

Abstract
Developing robust methodology for the sustainable production of red blood cells in vitro is essential for providing an alternative source of clinical-quality blood, particularly for individuals with rare blood group phenotypes. Immortalized erythroid progenitor cell lines are the most promising emergent technology for achieving this goal. We previously created the erythroid cell line BEL-A from bone marrow CD34+ cells that had improved differentiation and enucleation potential compared to other lines reported. In this study we show that our immortalization approach is reproducible for erythroid cells differentiated from bone marrow and also from far more accessible peripheral and cord blood CD34+ cells, consistently generating lines with similar improved erythroid performance. Extensive characterization of the lines shows them to accurately recapitulate their primary cell equivalents and provides a molecular signature for immortalization. In addition, we show that only cells at a specific stage of erythropoiesis, predominantly proerythroblasts, are amenable to immortalization. Our methodology provides a step forward in the drive for a sustainable supply of red cells for clinical use and for the generation of model cellular systems for the study of erythropoiesis in health and disease, with the added benefit of an indefinite expansion window for manipulation of molecular targets.
Keywords: erythroid, immortalized, cell lines, cord blood, bone marrow, peripheral blood, proteomics
Graphical abstract
Here Daniels et al. demonstrate reproducible immortalization of erythroid cells derived from bone marrow and peripheral and cord blood CD34+ cells and reveal a molecular signature of immortalization. This methodology provides a step toward the sustainable production of red cells for clinical use and for the generation of cellular model systems.
Introduction
The restricted availability of blood for transfusion presents a global healthcare problem. As a result, much work has focused on developing an alternative source of clinical-quality red blood cells, particularly for hard-to-match patient groups. This includes individuals with rare blood group phenotypes and chronically transfused patients, such as those with sickle cell disease, β-thalassemia, and some cancers, who can develop an alloimmune reaction, necessitating more extensively blood group-matched donors. Cultured red blood cells provide such an alternative and have additional potential advantages over donor blood, including reduced risk of disease transmission and the production of nascent red cells with improved survival rates in circulation.1
To date the most efficient stem cells for in vitro culture of erythroid cells are adult peripheral blood (PB) and umbilical cord blood (CB) hematopoietic stem cells (HSCs). However, although both types of HSCs can be differentiated to mature erythroid cells,1, 2, 3 they possess a limited proliferative capacity, restricting the number of red cells that can be obtained from each HSC donation.
In recent years immortalized erythroid cell lines have emerged as an alternative approach for the production of cultured red cells.4, 5, 6, 7, 8 These lines provide an unlimited supply of early erythroid cells with only minimal culture required to generate the final product, potentially enabling a sustainable supply of reticulocytes for clinical use. In addition, such lines serve as invaluable tools for the study of erythropoiesis in health and disease, providing an indefinite expansion window for manipulation of molecular targets.
In 2017 we reported the first human immortalized adult erythroid line (BEL-A) from adult bone marrow (BM) CD34+ cells.9 The line was also the first to closely recapitulate normal adult erythropoiesis. The cells express levels of adult globin equivalent to primary adult erythroid cells and enucleate to form reticulocytes that are functionally identical to those from primary adult erythroid cell cultures. Enucleation capacity of BEL-A was further improved by subsequent adjustment of the differentiation protocol to give a rate of ∼40%.10 The unlimited proliferative window and ease of molecular manipulation, combined with unparalleled enucleation capacity, have enabled the application of BEL-A for demonstrating proof-of-principle production of reticulocytes with increased transfusion compatibility,11 identifying the molecular basis of the MAM blood group antigen,12 and the study of surface protein requirements for Plasmodium falciparum invasion.13
However, isolation of BM CD34+ cells is highly invasive, so the ability to generate lines from more accessible PB and CB CD34+ cells is essential for opening up wider applications of the technology, such as creating lines from individuals with specific and rare blood group phenotypes and from patients with erythroid diseases for research purposes.
Despite both PB and BM CD34+ cells originating from the same source, differences have been detected in their characteristics, including surface marker expression14 and suitability for allogenic transplant,15 which may impact immortalization potential and phenotype of resultant lines. Although erythroid cells generated from CB express an increased proportion of fetal hemoglobin (HbF), which possesses an oxygen dissociation profile less optimal for adult circulation, individuals with genetic variants leading to heredity persistence of fetal globin (up to 30% HbF in adulthood) do not suffer any clinical consequences.16 Indeed, such observations have led to the ongoing development of therapeutic strategies to increase HbF levels in patients suffering from adult hemoglobinopathies, such as β-thalassemia and sickle cell disease.17 Therefore, lines generated from CB CD34+ cells also have potential for clinical application, with a particular advantage being their accessibility via human CB banks worldwide, representing a wider variety of donor ethnicities and genetic backgrounds than from blood donor banks for seeking an optimal match for chronically transfused patients.18,19
Previous studies describing the generation of immortalized erythroid lines from CB and PB CD34+ cells have resulted in lines with very poor enucleation capacity (HUDEP-1, HUDEP-2, HUDEP-3;4 EJ8), that display a globin expression profile differing from the primary cell source (HUDEP-24), or that require a stromal cell co-culture system for differentiation that is not appropriate for scale up or clinical application (iE6), as well as confounding enucleation efficiency determination due to removal of dead and dying nucleated erythroblasts by stromal cells. We therefore sought to determine whether our immortalization approach could generate lines from CB and PB CD34+ cells with the same improved erythroid phenotype as BEL-A in our feeder-free culture system and that also closely represent the phenotype of their parental cell origin.
In addition, all reported erythroid lines to date have been generated by immortalization of cells at predominantly the proerythroblast stage of erythroid differentiation. However, immortalization of earlier cells when more plastic, or at later stages of erythroid differentiation, may result in lines with further improved enucleation capacity.
In this study, using our BEL-A immortalization methodology,9 we demonstrate that multiple cell lines immortalized at different time points in culture from the same donor BM CD34+ cells all exhibit a predominantly proerythroblast morphology and invariant phenotype regardless of timing of induced HPV16-E6 and E7 expression. Thus, only proerythroblasts and, to a much lesser extent, early basophilic erythroblasts are amenable to immortalization. The data also demonstrate reproducibility of the BEL-A immortalization approach for BM CD34+ cells. We next use our methodology to successfully generate lines from the far more easily accessible CB (BEL-C) and PB (BEL-P) CD34+ cells, with similarly improved erythroid maturation and enucleation potential as BEL-A and the above BM lines. Extensive characterization of the lines, including differentiation, globin expression, and proteomics, showed them to recapitulate their primary cell source (BEL-C has a fetal phenotype, BEL-P has an adult phenotype), demonstrating the reproducibility of our immortalization approach across multiple stem cell sources and that immortalization does not alter the intrinsic and fundamental erythroid characteristics of the cells. Finally, interrogation of proteomic datasets across multiple lines provides a signature for HPV16 E6/E7 immortalization. These findings pave the way for use of this technology to generate cell lines under good manufacturing practice as a sustainable supply of red cells for transfusion and diagnostics, as well as creation of disease model systems to investigate underlying molecular defects and to trial novel therapeutics.
Results
Timing of initiation of HPV16 E6 and E7 expression during line establishment does not impact characteristics of resultant cell line
The original BEL-A line was generated via transduction of BM CD34+ cells with a Tet-inducible HPV16-E6/E7 expression system4 on day 1 of culture, with cells transferred to expansion medium containing doxycycline to induce E6/E7 expression on day 5.9 This resulted in a cell line comprising predominantly proerythroblasts with a small number of basophilic erythroblasts.9
We speculated that the timing of initiation of E6 and E7 expression could impact the properties of the line developed, with the potential to generate cell lines with further improved terminal differentiation and enucleation potential, for example, if cells were immortalized at a later stage of differentiation. To investigate this possibility, four immortalized lines were initiated from the same donor BM CD34+ cells. The CD34+ cells were incubated in the primary medium of our erythroid culture system and transduced with the Tet-inducible HPV16-E6/E7 construct on day 1 of culture, with transfer to expansion medium containing doxycycline on day 3, 5, 7, or 9 to induce E6 and E7 expression, referred to as the Tet-on day (td; schematic of experimental design shown in Figure 1A). The composition of the cell population on these days with respect to stage of differentiation is also shown in Figure 1A. The resulting erythroid lines, td3, td5, td7, and td9, were kept in expansion culture for over 6 months before being frozen for storage as the lines were established. The lines showed mean doubling times between 19 and 25 h (Figure 1B) and consistently high viability (>98% on average) (Figure 1C), with their expansion rate unchanged over time or after freeze-thawing and further culture (Figure S1A). The oldest cells cultured to date for td3, td5, td7, and td9 were 202, 203, 184, and 188 days post CD34+ cell thawing, respectively. Surprisingly, despite the differing compositions of erythroid cell maturation stages present at each of the four time points (Figure 1A), all four lines exhibited a predominantly proerythroblast morphology, with small numbers of early basophilic erythroblasts (Figure 1D; td3 86% ± 4%, td5 84% ± 5%, td7 83% ± 6%, td9 88% ± 7% proerythroblasts). Hence, although for example at day 9 the predominant cell types present were basophilic (51%) and polychromatic (22%) erythroblasts, with only 23% proerythroblasts, it was still predominantly proerythroblasts that became immortalized. At day 3 there were no proerythroblasts detected, the majority of cells being early progenitors with a small number (24%) of pre-proerythroblasts. However, the resultant cell line (td3) again comprised predominantly proerythroblasts, E6 and E7, although expressed earlier, clearly not taking effect until the cells had differentiated to this stage. It therefore appears that only proerythroblasts and early basophilic erythroblasts are amenable to immortalization, at least with this technique.
Figure 1.

Only early erythroid cells, predominantly proerythroblasts, display efficient immortalization potential
(A) schematic of the td3, td5, td7, and td9 cell line initiation protocol. Vertical bars depict morphology data obtained from cytospins of BM CD34+ cell erythroid culture at days 3, 5, 7, and 9 of differentiation. Pre-proE, pre-proerythroblast; ProE, proerythroblast; BasoE, basophilic erythroblast; PolyE, polychromatic erythroblast; OrthoE, orthochromatic erythroblast; Retic, reticulocyte. (B–E) characterization of the td3, td5, td7, and td9 cell lines during the undifferentiated stage. (B and C) doubling time (B) and percentage viability (C) determined by trypan blue exclusion assay of established cell lines from days 152–198 of expansion culture. Data are shown as mean ± standard deviation (SD). (D) representative images from cytospins of cell line expansion cultures. Scale bars, 20 μm. (E) expression of GPA, band 3, CD36, α4-integrin, and CD71 in the undifferentiated cell lines as determined by flow cytometry (n = 3) with MFI frequency distributions normalized to mode. Histograms are representative of three independent experiments.
To further investigate the maturation stage of the immortalized lines, flow cytometry was performed for cell surface maker proteins known either to increase (glycophorin A [GPA], band 3) or to decrease (CD36, α4-integrin, CD71) during erythroid differentiation.20 Consistent with the observed morphology, all four lines showed a similar surface marker expression pattern (Figure 1E).
Next we compared the behavior of the four lines during differentiation by transferring expanding cells to our erythroid culture system.11 Although td3 and td5 trended toward a ∼2-fold greater expansion rate during differentiation, only td3 versus td9 at day 8 and day 14 (p = 0.03 and p = 0.005, respectively) reached significance (Figure 2Ai). There were no significant differences in viability detected (Figure 2Aii), with a decline in viability in all lines from day 8, as found with the original BEL-A line, whereby cells that fail to enucleate enter apoptosis.10
Figure 2.

Characterization of td3, td5, td7, and td9 cell lines during differentiation
Expanding cells (day 0) were transferred to erythroid differentiation medium and samples taken at time points throughout differentiation. (A) cumulative fold expansion (i), percentage viability (ii), and percentage enucleation (iii) of the differentiating td3, td5, td7, and td9 cell lines. (B) percentage of erythroid cell types present during differentiation of the cell lines. ProE, proerythroblast; BasoE, basophilic erythroblast; PolyE, polychromatic erythroblast; OrthoE, orthochromatic erythroblast; Retic, reticulocyte. (C) representative images from cytospins of these cultures at days 0, 4, 8, and 12 of differentiation. Scale bars, 20 μm. Arrowheads indicate the following cell types: white, ProE; blue, BasoE; orange, PolyE; black, OrthoE; red, Retic. (D) MFI of GPA, band 3, CD36, and α4-integrin in the differentiating cell lines as determined by flow cytometry. Differentiation cultures were initiated between 154 and 200 days of continual expansion since the source CD34+ cells were thawed. Data are shown as mean ± SD, n = 3.
Morphological analysis showed that the differentiation potential of all four lines was also similar (Figures 2B and 2C), with only slight differences in differentiation rates at early time points. However, such discrete, defined stages by morphological analysis are selected within a continuum of differentiation, with further analysis by flow cytometry revealing no significant difference in levels of GPA, band 3, CD36, and α4-integrin (Figure 2D) at any time point, indicating a high level of similarity between the lines. There was also no significant difference in enucleation rates (Figure 2Aiii). The differentiation potential and rate for cells from the same line at different time points post immortalization were consistent (Figure 2), with cells at days 200, 199, 182, and 186 for td3, td5, td7, and td9, respectively, the oldest differentiated to date. Western blot analysis of differentiated cells confirmed an adult globin expression profile for all four lines equivalent to that seen for BEL-A and for primary adult erythroid cells, with high levels of β-globin and negligible γ-globin (Figure S2).
Overall, these results demonstrate that immortalizing populations of erythroid cells by transduction of HPV16 E6/E7 at progressively later stages of differentiation culture does not result in a line comprising correspondingly later-stage cells. Instead, only proerythroblasts, and to a lesser extent early basophilic erythroblasts, are amenable to immortalization, with the four lines generated exhibiting similar characteristics and behavior. The data also demonstrate reproducibility of the BEL-A immortalization approach for BM CD34+ cells.
The BEL-A immortalization protocol is reproducible for generating lines from more accessible cord and adult peripheral blood stem cells
Next we wanted to determine whether we could generate lines with similar recapitulation of erythropoiesis from the more accessible CB and PB CD34+ cells and that are also representative of their parental stem cell type (that is, with a fetal and adult phenotype, respectively).
The data above reveal no difference in the phenotype of lines created at different time points in culture, albeit with a possible expansion advantage of lines made at earlier time points. Moreover, for generation of the original BEL-A cell line, HPV16-E6 and E7 expression was induced at an early time point (day 5).9 Therefore, an equivalent protocol was used to attempt generation of immortalized lines from CB and PB stem cells.
Lines from both CB (BEL-C; Bristol Erythroid Line Cord) and PB (BEL-P; Bristol Erythroid Line Peripheral Blood) CD34+ cells were successfully established, with continued expansion in culture for over 6 months before being frozen for storage. Expansion rates for the lines did not change over time or after freeze-thawing and further culture (Figure S1B). The oldest cells cultured to date were 183 and 181 days post CD34+ cell thawing for BEL-C and BEL-P, respectively. BEL-C and BEL-P showed a predominantly proerythroblast morphology similar to BEL-A and additional cell lines above (Figure 3A; BEL-C 80% ± 4%, BEL-P 77% ± 5% proerythroblasts), exhibited mean doubling times of 26 h (Figure 3B), and maintained high viability during expansion (average >97%) (Figure 3C). There was no change in phenotype or morphology of the cells over time, and cells re-established efficiently in culture after freeze-thawing. When induced to differentiate, both lines displayed broadly similar expansion, viability, and enucleation profiles, with maximum enucleation rates of ∼26% on day 14 (Figure 3D). Leaving the cells in culture for longer did not result in increased yields of reticulocytes, the remaining orthochromatic erythroblasts that failed to enucleate entering apoptosis, as seen with BEL-A.10 The differentiation potential and rate of cells from the same line at different time points post immortalization were consistent (Figure 3), with cells for BEL-C and BEL-P at days 128 and 140, respectively, the oldest differentiated to date.
Figure 3.

Characterization of erythroid cell lines derived from PB and CB CD34+ cells
(A–C) characterization of the CB-derived (BEL-C) and PB-derived (BEL-P) cell lines during the undifferentiated stage. (A) representative images from cytospins of BEL-C and BEL-P expansion cultures. Scale bars, 20 μm. (B and C) doubling time (B) and percentage viability (C) determined by trypan blue exclusion assay of established cell lines from days 92–146 of expansion culture. (D) cumulative fold expansion (i), percentage viability (ii), and percentage enucleation (iii) of the differentiating BEL-C and BEL-P cell lines (n = 3). Differentiation cultures were initiated between 100 and 140 days of continual expansion since the source CD34+ cells were thawed. Data are shown as mean ± SD.
Molecular karyotyping of immortalized cell lines
As potential model systems, it is important to determine the extent of karyotype abnormalities in immortalized cell lines, especially when considering genome editing applications. To assess this, molecular karyotyping of established BEL-C and BEL-P cells, alongside samples of the td3, td5, td7, and td9 lines (all >150 days of continuous culture) was performed by single-nucleotide polymorphism array (SNP-array) analysis. As has been previously observed for both BEL-A10 and HUDEP-2,21,22 which were immortalized with the same HPV-16 E6 and E7 expression system, all lines showed a number of whole or partial trisomies, with some lines exhibiting whole or partial chromosome loss (detailed in Table S1). The average chromosome numbers in the lines ranged from 46 to 51 based on a threshold of 30% abnormality frequency determined by LogR ratio (BEL-C, 46 XY; BEL-P 51 XX; td3, 50 XX; td5, 50 XX; td7, 49 XX; td9, 48 XX). The most prevalent trisomies detected at the 30% threshold were of chromosomes 8 (all lines) and 19 (BEL-P, td3, td5, and td9) (Figure S3).
The BEL-C and BEL-P lines recapitulate phenotype of respective primary stem cell sources
As well as demonstrating successful immortalization of CB- and PB-derived erythroid cells with the BEL-A protocol, along with efficient erythroid differentiation, we wanted to determine whether the lines model their primary stem cell source in terms of erythroid surface marker, transcription factor, and globin subunit expression. To compare differentiation rates and profiles, cultures were initiated both from expanding BEL-C and BEL-P cells and from CB and PB CD34+ cells in parallel. Cell samples were taken at regular intervals for analysis, which for the primary cell cultures was from day 5 as this is the time point when HPV16 E6 and E7 expression was induced in the lines (i.e., is equivalent to the undifferentiated BEL-C and BEL-P cells).
Differentiation potential of BEL-C and BEL-P compared with primary CB and PB erythroid cells
Morphological analysis shows that both BEL-C and BEL-P undergo erythroid differentiation in line with that of the respective CB and PB primary cell cultures, albeit with less heterogeneity in erythroid cell types during mid-differentiation (Figures 4A and 4B). BEL-C differentiation cultures show an increased proportion of early-stage erythroid cells during mid-differentiation compared to BEL-P, with significantly more basophilic erythroblasts at days 6 and 8 (p = 0.001 and p = 0.006, respectively). This is consistent with the profile for CB and PB primary cell cultures, where a similar prevalence of early-stage cells is seen in CB compared to PB cultures, with a significant difference in the proportion of basophilic erythroblasts at day 13 (p = 0.02).
Figure 4.

Comparison of differentiating BEL-C and BEL-P cell lines with erythroid cell differentiation from respective cord blood and adult peripheral blood CD34+ cells
(A) Percentage of erythroid cell types present during differentiation of BEL-C, CB, BEL-P, and PB cells during erythroid differentiation. Pre-proE, pre-proerythroblast; ProE, proerythroblast; BasoE, basophilic erythroblast; PolyE, polychromatic erythroblast; OrthoE, orthochromatic erythroblast; Retic, reticulocyte. (B) representative images from cytospins of these cultures. Scale bars, 20 μm. Arrowheads indicate the following cell types: green, Pre-proE; white, ProE; blue, BasoE; orange, PolyE; black, OrthoE; red, Retic. Data are shown as mean ± SD (n = 3). (C) flow cytometry analysis of band 3 versus α4-integrin and GPA versus CD36 cell surface expression of BEL-C, BEL-PO, CB, and PB cells during erythroid differentiation. Plots are representative of three independent cultures.
For more detailed analysis of differentiation, the inverse expression of GPA versus CD36 and band 3 versus α4-integrin was measured by flow cytometry (Figure 4C). BEL-C and BEL-P exhibited a surface marker profile throughout differentiation similar to their primary cell counterparts, albeit with reduced heterogeneity and a lack of cells with a more immature immunophenotype at the early time points (most clearly seen by the CD36 and GPA double negative population present at day 5 and day 7 in the CB and PB cultures). In addition, a greater prevalence of cells with a more immature cell surface marker expression during differentiation is apparent in the BEL-C and CB cultures compared to the BEL-P and PB cultures, respectively, most clearly seen in the band 3 versus α4-integrin plots (Figure 4C) and band 3 median fluorescence intensity (MFI) (Figure S4), in line with the earlier erythroid cell types observed from morphological analysis during mid-differentiation (Figure 4A). Interestingly, these data also show that although there is heterogeneous surface marker expression in the CB and PB cultures at the time point of immortalization (i.e., day 5, Figure 4C) only a subpopulation of these cells are immortalized, represented by the surface marker profile of the undifferentiated cell lines (day 0, Figure 4C), providing further evidence for the preferential immortalization of a distinct proerythroblast/early basophilic erythroblast population.
Comparison of globin expression profiles of BEL-C and BEL-P cell lines with CB and PB primary cells
To investigate whether the globin expression profiles of the BEL-C and BEL-P lines reflect their stem cell origin, samples of mature erythroid cells were collected from late-stage cultures of the lines and primary cells for reverse-phase high-performance liquid chromatography (RP-HPLC) and western blot analysis. As can be seen in Figures 5A–5C, erythroid cells differentiated from CB CD34+ cells express predominantly γ- but with some β-globin, reflecting heterogeneity in the population due to the developmental globin switch (from γ- to β-globin), which commences just prior to birth and continues to ∼6 months of age, and the presence of CD34+ cells originating from both fetal liver and BM.23 In contrast, erythroid cells differentiated from PB CD34+ cells express β-globin, with very low levels of γ-globin (Figures 5A–5C). Accordingly, the BEL-P line expresses almost exclusively β-globin, the lower level of γ-globin compared to the PB-derived erythroid cells (more clearly seen by western blot; Figure 5C) likely due to variation between the donors. Similarly, the BEL-C line expresses predominantly γ-globin, with very low levels of β-globin (Figures 5A–5C), indicating a fetal phenotype. Differences in the γ-globin:β-globin ratio between BEL-C and primary CB erythroid cells may also reflect donor variation, or circumstantial immortalization of cells of fetal liver origin expressing predominantly γ-globin in the original CB sample. Hence, the two lines represent the desired fetal and adult phenotypes in terms of globin expression profile.
Figure 5.

Globin and erythroid transcription factor expression in BEL-C, BEL-P, CB, and PB cells
(A and B) RP-HPLC of BEL-C and BEL-P cells at day 10 of differentiation and CB and PB cells at day 17 of differentiation. (A) representative RP-HPLC trace. The injection peak is identified along with peaks for β-globin (β), Gγ-globin (Gγ), α-globin (α), and Aγ-globin (Aγ); δ-globin expression is below the detection limit. (B) quantification of RP-HPLC data showing percentage of α-, β-, and γ-globin (Gγ + Aγ) subunits detected. Data are shown as mean ± SD (n = 3). (C) western blots of lysates obtained from BEL-C and BEL-P erythroid cells at day 10 of differentiation and CB and PB erythroid cells at day 17 of differentiation incubated with antibodies to α-, β-, and γ-globin. α-globin was used as a protein loading control. (D) densitometry analysis from western blots of lysates obtained from early and mid-differentiation cultures (day 0/4 for BEL-C and BEL-P and day 5/9 for CB and PB for early/mid-differentiation, respectively) incubated with antibodies to BCL11A, GATA1, and KLF1 normalized to β-actin. Data are shown as mean ± SD (n = 2). ∗p < 0.05 Welch’s t test (all other relevant cell line versus primary cell comparisons are statistically non-significant).
In addition, we looked at the expression of key erythroid transcription factors, in particular those involved in the regulation of globin expression, at early and mid-stage differentiation by western blot along with densitometry analysis, selecting time points to match cells at as similar stages of maturation as possible. A representative western blot is shown in Figure S5. There was no statistically significant difference in the level of GATA1 and KLF1 protein in BEL-C and BEL-P compared to CB- and PB-derived erythroid cells, respectively (Figure 5D), or in the level of BCL11A between BEL-P and PB-derived erythroid cells. In contrast, the level of BCL11A was significantly lower in BEL-C compared to CB-derived cells (p = 0.04; Figure 5D). As BCL11A inhibits γ-globin expression,24 this is in line with the predominance of γ-globin expression and much lower level of β-globin in BEL-C. Such a correlation between BCL11A and γ-globin is supported by erythroid cells differentiated from CB CD34+ cells, whereby those with lower levels of γ-globin have higher levels of BCL11A (Figure S6), also demonstrating variation in levels between donor samples.
Comparative proteomic analysis of BEL-C and BEL-P cell lines with CB and PB primary erythroid cells
Finally, for a comprehensive comparative analysis of expanding BEL-C and BEL-P with their primary erythroid cell equivalents, we performed tandem mass tag (TMT) liquid chromatography-tandem mass spectrometry (LC-MS/MS) quantitative proteomics.
To obtain CB and PB erythroid cells stage-matched to expanding BEL-C and BEL-P, CD34+ cells were initially cultured in our primary erythroid medium to induce differentiation, followed by a further 2 days in expansion medium (Figure S7), the period in expansion medium ensuring that any differences in the proteome detected were not due to differences in the culture medium the cells were exposed to. Cells were obtained from 3 independent cultures of BEL-C and BEL-P cell lines and 2 independent cultures of pooled CB and pooled PB CD34+ samples, the pooled samples to quench differences in protein levels due to variation between individuals rather than between lines and primary cells. Data for BEL-C were compared to those for CB, and data for BEL-P were compared to those for PB.
LC-MS/MS of peptides derived from whole cell lysates gave quantitative data for 8,367 proteins for BEL-C versus CB comparison and 8,368 proteins for BEL-P versus PB comparison. Protein abundances were normalized to total protein abundance, ensuring that any differences in individual protein abundance detected were not due to differences in the total protein input between samples. Heatmaps of these data showed a very similar overall pattern of normalized protein abundances between the BEL-C and CB samples (Figure 6A, left) and the BEL-P and PB samples (Figure 6A, right).
Figure 6.

TMT LC-MS/MS comparative proteomic analysis of undifferentiated BEL-C and BEL-P with stage-matched equivalent primary cells
(A) heatmaps of proteomic data from BEL-C and CB samples (left) and BEL-P and PB samples (right) presented as log2 normalized abundance values. Yellow indicates high protein expression level; purple indicates low protein expression level. Lane labels indicate samples collected from independent cultures. (B) volcano plots of the proteomic data of all culture repeats from BEL-C and CB samples (left) and BEL-P and PB samples (right) depicting −log10 p value and log2 fold change for each protein hit that had data available from all repeats. p < 0.05 and Q < 0.05 (FDR) thresholds are indicated by red dashed lines. Percentages indicate the proportion of protein hits that showed a >2-fold difference in abundance between the cell line and the primary cells and also passed the FDR threshold. (C) levels of globins and other key fetal/adult proteins in undifferentiated BEL-C and BEL-P cells with stage-matched equivalent primary cells. Data from TMT LC-MS/MS comparative proteomic analysis are abundance values normalized to total protein shown as mean ± SD (n = 2 for CB and PB and n = 3 for BEL-C and BEL-P). ▲Globin abundances are further normalized to α-globin abundance. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. p values represent results from Welch’s ANOVA performed on log2 normalized data.
To display the frequency and magnitude of differences in the proteome of each cell line and primary erythroid cell equivalent, volcano plots were generated depicting −log10 p value and log2 fold change for each protein hit (Figure 6B). When a false discovery rate (FDR) threshold for significance was applied, only 2.06% of proteins were significantly ≥2-fold lower and 1.65% significantly ≥2-fold higher in abundance in BEL-C compared to CB (Figure 6B, left), and only 1.96% of proteins were significantly ≥2-fold lower and 1.40% significantly ≥2-fold higher in abundance in BEL-P compared to PB (Figure 6B, right).
To further determine whether BEL-C and BEL-P model their primary stem cell source, i.e., a fetal phenotype for BEL-C and an adult phenotype for BEL-P, we interrogated the datasets for proteins known to be differentially expressed between fetal and adult erythroid cells. Although at an early time point for complete globin expression, the profile of β- and γ-globin (Figure 6C) mirrored that of the differentiated cells shown earlier (Figures 5A–5C). In addition, the levels of embryonic ε- and ζ-globins were higher in BEL-C than in CB-derived erythroid cells (Figure 6C), again indicating that this line has a more fetal globin phenotype. Expression of these embryonic globin subunits is also repressed by BCL11A,24, 25, 26 so these data reflect the lower BCL11A levels observed in BEL-C. As expected ε- and ζ-globin were at a very low level in BEL-P and PB-derived erythroid cells (Figure 6C). Analysis of other proteins known to be at a higher level in fetal compared to adult erythroid cells (IGF2BP1 and IGF2BP3,27 LIN28B28) or at a lower level (BCL11A24 and LRF29) further confirmed the fetal erythroid phenotype of BEL-C and adult phenotype of BEL-P (Figure 6C); IGF2BP1, IGF2BP3, and LIN28B were significantly higher in BEL-C than in BEL-P, and BCL11A and LRF were significantly higher in BEL-P than BEL-C. Interestingly, IGF2BP1, IGF2BP3, and LIN28B were significantly lower in CB-derived erythroid cells compared to BEL-C and BCL11A and LRF significantly higher, supporting the globin profile and existence of a heterogeneous population of both fetal and adult-like cells in CB samples due to timing of the developmental globin switch, as mentioned above. This mixed population also explains why BCL11A and LRF, typically increased in adult erythroid cells, are significantly higher in BEL-P compared to BEL-C but not compared to CB (Figure 6C). The relative levels of IGF1BP1, IGF2BP3, and LIN28B were confirmed by western blot (Figure S8).
Finally, to determine whether the difference in proteome between the lines and respective primary cells is attributable to HPV16 E6 and E7 expression, and thus revealing the mechanism for immortalization, we compared the ≥2-fold differentially expressed proteins across proteomic datasets for four lines. 43% of the proteins were common between BEL-C and BEL-P, and of these 56% were also ≥2-fold different in BEL-A compared to primary cells. 86% of the changes common between these three lines were similarly ≥2-fold different in an additional line made from BM CD34+ cells with the same methodology (the latter datasets unpublished). This increasing commonality demonstrates the need for analysis across several cell lines to generate a list of protein changes that can be confidently attributed to HPV16 E6/E7 immortalization rather than other factors, for example, donor variation in the cells used for line generation compared to the primary cell cultures. Proteins that differed in abundance by ≥2-fold between the cell lines and respective primary cells across all 4 datasets are shown in Tables S2 and S3. Analysis of these differentially expressed proteins for overrepresented biological pathways using the Reactome Pathway Knowledgebase30 revealed multiple pathways related to cell cycle regulation, including evasion of cellular senescence, G1 phase, and G1/S phase transition (Table 1), highlighting their association with HPV16 E6/E7. When considering the proteins featured within the top 20 pathways identified, four cell cycle proteins appeared most frequently: CDK4 (18/20), CDK6 (16/20), p16INK4A (16/20), and Rb (9/20). On average across all four lines, CDK4 was increased 3-fold, p16INK4A was increased 6-fold, CDK6 was decreased 8-fold, and Rb was decreased 3-fold compared to primary cells, providing a molecular signature of these altered cell cycle proteins (Figure 7).
Table 1.
Altered biological pathways in immortalized erythroid cell lines compared to equivalent primary cells
| Pathway name |
|---|
| Oncogene-Induced Senescence |
| Diseases of Cellular Response to Stress |
| Diseases of Cellular Senescence |
| Cellular Senescence |
| Evasion of Oxidative Stress-Induced Senescence Due to p16INK4A Defects |
| Evasion of Oncogene-Induced Senescence Due to p16INK4A Defects |
| Evasion of Oxidative Stress-Induced Senescence Due to Defective p16INK4A Binding to CDK4 and CDK6 |
| Evasion of Oncogene-Induced Senescence Due to Defective p16INK4A Binding to CDK4 and CDK6 |
| Oxidative Stress-Induced Senescence |
| Cyclin D-Associated Events in G1 |
| G1 Phase |
| Evasion of Oxidative Stress Induced Senescence Due to Defective p16INK4A Binding to CDK4 |
| Evasion of Oncogene-Induced Senescence Due to Defective p16INK4A Binding to CDK4 |
| Aberrant Regulation of Mitotic G1/S Transition in Cancer Due to RB1 Defects |
| Defective Binding of RB1 Mutants to E2F1,(E2F2, E2F3) |
| Formation of Senescence-Associated Heterochromatin Foci (SAHF) |
| Apoptotic Execution Phase |
| Cellular Responses to External Stimuli |
| Mitotic G1 Phase and G1/S Transition |
| Diseases of Mitotic Cell Cycle |
Pathways generated by input of proteins where abundance differed by ≥2-fold in all of the BEL-C, BEL-P, BEL-A, and BM cell line datasets into the Reactome Pathway Knowledgebase. The top 20 pathways with false discovery rate of Q < 0.05 are shown.
Figure 7.
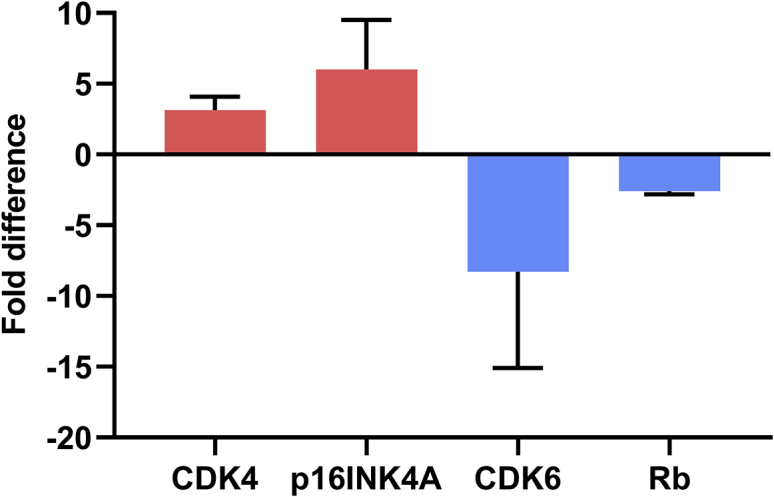
Signature cell cycle protein alterations in immortalized erythroid cell lines compared to primary cells
Average fold differences in key cell cycle proteins obtained from proteomic data of four immortalized erythroid cell lines (BEL-C, BEL-P, BEL-A, and BM) compared to equivalent primary cells. Data are shown as mean ± SD (n = 4).
Overall, the data show that the BEL-C and BEL-P cell lines provide representative cellular systems of fetal and adult erythroid cells, respectively, in terms of their proteome, globin expression, and differentiation profile. Moreover, the data provide a molecular signature for immortalization.
Discussion
In this study we show that our immortalization approach used to create BEL-A9 is reproducible for erythroid cells differentiated from BM and importantly also from CB and PB CD34+ cells, consistently generating lines with similar improved erythroid performance.
We also show that, at least with our approach, only erythroid cells at a discrete stage of differentiation are amenable to immortalization, namely proerythroblasts and to a lesser extent early basophilic erythroblasts. Efficiency of proerythroblasts and failure of later-stage erythroid cells to immortalize may be attributable to a lower proportion of HPV16-E6 and E7 target proteins as differentiation proceeds, reducing the efficiency of E6 and E7 activity, as the total protein content of erythroblasts decreases during differentiation.31 Failure of earlier cell stages to immortalize in these cultures, particularly HSCs, may be for this same reason, HSCs having lower levels of protein synthesis than lineage-restricted hematopoietic cell types.32 Alternatively, this may relate to the observed instability of these progenitors that do not exist as stable states but as constantly changing entities,33,34 severely reducing the window to immortalize specific erythroid lineage progenitors. Although we cannot exclude the possibility that some later-stage erythroid cells do get immortalized but are outcompeted during line establishment because of slower division rates, morphological analysis of our lines during this period showed very few such cells, suggesting this is unlikely.
The lines reported here all have improved enucleation potential/rates compared to that of other erythroid lines in the literature,5 even those generated from the same stem cell sources and with the identical construct (HUDEP lines;4 BMDEP-1 lines7 and EJ line8). However, as previously discussed,10 the measure of reticulocyte yield, rather than rate as routinely used, provides a more transparent and accurate assessment of the reticulocyte productivity of a line, as it accounts for expansion potential and cell death (which for many lines can be substantial) during differentiation; as more nucleated cells die, the proportion of reticulocytes (and thus the apparent enucleation rate) increases, compounded by the use of co-culture with stromal cells that remove dead or dying erythroblasts. For HUDEP-2, generated from CB CD34+ cells, we have previously calculated the yield under identical culture conditions,10 which is ∼50% lower than for BEL-C. Reticulocyte yield of the EJ line, generated from PB CD34+ cells, is 10 times lower than for BEL-P.8 Viability data were not available for the BMDEP-1 lines, and reticulocyte values were not available for the other HUDEP lines, although they were reported as very low.4 Nevertheless, the enucleation rate of BEL-C and BEL-P is lower than for our original line, BEL-A (∼26% versus ∼40%). This could be due to variation in how the immortalization mechanism affects cells during creation of the different lines or due to donor variation. Notwithstanding, the enucleation rate of BEL-A is lower than that of non-immortalized erythroid cells differentiated from adult CD34+ cells,1,3 the basis for which is presently under investigation and may also inform variation between lines.
Extensive characterization and analyses of BEL-C and BEL-P show them to accurately recapitulate their primary cell equivalents in terms of differentiation profile, globin expression, and proteome, with ≤2% of proteins significantly upregulated or downregulated. Moreover, from analysis of consistent differences in the proteome identified across BEL-C, BEL-P, BEL-A, and an additional erythroid line made from BM CD34+ cells, we reveal a molecular signature of differences in key cell cycle proteins CDK4, CDK6, P16INK4A, and Rb as a result of immortalization by HPV16 E6 and E7.
Rb is the primary target of E735 and can induce its degradation via the ubiquitin-proteasome pathway,36 in keeping with the lower levels observed in the immortalized lines. HPV16 E7 has also been shown to promote p16INK4A expression in cervical carcinoma lines, with p16INK4A required for cell survival.37 In addition, as p53 is necessary for p16INK4A repression,38 it is likely that the decreased level of p53 in the immortalized lines (due to degradation of p53 by E639,40) contributes to increased p16INK4A expression. Both CDK4 and CDK6 are activated by D-type cyclins to drive cell cycle progression from G1 to S phase.41 In line with the increased levels seen in the immortalized lines, CDK4 is known to be increased in many tumor types.42, 43, 44, 45 In contrast to the generally increased expression pattern seen in cancer,46 CDK6 is reduced in the immortalized lines, potentially compensating for increased CDK4 levels. As CDK6 has been previously reported to counteract the p53-mediated apoptotic response during immortalization,47 it could be that high levels of CDK6 in the lines are not required, as p53 is already being degraded by E6. Although these changes are associated with continual self-renewal, they do not alter the fundamental erythroid biology of the lines or impair their ability to differentiate upon removal of doxycycline and therefore loss of E6 and E7.
In addition to providing novel data on proteome changes associated with HPV16 E6/E7 and thus the mechanism of immortalization, the molecular signature of altered cell cycle proteins identified offers a valuable resource for identifying alternative methods of immortalization of erythroid progenitors that do not rely on viral oncoproteins. One possible strategy is the overexpression of CDK4, which has previously been utilized to generate keratinocyte and myoblast immortalized cell lines in combination with hTERT expression.48, 49, 50, 51 Such more targeted approaches may yield erythroid lines with further improved characteristics by only generating the minimal number of alterations necessary for immortalization, avoiding the potentially broader impact of viral oncogene expression.
Overall, these data expand the application of our immortalization methodology and provide a significant step forward in the drive for a sustainable supply of red cells with specific blood group phenotypes for therapeutics and diagnostics using PB, CB, or BM CD34+ cells. In addition, the methodology can be applied to create lines with disease phenotypes for investigating underlying molecular defects and as drug screening platforms.52 In particular, BEL-C and BEL-P provide valuable research tools with which to evaluate novel approaches for conversion of fetal to adult and adult to fetal globins in treatment of globin gene disorders, such as induction of fetal globin in patients with sickle cell disease and β-thalassemia.
Materials and methods
Immortalized line generation and culture
Cell lines were generated as previously described unless otherwise stated.9 Briefly, 5 × 105 to 1 × 106 cryopreserved BM, CB, or PB CD34+ cells (STEMCELL Technologies, Inc.) were recovered in erythroid differentiation medium (Iscove’s medium with stable glutamine [Merck] containing 3% [v/v] AB serum [Merck], 2% [v/v] fetal bovine serum [HyClone], 10 μg⋅mL−1 insulin [Merck], heparin 3 U⋅mL−1 [Merck], 200 μg⋅mL−1 holo-transferrin [Sanquin, the Netherlands], and 3 U⋅mL−1 erythropoietin [EPO] [Roche]) supplemented with 1 ng⋅mL−1 interleukin-3 (IL-3) and 10 ng⋅mL−1 stem cell factor (SCF) (both R&D Systems) (primary medium) for 24 h, before being transduced with the lentiviral vector CSIV-TRE-HPV16-E6/E7-UbC-KT and maintained in primary medium for a further 4 days (or until day 3, 5, 7, or 9 in the case of the td cell lines). To induce expression of HPV16-E6 and E7, cells were transferred to expansion medium (StemSpan SFEM [STEMCELL Technologies, Inc.] containing 50 ng⋅mL−1 SCF, 3 U⋅mL−1 EPO, 1 μM dexamethasone, and 1 μg⋅mL−1 doxycycline) and maintained in this medium for at least 6 months before cryopreservation for long-term storage.
Differentiation of cell lines was performed as previously described.11 In brief, expanding cells were transferred to primary medium supplemented with 1 μg⋅mL−1 doxycycline for 4 days and for a further 4 days without doxycycline. Cells were then transferred to erythroid differentiation medium supplemented with holo-transferrin to a final concentration of 500 μg⋅mL−1.
Erythroid differentiation culture of BM-, CB-, and PB-derived CD34+ cells
Erythroid differentiation of CD34+ cells was performed as previously described.3 In brief, CD34+ cells were seeded in primary medium at a density of 1–2 × 105 cells⋅mL−1. On day 7, cells were transferred to differentiation medium supplemented with 10 ng⋅mL−1 SCF and from days 11 to 21 to differentiation medium with additional holo-transferrin to a final concentration of 500 μg⋅mL−1.
Flow cytometry
Aliquots of 2 × 105 cells were incubated with primary antibodies in PBS containing 1% (w/v) BSA (Park Scientific Ltd) and 2 mg⋅mL−1 glucose (PBS-AG), followed by secondary antibodies (along with conjugate antibodies when appropriate). Cells were analyzed on a BD LSR Fortessa flow cytometer. Propidium iodide was used to exclude non-viable cells from analyses. From mid-differentiation onward (day 6 for cell lines and day 9 for primary erythroid cultures) cells were incubated with 5 μg⋅mL−1 Hoechst 33342 nucleic acid stain (Merck) to distinguish the erythroblast and reticulocyte populations. Data were analyzed with FlowJo v.10.6.1 (FlowJo LLC).
TMT labeling, mass spectrometry, and data analysis
TMT LC-MS/MS comparative proteomic experiments were performed as previously described11 with the following adaptations. Aliquots of 100 μg of protein from whole cell lysates were digested with trypsin (2.5 μg trypsin, 37°C overnight) and labeled with TMT 11-Plex reagents according to the manufacturer’s protocol (Thermo Fisher Scientific) before labeled samples were pooled.
Data analyses were performed on log2 normalized abundances with an un-paired, 2-tailed, heteroscedastic Student’s t test in Microsoft Excel. A 5% FDR threshold for each comparison was calculated with R Studio. For data visualization, heatmaps with Ward’s cluster analysis and volcano plots were generated with R Studio.
Statistical analysis
For statistical analysis of >2 groups, after Shapiro-Wilk normality testing Welch’s ANOVA with Dunnett’s multiple comparison testing was performed. For analysis of 2 groups, a Welch’s t test was performed. In both cases, statistical tests were performed in GraphPad Prism 8.
Acknowledgments
The study was supported by NHS Blood and Transplant (NHSBT) and the NIHR Blood and Transplant Research Unit (NIHR BTRU) in Red Cell Products (IS-BTU-1214-10032). The views expressed are those of the authors and not necessarily those of the NHS, the NIHR, or the Department of Health and Social Care. The study was also supported by MRC grant MR/S021140/1. The authors would like to thank Dr. Kate Heesom, Director of the Bristol University Proteomic Facility, UK for performing mass spectrometry, the University of Bristol Flow Cytometry Facility for use of equipment, and the Wellcome Trust Clinical Research Facility at the University of Edinburgh, UK for performing molecular karyotyping. IBGRL antibodies were kindly supplied by Mr. Jonathan Dixey, IBGRL, Filton, Bristol, UK. The IGF2BP1 antibody was a gift from Professor Stefan Hüttelmaier, Martin Luther University, Germany.
Author contributions
J.F. conceived and supervised study. Experiments were conceived and designed by J.F. and D.E.D. with contribution from K.T. The BEL-P cell line was generated by D.E.D. and K.T., BEL-C by D.E.D., and BEL-A td3, 5, 7, and 9 lines by R.E.G. and N.C. with input from J.F., K.T., and D.J.A. D.E.D. and D.C.J.F. conducted the majority of experiments, analyzed data, and prepared figures. R.E.G., K.T., N.C., K.A.M., T.A., K.E.M., I.F.-V., and D.R.J. conducted experiments. P.A.L. performed proteomics analysis. M.C.W. carried out the HPLC experiments and contributed to proteomics analysis. K.A.M. contributed to proteomics analysis. M.A.C. analyzed molecular karyotyping data. R.K. and Y.N. provided the HPV16 E6 and E7 construct. D.E.D. and J.F. wrote the manuscript. D.J.A. edited the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2021.06.002.
Supplemental information
References
- 1.Kupzig S., Parsons S.F., Curnow E., Anstee D.J., Blair A. Superior survival of ex vivo cultured human reticulocytes following transfusion into mice. Haematologica. 2017;102:476–483. doi: 10.3324/haematol.2016.154443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Timmins N.E., Athanasas S., Günther M., Buntine P., Nielsen L.K. Ultra-high-yield manufacture of red blood cells from hematopoietic stem cells. Tissue Eng. Part C Methods. 2011;17:1131–1137. doi: 10.1089/ten.TEC.2011.0207. [DOI] [PubMed] [Google Scholar]
- 3.Griffiths R.E., Kupzig S., Cogan N., Mankelow T.J., Betin V.M.S., Trakarnsanga K., Massey E.J., Lane J.D., Parsons S.F., Anstee D.J. Maturing reticulocytes internalize plasma membrane in glycophorin A-containing vesicles that fuse with autophagosomes before exocytosis. Blood. 2012;119:6296–6306. doi: 10.1182/blood-2011-09-376475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kurita R., Suda N., Sudo K., Miharada K., Hiroyama T., Miyoshi H., Tani K., Nakamura Y. Establishment of immortalized human erythroid progenitor cell lines able to produce enucleated red blood cells. PLoS ONE. 2013;8:e59890. doi: 10.1371/journal.pone.0059890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hirose S., Takayama N., Nakamura S., Nagasawa K., Ochi K., Hirata S., Yamazaki S., Yamaguchi T., Otsu M., Sano S. Immortalization of erythroblasts by c-MYC and BCL-XL enables large-scale erythrocyte production from human pluripotent stem cells. Stem Cell Reports. 2013;1:499–508. doi: 10.1016/j.stemcr.2013.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang X., Shah S., Wang J., Ye Z., Dowey S.N., Tsang K.M., Mendelsohn L.G., Kato G.J., Kickler T.S., Cheng L. Extensive ex vivo expansion of functional human erythroid precursors established from umbilical cord blood cells by defined factors. Mol. Ther. 2014;22:451–463. doi: 10.1038/mt.2013.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kurita R., Funato K., Abe T., Watanabe Y., Shiba M., Tadokoro K., Nakamura Y., Nagai T., Satake M. Establishment and characterization of immortalized erythroid progenitor cell lines derived from a common cell source. Exp. Hematol. 2019;69:11–16. doi: 10.1016/j.exphem.2018.10.005. [DOI] [PubMed] [Google Scholar]
- 8.Scully E.J., Shabani E., Rangel G.W., Grüring C., Kanjee U., Clark M.A., Chaand M., Kurita R., Nakamura Y., Ferreira M.U., Duraisingh M.T. Generation of an immortalized erythroid progenitor cell line from peripheral blood: A model system for the functional analysis of Plasmodium spp. invasion. Am. J. Hematol. 2019;94:963–974. doi: 10.1002/ajh.25543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Trakarnsanga K., Griffiths R.E., Wilson M.C., Blair A., Satchwell T.J., Meinders M., Cogan N., Kupzig S., Kurita R., Nakamura Y. An immortalized adult human erythroid line facilitates sustainable and scalable generation of functional red cells. Nat. Commun. 2017;8:14750. doi: 10.1038/ncomms14750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Daniels D.E., Downes D.J., Ferrer-Vicens I., Ferguson D.C.J., Singleton B.K., Wilson M.C., Trakarnsanga K., Kurita R., Nakamura Y., Anstee D.J., Frayne J. Comparing the two leading erythroid lines BEL-A and HUDEP-2. Haematologica. 2020;105:e389–e394. doi: 10.3324/haematol.2019.229211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hawksworth J., Satchwell T.J., Meinders M., Daniels D.E., Regan F., Thornton N.M., Wilson M.C., Dobbe J.G., Streekstra G.J., Trakarnsanga K. Enhancement of red blood cell transfusion compatibility using CRISPR-mediated erythroblast gene editing. EMBO Mol. Med. 2018;10:e8454. doi: 10.15252/emmm.201708454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thornton N., Karamatic Crew V., Tilley L., Green C.A., Tay C.L., Griffiths R.E., Singleton B.K., Spring F., Walser P., Alattar A.G. Disruption of the tumour-associated EMP3 enhances erythroid proliferation and causes the MAM-negative phenotype. Nat. Commun. 2020;11:3569. doi: 10.1038/s41467-020-17060-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Satchwell T.J., Wright K.E., Haydn-Smith K.L., Sánchez-Román Terán F., Moura P.L., Hawksworth J., Frayne J., Toye A.M., Baum J. Genetic manipulation of cell line derived reticulocytes enables dissection of host malaria invasion requirements. Nat. Commun. 2019;10:3806. doi: 10.1038/s41467-019-11790-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bender J.G., Unverzagt K.L., Walker D.E., Lee W., Van Epps D.E., Smith D.H., Stewart C.C., To L.B. Identification and comparison of CD34-positive cells and their subpopulations from normal peripheral blood and bone marrow using multicolor flow cytometry. Blood. 1991;77:2591–2596. [PubMed] [Google Scholar]
- 15.Bensinger W.I. Allogeneic transplantation: peripheral blood vs. bone marrow. Curr. Opin. Oncol. 2012;24:191–196. doi: 10.1097/CCO.0b013e32834f5c27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thein S.L., Menzel S. Discovering the genetics underlying foetal haemoglobin production in adults. Br. J. Haematol. 2009;145:455–467. doi: 10.1111/j.1365-2141.2009.07650.x. [DOI] [PubMed] [Google Scholar]
- 17.Vinjamur D.S., Bauer D.E., Orkin S.H. Recent progress in understanding and manipulating haemoglobin switching for the haemoglobinopathies. Br. J. Haematol. 2018;180:630–643. doi: 10.1111/bjh.15038. [DOI] [PubMed] [Google Scholar]
- 18.Broxmeyer H.E., Lee M.-R., Hangoc G., Cooper S., Prasain N., Kim Y.-J., Mallett C., Ye Z., Witting S., Cornetta K. Hematopoietic stem/progenitor cells, generation of induced pluripotent stem cells, and isolation of endothelial progenitors from 21- to 23.5-year cryopreserved cord blood. Blood. 2011;117:4773–4777. doi: 10.1182/blood-2011-01-330514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rao M., Ahrlund-Richter L., Kaufman D.S. Concise review: Cord blood banking, transplantation and induced pluripotent stem cell: success and opportunities. Stem Cells. 2012;30:55–60. doi: 10.1002/stem.770. [DOI] [PubMed] [Google Scholar]
- 20.Hu J., Liu J., Xue F., Halverson G., Reid M., Guo A., Chen L., Raza A., Galili N., Jaffray J. Isolation and functional characterization of human erythroblasts at distinct stages: implications for understanding of normal and disordered erythropoiesis in vivo. Blood. 2013;121:3246–3253. doi: 10.1182/blood-2013-01-476390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moir-Meyer G., Cheong P.L., Olijnik A.-A., Brown J., Knight S., King A., Kurita R., Nakamura Y., Gibbons R.J., Higgs D.R. Robust CRISPR/Cas9 Genome Editing of the HUDEP-2 Erythroid Precursor Line Using Plasmids and Single-Stranded Oligonucleotide Donors. Methods Protoc. 2018;1:28. doi: 10.3390/mps1030028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vinjamur D.S., Bauer D.E. Growing and Genetically Manipulating Human Umbilical Cord Blood-Derived Erythroid Progenitor (HUDEP) Cell Lines. Methods Mol. Biol. 2018;1698:275–284. doi: 10.1007/978-1-4939-7428-3_17. [DOI] [PubMed] [Google Scholar]
- 23.Stamatoyannopoulos G. Control of globin gene expression during development and erythroid differentiation. Exp. Hematol. 2005;33:259–271. doi: 10.1016/j.exphem.2004.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sankaran V.G., Menne T.F., Xu J., Akie T.E., Lettre G., Van Handel B., Mikkola H.K.A., Hirschhorn J.N., Cantor A.B., Orkin S.H. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science. 2008;322:1839–1842. doi: 10.1126/science.1165409. [DOI] [PubMed] [Google Scholar]
- 25.Trakarnsanga K., Wilson M.C., Lau W., Singleton B.K., Parsons S.F., Sakuntanaga P., Kurita R., Nakamura Y., Anstee D.J., Frayne J. Induction of adult levels of β-globin in human erythroid cells that intrinsically express embryonic or fetal globin by transduction with KLF1 and BCL11A-XL. Haematologica. 2014;99:1677–1685. doi: 10.3324/haematol.2014.110155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu N., Hargreaves V.V., Zhu Q., Kurland J.V., Hong J., Kim W., Sher F., Macias-Trevino C., Rogers J.M., Kurita R. Direct Promoter Repression by BCL11A Controls the Fetal to Adult Hemoglobin Switch. Cell. 2018;173:430–442.e17. doi: 10.1016/j.cell.2018.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Vasconcellos J.F., Tumburu L., Byrnes C., Lee Y.T., Xu P.C., Li M., Rabel A., Clarke B.A., Guydosh N.R., Proia R.L., Miller J.L. IGF2BP1 overexpression causes fetal-like hemoglobin expression patterns in cultured human adult erythroblasts. Proc. Natl. Acad. Sci. USA. 2017;114:E5664–E5672. doi: 10.1073/pnas.1609552114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee Y.T., de Vasconcellos J.F., Yuan J., Byrnes C., Noh S.-J., Meier E.R., Kim K.S., Rabel A., Kaushal M., Muljo S.A., Miller J.L. LIN28B-mediated expression of fetal hemoglobin and production of fetal-like erythrocytes from adult human erythroblasts ex vivo. Blood. 2013;122:1034–1041. doi: 10.1182/blood-2012-12-472308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Masuda T., Wang X., Maeda M., Canver M.C., Sher F., Funnell A.P.W., Fisher C., Suciu M., Martyn G.E., Norton L.J. Transcription factors LRF and BCL11A independently repress expression of fetal hemoglobin. Science. 2016;351:285–289. doi: 10.1126/science.aad3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jassal B., Matthews L., Viteri G., Gong C., Lorente P., Fabregat A., Sidiropoulos K., Cook J., Gillespie M., Haw R. The reactome pathway knowledgebase. Nucleic Acids Res. 2020;48(D1):D498–D503. doi: 10.1093/nar/gkz1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gautier E.F., Ducamp S., Leduc M., Salnot V., Guillonneau F., Dussiot M., Hale J., Giarratana M.C., Raimbault A., Douay L. Comprehensive Proteomic Analysis of Human Erythropoiesis. Cell Rep. 2016;16:1470–1484. doi: 10.1016/j.celrep.2016.06.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Signer R.A.J., Magee J.A., Salic A., Morrison S.J. Haematopoietic stem cells require a highly regulated protein synthesis rate. Nature. 2014;509:49–54. doi: 10.1038/nature13035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Laurenti E., Göttgens B. From haematopoietic stem cells to complex differentiation landscapes. Nature. 2018;553:418–426. doi: 10.1038/nature25022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gillespie M.A., Palii C.G., Sanchez-Taltavull D., Shannon P., Longabaugh W.J.R., Downes D.J., Sivaraman K., Espinoza H.M., Hughes J.R., Price N.D. Absolute Quantification of Transcription Factors Reveals Principles of Gene Regulation in Erythropoiesis. Mol. Cell. 2020;78:960–974.e11. doi: 10.1016/j.molcel.2020.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chellappan S., Kraus V.B., Kroger B., Munger K., Howley P.M., Phelps W.C., Nevins J.R. Adenovirus E1A, simian virus 40 tumor antigen, and human papillomavirus E7 protein share the capacity to disrupt the interaction between transcription factor E2F and the retinoblastoma gene product. Proc. Natl. Acad. Sci. USA. 1992;89:4549–4553. doi: 10.1073/pnas.89.10.4549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boyer S.N., Wazer D.E., Band V. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res. 1996;56:4620–4624. [PubMed] [Google Scholar]
- 37.McLaughlin-Drubin M.E., Park D., Munger K. Tumor suppressor p16INK4A is necessary for survival of cervical carcinoma cell lines. Proc. Natl. Acad. Sci. USA. 2013;110:16175–16180. doi: 10.1073/pnas.1310432110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leong W.F., Chau J.F., Li B. p53 Deficiency leads to compensatory up-regulation of p16INK4a. Mol. Cancer Res. 2009;7:354–360. doi: 10.1158/1541-7786.MCR-08-0373. [DOI] [PubMed] [Google Scholar]
- 39.Werness B.A., Levine A.J., Howley P.M. Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science. 1990;248:76–79. doi: 10.1126/science.2157286. [DOI] [PubMed] [Google Scholar]
- 40.Scheffner M., Werness B.A., Huibregtse J.M., Levine A.J., Howley P.M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 1990;63:1129–1136. doi: 10.1016/0092-8674(90)90409-8. [DOI] [PubMed] [Google Scholar]
- 41.Sherr C.J., Roberts J.M. Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 2004;18:2699–2711. doi: 10.1101/gad.1256504. [DOI] [PubMed] [Google Scholar]
- 42.Lindberg D., Hessman O., Akerström G., Westin G. Cyclin-dependent kinase 4 (CDK4) expression in pancreatic endocrine tumors. Neuroendocrinology. 2007;86:112–118. doi: 10.1159/000106762. [DOI] [PubMed] [Google Scholar]
- 43.Fang W., Li X., Jiang Q., Liu Z., Yang H., Wang S., Xie S., Liu Q., Liu T., Huang J. Transcriptional patterns, biomarkers and pathways characterizing nasopharyngeal carcinoma of Southern China. J. Transl. Med. 2008;6:32. doi: 10.1186/1479-5876-6-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Poomsawat S., Buajeeb W., Khovidhunkit S.O., Punyasingh J. Alteration in the expression of cdk4 and cdk6 proteins in oral cancer and premalignant lesions. J. Oral Pathol. Med. 2010;39:793–799. doi: 10.1111/j.1600-0714.2010.00909.x. [DOI] [PubMed] [Google Scholar]
- 45.Wu A., Wu B., Guo J., Luo W., Wu D., Yang H., Zhen Y., Yu X., Wang H., Zhou Y. Elevated expression of CDK4 in lung cancer. J. Transl. Med. 2011;9:38. doi: 10.1186/1479-5876-9-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nebenfuehr S., Kollmann K., Sexl V. The role of CDK6 in cancer. Int. J. Cancer. 2020;147:2988–2995. doi: 10.1002/ijc.33054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bellutti F., Tigan A.-S., Nebenfuehr S., Dolezal M., Zojer M., Grausenburger R., Hartenberger S., Kollmann S., Doma E., Prchal-Murphy M. CDK6 Antagonizes p53-Induced Responses during Tumorigenesis. Cancer Discov. 2018;8:884–897. doi: 10.1158/2159-8290.CD-17-0912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Piboonniyom S.O., Duensing S., Swilling N.W., Hasskarl J., Hinds P.W., Münger K. Abrogation of the retinoblastoma tumor suppressor checkpoint during keratinocyte immortalization is not sufficient for induction of centrosome-mediated genomic instability. Cancer Res. 2003;63:476–483. [PubMed] [Google Scholar]
- 49.Stadler G., Chen J.C., Wagner K., Robin J.D., Shay J.W., Emerson C.P.J., Jr., Wright W.E. Establishment of clonal myogenic cell lines from severely affected dystrophic muscles - CDK4 maintains the myogenic population. Skelet. Muscle. 2011;1:12. doi: 10.1186/2044-5040-1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thorley M., Duguez S., Mazza E.M.C., Valsoni S., Bigot A., Mamchaoui K., Harmon B., Voit T., Mouly V., Duddy W. Skeletal muscle characteristics are preserved in hTERT/cdk4 human myogenic cell lines. Skelet. Muscle. 2016;6:43. doi: 10.1186/s13395-016-0115-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ramirez R.D., Sheridan S., Girard L., Sato M., Kim Y., Pollack J., Peyton M., Zou Y., Kurie J.M., Dimaio J.M. Immortalization of human bronchial epithelial cells in the absence of viral oncoproteins. Cancer Res. 2004;64:9027–9034. doi: 10.1158/0008-5472.CAN-04-3703. [DOI] [PubMed] [Google Scholar]
- 52.Trakarnsanga K., Tipgomut C., Metheetrairut C., Wattanapanitch M., Khuhapinant A., Poldee S., Kurita R., Nakamura Y., Srisawat C., Frayne J. Generation of an immortalised erythroid cell line from haematopoietic stem cells of a haemoglobin E/β-thalassemia patient. Sci. Rep. 2020;10:16798. doi: 10.1038/s41598-020-73991-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.