) housed at the Guarulhos Municipal Zoo, Guarulhos, São Paulo state, weighed the individual, and photographed it. Zoological records state that the individual originates from São Jose dos Campos, SP (Fig. 1). Based on previous phenotypic and morphological description of the species7,8,17, we initially identified the individual as C. aurita. The individual had the yellow ear tufts characteristic of C. aurita, as well as yellow pelage above the forehead, and other yellow patches around the facial region17. The overall pelage of the individual was black, with yellow pelage prominently visible, but intermixed with darker pelage at the hands. As can be seen from Fig. 1, the phenotype of C. aurita is distinct from that of C. jacchus and C. penicillata, which are commonly found as invasive, exotic species in the natural range of C. aurita. Morphologically, the individual was within the expected body weight range for C. aurita (approximately 450g)8. Other Callithrix species tend to weight relatively much less (200-300g)8.
Complete mitogenome reconstruction
After direct ONT sequencing of genomic DNA (gDNA) from individual BJT022, we successfully determined a full mitogenome using the genomic skimming/minION approach. ONT sequence reads were first filtered for quality and to remove sequencing adapters. Then ONT mtDNA reads were identified with the BLAST-based mtBlaster script5, with the C. aurita (GenBank Accession MN787075) mitogenome as the reference genome. Table 1 shows overall characteristics for gDNA ONT reads as well as ONT mtDNA reads. The final length of the reconstructed ONT-derived mitogenome was 16,181 base pairs (bps) with an average coverage of 9x. Annotation of the ONT-derived mitogenome with the CHLOROBOX web server19 showed that the mitogenome contained 37 mitochondrial genes (13 protein– coding genes, 22 tRNAs, and two rRNAs), as shown in Supplementary Figure 1.
Table 1.
Metrics for BJT022 minION mitogenome assembly.
| Information | Result |
|---|---|
| Total number of genomic reads | 50553 |
| Mean genomic read length | 3208.31 |
| Mean genomic read quality | 12.2 |
| Average read length N50 | 4669.63 |
| Number of mtDNA Reads | 60 |
| Average mtDNA read length (bp) | 3866.75 |
| Average mtDNA base quality (phred score) | 20.44 |
| Average mtDNA sequencing depth | 9x |
| Final mtDNA genome length (bp) | 16181 |
We assessed accuracy of the reconstructed ONT-derived mitogenome by comparing the resulting sequence with that of a Sanger-sequenced mitogenome from the same individual. To note, the Sanger sequence mitogenome was missing 119 bases that comprised the entire tRNA-Phe and part of the 12S rRNA region. Nonetheless, pairwise-alignment of the Sanger and ONT-derived sequences of the BJT022 mitogenome showed 99.4% concordance, as illustrated in the dot plot in Fig. 2. Sites of non-concordant bases between the two sequences accounted for 0.2% of overall alignment sites. Then sites with missing bases, mostly within long homopolymer runs within the ONT sequence, accounted for 0.4% of all alignment sites. To finalize the mitogenome assembly of BJT022, we made a consensus sequence by using the Sanger sequence to correct for non-concordant sites and homopolymer runs, and the ONT sequence to fill the 119 bases missing from the Sanger sequence. This consensus alignment had a nucleotide composition of A 32.6%, T 27.0%, C 27.0%, and G 13.4%.
Figure 2.
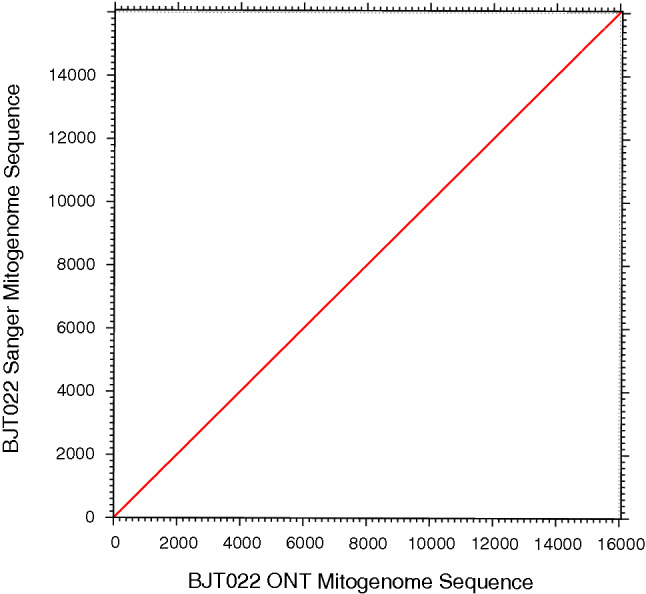
Dot plot of pairwise alignment of the mionION and Sanger sequence Callithrix mitogenome of marmoset BJT022. The alignment was produced in MAFFT.
Phylogenetic reconstruction
We obtained a well supported Maximum Likelihood (ML) phylogeny (Fig. 3, Supplementary Figure 2) that combined the new BJT022 consensus mitogenome sequence with previously available Callithrix and other New World primate mitogenome sequences7. As we obtained a phylogeny with a highly similar topology and levels of statistical support to that of Malukiewicz et al. (2021)7, we used the same phylogenetic clade designations for Callithrix mitogenomic haplotypes as in that previous study. In examining the C. aurita clade within the resulting phylogeny, two distinct geographical clusters appear. One cluster is composed of two haplotypes originating from Rio de Janeiro state (BJT107, BJT115), and the other cluster is composed of a mix of haplotypes originating from Minas Gerais and São Paulo states (following provenance information in Malukiewicz et al. (2021)7).
Figure 3.

ML tree showing phylogenetic clustering of the Callithrix mitogenome of marmoset BJT022 within the C. penicillata Caatinga clade (complete tree with outgroups is presented in Supplementary Figure 2). The BJT022 mitogenome is highlighted at the tree tips in purple along with two other mitogenomes of marmosets with C. aurita phenotypes from Malukiewicz et al. (2021)7 that were incongruent with their mitogenomic lineages. Categorization of major Callithrix mitogenomic clades follow that of Malukiewicz et al. (2021)7.
In our phylogeny, the mitogenomic lineage of individual BJT022 unexpectedly clustered not within the C. aurita clade, but rather with the C. penicillata Caatinga clade. Additionally, two mitogenome lineages from Malukiewicz et al. (2021)7 that originated from marmosets with C. aurita phenotypes phylogenetically clustered within the C. jacchus clade in Fig. fig:tree and in Malukiewicz et al. (2021)7. Thus, these two studies show three cases of cryptic hybrids (BJT020, BJT022, and BJT079) whose C. aurita phenotypes are discordant with their jacchus group mitogenomic lineages.
Callithrix mitochondrial genetic diversity
We used Callithrix mitogenome sequences from Malukiewicz et al. (2021)7 to calculate diversity indexes of four Callithrix species (C. aurita, C. jacchus, C. geoffroyi, and C. penicillata), for which haplotypes were available for three or more individuals per species (Table 2). Sequences from sampled marmosets with discordant species phenotypes and mitogenomic phylogenetic clustering were excluded from these analyses. Relative to the other four species, C. penicillata showed much higher values across all diversity indexes (Table 2). Callithrix jacchus showed among the highest haplotype diversity values, but then had the lowest values for all other diversity indexes. Diversity indexes for C. aurita generally fell in the middle relative to values of the other species. This was also the case for C. geoffroyi diversity indexes.
Table 2.
Callithrix diversity measures.
| Species | Sequences | Haplotypes | Haplotype diversity | Nucleotide diversity | Theta | Variable sites |
|---|---|---|---|---|---|---|
| C. aurita | 6 | 4 | 0.87 | 0.00315 | 44.67 | 102 |
| C. geoffroyi | 7 | 4 | 0.81 | 0.00535 | 78.37 | 190 |
| C. jacchus | 9 | 7 | 0.94 | 0.00109 | 23.92 | 65 |
| C. penicillata | 8 | 7 | 0.96 | 0.01166 | 162.75 | 414 |
Discussion
Feasibility of genomic skimming on the ONT minION sequencer
The term ’genomic skimming’ was first coined by in 2012 by Straub et al. (2012)20 as a way to utilize shallow sequencing of gDNA to obtain relatively deeper coverage of high-copy portions of the genome, including mitogenomes. In combining genomic skimming with ONT long-read sequencing, we successfully reconstructed a complete marmoset mitogenome without the need for prior PCR enrichment, using standard molecular biology equipment, and a compact portable sequencer that connects to a laptop computer. Preparation of genetic material for ONT sequencing in this study took less than a full day, and sequencing reads were available within 48 hours. Although the coverage of our reconstructed ONT mitogenome was low-medium (9x) and one of the largest sources of error for the ONT reads were missing reads in long homopolymer runs, the ONT data showed a high degree of concordance with gold standard mtDNA Sanger sequencing reads for the same individual. Hence, this work along with a number of previous studies (e.g.,5,21), highlights ONT-based genomic skimming as holding great potential for enhancing mitogenomic and diversity studies of data-deficient and/or non-model organisms.
A major challenge in ONT sequencing is the relatively high sequencing error (5%-15%), but the application of computational ’polishing’ significantly reduces errors of raw ONT data (e.g.22). Another challenge with ONT methodologies is the large amount of input DNA needed for sequencing relative to other types of methods, particularly PCR and Sanger sequencing. Multiplexing samples onto the same flow cell is one way to reduce the required amount of per sample DNA, and currently ONT chemistry allows for up to 24 individual gDNA samples to be multiplex per flow cell. Another option to improve mitogenome coverage from genome skimming shotgun data, especially for sensitive applications is to use sample preparation approaches that specifically enrich for mtDNA (e.g., https://www.protocols.io/view/isolation-of-high-quality-highly-enriched-mitochon-mycc7sw).
It is important to point out that our approach represents a starting point from which methodological aspects could be adjusted to further improve and modify our protocol. An important consideration for long-read sequencing is access to high-quality DNA which is not degraded. For marmosets especially, another consideration for input DNA is whether chimerism could bias genomic analysis or not, as levels of chimerism vary between marmoset biological tissues. Marmosets usually give birth to twins that are natural hematopoietic chimeras due to cellular exchange from placental vascular anastomoses during early fetal development23–25. This chimerism may result in the presence of up to 4 alleles of a single-copy genomic locus within a single individual. In marmosets, skin shows some of the lowest amounts of chimerism while blood is highly chimeric24–26. Depending on project design, high levels of chimerism can bias base calling of nuclear genome derived sequence reads, but this is less of a concern for mitogenomic studies as mtDNA is haploid and transmitted maternally.
In this work, we obtained DNA from a ear skin biopsy, but this represents a minimally invasive source of genetic material. As an epidermal tissue, buccal swabs are a relatively less invasive source of low-chimerism epidermal DNA. Recently, urine has also been shown to be a non-invasive source of high-quality DNA27, but the amount of chimerism is currently not known for marmoset urine. Urine represents a potentially non-invasive genetic tissue which could be combined with genomic skimming of highly endangered non-model organisms, particularly within captive settings.
Callithrix aurita and anthropogenic marmoset hybridization
Our original aim in this work was to reconstruct the mitogenome of the endangered buffy-tufted-ear marmoset with a PCR-free ’genomic skimming’ approach with minimal technical requirements. We successfully reconstructed the full mitogenome from a captive individual possessing a C. aurita phenotype, but the mitogenomic lineage showed unexpected discordance with this phenotype. While we expected the mitogenome of the sampled individual to be that of C. aurita, instead the sampled individual possessed a C. penicillata mitogenomic lineage. Our results also represent the first ever known instance of one-way genetic introgression from C. penicillata into C. aurita, and indicate that our sampled marmoset was actually a cryptic C. aurita x C. penicillata hybrid.
Although a number of scenarios could explain the phenotypic-genotypic discordance we uncovered in individual BJT022, this case is likely the result of relatively recent anthropogenic hybridization between a C. penicillata female and C. aurita male. Callithrix species are naturally allo- and parapatric, and natural hybridization occurs between marmoset species under secondary contact8. Past natural genetic introgression between C. aurita and C. penicillata would most likely have occurred in the natural contact zone between these species that exists in the transitional areas between the Cerrado and Atlantic Forest Biomes of southeastern Brazil. Because C. penicillata mitogenomic clades tend to be well defined by their biogeographic origin7, for past, natural introgresssion of C. penicillata into C. aurita, we would expect haplotype BJT022 to have grouped with the C. penicillata Atlantic Forest/Cerrado clade. However, that is not the case, as the BJT022 haplotype grouped instead within the C. penicillata Caatinga Clade. There is a relatively large geographic separation between the Caatinga biome of northeastern Brazil and the portion of the southeastern Brazilian Atlantic Forest that houses the natural region of C. aurita. This wide geographic gap highly reduces the possibility of past natural interbreeding between Caatinga populations of C. penicillata and any C. aurita population.
We could also consider incomplete lineage sorting to explain the phylogenetic position of the BJT022 mitogenomic haplotype as reflecting a C. aurita mitogenome that sorted within a C. penicillata phylogenetic clade instead of a C. aurita clade. Overall, we see strong consistency in grouping patterns of mitogenomic haplotypes within their expected Callithrix phylogenetic clades. Further, C. aurita and the jacchus marmoset subgroup (C. geofforyi, C. kuhlii, C. jacchus, C. penicillata) diverged about 3.54 million years ago7, leaving relatively more time for mitochondrial lineage sorting between C. aurita and the jacchus group than among jacchus group species. While incomplete lineage sorting has indeed been used to explain C. penicillata and C. kuhlii polyphyly7, we still do see clear grouping patterns of C. kuhlii and C. penicillata mitogenomic clades according to their species of origin. Therefore, the strong tendency for Callithrix mitogenomic lineages to group within their expected clades reduces the likelihood of incomplete lineage sorting of mitogenomic lineages between C. aurita and the jacchus group.
The similarity of the case of BJT022 to other likely instances of anthropogenic Callithrix hybridization provide further support for BJT022 representing anthropogenic interbreeding between C. aurita and C. penicillata. Callithrix penicillata and C. jacchus have been introduced into the native range of C. aurita in southeastern Brazil largely as a result of the illegal pet trade and subsequent releases of exotic marmosets into forest fragments7,8. Malukiewicz et al. (2021)7 recently found evidence of genetic introgression from of exotic C. jacchus into C. aurita within the metropolitan area of the city of São Paulo. A cryptic C. aurita hybrid sampled by Malukiewicz et al.7 originates from the municipality of Mogi das Cruzes, which lies in the eastern portion of metropolitan São Paulo8,28. Following zoological records, BJT022 originated from the municipality of São Jose dos Campos, which also lies in the eastern portion of metropolitan São Paulo. These cryptic hybrids also likely represent an advanced stage of anthropogenic hybridization between native C. aurita and exotic jacchus group species. First generation and early generation aurita and jacchus group marmoset hybrids are known to possess a distinct “koala bear” appearance10,11,29. As this is not the phenotype seen for BJT022 and the cryptic C. aurita hybrids from Malukiewicz et al.7, this observation suggests that these cases of anthropogenic hybridization arose through backcrossing of an earlier non-cryptic C. aurita x Callithrix sp. hybrid with C. aurita. Eventually these backcrosses led to the genomic capture of introgressed jacchus group mitogenome lineages by the C. aurita populations of the eastern portion of the São Paulo metropolitan area.
The above results are alarming since they suggest that genetic introgression is underway from exotic, invasive marmosets to the endangered, native marmosets of southeastern Brazil. At this time, it is not possible to determine how board this pattern is at the geographic, genomic and species levels, and whether introgression is only unidirectional and exactly which exotic and native species are involved. Specifically for C. aurita, unidirectional genetic introgression from invasive marmosets as well as cryptic hybridization is worrying due to the species’ threatened conservation status. A small number of captive facilities around southeastern Brazil are currently breeding captive C. aurita for eventual reintroductions into the wild8,12. Individuals within these captive populations should be confirmed both genetically and phenotypically as not being of hybrid origin, as to avoid introducing exogenous genetic material into the captive population and subsequently into the wild. Additionally, further genetic information is needed for wild C. aurita populations to not only characterize diversity within the species, but also to better assess the occurrence of hybridization between exotic and native marmosets in southeastern Brazil. This information is critical for defining genetic diversity of C. aurita and maintaining species genetic integrity in the wild and captivity.
Utility of mitogenomics for evolutionary and conservation studies of Callithrix aurita and other marmosets
The buffy-tufted-ear marmoset is not only critically endangered but also highly data-deficient in terms of genetic information. The limited number of genetic studies involving C. aurita have used the mtDNA control region13,15, COI10, and the mitogenome7 for phylogenetic study of Callithrix mtDNA lineages, species identification, and detection of hybridization. The phylogenies obtained by us and Malukiewicz et al. (2021)7 do show some geographical separation between C. aurita mitogenome haplotypes originating from different portions of the species’ natural range. Our calculation of Callithrix mtDNA diversity indexes based on data from Malukiewicz et al. (2021)7 show that diversity in C. aurita is still comparable to that of other Callithrix species. However, a large sampling effort of C. aurita in terms of individual numbers and across the species range is needed for accurate determination of current levels of species standing genetic variation. Additionally, surveys should be conducted of the standing genetic variation levels of the captive C. aurita population. These data are crucial for understanding anthropogenic impacts on the species as well for making appropriate decisions for species conservation.
The application of genomic skimming based on portable ONT long-read technology can be applied to address several of these knowledge gaps for C. aurita. First, with large-scale sampling of wild and captive C. aurita, genetic diversity estimates, demographic history, and other evolutionary analyses can be calculated relatively easily from mitogenomic data. Given the relatively fast turnaround time to obtain sequencing data from the minION, such data could be quickly obtained for a primate as highly endangered as C. aurita, without weeks or months long wait times for sequencing data. Laboratory setup of the minION also does not require any additional special equipment, which also makes genomic work with highly endangered species as C. aurita accessible for investigators under relatively constrained budgets.
Callithrix aurita’s sister species Callithrix flaviceps faces a similar plight as C. aurita, but with an adult population estimated to be at about 2000 adult individuals30. Currently there are also plans to breed C. flaviceps in captivity for eventual wild reintroduction, but currently there is, to our knowledge, no genetic data available for this species. Thus, the same sort of sampling and research efforts are needed for C. flaviceps as for C. aurita, perhaps even more urgently for the former species given its smaller population. As such, C. flaviceps is a good candidate case for the adaptation of techniques such as genomic skimming and low-cost desktop sequencing to rapidly increase genomic resources for a non-model species for conservation and evolutionary studies.
In the case of marmosets, while mitogenomics shows great potential for usage in evolutionary and conservation studies, we strongly urge against sole use of mtDNA markers for identification of species and hybrids. As the results of this study, as well as that of Malukiewicz et al. (2021)7 clearly show, cryptic hybrids can easily be mistaken for species, and had we only depended on mtDNA results we would have misidentified three cryptic Callithrix hybrids as C. jacchus and C. penicillata. Instances of cryptic hybrids have also been shown among natural C. jacchus x C. penicillata hybrids25. All of these instances underline the need to use several lines of evidence for taxanomic identification of marmoset individuals, particularly due to widespread anthropogenic hybridization among marmosets. We used a combination of phenotypic and mitochondrial data to classify the sampled individual BJT022 as a cryptic hybrid. As mitochondrial DNA is maternally transmitted, it is also not possible to genetically identify the paternal lineage of hybrids without further use of autosomal or Y-chromosome genetic markers. When ever phenotypic data are available, these data should be used jointly with molecular data for identification or classification of a marmoset individual as belonging to a specific species or hybrid type. Indeed, the integrated use of phenotypic and molecular approaches will lead to a better understand the phenomena that involve hybridization processes31.
Conclusion
Brazilian legal instruments that protect C. aurita consider hybridization a major threat to the survival of this species8,12. In this report, we have uncovered the first known case of cryptic hybridization between C. aurita and C. penicillata, which may represent a larger trend of genetic introgression from exotic into native marmosets in southeastern Brazil. Our findings are based on the combination of two recent innovations in the field of genomics, that of genomic skimming and portable long-read sequencing on the ONT minION. Given that C. aurita is still very deficient for genetic data, our approach provides a substantial advance in making more genomic data available for one of the world’s most endangered primates. Genomic skimming based on ONT sequencing can be integrated easily with phenotypic and other genetic data to quickly make new information accessible on species biodiversity and hybridization. Such data can then be utilized within the legal Brazilian framework to protect endangered species like C. aurita. More specifically, rapid access to emerging biological information on such species leads to more informed decisions on updating or modifying legal actions for protecting endangered fauna. The ONT genomic skimming approach we present here can be further utilized and optimized to more rapidly generate genomic information without the need for specialized technological infrastructure nor the need for a priori genomic information.
Methods
Sampling
A skin biopsy was sampled from a single adult male marmoset with a Callithrix aurita phenotype at the Guarulhos Municipal Zoo, Guarulhos, Sao Paulo, Brazil following the procedure described in Malukiewicz et al. (2017, 2021)7,16. Tissues were collected under the approval of the ASU Institutional Animal Care and Use Committee Animals (ASU IACUC, protocols #11-1150R, 15-144R) and Brazilian Environmental Ministry (SISBIO protocols #47964-2 and #28075-2). Biological tissue sampling complied with all institutional, national, and international guidelines. The sample is registered in the Brazilian SISGEN database under number A18C1CE. This study is reported in accordance to ARRIVE guidelines (https://arriveguidelines.org/resources/questionnaire).
Sequencing
The mitochondrial genome of the sampled marmoset was reconstructed using a previously published approach based on long range polymerase chain reaction (L-PCR) and Sanger sequencing following Malukiewicz et al. (2017)16 and Malukiewicz et al. (2021)7. Then the Oxford Nanopore minION portable sequencer was used in-house for sequencing of the same sample on two R9.4 (Oxford Nanopore FLO-MIN106) flow cells. DNA was prepared following manufacturer instructions for the Ligation Sequencing Kit 1D (Oxford Nanopore SQK-LSK108). Briefly, for each minION sequencing run, 1000 ng of input genomic DNA was used, which was quantified prior with a Qubit 3.0 dsDNA BR assay. The genomic DNA was adjusted to a volume of 46 ul with nuclease– free water (NFW) and then transferred to a Covaris g-Tube for fragmentation into 8 kilobase (kb) fragments. The g-Tube was centrifuged in a minicentrifuge for 1 minute, inverted, and then spun again for 1 minute. One ul of the fragmented DNA was removed to check the fragment size via gel electrophoresis and quantification with the Qubit 3 dsDNA BR assay. End repair and dA- tailing of the fragmented DNA was done with the NEBNext Ultra II End Repair/dA- Tailing Module (NEB E7546S). A reaction mix was made by adding 7 ul of NEB Ultra II end-prep reaction buffer, 3 ul of NEB Ultra II enzyme mix, and 5 ul of NFW to 45 ul of fragmented DNA, and the reaction was incubated at 20C for 5 min and 65C for 5 min. The reaction was cleaned up with a 1 volume (60 ul) of AMPure XP beads, and the cleaned DNA was eluted in 31 ul NFW. A 1 ul aliquot was quantified via the Qubit 3 dsDNA BR assay to check that at least 700 ng of DNA were retained. Adapter ligatation was done by mixing 30 ul of end-prepped DNA with 20 ul SQK-LSK108 adapter Mix, and 50 ul NEB Blunt/TA Master Mix (NEB M0367S). The mix was flicked and then incubated at room temperature for 10 min. The adapter-ligated DNA was cleaned up by adding a 0.4 volume (40 ul) of AMPure XP beads, followed by resuspension of the bead pellet in 140 ul ABB buffer (from SQK-LSK108). The beads were then resuspended in 15 ul SQK-LSK108 ELB buffer, incubated at room temperature for 10 min, pelleted again, and then the eluate was transferred to a new tube. One ul of the eluate was quantified by Qubit to check that at least 430 ng of DNA were retained.
Each MinION R9.4 flow cells was primed prior sequencing by loading 800 ul of priming buffer (48% v/v SQK-LSK108 RBF buffer in NFW) into the flow cell priming port, waiting 5 minutes, and then lifting the SpotON sample port cover. Then another 200 ul of priming mix were loaded into the flow cell priming port. Sequencing libraries for both runs were prepared by adding 35 ul RBF buffer, 25.5 ul of Library Loading Beads from the Oxford Nanopore Libary Loading Bead kit (EXP-LLB001), and 2.5 ul NFW to 12 ul of the presequencing mix. A volume of 75 ul of library was loaded drop by drop onto the SpotON sample port and capillary action pulled the library into the flow cell. MinKNOW software 2.2 was used to set up two 48 hours MinION sequencing runs on a MacBook Pro laptop computer and read base calling of resulting reads in FAST5 format and conversion to FASTQ format was done with Guppy 2.1.2. Guppy also generated a series of quality control files which were used to assess the performance of both sequencing runs. Both MinKNOW and Guppy are only available to ONT customers via their community site (https://community.nanoporetech.com).
ONT sequencing data analysis and mitogenome reconstruction and annotation
To first filter FASTQ reads output by Guppy, we first used PORECHOP 0.2.432 to remove sequencing adapters and with NANOFILT 2.7.133 remove low quality reads (Q<7). Then we identified minION reads representing the mitogenome with the BLAST-based mtBlaster script5, using the mitogenome of C. aurita (GenBank Accession MN787075) as the reference genome. MtBlaster output was assembled de novo into a full mitogenome with the FLYE 2.8.234,35 assembler and polisher for single molecule sequencing reads such as those produced by the minION. The command line script for the above steps is available at https://github.com/Callithrix-omics/aur_nano.
The assembled minION mitogenome was aligned to the Sanger sequencing results with the MAFFT 7.475 online server (https://mafft.cbrc.jp/alignment/server/) with default settings for comparison of the two mitogenomes. The alignment was visually inspected in MESQUITE36, and merged manually for a final consensus sequence. The reconstructed mitogenome sequence was annotated using the CHLOROBOX web server19 with the following parameters: “circular sequence” option, mitochondrial DNA, tRNAscan-SE v2.0 in “Mammalia Mitochondrial tRNAs” mode was enabled, and Server References from NCBI were selected including all RefSeqs for Callithrix.
Mitogenome phylogenetic and diversity analyses
To determine the lineage of the reconstructed mitogenome, we combined this sequence with other Callithrix mitogenomes7, and used other New World primate GenBank sequences as outgroups. Accession numbers of all sequences used are listed in Table S1. We kept mitochondrial genomes in their entirety, but trimmed part of tRNA-Phe, 12s rRNA and the control region to accommodate the length of all utilized sequences. All mitogenomes were aligned using the online MAAFT server with default settings. A maximum likelihood phylogeny of all sequences was generated with the IQTREE 1.6.1237 online server http://iqtree.cibiv.univie.ac.at with default setting which equated to the command “iqtree -s input_alignment.fasta -m TEST -bb 1000 -alrt 1000.” We used the optimal substitution model (TPM2u+F+I+G4) as calculated automatically with ModelFinder38 in IQ-TREE under the Bayesian Information Criterion (BIC). We performed the ML analysis in IQ-TREE with 1,000 ultrafast bootstrap (BS) replications39. DNASp 6.12.0340 was used for population genomic analysis to calculate standard diversity indices.
Supplementary information
Acknowledgements
This work was supported by a Brazilian CNPq Jovens Talentos Postdoctoral Fellowship (302044/2014-0), a Brazilian CNPq DCR grant (300264/2018-6), an American Society of Primatologists Conservation Small Grant, a Marie-Curie Individual Fellowship (AMD-793641-4) and an International Primatological Society Research Grant for JM. This work was also supported by an Arizona State University SOLS/OKED Research Investment grant to RAC, AV, MAW, and JM, as well as an Arizona State University RTI Postdoctoral Grant to JM. Additional internal funding from the German Primate Center to LW and JM also supported this work. The funding agencies had no roles in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We would like to thank the Golden Lion Tamarin Association, CEMAFAUNA, the Guarulhos Municipal Zoo, the Beagle Lab at UFV, and many biologists, field technicians, veterinarians, and other individuals that made this research possible.
Author contributions
J.M. formulated the idea for the study, collected samples, obtained funding, conducted wet and dry laboratory work, and wrote the original manuscript. R.A.C., A.V., and M.A.W. provided study guidance, obtained funding, and provided logistical support. J.A.D. provided study guidance, and logistical support. C.S.I. gave access and provided logistical support to collect samples from animals kept at Guarulhos Zoo. P.A.N. provided study guidance, and logistical support. L.C.M.P. provided study guidance, and logistical support. C.R.R.M. and D.Z. were major contributors in writing the manuscript. D.L.S. performed DNA extractions, methodological optimization, and carried out PCR. A.C.S. was a major contributor in writing the manuscript, provided study guidance, and logistical support. L.W. and C.R. gave significant guidance and input in the development of this study. All authors read and approved the final manuscript.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-021-96404-6.
References
- 1.Dodsworth S. Genome skimming for next-generation biodiversity analysis. Trends Plant Sci. 2015;20:525–527. doi: 10.1016/j.tplants.2015.06.012. [DOI] [PubMed] [Google Scholar]
- 2.Richter S, Schwarz F, Hering L, Böggemann M, Bleidorn C. The utility of genome skimming for phylogenomic analyses as demonstrated for glycerid relationships (Annelida, Glyceridae) Genome Biol. Evol. 2015;7:3443–3462. doi: 10.1093/gbe/evv224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Trevisan B, Alcantara DM, Machado DJ, Marques FP, Lahr DJ. Genome skimming is a low-cost and robust strategy to assemble complete mitochondrial genomes from ethanol preserved specimens in biodiversity studies. PeerJ. 2019;7:e7543. doi: 10.7717/peerj.7543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johri, S. et al. ‘Genome skimming’ with the MinION hand-held sequencer identifies CITES-listed shark species in India’s exports market. Sci. Rep.9, 10.1038/s41598-019-40940-9 (2019). [DOI] [PMC free article] [PubMed]
- 5.Franco-Sierra ND, Díaz-Nieto JF. Rapid mitochondrial genome sequencing based on oxford Nanopore sequencing and a proxy for vertebrate species identification. Ecol. Evol. 2020;10:3544–3560. doi: 10.1002/ece3.6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rylands A, Coimbra-Filho R, Mittermeier AF. The systematics and distributions of the marmosets (Callithrix, Callibella, Cebuella, and Mico) and Callimico (Callimico) (callitrichidae, primates) In: Ford S, Porter L, Davis L, editors. The smallest anthropoids: the marmoset/Callimico radiation. New York, N.Y.: Oxford University Press; 2009. [Google Scholar]
- 7.Malukiewicz, J. et al. Mitogenomic phylogeny of Callithrix with special focus on human transferred taxa. BMC Genom.22. 10.1186/s12864-021-07533-1 (2021). [DOI] [PMC free article] [PubMed]
- 8.Malukiewicz, J. et al. An introduction to the Callithrix genus and overview of recent advances in marmoset research. Preprints. 10.20944/preprints202011.0256.v2 (2020). [DOI] [PubMed]
- 9.de Melo, F. et al.Callithrix aurita (amended version of 2020 assessment). The IUCN red list of threatened species 2021. Tech. Rep., IUCN (2021). 10.2305/IUCN.UK.2021-1.RLTS.T3570A191700629.en. Downloaded on 15 April 2021.
- 10.Carvalho, R. Conservação do saguis-da-serra-escuro (Callithrix aurita (Primates))– Analise molecular e colormetrica de populaçães do gênero Callithrix e seus híbridos. Ph.D. thesis, Universidade do Estado do Rio de Janeiro. 2015 (2015).
- 11.Malukiewicz, J. A review of experimental, natural, and anthropogenic hybridization in Callithrix marmosets. Int. J. Primatol. 72–98 (2019).
- 12.Carvalho R, et al. A tiny primate on the edge of extinction in the brazilian atlantic forest. neotropical primates. Neotropical Primates. 2018;24:1–8. [Google Scholar]
- 13.Tagliaro CH, Schneider MP, Schneider H, Sampaio IC, Stanhope MJ. Marmoset phylogenetics, conservation perspectives, and evolution of the mtDNA control region. Mol. Biol. Evol. 1997;14:674–684. doi: 10.1093/oxfordjournals.molbev.a025807. [DOI] [PubMed] [Google Scholar]
- 14.Sena L, et al. Mitochondrial COII gene sequences provide new insights into the phylogeny of marmoset species groups (callitrichidae, primates) Folia Primatol. 2002;73:240–251. doi: 10.1159/000067456. [DOI] [PubMed] [Google Scholar]
- 15.Malukiewicz J, et al. Hybridization effects and genetic diversity of the common and black-tufted marmoset (Callithrix jacchus and Callithrix penicillata) mitochondrial control region. Am. J. Phys. Anthropol. 2014;155:522–536. doi: 10.1002/ajpa.22605. [DOI] [PubMed] [Google Scholar]
- 16.Malukiewicz J, Hepp CM, Guschanski K, Stone AC. Phylogeny of the jacchus group of Callithrix marmosets based on complete mitochondrial genomes. Am. J. Phys. Anthropol. 2017;162:157–169. doi: 10.1002/ajpa.23105. [DOI] [PubMed] [Google Scholar]
- 17.Hershkovitz P. Living new world monkeys (Platyrrhini): With an introduction to primates. Chicago, IL: University of Chicago Press; 1977. [Google Scholar]
- 18.R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria (2020).
- 19.Tillich M, et al. GeSeq – versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017;45:W6–W11. doi: 10.1093/nar/gkx391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Straub SCK, et al. Navigating the tip of the genomic iceberg: Next-generation sequencing for plant systematics. Am. J. Bot. 2012;99:349–364. doi: 10.3732/ajb.1100335. [DOI] [PubMed] [Google Scholar]
- 21.Baeza, J. A. Yes, we can use it: a formal test on the accuracy of low-pass nanopore long-read sequencing for mitophylogenomics and barcoding research using the caribbean spiny lobster Panulirus argus. BMC Genomics21, 10.1186/s12864-020-07292-5 (2020). [DOI] [PMC free article] [PubMed]
- 22.Rang, F. J., Kloosterman, W. P. & de Ridder, J. From squiggle to basepair: computational approaches for improving nanopore sequencing read accuracy. Genom. Biol.19, 10.1186/s13059-018-1462-9 (2018). [DOI] [PMC free article] [PubMed]
- 23.Benirschke K, Brownhill LE. Further observations on marrow chimerism in marmosets. Cytogenet. Genome Res. 1962;1:245–257. doi: 10.1159/000129734. [DOI] [PubMed] [Google Scholar]
- 24.Sweeney CG, Curran E, Westmoreland SV, Mansfield KG, Vallender EJ. Quantitative molecular assessment of chimerism across tissues in marmosets and tamarins. BMC Genomics. 2012;13:98. doi: 10.1186/1471-2164-13-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Malukiewicz J, et al. Natural and anthropogenic hybridization in two species of eastern brazilian marmosets (Callithrix jacchus and C. penicillata) PLOS ONE. 2015;10:e0127268. doi: 10.1371/journal.pone.0127268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ross, C. N., French, J. A. & Ortí, G. Germ-line chimerism and paternal care in marmosets (Callithrix kuhlii). Proc. Natl. Acad. Sci.104, 6278–6282. 10.1073/pnas.0607426104 (2007). https://www.pnas.org/content/104/15/6278.full.pdf. [DOI] [PMC free article] [PubMed]
- 27.Ozga AT, et al. Urine as a high-quality source of host genomic DNA from wild populations. Mol. Ecol. Resour. 2020;21:170–182. doi: 10.1111/1755-0998.13260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Silva F, et al. A survey of wild and introduced marmosets (Callithrix: Callitrichidae) in the southern and eastern portions of the state of minas gerais, Brazil. Primate Conserv. 2018;32:1–8. [Google Scholar]
- 29.Coimbra-Filho R, Pissinatti A, Rylands A. Experimental multiple hybridism and natural hybrids among Callithrix species from eastern brazil. In: Rylands A, editor. Marmosets and tamarins: Systematics, behaviour, and ecology. New York, N.Y.: Oxford University Press; 2009. pp. 95–120. [Google Scholar]
- 30.de Melo, F. et al.Callithrix flaviceps (amended version of 2020 assessment). Tech. Rep. 2, The IUCN Red List of Threatened Species 2021: e.T3571A191700879 (2021). Downloaded on 02 April 2021.
- 31.Gaubert, P., Taylor, P. J., Fernandes, C. A., Bruford, M. W. & Veron, G. Patterns of cryptic hybridization revealed using an integrative approach: a case study on genets (Carnivora, Civerridae, Genetta spp.) from the southern African subregion. Biol. J. Linn. Soc.86, 11–33. 10.1111/j.1095-8312.2005.00518.x (2005).
- 32.Wick, R. R., Judd, L. M., Gorrie, C. L. & Holt, K. E. Completing bacterial genome assemblies with multiplex MinION sequencing. Microb. Genom.3, 10.1099/mgen.0.000132 (2017). [DOI] [PMC free article] [PubMed]
- 33.Coster WD, D’Hert S, Schultz DT, Cruts M, Broeckhoven CV. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics. 2018;34:2666–2669. doi: 10.1093/bioinformatics/bty149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lin Y, et al. Assembly of long error-prone reads using de Bruijn graphs. Proc. Natl. Acad. Sci. 2016;113:E8396–E8405. doi: 10.1073/pnas.1604560113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kolmogorov M, Yuan J, Lin Y, Pevzner PA. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019;37:540–546. doi: 10.1038/s41587-019-0072-8. [DOI] [PubMed] [Google Scholar]
- 36.Maddison, W. & Maddison, D. Mesquite: a modular system for evolutionary analysis. (2019). Last accessed on 14 April 2021.
- 37.Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2014;32:268–274. doi: 10.1093/molbev/msu300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods. 2017;14:587–589. doi: 10.1038/nmeth.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schrempf D, Minh BQ, von Haeseler A, Kosiol C. Polymorphism-aware species trees with advanced mutation models, bootstrap, and rate heterogeneity. Mol. Biol. Evol. 2019;36:1294–1301. doi: 10.1093/molbev/msz043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rozas J, et al. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017;34:3299–3302. doi: 10.1093/molbev/msx248. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.