

Abstract
Background and Aims:
Human TM6SF2 variant rs58542926 is associated with nonalcoholic fatty liver disease (NAFLD) and hepatocellular cancer (HCC). However, conflicting reports in germline Tm6sf2 knockout mice suggest no change or decreased VLDL secretion and either unchanged or increased hepatic steatosis, with no increased fibrosis. We generated liver specific Tm6Sf2 knockout mice (Tm6 LKO) to study VLDL secretion and the impact on development and progression of NAFLD.
Approach and Results:
Two independent lines of Tm6 LKO mice exhibited spontaneous hepatic steatosis. Targeted lipidomic analyses showed increased triglyceride (TG) species whose distribution and abundance phenocopied findings in mice with liver specific deletion of microsomal triglyceride transfer protein. VLDL TG secretion was reduced, with small, underlipidated particles and unchanged or increased APOB. Liver-specific adeno-associated viral (AAV8-TBG) rescue using either wild type (WT) or mutant E167K-Tm6 reduced hepatic steatosis and improved VLDL secretion. Tm6 LKO mice fed a high milk-fat diet for 3 weeks exhibited increased steatosis and fibrosis and those phenotypes were further exacerbated when mice were fed fibrogenic, high fat/fructose diets for 20 weeks. In two models of HCC, either neonatal mice injected with streptozotocin (NASH/STAM) and high fat fed or with diethylnitrosamine (DEN) injection plus fibrogenic diet feeding, Tm6 LKO mice exhibited increased steatosis, greater tumor burden and increased tumor area versus Tm6 flox controls. Additionally, DEN-injected and fibrogenic diet fed Tm6 LKO mice administered WT Tm6 or E167K-mutant Tm6 AAV8 revealed significant tumor attenuation, with tumor burden inversely correlated with Tm6 protein levels.
Conclusions:
Liver-specific Tm6sf2 deletion impairs VLDL secretion, promoting hepatic steatosis, fibrosis and accelerated development of HCC, which was mitigated with AAV8- mediated rescue.
Keywords: VLDL, Endoplasmic reticulum, NAFLD, Genetics
The relentless global increase in obesity has produced a parallel increase in important comorbidities, including type 2 diabetes and nonalcoholic fatty liver disease (NAFLD), which is now the most prevalent liver disease worldwide (1). The incidence and progression of NAFLD, however, is influenced by both environmental and genetic factors (2–4) which has focused interest on identifying factors that would merit early intervention to prevent the development of progressive disease including nonalcoholic steatohepatitis (NASH) with fibrosis and also hepatocellular cancer (HCC). Among the genetic risk factors, defects in the assembly and secretion of hepatic VLDL -- including mutations in either of two gatekeeper genes, APOB and MTTP, encoding the structural VLDL protein apolipoproteinB (apoB) or the endoplasmic reticulum (ER) protein microsomal triglyceride transfer protein (Mttp) respectively -- produce hepatic steatosis and NAFLD that may progress to fibrosis with development of HCC (5, 6). Among the distinctive characteristics of NAFLD accompanying APOB and MTTP defects are that patients are not typically obese, do not exhibit type 2 diabetes and, most distinctively, manifest very low plasma cholesterol and triglyceride (TG) levels as a result of impaired VLDL secretion (6).
A search for other genetic causes of NAFLD led to the identification by three groups of an exonic variant in TM6SF2, p.E167K (c449>T, rs58542926) which was associated with hepatic steatosis and reduced plasma cholesterol and TG levels (7–9). Those studies and others (10) demonstrated that subjects with the rs58542926 variant exhibited reduced VLDL secretion, hepatic steatosis and increased susceptibility to progressive NAFLD, including NASH with fibrosis and increased incidence of HCC (11–14). Those findings, replicated in most but not all studies (15), have driven intense interest in understanding the mechanisms and pathways underlying the development and progression of NAFLD in subjects harboring the TM6SF2 variant. Because the rs58542926 variant likely functions as a loss-of-function allele (16), attempts to replicate key features of the human phenotypes in preclinical models have focused on germline knockout and liver specific transgenic or viral-mediated overexpression of Tm6sf2 (17, 18). However, the phenotypes observed in Tm6sf2 knockout mice have been inconsistent, with one report of germline deletion showing spontaneous hepatic steatosis and defective VLDL secretion (18), while another report showed no increase in baseline hepatic steatosis and increased serum TG, even when those Tm6sf2 knockout mice were fed a high fat diet (17). Yet another study showed that adeno-associated viral (AAV) overexpression of mouse TM6SF2 produced defective VLDL secretion and paradoxically recapitulated phenotypes observed in Tm6sf2 knockout mice (19), while liver transgenic overexpression of human TM6SF2 resulted in increased plasma cholesterol levels and increased hepatic cholesterol and TG content (17).
Because of those inconsistencies and because our prior findings in which mice with intestine-specific Mttp deletion demonstrated compensatory increases in hepatic VLDL secretion (20), we reasoned that examination of a distinctive loss-of-function phenotype for hepatic Tm6sf2 required development of a liver-specific knockout allele. Here we report findings in two independent lines of mice engineered with CRISPR-Cas9 to produce liver-specific Tm6sf2 deletion (Tm6 LKO). Our findings showed that Tm6 LKO mice manifest hepatic steatosis with decreased hepatic VLDL TG secretion yet similar or increased APOB secretion, consistent with observations from one of those germline knockout mice (18). In addition, we show that Tm6 LKO mice exhibit progressive NAFLD with fibrosis when fed fibrogenic diets and, using two different models of experimental hepatocarcinogenesis, display accelerated and more extensive HCC. Finally, we demonstrate that adeno-associated viral (AAV8) rescue with wild type (WT) TM6SF2, but not AAV8 encoding LacZ, mitigates HCC in Tm6 LKO mice.
METHODS
Animals:
All animal studies were approved by the Washington University Institutional Animal Care and Use Committee (IACUC #20180266, 20190028). Mice were housed in ventilated cages on a 12 hour light/dark cycle with corncob bedding, and ad libitum access to rodent chow (PicoLab Rodent Diet 20, LabDiet, St Louis, MO) and water unless otherwise noted. Mttp-LKO and Apobec1 knockout mice (C57BL/6J congenic) were characterized previously (21, 22)
Conditional Tm6sf2 knockout mice were generated by the Genome Engineering and iPSC Center (Washington University) using CRISPR-Cas9 mediated insertion of Lox P recombination sites into intronic regions flanking Exon 2 (Figure 1A). Guide RNAs were selected, tested for off target effects, and injected with Cas9 mRNA into B6/CBA embryos (#100011, The Jackson Laboratory, Bar Harbor, ME). Chimeric founders were identified by deep sequencing and bred to C57BL/6J (#000664, Jax) mice to generate two separate lines of floxed Tm6sf2 mice (hereafter Tm6 Flox mice) from independent chimeric founders. Integrity of the lox P sites was confirmed by sequencing and Cre-mediated recombination, and the sequence of Exon 2 was verified. Each Tm6 flox strain was bred to Albumin CreTg mice (#003574, Jax) to generate 2 lines with liver specific deletion (LKO) of Tm6sf2. Because our initial studies revealed both lines of Tm6 flox/LKO mice exhibit similar phenotypes, only data from one line will be presented, with reproducibility noted in the text or figure legend. Mice are C57BL/6J:CBA, with non-transgenic floxed mice of the same gender used as controls.
Figure 1:
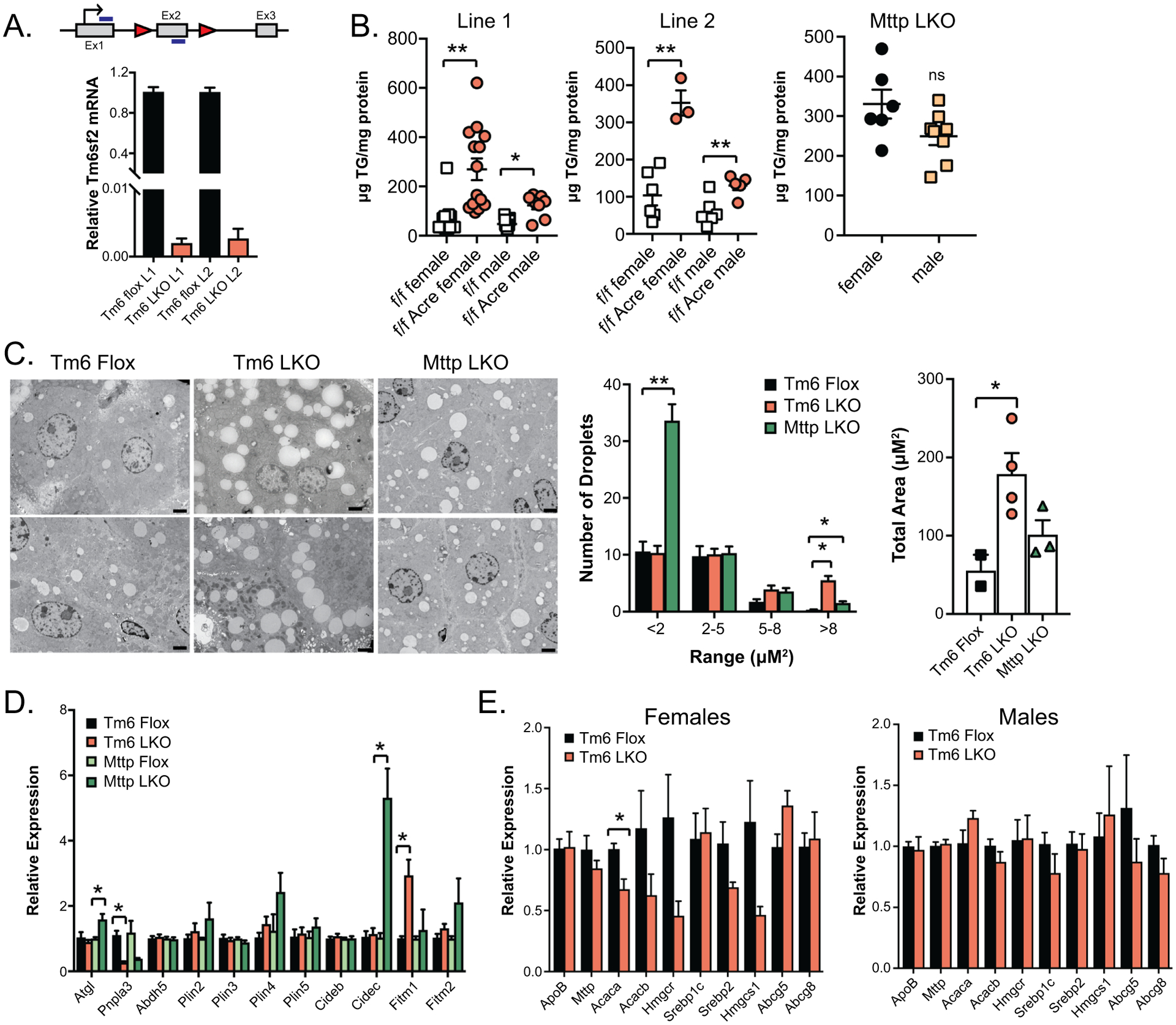
Targeting strategy and baseline characterization of Tm6 LKO mice. A. Schematic diagram of Tm6sf2 gene showing the location of Lox P recombination sites (triangles) in intronic regions surrounding Exon 2 (Ex2). Also shown is the translation initiation site (arrow) and the location of primers used to detect Tm6sf2 mRNA (blue bars). Lower panel: Relative expression of Tm6sf2 mRNA in the liver of two distinct conditional knock out lines of Tm6 Flox and Tm6 LKO mice (L1, L2; n=5–7 females/group). B. Hepatic triglyceride levels in male and female, Tm6 flox and Tm6 LKO mice from 2 distinct Tm6sf2 LKO lines (left, middle). Right panel shows hepatic triglyceride levels in male and female Mttp LKO mice for comparison. All mice are 12–14 weeks of age, fed chow diet. C. Left: Representative TEM images of intracellular lipid droplets in chow fed Tm6 Flox, Tm6 LKO and Mttp LKO liver tissue (2000x, scale bar = 2μM). Middle: Quantitation of lipid droplet size distribution in Tm6 Flox (n=2), Tm6 LKO (n=4) and Mttp LKO (n=3) female mice (6 images/mouse). Right: Average total lipid droplet area in 6 images/animal. D. Relative gene expression of lipid droplet associated proteins in Tm6 Flox, Tm6 LKO, Mttp Flox and Mttp LKO liver (chow diet females, n=4/genotype), with expression normalized to respective flox control samples. E. Baseline expression of genes involved in TG secretion, fibrogenesis, and inflammation in male and female Tm6 LKO and control mice (n=5/genotype, 12–14 weeks, chow diet). For all panels, * indicates p<0.05, ** indicates p<0.01.
HCC models.
STAM (23):
Streptozotocin (AdipoGen Life Sciences, AG-CN2-0046) was administered on postnatal day 2 (0.4mg/mouse, subcutaneous) and male mice were weaned to a 60% lard diet (TD.06414, Envigo) (23). Blood glucose was checked at 6 and 9 weeks of age (Glucocard Vital glucometer) and animals with consistently low glucose (<400mg/dL) were excluded. Tissue and serum were collected at 12 or 18 weeks of age following a 4h fast and 2h after injection of BrdU (200μl/IP, 18mg/ml bromodeoxyuridine, 1.8mg/ml fluorodeoxyuridine). DEN/TFF: Male mice were injected with diethyl nitrosamine (DEN, #N0258, Sigma-Aldrich) at P14 (25mg/kg, IP) (24) and fed high transfat, fructose diet (TFF TD.06303, Envigo) (25). Serum and tissue were collected at 4 or 7 months of age and 2h after injection of BrdU. In some studies, mice were injected with AAV at 2–4 weeks of age (IP) as indicated. For all HCC studies, animals were weighed weekly and monitored for signs of distress.
Additional reagent information and experimental protocols including details of dietary studies, electron microscopy, histological assessment and untargeted lipidomics are provided in Supplemental Methods.
Statistics:
Data were analyzed using GraphPad Prism 7.02 by unpaired t-test or Mann-Whitney test and are presented as mean ± standard error of mean (SEM). For multigroup comparisons, statistical differences were performed by one-way ANOVA and Tukey’s multiple comparison post-hoc test. The level of significance was set at p<0.05 and indicated using *p<0.05; **p<0.01.
RESULTS
Spontaneous hepatic steatosis in Tm6 LKO mice:
Tm6sf2 mRNA was significantly (>95%) decreased in both lines of chow fed Tm6 LKO mice (L1, L2 Figure 1A) compared to controls. Hepatic TG was increased in 3 month old Tm6 LKO mice, females greater than males (Figure 1B), with no difference in hepatic cholesterol and only minor differences in FFA levels (Supplemental Table 1). Hepatic TG content in both lines of female Tm6 LKO mice was similar to levels observed in mice with liver specific Mttp deletion (Mttp LKO, Figure 1B) (21), but no sexual dimorphism was observed in Mttp LKO mice. Tm6 mRNA abundance was ~50% higher in female wild type liver (Supplemental Figure 1A), which might contribute to the dimorphic effects of Tm6sf2 deletion. A similar sexual dimorphism in TG content was found in primary hepatocytes from adult male and female Tm6 LKO mice (Supplemental Figure 1B). Hepatic steatosis was noted as early as 21 days in Tm6 LKO mice (Supplemental Figure 1C). In general, we found no dramatic differences in body or liver weight, serum or hepatic lipids in either of the LKO lines (Supplemental Table 1), although serum TG was lower in adult Tm6 LKO mice (Tm6 Flox, 45.9 ± 2.8 mg/dL; Tm6 LKO, 31.5 ± 2.7; n=26–30/genotype, sexes combined; p<0.001. See Supplemental Table 1 for individual gender data).
Distinct changes in Lipid droplet (LD) abundance and size in Tm6 LKO and Mttp LKO mice:
Transmission EM revealed increased numbers of large droplets in Tm6 LKO mice but no change in small LDs compared to controls (Figure 1C). By contrast, the abundance of small LDs increased in Mttp LKO mice, with only a slight increase in the number of LDs > 5μM2. Total calculated LD area (Figure 1C, right panel) was increased in both Tm6 LKO and Mttp LKO mice compared to controls, with overall droplet area lower in Mttp LKO vs Tm6 LKO. We also surveyed the expression of LD proteins by genotype. Expression of Fitm1 was increased, while Pnpla3 mRNA abundance was significantly reduced in Tm6 LKO mice (Figure 1D), consistent with findings in germline Tm6sf2 knockout mice (18). However, despite the presence of large LDs in Tm6 LKO mice, mRNA expression of perilipins and other LD proteins were unaltered compared to Tm6 Flox mice, while Mttp LKO mice demonstrated increased expression of Cidec/Fsp27 and Plin 4, with increased expression of Atgl/Pnpla2 and reduced expression of Pnpla3 compared to Tm6 Flox mice (Figure 1D). Moreover, we found no compensatory alteration in the expression of genes related to hepatic VLDL production, including ApoB and Mttp, but subtle changes in expression of genes related to cholesterol metabolism, particularly in female Tm6 LKO mice (Figure 1E, compare left and right panels).
Overlapping changes in untargeted lipidomic profiles in Tm6 LKO and Mttp LKO mice:
All major TG and DAG species were significantly increased in livers of both Tm6 and Mttp LKO mice with similar relative abundance of each species in both knockout genotypes (Figure 2A–B). A similar increase in abundance of all TG species was also observed in Tm6 LKO and Mttp LKO mice fed a high transfat fructose (TFF) diet (Supplemental Figure 2). The profile of FFA species was also similar among genotypes (Figure 2C). Subtle changes were observed in the abundance of several minor PC species, while the abundance of several lysoPC species (16:0, 18:1, 18:2) was decreased in both Mttp and Tm6 LKO genotypes (Figure 2D). Lipidomic analysis of serum TG species reflects reduced abundance of almost all TG species in Mttp LKO mice, with a similar, albeit less dramatic reduction in Tm6 LKO mice (Figure 2E), consistent with a defect in VLDL secretion in both genotypes. To investigate if alterations in fatty acid metabolism contribute to steatosis in Tm6 LKO mice, we surveyed the expression of genes related to TG synthesis and fatty acid oxidation. Expression of mRNAs related to hepatic de novo lipogenesis and desaturation was reduced in both Tm6 LKO and Mttp LKO mice (Figure 2F). Expression of genes related to fatty acid β-oxidation (Cpt1a, Acadm, Acox1) were unchanged in Tm6 LKO mice (Figure 2F), although serum β-hydroxybutyrate levels showed a trend to increase in Tm6 LKO mice (Tm6 Flox, 0.270 ± 0.021 mm; Tm6 LKO 0.330 ± 0.030, p=0.113, n=13–14, genders combined. See Supplemental Table 1 for data by gender) suggesting a moderate increase in ketogenesis in the setting of increased steatosis. In comparison, Mttp-LKO mice displayed both an increase in serum ketones (Mttp Flox, 0.350 ± 0.019 mm; Mttp-LKO, 0.459 ± 0.048, n=9–10, p=0.043) and FAO gene expression (Figure 2F). Together these data suggest that increased TG content in Tm6 LKO mice is primarily due to decreased secretion, not increased lipogenesis or reduced fatty acid oxidation.
Figure 2:
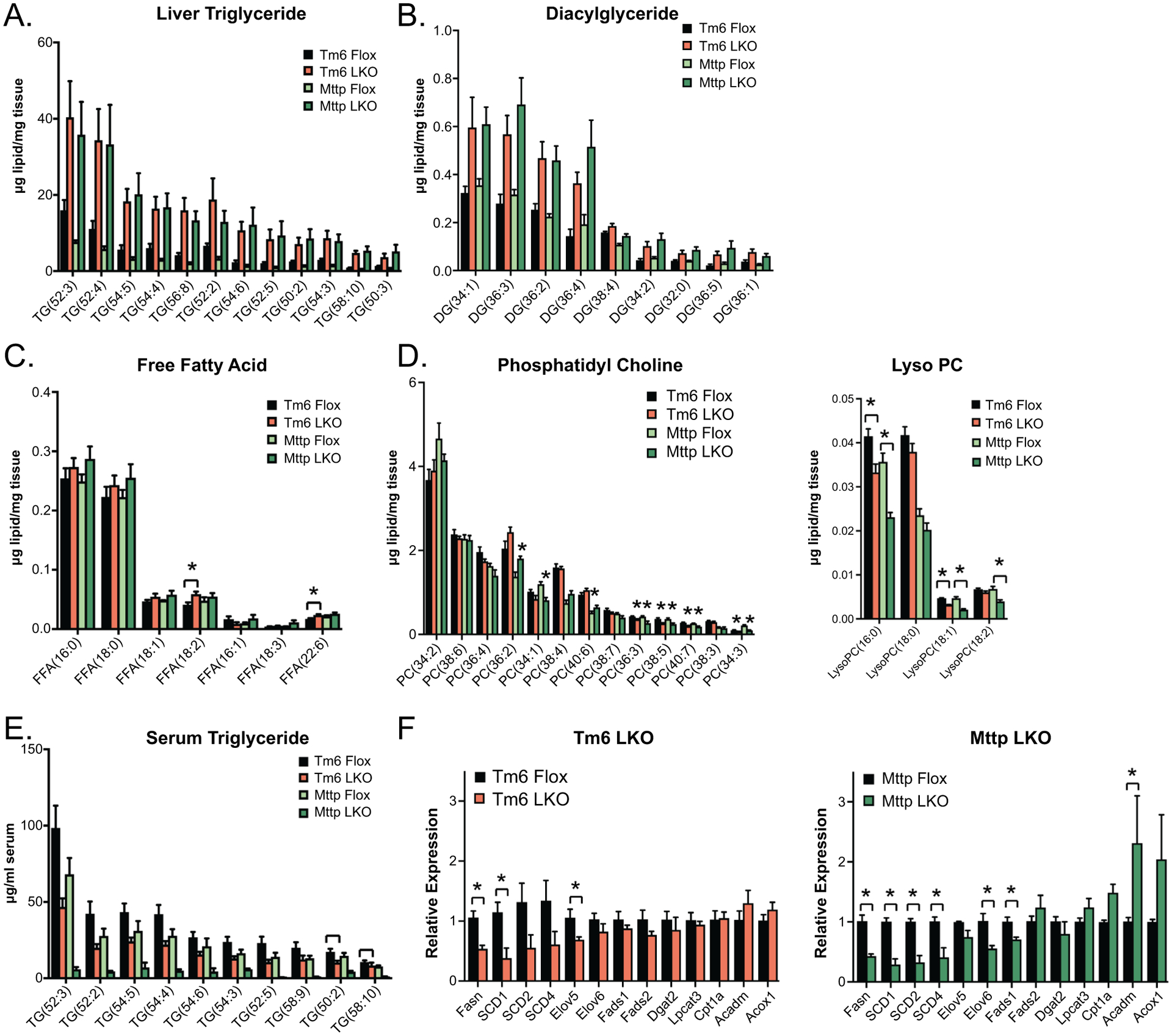
Untargeted lipidomic characterization of indicated genotypes. A. Levels of major triglyceride species in Tm6 Flox, Tm6 LKO, Mttp Flox and Mttp LKO liver tissue. All TG species shown are significantly more abundant in mice with liver specific deletion of Tm6sf2 or Mttp versus respective control livers. N= 5–6/genotype, chow diet fed females, 12–15 weeks of age. Species shown represent >85% percent of total TG species peak area. B. Diacylglyceride species in Tm6 Flox, Tm6 LKO, Mttp Flox and Mttp LKO liver tissue. All species shown are significantly more abundant in Tm6 LKO and Mttp LKO liver tissue versus respective control livers. C. Free fatty acid species in above genotypes. Significant differences are indicated. D. Phosphatidyl choline (left panel) and lyso-phosphatidylcholine (Lyso PC) species E. Abundance of triglyceride species in serum of Tm6 Flox, Tm6 LKO, Mttp Flox and Mttp LKO mice following a 4h fast (chow diet, females). . F. Expression of genes related to fatty acid synthesis, β-oxidation and modification in Tm6 LKO and Mttp LKO liver (n=4 females/genotype, chow diet). For all panels, data are presented as mean ± SEM; asterisks indicate p<0.05 and ns indicates not significant.
Altered hepatic VLDL secretion kinetics and composition in Tm6 LKO mice:
Chow fed Tm6 LKO mice revealed decreased hepatic TG secretion compared to Tm6 flox controls (Figure 3A) with similar abundance of APOB isoforms in the two genotypes (Right panel, Figure 3A). Note that hepatic VLDL secretion was virtually eliminated in Mttp LKO mice (Figure 3A), as observed earlier (26). FPLC fractionation to separate nascent lipoprotein particles by size revealed reduced TG in VLDL fractions from Tm6 LKO mice, and a slight shift (peak 29–31) in distribution of TG, cholesterol and APOB protein (Figure 3B), but no decrease in the total amount of APOB in Tm6 LKO mice (lower panel), consistent with secretion of smaller, underlipidated VLDL particles. VLDL particles were then isolated by density centrifugation and examined by EM, which confirmed increased abundance of small particles and a corresponding decrease in larger particles in Tm6 LKO mice (Figure 3C). In vitro studies using isolated primary hepatocytes showed no difference in TG synthesis in Tm6 LKO hepatocytes, following [3H] oleate incorporation (Figure 3D, left panel), and no decrease in lipogenesis (acetate incorporation, data not shown), but a ~75% reduction in TG secretion (right) in Tm6 LKO hepatocytes. Importantly, as suggested by in vivo studies, secretion of APOB was increased in Tm6 LKO hepatocytes, with no difference in APOB synthesis (Figure 3E). Taken together these data demonstrate that Tm6-deficient hepatocytes synthesize and secrete APOB containing VLDL particles that are smaller and contain less TG than WT mice.
Figure 3:
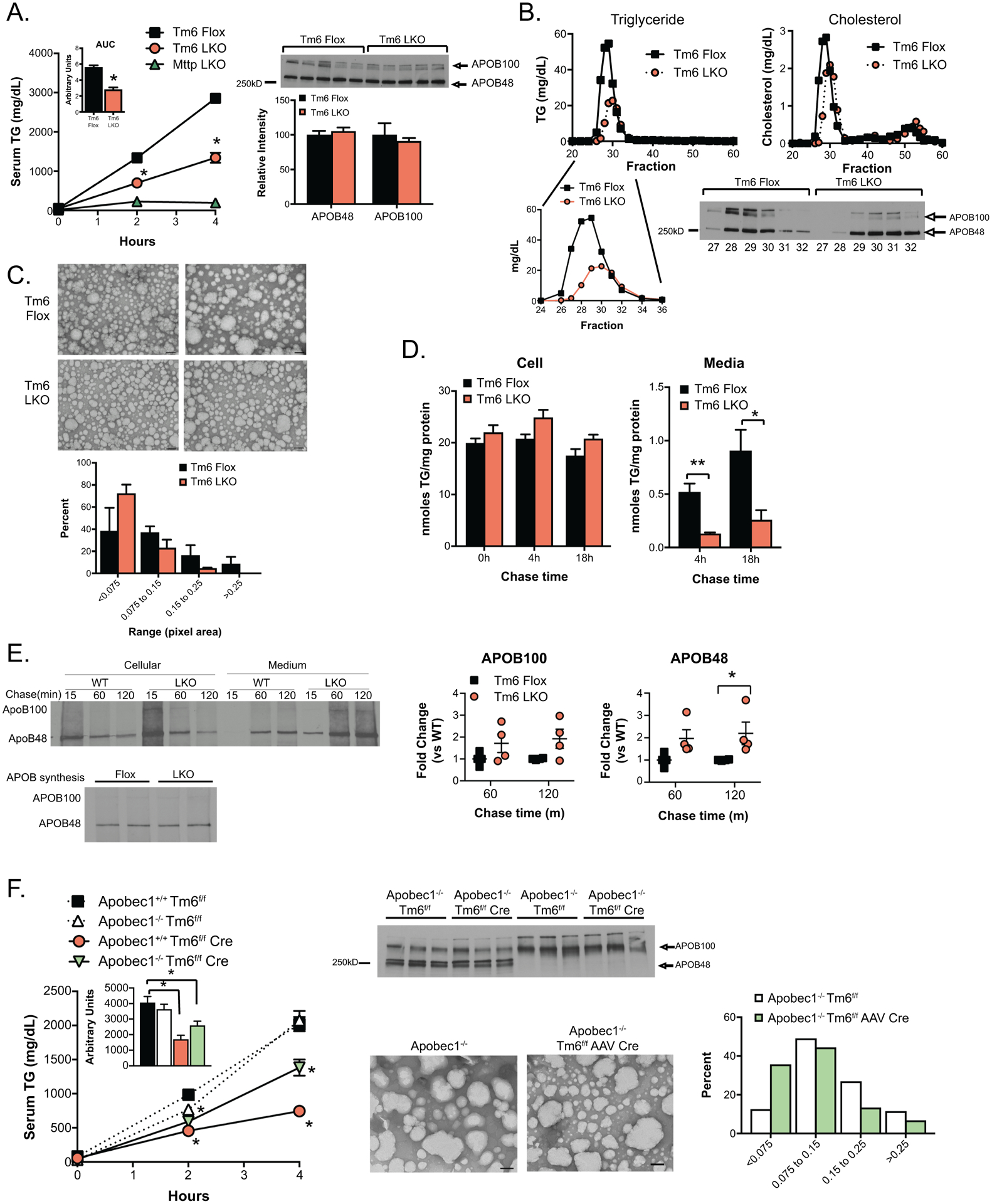
VLDL secretion characteristics of Tm6 LKO mice. A. Serum triglyceride levels at 0, 2 and 4 hours after injection of Pluronic −127 in Tm6 Flox, Tm6 LKO, and Mttp LKO mice (n=4–5/genotype). Inset shows average area under curve for Tm6 Flox and LKO genotypes. Right: Western blot of APOB protein in 4h serum of individual Tm6 Flox and Tm6 LKO mice, with densitometric quantification shown below. B. Serum from 4h bleed (Panel A) was pooled by genotype and fractionated by FPLC to separate lipoprotein particles. Column fractions were assayed for triglyceride and cholesterol to identify VLDL- and HDL-containing fractions (Fractions 27–32 and 48–55, respectively). Lower panel: Western blot showing APOB100 and APOB48 protein in VLDL FPLC fractions. C. Size distribution of negatively stained VLDL particles isolated from Tm6 Flox and Tm6 LKO mice. Serum was pooled from 3–4 mice per genotype 4h after Pluronic injection and fractionated by density centrifugation. Data are presented as percent of total droplets in each size range and was generated from 2 independent isolations per genotype. Representative images of negatively stained VLDL particles are shown (50,000x; scale bar=100nm). D. Synthesis and secretion of [3H]-labeled triglyceride in isolated primary hepatocytes. Left: Cellular [3H]-TG levels in Tm6 Flox and LKO hepatocytes at 0h, 4h and 18 hours after labeling, normalized to cellular protein. Right: [3H]-TG in media collected 4 or 18 hours after labeling. E. Synthesis and secretion of APOB in primary hepatocytes. Top left: Newly synthesized APOB100 and APOB48 in cells or medium 15, 60 or 120 minutes after pulse labeling of Tm6 Flox (WT) and LKO hepatocytes. Bottom left: APOB synthesis in isolated hepatocytes. Right: Quantitation of APOB100 and APOB48 secretion in 3 independent experiments, normalized to APOB secretion in WT hepatocytes in each experiment. F. Effect of Tm6sf2 deletion on VLDL secretion and particle size in APOB100 and APOB48 mice. Left: Serum triglyceride in HMFD-fed Apobec1+/+ Tm6f/f, Apobec1−/- Tm6f/f, Apobec1+/+ Tm6 f/f Cre and Apobec1−/- Tm6f/f Cre mice at 0, 2 and 4 hours after Pluronic F-127 injection. Deletion of Tm6sf2 was induced by AAV Cre in Apobec1−/- mice and with Alb CreTg in Apobec1+/+ mice. Tm6f/f mice received control AAV (null or LacZ). N=4–5 animals/group, mixed genders. Middle: Serum from 4 hour timepoint was analyzed by western blot analysis to monitor levels of APOB100 and APOB48 protein, and by density centrifugation to examine VLDL particle size in Apobec1−/- Tm6f/f and Apobec1−/- Tm6f/f AAV Cre mice fed chow diet. Representative images of negatively stained VLDL particles are shown (50,000x, scale bar=100nm), with quantitative size distribution presented as percent of total droplets in each size range (right). For all panels, asterisks indicate p<0.05 versus Tm6 Flox controls.
Because APOB and VLDL TG secretion in Tm6 LKO mice occurs predominantly with APOB48 (Figure 3A) while VLDL secretion in humans is exclusively associated with APOB100 production (27), we generated a compound line of Tm6 LKO mice (Apobec1 −/− Tm6f/f AAV Cre) where only APOB100 is produced (22). Interestingly, the effect of Tm6sf2 deletion on VLDL secretion was significantly less dramatic in Apobec1–/– (APOB100) only mice compared to wild-type (Apobec1+/+) mice, both in mice fed a chow diet (Supplemental Figure 3A) and in mice fed a high milk fat diet (HMFD) for 3 weeks (Figure 3F). As observed in mice fed a chow diet (Figure 3A), levels of APOB protein in serum were not markedly changed with Tm6sf2 deletion (Figure 3F, middle panel), suggesting secretion of smaller VLDL particles. Consistent with this, we observed reduced VLDL size in Apobec1−/- Tm6f/f AAV Cre mice compared to Apobec1–/– mice, using both density gradient centrifugation (Figure 3F, right) and FPLC fractionation (Supplemental Figure 3B). These findings suggest that VLDL secretion is impaired in Tm6 LKO mice, but with greater impact in mice with both APOB100 and APOB48. These findings differ from studies with hepatic Mttp deletion where secretion of APOB100 containing VLDL is more severely impaired (28).
Impact of high fat feeding on hepatic lipid secretion, steatosis and fibrosis in Tm6 LKO mice:
Hepatic TG was significantly increased in both male and female Tm6 LKO mice fed HMFD (Figure 4A), with no difference in body weight or serum glucose by genotype (Supplemental Table 2). AAV rescue with exogenous Tm6sf2 – both WT and E167K mutant variants containing carboxy-terminal FLAG tag - resulted in similar levels of Tm6 mRNA, but only WT Tm6 protein was detectable by anti-FLAG western blot (Figure 4B), consistent with the Tm6 E167K mutation yielding an unstable protein (16, 19). Despite this, AAV8 rescue with either WT or mutant Tm6 reduced steatosis to levels comparable to Tm6 Flox mice (Figure 4C, left) and reduced intracellular LD size (Figure 4C, right). In contrast, VLDL secretion was only partially restored upon AAV8 rescue both in HMFD-fed mice (Figure 4D), and in mice fed a chow diet (Supplemental Figure 4A), and although AAV8 Tm6 (WT or mutant) increased the number of large VLDL particles, neither isoform fully restored VLDL size distribution (Supplemental Figure 4B). Expression of genes related to FA synthesis (FASN, SCD1) was increased in mice with AAV8 Tm6 rescue (Figure 4E). These findings show a disconnect between levels of hepatic steatosis and VLDL secretion and imply either that there are additional compensatory mechanisms that account for the reduced hepatic steatosis or that the in-vivo measures of VLDL export are insufficiently sensitive to discern an adequate threshold for effective rescue. We observed early, relatively mild hepatic fibrosis following high fat feeding (Figure 4F), with a trend to increased fibrogenic mRNA expression (Supplemental Figure 4C). AAV8 rescue with either WT or E167K Tm6 reduced fibrosis in Tm6 LKO mice (Figure 4F).
Figure 4:
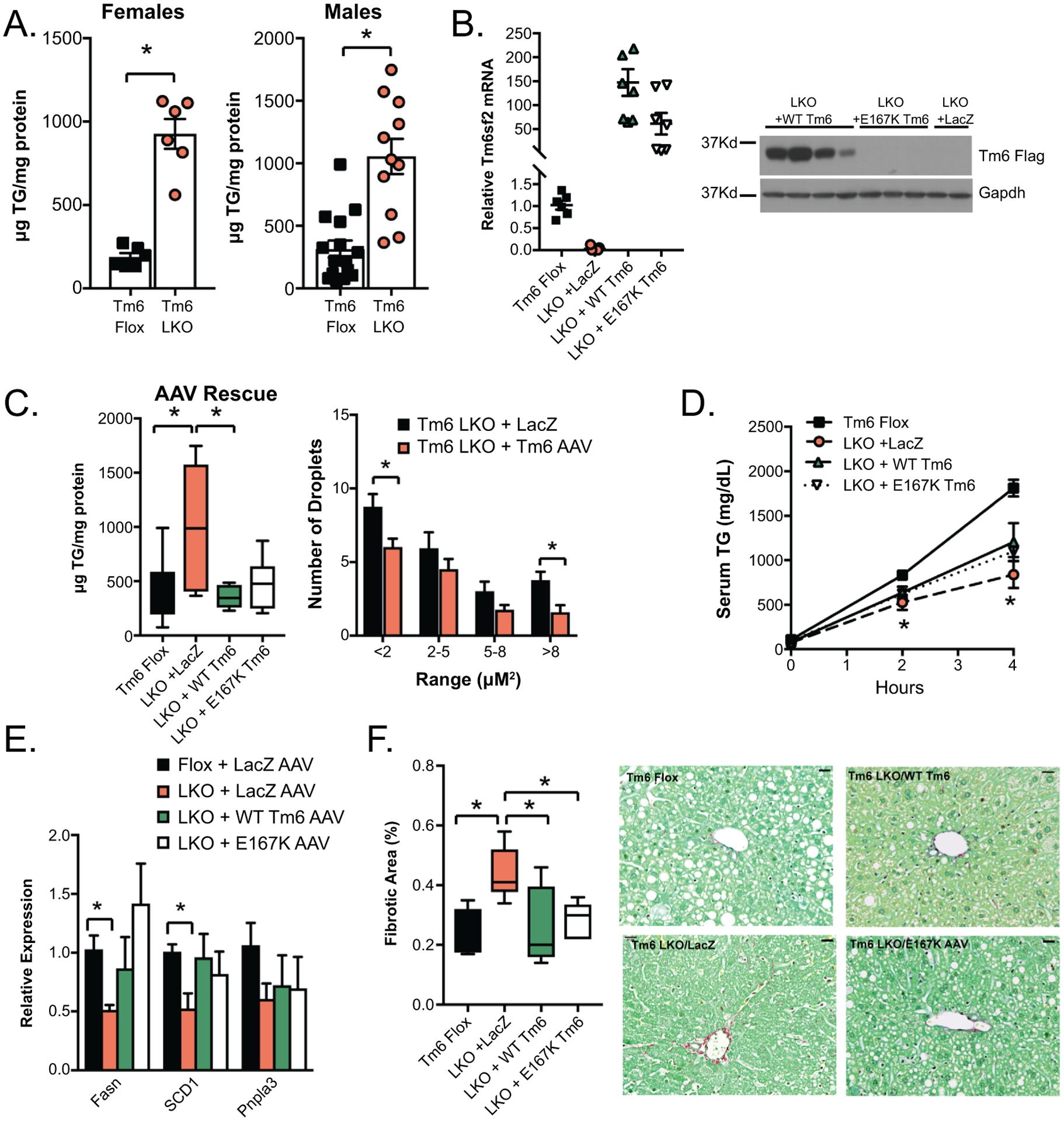
Impact of high fat diet on hepatic lipid content of Tm6 LKO mice and rescue with AAV8 Tm6sf2. A. Hepatic triglyceride content in male and female Tm6 Flox and Tm6 LKO mice fed HMFD for 3 weeks (female, n=6/genotype; male, n= 11–15). A similar phenotype was found using Tm6 LKO line 2. B. AAV-mediated expression of Flag Tm6sf2. Left: Relative expression of Tm6 mRNA in livers of Tm6 LKO mice (n=6–8/group) transduced with LacZ AAV, WT Tm6 AAV, or E167K AAV, normalized to expression of endogenous Tm6sf2 in Tm6 Flox livers. All groups are significantly different compared to flox controls. Right: Western blot showing expression of Flag tagged Tm6 in AAV transduced liver tissue using anti-Flag antibody. Expression of Gapdh protein is shown as a loading control. C. Left: Hepatic TG content in liver of HMFD- fed Tm6 Flox and Tm6 LKO mice transduced with AAV constructs (n=6–10 males/group). Right: Quantitation of lipid droplet size in Tm6 LKO mice transduced with Lac Z or WT Tm6 AAV. D. VLDL secretion in HMFD- fed male and female Tm6 Flox and Tm6 LKO mice transduced with Lac Z, Tm6 WT, and E167K AAV (n=3–4/group). E. Expression of lipogenic genes in Tm6 Flox and AAV-transduced LKO mice fed HMFD (n=5–7 animals/group). F. Sirius red stained fibrotic area in Tm6 Flox and Tm6 LKO mice transduced with AAV (n=5–6/group). Representative images are shown (400x magnification, scale bar = 20μM). For all panels, asterisks indicate p<0.05.
To more fully characterize the effect of Tm6 deletion on hepatic fibrosis we turned to two established, long term dietary fibrogenic models. First, mice were fed a high transfat, fructose (TFF) diet for 20 weeks (21). Both male and female Tm6 LKO mice displayed increased hepatic steatosis (Figure 5A), with increased fibrogenic mRNA expression (Figure 5B) and increased Sirius red staining (Figure 5C), but no difference in body weight compared to controls (Supplemental Table 2). In the second model, mice were fed a palm oil diet containing fructose and cholesterol (PFC diet) for 26 weeks (29). Both hepatic triglyceride and cholesterol were increased in Tm6 LKO mice (Figure 5D, Supplemental Table 2), with a corresponding increase in fibrotic area, assessed using both Sirius staining and Second Harmonic Generation microscopy (30, 31), with increased expression of fibrogenic mRNAs (Figure 5 E–F). Tm6 LKO mice had increased liver/body ratio, but no difference in body weight or serum glucose compared to controls (Supplemental Table 2).
Figure 5:
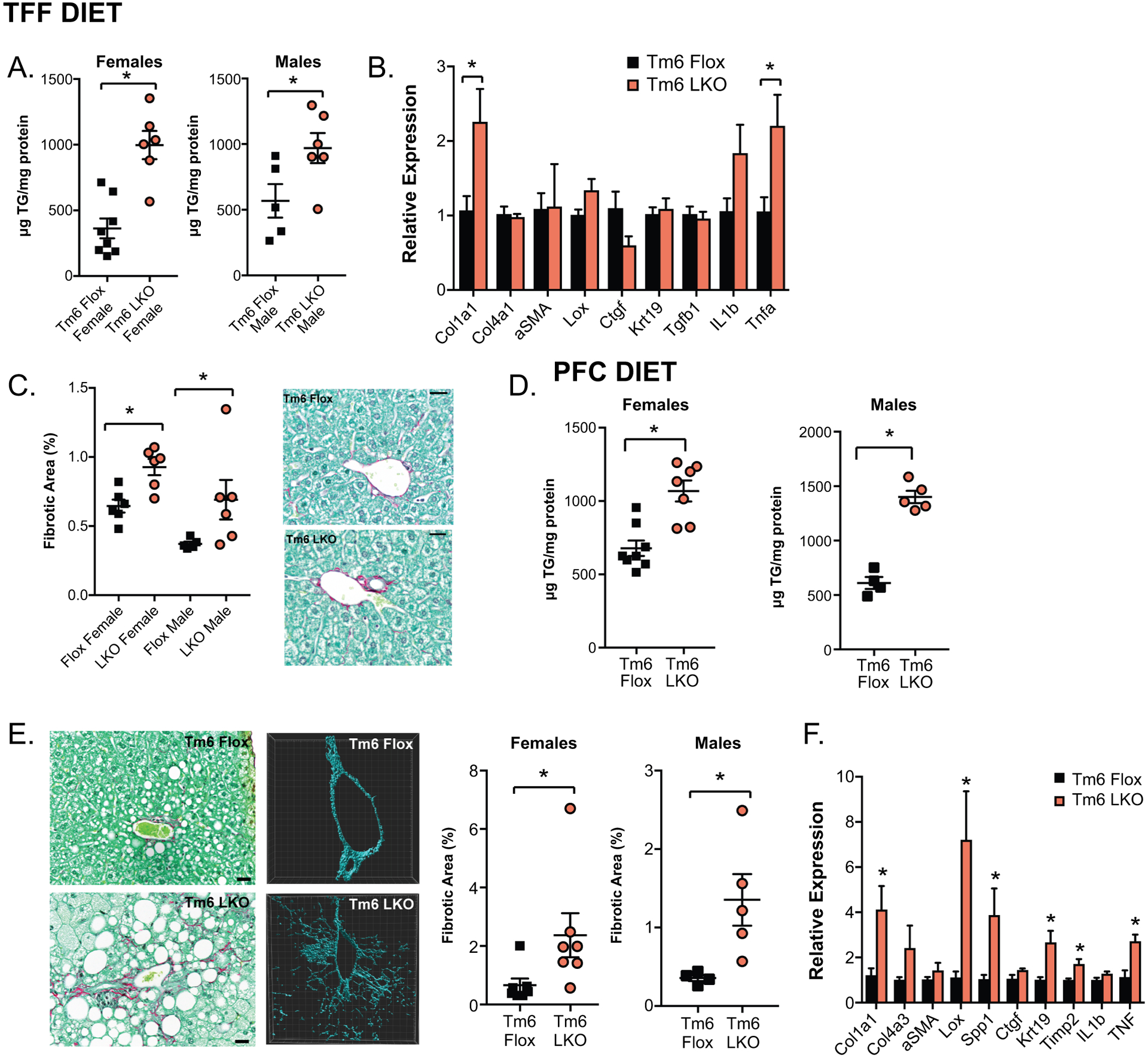
Impact of high fat, fibrogenic diet on Tm6 LKO mice. A. Hepatic triglyceride in male and female (left) Tm6 Flox and Tm6 LKO mice fed a transfat fructose (TFF) diet for 20 weeks (n=5–8/group). Similar findings were obtained using an independent line of Tm6 LKO mice (not shown) B. Fibrogenic and inflammatory gene expression in female mice fed TFF diet (n=5–6/genotype). C. Sirius red stained fibrotic area in male and female mice fed TFF diet (n=5–6/group), with representative images shown (40x, scale bar= 20μm). D. Hepatic triglyceride in male and female (left) mice fed a palm oil/ fructose/ cholesterol (PFC) containing diet for 20 weeks (n=5–8/group). E. Representative images of periportal fibrosis in PFC fed femaleTm6 Flox and LKO livers using Sirius red staining and second-harmonic generation microscopy (middle) to visualize collagen fibers. Fibrotic area (right) was quantitated from the Sirius red stained sections (n=7/genotype, scale bar = 20μm). F. Fibrogenic and inflammatory gene expression in livers of female Tm6 Flox and LKO mice fed PFC diet (n=5/genotype). For all panels, asterisks indicate p<0.05 versus flox controls.
Because fibrosis was most extensive in livers of mice fed the cholesterol enriched PFC diet, we measured total and free cholesterol in livers of mice fed various fibrogenic diets. Cholesterol levels were significantly higher in female Tm6 LKO mice fed HMFD and in Tm6 LKO males fed PFC compared to respective controls, with a trend to increase in all Tm6 LKO mice fed TFF diet and Tm6 LKO females fed PFC diet (Supplemental Table 2). Overall, we found that tissue cholesterol levels, both free and total, correlated with cholesterol content of the diet, and in general with levels of fibrosis in both Tm6 Flox and LKO mice. Together, these findings show that increased hepatic steatosis in Tm6 LKO mice is accompanied by increased fibrosis, as inferred from several different dietary models.
Altered susceptibility to HCC in Tm6 LKO mice:
Recent studies suggest that the rs58542926 Tm6sf2 variant may correlate not only with increased NAFLD but may also be linked to an increased incidence of HCC (11–13). To this end, we examined whether Tm6 LKO mice are more susceptible to HCC (initiation, progression) using two mouse tumorigenesis models. First, we employed a STAM model (32), in which mice develop steatosis and NASH by 12 weeks and HCC by 18 weeks. In our hands, mice that did not exhibit high blood glucose (>400mg/dL) at 6 weeks (~50% of flox mice) had smaller livers and significantly fewer tumors (Supplemental Figure 5). We therefore confined our observations to mice that met the threshold of hyperglycemia at 6 weeks of age. We found that STAM Tm6 LKO mice manifest increased hepatic steatosis at 12 weeks (Figure 6A), with increased numbers and size of visible tumors (Figure 6B), along with increased dysplasia and proliferation (Figure 6C). Tm6 LKO mice had increased body weight and liver weight at sacrifice (Supplemental Table 2), but no difference in serum glucose, ALT, or AST by genotype (ALT: STAM Tm6 flox, 137.6 ± 31.1 IU/L; STAM Tm6 LKO, 145.3 ± 30.4; AST: STAM Tm6 Flox, 66.9 ± 10.8 U/L; STAM Tm6 LKO, 74.6 ± 12.6; n=8–9/genotype) although ALT/AST levels were significantly increased in STAM animals compared to chow controls (Supplemental Table 1), consistent with increased NAFLD activity score (NAS). At 18 weeks, Tm6 LKO mice again exhibited increased steatosis (Figure 6D), with significantly increased body and liver weight (Supplemental Table 2). Tm6 LKO mice had increased tumor burden compared to Flox controls, assessed both visually at sacrifice and histologically by measurement of tumor area in H&E stained sections (Figure 6E–F).
Figure 6.
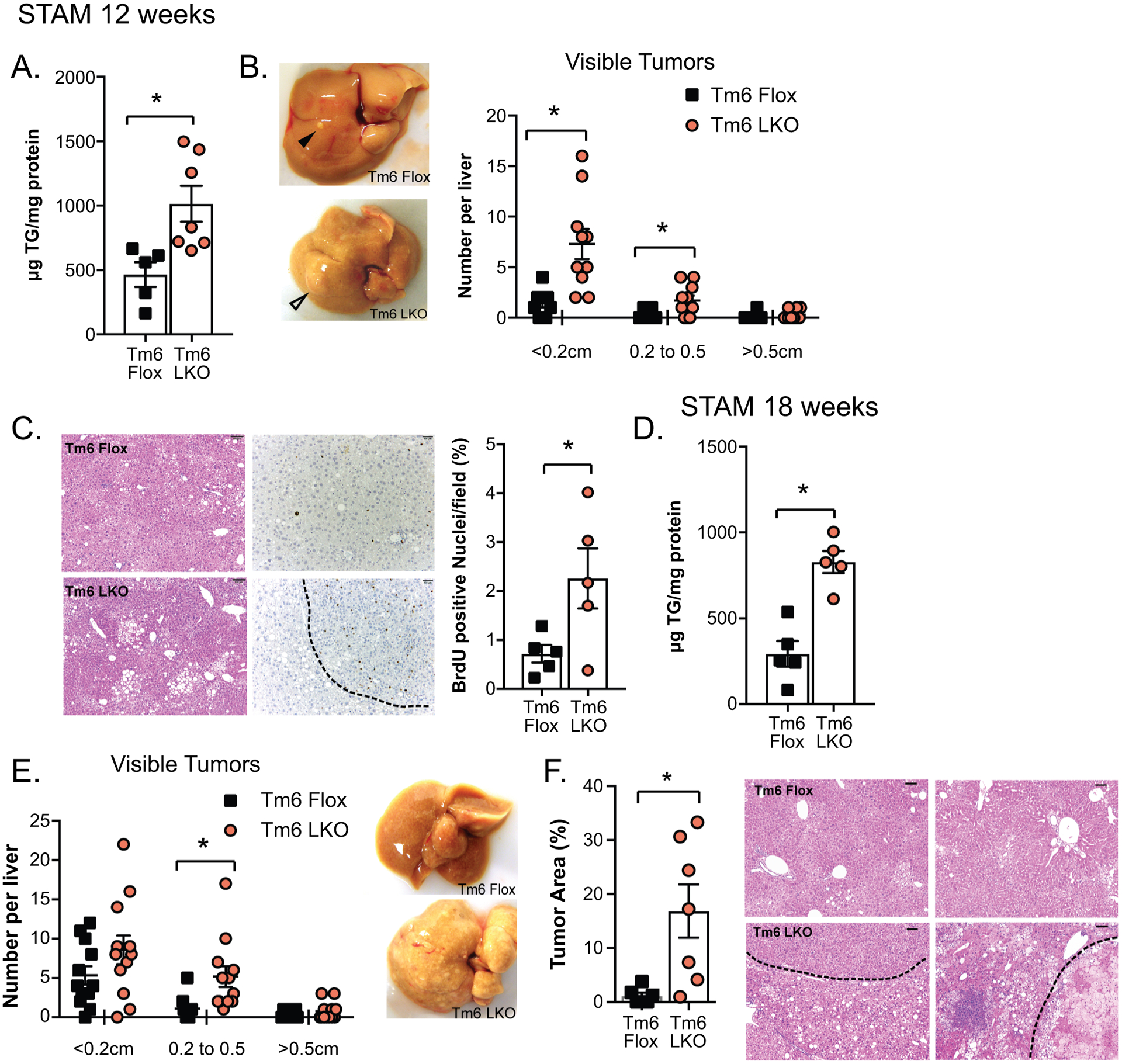
Effect of STAM model on hepatic tumorigenesis in Tm6 LKO mice. A. Hepatic triglyceride content in STAM model Tm6 Flox and Tm6 LKO mice at 12 weeks of age (n=5–7mice/genotype). B. Distribution of visible tumors in Tm6 Flox and LKO STAM mice at 12 weeks (n=10–12/genotype). Representative images are shown (right). C. H&E staining (left) and BrdU immunohistochemistry in STAM liver tissue (12 weeks) showing regions of dysplasia and proliferation (H&E images at 100x, scale bar = 100μM; BrdU images at 200x, scale bar= 50μM). Right panel shows BrdU positive nuclei, expressed as percent of total hepatocyte nuclei, per field. Nuclei were counted in at least 10 images per animal, taken from 3–4 distinct pieces of tissue. D. Hepatic triglyceride content in STAM model mice at 18 weeks of age (n=5/genotype). E. Distribution of visible tumors at 18 weeks of age. N=12/genotype. F. Tumor area expressed as a percent of total tissue area (n=6–7 mice/genotype, with 10–12 images per animal). Representative images are shown (100x, scale bar=100μM).
To investigate whether there is a correlation between tumorigenesis and inflammation, we surveyed the expression of several inflammatory genes (Il1b, Ccl2, Cxcl10 and Cd44) in control (chow diet) mice versus STAM, at several time points after streptozotocin (8, 12 and 18 weeks) and found increased expression of these genes in Tm6 LKO livers, particularly at the later time points, with a trend to increased expression at 8 weeks (Supplemental Figure 6A). F4/80 immunostaining at 12 weeks showed increased macrophage infiltration in a subset of Tm6 LKO livers (Supplemental Figure 6B). In contrast, hepatic fibrosis was not different by genotype, either histologically or in fibrogenic mRNA expression (Supplemental Figure 6C). These findings suggest that inflammation tends to be a feature of tumorigenic progression at later time points but further work will be required in order to determine the signaling pathways involved.
We also assessed HCC susceptibility using a second model in which male mice received postnatal injection of DEN followed by high fat, fructose feeding to promote tumorigenesis. At 4 months, Tm6 LKO mice again exhibited increased liver size and steatosis compared to Tm6 flox mice (Figure 7A), with a trend to increased tumor burden (Figure 7B). Hepatocyte BrdU incorporation was significantly increased in Tm6 LKO livers, suggesting increased proliferation (Figure 7C), though we observed minimal changes in proliferative gene expression (Supplemental Figure 6D). At 7 months, differences in liver size and steatosis by genotype were more dramatic (Figure 7D), with a significant increase in the number of visible tumors in each size range in Tm6 LKO mice (Figure 7E). Tumor area (H&E stained sections) was markedly increased, with increased proliferation as evidenced by BrdU incorporation (Figure 7F).
Figure 7.
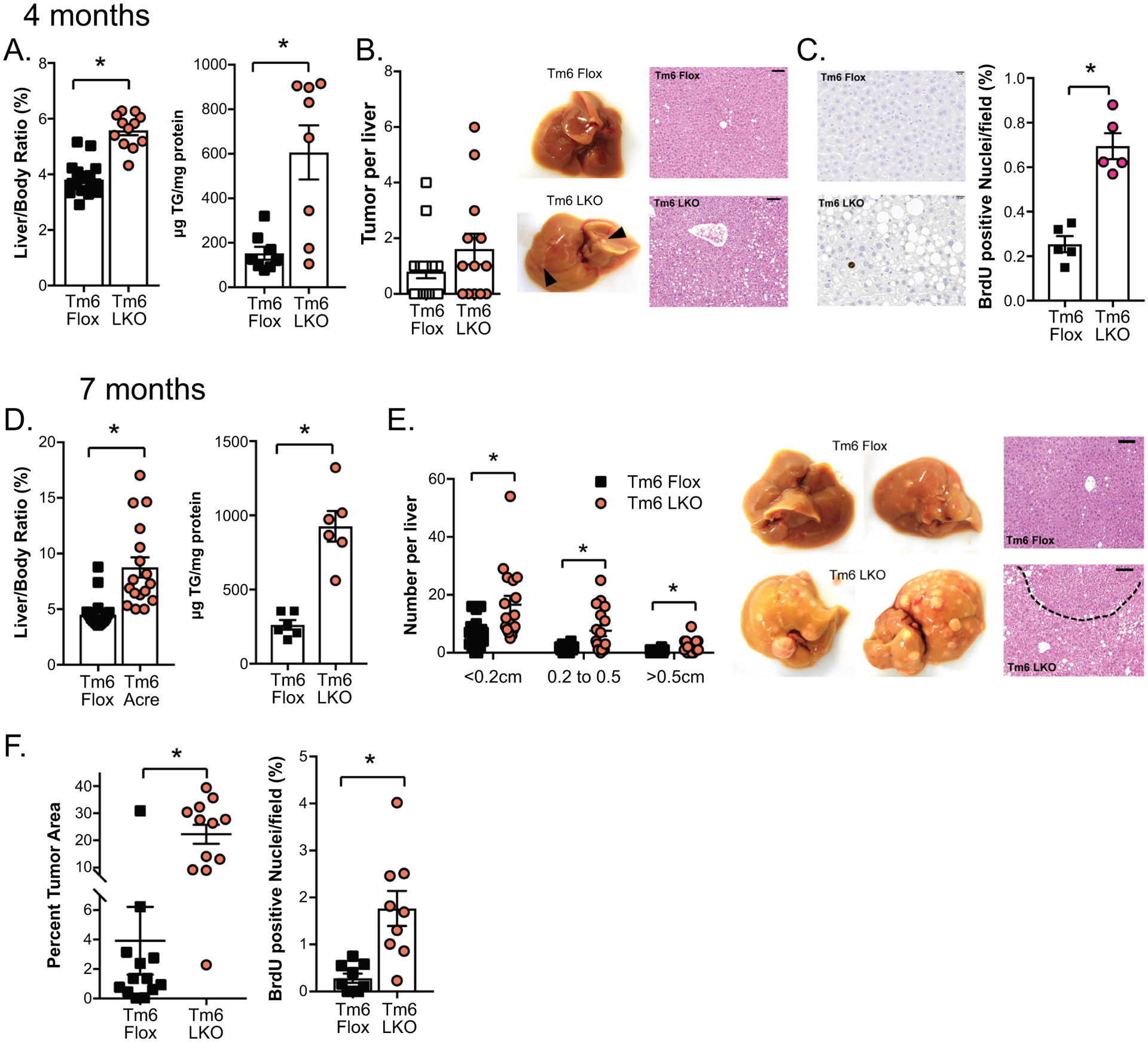
Effect of DEN induced tumorigenesis in Tm6 LKO mice. A. Liver size and triglyceride content in Tm6 Flox and Tm6 LKO mice in the DEN high fat diet model at 4 months of age (n=20 flox, 13 LKO for liver size; n=8/genotype for TG). B. Average number of visible tumors at 4 months (n=13–20/genotype), with representative livers and H&E images shown (100x, scale bar = 100μM). C. BrdU incorporation in liver tissue of DEN treated animals at 4 months. Left: BrdU images at 400x, scale bar=20μM. Right: Quantitation of BrdU positive hepatocytes, expressed as a percent of total hepatocyte nuclei per field. D. Liver size and triglyceride content of mice in DEN high fat model at 7 months of age (n=30 flox, 23 LKO for liver size; n=6/genotype for TG). E. Size distribution of visible tumors in DEN high fat mice at 7 months, with representative livers and H&E images shown (100x, scale bar=100μm). F. Quantitation of tumor area and BrdU positive nuclei in Tm6 Flox and Tm6 LKO mice in DEN high fat model at 7 months. For all panels, asterisks indicate p<0.05 vs controls.
HCC initiation and progression following AAV8 mediated rescue of Tm6:
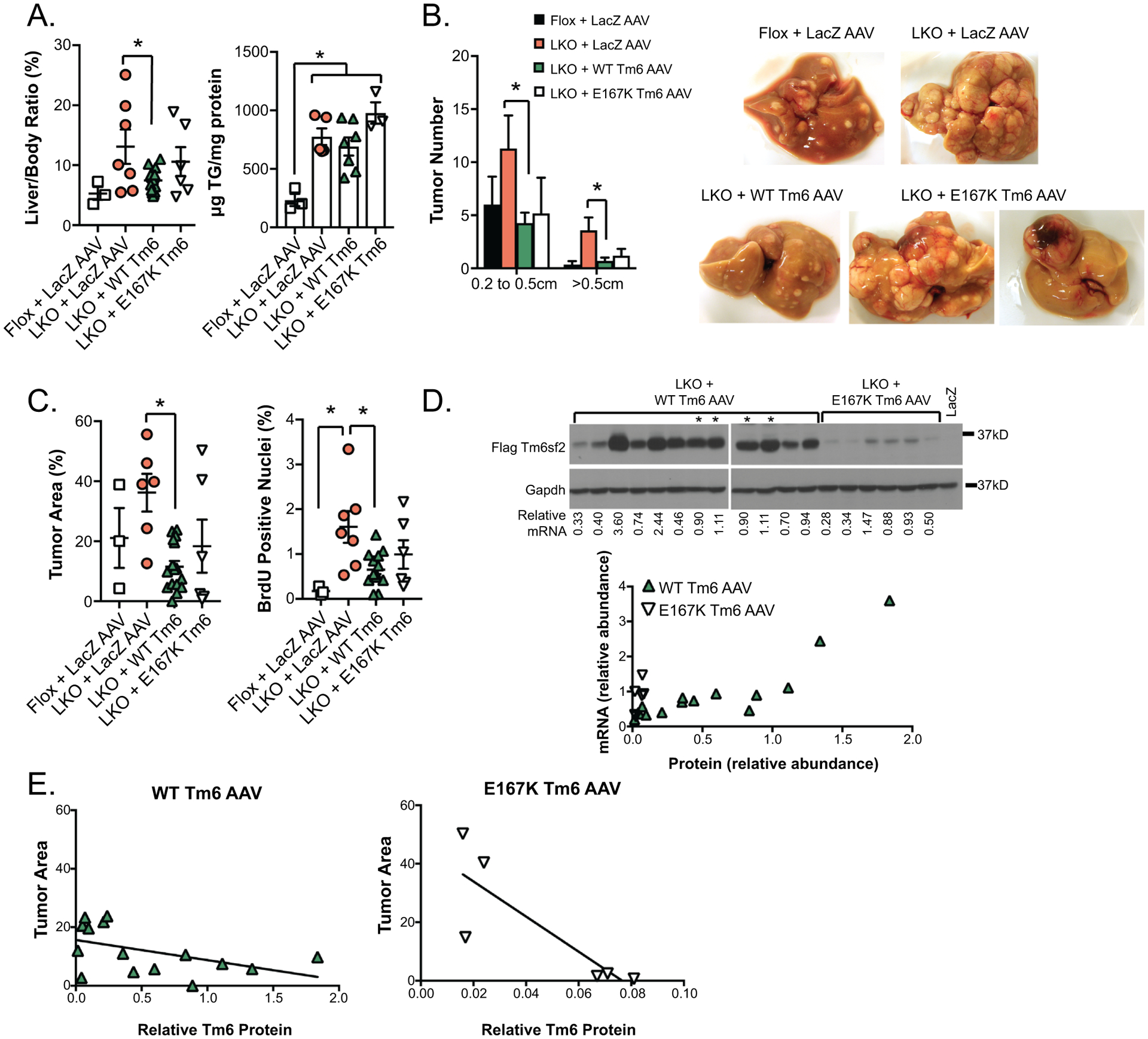
Based on our observations above regarding rescue of the hepatic steatosis phenotype with exogenous rescue, we asked whether long term expression of exogenous Tm6 could reduce DEN-induced tumor burden or progression in Tm6 LKO mice. As an additional control for these experiments, we found that transduction with AAV8-LacZ into Tm6 Flox or Tm6 LKO mice appeared to increase tumorigenesis, with increased liver size and overall tumor burden (compare Figure 8A vs Figure 7D). In comparison, Tm6 LKO mice transduced with WT Tm6 AAV had dramatically decreased liver size and tumor burden compared to LacZ AAV-injected Tm6 LKO mice (Figure 8A–C), but no reduction in hepatic TG content (Figure 8A, right panel). We observed an intermediate phenotype in Tm6 LKO mice transduced with AAV E167K particles (Figure 8B, C) where some mice had few tumors while others exhibited a significant tumor burden. To explain this observation, we examined Tm6 mRNA and protein expression in mice transduced with either WT or mutant AAV (Figure 8D) and found that protein expression was greatly, albeit variably, reduced in Tm6 LKO mice transduced with the E167K compared to the WT AAV8 construct, and independent of mRNA expression levels. Tumor burden in Tm6 LKO mice receiving either AAV8 WT or E167K particles was inversely correlated with expression of Tm6 protein (Figure 8E). These findings suggest that even low levels of Tm6 expression following AAV rescue mitigates tumor development and progression in DEN-treated Tm6 LKO mice.
Figure 8.
Rescue of DEN induced hepatic tumorigenesis with AAV8 delivery of Tm6sf2.
A. Liver size (left, normalized to body weight) and hepatic TG content in DEN high fat diet mice injected with AAV. N=3 Tm6 Flox + LacZ AAV, n=6–10 Tm6 LKO mice + AAV, sacrificed at 7 months. B. Distribution of larger visible tumors at 7 months. Tumors smaller than 2mm were too numerous to precisely count. Representative images of livers are shown, including livers of 2 Tm6 LKO + E167K AAV to show variability. C. Average tumor area (left) and BrdU incorporation in DEN high fat diet Tm6 Flox and Tm6 LKO mice + AAV at 7 months. D. Western blot with anti-FLAG antibody showing expression of Tm6sf2 protein in livers of DEN high fat Tm6 LKO mice injected with WT Tm6 or E167K Tm6 AAV. Gapdh protein is shown as a loading control. Relative exogenous Tm6sf2 mRNA levels are shown below the blot, with expression of both protein and mRNA normalized to expression in 2 reference samples (marked with asterisks on western blot, and loaded on both gels). In reference samples, exogenous Tm6sf2 mRNA is expressed at levels that are 60- and 74-fold higher (respectively) than endogenous Tm6sf2 (data not shown). Lower panel shows direct correlation between abundance of Tm6sf2 mRNA and protein in mice receiving WT AAV (closed triangles), but not in mice expression E167K AAV (open triangles). E. Inverse correlation between Tm6sf2 protein expression and tumor area in both Tm6 LKO + WT Tm6 AAV (left) and Tm6 LKO + E167K Tm6 AAV.
DISCUSSION
The current findings establish an unequivocal role for liver-specific deletion of Tm6sf2 in decreasing hepatic VLDL secretion and further show that Tm6 LKO mice exhibit increased fibrosis when challenged with high fat diets and exhibit accelerated development and progression of HCC. Our findings bring clarity to unresolved issues regarding loss-of-function alleles for Tm6sf2, where earlier reports from siRNA knockdown (7–9) found impaired VLDL secretion in mouse hepatocytes, suggesting that genetic deletion strategies would reveal an unambiguous loss-of-function phenotype. However, one report of germline Tm6sf2 deletion found no hepatic steatosis in knockout mice fed either chow or high fat diets (17), while another report demonstrated impaired VLDL secretion and hepatic steatosis (18), findings similar to those reported here. In addition, as alluded to above, none of those earlier studies undertook to examine the longer-term consequences of decreased Tm6sf2 expression, specifically the development of progressive features of NAFLD (inflammation, fibrosis) and the development of HCC. These are important questions in light of the findings that, among subjects with histologically confirmed NAFLD, those harboring the TM6SF2 rs58542926 variant exhibit greater steatosis, inflammation, ballooning and fibrosis even after adjusting for obesity, diabetes and age (14, 16, 33). In addition, other studies demonstrate that the TM6SF2 variant predisposes subjects to HCC in the setting of NAFLD (34) and alcohol use disorder associated liver disease (13). Accordingly, these findings have particular relevance because HCC is increasingly recognized as a complication of NAFLD (35).
Among the strengths of this study are that we undertook CRISPR deletion of Tm6sf2 and developed two distinct lines of liver-specific Tm6 knockout mice, characterized the loss-of-function phenotype in both sexes and confirmed our observations regarding steatosis and fibrosis using multiple different diets. Our finding that hepatic fibrosis in some of the dietary models tended to align with increased hepatic total and free cholesterol in Tm6 LKO mice is in agreement with other studies implicating cholesterol accumulation in the progression of murine NASH (36, 37). In addition, we confirmed our findings regarding HCC development using two different models of tumorigenesis. We believe this approach brings experimental rigor to our conclusions, several elements of which merit expanded discussion. We undertook to compare the phenotypes associated with impaired VLDL secretion caused by inactivation of Mttp (Mttp LKO) in which there is an almost complete block in VLDL assembly within the ER lumen (26) to the partial (~50%) impairment observed in Tm6 LKO mice. Those findings demonstrate subtle differences between the genotypes, including a shift to accumulation of smaller LDs and enhanced β-oxidation in Mttp LKO mice, though overall, we observed similar patterns of both triglyceride and other hepatic lipid species accumulation with inactivation of either Mttp or Tm6sf2. In attempting to reconcile the shift in LDs we examined expression of LD associated genes. We observed decreased mRNA abundance for Pnpla3 in both LKO genotypes, increased Cidec in Mttp LKO mice and increased Fltm1 in Tm6 LKO mice, the functional impact of which will require further study. However, we found no difference in Cideb mRNA expression in either LKO genotype. This was an important consideration because previous studies demonstrated that Cideb knockout mice exhibit impaired lipidation of hepatic VLDL with null mice secreting smaller, underlipidated particles but no change in APOB secretion (38). Accordingly, we conclude that the phenotypes observed in Tm6 LKO mice are unlikely to be explained by loss of Cideb. However, it is possible that Tm6sf2 deletion impairs the ability of nascent APOB to interact with ER membrane proteins (including Cideb) in the dynamic remodeling processes of VLDL assembly.
Our findings hint at there being a functional “threshold” on VLDL secretion capacity and hepatic steatosis where partial impairment, as observed in Tm6 LKO mice, produces an indistinguishable pattern of steatosis as observed in Mttp LKO mice, which exhibit almost complete impairment of VLDL secretion. This discrepancy may be linked to differences in the upregulation of FA oxidation in Mttp LKO vs Tm6 LKO mice. Our findings show several important distinctions between hepatic Tm6sf2 and Mttp deletion. Liver specific deletion of Tm6sf2 impacts APOB48 VLDL secretion more dramatically than APOB100 secretion in both chow and high fat diet fed mice (Figure 3F), yet still results in secretion of smaller VLDL particles in the background of either APOB100 or APOB48. Previous studies have shown that Mttp is involved in lipidating nascent APOB [reviewed in (39)] and that impaired expression or function of Mttp produces a disproportionate effect on APOB100 stability and secretion from hepatoma cells (28) and conditional lethality in intestinal Mttp knockout mice crossed into an APOB100-only background (40). Those earlier findings, considered with our current observations, suggest that Tm6sf2 functions in hepatic VLDL assembly at a point downstream of Mttp when the nascent APOB protein has been initially lipidated and a primordial VLDL particle is being formed. We would predict that combined deletion of both Mttp and Tm6sf2 would completely eliminate VLDL secretion, but that prediction is yet to be tested.
We undertook AAV8 mediated rescue of the Tm6 LKO phenotype, with an a priori expectation that the WT protein would rescue the defect in VLDL secretion and reverse the hepatic steatosis in Tm6 LKO mice. However, this prediction was not entirely fulfilled. Administration of either WT or mutant Tm6 reduced hepatic steatosis in high fat diet fed Tm6 LKO mice and reduced the mild fibrosis occurring in this model (Figure 4), yet produced almost no effect on VLDL secretion. Among the possibilities to explain this discrepancy is that our AAV8 rescue results in high levels of Tm6 protein, likely exceeding endogenous levels by orders of magnitude, which may be associated with a “dominant negative” effect on VLDL secretion as implied by other gain-of-function approaches (19). This explanation may at least partially account for the observation that mice with transgenic overexpression of WT Tm6sf2 appeared to develop hepatic steatosis with overexpression of cholesterogenic pathway genes (17).
Our findings highlight another important question in the function of Tm6sf2. Why does the mutant protein reduce hepatic steatosis and mitigate tumor development? We suspect, as previously noted, that the mutant protein is unstable because mRNA expression levels of (exogenous) WT and mutant Tm6 mRNAs were comparable (and considerably higher than endogenous), yet the mutant protein product was barely and variably detectable (Figures 4, 8). We speculate that the partial rescue of the tumorigenic phenotype in Tm6 LKO mice receiving AAV8 E167K Tm6 reflects low levels of functional Tm6 mutant protein, again suggesting a minimal “threshold” effect for rescue. However, as alluded to above, the mechanisms by which even low levels of Tm6sf2 mitigate tumor development in DEN treated Tm6 LKO mice remain to be explored. Likewise, the observation that tumor mitigation following AAV8 rescue appears independent of an effect on hepatic steatosis in those Tm6 LKO mice is also unexplained. We suspect that resolution of these unanswered issues will require an alternative genetic approach, similar to the more nuanced approaches taken with resolving the function of Pnpla3, where mice with germline deletion of Pnpla3 did not develop hepatic steatosis (41, 42) but a knockin allele with the variant Pnpla3 permitted elucidation of a pathogenic phenotype (43). Those and other questions regarding the function of Tm6sf2 as a genetic regulator of lipid export will be the focus of future reports.
Supplementary Material
Acknowledgements:
The authors acknowledge the Digestive Disease Research Core Center (DDRCC) at Washington University for assistance with histological analysis and imaging, the Genome Engineering and iPSC Center (GeiC) at Washington University for gRNA design and validation, and the Mouse Genetics core at Washington University for assistance with generation of conditional Tm6sf2 mice. We acknowledge William Moritz and Kamy Chong for technical assistance, and the Center for Cellular Imaging (WUCCI) for assistance with electron microscopy.
Financial support: This work was supported by grants from the Medical Research Council MC UP A90 1006, MC PC 13030 (to ZH, JLG) and National Institutes of Health grants DK-119437, DK-112378, Washington University Digestive Diseases Research Core Center P30 DK-52574 (to NOD). POB, GWS and JAJF are supported by the Washington University Center for Cellular Imaging (WUCCI) which is funded in part by Washington University School of Medicine, The Children’s Discovery Institute of Washington University and St. Louis Children’s Hospital (CDI-CORE-2015-505 and CDI-CORE-2019-813), the Foundation for Barnes-Jewish Hospital (3770), the Washington University Diabetes Research Center (DK020579), the Washington University Rheumatic Diseases Research Resource-based Center (AR073752) and Siteman Cancer Center of Barnes-Jewish Hospital and Washington University School of Medicine (CA091842).
ABBREVIATIONS:
- AAV8
adeno-associated virus, serotype 8
- APOB
apolipoproteinB
- BSA
bovine serum albumin
- BrdU
bromodeoxyuridine
- DAG
diacylglycerol
- DEN
diethyl nitrosamine
- EM
electron microscopy
- ER
endoplasmic reticulum
- FA
fatty acid
- FPLC
fast pressure liquid chromatography
- HCC
hepatocellular carcinoma
- HMFD
high milkfat diet
- LD
lipid droplet
- LysoPC
lysophosphatidylcholine
- MTTP
microsomal triglyceride transfer protein
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- PC
phosphatidylcholine
- PFC
palm oil, fructose, cholesterol diet
- TFF
transfat fructose diet
- TG
triglyceride
- TM6SF2
transmembrane 6 superfamily 2
- VLDL
very low density lipoprotein
- WT
wild type
REFERENCES
- 1.Cotter TG, Rinella M. Nonalcoholic Fatty Liver Disease 2020: The State of the Disease. Gastroenterology 2020;158:1851–1864. [DOI] [PubMed] [Google Scholar]
- 2.Sookoian S, Pirola CJ, Valenti L, Davidson NO. Genetic Pathways in Nonalcoholic Fatty Liver Disease: Insights From Systems Biology. Hepatology 2020;72:330–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Trepo E, Valenti L. Update on NAFLD genetics: From new variants to the clinic. J Hepatol 2020;72:1196–1209. [DOI] [PubMed] [Google Scholar]
- 4.Krawczyk M, Liebe R, Lammert F. Toward Genetic Prediction of Nonalcoholic Fatty Liver Disease Trajectories: PNPLA3 and Beyond. Gastroenterology 2020;158:1865–1880 e1861. [DOI] [PubMed] [Google Scholar]
- 5.Di Filippo M, Moulin P, Roy P, Samson-Bouma ME, Collardeau-Frachon S, Chebel-Dumont S, Peretti N, et al. Homozygous MTTP and APOB mutations may lead to hepatic steatosis and fibrosis despite metabolic differences in congenital hypocholesterolemia. J Hepatol 2014;61:891–902. [DOI] [PubMed] [Google Scholar]
- 6.Welty FK. Hypobetalipoproteinemia and abetalipoproteinemia: liver disease and cardiovascular disease. Curr Opin Lipidol 2020;31:49–55. [DOI] [PubMed] [Google Scholar]
- 7.Holmen OL, Zhang H, Fan Y, Hovelson DH, Schmidt EM, Zhou W, Guo Y, et al. Systematic evaluation of coding variation identifies a candidate causal variant in TM6SF2 influencing total cholesterol and myocardial infarction risk. Nat Genet 2014;46:345–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kozlitina J, Smagris E, Stender S, Nordestgaard BG, Zhou HH, Tybjaerg-Hansen A, Vogt TF, et al. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2014;46:352–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mahdessian H, Taxiarchis A, Popov S, Silveira A, Franco-Cereceda A, Hamsten A, Eriksson P, et al. TM6SF2 is a regulator of liver fat metabolism influencing triglyceride secretion and hepatic lipid droplet content. Proc Natl Acad Sci U S A 2014;111:8913–8918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pirola CJ, Sookoian S. The dual and opposite role of the TM6SF2-rs58542926 variant in protecting against cardiovascular disease and conferring risk for nonalcoholic fatty liver: A meta-analysis. Hepatology 2015;62:1742–1756. [DOI] [PubMed] [Google Scholar]
- 11.Donati B, Dongiovanni P, Romeo S, Meroni M, McCain M, Miele L, Petta S, et al. MBOAT7 rs641738 variant and hepatocellular carcinoma in non-cirrhotic individuals. Sci Rep 2017;7:4492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stickel F, Buch S, Nischalke HD, Weiss KH, Gotthardt D, Fischer J, Rosendahl J, et al. Genetic variants in PNPLA3 and TM6SF2 predispose to the development of hepatocellular carcinoma in individuals with alcohol-related cirrhosis. Am J Gastroenterol 2018;113:1475–1483. [DOI] [PubMed] [Google Scholar]
- 13.Yang J, Trepo E, Nahon P, Cao Q, Moreno C, Letouze E, Imbeaud S, et al. PNPLA3 and TM6SF2 variants as risk factors of hepatocellular carcinoma across various etiologies and severity of underlying liver diseases. Int J Cancer 2019;144:533–544. [DOI] [PubMed] [Google Scholar]
- 14.Liu YL, Reeves HL, Burt AD, Tiniakos D, McPherson S, Leathart JB, Allison ME, et al. TM6SF2 rs58542926 influences hepatic fibrosis progression in patients with non-alcoholic fatty liver disease. Nat Commun 2014;5:4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wong VW, Wong GL, Tse CH, Chan HL. Prevalence of the TM6SF2 variant and non-alcoholic fatty liver disease in Chinese. J Hepatol 2014;61:708–709. [DOI] [PubMed] [Google Scholar]
- 16.Sookoian S, Castano GO, Scian R, Mallardi P, Fernandez Gianotti T, Burgueno AL, San Martino J, et al. Genetic variation in transmembrane 6 superfamily member 2 and the risk of nonalcoholic fatty liver disease and histological disease severity. Hepatology 2015;61:515–525. [DOI] [PubMed] [Google Scholar]
- 17.Fan Y, Lu H, Guo Y, Zhu T, Garcia-Barrio MT, Jiang Z, Willer CJ, et al. Hepatic Transmembrane 6 Superfamily Member 2 Regulates Cholesterol Metabolism in Mice. Gastroenterology 2016;150:1208–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smagris E, Gilyard S, BasuRay S, Cohen JC, Hobbs HH. Inactivation of Tm6sf2, a Gene Defective in Fatty Liver Disease, Impairs Lipidation but Not Secretion of Very Low Density Lipoproteins. J Biol Chem 2016;291:10659–10676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ehrhardt N, Doche ME, Chen S, Mao HZ, Walsh MT, Bedoya C, Guindi M, et al. Hepatic Tm6sf2 overexpression affects cellular ApoB-trafficking, plasma lipid levels, hepatic steatosis and atherosclerosis. Hum Mol Genet 2017;26:2719–2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xie Y, Newberry EP, Young SG, Robine S, Hamilton RL, Wong JS, Luo J, et al. Compensatory increase in hepatic lipogenesis in mice with conditional intestine-specific Mttp deficiency. J Biol Chem 2006;281:4075–4086. [DOI] [PubMed] [Google Scholar]
- 21.Newberry EP, Xie Y, Kennedy SM, Graham MJ, Crooke RM, Jiang H, Chen A, et al. Prevention of hepatic fibrosis with liver microsomal triglyceride transfer protein deletion in liver fatty acid binding protein null mice. Hepatology 2017;65:836–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hirano K, Young SG, Farese RV Jr., Ng J, Sande E, Warburton C, Powell-Braxton LM, et al. Targeted disruption of the mouse apobec-1 gene abolishes apolipoprotein B mRNA editing and eliminates apolipoprotein B48. J Biol Chem 1996;271:9887–9890. [DOI] [PubMed] [Google Scholar]
- 23.Takakura K, Koido S, Fujii M, Hashiguchi T, Shibazaki Y, Yoneyama H, Katagi H, et al. Characterization of non-alcoholic steatohepatitis-derived hepatocellular carcinoma as a human stratification model in mice. Anticancer Res 2014;34:4849–4855. [PubMed] [Google Scholar]
- 24.Heindryckx F, Colle I, Van Vlierberghe H. Experimental mouse models for hepatocellular carcinoma research. Int J Exp Pathol 2009;90:367–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen A, Tang Y, Davis V, Hsu FF, Kennedy SM, Song H, Turk J, et al. Liver fatty acid binding protein (L-Fabp) modulates murine stellate cell activation and diet-induced nonalcoholic fatty liver disease. Hepatology 2013;57:2202–2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Raabe M, Veniant MM, Sullivan MA, Zlot CH, Bjorkegren J, Nielsen LB, Wong JS, et al. Analysis of the role of microsomal triglyceride transfer protein in the liver of tissue-specific knockout mice. J Clin Invest 1999;103:1287–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davidson NO, Shelness GS. APOLIPOPROTEIN B: mRNA editing, lipoprotein assembly, and presecretory degradation. Annu Rev Nutr 2000;20:169–193. [DOI] [PubMed] [Google Scholar]
- 28.Kulinski A, Rustaeus S, Vance JE. Microsomal triacylglycerol transfer protein is required for lumenal accretion of triacylglycerol not associated with ApoB, as well as for ApoB lipidation. J Biol Chem 2002;277:31516–31525. [DOI] [PubMed] [Google Scholar]
- 29.Wang X, Zheng Z, Caviglia JM, Corey KE, Herfel TM, Cai B, Masia R, et al. Hepatocyte TAZ/WWTR1 Promotes Inflammation and Fibrosis in Nonalcoholic Steatohepatitis. Cell Metab 2016;24:848–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chang PE, Goh GBB, Leow WQ, Shen L, Lim KH, Tan CK. Second harmonic generation microscopy provides accurate automated staging of liver fibrosis in patients with non-alcoholic fatty liver disease. PLoS One 2018;13:e0199166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gailhouste L, Le Grand Y, Odin C, Guyader D, Turlin B, Ezan F, Desille Y, et al. Fibrillar collagen scoring by second harmonic microscopy: a new tool in the assessment of liver fibrosis. J Hepatol 2010;52:398–406. [DOI] [PubMed] [Google Scholar]
- 32.Saito K, Uebanso T, Maekawa K, Ishikawa M, Taguchi R, Nammo T, Nishimaki-Mogami T, et al. Characterization of hepatic lipid profiles in a mouse model with nonalcoholic steatohepatitis and subsequent fibrosis. Sci Rep 2015;5:12466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dongiovanni P, Petta S, Maglio C, Fracanzani AL, Pipitone R, Mozzi E, Motta BM, et al. Transmembrane 6 superfamily member 2 gene variant disentangles nonalcoholic steatohepatitis from cardiovascular disease. Hepatology 2015;61:506–514. [DOI] [PubMed] [Google Scholar]
- 34.Pelusi S, Baselli G, Pietrelli A, Dongiovanni P, Donati B, McCain MV, Meroni M, et al. Rare Pathogenic Variants Predispose to Hepatocellular Carcinoma in Nonalcoholic Fatty Liver Disease. Sci Rep 2019;9:3682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dongiovanni P, Romeo S, Valenti L. Hepatocellular carcinoma in nonalcoholic fatty liver: role of environmental and genetic factors. World J Gastroenterol 2014;20:12945–12955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kerr TA, Davidson NO. Cholesterol and nonalcoholic fatty liver disease: renewed focus on an old villain. Hepatology 2012;56:1995–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tomita K, Teratani T, Suzuki T, Shimizu M, Sato H, Narimatsu K, Okada Y, et al. Free cholesterol accumulation in hepatic stellate cells: mechanism of liver fibrosis aggravation in nonalcoholic steatohepatitis in mice. Hepatology 2014;59:154–169. [DOI] [PubMed] [Google Scholar]
- 38.Ye J, Li JZ, Liu Y, Li X, Yang T, Ma X, Li Q, et al. Cideb, an ER- and lipid droplet-associated protein, mediates VLDL lipidation and maturation by interacting with apolipoprotein B. Cell Metab 2009;9:177–190. [DOI] [PubMed] [Google Scholar]
- 39.Hussain MM, Rava P, Walsh M, Rana M, Iqbal J. Multiple functions of microsomal triglyceride transfer protein. Nutr Metab (Lond) 2012;9:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xie Y, Luo J, Kennedy S, Davidson NO. Conditional intestinal lipotoxicity in Apobec-1−/− Mttp-IKO mice: a survival advantage for mammalian intestinal apolipoprotein B mRNA editing. J Biol Chem 2007;282:33043–33051. [DOI] [PubMed] [Google Scholar]
- 41.Chen W, Chang B, Li L, Chan L. Patatin-like phospholipase domain-containing 3/adiponutrin deficiency in mice is not associated with fatty liver disease. Hepatology 2010;52:1134–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Basantani MK, Sitnick MT, Cai L, Brenner DS, Gardner NP, Li JZ, Schoiswohl G, et al. Pnpla3/Adiponutrin deficiency in mice does not contribute to fatty liver disease or metabolic syndrome. J Lipid Res 2011;52:318–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.BasuRay S, Smagris E, Cohen JC, Hobbs HH. The PNPLA3 variant associated with fatty liver disease (I148M) accumulates on lipid droplets by evading ubiquitylation. Hepatology 2017;66:1111–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.