

Abstract
Cardiac muscle contraction is driven by the molecular motor myosin, which uses the energy from ATP hydrolysis to generate a power stroke when interacting with actin filaments, although it is unclear how this mechanism is impaired by mutations in myosin that can lead to heart failure. We have applied a fluorescence resonance energy transfer (FRET) strategy to investigate structural changes in the lever arm domain of human β-cardiac myosin subfragment 1 (M2β-S1). We exchanged the human ventricular regulatory light chain labeled at a single cysteine (V105C) with Alexa 488 onto M2β-S1, which served as a donor for Cy3ATP bound to the active site. We monitored the FRET signal during the actin-activated product release steps using transient kinetic measurements. We propose that the fast phase measured with our FRET probes represents the macroscopic rate constant associated with actin-activated rotation of the lever arm during the power stroke in M2β-S1. Our results demonstrated M2β-S1 has a slower actin-activated power stroke compared with fast skeletal muscle myosin and myosin V. Measurements at different temperatures comparing the rate constants of the actin-activated power stroke and phosphate release are consistent with a model in which the power stroke occurs before phosphate release and the two steps are tightly coupled. We suggest that the actin-activated power stroke is highly reversible but followed by a highly irreversible phosphate release step in the absence of load and free phosphate. We demonstrated that hypertrophic cardiomyopathy (R723G)- and dilated cardiomyopathy (F764L)-associated mutations both reduced actin activation of the power stroke in M2β-S1. We also demonstrate that both mutations alter in vitro actin gliding in the presence and absence of load. Thus, examining the structural kinetics of the power stroke in M2β-S1 has revealed critical mutation-associated defects in the myosin ATPase pathway, suggesting these measurements will be extremely important for establishing structure-based mechanisms of contractile dysfunction.
Significance
Mutations in human β-cardiac myosin are known to cause various forms of heart disease, although it is unclear how the mutations lead to contractile dysfunction and pathogenic remodeling of the heart. In this study, we investigated two mutations with opposing phenotypes and examined their impact on ATPase cycle kinetics, structural changes associated with the myosin power stroke, and ability to slide actin filaments. We found that both mutations impair actin activation of the myosin power stroke and actin filament sliding in the presence and absence of load. Our results provide a structural basis for how mutations disrupt molecular-level contractile dysfunction.
Introduction
The myosin superfamily shares a highly conserved ATPase cycle that efficiently converts the energy from ATP hydrolysis into mechanical force (1,2). One central question that continues to be debated in the field is how specific steps in the ATPase cycle are coupled to force generation. It is well understood that when myosin containing the hydrolyzed products in its active site (ADP and Pi) binds to actin, this triggers structural rearrangements in the myosin motor that result in force generation and product release (3, 4, 5). However, the precise timing, as well as the sequence of events associated with force generation and product release, still remains elusive. Characterizing the structural and biochemical coupling between those critical steps is crucial to understand the myosin energy transduction mechanism.
The lever arm hypothesis suggests that force generation occurs when structural changes in the active site and actin-binding regions are communicated to the lever arm domain, a long α-helix that extends from the motor domain that is stabilized by calmodulin-like light chains (3). The lever arm is known to undergo an ∼70° rotation that can produce force when myosin is tightly bound to actin. The lever arm rotation is thought to be coupled to actin-activated product release, especially phosphate release, which can be accelerated more than 1000-fold in the presence of actin (6). Different experimental techniques have been applied to study the structural and biochemical coupling in myosin, including single-molecule mechanics, x-ray crystallography, fluorescence spectroscopy, and cryo-electron microscopy (7, 8, 9, 10, 11, 12, 13). Additionally, fluorescence resonance energy transfer (FRET) is a powerful approach to monitor the structural dynamics of lever arm rotation by engineering site-directed probes into the myosin molecule. FRET has been successfully utilized in several myosin isoforms including myosin V, skeletal muscle myosin, and bovine cardiac myosin to investigate the structural kinetics of the lever arm rotation (14, 15, 16, 17, 18, 19). Specifically, FRET studies have monitored the macroscopic rate constant associated with actin-activated lever arm rotation, which comprises several reaction steps including attachment to actin, the transition from weak to strong actin binding, and rotation of the lever arm (referred to as actin-activated power stroke hereafter). Therefore, although the lever arm rotation associated with the power stroke occurs on a microsecond timescale (11), as with many conformational changes in proteins (3), the macroscopic rate constant associated with the actin-activated power stroke occurs on a millisecond timescale and can be measured with conventional stopped-flow methods. The previous FRET results demonstrated that different myosin isoforms could display different biochemical properties of the actin-activated power stroke, possibly tuning them for a specific physiological role. For example, the actin-activated power-stroke rate constants measured in myosin V and skeletal muscle myosin are much faster than bovine cardiac myosin (14,15,17,19). Therefore, it is important to perform FRET measurements of the actin-activated power stroke in human β-cardiac myosin to better understand its force-generating properties, which will provide fundamental insights into understanding cardiac myosin motor performance in health and disease.
Currently, more than 300 pathogenic single missense mutations have been reported clinically in the gene MYH7, encoding human β-cardiac myosin heavy chain (M2β) (20). Mutations in MYH7 are commonly associated with hypertrophic cardiomyopathy (HCM) and dilated cardiomyopathy (DCM), although detailed molecular mechanisms of those mutations are still unclear. Understanding the molecular basis of disease-causing mutations will shed light on elucidating the structural and biochemical coupling between subdomains within the myosin molecule, as well as identifying potential therapeutic interventions. R723G and F764L are cardiomyopathy mutations both located in the converter domain, a region important for communication between the active site and lever arm, of M2β and clinically associated with HCM and DCM, respectively (21,22). The converter domain is a hot spot for cardiomyopathy mutations in M2β and is also the load-sensing region that undergoes most of the elastic distortion during the power stroke (23,24). Therefore, mutations in the converter domain are hypothesized to be prone to alter the force-generating properties of M2β.
The biochemical impact of the F764L mutation in human M2β was well characterized together by previous work and our recent publication (25, 26, 27, 28, 29). Overall, F764L causes slightly reduced motor properties in M2β subfragment 1 (S1), containing the motor domain and entire light-chain-binding region, by slowing the actin-activated phosphate release and ADP release steps (25). Kinetic simulation results demonstrate that the duty ratio (fraction of ATPase cycle that myosin is in the force-generating states) is slightly decreased or not significantly altered by F764L in unloaded conditions (25,26). The R723G mutation has been studied in human soleus muscle biopsy samples and isolated human cardiomyocytes (30, 31, 32). Interestingly, Kraft et al. (33) reported that the R723G mutation caused a reduced maximal force and similar Ca2+ sensitivity in isolated human cardiomyocytes but higher maximal force and lower Ca2+ sensitivity in human soleus muscle fibers. Kawana et al. (34) characterized the R723G mutant using the M2β-shortS1 construct (containing the motor domain and essential light-chain-binding region). Interestingly, the HCM R723G mutation causes a decrease in maximal ATPase activity and intrinsic force but a 5–10% increase in actin sliding velocity. The impact of the R723G mutation on the detailed kinetics of key steps in the myosin ATPase was recently characterized by Vera et al. (35). They demonstrated that R723G increased the ADP release rate constant and weakened the ATP binding affinity, which resulted in a slight decrease in the duty ratio.
In this study, we successfully monitored the structural kinetics of the actin-activated power stroke using site-specific FRET probes engineered into human M2β-S1. Our data suggest that human cardiac myosin has a relatively slow actin-activated power stroke similar to that measured in bovine cardiac myosin, and the actin-activated power stroke is tightly coupled to the phosphate release step. Our results clearly support a model in which the actin-activated power stroke occurs before phosphate release. In addition, we examined the impact of the R723G mutation on the key steps of the myosin ATPase cycle with transient kinetic measurements and found that R723G causes an increase in the ADP release rate constant. We further examined the effect of R723G and F764L mutations on the actin-activated power stroke. Despite the different clinical phenotypes observed in patients, both R723G and F764L cause a significant decrease in actin activation of the power stroke. We also demonstrated the R723G and F764L mutants alter actin filament gliding in the in vitro motility assay and the sensitivity to load. Our results provide important insight into the molecular mechanisms of cardiomyopathy mutations and the structural basis of the actin-activated power stroke in cardiac myosin.
Materials and methods
Reagents
Cy3ATP and Cy3ADP were purchased from Jena Bioscience (1 mM stock; Jena, Germany). Alexa Fluor 488 C5 Maleimide powder was purchased from Invitrogen (Carlsbad, CA) and dissolved in dimethyl sulfoxide (DMSO). The 2′-deoxy-ADP labeled with N-methylanthraniloyl at the 3′-ribose position (mantADP) was purchased from Jena Biosciences. ATP and ADP were prepared from powder (MilliporeSigma, Burlington, MA) and concentrations were determined by absorbance at 259 nm (ε259 = 15,400 M−1 cm−1). The fluorescently labeled phosphate binding protein (MDCC-PBP) was prepared as described (36). MOPS 20 buffer (10 mM MOPS, 20 mM KCl, 1 mM EGTA, 1 mM MgCl2, and 1 mM dithiothreitol [DTT],pH 7.0) was used for all solution experiments.
Protein construction, expression, and purification
We cloned the complementary DNA (cDNA) of M2β-S1 (841 aa) into a pShuttle vector that contains a C-terminal Avi tag and N-terminal FLAG tag (DYKDDDDK). The R723G and F764L point mutations were introduced using Quikchange site-directed mutagenesis (Stratagene, La Jolla, CA). Recombinant adenovirus was produced and used to infect C2C12 cells as previously described (25,37). Cells were harvested 7–10 days after infection and purified with FLAG affinity chromatography. Endogenous mouse regulatory light chain (RLC) was removed from M2β-S1 using an on-column stripping method described previously (38). Briefly, M2β-S1 was bound to the FLAG column, by first flowing through cell lysate and washing the resin with wash buffer (10 mM Tris pH 7.5, 200 mM KCl, 1 mM EGTA, 1 mM EDTA, 2 mM MgCl2, 2 mM ATP, 1 mM DTT, 0.01 mg/mL aprotinin, 0.01 mg/mL leupeptin, and 1 mM phenylmethylsulfonyl fluoride [PMSF]), and then incubated with stripping buffer (20 mM Tris pH 7.5, 0.5% Triton X-100, 200 mM KCl, 5 mM CDTA, 2 mM ATP, 1 mM DTT, 0.01 mg/mL aprotinin, 0.01 mg/mL leupeptin, and 1 mM PMSF) for 1 h, followed by incubation with 20 μM Alexa 488-labeled human RLC (A488RLC) in wash buffer to allow the exchange to occur. The exchanged M2β-S1 was washed again to remove exogenous A488RLC and eluted with FLAG peptide. About 0.5–1 mg of protein was obtained per 20 plates of C2C12 cells. M2β-S1 used for the in vitro motility assays was biotinylated with BirA (10 μg/mL, Avidity Biosciences, La Jolla, CA) as described, followed by ammonium sulfate precipitation (25). An N-terminally His-tagged version of the human ventricular cardiac regulatory light chain (hRCL) with a single reactive cysteine at position 105 was expressed using the pET15b vector in Escherichia coli cells. The cell lysate was collected and hRLC purified with Talon-Affinity chromatography (His tag). The purified hRLC was labeled with 5 molar excess Alexa Fluor 488 overnight at 4°C and dialyzed into wash buffer to remove excess dye. The labeled A488RLC was snap frozen in liquid nitrogen and kept at −80°C until use. Actin was purified using acetone powder from rabbit skeletal muscle (39) and labeled with pyrene iodoacetamide when needed (40). All proteins were dialyzed in MOPS 20 buffer overnight before experiments.
Steady-state ATPase measurements
0.1 μM M2β-S1 in the presence of varying actin concentrations (0, 5, 10, 20, 40, and 60 μM) was examined in actin-activated ATPase experiments using an NADH-coupled ATP regenerating system as previously described (41,42). Data were acquired for 200 s (0.2 s intervals) at 25°C using an Applied Photophysics (Surrey, UK) stopped-flow device. The ATPase rate was plotted as a function of actin concentration and fitted to the Michaelis-Menten equation to determine the kcat and KATPase. Results of wild-type (WT) and two mutants were reported as the average of three to six protein preparations. Statistical analysis was done with unpaired Student’s t-tests to determine the impact of A488RLC exchange and compare each mutant with WT.
In vitro motility
The actin sliding velocity was measured as previously described (25). 1% nitrocellulose-coated coverslips were incubated with streptavidin (0.1 mg/mL) and blocked with bovine serum albumin (2 mg/mL). Biotinylated M2β-S1 WT or R723G or F764L was attached to the surface at 0.24 μM loading concentration. Sheared unlabeled actin (2 μM) followed by 2 mM ATP were added to block the dead myosin heads on the surface. The activation buffer containing MOPS 20 buffer, 0.35% methylcellulose, 0.45 mM phosphoenolpyruvate, 45 units/mL pyruvate kinase, 0.1 mg/mL glucose oxidase, 5 mg/mL glucose, 0.018 mg/mL catalase, and 2 mM ATP was added right before video acquisition. Alexa 488 or Rhodamine 555 phalloidin-labeled actin was visualized with a Nikon TE2000 fluorescence microscope (Nikon, Tokyo, Japan). Videos were collected for 2 min at a 1 s frame rate. The velocities were either manually analyzed by tracking actin filaments using MTrackJ in ImageJ (43) or processed by the program FAST (44). Unpaired Student’s t-tests were performed to compare each mutant with WT and the impact of A488RLC exchange.
In the loaded in vitro motility assay, the M2β-S1 motor-dead mutant (E466A) protein was mixed with WT, R723G, or F764L before being added to the coverslip, and the total amount of M2β-S1 loaded remained constant at 0.6 μM. Added load in the assay was estimated by using the equations outlined in Greenberg and Moore (45) and the myosin surface density estimated with NH4 ATPase assays (41). Relative velocities were normalized to the velocity at 0% motor dead to compare the relative decrease in velocity as a function of load.
Transient kinetic measurements
An Applied Photophysics stopped-flow device equipped with an excitation monochromator, 1.2 ms dead-time, and a 9.3 nm band pass was used for all transient kinetic experiments. Tryptophan fluorescence was excited at 290 nm and monitored with a 320 nm long-pass emission filter. The mant fluorescence was examined with 290 nm excitation and a 395 nm long-pass emission filter. Pyrene actin fluorescence was monitored with 365 nm excitation and a 395 nm long-pass emission filter. MDCC-PBP was monitored with a 380 nm excitation and a 425 nm long-pass emission filter. Fluorescence transients were fitted to the sum of exponentials using the stopped-flow program or GraphPad Prism. For example, the following function was used to fit fluorescence enhancements: F(t) = F∞ + , where F(t) is the fluorescence as a function of time t, F∞ is the intensity at infinity, Ai is the amplitude, ki is the observed rate constant characterized by the i-th transition, and n is the total number of observed transitions. To fit fluorescence enhancement transients that contain a lag phase, a double-exponential function was used, but with the lag component given a positive amplitude (46,47).
FRET measurements
The steady-state FRET was measured by exciting the sample at 460 nm and measuring the fluorescence emission as a function of wavelength (480–650 nm) using a PTI spectrofluorometer equipped with excitation and emission monochromators and a Peltier temperature control cuvette holder. Stopped-flow FRET was examined by monitoring the change in donor fluorescence (Alexa 488) using an excitation wavelength of 470 nm and measuring the emission with an interference filter (500–525 nm), which eliminated background fluorescence from Cy3ATP or Cy3ADP. The steady-state FRET efficiency (E) was calculated by examining donor quenching using the following equation (48,49),
where FDA is the donor fluorescence intensity in the presence of acceptor, FD the donor fluorescence intensity in the absence of acceptor, and fD is the fractional labeling with donor. The fraction of M2β-S1 with Cy3 nucleotide bound (fD) was determined from the KD, estimated from the stopped-flow measurements (Fig. 1). The distance (r) between the donor and acceptor was calculated based on the equation below (49),
where the Förster distance (R0), the distance at which energy transfer is 50% efficient, was determined to be 67 Å (17). Unpaired Student’s t-tests were performed to compare differences between the mutants and WT in the FRET distance in each nucleotide state.
Figure 1.

Cy3-labeled nucleotide binding to M2β-S1. The FRET change observed upon Cy3ATP or Cy3ADP binding to M2β-S1 was monitored by mixing 1 μM M2β-S1 A488RLC with varying concentrations of fluorescent nucleotide and monitoring the decrease in donor fluorescence. Fluorescence transients were best fitted by a double-exponential function. (A) The observed fast phase rate constant for Cy3ATP binding to M2β-S1 was linearly dependent on Cy3ATP concentration, which allowed determination of the second-order binding constant. (B) Similarly, the observed fast phase rate constant for Cy3ADP binding to M2β-S1 was also linearly dependent on Cy3ADP concentration. (C) The amplitude of the FRET signal in the Cy3ATP and Cy3ADP experiments was plotted as a function of nucleotide concentration and was fitted to a hyperbolic function to compare the relative amplitudes of the FRET signal (Cy3ATP, AMax = 0.69 ± 0.04; Cy3ADP, AMax = 0.43 ± 0.04). (D) Representative fluorescent transients from the Cy3ATP and Cy3ADP binding experiments fitted to a double-exponential function (Cy3ATP, kFast = 8.8 ± 0.1 s−1, AFast = 0.52; Cy3ADP, kFast = 8.4 ± 0.1 s−1, AFast = 0.54).
Kinetic modeling and duty ratio
The fluorescence transients of actin-activated power stroke and phosphate release were fitted to kinetic models as shown in Scheme 1 using Kintek Explorer (50,51). The forward and reverse rates constants used for modeling the steady-state ATPase and specific transient kinetics steps are listed in Table S3. The duty ratio was estimated using the following equation (52):
where kH is the maximal rate of ATP hydrolysis measured by intrinsic tryptophan fluorescence, k′PW is the maximal rate of the fast power-stroke rate constant, and k′−D is the ADP release rate constant monitored with pyrene actin fluorescence.
Scheme 1.

Overall actomyosin ATPase pathway (dotted line highlights actin-activated power-stroke steps).
Results
Purification and preparation of cardiac myosin constructs
M2β-S1 proteins were expressed and purified in C2C12 cells as previously described (25). Purified M2β-S1 contained the S1 fragment of human cardiac myosin heavy chain (amino acids 1–841) and associated mouse ELCs and RLCs, which are endogenously expressed in C2C12 cells. Our previous work determined the sequences of the two mouse light chains using liquid chromatography-tandem mass spectrometry (LC-MS/MS) and demonstrated their degree of homology compared to the human isoforms (41). To examine the structural kinetics of the actin-activated power stroke by FRET, we exchanged the mouse RLC with A488RLC (labeled at V105C) during the M2β-S1 purification process (see Materials and methods for details). For all M2β-S1 WT, R723G, and F764L constructs, nonexchanged M2β-S1 protein was prepared in parallel with the exchanged protein and served as a control in all FRET experiments. Human RLC labeling and exchange efficiencies were determined by examining Alexa 488 concentration by absorbance and utilizing fluorescence gel quantitation methods (Fig. S1). Briefly, M2β-S1 A488RLC complexed with actin was pelleted by ultracentrifugation and then released with ATP, the amount of A488RLC in the supernatant was quantified using an in-gel standard curve with an A488RLC standard. The total concentration of M2β-S1 A488RLC in the supernatant was determined by Bradford. We found that the stoichiometry of A488RLC to M2β-S1 was close to 1:1 (1:1 ± 0.2, for WT M2β-S1 A488RLC, N = 3).
Steady-state motor properties of RLC-exchanged constructs
We first examined the impact of exchanging A488RLC onto M2β-S1 with steady-state ATPase and in vitro motility assays. The actin-activated ATPase activity was plotted as a function of actin concentration and fitted to a Michaelis-Menten equation to determine the maximal rate (kcat) and actin concentration as which ATPase in one-half maximal (KATPase) (Fig. S2; Table S1). The maximal rate was similar in M2β-S1 A488RLC (4.4 ± 0.6 s−1) compared with nonexchanged controls (5.4 ± 1.3 s−1) (Fig. S2 A; Table S1). The average sliding velocity was examined in the in vitro motility assay, and velocity values were analyzed manually. The M2β-S1 A488RLC displays a slightly slower sliding velocity compared with the M2β-S1 (p ≤ 0.0001) (Fig. S2 B; Table S1). We hypothesized the difference could be due to the different RLC isoforms. We further examined the actin-activated ATPase and sliding velocity of M2β-S1 exchanged with unlabeled human RLC (V105C), and we found a slight decrease in actin sliding velocity (p = 0.0014) and no change in maximal ATPase rate (5.3 ± 0.9 s−1) (Table S1). Overall, we conclude that the exchange of labeled or unlabeled human RLC onto M2β-S1 only causes minor changes in in vitro motility and no significant changes in the steady-state ATPase properties of M2β-S1.
Steady-state and transient FRET measurements
Previously, Rohde et al. (17) demonstrated a method of measuring the actin-activated power stroke using Cy3-labeled nucleotides and A488RLC in bovine cardiac myosin. In this work, we used a similar approach, and successfully monitored the FRET signal associated with the actin-activated power stroke in human M2β-S1 A488RLC. Our transient kinetic results are interpreted in the context of Scheme 1, which is similar to previous studies (25). We observed different FRET efficiencies between Cy3ATP (0.48 ± 0.01) and Cy3ADP (0.40 ± 0.02) when examining 0.5 μM M2β-S1 A488RLC in the presence of 1 μM Cy3-labeled nucleotide under equilibrium conditions (corrected for fraction bound, see Materials and methods) (Fig. 2; Table 1). We calculated the average distance change between donor and acceptor in the Cy3ATP and Cy3ADP conditions (ΔFRET = 3.8 ± 1.0 Å), which is similar to the distance change expected from structural modeling in the pre- and post-power stroke states (estimated distances of 84.3 and 86.9 Å, respectively) (Fig. 3; Table 1). Direct binding of Cy3ADP or Cy3ATP was monitored by mixing 1 μM M2β-S1 A488RLC with varying concentrations of Cy3ATP (0.1–2.5 μM) or Cy3ADP (0.5–5 μM) (Fig. 1, A and B) in the stopped-flow apparatus. The second-order binding constants were similar to published values for ATP and ADP (Cy3ATP, 3.36 ± 0.16 μM−1 s−1; Cy3ADP, 3.43 ± 0.28 μM−1 s−1), respectively (25,53). Consistent with steady-state FRET, the maximal amplitude of the FRET change was larger with Cy3ATP (0.69 ± 0.04) than Cy3ADP (0.43 ± 0.04) (Fig. 1 C), indicating our FRET method monitors average lever arm position as a function of nucleotide state.
Figure 2.
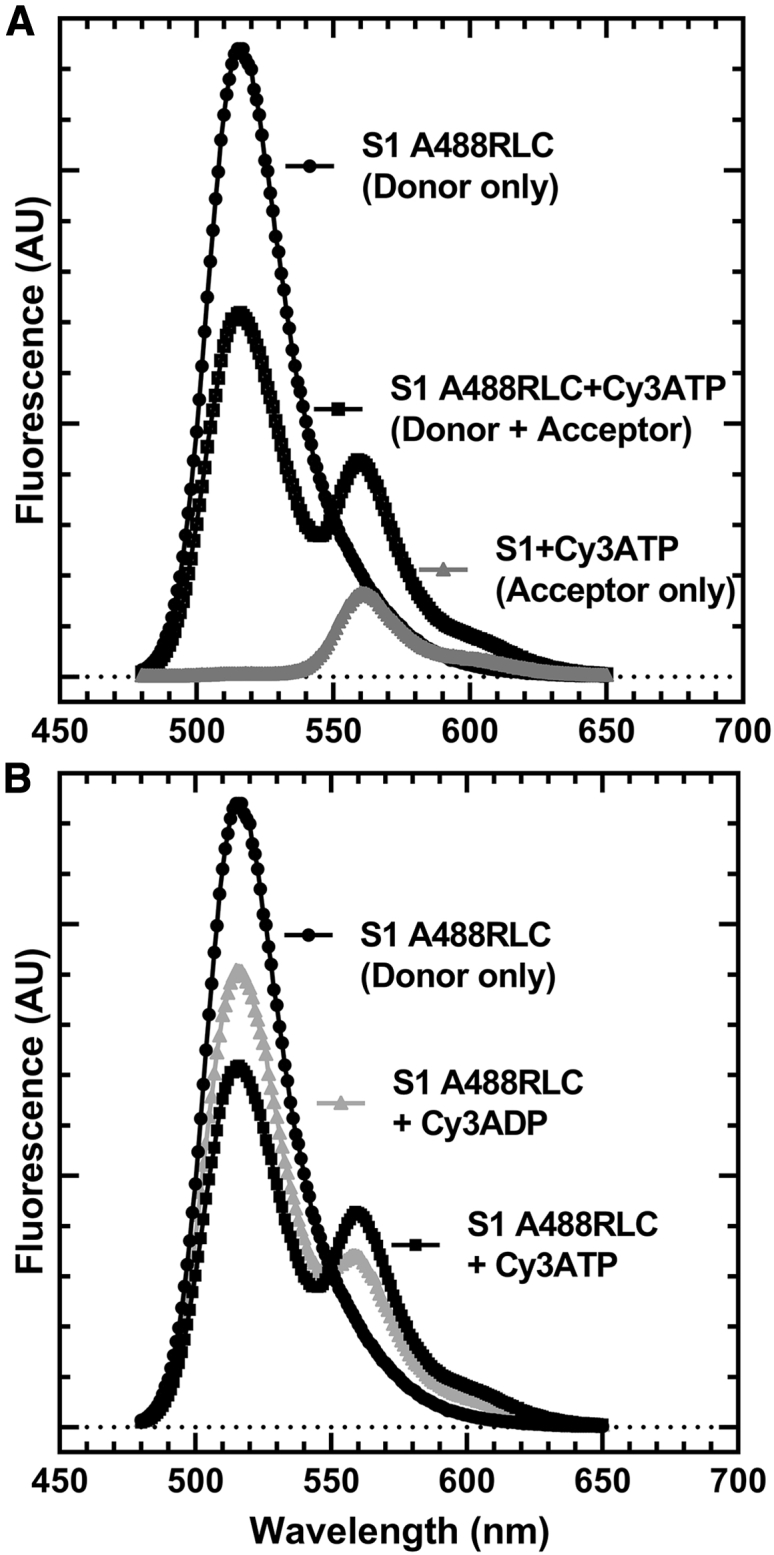
Examination of steady-state FRET as a function of nucleotide state. Representative wavelength scans used to perform FRET efficiency measurements of M2β-S1 A488 RLC with Cy3ATP and Cy3ADP. (A) The fluorescence spectrum of donor-acceptor, donor only, and acceptor only is shown. (B) The fluorescence spectrum of donor only compared with donor-acceptor and comparing Cy3ATP and Cy3ADP. The FRET efficiency was calculated after correcting for the fraction of Cy3 nucleotide bound, using the KD estimated from stopped-flow measurements (Fig. 1C). A summary of the FRET efficiency measurements and associated distances is shown in Table 1.
Table 1.
Steady-state FRET efficiency in different nucleotide states
| FRET parameter, N = 6, ± SEM | WT | R723G | F764L |
|---|---|---|---|
| Cy3ATP FRET efficiency | 0.48 ± 0.01 | 0.45 ± 0.01 | 0.48 ± 0.01 |
| Cy3ADP FRET efficiency | 0.40 ± 0.02 | 0.36 ± 0.02 | 0.35 ± 0.02 |
| Cy3ATP FRET distance (Å) | 67.9 ± 0.5 | 69.3 ± 0.5 | 67.9 ± 0.5 |
| Cy3ADP FRET distance (Å) | 71.7 ± 1.0 | 73.7 ± 1.1 | 74.5 ± 1.1 |
| ΔFRET (Å)a | 3.8 ± 1.0 | 4.4 ± 1.1 | 6.6 ± 1.1 |
Difference between the distances determined in the presence Cy3ATP and Cy3ADP.
Figure 3.

Structural alignment of M2β-S1 in pre- and post-power-stroke conformations. The pre- (blue) and post-power-stroke (orange) homology structures of M2β-S1 were aligned using the structural alignment feature in the program Chimera. Essential light chain (ELC) is shown in tan, regulatory light chain (RLC) is shown in magenta, cysteine mutation introduced at residue 105 (V105C) in the RLC is shown in green, and the residue Asn238 (N238) in the active site, which is proposed to be close to the Cy3 fluorophore, is shown in cyan. The distances between N238 and V105C were determined using the program Chimera. The M2β-S1 pre- and post-power-stroke homology structures are acquired from the Spudich Lab website (http://spudlab.stanford.edu/homology-models).
Structural kinetics of lever arm rotation during the actin-activated power stroke
A sequential-mix stopped-flow apparatus was used to measure the kinetics of lever arm rotation during the actin-activated power stroke (see Scheme 1 for the actin-activated power stroke steps highlighted with a dotted line). M2β-S1 A488RLC (0.25 μM) was first mixed with 0.2 μM Cy3ATP, aged for 10 s to allow ATP hydrolysis to occur, and then mixed with varying concentrations of actin (5–30 μM) (Fig. 4 A). The traces (Fig. 4 B) were best fitted to a double-exponential function. The rate constants of the two phases (fast and slow) at different actin concentrations were fitted to a hyperbolic function. We determined that the fast phase of the actin-activated power stroke has a maximal rate of 17.2 ± 4.8 s−1 (Table 2). The slow phase was also actin dependent but did not fit well to a hyperbolic function (rate constant of 1.5 s−1 at 30 μM actin). The relative amplitude of the fast phase was 60% of the fluorescence signal at 30 μM actin. To further characterize the fast and slow phases of the actin-activated power stroke, we repeated our measurements at 30 μM actin at different temperatures and plotted the rate constants together with temperature-dependent ATPase measurements (Fig. 4 C). We found the fast phase of the actin-activated power stroke was faster than the ATPase values and the slow phase slower than the ATPase rate at all temperatures examined. The Eyring plot comparing the fast and slow phases with ATPase values (Fig. 4 D) showed that the three rate constants share a similar temperature dependence (slope, kFast, −17.03 ± 0.76; kSlow, −18.25 ± 1.85; ATPase, −15.53 ± 0.51).
Figure 4.

Measurements of actin activation of the power stroke in M2β-S1. The rate constants of the actin-activated power stroke were measured by monitoring the fluorescence enhancement of Alexa 488 during actin-activated product release. Sequential-mix stopped-flow experiments were performed by mixing 0.25 μM M2β-S1 A488RLC with 0.2 μM Cy3ATP, aged for 10 s for hydrolysis to occur, and then mixed with varying concentrations of actin (5–30 μM). The fluorescence transients were best fitted by a double-exponential function. (A) The rate constants of the fast phase were plotted as a function of actin concentration and fitted to a hyperbolic function. The slow phase was linearly dependent on actin concentration and has a rate of 1.5 s−1 at 30 μM actin. Data points at each actin concentration represent the average ± SEM of three to seven experiments from separate protein preparations. (B) Representative fluorescence transient in the presence of 30 μM actin (average of two transients) is shown fitted to a double-exponential function. The donor-only and acceptor-only controls are shown for comparison. (C) Experiments in (A) were performed as a function of temperature, and the rate constants of the fast and slow phases of the power stroke were plotted together with the actin-activated ATPase activity (10–35°C). (D) Eyring plots of the power-stroke rate constants and corresponding ATPase activity demonstrate the temperature dependence of the different rate constants (slopes, kFast, −17.03 ± 0.76; kSlow, −18.25 ± 1.85; ATPase, −15.53 ± 0.51).
Table 2.
Summary of steady-state and transient kinetic results comparing WT and mutants
| WT | R723G | F764L | |
|---|---|---|---|
| Steady-state ATPase values (± SEM), N = 3–6 | |||
| v0 (s−1) | 0.02 ± 0.01 | 0.03 ± 0.01 | 0.02 ± 0.01 |
| kcat (s−1) | 8.3 ± 1.1 | 6.1 ± 1.5 | 6.4 ± 1.4 |
| KATPase (μM) | 78 ± 16 | 60 ± 26 | 70 ± 25 |
| In vitro motility value (± SEM), N = 90 | |||
| Mean velocity (nm/s) | 1591 ± 16 | 1699 ± 17∗ | 1425 ± 16∗ |
| Rate/equilibrium constants (± SEM) | WTa | R723G | F764La |
| ATP binding/hydrolysis (myosin), N = 2b | |||
| K0.5 (μM) | 48 ± 6 | 48 ± 9 | 79 ± 6 |
| K1Tk+2T (μM−1 s−1) | 2.8 ± 0.1 | 3.9 ± 0.1 | 1.9 ± 0.1 |
| k+H + k-H (s−1) | 182 ± 6 | 231 ± 12 | 166 ± 6 |
| ATP binding (actomyosin), N = 3c | |||
| K′1Tk′+2T (μM−1 s−1) | 9.6 ± 0.2 | 8.2 ± 0.1 | 9.4 ± 0.2 |
| K0.5 (μM) | 31 ± 9 | 42 ± 22 | 24 ± 8 |
| k′+2T (s−1) | 512 ± 47 | 668 ± 121 | 409 ± 42 |
| Actin-activated Pi release at 30 μMd | |||
| k′+Pi (fast, s−1) | 20.7 ± 0.9 | ND | 16.4 ± 1.7 |
| k′+Pi (slow, s−1) | 1.86 ± 0.02 | ND | 1.68 ± 0.01 |
| Actin-activated power stroke, N = 3–4e | |||
| k′+PW (fast phase, maximal rate, s−1) | 17.2 ± 4.8 | 10.8 ± 4.1 | 8.7 ± 4.5 |
| K0.5, fast (μM) | 48 ± 33 | 29 ± 19 | 29 ± 26 |
| k′+PW (slow phase, at 30 μM actin, s−1) | 1.5 ± 0.3 | 0.9 ± 0.2 | 0.8 ± 0.3 |
| Actomyosin ADP releasef | |||
| k′+D (s−1), N = 3–5f | 332 ± 57 | 380 ± 55 | 290 ± 46 |
| k′+D (s−1), N = 3c | 174 ± 12 | 218 ± 15∗ | 132 ± 16∗ |
∗p < 0.05. ND, not determined.
Values acquired from Tang et al. (25) except for the power-stroke parameters.
Intrinsic tryptophan fluorescence.
Pyrene actin.
MDCC-PBP fluorescence.
FRET.
mant-ADP fluorescence.
Temperature dependence of actin-activated phosphate release and power stroke
We performed sequential-mix experiments as described above to measure actin activation of phosphate release and the power stroke under identical conditions in the presence of 30 μM actin at three temperatures: 25, 30, and 35°C (Fig. 5; Table S2). In phosphate release experiments, a lag was observed at 25°C (8.2 ± 0.7 s−1) and 30°C (21.6 ± 6.2 s−1), with an observed Pi release rate of 3.0 ± 0.1 and 7.1 ± 0.1 s−1, respectively. The fast phase of the power stroke was very similar to the lag in the Pi release measurements (7.3 ± 0.3 s−1 at 25°C and 22.0 ± 1.4 s−1 at 30°C), indicating that Pi release was rate limited by the power stroke at these two temperatures. No lag was observed in the Pi release experiments at 35°C, and the Pi release rate constant (19.2 ± 0.1 s−1) was nearly 50% slower than the fast power-stroke rate constant (36.5 ± 1.3 s−1).
Figure 5.

Temperature dependence of actin-activated power stroke and phosphate release in M2β-S1. The phosphate binding protein (MDCC-PBP) was used to monitor the phosphate release step using sequential-mix experiments similar to that described in Fig. 4. 1–2 μM M2β-S1 A488RLC was mixed with substoichiometric ATP, aged for 10 s, and then mixed with 30 μM actin and MDCC-PBP. (A) Representative fluorescence transients (average of two to three normalized transients) of the power stroke (blue) and Pi release (red) in the presence of 30 μM actin are shown at 25°C. (B) A comparison of the Pi release transient (first 0.25 s) fitted to a single exponential function with (χ2 = 0.12) and without (χ2 = 0.29) a lag demonstrates an improved fit with the lag. The inset demonstrates the fit to the entire time course. A comparison of the power stroke and Pi release transients measured at (C) 30°C and (D) 35°C is given. The power-stroke experiments were best fitted to a double-exponential function at all temperatures. The Pi release experiments were best fitted by a lag followed by a single exponential fluorescence increase at 25 and 30°C, whereas the transients were single exponential at 35°C. (25°C, kFast = 7.3 ± 0.3 s−1, kSlow = 1.3 ± 0.1 s−1, kLag = 8.2 ± 0.7 s−1, kPi = 3.0 ± 0.1 s−1, AFast = 0.5; 30°C, kFast = 22 ± 1.4 s−1, kSlow = 3.7 ± 0.2 s−1, kLag = 21.6 ± 6.2 s−1, kPi = 7.1 ± 0.1 s−1, AFast = 0.5; 35°C, kFast = 36.5 ± 1.3 s−1, kSlow = 5.0 ± 0.2 s−1, no lag, kPi = 19.2 ± 0.1 s−1, AFast = 0.7).
Impact of converter domain mutations on steady-state motor properties
We previously reported the steady-state and transient kinetic measurements of M2β-S1 F764L in our recent work (25). In this work, we compared the steady-state motor properties of the two mutants with WT in parallel. Both R723G (6.1 ± 1.5 s−1) and F764L (6.4 ± 1.4 s−1) have slightly slower maximal ATPase rates compared with WT (8.3 ± 1.1 s−1), but the difference is not significant (Fig. 6 A; Table 2). The average sliding velocities in the in vitro motility assay were determined by pooling together 90 filaments from three protein preparations (30 each) at 0.24 μM loading concentration (Fig. 6 B). M2β-S1 R723G shows a 7% increase in average sliding velocity (1699 ± 17 nm/s, p < 0.0001) compared with WT (1591 ± 16 nm/s), whereas F764L causes about a 10% decrease in average sliding velocity (1425 ± 16 nm/s, p < 0.0001) (Table 2).
Figure 6.

Steady-state ATPase and in vitro motility of M2β-S1 WT, R723G, and F764L. (A) The actin-activated ATPase activity of purified M2β-S1 WT, R723G, and F764L was determined as a function of actin in MOPS 20 buffer at 25°C. The data points represent the average ± SEM from three to six protein preparations. (B) The sliding velocity of M2β-S1 WT, R723G, and F764L was measured (90 filaments in total, 30 filaments from each protein preparation) at 0.24 μM loading concentration and analyzed manually, and velocities were pooled together to determine the average velocity. The black bar represents the mean ± SD. Unpaired Student’s t-tests were done to compare each mutant with WT (∗∗∗p < 0.0001).
Transient kinetic measurements
We compared the transient kinetic parameters of M2β-S1 R723G and F764L mutants with WT (Table 2). The rate constants of WT and F764L were published in our previous work (25). We examined the ATP binding and hydrolysis steps in R723G by monitoring tryptophan fluorescence. An enhancement in fluorescence was observed upon mixing 1 μM M2β-S1 R723G with varying concentrations of ATP (2.5–1000 μM) (Fig. S3). The tryptophan fluorescence traces were best fitted to a double-exponential function with relative amplitude of the fast phase that was ∼60% of the signal at saturating ATP concentrations. The fast phase rate constants were hyperbolically dependent on ATP concentration, which allowed us to determine the maximal rate of ATP hydrolysis (231 ± 12 s−1) and ATP concentration dependence (K0.5 = 48 ± 9 μM) (Table 2). The second-order ATP binding constant in R723G determined by the linear dependence at low ATP concentrations was 3.9 ± 0.1 μM−1 s−1. We also measured ATP-induced dissociation from actomyosin by mixing a M2β-S1/pyrene actin complex with varying concentrations of ATP (2–125 μM) (Fig. S4). The pyrene fluorescence traces were best fitted to a double-exponential function. The rate constant of the fast phase (amplitude was ∼90% of the signal) plotted as a function of ATP concentration was fitted to a hyperbolic equation kobs = K′1k′+2T × [ATP]/(1 + K′1 × [ATP]) (25), which allowed us to determine the maximal rate k′+2T = 668 ± 121 s−1 and second-order binding constant for ATP K′1k′+2T = 8.2 ± 0.1 μM (Table 2). The ADP release rate constant was measured by mixing an actomyosin-mantADP complex with 1 mM ATP (data not shown). The fluorescence transients from at least three separate preparations were fitted to a single exponential function, and the average and standard errors (SEMs) were compared with WT and F764L (Table 2). The ADP release rate constant was also examined by mixing M2β-S1 R723G-pyrene actin-ADP complex (0.5 μM M2β-S1 R723G, 0.5 μM pyrene actin, and 50 μM ADP) with varying concentrations of ATP (10–2000 μM). The data were fitted to a hyperbolic function, and the maximal rate was defined as the ADP release rate constant as previously described (Table 2; (25)). Overall, the rate constants measured for R723G were quite similar to WT except for the ADP release rate constant. We did not observe evidence for population of an off-pathway intermediate in the ADP release experiments as was reported in other myosins (54). The release of mantADP from acto-M2β-S1 was faster than the measurements of ADP release from pyrene acto-M2β-S1, which we also reported in our previous work (41), and is likely due to a reduced affinity for the mant-labeled nucleotide. Compared to WT M2β-S1, ADP release from pyrene acto-M2β-S1 was 25% faster in R723G and 24% slower in F764L, which follows a similar trend to the more moderate changes observed in the in vitro motility experiments (7% faster and 10% slower, respectively).
Impact of mutations on actin-activated power stroke
The steady-state FRET efficiency with Cy3ATP or Cy3ADP was determined for WT, R723G, and F764L using M2β-S1 A488RLC constructs. No significant difference in FRET efficiency or ΔFRET (difference between Cy3ADP and Cy3ATP conditions) was found between R723G or F764L and WT (Fig. 7; Table 1). Measurements of the fast and slow actin-activated power-stroke rate constants showed that R723G causes a 37% decrease in the maximal rate of the fast phase (10.8 ± 4.1 s−1), whereas the decrease was slightly larger (50%) for F764L (8.7 ± 4.5 s−1) (Fig. 8; Table 2). Student’s t-tests were performed at each actin concentration to compare each mutant with WT, and significant differences were found at 30 μM actin in R723G and 20 and 30 μM actin in F764L. The slow phase in the actin-activated power-stroke experiments was also actin-concentration dependent for R723G and F764L and was slower in both R723G (0.9 ± 0.2 s−1) and F764L (0.8 ± 0.3 s−1) compared with WT (1.5 ± 0.3 s−1) at 30 μM actin (Table 2). In both mutants, the relative amplitudes of the fast and slow phases were similar to WT. We estimated the duty ratio for the mutant and WT constructs using an equation that assumes the hydrolysis and actin-activated power-stroke rate constants control the transition into the strong binding states and the rate of ADP release from actomyosin controls the transition into the weak-binding states (see Materials and methods). We found that compared to WT M2β-S1 (∼0.08), the estimated duty ratio is decreased in R723G (∼0.05) and F764L (∼0.06).
Figure 7.

Impact of R723G and F764L mutants on lever arm conformation monitored by FRET. Measurements of Cy3ATP and Cy3ADP binding to M2β-S1 were examined as described in Fig. 1. The amplitude of the FRET change was plotted as a function of Cy3ATP or Cy3ADP concentration and fitted to a hyperbolic function for (A) R723G and (B) F764L. (C) Summary of steady-state FRET efficiency determined from measuring the emission spectra of 0.5 μM M2β-S1 in the presence of 1 μM Cy3ATP or Cy3ADP in WT, R723G, and F764L (average of six repeats from three protein preparations). The black bar represents the mean ± SD. Unpaired Student’s t-tests were performed to compare each mutant with WT, and no significant differences were found between the mutants and WT.
Figure 8.

Impact of R723G and F764L mutants on the power-stroke rate constant in M2β-S1. Sequential-mix stopped-flow experiments were performed as described in Fig. 4. The rate constants of the fast phase were plotted as a function of actin concentration and fitted to a hyperbolic function to determine the maximal rate of power stroke for (A) R723G, 10.8 ± 4.1 s−1 or (B) F764L, 8.7 ± 4.5 s−1. The slow phase was also actin-concentration dependent for both mutants, with a rate of 0.9 ± 0.2 s−1 for R723G and 0.8 ± 0.3 s−1 for F764L at 30 μM actin. Data points at each actin concentration represent the average ± SEM of three experiments from separate protein preparations. The fast phase of the power stroke for WT is replotted from Fig. 4 for comparison with the mutants. Student’s t-tests were performed at each actin concentration to compare each mutant with WT, and significant differences were found at 30 μM actin in R723G and 20 and 30 μM actin in F764L (∗p < 0.05).
Impact of converter domain mutations on mechanosensitivity
Additionally, we performed loaded in vitro motility assays to measure the sliding velocity in the presence of an increasing amount of tethering load induced by the presence of a motor-dead version of M2β-S1. We used a motor-dead mutant (E466A) M2β-S1, which is known to inhibit ATP hydrolysis in myosins (55) and movement of actin filaments in the motility assay. The advantage of using M2β-S1 E466A is that it has the same size and surface attachment strategy as the M2β-S1 constructs examined. Based on calculations outlined in Greenberg and Moore (45), we converted the motor-dead density into tethering load and plotted the data as a force-velocity curve. Because the data set lacks data points at higher loads, we were not able to fit the data to the force-velocity relationship. This is likely because E466A is predicted to have relatively weak affinity for actin in the presence of ATP (Kd ∼30 μM) (55). We also plotted relative velocity (normalized to 0% motor dead) as a function of tethering load for comparison. The relative reduction in sliding velocity was more pronounced in R723G and F764L compared to WT at the highest loads examined (∼300 pN) (based on three separate protein preparations) (Fig. 9).
Figure 9.

Loaded in vitro motility of M2β-S1 WT, R723G, and F764L. The impact of a tethering load imposed by the presence of M2β-S1 E466A (motor dead) was examined in WT, R723G, and F764L M2β-S1 from three different protein preparations. The total concentration of M2β-S1 loaded onto the motility surface was kept constant at 0.6 μM. Velocities were analyzed by the program FAST (44). (A) The sliding velocity was expressed relative to 0% motor dead and plotted as a function of the fraction of motor dead present. (B) The sliding velocity was plotted as a function of the tethering load produced by the presence of motor-dead myosin and converted to force (piconewtons) as described in the Materials and methods. (N = 6, error bars represent SEM). Unpaired Student’s t-tests were performed to compare each mutant with WT at each [motor dead]% (∗p < 0.05).
Discussion
In this study, we successfully utilized a FRET technique to monitor the structural kinetics of lever arm rotation (actin-activated power stroke) in human cardiac myosin. We determined the actin-activated power-stroke rate constants in human cardiac myosin using sequential-mix stopped-flow experiments, and the values agree well with the previous measurements in bovine cardiac myosin. We demonstrated clear evidence that the actin-activated power stroke gates Pi release in human cardiac myosin; the observed lag associated with Pi release was equivalent to the actin-activated power-stroke rate constant at lower temperatures (25 and 30°C). At near-physiological temperature (≥35°C), the power stroke was accelerated such that no lag in Pi release was observed. We also measured the actin-activated power-stroke rate constants in two mutants, one associated with HCM and another associated with DCM, and measured their ability to slide actin filaments in the in vitro motility assay. Surprisingly, both mutants slowed actin activation of the power stroke as well as altered in vitro actin gliding in the presence and absence of load. Thus, our measurements have revealed critical mutation-specific defects that provide important insights into the molecular mechanisms of inherited cardiomyopathies.
The actin-activated power stroke in human cardiac myosin
In the actin-activated power-stroke measurements, two phases of the FRET transients were observed: a fast phase with a maximal rate constant of 17.2 ± 4.8 s−1 and a slow phase with a rate constant of 1.5 ± 0.3 s−1 at 30 μM actin. By plotting the fast and slow phase rate constants together with the ATPase rates in the presence of 30 μM actin at different temperatures (Fig. 4 B), we found the fast power-stroke rate constant was faster than the ATPase rate at all temperatures examined, and the slow phase was slightly slower than the ATPase rate. Thus, we concluded that the fast phase rate constant represents the macroscopic rate constant associated with actin activation of lever arm rotation (power stroke). The slow phase may represent the flux through the actin-detached pathway or prehydrolysis intermediates that transition through ATP hydrolysis weakly bound to actin, which has been suggested in previous studies (17,36).
The maximal rate of the actin-activated power stroke in human cardiac myosin determined in the current study (17.2 s−1) agrees well with the power-stroke measurement in bovine cardiac myosin (15.3 s−1) using a similar FRET technique (17). However, Woody et al. (11) recently reported a much faster working-stroke rate constant, >700 s−1, determined in an ultrafast-force-clamp-equipped single-molecule optical trapping experiment. This technique also allowed direct examination of the transition for weakly to strongly attached myosin heads. They monitored the actomyosin attachment duration as well as the detachment rate of human β-cardiac myosin heavy meromyosin under loaded conditions ranging from 1.5 to 4.5 pN, and the rate of the working stroke was found to be extremely fast (700–5250 s−1) and increased with load. The mechanical transients observed by optical trapping were characterized by force-generating power strokes that were sometimes followed by power-stroke reversals and rapid detachment from actin. The power-stroke reversals were also increased by the presence of load.
It is well established that myosin, with the hydrolyzed products in its active site, first associates weakly with actin and then transitions into a strong actin-attached state to perform the power stroke. Woody et al. have proposed a pre-force-generating actin-attached state that is populated immediately before myosin transitions into the strong actomyosin state (11,56,57). The pre-force-generating state is highly reversible (can rapidly detach from actin), but it is proposed to be stereospecific in nature (11). Consequently, we developed a kinetic model to evaluate our transient kinetic results in light of the recent optical trapping results. Our modeling suggests that the actin-activated power stroke is highly reversible and followed by a more rapid irreversible phosphate release step (Scheme 2 A) under the conditions of our experiments with no load and very low inorganic phosphate concentrations. The model is compatible with the slower actin-activated power-stroke data (∼17 s−1) in our work and the extremely rapid working-stroke data (≥700 s−1) from the optical trapping studies, as the working-stroke rate constant measured in the optical trapping assay is thought to be an ensemble of the forward and reverse rate constants. A highly reversible conformational change before the power stroke, such as the transition from the weak actin-binding to the pre-force-generating intermediate discussed above, is also a possible mechanism (Scheme 2 B). The ability to examine structural changes in the actin-binding region during the actin-activated power stroke in cardiac myosin will be crucial for evaluating this model. The implications for a highly reversible actin-activated power stroke in human cardiac myosin are that factors that impact the forward or reverse rate constants can significantly alter the ATPase kinetics. Indeed, the actin regulatory proteins troponin and tropomyosin enhance ATPase activity and accelerate the rate of attachment to actin in skeletal or cardiac muscle myosin (58,59), although it is unclear how they impact the power stroke.
Scheme 2.

Actin-activated power-stroke mechanisms.
The sequence of events of the power stroke and Pi release in cardiac myosin
We observed a lag in the Pi release measurements that was similar to the actin-activated power-stroke rate constant measured by FRET at 25 and 30°C (Fig. 5; Table S2). These results provide strong evidence that the actin-activated power stroke occurs before Pi release in human cardiac myosin. To confirm this conclusion, we fitted our experimental data to the proposed kinetic model of the ATPase cycle described above. Our kinetic simulation of the actin-activated power stroke and Pi release fluorescence transients are best fitted to a model with an actin-activated power stroke that occurs first and is followed by Pi release (Fig. S5, A and B; Table S3). Thus, our results are consistent with a model in which the actin-activated power stroke rate-limits the release of Pi and causes the observed lag in the Pi release transients. We found that at near-physiological temperature (35°C), the lag disappeared, suggesting that the actin-activated power stroke is no longer slow enough to cause a lag in Pi release. Interestingly, the rate of actin attachment measured with pyrene actin was found to be highly temperature dependent in skeletal muscle myosin (60), suggesting that the acceleration of actin attachment may explain the faster actin-activated power stroke observed at higher temperatures. Our proposed kinetic model (including most steps in the ATPase cycle) was also used to simulate the steady-state actin-activated ATPase data, and it matches well with our experimental results (Fig. S5 C; Table S3; (25)). We acknowledge that there are alternative kinetic schemes that may explain our FRET and phosphate release results (e.g., models in which Pi release is released before lever arm rotation), but we favor the proposed model because it is also consistent with the results in the literature.
Woody et al. (11) observed no effect on the rate of the ensemble average displacements with added 10 mM free Pi, also suggesting that Pi release occurs after the power stroke. Previous work found a two-step power stroke in myosin V, one faster step before Pi release and one slower step before ADP release (15). In both bovine (17) and human cardiac myosin, two phases were observed, with the second slower phase proposed to be caused by noncanonical pathways. Nevertheless, the observed fast phase of the actin-activated power stroke that occurs before Pi release in cardiac myosin is significantly slower than that observed in myosin V and skeletal muscle myosin (14,15,19). We suggest this could be due to the more rapid rate of attachment to actin, especially in myosin V. Indeed, myosin V displays a more rapid rate of attachment to actin compared to skeletal muscle myosin in the absence of nucleotide (60). Cardiac muscle is known to have a slow rate of force development compared with skeletal muscle myosin at high Ca2+ concentrations, which suggests our actin-activated power-stroke measurements may correlate with the slower force development observed in cardiac muscle (61). However, the rate of Pi release is thought to correlate with force development in muscle fiber experiments, in which the Pi release rate constant is equal to the sum of the forward and reverse rate constants (62,63). Consequently, increases in phosphate concentration are known to enhance the rate of force development in muscle fiber experiments. Thus, the slowed actin-activated power stroke in cardiac myosin, which occurs before Pi release, may indirectly slow Pi release and force development. The slower rate of force development may be crucial for tuning cooperative activation of the thin filament in cardiac muscle. Interestingly, activation of cardiac muscle is less dependent on Ca2+ concentrations compared with skeletal muscle (64). Force development may be an important regulatory step in cardiac muscle, which can be modified by additional Ca2+-independent thick-thin filament processes such as interactions with myosin binding protein C.
Impact of mutations on the actin-activated power stroke
Despite their clinical HCM and DCM phenotypes, both R723G and F764L mutations cause a significant decrease in the actin-activated power-stroke rate constant. Because the actin-activated power stroke includes both the actin-binding and lever arm rotation steps, it is possible that the mutations could impact either of these steps. Most models suggest that lever arm rotation and the weak to strong actin-binding transition occur simultaneously (11,56). Thus, we propose that the slowed actin-activated power stroke is associated with an overall reduction in the rate of transition into the strongly bound states. We have calculated the duty ratio assuming the actin-activated power stroke represents the transition into the strong binding states and the ADP release rate constant represents transition out of the strong binding states. Our results suggest that both mutants have a reduced duty ratio under unloaded conditions, which is consistent with the previous kinetics studies (26,35).
Structural analysis of the local environment of R723G revealed that it is a highly conserved arginine residue located at an extended loop in the converter domain and is close to the essential light chain (ELC) in the pre-power-stroke state (Fig. S6). The positive charge of the Arg723 side chain forms a salt bridge with the negatively charged side chain of glutamic acid at residue 136 in the ELC. Altering this interaction in myosin V impacts the ATP hydrolysis and recovery-stroke rate constants (14,65). The R723G mutation abolishes the charge-charge interaction between the converter domain and ELC, which may weaken the allosteric coupling between the two domains. As described in our recent work, Phe764 is located in a hydrophobic patch between the relay helix and the converter domain. The conversion of leucine to phenylalanine at residue 764 likely weakens the hydrophobic interaction and increases the flexibility of the local environment, which may decrease the communication efficiency between the converter domain and the active site. Overall, we predict that both mutations would destabilize the pre-power-stroke state and disrupt communication between subdomains within the motor, which may contribute to its impairment of the structural kinetics of lever arm rotation during the actin-activated power stroke.
We observed no significant difference in the steady-FRET efficiency in either R723G or F764L, indicating similar pre- and post-power-stroke conformations of the lever arm in both R723G and F764L in the absence of actin (Table 1). It would be interesting to investigate further whether the two mutations shift the mole fraction of pre- and post- power-stroke structural states in the presence and absence of actin, which can be determined with time-resolved FRET (14). In a previous study, we examined the corresponding mutations in myosin V using time-resolved FRET (14). The F764L mutation (F750L in myosin V) did decrease the mole fraction of the pre-power-stroke state in the presence of ATP, but the R723G (R712G in myosin V) did not. In the same study, the R712G mutation slowed the recovery stroke, whereas F750L enhanced it, and both mutants did not alter the actin-activated power stroke. Thus, destabilizing the pre-power-stroke state does not necessarily accelerate the transition into the post-power-stroke state, suggesting that the recovery- and power-stroke transitions are governed by a complex set of allosteric interactions.
Impact of mutations on actin gliding in the presence and absence of load
The mutations slightly altered the in vitro actin gliding velocity in the absence of load, which correlated with their changes in ADP release kinetics. The unloaded sliding velocity is 7% faster in R723G, and 10% slower in F764L compared with WT, respectively. The faster ADP release rate constant (218 ± 15 s−1) observed in R723G generally correlates with its faster unloaded sliding velocity, and the slower ADP release rate constant in F764L (132 ± 16 s−1) also correlates with its slower sliding velocity compared with WT (174 ± 12 s−1) (Table 2).
The converter domain is the load-sensing region of the myosin motor that is thought to communicate external load to the nucleotide binding pocket, which mediates ADP release kinetics (12,66, 67, 68, 69). Variations in the converter domain of myosin have been shown to modulate the contractile properties of various muscle types (70). Interestingly, both converter domain mutations R723G and F764L showed a reduced capability to overcome the tethering loads, only at the highest loads measured. In our previous study that examined the two corresponding mutations (R712G and F750L) in myosin V, an even larger difference was observed in the ability of the mutations to overcome resistive load (e.g., assessed by the number of filaments moving as a function of percent motor dead on the surface). Furthermore, R723G was found to cause a reduction in intrinsic force measured with single-molecule optical trapping studies (34,35), and thus could also contribute to the observed reduction in gliding velocity in the presence of load. The results obtained in this study were performed at low and intermediate loads, which prevented us from fitting our data to the force-velocity and force-power relationships and obtaining estimates of ensemble force and maximal power. Nevertheless, our work does demonstrate that both mutants displayed a larger relative decrease in velocity at the highest loads measured, indicating disrupted load sensitivity.
Potential disease-causing mechanisms for R723G and F764L
The R723G mutation causes a reduced actin-activated power-stroke rate constant, reduced intrinsic force, lower duty ratio, faster unloaded in vitro gliding velocity, and greater sensitivity to load. All the changes together indicate that R723G impairs motor contractility, which contrasts with the proposed hypercontractility hypothesis of HCM mutations (71). The F764L mutation has a reduced actin-activated power stroke, reduced duty ratio, depressed unloaded actin sliding velocity, and enhanced load sensitivity which agrees with the hypocontractility hypothesis for DCM mutations. However, the changes in many of these parameters are relatively small.
A recent “mesa” theory proposes that residues located on a plateau-like surface within the cardiac myosin motor domain are important for forming an autoinhibited structure called the interacting-heads motif (IHM) (72, 73, 74). Myosin heads in the IHM contain head-head and head-tail interactions that are proposed to cause a fivefold slower ATP turnover rate, referred to as the super-relaxed (SRX) state, and inhibit interactions with actin until they transition out of the SRX state. Mutations located on the mesa surface, including R723G, may destabilize the IHM structure and increase the number of active heads on thick filaments (34,72). Therefore, the R723G mutation may increase the total number of available myosin heads on the thick filaments and thus increase overall isometric force produced in muscle, despite its reduced intrinsic motor properties. Phe764 is not predicted to be at the interface mediating the formation of the IHM but may still indirectly promote the SRX state by stabilizing a conformation required for entry into the IHM (72). Interestingly, cardiac tissue from Myh6764/764 mice demonstrated a slightly stabilized SRX state based on single ATP turnover studies (75).
Conclusions
In conclusion, our work suggests the actin-activated power stroke is highly reversible and may be an important regulatory step in the human cardiac myosin ATPase cycle. In addition, cardiomyopathy mutants that are associated with HCM and DCM phenotypes can directly alter the actin-activated power-stroke rate constant, a key step in the force-generating ATPase cycle of myosins. Future single-molecule optical trapping experiments to characterize the impact of these mutants under loaded conditions could shed light on the importance of load sensitivity in HCM and DCM. In addition, examining the impact of these two converter domain mutations on the stability of the SRX state with human β-cardiac myosin heavy meromyosin constructs, using single turnover and electron microscopy experiments, will be extremely revealing. Overall, our results provide evidence that examining the impact of mutations on important conformational changes can uncover specific structural defects and may lead to novel insight into disease mechanisms and therapeutic interventions.
Acknowledgments
This work was supported by a NIH grant to C.M.Y. (HL127699) and an AHA grant to W.T. (19PRE34380569).
Editor: David Thomas.
Footnotes
Supporting material can be found online at https://doi.org/10.1016/j.bpj.2021.04.007.
Author contributions
W.T. and C.M.Y. designed and performed experiments. J.G., W.C.U., and R.D. generated key reagents and provided technical support. The text was co-written by W.T. and C.M.Y. and approved by all authors.
Supporting material
References
- 1.Geeves M.A. Review: the ATPase mechanism of myosin and actomyosin. Biopolymers. 2016;105:483–491. doi: 10.1002/bip.22853. [DOI] [PubMed] [Google Scholar]
- 2.Málnási-Csizmadia A., Kovács M. Emerging complex pathways of the actomyosin powerstroke. Trends Biochem. Sci. 2010;35:684–690. doi: 10.1016/j.tibs.2010.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sweeney H.L., Houdusse A. Structural and functional insights into the myosin motor mechanism. Annu. Rev. Biophys. 2010;39:539–557. doi: 10.1146/annurev.biophys.050708.133751. [DOI] [PubMed] [Google Scholar]
- 4.Preller M., Manstein D.J. Myosin structure, allostery, and mechano-chemistry. Structure. 2013;21:1911–1922. doi: 10.1016/j.str.2013.09.015. [DOI] [PubMed] [Google Scholar]
- 5.Llinas P., Isabet T., Houdusse A. How actin initiates the motor activity of myosin. Dev. Cell. 2015;33:401–412. doi: 10.1016/j.devcel.2015.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lymn R.W., Taylor E.W. Mechanism of adenosine triphosphate hydrolysis by actomyosin. Biochemistry. 1971;10:4617–4624. doi: 10.1021/bi00801a004. [DOI] [PubMed] [Google Scholar]
- 7.Veigel C., Schmidt C.F. Moving into the cell: single-molecule studies of molecular motors in complex environments. Nat. Rev. Mol. Cell Biol. 2011;12:163–176. doi: 10.1038/nrm3062. [DOI] [PubMed] [Google Scholar]
- 8.Robert-Paganin J., Pylypenko O., Houdusse A. Force generation by myosin motors: a structural perspective. Chem. Rev. 2020;120:5–35. doi: 10.1021/acs.chemrev.9b00264. [DOI] [PubMed] [Google Scholar]
- 9.Mentes A., Huehn A., Sindelar C.V. High-resolution cryo-EM structures of actin-bound myosin states reveal the mechanism of myosin force sensing. Proc. Natl. Acad. Sci. USA. 2018;115:1292–1297. doi: 10.1073/pnas.1718316115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shuman H., Greenberg M.J., Ostap E.M. A vertebrate myosin-I structure reveals unique insights into myosin mechanochemical tuning. Proc. Natl. Acad. Sci. USA. 2014;111:2116–2121. doi: 10.1073/pnas.1321022111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Woody M.S., Winkelmann D.A., Goldman Y.E. Single molecule mechanics resolves the earliest events in force generation by cardiac myosin. eLife. 2019;8:e49266. doi: 10.7554/eLife.49266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wulf S.F., Ropars V., Schröder R.R. Force-producing ADP state of myosin bound to actin. Proc. Natl. Acad. Sci. USA. 2016;113:E1844–E1852. doi: 10.1073/pnas.1516598113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Greenberg M.J., Lin T., Ostap E.M. Mechanochemical tuning of myosin-I by the N-terminal region. Proc. Natl. Acad. Sci. USA. 2015;112:E3337–E3344. doi: 10.1073/pnas.1506633112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gunther L.K., Rohde J.A., Yengo C.M. Converter domain mutations in myosin alter structural kinetics and motor function. J. Biol. Chem. 2019;294:1554–1567. doi: 10.1074/jbc.RA118.006128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Trivedi D.V., Muretta J.M., Yengo C.M. Direct measurements of the coordination of lever arm swing and the catalytic cycle in myosin V. Proc. Natl. Acad. Sci. USA. 2015;112:14593–14598. doi: 10.1073/pnas.1517566112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rohde J.A., Roopnarine O., Muretta J.M. Mavacamten stabilizes an autoinhibited state of two-headed cardiac myosin. Proc. Natl. Acad. Sci. USA. 2018;115:E7486–E7494. doi: 10.1073/pnas.1720342115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rohde J.A., Thomas D.D., Muretta J.M. Heart failure drug changes the mechanoenzymology of the cardiac myosin powerstroke. Proc. Natl. Acad. Sci. USA. 2017;114:E1796–E1804. doi: 10.1073/pnas.1611698114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muretta J.M., Petersen K.J., Thomas D.D. Direct real-time detection of the actin-activated power stroke within the myosin catalytic domain. Proc. Natl. Acad. Sci. USA. 2013;110:7211–7216. doi: 10.1073/pnas.1222257110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muretta J.M., Rohde J.A., Thomas D.D. Direct real-time detection of the structural and biochemical events in the myosin power stroke. Proc. Natl. Acad. Sci. USA. 2015;112:14272–14277. doi: 10.1073/pnas.1514859112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moore J.R., Leinwand L., Warshaw D.M. Understanding cardiomyopathy phenotypes based on the functional impact of mutations in the myosin motor. Circ. Res. 2012;111:375–385. doi: 10.1161/CIRCRESAHA.110.223842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Enjuto M., Francino A., Ballesta A.M. Malignant hypertrophic cardiomyopathy caused by the Arg723Gly mutation in beta-myosin heavy chain gene. J. Mol. Cell. Cardiol. 2000;32:2307–2313. doi: 10.1006/jmcc.2000.1260. [DOI] [PubMed] [Google Scholar]
- 22.Kamisago M., Sharma S.D., Seidman C.E. Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N. Engl. J. Med. 2000;343:1688–1696. doi: 10.1056/NEJM200012073432304. [DOI] [PubMed] [Google Scholar]
- 23.Baumketner A. The mechanism of the converter domain rotation in the recovery stroke of myosin motor protein. Proteins. 2012;80:2701–2710. doi: 10.1002/prot.24155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bloemink M.J., Melkani G.C., Geeves M.A. The relay/converter interface influences hydrolysis of ATP by skeletal muscle myosin II. J. Biol. Chem. 2016;291:1763–1773. doi: 10.1074/jbc.M115.688002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tang W., Unrath W.C., Yengo C.M. Dilated cardiomyopathy mutation in the converter domain of human cardiac myosin alters motor activity and response to omecamtiv mecarbil. J. Biol. Chem. 2019;294:17314–17325. doi: 10.1074/jbc.RA119.010217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ujfalusi Z., Vera C.D., Leinwand L.A. Dilated cardiomyopathy myosin mutants have reduced force-generating capacity. J. Biol. Chem. 2018;293:9017–9029. doi: 10.1074/jbc.RA118.001938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Debold E.P., Schmitt J.P., Warshaw D.M. Hypertrophic and dilated cardiomyopathy mutations differentially affect the molecular force generation of mouse alpha-cardiac myosin in the laser trap assay. Am. J. Physiol. Heart Circ. Physiol. 2007;293:H284–H291. doi: 10.1152/ajpheart.00128.2007. [DOI] [PubMed] [Google Scholar]
- 28.Schmitt J.P., Debold E.P., Seidman J.G. Cardiac myosin missense mutations cause dilated cardiomyopathy in mouse models and depress molecular motor function. Proc. Natl. Acad. Sci. USA. 2006;103:14525–14530. doi: 10.1073/pnas.0606383103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Palmer B.M., Schmitt J.P., Maughan D.W. Elevated rates of force development and MgATP binding in F764L and S532P myosin mutations causing dilated cardiomyopathy. J. Mol. Cell. Cardiol. 2013;57:23–31. doi: 10.1016/j.yjmcc.2012.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kirschner S.E., Becker E., Kraft T. Hypertrophic cardiomyopathy-related beta-myosin mutations cause highly variable calcium sensitivity with functional imbalances among individual muscle cells. Am. J. Physiol. Heart Circ. Physiol. 2005;288:H1242–H1251. doi: 10.1152/ajpheart.00686.2004. [DOI] [PubMed] [Google Scholar]
- 31.Köhler J., Winkler G., Kraft T. Mutation of the myosin converter domain alters cross-bridge elasticity. Proc. Natl. Acad. Sci. USA. 2002;99:3557–3562. doi: 10.1073/pnas.062415899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seebohm B., Matinmehr F., Kraft T. Cardiomyopathy mutations reveal variable region of myosin converter as major element of cross-bridge compliance. Biophys. J. 2009;97:806–824. doi: 10.1016/j.bpj.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kraft T., Witjas-Paalberends E.R., van der Velden J. Familial hypertrophic cardiomyopathy: functional effects of myosin mutation R723G in cardiomyocytes. J. Mol. Cell. Cardiol. 2013;57:13–22. doi: 10.1016/j.yjmcc.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 34.Kawana M., Sarkar S.S., Spudich J.A. Biophysical properties of human β-cardiac myosin with converter mutations that cause hypertrophic cardiomyopathy. Sci. Adv. 2017;3:e1601959. doi: 10.1126/sciadv.1601959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vera C.D., Johnson C.A., Leinwand L.A. Myosin motor domains carrying mutations implicated in early or late onset hypertrophic cardiomyopathy have similar properties. J. Biol. Chem. 2019;294:17451–17462. doi: 10.1074/jbc.RA119.010563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.White H.D., Belknap B., Webb M.R. Kinetics of nucleoside triphosphate cleavage and phosphate release steps by associated rabbit skeletal actomyosin, measured using a novel fluorescent probe for phosphate. Biochemistry. 1997;36:11828–11836. doi: 10.1021/bi970540h. [DOI] [PubMed] [Google Scholar]
- 37.Winkelmann D.A., Forgacs E., Stock A.M. Structural basis for drug-induced allosteric changes to human β-cardiac myosin motor activity. Nat. Commun. 2015;6:7974. doi: 10.1038/ncomms8974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nag S., Trivedi D.V., Spudich J.A. The myosin mesa and the basis of hypercontractility caused by hypertrophic cardiomyopathy mutations. Nat. Struct. Mol. Biol. 2017;24:525–533. doi: 10.1038/nsmb.3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pardee J.D., Spudich J.A. Purification of muscle actin. Methods Enzymol. 1982;85:164–181. doi: 10.1016/0076-6879(82)85020-9. [DOI] [PubMed] [Google Scholar]
- 40.Pollard T.D. Polymerization of ADP-actin. J. Cell Biol. 1984;99:769–777. doi: 10.1083/jcb.99.3.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Swenson A.M., Tang W., Yengo C.M. Omecamtiv mecarbil enhances the duty ratio of human β-cardiac myosin resulting in increased calcium sensitivity and slowed force development in cardiac muscle. J. Biol. Chem. 2017;292:3768–3778. doi: 10.1074/jbc.M116.748780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.De La Cruz E.M., Sweeney H.L., Ostap E.M. ADP inhibition of myosin V ATPase activity. Biophys. J. 2000;79:1524–1529. doi: 10.1016/S0006-3495(00)76403-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meijering E., Dzyubachyk O., Smal I. Methods for cell and particle tracking. Methods Enzymol. 2012;504:183–200. doi: 10.1016/B978-0-12-391857-4.00009-4. [DOI] [PubMed] [Google Scholar]
- 44.Aksel T., Choe Yu E., Spudich J.A. Ensemble force changes that result from human cardiac myosin mutations and a small-molecule effector. Cell Rep. 2015;11:910–920. doi: 10.1016/j.celrep.2015.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Greenberg M.J., Moore J.R. The molecular basis of frictional loads in the in vitro motility assay with applications to the study of the loaded mechanochemistry of molecular motors. Cytoskeleton (Hoboken) 2010;67:273–285. doi: 10.1002/cm.20441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shimizu T., Johnson K.A. Kinetic evidence for multiple dynein ATPase sites. J. Biol. Chem. 1983;258:13841–13846. [PubMed] [Google Scholar]
- 47.Johnson K.A., Shimizu T., Holzbaur E.L. Kinetic evidence for multiple dynein heads: modeling the dissociation reaction. J. Biol. Chem. 1983;258:13847–13848. [PubMed] [Google Scholar]
- 48.Jacobs D.J., Trivedi D., Yengo C.M. Kinetics and thermodynamics of the rate-limiting conformational change in the actomyosin V mechanochemical cycle. J. Mol. Biol. 2011;407:716–730. doi: 10.1016/j.jmb.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lakowicz J.R. Springer; Boston, MA: 2006. Principles of Fluorescence Spectroscopy. [Google Scholar]
- 50.Johnson K.A., Simpson Z.B., Blom T. Global kinetic explorer: a new computer program for dynamic simulation and fitting of kinetic data. Anal. Biochem. 2009;387:20–29. doi: 10.1016/j.ab.2008.12.024. [DOI] [PubMed] [Google Scholar]
- 51.Johnson K.A., Simpson Z.B., Blom T. FitSpace explorer: an algorithm to evaluate multidimensional parameter space in fitting kinetic data. Anal. Biochem. 2009;387:30–41. doi: 10.1016/j.ab.2008.12.025. [DOI] [PubMed] [Google Scholar]
- 52.Forgacs E., Sakamoto T., White H.D. Switch 1 mutation S217A converts myosin V into a low duty ratio motor. J. Biol. Chem. 2009;284:2138–2149. doi: 10.1074/jbc.M805530200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu Y., White H.D., Forgacs E. Omecamtiv Mecarbil modulates the kinetic and motile properties of porcine β-cardiac myosin. Biochemistry. 2015;54:1963–1975. doi: 10.1021/bi5015166. [DOI] [PubMed] [Google Scholar]
- 54.Hannemann D.E., Cao W., De La Cruz E.M. Magnesium, ADP, and actin binding linkage of myosin V: evidence for multiple myosin V-ADP and actomyosin V-ADP states. Biochemistry. 2005;44:8826–8840. doi: 10.1021/bi0473509. [DOI] [PubMed] [Google Scholar]
- 55.Yengo C.M., De la Cruz E.M., Sweeney H.L. Kinetic characterization of the weak binding states of myosin V. Biochemistry. 2002;41:8508–8517. doi: 10.1021/bi015969u. [DOI] [PubMed] [Google Scholar]
- 56.Gunther L.K., Rohde J.A., Yengo C.M. FRET and optical trapping reveal mechanisms of actin activation of the power stroke and phosphate release in myosin V. J. Biol. Chem. 2020;295:17383–17397. doi: 10.1074/jbc.RA120.015632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sun M., Rose M.B., Yengo C.M. Characterization of the pre-force-generation state in the actomyosin cross-bridge cycle. Proc. Natl. Acad. Sci. USA. 2008;105:8631–8636. doi: 10.1073/pnas.0710793105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McKillop D.F., Geeves M.A. Regulation of the interaction between actin and myosin subfragment 1: evidence for three states of the thin filament. Biophys. J. 1993;65:693–701. doi: 10.1016/S0006-3495(93)81110-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Johnson D., Angus C.W., Chalovich J.M. Stepwise C-terminal truncation of cardiac troponin T alters function at low and saturating Ca2. Biophys. J. 2018;115:702–712. doi: 10.1016/j.bpj.2018.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Coureux P.D., Wells A.L., Houdusse A. A structural state of the myosin V motor without bound nucleotide. Nature. 2003;425:419–423. doi: 10.1038/nature01927. [DOI] [PubMed] [Google Scholar]
- 61.Regnier M., Martin H., Clemmens E. Cross-bridge versus thin filament contributions to the level and rate of force development in cardiac muscle. Biophys. J. 2004;87:1815–1824. doi: 10.1529/biophysj.103.039123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hibberd M.G., Dantzig J.A., Goldman Y.E. Phosphate release and force generation in skeletal muscle fibers. Science. 1985;228:1317–1319. doi: 10.1126/science.3159090. [DOI] [PubMed] [Google Scholar]
- 63.Takagi Y., Shuman H., Goldman Y.E. Coupling between phosphate release and force generation in muscle actomyosin. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2004;359:1913–1920. doi: 10.1098/rstb.2004.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hancock W.O., Huntsman L.L., Gordon A.M. Models of calcium activation account for differences between skeletal and cardiac force redevelopment kinetics. J. Muscle Res. Cell Motil. 1997;18:671–681. doi: 10.1023/a:1018635907091. [DOI] [PubMed] [Google Scholar]
- 65.De La Cruz E.M., Wells A.L., Ostap E.M. Actin and light chain isoform dependence of myosin V kinetics. Biochemistry. 2000;39:14196–14202. doi: 10.1021/bi001701b. [DOI] [PubMed] [Google Scholar]
- 66.Greenberg M.J., Arpağ G., Ostap E.M. A perspective on the role of myosins as mechanosensors. Biophys. J. 2016;110:2568–2576. doi: 10.1016/j.bpj.2016.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sung J., Nag S., Spudich J.A. Harmonic force spectroscopy measures load-dependent kinetics of individual human β-cardiac myosin molecules. Nat. Commun. 2015;6:7931. doi: 10.1038/ncomms8931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Reconditi M., Linari M., Lombardi V. The myosin motor in muscle generates a smaller and slower working stroke at higher load. Nature. 2004;428:578–581. doi: 10.1038/nature02380. [DOI] [PubMed] [Google Scholar]
- 69.Piazzesi G., Lucii L., Lombardi V. The size and the speed of the working stroke of muscle myosin and its dependence on the force. J. Physiol. 2002;545:145–151. doi: 10.1113/jphysiol.2002.028969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Swank D.M., Knowles A.F., Bernstein S.I. The myosin converter domain modulates muscle performance. Nat. Cell Biol. 2002;4:312–316. doi: 10.1038/ncb776. [DOI] [PubMed] [Google Scholar]
- 71.Spudich J.A. Hypertrophic and dilated cardiomyopathy: four decades of basic research on muscle lead to potential therapeutic approaches to these devastating genetic diseases. Biophys. J. 2014;106:1236–1249. doi: 10.1016/j.bpj.2014.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Alamo L., Ware J.S., Padrón R. Effects of myosin variants on interacting-heads motif explain distinct hypertrophic and dilated cardiomyopathy phenotypes. eLife. 2017;6:e24634. doi: 10.7554/eLife.24634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hooijman P., Stewart M.A., Cooke R. A new state of cardiac myosin with very slow ATP turnover: a potential cardioprotective mechanism in the heart. Biophys. J. 2011;100:1969–1976. doi: 10.1016/j.bpj.2011.02.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Spudich J.A. The myosin mesa and a possible unifying hypothesis for the molecular basis of human hypertrophic cardiomyopathy. Biochem. Soc. Trans. 2015;43:64–72. doi: 10.1042/BST20140324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Toepfer C.N., Wakimoto H., Seidman C.E. Hypertrophic cardiomyopathy mutations in MYBPC3 dysregulate myosin. Sci. Transl. Med. 2019;11:eaat1199. doi: 10.1126/scitranslmed.aat1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.