

SUMMARY
A fraction of mature T cells can be activated by peripheral self-antigens, potentially eliciting host autoimmunity. We investigated homeostatic control of self-activated T cells within unperturbed tissue environments by combining high-resolution multiplexed and volumetric imaging with computational modeling. In lymph nodes, self-activated T cells produced IL-2, which enhanced local regulatory T cell (Treg) proliferation and inhibitory functionality. The resulting micro-domains reciprocally constrained inputs required for damaging effector responses, including CD28 co-stimulation and IL-2 signaling, constituting a negative feedback circuit. Due to these local constraints, self-activated T cells underwent transient clonal expansion, followed by rapid death (“pruning”). Computational simulations and experimental manipulations revealed the feedback machinery’s quantitative limits: modest reductions in Treg micro-domain density or functionality produced non-linear breakdowns in control, enabling self-activated T cells to subvert pruning. This fine-tuned, paracrine feedback process not only enforces immune homeostasis but also establishes a sharp boundary between autoimmune and host-protective T cell responses.
Keywords: Immune Homeostasis, Autoimmunity, Regulatory T Cells, Quantitative Tissue Imaging, Feedback Control, Computational Modeling, IL-2, IL-2Rα, CTLA-4, Apoptosis
Graphical Abstract
A combination of imaging and modeling approaches identifies a paracrine feedback mechanism that enables regulatory T cells to eliminate highly self- reactive T cells, thereby dampening autoimmunity.
INTRODUCTION
To promote host defense, αβ-T lymphocytes undergo clonal expansion and differentiate into effectors that produce inflammatory cytokines or cytotoxins. This process is initiated by productive interactions between the T cell receptor (TCR) and antigenic peptides bound to major histocompatibility complex-encoded molecules (pMHCs), which are displayed by antigen presenting cells (APCs). Due to a quasi-random gene recombination process, each T cell harbors a structurally unique TCR with distinct binding specificity. The net result is a repertoire of cells that can recognize and respond to a broad range of peptides (Vrisekoop et al., 2014). While this receptor diversity protects the host against disparate threats, it also introduces significant risk: many TCRs generated during lymphocyte development will react strongly to self-pMHCs (Xing and Hogquist, 2012). Because no genetic mechanisms exist to limit this self-reactivity, T cell-extrinsic control systems are required to prevent autoimmunity.
Initial control occurs within the thymus, filtering developing T cells based on their reactivity toward self-pMHCs. T cells that react modestly receive survival signals and mature (“positive selection”), whereas those that fail to react sufficiently undergo apoptosis (“death by neglect”). Many developing T cells are also eliminated by apoptosis if they react too strongly (“negative selection”). Although efficient, the latter process is not absolute: some overtly self-reactive T cells escape elimination, mature, and exit the thymus (Klein et al., 2014; Malhotra et al., 2016).
Incomplete negative selection necessitates auxiliary control within secondary lymphoid organs (SLOs), critical hubs that initiate T cell effector responses (Baptista et al., 2019). In the absence of infection or malignancy, APCs within SLOs — predominantly dendritic cells (DCs) — display an abundance of self-pMHCs that could stimulate ineffectively filtered T cells, potentially generating tissue-damaging effectors. To prevent this outcome, SLOs contain specialized CD4+ T cells, known as Tregs, which restrain T cell responses to self-antigens (Fontenot et al., 2003; Hori et al., 2003; Khattri et al., 2003). Notably, loss of Tregs precipitates rapid autoimmunity in mice and humans (Bennett et al., 2001; Brunkow et al., 2001; Fontenot et al., 2003, 2005; Kim et al., 2007).
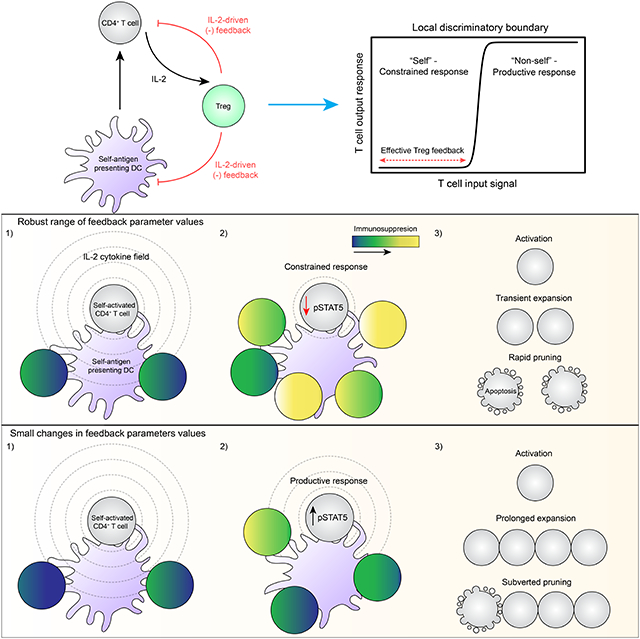
Two models could explain Treg operations in SLOs: 1) preventing T cell activation toward self-pMHC or 2) constraining T cells in response to their activation by self-pMHC. The latter constitutes a negative feedback design, which often confers a system with robustness towards noise (Becskei and Serrano, 2000). In this scenario, T cell activation by self-pMHC would represent “noise” sensed by Tregs and subsequently suppressed, restoring the system to a steady state. Although Tregs can prevent T cell activation, supporting model 1, this form of proactive control is incomplete (Akkaya et al., 2019; Tadokoro et al., 2006; Tang et al., 2006). Indeed, we recently documented activated CD4+ T cells that produced the cytokine interleukin 2 (IL-2) under homeostatic conditions (Liu et al., 2015).
The negative feedback model is conceptually attractive but lacks experimental support. Tregs possess multiple molecular mechanisms that could constrain T cells activated by self-antigens (“self-activated T cells”) (Shevach, 2009). Whether these mechanisms operate concurrently, sequentially, or opportunistically is unknown. Moreover, the terminal fate of a self-activated T cell constrained by a potential feedback process is difficult to predict. Lastly, if Tregs do exert feedback, the margin for error before a self-activated T cell escapes control requires quantitative assessment. A framework integrating these distinct features is lacking, precluding the development of models that can accurately predict the emergence of autoimmune disease (Marson et al., 2015).
By combining quantitative tissue imaging and computational modeling, we identified a negative feedback circuit that limits incipient autoimmunity. Self-activated CD4+ T cells secreted IL-2, which triggered paracrine signaling in neighboring Tregs. As a result, Tregs increased in local density and functionality, constraining inputs required to sustain self-activated T cell proliferation and survival. This feedback machinery is fine-tuned by the local density and functionality of Tregs surrounding an activated CD4+ T cell. Our findings reveal an extra-thymic filtering process that continuously removes self-activated T cells from the host, ultimately preserving immune homeostasis.
RESULTS
A small fraction of CD4+ T cells are continually activated by self-antigens in healthy hosts
We previously identified rare CD4+ T cells producing IL-2 in healthy hosts, suggesting that Tregs likely operate in a negative feedback mode, rather than pre-empting responses against self (Liu et al., 2015). However, this cytokine measurement underestimates the number of self-activated T cells due to limited detection sensitivity. As an alternative, we exploited the fact that T cells express inhibitory receptors, particularly programmed cell death protein 1 (PD-1), in response to sufficient TCR signaling (Hochweller and Anderton, 2005). We combined immunostaining, confocal microscopy, and computer vision techniques to assess PD-1 expression in lymph nodes (LNs) of healthy C57BL/6 mice (Figure 1SA-C) (Gerner et al., 2012). To distinguish between CD4+ Foxp3+ Tregs (Tregs) and conventional CD4+ Foxp3− T cells (CD4+ T cells), we included antibodies directed against CD3, CD4 and Foxp3 (Figure 1A, Figure S1B). Among paracortical CD4+ T cells, PD-1 expression ranged from undetectable to highly enriched on individual cells (Figure 1A & B, Figure S1C).
Figure 1. Tregs selectively accumulate around self-activated T cells, forming micro-domains.

(A) Left side: PD-1 expression variation among paracortical CD4+ T cells. Dashed yellow line: popliteal LN paracortex. White box: region of interest (ROI). Right side: Magnified ROI. XZ optical slices highlight cell of interest (white arrow). Scale bars = 200 μm (left) and 20 μm (right) (B) In situ quantification of A. TRAP = anti-CD62L Ab + FTY720. n = 3-4 animals. Data are from 2 independent experiments. (C) Spatial density function of Tregs shown in A. White dots: PD-1+ CD4+ T cells within the paracortex. Scale bar = 200 μm. (D) Local Treg densities surrounding PD-1+ CD4+ T cells or randomly sampled PD-1− CD4+ T cells. Solid lines: local regressions determined by the LOESS method. Error bars: 95% confidence intervals. Data are representative of 3 independent experiments.
To test whether the PD-1 expression was due to TCR-mediated stimulation, we injected animals with a blocking directed against I-Ab, the MHC class II molecule in C57BL/6 mice (Bhattacharya et al., 1981). Because TCR-MHCII interactions also control CD4+ T cell transit times through the LN, we first trapped T cells in the LN using CD62L blocking antibodies and FTY720 (Gallatin et al., 1983; Mandl et al., 2012; Matloubian et al., 2004). While trapping alone did not affect PD-1 expression, trapping combined with I-Ab blocking antibodies reduced PD-1 expression by ~ 45% in CD4+ T cells and ~50% in Tregs after 24 hours (Figure 1B, Figure S1D).
Exogenous food proteins, commensal organisms, or self-proteins could account for CD4+ T cell stimulation in the absence of infection. To examine the first two possibilities, we harvested skin draining LNs, which lack presentation of food antigens, from germ-free (GF) mice, which lack commensal organisms. When compared to equivalent LNs from specific-pathogen-free (SPF) mice, PD-1 expression was similar in CD4+ T cells (Figure 1B). Thus, PD-1 expression marked CD4+ T cells activated by self-antigens during homeostasis.
Tregs form densely packed micro-domains around self-activated T cells
The negative feedback model requires Tregs and self-activated T cells to reside near one another. Therefore, we assessed the spatial relationship between both cell types in the LN paracortex (Figure 1A). We first discretized PD-1 expression, setting a threshold that encompassed the top 2% of the distribution (“PD-1+”) (Figure 1B, Figure S1C). We then computed a spatial density estimation, which revealed multiple Treg “micro-domains” throughout the paracortex. Notably, self-activated PD-1+ CD4+ T cells localized within these Treg-dense micro-domains (Figure 1C). To quantify this observation, we treated each PD-1+ CD4+ T cell as an origin point and computed the surrounding Treg density in three-dimensional space (λTreg). Compared to randomly sampled PD-1− CD4+ T cells, λTreg was selectively enriched around PD-1+ CD4+ T cells, both in SPF and GF animals (Figure 1D, Figure S1E).
Paracrine IL-2 signaling enhances local Treg functionality, forming a negative feedback circuit
To exert negative feedback, Tregs in micro-domains should alter their functional state in response to signals from self-activated T cells. This prediction was tested using multiplexed imaging to simultaneously measure cell-to-cell variation across parameters previously implicated in Treg function (Figure 2A) (Buszko and Shevach, 2020). The measurements were then subjected to unsupervised hierarchical clustering, which revealed four groups of cells (Figure 2B). Cluster 1 comprised Tregs greater than one standard deviation (SD) below the mean of most parameter distributions (“non-activated”). By contrast, clusters 3 and 4 generally comprised cells with inverse phenotypes (“activated”), likely corresponding to “effector Tregs” (Levine et al., 2014; Smigiel et al., 2014; Vahl et al., 2014). Cluster 2 was more heterogeneous. Consistent with the negative feedback model, Tregs within clusters 3 and 4, but not 1 and 2, were selectively enriched near PD-1+ CD4+ T cells compared to randomly sampled PD-1− CD4+ T cells (Figure 2C).
Figure 2. Paracrine IL-2 signaling initiates local Treg feedback.

(A) Multiplexed imaging of Treg micro-domains in inguinal LNs. Left panel: Treg micro-domain. Arrows: PD-1+ CD4+ T cell. Scale bar = 20 μm. Right panel: Image gallery. Inner and outer zones (yellow dashed lines) depict Tregs inside and outside of the micro-domain, respectively. White dashed circle: PD-1+ CD4+ T cell. (B) Unsupervised hierarchical clustering of paracortical Tregs (k = 3477 cells) during homeostasis. Representative of 3 independent experiments (C) Tregs from each cluster in B residing within 40 μm of PD-1+ CD4+ T cells (k = 260 cells) or randomly sampled PD-1− CD4+ T cells (k = 260 cells). ***p < 0.0001 determined using a two-way ANOVA with the Tukey correction (D) Log2 fold-change in local Treg Foxp3, CTLA-4, and IL-2Rα expression 24h post-IL2 blocking Abs. Dashed red line: isotype control condition. Individual cells pooled from n = 3 animals. Data are mean ± 95% confidence intervals derived using non-parametric bootstrapping. ***p < 0.0001 determined using a one-tailed Student’s T test (E) pSTAT5 signal in Foxp3DTR+/+ animals treated with DTX for 7h. Inner dashed circle: PD-1+ CD4+ T cell. Outer dashed circle: Treg micro-domain. Scale bar = 20 μm. (F) PDFs of pSTAT5+ Tregs in the popliteal LN paracortex 7h after injecting PBS (left) or DTX (right). White dots: pSTAT5+ CD4+ T cells. Scale bar = 250 μm (G) pSTAT5+ CD4+ T cell (left) or pSTAT5+ Treg (right) frequencies in the popliteal LN paracortex. Data are mean ± SEM. Each dot represents an individual mouse pooled from two independent experiments. p values determined using an unpaired, two-tailed t test. (H) Single-cell PD-1 expression in paracortical Foxp3− CD4+ IL-2 responders (pSTAT5+) or non-responders (pSTAT5−) 7h post-DTX. n = 4 animals. Data are from 2 independent experiments.
Among candidate signals that could drive a negative feedback response from Tregs, IL-2 stood out: CD4+ T cells rapidly produce this cytokine following activation, Tregs require IL-2 for optimal function yet paradoxically do not produce it, and rare CD4+ T cells are the predominant source of IL-2 during homeostasis (Almeida et al., 2006; Amado et al., 2013; Fontenot et al., 2005; Furtado et al., 2002; Liu et al., 2015; Setoguchi et al., 2005). To explore whether IL-2 secreted by self-activated T cells initiated Treg feedback, we first examined if CD4+ PD-1+ T cells produced IL-2 in vitro. Indeed, a significant fraction of activated CD4+ T cells expressed PD-1 while simultaneously secreting cytokine (Figure S1F & G). Endogenous CD4+ IL-2+ T cells also exhibited elevated PD-1 expression in situ (Figure S1H & I). Most importantly, CD4+ PD-1+ T cells in LNs were often surrounded by Tregs containing pSTAT5, a hallmark of signaling downstream of the IL-2 receptor complex (Figure 2B, Figure S1H & I). Thus, many CD4+ PD-1+ T cells were producing or had recently produced IL-2 in LNs of healthy hosts.
Upon sensing paracrine IL-2, neighboring Tregs should enhance their local functionality, accounting in part for their unique phenotypes in micro-domains. We tested this possibility by administering IL-2 blocking antibodies (Spangler et al., 2015). After 24h, we observed a modest 34%, 36%, 41% reduction in global Treg Foxp3, CTLA-4, and IL-2Rα expression, respectively, suggestive of partially reduced suppressive functionality (Figure S1J). However, the Tregs in micro-domains surrounding PD-1+ CD4+ T cells were disproportionately affected as local Treg Foxp3, CTLA4, and IL-2Rα expression fell by 60%, 67%, and 62%, respectively (Figure 2D). These observations demonstrated that IL-2 secreted from self-activated T cells enhanced local Treg function.
To sustain high level IL-2 production, activated CD4+ T cells require signaling through CD28, a costimulatory receptor that binds CD80 and CD86 on DCs. Tregs within micro-domains should limit these interactions due to enhanced expression of CTLA-4, which outcompetes CD28 for both costimulatory ligands (Figure 2A-C) (Collins et al., 2002; Qureshi et al., 2011). Tregs in micro-domains should also limit autocrine and paracrine IL-2 signaling in self-activated T cells due to enhanced expression of IL-2Rα, which greatly increases binding affinity for the cytokine (Feinerman et al., 2010). Disrupting micro-domain functionality should, therefore, increase IL-2 production and sensing in self-activated T cells. We tested this hypothesis using animals in which Tregs expressed the human diphtheria toxin receptor (DTR) (Foxp3 DTR+/+) (Kim et al., 2007). This system enabled acute Treg perturbation since diphtheria toxin (DTX) selectively inhibits protein synthesis in cells expressing the DTR (Honjo et al., 1969). Seven hours after injecting DTX, Treg CTLA-4 and IL-2Rα expression declined by 47% and 32%, respectively, without grossly altering Treg cell number (Figure S1K, L). Concomitantly, we observed a ~14-fold increase in pSTAT5+ CD4+ T cells, an effect prevented by IL-2 blocking Abs (Figure 2E-G, Figure S1M). These emerging IL-2 “responders” were highly enriched in PD-1 expression (Figure 2H). Animals treated with DTX also displayed a ~4-fold increase in pSTAT5+ Tregs, an effect once again prevented by IL-2 blocking Abs (Figure 2E-G). This observation was consistent with enhanced IL-2 production, presumably saturating neighboring Tregs and thereby increasing the range of cytokine diffusion throughout the LN (Oyler-Yaniv et al., 2017). Overall, these data revealed a paracrine feedback process that rapidly constrained IL-2 production and sensing by self-activated T cells.
Self-activated T cells proliferate despite local IL-2 constraints but are rapidly pruned from the host
To determine the fate of self-activated T cells, we synchronized T cell activation and tracked responses over time using an adoptive transfer system. Healthy BALB/c mice were injected with Rag2−/− TxA23 cells — transgenic CD4+ T cells that recognize an endogenous self-peptide derived from the gastric K+/H+ ATPase. The peptide is presented constitutively by DCs within the gastric LN of BALB/c mice (Scheinecker et al., 2002). Importantly, low numbers of transferred TxA23 cells only induce autoimmune gastritis in the absence of recipient Tregs (McHugh et al., 2001; Suri-Payer et al., 1998).
Within the gastric LN, TxA23 cells, but not co-transferred WT cells, upregulated PD-1 over time, consistent with TCR-mediated activation despite functional Tregs. However, most TxA23 cells lacked pSTAT5 expression, analogous to polyclonal self-activated T cells in C57BL/6 mice (Figure S2A-C, Figures 1, Figure 2). Next, we imaged the entire gastric LN at single-cell resolution using Clearing enhanced 3D (Ce3D) (Li et al., 2017). This method allowed us to study physiological numbers of TxA23 and WT cells in situ, limiting artefacts due to intraclonal competition (Figure 3A, B) (Hataye et al., 2006; Jenkins and Moon, 2012). We initially allowed a small number of TxA23 and WT cells to enter the gastric LN for 24h, then blocked further entry and exit using anti-CD62L and FTY720, respectively, confining cells to a single location for accurate enumeration. 24h post-transfer, ~18 ± 4 TxA23 cells and ~20 ± 3 WT cells were observed within the entire gastric LN (Figure 3A, Movie S1). Surprisingly, by 72h post-transfer, TxA23 cells selectively increased in number by 20-fold, and many showed increased physical volume, a prerequisite to division. We also observed rare clusters of these cells scattered throughout the gastric LN, consistent with local proliferation (Figure 3A, B, Movie S2). However, such expansion was relatively short-lived; TxA23 cell numbers fell significantly by 120h post-transfer and returned to an average of ~31 ± 7 by 192h post-transfer (Figure 3A, Movie S3). These data revealed that self-activated TxA23 cells underwent fleeting proliferation followed by rapid death, a process we refer to as “pruning”.
Figure 3. Self-activated T cells proliferate despite local IL-2 constraints but are rapidly pruned from the host.

(A) Top panel: experimental schematic. Bottom panel: absolute number of cells per gastric LN. Each dot represents a LN from a single mouse. Local regressions with 95% confidence intervals are shown for each cell type over time. Gastric LNs from n = 3-8 mice were imaged at each time point. Data are pooled from 2-3 independent experiments (B) Ce3D imaging of the gastric LN at 72h post-transfer. Insets: magnified ROIs (white dashed boxes). Scale bars = 100 μm and 20 μm (Inset) (C) In situ quantification of TxA23 cell proliferation at 96h post-transfer without FTY-720 and anti-CD62L antibodies. A Gaussian mixture model (GMM) was fit to the CellTracker DeepRed distribution. k = 552 TxA23 cells pooled from gastric LNs of n = 4 animals. Data are from two independent experiments. Inset: fraction of TxA23 cells within each mixture component. Data are Mean ± 95% confidence intervals (D) Ki67 expression in paracortical CD4+ T cells. Left side: PD-1+ CD4+ T cells surrounded by high densities of Tregs. Right side: Magnified image gallery of the ROI (white dashed box). Arrows: PD-1+ CD4+ T cells enriched in Ki67. Scale bar = 40 μm and 20 μm (ROI). (E) Ki67 versus PD-1 expression in paracortical CD4+ T cells of GF C57BL/6 nice. Data are representative of 3 independent experiments. (F) Enriched active caspase 3 expression in a PD-1+ CD4+ T cell. Inset: magnified ROI (white dashed box). Scale bars = 20 μm and 5 μm (inset). (G) IL-2:S4B6-1 innunoconplex schematic. (H) In situ quantification of active caspase 3+ cells. Data are mean ± SEM. Each dot represents an individual nouse pooled from 2 independent experiments. p values determined using a one-way ANOVA with the Tukey correction.
To ensure that confining TxA23 cells to the gastric LN did not artefactually promote proliferation, we repeated the experiment in the absence of anti-CD62L and FTY-720. Transferred cells were pre-labelled with fluorescent dyes, providing a means to quantify cell division in situ (Figure S3A-C). 48h post-transfer, two TxA23 cell populations were observed in the gastric LN: 60% were undivided, while 40% were a product of one division (Figure S3B). At 96h post-transfer, TxA23 cell proliferation increased further; 18% of cells were undivided while the remainder were the product of 1 (19%), 2 (34%) or 3 (29%) rounds of cell division (Figure 3C, Figure S3C). In comparison, 100% of WT cells were undivided at both timepoints (Figure S3D, E). Proliferation was not a unique property of TxA23 cells as polyclonal CD4+ T cells enriched in PD-1 exhibited high expression of Ki67, a cell-cycle associated protein (Figure 3D & E).
Because C57BL/6 animals generally fail to develop autoimmunity, many PD-1+ CD4+ T cells should be eliminated by pruning, analogous to TxA23 cells. We assessed this possibility in situ by immunostaining for active caspase 3, the effector protease underlying apoptosis (Nicholson et al., 1995). Compared to PD-1− CD4+ T cells, PD-1+ CD4+ T cells were highly enriched in active caspase 3, consistent with ongoing pruning (Figure 3F-H).
Given IL-2’s role in enhancing T cell proliferation and survival, Treg micro-domains likely promote pruning by restricting IL-2 sensing in self-activated T cells (Akbar et al., 1993, 1996). Therefore, we administered the IL-2:S4B6-1 immunocomplex — which engages IL2-Rβ and IL-2Rγ, but not IL-2Rα — to force IL-2 signaling in self-activated T cells, despite Tregs (Boyman et al., 2006; Spangler et al., 2015). This complex reduced activated caspase 3 expression by ~50% in PD-1+ CD4+ T cells, but not PD-1− CD4+ T cells (Figure 3G &H). IL-2-S4B61 also enhanced the absolute number of TxA23 cells in the gastric LN by ~4-fold at 72h post-transfer, consistent with increased proliferation and survival, while selectively increasing the number and size of TxA23 cell clusters (Figure S3F-J). These results supported a model in which Treg micro-domains rapidly constrained IL-2 sensing in self-activated T cells, ultimately shaping their division number and “time to death” (Heinzel et al., 2017; Marchingo et al., 2014).
IL-2 enhances local Treg proliferation, promoting micro-domain formation
We explored whether IL-2 contributed to micro-domain formation itself by monitoring TxA23 cell activation along with local Treg signaling state and density over time. 12.5h post-transfer, we observed enrichment of pSTAT5+ Tregs (λpSTAT5+ Treg) surrounding TxA23 but not WT cells (Figure 4A), implying that activated TxA23 cells secreted IL-2 sensed by proximal Tregs. However, this early wave of IL-2 consumption preceded the formation of true micro-domains since the local Treg density (λTreg), irrespective of pSTAT5 status, remained low (Figure 4A).
Figure 4. Treg micro-domain formation is part of the IL-2-driven feedback process.

(A) 1x105 TxA23 cells and WT cells were co-transferred into BALB/c recipients. Local densities of pSTAT5+ Tregs (λpSTAT5+Treg - top panel) and total Tregs (λTreg – bottom panel) surrounding individual TxA23 cells (nagenta) or WT cells (green). Local regressions with 95% confidence intervals determined by the LOESS method. k = 25-150 cells pooled from gastric LN sections of n = 4-6 animals at each tine point. Data are from 2 independent experiments. (B) pSTAT5 signal in Tregs surrounding a WT cell (top panel) or TxA23 cell (bottom panel) in the sane gastric LN 24h post-transfer. Scale bar = 20 μm. (C) Adoptive transfers performed as in Figure 3A while co-injecting IL-2 blocking Abs or isotype controls. Gastric LNs harvested 24h post-transfer. Scale bar = 20 μm (D) Maximum deviation in λTreg (Δ λTreg) between TxA23 vs. WT cells 24h after injecting IL-2 blocking Abs or isotype controls (E) Δ λTreg between PD1+ CD4+ T cells vs. randomly sampled PD-1− CD4+ T cells 24h after injecting C57BL/6 nice with IL-2 blocking Abs or isotype controls. For D and E, 95% confidence intervals were derived by non-parametric bootstrapping. Data are from 2 independent experiments.
By 24h post-transfer, λpSTAT5+ Treg began to decay, suggesting that TxA23 cells now produced less IL-2 due to Treg-mediated feedback. In accord, λTreg increased simultaneously and selectively near TxA23 cells (Figure 4A and B). By 48h post-transfer, λpSTAT5+ Treg for TxA23 cells returned to near baseline levels, indicating that the response had largely subsided, while λTreg remained partially elevated compared to WT cells (Figure 4A).
Next, we repeated the experiment but co-injected IL-2 blocking antibodies (Spangler et al., 2015). After 24h, the blocking antibodies eliminated the pSTAT5 signal within Tregs without significantly altering their global density (Figure S4A & B). However, we observed a preferential reduction in the local Treg density surrounding TxA23 cells (Figure 4C & D). These findings also held true in C57BL/6 mice: IL-2 blocking antibodies preferentially reduced the local Treg density surrounding PD-1+ CD4+ T cells (Figure 4E).
Many Tregs in micro-domains expressed Ki67 and exhibited increased volume, suggesting that paracrine IL-2 signaling facilitated local Treg proliferation, giving rise to micro-domains (Figure 2A-C) (Setoguchi et al., 2005). To better visualize this pattern, we computed local Treg densities weighted to local Treg Ki67 expression levels, revealing highly proliferative micro-domains within the paracortex (Figure 5A). Correlating Treg Ki67 expression with intercellular distance further revealed that Tregs most enriched in Ki67 were highly clustered. These results could not be explained by chance as the observed probability distribution did not fit a random permutation null model (Figure 5B).
Figure 5. Nascent micro-domains are a product of localized Treg proliferation.

(A) Perspective plot illustrating the local Treg density weighted to local Treg Ki67 expression within a popliteal LN section. Dashed white lines: B cell follicles (B) Ki67 expression in individual Tregs as a function of intercellular distance. The experimentally derived probability distribution (black line) was compared to a random permutation null model. Upper and lower pointwise envelopes (shaded purple regions) are shown for 99 Monte Carlo simulations. Dashed purple line represents the average of the simulations. p = 0.02 at a distance of 5 μm using a two-sided Monte Carlo test. Data is representative of 3 independent experiments. (C) Fluorescent protein recombination frequencies in Tregs 14 days following tamoxifen. Each dot represents an individual animal. (D) Clonal Treg clusters (dashed white circles) in Foxp3CreERT2+/−-Confettifl/− mice. White traces: individual Tregs of interest expressing Foxp3. Scale bars = 20 μm. (E) Local density of Tregs expressing the same FP within 40 μm of one another. Observed FP frequencies were permuted at random across paracortical Treg positions. Points that reside on the dashed black line (slope = 1) are explained by chance. LNs from 4 representative mice were quantified and pooled from 2 independent experiments.
If nascent micro-domains are a product of local proliferation, then the constituent Tregs should display clonality. We therefore generated Foxp3CreERT2+/−-Confettifl/− animals in which Tregs stochastically express distinct fluorescent proteins (FPs) upon administrating tamoxifen (Figure S4C) (Livet et al., 2007; Rubtsov et al., 2010). Micro-domain clonality was subsequently assessed based on Treg clusters expressing the same fluorescent protein (FP). Low gene recombination frequencies made it unlikely for single-FP clusters to arise by chance (Figure 5C).
We observed co-localized Treg clusters expressing either CFP, YFP or RFP near CD4+ PD-1+ T cells in the LN paracortex, consistent with local expansion (Figure 5D and E, Figure S4D). Such clusters could not be explained by chance as the observed density of single FP-expressing Tregs failed to fit a random permutation null model (Figure 5E). These results revealed that IL-2 secreted by self-activated T cells enhanced local Treg proliferation, forming micro-domains of activated Tregs that reciprocally constrained self-activated T cells.
A multiscale computational model predicts the feedback machinery’s quantitative limits
Our data revealed a feedback control system that constrained weakly activated T cells, halting incipient autoimmune responses. To assess the feedback machinery’s robustness — the property of functional invariance towards internal parameter variation — we built a multiscale computational model that incorporated our experimental observations and data from the literature (Figure 6A, Table S1 & S2). Importantly, we simulated internal parameter variation by sampling combinations of parameter values from their homeostatic ranges (Table S1).
Figure 6. A computational model predicts that modest reductions in micro-domain size or functionality enable self-activated T cells to respond to IL-2.

(A) Schematic illustrating multiscale model dynamics (B) Distribution of pSTAT5CD4-max values from 20000 dynamic simulations. Inset: log10 scale. Dashed red line: discrete threshold for parameter configurations within the top 5% of the distribution (C) Two-dimensional visualization of sampled parameter space using t-SNE. Each dot represents a parameter configuration. Colored dots represent configurations with pSTAT5CD4-max values within the top 5% of the distribution (D) Heatmap illustrating individual parameters (columns) and their standard scores (Z-score) within each configuration (rows) from the top 5% of the pSTAT5CD4-max distribution. (E) Global variable importance (GVI) of dynamical model parameters (y-axis) fitted by the RFML regression model using “combined data” (see Figure S5B). (F) and (G) Percentage of active, inactive, and susceptible configurations in the parameter space following indicated perturbations. (H) Susceptible configurations were pooled from each perturbation in G and visualized within parameter space (red dots). (I) GVI of the RFML classification model comparing susceptible and active configurations. (J) Pearson correlation matrix of individual parameters in susceptible configurations.
We simulated CD4+ T cell activation by an antigen-bearing DC, along with Treg imposed signaling constraints that evolved over time (Figure 2D, Figure 4A, Figure 6A). We then measured the maximum pSTAT5 signal within the CD4+ T cell (pSTAT5CD4-max) at any time point following activation (up to 120h) as a readout of escape from Treg control. To assess the impact of individual parameters on the pSTAT5CD4-max, we applied a new computational framework, termed Machine learning of Parameter-Phenotype Analysis (MAPPA) (Park et al., 2019). MAPPA analyzed 20,000 dynamic simulations, each initialized with a combination of biologically plausible parameter values (“configurations”). The majority of parameter configurations generated a low pSTAT5CD4-max, consistent with the nearly negligible frequency of pSTAT5+ CD4+ T cells in LNs of healthy animals (Figure 2F & G, Figure 6B & C). However, the few configurations that did produce a large pSTAT5CD4-max — referred to as “active” configurations — showed characteristic features, such as elevated co-stimulatory molecules on DCs ([CD80/86]) and low local Treg densities (Tregλ) (Figure 6D, Table S1).
MAPPA subsequently built a random forest (RF) machine-learning (ML) regression model that mapped parameter configurations to pSTAT5CD4-max with high predictive accuracy (r = 0.95) (Figure S5A, B). This statistical model determined which parameters were most critical in predicting pSTAT5CD4-max and ranked Tregλ second highest in terms of “variable importance” (Figure 6E, Figure S5C). Accordingly, new dynamical simulations showed that increasing or decreasing Tregλ by two-fold significantly impacted the number of active configurations (Figure 6E, F).
We proceeded to examine dynamic trajectories of IL-2 secretion, IL-2Rα expression, and pSTAT5 signaling in CD4+ T cells as Tregλ was lowered linearly down to 50% (Figure S5D-F). Decreasing the local Treg density increased IL-2 secretion duration and amplitude linearly (Figure S5D). By comparison, IL-2Rα expression and pSTAT5 signaling exhibited non-linearities: both parameter values increased sharply after a 40% reduction in the local Treg density (Figure S5E & F). Thus, modest reductions in micro-domain size shifted specific configurations from inactive to active.
The model implied that high Treg densities compensated for reduced Treg functionality in microdomains, preventing self-activated CD4+ T cells from responding to IL-2 (Figure 6D-F). To explore the limits of this compensation, we simulated disrupting local Treg functions by setting critical parameters outside of their homeostatic range, including Treg’s ability to strip co-stimulatory ligands or sense IL-2. Such perturbations can arise naturally due to genetic variation. For instance, two heterozygous variants have been identified in humans that reduce cell-surface CTLA-4 or IL-2Rβ expression by ~50%. Many patients expressing the CTLA-4 variant develop multi-organ autoimmune disease, primarily attributed to defective Treg function, while those expressing the IL-2Rβ variant appear stable (Kuehn et al., 2014; Schubert et al., 2014; Zhang et al., 2019). To test the impact of these perturbations on self-activated T cell control, we ran 20,000 new simulations, selectively reducing each protein by 50% in Tregs. As shown in Figure 6G, reducing CTLA-4 or IL-2Rβ by 50% resulted in 783 (3.9%) or 85 (0.4%) new configurations with a large pSTAT5CD4-max, respectively. We refer to these configurations as “susceptible” since they only become active upon perturbing local Treg functionality (Figure 6G & H).
To determine why the Treg CTLA-4 variant revealed significantly more susceptible configurations than the IL-2Rβ variant, we again examined dynamic trajectories. A 50% reduction in Treg CTLA-4 but not IL-2Rβ modestly increased IL-2 secretion duration and amplitude due to increased co-stimulation, leading to a non-linear amplification of IL-2Rα and pSTAT5 expression in the CD4+ T cell (Figure S5G-I). Thus, our model predicts that time-integrated (“total”) IL-2 secretion is critical in determining whether an activated T cell exceeds local micro-domain constraints, even when Treg density is high. These in silico data begin to explain why patients harboring heterozygous variants in CTLA-4 but not IL-2Rβ often develop autoimmunity.
Within the parameter space, susceptible configurations resided near those that were active, suggesting that the former were on the verge, but fell just short of breaking down during homeostasis (Figure 6H). MAPPA revealed that Tregλ was by far the most important parameter separating these two groups, with susceptible configurations generally exhibiting ~2-fold higher Tregλ than active configurations (Figure 6I and Figure S5J-L). Thus, elevated Treg densities in susceptible configurations were often sufficient to constrain self-activated T cells as long as the other parameter values fell within homeostatic ranges. However, modest secondary hits that affected local Treg functionality (e.g., reduction in CTLA-4 expression) made these regions active, enabling self-activated T cells to escape control.
To understand why susceptible configurations were on the verge of breaking down, we computed a correlation matrix, which revealed critical dependencies between parameters (Figure 6J). For instance, we observed a positive correlation between Tregλ and [CD80/86], indicating that high local Treg densities could be overridden by high levels of co-stimulatory ligands on DCs. A similar relationship emerged between Tregλ and the amount of antigen displayed on DCs ([Antigen]). Conversely, we observed a negative correlation between Tregλ and TCRoff-rate, indicating that high local Treg densities were also offset by activated CD4+ T cells with low pMHC-TCR off-rates (Figure 6J). These data illustrated how variation in baseline conditions altered the risk of self-activated T cells escaping control following modest changes to Treg functionality.
Modest reductions in Treg micro-domain size or functionality promote non-linear breakdowns in self-activated T cell control
To test our modeling predictions in vivo, we manipulated specific parameters underlying the feedback machinery (Figure 7A). To control micro-domain size, we employed Foxp3CreERT+/+-TCR-αfl/fl mice to selectively delete the TCR alpha chain in Tregs using tamoxifen (Levine et al., 2014) (Figure S6A). Loss of the Treg TCR caused micro-domains to dissipate without significantly affecting Treg numbers, while increasing paracortical pSTAT5+ CD4+ T cell frequency by ~4-fold (Figure 7B, Figure S6B & C). These IL-2 responders exhibited low densities of surrounding Tregs and expressed high levels of PD-1 and IL-2Rα (Figure 7C, Figure S6C & H). In parallel, paracortical pSTAT5+ Tregs increased by ~two-fold, suggestive of enhanced IL-2 production by self-activated T cells (Figure 7B). These observations demonstrated that Treg-mediated feedback failed in the absence of micro-domains, consistent with previous findings (Liu et al., 2015).
Figure 7. Modest reductions in Treg micro-domain size or functionality promote non-linear breakdowns in control.

(A) Schematic depicting inducible systems used to manipulate Treg parameters in vivo. (B) Log2 fold-changes in paracortical pSTAT5+ CD4+ T cells or pSTAT5+ Treg frequencies. Individual dots represent individual mice. p values determined using Welch’s two-sided t test comparing the treated animals to their respective controls (dashed red line) (C) PD-1 expression in IL-2 responders (pSTAT5+) vs. non-responders (pSTAT5−). Single-cell distributions pooled from n = 3-5 animals (D) 60% WT + 40% Foxp3-DTR+/+ chimeras were injected with DTX. Micrographs highlight a pSTAT5+ PD-1+ CD4+ T cell (dashed white circle) surrounded by a low density of Tregs. Scale bars = 10 μm. (E) Foxp3CreERT2+/+-CTLA-4+/fl were injected with tamoxifen. Micrographs highlight a PD-1+ CD4+ T cell exhibiting high pSTAT5 signal (dashed white circle), despite high local Treg density. Scale bars = 10 μm. (F) Log2 fold-change in active caspase 3+ CD4+ T cells in the paracortex. Data were normalized to respective vehicle controls (dashed red line). Each dot represents an individual mouse. p values determined using a one-way ANOVA with the Tukey correction. (G) pSTAT5 signal in 5C.C7 cells activated by the partial agonist (T102S) or the agonist (MCC(88-103)) at 48h post-injection. Dashed circles: 5C.C7 cells of interest. Scale bar = 10 μm. (H) Quantification of G. Each dot represents an individual mouse from two independent experiments. Data are mean ± SEM. p value determined using a two-tailed, Student’s t test.
The computational model predicted that the impact of micro-domain size on self-activated T cells was non-linear: reductions of less than 40% had only minor effects while those at or above 40% significantly increased IL-2 sensing (Figure S5D-F). To test these predictions, we sub-lethally irradiated WT animals, reconstituting their hematopoietic system with different ratios of bone marrow from WT and Foxp3DTR+/+ mice (Figure S6D & E). After 10-weeks, we selectively ablated the Foxp3DTR+/+ Tregs by administering DTX for two days (Figure S6F). A 15% reduction in Treg density had little effect on the system whereas a 40% reduction substantially increased the frequency of pSTAT5+ CD4+ T cells (~3-fold). These IL-2 responders were highly enriched in PD-1, consistent with activation by self-pMHC (Figure 7B-D). The model also predicted that reducing micro-domain size would increase IL-2 production by self-activated T cells (Figure S5D). Accordingly, we observed a ~two-fold increase in paracortical pSTAT5+ Tregs following a 40% reduction in Treg density (Figure 7B).
To simulate secondary hits that might convert potential susceptible configurations to active configurations, we manipulated micro-domain functionality by generating Foxp3CreERT+/+-CTLA-4+/fl and Foxp3CreERT+/− -IL2Rβ+/fl animals. In these mice, a single CTLA-4 or IL-2Rβ allele is disrupted in Tregs following tamoxifen administration (Figure S6G). pSTAT5+ CD4+ T cells increased by ~4- and ~2-fold upon disrupting Treg CTLA-4 or IL-2Rβ, respectively, supporting our predictions in silico (Figure 6G, Figure 7B, and Figure S5G-I). As expected, IL-2 responders exhibited high expression of PD-1 and IL-2Rα (Figure 7C and Figure S6H). Our simulations further predicted that perturbing Treg CTLA-4 but not IL-2Rβ would enhance IL-2 production by self-activated T cells (Figure S5G-I). Consistent with this possibility, we observed a ~2.5-fold increase in paracortical pSTAT5+ Tregs after deleting one copy of CTLA-4. By contrast, deleting one copy of Treg IL-2Rβ decreased the frequency of paracortical pSTAT5+ Tregs by ~two-fold, with a less severe effect on self-activated T cell control (Figure 7B).
Enhanced Treg IL-2 sensing coincided with elevated paracortical Treg frequencies in Foxp3CreERT+/+-CTLA-4+/fl animals treated with tamoxifen, likely due to enhanced proliferation (Figure S7A). This effect was not observed upon perturbing one copy of IL-2Rβ. However, many micro-domains still failed to prevent self-activated T cell escape in the CTLA-4+/− condition (Figure 7B and E). Thus, increasing local Treg density alone did not sufficiently compensate for reduced Treg functionality, specifically in terms of CTLA-4 (Figure S7B). This finding is in keeping with the fact that patients harboring heterozygous CTLA-4 variants also exhibit elevated Treg frequencies but often develop autoimmunity (Kuehn et al., 2014; Schubert et al., 2014).
Increased IL-2 sensing in self-activated T cells should impair their pruning. To examine this hypothesis, we chose two of the five inducible systems and assessed active caspase 3 expression. In either the 60% WT + 40% Foxp3DTR+/+ chimeras treated with DTX or Foxp3CreERT+/−-IL-2Rβ+/fl animals treated with tamoxifen, we observed a ~50% reduction in the frequency of apoptotic PD-1+ CD4+ T cells, but not PD-1− CD4+ T cells compared to vehicle controls (Figure 7F). These results demonstrated that modest perturbations to the feedback machinery enabled some self-activated T cells to escape control.
Local Treg feedback control gives rise to a sharp boundary between autoimmune and host-protective T cell responses
The Treg feedback system must constrain autoimmune T cell responses while permitting host-protective responses during times of infection. To explore how such discrimination occurs, we again turned to our modeling studies. MAPPA predicted that T cells in active and susceptible configurations generally exhibited reduced TCR off-rates than those in inactive configurations (Figure S7C). Thus, T cells stimulated by ligands with high affinities for the TCR — characteristic of foreign pathogens — might overcome Treg-mediated feedback (Vrisekoop et al., 2014). We examined this prediction by injecting healthy B10.A mice with transgenic Rag2−/− 5C.C7 cells. 5C.C7 T cells recognize an exogenous peptide derived from the foreign moth cytochrome c protein (MCC88-103) and presented by the MHC class II molecule, I-Ek (Fink et al., 1986).
We first employed a variant of the naturally occurring peptide that contains a threonine to serine substitution at position 102 (T102S) (Figure 7G). This peptide in complex with I-Ek exhibits a high off-rate for the 5C.C7 TCR, serving as a partial agonist that simulates a weak but stimulatory self-pMHC (Corse et al., 2010; Reay et al., 1994). Following subcutaneous injection of T102S, many transferred 5C.C7 cells displayed signs of TCR-mediated activation in the draining popliteal LN (Figure S7D). However, despite being stimulated by a foreign antigen, the behavior of these cells mimicked self-activated T cells, with an early wave of IL-2 sensing in proximal Tregs, followed by an increase in local Treg density (Figure S7E). Most importantly, the majority of 5C.C7 cells stimulated by T102S failed to phosphorylate STAT5 at any measured time points (Figure 7G and H, Figure S7F). These findings implied that Tregs were equally capable of constraining T cell responses toward self-antigens and weak foreign antigens (Pace et al., 2012).
We then repeated the experiment with an agonistic peptide, MCC(88-103), which, in complex with I-Ek, exhibits a low off-rate for the 5C.C7 TCR (Figure 7G). 48h post-injection, we observed a ~5-fold increase in transferred 5C.C7 cells expressing pSTAT5, consistent with their ability to respond to IL-2 even in the presence of Tregs (Figure 7H). Both λpSTAT5+ Treg and λTreg were also lower near cells stimulated with MCC(88-103) than T102S at later points in time, suggesting that some 5C.C7 cells outcompeted Tregs for IL-2 when engaging the agonist, limiting micro-domain growth (Figure S7E). This increase in 5C.C7 cell IL-2 sensing correlated with enhanced proliferation (Figure S7G & H). These results demonstrated that increasing TCR ligand strength alone enabled a fraction of activated T cells to escape Treg control.
DISCUSSION
Here we applied new imaging and computational approaches to study the interactions between self-activated T cells and Tregs at physiological numbers in situ. Our data reveal a paracrine feedback process initiated following CD4+ T cell activation. T cells activated by self-antigens or weak foreign antigens produce IL-2 and transiently expand, only for the progeny to die rapidly. Small changes (< two-fold) in the local Treg density or expression of critical molecules, such as CTLA-4, enable self-activated T cells to exceed feedback control and subvert pruning. These findings provide an integrated view of how immune homeostasis is maintained and emphasize how subtle shifts in immune cell parameter values can initiate autoimmune states (Simeonov & Wong et al., 2020).
Our approaches revealed ongoing pruning of self-activated T cells, building upon pioneering work from Lenardo and colleagues (Pandiyan et al., 2007). This process depends on at least two distinct Treg mechanisms operating within microdomains to rapidly limit IL-2 production and sensing by self-activated T cells (Pandiyan et al., 2007; Qureshi et al., 2011). Additional forms of control are also likely to function within these regions (Buszko and Shevach, 2020). While seemingly diverse, many Treg activities can constrain IL-2 production or sensing, a focus likely due to this cytokine’s multifactorial role in scaling immune responses (Liao et al., 2011). Moreover, since IL-2 is a soluble mediator, it can permeate throughout SLOs, potentially promoting cooperativity between distant T cells (Butler et al., 2013). Thus, Treg micro-domains must control the length scale over which this cytokine diffuses, limiting paracrine communication between multiple self-activated T cells (Oyler-Yaniv et al., 2017).
The present findings further emphasize that Tregs fail to prevent a fraction of T cells from being activated by self-antigens (Liu et al., 2015). While pro-active mechanisms, such as continuous stripping of pMHC complexes, may reduce the overall number of self-activation events, they are insufficient to maintain immune homeostasis (Akkaya et al., 2019; Qureshi et al., 2011). Instead, feedback control is essential for constraining the ~2–4% of CD4+ T cells that are continually activated in SLOs of healthy hosts.
Tregs exert this feedback control in a time-delayed manner. This design reoccurs throughout biology, conferring systems with unique dynamical properties. In response to an input stimulus, time-delayed negative feedback can lead to a pulsed output followed by adaptation, a process in which a system returns to its initial conditions (Ferrell, 2016). In biochemical networks, delays of this type most often occur due to the time necessary for de novo synthesis of the feedback molecules themselves (Alon, 2007). In the case of Tregs, the rates at which they proliferate and differentiate within micro-domains could serve as the delay, accounting for transient self-activated T cell proliferation (pulsed output), followed by rapid pruning (adaptation). A time delay would also permit host-protective immune responses during infection since T cells activated by high-affinity foreign antigens would have a critical time window during which they could override the slow rise in negative feedback. We suggest this override occurs in part due to the scaling of IL-2Rα expression and IL-2 production with increasing TCR ligand strength, allowing activated T cells to outcompete and saturate Tregs (Allison et al., 2016). The net result would be a pulsed output of higher amplitude or longer duration prior to adaptation.
The ability of individual micro-domains to constrain a self-activated T cell is tied to the density and functional state of constituent Tregs. These two parameters are fine-tuned by the degree of local IL-2 consumption, along with TCR and likely co-stimulatory receptor-mediated signaling. Different combinations of these three input signals should produce local Treg variation, likely accounting for micro-domain heterogeneity. This view explains previous observations whereby ablating the TCR, CD28 co-stimulatory receptor, or IL-2 receptor complex in Tregs alters their phenotype and promotes autoimmune disease (Chinen et al., 2016; Levine et al., 2014; Vahl et al., 2014; Zhang et al., 2013). Thus, no single input signal is sufficient for Tregs to maintain immune homeostasis. This concept also parallels the behavior of conventional T cells for which summation of the same signals dictates proliferation, differentiation, and survival (Heinzel et al., 2017; Marchingo et al., 2014; Tubo et al., 2013). These observations imply that competing signal integration processes operate between Tregs and self-activated T cells, resulting in a localized, multi-input “tug of war” (Höfer et al., 2012).
The Treg feedback machinery displayed robustness within a narrow range of parameter values. These findings are in line with the fact that specific monoallelic mutations, such as those affecting CTLA-4, can promote autoimmune disorders in humans (Kuehn et al., 2014; Schubert et al., 2014). This behavior is characteristic of systems governed by a sharp decision boundary. On either side, a system will converge to qualitatively distinct states (e.g., homeostasis or autoimmunity), in part due to non-linear amplification steps that reinforce stable “fixed points” (Altan-Bonnet and Germain, 2005; Busse et al., 2010). Our findings indicate that the local density and immunosuppressive capacity of Tregs in a micro-domain sets the decision boundary for individual T cell responses. While the particular non-linear components remain to be determined, such considerations suggest that breakdowns in immune homeostasis need not arise solely from a single, severe insult, such as a loss or gain of function mutation. Rather, a combination of subtle immune imbalances may erroneously tip rare self-activated T cells across a decision boundary, leading to small, but iterative bursts of tissue damage that culminate in disease over time (Marson et al., 2015). These imbalances could stem from disease-associated genetic variants that only modestly impact the expression kinetics or quantity of proteins implicated in Treg, DC, or mature T cell function. Modern computational algorithms have identified several candidate variants of this class predominantly within non-coding regions of the genome (e.g., expression quantitative trait loci) (Farh et al., 2015; Simeonov et al., 2017). Additionally, inflammatory signals or clinical immunotherapies may transiently shift the system outside of its robust parameter range, enabling some self-activated T cells to escape Treg feedback control (June et al., 2017; Sallusto et al., 1995; Tang et al., 2008; Termeer et al., 2002).
The integrative framework established in this study revealed new insights into the maintenance of immunological homeostasis. By studying the interplay between Tregs and activated T cells within intact tissue environments, we uncovered a paracrine feedback process that restrained early autoimmune and weak foreign responses. The quantitative features of this control system are highly relevant for understanding the emergence of autoimmune disorders, facilitating successful tumor immunotherapy, and enabling therapeutic manipulation of Tregs.
LIMITATIONS OF THE STUDY
The specificity of polyclonal self-activated T cells identified in this study remains unknown. Future work is required to identify the precise TCR-pMHC combinations that promote T cell activation during homeostasis. In addition, although we refer to polyclonal CD4+ PD-1+ T cells as “self-activated”, some of these cells may in fact be stimulated by peptides derived from endogenous retroviruses co-existing with the host (i.e., stably integrated into the mouse genome). We suscept that T cell responses against such peptides are constrained by Treg feedback control, analogous to responses against bonafide self-peptides. This concept warrants future investigation. Finally, our study explored conditions during which self-activated T cells escape Treg feedback control and pruning in SLOs. However, we did not track the behavior and fate of these escapees within peripheral tissues. Future work is required to determine how much tissue damage each escapee can impart. It will also be essential to determine the rate at which escapees must enter peripheral tissues to initiate clinically relevant autoimmunity.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Harikesh S. Wong (harikesh.wong@nih.gov)
Materials Availability
Mouse lines generated in this study are available upon request
Data and Code Availability
The code supporting the current study has been deposited on Github
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
The following mouse strains were used throughout this study:
C57BL/6J – The Jackson Laboratories, Cat#: 000664
C57BL/6J.Ly5a – NIAID-Taconic exchange program, Cat#: 8478
Germ Free C57BL/6 – NIAID Microbiome Program gnotobiotic animal facility
C57BL/6Ubi-GFP+/+ (C57BL/6-Tg(UBC-GFP)30Scha/J) – The Jackson Laboratories, Cat#: 004353
BALB/cJ – The Jackson Laboratories, Cat#: 000651
BALB/c CD45.1/.1 (CByJ.SJL(B6)-Ptprca/J) – The Jackson Laboratories, Cat#: 006584
BALB/c-Ubi-GFP+/+ (CByJ.B6-Tg(UBC-GFP)30Scha/J) – The Jackson Laboratories, Cat#: 007076
IL2Rβfl/fl (B6.129S1-Il2rbtm1Ukl/J) – The Jackson Laboratories, Cat#: 029657
R26R-Confetti (B6.129P2-Gt(ROSA)26Sortm1(CAG-Brainbow2.1)Cle/J) – The Jackson Laboratories, Cat#: 017492
Foxp3CreERT2 (Foxp3tm9(EGFP/cre/ERT2)Ayr/J) – The Jackson Laboratories, Cat#: 016961
Foxp3DTR+/+ (B6.129(Cg)-Foxp3tm3(DTR/GFP)Ayr/J) – The Jackson Laboratories, Cat#: 016958
CTLA4fl/fl – Kindly provided by Dr. Arlene Sharpe (Harvard Medical School, Boston, MA)
5C.C7 Rag2−/− (B10.A/Ai-[tg]TCR-Cyt-5CC7-I-[ko]RAG2 N12) – NIAID-Taconic exchange program, Cat#: 000094
B10.A (B10.A-Cd45a(Ly5a)/NAi N5) – NIAID-Taconic exchange program, Cat#: 000031
A23 Rag2−/− – Kindly provided by Dr. David Margulies (NIAID, NIH, Bethesda, MD)
Foxp3CreERT2+/−-Confettifl/− – Generated F1 hybrids for this study (Foxp3CreERT2+/+ x R26R-Confetti)
Foxp3CreERT2+/−-IL2Rβ+/fl – Generated F1 hybrids for this study (Foxp3CreERT2+/+ x IL2Rβfl/fl)
Foxp3CreERT2+/+-CTLA4+/fl – Generated for this study (Foxp3CreERT2+/+ x Foxp3CreERT2+/−-CTLA-4fl/fl)
Foxp3CreERT2+/+-TCR-αfl/fl – Kindly provided by Dr. Alexander Rudensky (Memorial Sloan Kettering Cancer Center, New York, NY)
Mice were bred and maintained under specific pathogen-free conditions at an American Association for the Accreditation of Laboratory Animal Care (AAALAC)-accredited animal facility within the NIAID. Mice were housed in accordance with the procedures outlined in the NIH Guide for the Care and Use of Laboratory Animals. Unless otherwise stated, sex and age-matched littermates (6-12 weeks of age) were used for individual experiments. For experiments involving Foxp3CreERT2+/+-CTLA4+/fl and Foxp3CreERT2+/−-Confettifl/− animals, we used female mice exclusively. For all other experiments, male and female mice were used at equal ratios.
METHOD DETAILS
Adoptive T cell transfers
Peripheral LNs and spleens from donor mice were homogenized in ice cold PBS supplemented with 1% BSA and 2mM EDTA. Cell suspensions were subsequently filtered through a 70 μm cell strainer. CD4+ T cell isolation was achieved by magnetic bead-based negative selection following the manufacturer’s protocol (Miltenyi Biotec, Cat#: 130-104-454). Note that the standard cocktail of biotin-conjugated antibodies was supplemented with biotin-conjugated anti-IL-2R-α (clone 7D4, 5 μg/mL) to remove highly activated CD4+ T cells and Tregs. 5 x 104 or 1 x 105 CD4+ T cells were transferred into individual recipient mice by intravenous (i.v.) injection.
Where indicated, purified CD4+ T cells were labelled with one of the following fluorescent dyes prior to adoptive transfer: 1 μM CellTracker Deep Red, 1 μM CellTracker Green, 1 μM CellTrace Violet, or 1 μM CellTrace Yellow. Dyes were diluted in PBS and cell labelling was performed for 20 min at 22°C with gentle end-over-end rotation. Unbound dye was quenched by washing cells 2x in PBS supplemented with 1% BSA.
For long term tracking experiments, CD45.2+/+ donor T cells were transferred into congenic CD45.1+/+ recipients. After 24 h, recipient animals were administered 100 μg of anti-CD62L blocking antibody (clone Mel-14) and 20 μg of FTY720 (Sigma-Aldrich, Cat#: SML0700) daily by intraperitoneal (i.p.) injection for up to 7 consecutive days.
For experiments involving the IL-2:S4B61 complex, 0.5 μg of recombinant murine IL-2 was incubated with 5 μg of anti-IL-2 antibody (clone S4B61) in PBS for 20 minutes at room temperature. The complex was then injected into recipient animals at the indicated timepoints.
In some instances, recipient animals were administered 500 μg of I-Ab blocking antibody (clone M5/114).
IL-2 secretion assay
5C.C7 T cells were harvested from transgenic 5C.C7 Rag2−/− animals. In parallel, splenocytes were harvested from WT B10.A animals, labelled with 5 μM CellTrace Violet, and pulsed with indicated peptides for 6 h at 37°C. 3x105 splenocytes were then incubated with 1x105 5C.C7 cells in individual wells of V bottom 96 well plates. After 12, 24, 36, or 60 h, plates were centrifuged, and cells were washed in cold MACS buffer. Next, cells were incubated with the IL-2 catch reagent (Miltenyi Biotec, Cat#: 130-090-491) for 5 mins on ice and subsequently transferred to a deep well plate with warm RPMI. The plate was glue-sealed with plastic film and rotated for 45 min at 37°C. Plates were then centrifuged and cells were washed twice in MACS buffer prior to immunostaining with the PE-conjugated anti-IL-2 detection reagent (Miltenyi Biotec, Cat#: 130-090-491), FITC-conjugated anti-CD4 (clone RM4-5), and BV785-conjugated anti-PD-1 (clone J43). Fluorescence measurements were made using flow cytometry.
Laser scanning confocal microscopy
Digital images were acquired using an upright or inverted Leica TCS SP8 X spectral detection system (Leica) equipped with a pulsed white light laser, 4 Gallium-Arsenide Phosphide (GaAsP) Hybrid Detectors (HyDs), 1 photomultiplier tube (PMT), 40x (NA = 1.3) and 20x (NA = 0.75) oil immersion objective lenses, and a motorized stage. For tissue sections (18-50 μm), images were acquired using the 40x objective with a pixel size of 0.271-0.286 μm, z step size of 0.3-1 μm, and detector bit-depth of 12.
For cleared, whole organ imaging, acquisition was performed on the inverted system using the 20x objective with a 2x optical zoom, pixel size of 0.286 μm, z step size of 2 μm, and detector bit-depth of 8. In all experiments, image acquisition was controlled using LAS X software.
Tissue section preparation, processing and immunostaining
Relevant LNs were quickly harvested and fixed for 14h at 4°C in BD Cytoperm/Cytofix (BD Bioscience, Cat#: 554722) diluted 1/4 in PBS. LNs were washed 3x in PBS (10 min per wash), carefully trimmed of fat using a stereo dissection microscope and fine forceps, and dehydrated for 24 h in a 30% sucrose solution made in 0.1 M phosphate buffer. LNs were then embedded in optimal cutting temperature (O.C.T.) compound (Sakura Finetek, Cat#: 50-363-579), frozen on dry ice, and stored at −80°C. 18-50 μm sagittal LN sections were prepared using a cryostat (Leica) equipped with a Surgipath DB80LX blade (Leica, Cat#: 14035843497). Cryochamber and Specimen cooling was set to −17°C.
To detect endogenous IL-2 production, animals were euthanized and quickly subjected to cardiac perfusion fixation. Animals were perfused with 2% PFA at a rate of 5ml/min for 3 minutes. Relevant LNs were then harvested and post-fixed in 1% BD Cytoperm/Cytofix for 14h at 4°C. LN storage and preparation was performed as described above.
Tissue sections were adhered to Superfrost Plus microscopy slides (VWR, Cat#: 48311-703), permeabilized using 0.1% Triton X-100 for 10 min at 22°C, blocked in 5% mouse serum for 1 h at 22°C, and washed in PBS. Tissue sections were next incubated with directly conjugated primary antibodies (see STAR Methods for working concentrations) diluted in PBS for 15 h at 4°C. After washing 3x in PBS (10 min per wash) at 22°C, samples were mounted in Fluoromount-G (SouthernBiotech, Cat#: 0100-01), which was allowed to cure for a minimum of 14h at 22°C. All imaging was performed using No. 1.5 coverglass (VWR, Cat#: 48393-241). Combinations of the following organic fluorophores were used for immunostaining: Brilliant Violet 421, Brilliant Violet 480, Alexa Fluor 555, eFluor 570, Alexa Fluor 594, eFluor660, Alexa Fluor 647, Alexa Fluor 488, and Alexa Fluor 700.
For pSTAT5 immunostaining, fixed tissue sections were permeabilized in pre-chilled 100% methanol for 18 min at −20°C, washed extensively in PBS, blocked in 5% donkey serum for 1 h at 22°C, and washed further in PBS. Tissue sections were next incubated with unconjugated anti-pSTAT5 (clone C11C5, 1.715 μg/ml) diluted in PBS for 15 h at 4°C. Following washing in PBS at 22°C, tissue sections were incubated with either Alexa 488, Alexa 594, or Alexa 647-conjugated donkey anti-rabbit F(ab’)2 fragments (0.483 μg/ml, Jackson ImmunoResearch Laboratories, Inc, Cat#: 711-546-152 or 711-6060152) for 2h at 22°C. Sections were then washed 4x in PBS (10 min per wash) at 22°C prior to mounting in Fluoromount-G as described above.
Whole tissue preparation, immunostaining and clearing
Tissue preparation, immunostaining, and clearing was performed using a protocol adapted from Li et al (Li et al., 2017). In brief, relevant LNs were quickly harvested and fixed for 14h at 4°C in 1% paraformaldehyde. Following 3x washes in PBS (10 min per wash), LNs were incubated in a 100 mM glycine solution for 30 min to neutralize free aldehydes. Next, LNs were carefully trimmed of fat using a stereo dissection microscope and fine forceps. Individual LNs were then incubated in 1 mL of blocking buffer (PBS with 1% normal mouse serum, 1% bovine serum albumin, and 0.1% Triton X-100) at 22°C for ~16 h with shaking (600 rpm). For immunostaining, each LN was incubated in a 400 μL staining buffer (PBS with 0.1% Triton X-100 and diluted antibodies of interest) for 3 days at 37°C with shaking (220 rpm). The following directly conjugated primary antibodies were used: eFluor 570-conjugated anti-Foxp3 (clone FJK-16s, 2 μg/ml), Alexa 647-conjugated anti-CD45.2 (clone 104, 5 μg/ml), and Alexa 488-conjugated anti-GFP (polyclonal, 4 μg/ml). After immunostaining, each LN was washed in 1 mL of wash buffer (PBS with 0.1% Triton X-100) for 14 h at 37°C with shaking (220 rpm). A final wash was performed at 22°C for 4h with shaking (600 rpm) prior to clearing.
Tissue clearing was performed using Ce3D solution: 40% (v/v) N-methylacetamide (Sigma-Aldrich, Cat#: M26305), 80% (w/v) Histodenz (Sigma-Aldrich, Cat#: D2158), 0.1% (v/v) Triton X-100, and 0.5% (v/v) 1-thioglycerol (Sigma-Aldrich, Cat#: M1753). Individual LNs were incubated in 1 mL of Ce3D solution for 18-24 h at 22°C with gentle end-over-end rotation. Prior to imaging, a stereo microscope was used to position each cleared LN with its hilum facing up on a No. 1.5 glass bottom microwell dish (MatTek, Cat#: P35G-1.5-20-C). Vacuum grease was then applied around the perimeter of the LN, creating a trough that was filled with a small volume of Ce3D solution. The trough was sealed by gently pressing an 18 mm, round coverglass on top of the vacuum grease.
Inducible perturbations
Tamoxifen (Sigma-Aldrich, Cat#:10540-29-1) was dissolved in corn oil at a concentration of 20 mg/ml. Female Foxp3CreERT+/−-Confetti+/−, Foxp3CreERT2+/−-IL2Rβ+/fl, and Foxp3CreERT2+/+-CTLA-4+/fl animals were administered 100 μL of tamoxifen emulsion (~75 mg tamoxifen/kg) or corn oil control daily by i.p. injection for 4 consecutive days. For Foxp3CreERT+/−-Confetti+/− mice, LNs were harvested 14 days after the final injection whereas for Foxp3CreERT2+/−-IL2Rβ+/fl and Foxp3CreERT2+/−-CTLA-4+/fl mice, LNs were harvested 7 days after the final injection. Male and female Foxp3CreER2+/+-TCR-αfl/fl animals were injected with tamoxifen as described; LNs were harvested 5 days after the final injection.
500 ng of diphtheria toxin (Calbiochem, Cat# 322326) was diluted in PBS and administered to Foxp3-DTR+/+ by i.p. injection. PBS alone was used as a control. LNs were harvest 7h later. All LNs were prepared for imaging as described in “Tissue section preparation, processing, and immunostaining” section.
Mixed bone marrow chimeras
To ablate bone marrow, adult C57BL/6J.Ly5a recipient animals were exposed to two doses of 137-Cesium-generated, 6 Gy gamma rays. Each dose of radiation was separated by a 3 h rest period. Donor bone marrow was extracted from C57BL/6J, C57BL/6-Ubi-GFP+/+, and Foxp3-DTR+/+ animals, mixed at different ratios, and injected intravenously into irradiated recipients immediately following the second rest period. Recipient animals were then treated with neomycin or trimethoprim/sulfamethoxazole for 4 weeks. Experiments were performed 10 weeks after the initial irradiation. To assess global chimerism, eGFP expression was measured in Treg cells from blood samples by flow cytometry. To assess local chimerism in situ, eGFP expression was measured in LNs within regions of high and low Treg cell density by quantitative imaging.
Donor bone marrow was extracted from the femurs and tibias of C57BL/6J, C57BL/6-Ubi-GFP+/+, and Foxp3-DTR+/+ animals by PBS perfusion with a 21-gauge needle. The resulting cell suspensions were filtered through a 40 μm cell-strainer prior to i.v. injection.
Image processing and segmentation
Image files generated in LAS X software were converted into “.ims” files in Imaris software (Bitplane) and subjected to a 1 pixel Gaussian filter to reduce noise.
For tissue sections, segmentation of densely packed T cells was performed using a protocol modified from Li et al. (Li et al., 2017). This process involved creating artificial T cell nuclei, which in turn were subjected to segmentation algorithms. In brief, “.ims” files were initially imported into Fiji (Schindelin et al., 2012). Note that Fiji imports 12-bit images in a 16-bit format. The brightness and contrast of CD3 or, in some instances, CD4 channels were adjusted linearly to thinly demarcate the plasma membranes of T cells. Adjusted CD3 or CD4 channels were then converted into an 8-bit format, duplicated and inverted to create an inverted sum channel. Next, the original channel was subtracted from the inverted sum channel, producing a high-contrast inverted channel, which was subsequently binarized using the “Auto Local Thresholding” tool. We employed the median algorithm with a local domain radius value (in pixels) that approximated the radius of a T cell (~3.5 μm). Binarized images were then despeckled to remove noise and subtracted from the inverted sum channel to improve separation between artificial T cell nuclei. The final product was converted back to a 16-bit format, exported as a “.TIFF” image sequence, and imported into the original “.ims” file in Imaris.
Image segmentation was performed in Imaris using the “Surface Object Creation” module, which employs a seeded region growing, k-means, and watershed algorithm to define individual cells. Segmentation artefacts were excluded using a combination of sphericity and volume thresholds, as well as manual correction.
For experiments involving Foxp3CreERT2+/−-Confettifl/− animals treated with tamoxifen, FP signals were masked in Imaris to improve visual clarity.
Quantitative image and spatial analysis
Following T cell segmentation and surface creation (see “Image processing and segmentation”), the mean or summed voxel fluorescence intensity values per channel were assessed in Imaris. In certain instances, these fluorescence distributions were used to selectively visualize T cells with specific phenotypes by creating discrete thresholds using the “filter” tool. Individual T cell statistics, including XYZ positional data—which were based on cell centroids — fluorescence intensity values per channel, and volume measurements were exported as .csv files and imported into the R statistical environment for further analysis.
To estimate the extent of T cell division in situ, an automated computational method was employed. In brief, the mclust package was used to fit a finite Gaussian mixture model via the EM algorithm, which was initialized by hierarchical model-based agglomerative clustering. The optimal model and number of mixture components was then selected using the Bayesian Information Criterion (BIC) (Scrucca et al., 2016).
Unsupervised hierarchical clustering was performed using the pheatmap package. Cells were clustered based on their Pearson correlation in multidimensional space using Ward’s minimum variance method.
Segmented imaging data were treated as realizations of multivariate, marked point patterns in two-dimensional or three-dimensional space (Baddeley et al., 2015). Associated marks were both categorical and numerical in value. All nearest neighbor distances were calculated in three-dimensional space using the nncross.pp3 function in Spatstat. To visualize the probability density function of Tregs throughout the LN, we extracted the XY coordinates of these cells from thin 18 μm LN sections and performed a fixed-bandwidth kernel density estimation. The smoothing bandwidth (σ) was estimated by cross-validation using the bw.diggle function in the Spatstat package (Baddeley and Turner, 2005; Baddeley et al., 2015). In brief, σ was chosen to minimize the mean-square error criterion according to Berman and Diggle (Berman and Diggle, 1989; Diggle, 1985). The kernel density estimation was calculated using the stat_density2d function in the ggplot2 package.
To automatically detect TxA23 cell clusters, we employed an implementation of the density-based spatial clustering of applications with noise (DBSCAN) algorithm using the dbscan package. The epsilon neighborhood (eps) using a k-dimensional tree to find k-nearest neighbours among the Tregs and their distances. These distances were then plotted as a function of Tregs sorted by ascending distance in the data set, and the optimal eps was chosen based on the “knee” in the plot (i.e., the inflection point)
To visualize the empirical density (commonly referred to as intensity in spatial point patterns) of Tregs weighted to the expression of Ki67, we performed a fixed-bandwidth kernel estimate of the intensity function. This function computes the convolution of the isotropic Gaussian kernel with standard deviation, σ. σ was estimated as described above. The intensity function was estimated using XY coordinates from thin, 18 μm LN sections. The intensity value at a point u is:
where k is the Gaussian smooth kernel, e(xi) is an edge correction factor, and ωi are the Ki67 weights. The edge correction term, e(u) is defined as follows (Diggle, 2013; Jones, 1993):
where W is the observation window, which was defined using the clickpoly function in the Spatstat package.
To correlate Ki67 expression in Tregs with intercellular distance, we implemented Stoyan’s mark correlation function:
where E[0u] is the conditional expectation for two points, 0 and u, separated by r and M(0) and M(u)) represents the marks (i.e. the Ki67 values) associated with these two points. E(M * M′) is the expectation for random marks, M and M’, drawn independently from the marginal distribution of Ki67 values. Pointwise envelopes for Stoyan’s mark correlation function were computed using the envelope function in Spatstat. For the null hypothesis (H0), the observed Ki67 marks were permutated at random across the fixed positions of Tregs within the LN paracortex using the rlabel function.
A similar permutation approach was used to quantify Treg clusters in the Foxp3CreERT2+/−-Confettifl/− animals treated with tamoxifen. Single-FP Treg clusters were quantified in three-dimensional space using the pointdensity function in the dbscan package. We excluded GFP from downstream analysis due to nearly negligible expression in Tregs — a well-documented consequence of weak Cre recombinase expression (Snippert et al., 2010)
To calculate the local density of Tregs (λTreg) surrounding individual T cells, we employed a moving window analysis. In brief, we initially centered a 1 μm spherical shell on a T cell of interest. The shell was then advanced radially up to a distance of 100 μm; the number of Tregs was enumerated within each 1 μm interval. We then calculated the volume at each interval, but only included proportions of each shell that fell within the boundaries of the LN (i.e., edge-corrected volumes). These boundaries were determined by centering a complete sphere with a radius of 1 μm on the T cell of interest and expanding it up to a radius of 100 μm. To determine the intersections between the expanding sphere and the LN boundaries, we used a customized R wrapper function of the Boolean operation from the CGAL library. Next, the volumes of these intersections were computed using a customized R wrapper function of the volume operation from the libigl library. Finally, the volume of each shell was calculated by subtracting the volume of two adjacent spheres. After determining the edge-corrected volume of each shell, along with the number of Tregs within each shell, we applied a 5 μm moving window in which local Treg density was calculated; the window was advanced radially from the T cell of interest up to a distance of 100 μm. At each window position, we calculated the density of Tregs by dividing the summed Treg numbers by the summed volumes of 5 adjacent shells, accounting for noise due to uncertainty in determining the centroid and size of each segmented cell (https://github.com/pkm304/density_moving_window).
Dynamical modeling
We describe a detailed model of the interactions between three cell types: conventional CD4+ T cells (Tconvs), regulatory T cells (Tregs), and dendritic cells (DCs). The variables and parameters of the model can be found in Table S1 & S2.
1. TCR signaling (DC:TC1)
This component consists of kinetic proofreading of antigen recognition and the effective signaling strength that regulates downstream pathways, including production of IL-2, and IL-2Rα (François et al., 2013; Voisinne et al., 2015). To make the model description self-contained, we lay out the detailed derivation from (François et al., 2013).
1.1. Kinetic proofreading
1.1.1. Reactions
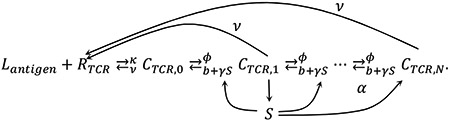 |
1.1.2. Equations
Corresponding equations are as bellow:
where j runs from 1 to N-1.
At steady states, the summation of equations for CTCR,j above leads to:
Assuming , we obtain
Other equations become
To solve for CTCR,j′s, we first obtain solutions of
as
Finally, CTCR,j can be expressed as
Since f(1) = −v < 0 < ϕ = f(0), it is always the case that r+ > 1 > r− > 0. To obtain a+ and a−, we manipulate the following two equations.
Finally, we obtain
Other variables at steady state can be obtained as,
Once SSHP1 is determined using , other variables follow from it.
2. Costimulatory signaling and inhibition by CTLA4 (DC:TC2 and TR:DC1)
This component describes binding kinetics of CD80∣CD86, CD28, and CTLA4 and the trans-endocytosis of CD80∣CD86 by CTLA4 (Collins et al., 2002; Jansson et al., 2005; Khailaie et al., 2017; van der Merwe et al., 1997; Qureshi et al., 2011; Sugár et al., 2017).
2.1. CD28-CD80∣CD86 binding kinetics
2.1.1. Reactions for both Tconvs and Tregs
We assumed the expression level of CD28 in the plasma membrane of Tconvs remains fixed. The cycling of CTLA4 is maintained at steady state for Tregs. Also, once the complexes of CTLA4 and CD80∣CD86 are internalized, the complexes are instantly degraded.
2.1.2. Equations
2.1.2.1. Tconvs
We assumed that the Tconv:DC engagement ceases before the considerable accumulation of CTLA4 by the Tconv. Thus, RCTLA4 = 0 for the whole time.
At steady state,
Ccostim was assumed to be the determinant of immune synapse formation. If Ccostim falls below a threshold value of 5 during the simulation or alternatively, if the simulation time exceeds 48 hours, the Tconv stops receiving TCR and costimulatory signals and disengages from the DC.
2.1.2.2. Tregs
We assumed that Tregs maintain a constant CTLA4 level in their cell membranes. This assumption slightly underestimates the actual value during active negative feedback regulation, given our data that IL-2 signaling increases CTLA4 expression by Tregs.
At steady states, given , , , and ,
which can be solved numerically for Ccostim and CCTLA4.
2.2. Detailed description of trans-endocytosis of CD80∣CD86 by Tregs
We further implemented the regulation of the trans-endocytosis efficiency by costimulation with the assumption that costimulation strength upregulates the trans-endocytosis efficiency. This assumption was based on the fact that CD28 signaling increases the adhesion of T cells to DCs and therefore, is likely to increase the efficiency of CTLA-4-mediated trans-endocytosis (Thauland et al., 2014).
where is the effective rate of endocytosis of CTLA4 and CCTLA4,tot is the total number of the CD80∣CD86:CTLA4 complexes in the immune synapses formed by Tregs and a DC.
3. Competition for IL-2 between a Tconv and Tregs (TC:I1, TC:I2, and TR:I1)
In this component, we describe the dynamics of IL2Ra production, IL-2 production, secretion, and diffusion, and IL-2-IL2R binding and endocytosis.
3.1. IL2 receptor production and IL-2-IL2 receptor binding dynamics
We follow the descriptions by: (Busse et al., 2010; Feinerman et al., 2010; Tkach et al., 2014; Voisinne et al., 2015)
3.1.1. Reactions
3.1.2. Equations
where
The rest of the differential equations are
Thus, at steady state,
3.2. IL2 production dynamics
We follow the descriptions by: (Tkach et al., 2014; Voisinne et al., 2015)
3.2.1. Reactions
3.2.2. Equations
where
for CD4 T cells (Lim et al., 2015).
The secretion rate of IL-2 is
3.3. IL-2 secretion, diffusion, and consumption dynamics
We integrated models from (Busse et al., 2010; Oyler-Yaniv et al., 2017; Shvartsman et al., 2001; Thurley et al., 2015) into a partial differential equation (PDE). Ordinary differential equations (ODEs) in the previous subsection are linked to the model component introduced here.
3.3.1. Reactions
3.3.2. Equations
The density of IL-2 is modeled through a reaction-diffusion equation.
where
Boundary conditions are
and
4. Spatiotemporal dynamics of Tregs and their internal states described by coupled partial differential equations.
In this component, we describe the spatial dynamics of Tregs and their internal states, resulting in coupled partial differential equations along with the equations in 3.3.2.
4.1. Treg movement
There are three possible mechanisms of the dynamical spatiotemporal regulation of Treg density: 1) chemo-attraction or chemo-repulsion due to cytokines or chemokines secreted by responding Tconvs or DCs (Chavanis, 2009; Rapp et al., 2019), 2) proliferation induced by IL-2 sensing (Amado et al., 2013), and 3) decrease of motility upon engagement with DCs (Thauland et al., 2014). All of these possibilities are taken into account as:
where U is the attractive potential describing the decrease of Treg motility due to the engagement with DCs and regulated by costimulation and pSTAT5 levels of Tregs (Chinen et al., 2016; Thauland et al., 2014):
is the carrying capacity estimated based on the physical volume of Tregs, and a is an arbitrary cytokine or chemokine inducing chemoattraction or chemorepulsion. Dtr is also assumed to be a function of r as
The boundary condition is
at r = ρ and ∞.
4.2. Time evolution of internal states of Tregs
The internal states of Tregs evolve over time upon receiving signals from secreted IL-2 or other sources such costimulatory ligands or pMHCII of DCs. We developed a general mathematical description of the internal dynamics of an entity described as a frequency using PDEs. In general, the number density of a molecular species R in Tregs is written as nR = R ∙ ntr, where R is the number of a molecular species per cell. Then, the time evolution of nR is described as
where fprod and fdeg are production and degradation rates of R, respectively. The suitable form this equation for implementing in the pdepe function of the MATLAB software is
where
Specifically, we implemented dynamics of mIL2Rα (mRNA of IL2Rα) and IL2Rα as
where
Model Parameters
Most of model parameters were obtained from existing previous literature (see Table S1 & S2 for detailed descriptions and references of parameters). While parameters related to ligand-receptor binding kinetics could be obtained from the literature, parameters related to Hill functions connecting upstream signals and downstream effects could not be determined since they are phenomenological descriptions and subject to cell-cell heterogeneities, even under shared environments. Initial conditions of model variables (such as the initial level of CD80∣CD86 or initial density of Tregs) can be considered as parameters externally provided. However, these values also vary significantly during homeostatic conditions. Therefore, simulating the model under a few fixed parameter combinations cannot account for phenotypic variations of the T cell response, leading us to apply the MAPPA framework outlined below.
MAPPA framework and simulation procedure
MAPPA is a computational framework that uses Random Forests to build quantitative maps between parameter space and phenotypic space for complicated models with analytical intractability (Park et al., 2019). This framework determines the contribution of individual parameters sampled throughout the plausible parameter space on a range of phenotypes, both at the global level (i.e., across all configurations in the parameter space), or local level (i.e., across specific configurations in the parameter space only). For our studies, the plausible parameter space was constructed using homeostatic parameter value ranges (see Table S1). We did not factor in the effects of inflammation on certain parameters (i.e., increased CD80∣86 ranges on a DC). Within these parameter ranges, we sampled 20,000 parameter combinations using the Sobol’ sampling scheme, which, when compared to a pseudorandom sampling scheme, ensures uniform coverage of high-dimensional space.
Simulations were conducted using the MATLAB software for each parameter combination. The simulation time was from 0 to 120 hours reflecting the typical time scale of the T cell response. The units of variables were set to be either copy numbers for intracellular species or copy numbers per um3 for extracellular species. The units of parameters were converted to those compatible with the units of variables. Matlab code is available on Github (https://github.com/pkm304/multiscale_t_cell_activation)
ODEs and PDEs were connected with boundary conditions at the interface of spatial compartments (Figure 6A). By utilizing the separation of time scales between gene expression dynamics occurring over hours and spatial dynamics of cell movement and IL-2 diffusion reaching stationary states over minutes, we were able to use the built-in ODE and PDE solvers (ode45 and pdepe, respectively) of MATLAB, rather than creating a new simulation scheme. We decomposed each simulation of ODEs and PDEs into 5-minute intervals, which is the optimal separation of time scales. For each 5-minute interval, the initial boundary conditions were based on the simulation results from the previous time interval.
QUANTIFICATION AND STATISTICAL ANALYSIS
No statistical methods were used to predetermine sample sizes. Statistical details for all experiments can be found in the Figure legends. Single-cell empirical distributions were compared using the two-tailed Kolmogorov Smirnov (KS) test. Two sample groups were compared with Welch’s two.sided t test. Multiple groups were compared using a one-way analysis of variance (ANOVA) test with the Tukey correction for multiple comparisons. Local regressions were determined using the locally estimated scatterplot smoothing (LOESS) method. The k values in each figure legend represent the number of cells while the n values represent the number of animals. All analysis was performed in the R statistical environment.
Supplementary Material
Figure S1, related toFigures 1 and 2. Paracrine IL-2 signaling enhances local Treg functionality, forming a negative feedback circuit topology that constrains IL-2 production and sensing in self-activated T cells (A) Representative fixed bandwidth KDE of segmented T cell positional coordinates. The resulting probability density function (PDF) was used to define the boundaries of the LN paracortex. Scale bar = 200 μm (B) Discrete gating to distinguish CD4+ T cells from Tregs within the LN paracortex. (C) In situ quantification of PD-1 expression in paracortical CD4+ T cells. Measurements were made in popliteal LNs of WT or PD-1−/− animals. The PDFs were determined by KDE, pooling cells from LNs of n = 2 animals per condition. Dashed red line represents the top 2% of the distribution in WT animals. (D) In situ quantification of PD-1 expression in paracortical CD4+ T cells and Tregs 24h after injecting an I-Ab blocking Ab or isotype control. T cells were initially trapped in the LN using a combination of CD62L blocking Abs and FTY-720. The PDFs were determined by KDE. n = 4 animals per condition from 2 independent experiments. (E) Local empirical densities of Tregs surrounding PD-1+ CD4+ T cells or randomly sampled PD-1− CD4+ T cells from the same LN paracortex in GF mice. Measurements were made in the inguinal LNs. Solid lines represent local regressions determined by the LOESS method. Error bars represent 95% confidence intervals. Data are representative of LNs from n = 5 animals from 2 independent experiments. (F) Quantification of IL-2 secretion in vitro using flow cytometry. Transgenic 5C.C7 Rag2−/− (5C.C7) cells were stimulated by splenocytes pulsed with cognate K3 peptide. At various timepoint post-stimulation, 5C.C7 cells were incubated with an IL-2 capture reagent and secreted cytokine was captured for 45 minutes. Representative scatterplot shows single-cell IL-2 and PD-1 measurements at the 12h timepoint. (G) The fraction of IL-2+ and PD-1+ 5C.C7 cells was determined at 12, 24, 36, and 60h post-stimulation. Data points are mean ± SEM from 2 independent experiments. Solid lines represent local regressions determined by the LOESS method. Shaded error bars represent 95% confidence intervals. (H) Detection of endogenous IL-2 in situ. Animals were euthanized and rapidly perfused with paraformaldehyde (see STAR Methods). Representative confocal micro-graph highlights two IL-2+ (red puncta) CD4+ T cells (dashed white lines) surrounded by pSTAT5+ Tregs. Inset – magnified XY optical slice illustrating a CD4+ IL-2+ T cell enriched in PD-1. Scale bars = 10 μm and 5 μm (inset). (I) Single-cell quantification of PD-1 expression on IL-2 producers and non-producers in situ. IL-2 producers were pooled from the popliteal LNs of n = 3 animals. Data are from 2 independent experiments. The p value was determined using a two-tailed KS test. (J) Log2 fold-change in global Treg Foxp3, CTLA-4, and IL-2Rα expression 24h after injecting α-IL2 blocking antibodies. Dashed red line represents the isotype control condition. Individual cells were pooled from n=3 animals. Data are mean ± 95% confidence intervals derived using non-parametric bootstrapping. ***p < 0.0001 determined using a one-tailed Student’s T test. (K) Log2 fold-change in global Treg Foxp3, CTLA-4, and IL-2Rα expression 7h after injecting Foxp3DTR+/+ with DTX. Dashed red line represents the PBS control condition. Data are mean ± 95% confidence intervals derived using non-parametric bootstrapping. ***p < 0.0001 determined using a one-tailed Student’s T test. (L) In situ quantification of Tregs as a percentage of total CD4+ T cells within the LN. Foxp3DTR +/+ mice were injected as in K. Data are mean ± SEM. Each dot represents an individual mouse pooled from 2 independent experiments. (M) In situ quantification of pSTAT5 expression among paracortical CD4+ T cells. Representative distribution is shown from a Foxp3DTR+/+ mouse injected with DTX for 7h. Dashed red line represents a discrete threshold of pSTAT5 expression (pSTAT5+) set at 3 times above the population median. Inset = zoomed tail of the distribution.
Figure S2, related toFigure 3. TxA23 cells are activated in the presence of Tregs but fail to respond IL-2. (A) 1x105 TxA23 cells and WT cells were differentially labeled with fluorescent dyes and co-transferred into healthy BALB/c recipients. Top panel: representative confocal micrograph highlights an area of the gastric LN containing both transferred cell types at 48h post-transfer. Bottom panel: magnified ROI (white dashed boxes) from the top panel. Scale bars = 20 μm and 10 μm (insets). (B) Quantification of PD-1 expression over time. PD-1 fluorescence intensity values were normalized to the median of co-transferred WT cells (dashed red line) at each time point. (C) In situ single-cell quantification of pSTAT5 signal within TxA23 cells compared to Tregs over time. Data are normalized to the median of TxA23 cells at 12.5 post-transfer. Dashed red line represents a discrete cut off for pSTAT5 signal. For B and C, k = 17-70 cells were pooled from gastric LN sections of n = 6-7 animals. Data are from 2 independent experiments.
Figure S3, related toFigure 3. TxA23 cells proliferate despite local IL-2 constraints. (A) TxA23 cell proliferation in the gastric LN, 48h post-transfer. Left: TxA23 cells, WT cells, and Tregs were visualized in an entire gastric LN volume using Ce3D imaging. Scale bar = 200 μm. Right: Zoomed insets highlighting heterogeneous fluorescence intensities of TxA23 cells. Scale bar = 20 μm. (B) Quantification of A. A GMM with unequal variance was fit to the distribution of CellTracker DeepRed using the EM algorithm and Bayesian information criterion (BIC) (see Figure 3C). The graph depicts the fraction of TxA23 cells within each mixture component. (C) TxA23 cell proliferation in the gastric LN, 96h post-transfer. Plot of the BIC values determining the best model (i.e., equal vs. unequal variance) and optimal number of mixture components. (D) and (E) CellTrace Violet distributions of co-transferred WT cells in the gastric LN at 48h and 96h post-transfer. For each timepoint, the fluorescence intensity values were normalized the median of the distribution. Model fitting was performed as in B and C. Both histograms fit a single component, consistent with 0 rounds of cell division. (F) Experimental schematic illustrating the timing of anti-CD62L blocking antibodies, FTY720, and IL-2:S4B61 immunocomplex injections in G-J (G) Ce3D imaging of an entire gastric LN at 72h post-transfer with the IL-2:S4B61 immunocomplex. Insets: magnified ROIs (white dashed boxes) highlighting multiple TxA23 cell clusters. Scale bars = 100 μm and 20 μm (Inset). (H) The absolute number of TxA23 or WT cells per gastric LN was determined by Ce3D imaging at the indicated time points. Each small dot represents a LN from a single mouse. Large dots and error bars represent mean ± SEM. **p < 0.001 determined using a two-tailed Student’s T test. (I) Quantification of WT and TxA23 cell clusters in the gastric LN at 72h post-transfer +/− IL-2:S4B61 immunocomplex using the DBSCAN algorithm (see STAR Methods). (J) Related to I. Quantification of the number of TxA23 cells per cluster using the DBSCAN algorithm.
Figure S4, related toFigures 4 and 5 Paracrine IL-2 signaling enhances local Treg proliferation, promoting micro-domain formation and growth (A) In situ quantification of pSTAT5 signal in Tregs. Violin and dot plots illustrate single-cell distributions in C57BL/6 animals injected with IL-2 blocking Abs or isotype controls. Popliteal LNs were assessed 24h after Ab injections. Dashed red line represents a discrete threshold for pSTAT5 expression. (B) Quantification of the global (i.e., mean) Treg density within the gastric LN of BALB/c mice. LNs were assessed 24h after injecting IL-2 blocking Abs or isotype controls. Each dot represents a density measurement from 20 μm LN section. Sections were pooled from LNs of n = 4 animals. (C) Quantification of Foxp3 expression in cells expressing fluorescent proteins 14 days after injecting tamoxifen into Foxp3CreERT2+/−-Confettifl/− mice. Each dot represents an individual animal. (D) Representative confocal micrographs illustrating Treg clonality near a CD4+ PD-1+ T cell. Dashed white circle highlights a CD4+ PD-1+ T cell of interest. Dashed white traces outline individual Tregs of interest and highlight Foxp3 expression. XZ optical slices highlight CD4 and PD-1 expression (arrow). Scale bar = 10 μm.
Figure S5, related toFigure 6A computational model predicts that modest reductions in micro-domain size or functionality can enable self-activated T cells to respond to IL-2. (A) and (B) Scatter plots showing the concordance between the independently simulated pSTAT5CD4-max (x-axis) and those predicted by a RFML regression model (y-axis) using out-of-bag (OOB) data (i.e., data not used for training). Two RFML models are shown: A is based on the initial simulations (“initial data”) only while B is based on initial data plus additional simulations enriched for parameter configurations with large pSTAT5CD4-max values (“combined data”), thus increasing prediction performance. Each r value represents the Pearson correlation coefficient between the predicted and simulated pSTAT5CD4-max values. Each dot represents the pSTAT5CD4-max for a specific parameter configuration. (C) Averaged local variable importance (LVI) of the parameter configurations above the cut-off for the pSTAT5CD4-max shown in Figure 6B. The LVI was calculated using the combined data model described above. “Permutation importance” (x-axis) represents the increase in prediction errors for the OOB data after permuting the indicated variables. (D), (E), and (F) Dynamical trajectories of IL-2 secretion, IL-2Rα protein copy number, and pSTAT5 signal in a CD4+ T cell as Tregλ is varied. The control condition represents an inactive configuration that was picked at random from parameter space. (G), (H) and (I) Dynamical trajectories as in D-F, but this time CTLA-4 or IL-2Rβ expression was reduced by 50% in Tregs. (J) Prediction performance of the RFML classification model comparing susceptible and active configurations. Performance was assessed using the receiver operator characteristic (ROC) and precision-recall curves; area under the curve (AUC) was used as the quantitative metric. (K) and (L) Distributions of values for the top two parameters (i.e Tregλ and TregContact_Baseline) in main Figure 6I and the calculated Tregλ-effective (Tregλ * TregContact_Baseline). Distributions are shown for active, inactive, and susceptible configurations.
Figure S6, related toFigure 7Modest reductions in Treg micro-domain size or functionality promote non-linear breakdowns in control, enabling self-activated T cells to subvert pruning (A) Quantification of cell-surface TCR-β in Tregs harvested from SLOs of Foxp3CreERT2+/+-TCR-αfl/fl. Measurements were made by flow cytometry 5 days after the final injection of corn oil (vehicle control) or tamoxifen. Discrete gating was performed on CD3, CD4, and Foxp3. (B) PDFs of Treg spatial coordinates overlaid with the positions of individual PD-1+ CD4+ T cells (black dots) in Foxp3CreERT2+/+-TCR-αfl/fl animals. Representative inguinal LNs 5 days after the final injection of corn oil (vehicle control) or tamoxifen. Scale bar = 200 μm (C) Local, empirical densities of Tregs surrounding individual PD-1+ CD4+ T cells within inguinal LNs of Foxp3CreERT2+/+-TCR-αfl/fl injected with corn oil (vehicle) or tamoxifen. Solid lines represent local regressions determined by the LOESS method. Error bars represent 95% confidence intervals. For A-C, data are representative of LNs from n = 5 animals from 2 independent experiments. (D) Quantification of chimerism 10 weeks after irradiation and bone marrow reconstitution of C57BL/6 mice. Irradiated recipient animals were injected with different ratios of WT and Foxp3DTR+/+ bone marrow. The DTR is fused to an eGFP (DTR-eGFP) in the Foxp3DTR+/+ strain. Measurements were made by flow cytometry using peripheral blood. Discrete gating was performed on CD3, CD4, and Foxp3. Percentages represent the mean ± SEM from LNs of n = 5 animals. (E) Representative confocal micrograph illustrates a clustered micro-domain of WT and DTR expressing Tregs. Popliteal LN sections were prepared from a 60% WT + 40% Foxp3DTR+/+ chimera that was not injected with DTX. Scale bar = 20 μm. (F) Local empirical densities of Tregs surrounding individual PD-1+ CD4+ T cells within the popliteal LN paracortex of 60% WT + 40% Ubi-GFP+/+ or 60% WT + 40% Foxp3DTR+/+ chimeras, both injected with DTX. Solid lines represent local regressions determined by the LOESS method. Error bars represent 95% confidence intervals. Data are representative of LNs from n = 3-5 animals per condition (G) In situ quantification of CTLA-4 and IL-2Rβ in Tregs from the indicated animals. Analysis was performed 7 days after the final injection of corn oil (vehicle) or tamoxifen. Measurements were made in the popliteal LNs. Data are representative of LNs from n = 4 animals per condition from 2 independent experiments. (H) PDFs of IL-2Rα expression in IL-2 responders (pSTAT5+) vs. non-responders (pSTAT5−) in each indicated experimental condition. Single-cell distributions were pooled from LNs of n = 3-4 animals for each KDE.
Figure S7, related toFigure 7. Local Treg feedback gives rise to a sharp discriminatory boundary between self and non-self (A) In situ quantification of paracortical Tregs frequencies in the indicated animal strains. *p < 0.05 determined using a two-tailed, Student’s t test (B) In situ quantification of Tregs within 40 μm of pSTAT5+ CD4+ T cells (i.e., emergent active configurations). Each dot represents an individual active configuration pooled from n = 3 animals. ***p < 0.0001 was determined using a two-tailed Kolmogorov-Smirnov test. (C) Distributions of TCR off-rates predicted by MAPPA in inactive, susceptible, and active configurations. (D) 5C.C7 and WT cells were differentially labelled with fluorescent dyes and co-transferred into healthy B10.A recipients. 24h post-transfer, 1 μg of T102S peptide was injected subcutaneously. Quantification of PD-1 expression post peptide injection at the indicated time points in the draining popliteal LN. PD-1 fluorescence intensity values were normalized to the median of co-transferred WT cells (dashed red line) at each time point. (E) Maximum deviation in λTreg (Δ λTreg) or λpSTAT5+Treg (ΔλpSTAT5+Treg) between 5C.C7 and WT cells over time. Animals were injected subcutaneously with 1 μg of MCC88-103 or T102S and popliteal LNs were assessed at the indicated time points. Colored curves represent local regressions with 95% confidence intervals determined by the LOESS method. (F) Quantification of pSTAT5 signal within 5C.C7 cells responding to T102S or MCC(88-103). Dashed red line represents discrete cutoff for pSTAT5 expression. For D, E, and F k = 24-240 cells pooled from 3 animals per timepoint. Data are from 2 independent experiments. (G) A GMM was fit to the normalized CellTracker DeepRed distributions of 5C.C7s as in Figure 3C. Fluorescence measurements were made in draining popliteal LNs using confocal microscopy 48h post peptide injection. The illustrated histogram is decomposed into its individual mixture components. (H) Fraction of 5C.C7 cells belonging to each mixture component. Nonparametric bootstrapping was used to determine the robustness of the model. Data are Mean ± 95% confidence intervals derived from 999 bootstrap samples. For G and H, k = 83 cells for the T102S condition and k = 175 cells for the MCC88-103 condition. Data are from two independent experiments.
Movie S1, related toFigure 3Ce3D imaging enables accurate localization and enumeration of rare CD4+ T cells. The entire volume of the gastric LN is shown 24h after co-transferring 5x104 TxA23 (orange) and WT cells (cyan) into healthy BALB/c recipients. Endogenous Foxp3 (purple) depicts Tregs.
Movie S2, related toFigure 3TxA23 cells proliferate in the presence of functional Tregs. Ce3D imaging of the entire gastric LN 72h after co-transferring 5x104 TxA23 (orange) and WT cells (cyan) into healthy BALB/c recipients. See Figure 3A for experimental details. Endogenous Foxp3 (purple) depicts Tregs.
Movie S3, related toFigure 3TxA23 cells are rapidly pruned from the host following proliferation. Ce3D imaging of the entire gastric LN 192h after co-transferring 5x104 TxA23 (orange) and WT cells (cyan) into healthy BALB/c recipients. See Figure 3A for experimental details. Endogenous Foxp3 (purple) depicts Tregs.
Highlights.
Rare T cells are activated by self-antigens to produce IL-2 in healthy hosts
Paracrine IL-2 enhances local Treg function and density, forming a feedback circuit
Local feedback constrains activated T cell signaling, curbing post-division survival
Small changes in feedback parameters cause non-linear deviations from homeostasis
ACKNOWLEDGEMENTS
We thank A.H. Sharpe and D.H. Margulies for providing the CTLA-4fl/fl and A23 Rag2−/− animals, respectively. D.D. was supported by a Medical Scientist Training Program grant from the National Institute of General Medical Sciences of the National Institutes of Health under award number: T32GM007739 to the Weill Cornell/Rockefeller/Sloan Kettering Tri-Institutional MD-PhD Program. We thank S. Grinstein for critical reading of the manuscript. We are grateful to all members of the LBS, both past and present, for their ongoing support. This work was supported by the intramural research program of the National Institute of Allergy and Infectious Diseases, NIH.
Footnotes
DECLARATIONS OF INTEREST
A.Y.R. is a co-founder of Sonoma Biotherapeutics; he is an SAB member and reports personal fees from Sonoma Biotherapeutics, RAPT Therapeutics, and Vedanta Biosciences, and holds an IP licensed to Takeda all outside the submitted work. All other authors declare no competing interests
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Akbar AN, Borthwick N, Salmon M, Gombert W, Bofill M, Shamsadeen N, Pilling D, Pett S, Grundy JE, and Janossy G (1993). The significance of low bcl-2 expression by CD45RO T cells in normal individuals and patients with acute viral infections. The role of apoptosis in T cell memory. J. Exp. Med 178, 427–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akbar AN, Borthwick NJ, Wickremasinghe RG, Panayoitidis P, Pilling D, Bofill M, Krajewski S, Reed JC, and Salmon M (1996). Interleukin-2 receptor common gamma-chain signaling cytokines regulate activated T cell apoptosis in response to growth factor withdrawal: selective induction of anti-apoptotic (bcl-2, bcl-xL) but not pro-apoptotic (bax, bcl-xS) gene expression. Eur. J. Immunol 26, 294–299. [DOI] [PubMed] [Google Scholar]
- Akkaya B, Oya Y, Akkaya M, Al Souz J, Holstein AH, Kamenyeva O, Kabat J, Matsumura R, Dorward DW, Glass DD, et al. (2019). Regulatory T cells mediate specific suppression by depleting peptide-MHC class II from dendritic cells. Nat. Immunol 20, 218–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allison KA, Sajti E, Collier JG, Gosselin D, Troutman TD, Stone EL, Hedrick SM, and Glass CK (2016). Affinity and dose of TCR engagement yield proportional enhancer and gene activity in CD4+ T cells. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida ARM, Zaragoza B, and Freitas AA (2006). Indexation as a novel mechanism of lymphocyte homeostasis: the number of CD4+CD25+ regulatory T cells is indexed to the number of IL-2-producing cells. J. Immunol 177, 192–200. [DOI] [PubMed] [Google Scholar]
- Alon U (2007). Network motifs: theory and experimental approaches. Nat. Rev. Genet 8, 450–461. [DOI] [PubMed] [Google Scholar]
- Altan-Bonnet G, and Germain RN (2005). Modeling T cell antigen discrimination based on feedback control of digital ERK responses. PLoS Biol. 3, e356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amado IF, Berges J, Luther RJ, Mailhé M-P, Garcia S, Bandeira A, Weaver C, Liston A, and Freitas AA (2013). IL-2 coordinates IL-2-producing and regulatory T cell interplay. J. Exp. Med 210, 2707–2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baddeley A, and Turner R (2005). spatstat: An R Package for Analyzing Spatial Point Patterns. Journal of Statistical Software 12, 1–42. [Google Scholar]
- Baddeley A, Rubak E, and Turner R (2015). Spatial Point Patterns: Methodology and Applications with R (Chapman and Hall/CRC Press, London: ). [Google Scholar]
- Baptista AP, Gola A, Huang Y, Milanez-Almeida P, Torabi-Parizi P, Urban JF, Shapiro VS, Gerner MY, and Germain RN (2019). The Chemoattractant Receptor Ebi2 Drives Intranodal Naive CD4+ T Cell Peripheralization to Promote Effective Adaptive Immunity. Immunity 50, 1188–1201.e6. [DOI] [PubMed] [Google Scholar]
- Becskei A, and Serrano L (2000). Engineering stability in gene networks by autoregulation. Nature 405, 590–593. [DOI] [PubMed] [Google Scholar]
- Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, Kelly TE, Saulsbury FT, Chance PF, and Ochs HD (2001). The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat. Genet 27, 20–21. [DOI] [PubMed] [Google Scholar]
- Berman M, and Diggle P (1989). Estimating Weighted Integrals of the Second-Order Intensity of a Spatial Point Process. Journal of the Royal Statistical Society. Series B (Methodological) 51, 81–92. [Google Scholar]
- Bhattacharya A, Dorf ME, and Springer TA (1981). A shared alloantigenic determinant on Ia antigens encoded by the I-A and I-E subregions: evidence for I region gene duplication. J. Immunol 127, 2488–2495. [PubMed] [Google Scholar]
- Boyman O, Kovar M, Rubinstein MP, Surh CD, and Sprent J (2006). Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science 311, 1924–1927. [DOI] [PubMed] [Google Scholar]
- Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, Wilkinson JE, Galas D, Ziegler SF, and Ramsdell F (2001). Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat. Genet 27, 68–73. [DOI] [PubMed] [Google Scholar]
- Busse D, de la Rosa M, Hobiger K, Thurley K, Flossdorf M, Scheffold A, and Höfer T (2010). Competing feedback loops shape IL-2 signaling between helper and regulatory T lymphocytes in cellular microenvironments. Proc. Natl. Acad. Sci. U.S.A 107, 3058–3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buszko M, and Shevach EM (2020). Control of regulatory T cell homeostasis. Current Opinion in Immunology 67, 18–26. [DOI] [PubMed] [Google Scholar]
- Butler TC, Kardar M, and Chakraborty AK (2013). Quorum sensing allows T cells to discriminate between self and nonself. Proc. Natl. Acad. Sci. U.S.A 110, 11833–11838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavanis P-H (2009). A stochastic Keller-Segel model of chemotaxis. ArXiv:0804.4425 [Cond-Mat]. [Google Scholar]
- Chinen T, Kannan AK, Levine AG, Fan X, Klein U, Zheng Y, Gasteiger G, Feng Y, Fontenot JD, and Rudensky AY (2016). An essential role for the IL-2 receptor in Treg cell function. Nat. Immunol 17, 1322–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins AV, Brodie DW, Gilbert RJC, Iaboni A, Manso-Sancho R, Walse B, Stuart DI, van der Merwe PA, and Davis SJ (2002). The interaction properties of costimulatory molecules revisited. Immunity 17, 201–210. [DOI] [PubMed] [Google Scholar]
- Corse E, Gottschalk RA, Krogsgaard M, and Allison JP (2010). Attenuated T Cell Responses to a High-Potency Ligand In Vivo. PLoS Biol 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotari JW, Voisinne G, Dar OE, Karabacak V, and Altan-Bonnet G (2013). Cell-to-Cell Variability Analysis Dissects the Plasticity of Signaling of Common γ Chain Cytokines in T Cells. Sci Signal 6, ra17–ra17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diggle P (1985). A Kernel Method for Smoothing Point Process Data. Journal of the Royal Statistical Society. Series C (Applied Statistics) 34, 138–147. [Google Scholar]
- Diggle PJ (2013). Statistical Analysis of Spatial and Spatio-Temporal Point Patterns, Third Edition (CRC Press; ). [Google Scholar]
- Duprez V, and Dautry-Varsat A (1986). Receptor-mediated endocytosis of interleukin 2 in a human tumor T cell line. Degradation of interleukin 2 and evidence for the absence of recycling of interleukin receptors. J. Biol. Chem 261, 15450–15454. [PubMed] [Google Scholar]
- Farh KK-H, Marson A, Zhu J, Kleinewietfeld M, Housley WJ, Beik S, Shoresh N, Whitton H, Ryan RJH, Shishkin AA, et al. (2015). Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 518, 337–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinerman O, Jentsch G, Tkach KE, Coward JW, Hathorn MM, Sneddon MW, Emonet T, Smith KA, and Altan-Bonnet G (2010). Single-cell quantification of IL-2 response by effector and regulatory T cells reveals critical plasticity in immune response. Mol Syst Biol 6, 437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson HA, Kugel JF, and Goodrich JA (2001). Kinetic and mechanistic analysis of the RNA polymerase II transcription reaction at the human interleukin-2 promoter. J. Mol. Biol 314, 993–1006. [DOI] [PubMed] [Google Scholar]
- Ferrell JE (2016). Perfect and Near-Perfect Adaptation in Cell Signaling. Cell Syst 2, 62–67. [DOI] [PubMed] [Google Scholar]
- Fink PJ, Matis LA, McElligott DL, Bookman M, and Hedrick SM (1986). Correlations between T-cell specificity and the structure of the antigen receptor. Nature 321, 219–226. [DOI] [PubMed] [Google Scholar]
- Fontenot JD, Gavin MA, and Rudensky AY (2003). Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol 4, 330–336. [DOI] [PubMed] [Google Scholar]
- Fontenot JD, Rasmussen JP, Gavin MA, and Rudensky AY (2005). A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat. Immunol 6, 1142–1151. [DOI] [PubMed] [Google Scholar]
- François P, Voisinne G, Siggia ED, Altan-Bonnet G, and Vergassola M (2013). Phenotypic model for early T-cell activation displaying sensitivity, specificity, and antagonism. Proc. Natl. Acad. Sci. U.S.A 110, E888–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furtado GC, Curotto de Lafaille MA, Kutchukhidze N, and Lafaille JJ (2002). Interleukin 2 signaling is required for CD4(+) regulatory T cell function. J Exp Med 196, 851–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallatin WM, Weissman IL, and Butcher EC (1983). A cell-surface molecule involved in organ-specific homing of lymphocytes. Nature 304, 30–34. [DOI] [PubMed] [Google Scholar]
- Gerner MY, Kastenmuller W, Ifrim I, Kabat J, and Germain RN (2012). Histo-cytometry: a method for highly multiplex quantitative tissue imaging analysis applied to dendritic cell subset microanatomy in lymph nodes. Immunity 37, 364–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hataye J, Moon JJ, Khoruts A, Reilly C, and Jenkins MK (2006). Naive and memory CD4+ T cell survival controlled by clonal abundance. Science 312, 114–116. [DOI] [PubMed] [Google Scholar]
- Heinzel S, Binh Giang T, Kan A, Marchingo JM, Lye BK, Corcoran LM, and Hodgkin PD (2017). A Myc-dependent division timer complements a cell-death timer to regulate T cell and B cell responses. Nat. Immunol 18, 96–103. [DOI] [PubMed] [Google Scholar]
- Hochweller K, and Anderton SM (2005). Kinetics of costimulatory molecule expression by T cells and dendritic cells during the induction of tolerance versus immunity in vivo. Eur J Immunol 35, 1086–1096. [DOI] [PubMed] [Google Scholar]
- Höfer T, Krichevsky O, and Altan-Bonnet G (2012). Competition for IL-2 between Regulatory and Effector T Cells to Chisel Immune Responses. Front Immunol 3, 268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honjo T, Nishizuka Y, and Hayaishi O (1969). Adenosine diphosphoribosylation of aminoacyl transferase II by diphtheria toxin. Cold Spring Harb. Symp. Quant. Biol 34, 603–608. [DOI] [PubMed] [Google Scholar]
- Hori S, Nomura T, and Sakaguchi S (2003). Control of regulatory T cell development by the transcription factor Foxp3. Science 299, 1057–1061. [DOI] [PubMed] [Google Scholar]
- Huang J, Brameshuber M, Zeng X, Xie J, Li Q, Chien Y, Valitutti S, and Davis MM (2013). A Single Peptide-Major Histocompatibility Complex Ligand Triggers Digital Cytokine Secretion in CD4+ T Cells. Immunity 39, 846–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansson A, Barnes E, Klenerman P, Harlén M, Sørensen P, Davis SJ, and Nilsson P (2005). A theoretical framework for quantitative analysis of the molecular basis of costimulation. J. Immunol 175, 1575–1585. [DOI] [PubMed] [Google Scholar]
- Jenkins MK, and Moon JJ (2012). The role of naive T cell precursor frequency and recruitment in dictating immune response magnitude. J. Immunol 188, 4135–4140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MC (1993). Simple boundary correction for kernel density estimation. Stat Comput 3, 135–146. [Google Scholar]
- June CH, Warshauer JT, and Bluestone JA (2017). Is autoimmunity the Achilles’ heel of cancer immunotherapy? Nat. Med 23, 540–547. [DOI] [PubMed] [Google Scholar]
- Khailaie S, Rowshanravan B, Robert PA, Walker LSK, Sansom DM, and Meyer-Hermann M (2017). Quantitative characterization of CTLA4 trafficking and turnover using a combined in vitro and in silico approach. BioRxiv 106898. [Google Scholar]
- Khattri R, Cox T, Yasayko S-A, and Ramsdell F (2003). An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat. Immunol 4, 337–342. [DOI] [PubMed] [Google Scholar]
- Kim JM, Rasmussen JP, and Rudensky AY (2007). Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat. Immunol 8, 191–197. [DOI] [PubMed] [Google Scholar]
- Kitz A, Singer E, and Hafler D (2018). Regulatory T Cells: From Discovery to Autoimmunity. Cold Spring Harb Perspect Med 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein L, Kyewski B, Allen PM, and Hogquist KA (2014). Positive and negative selection of the T cell repertoire: what thymocytes see (and don’t see). Nat. Rev. Immunol 14, 377–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehn HS, Ouyang W, Lo B, Deenick EK, Niemela JE, Avery DT, Schickel J-N, Tran DQ, Stoddard J, Zhang Y, et al. (2014). Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science 345, 1623–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine AG, Arvey A, Jin W, and Rudensky AY (2014). Continuous requirement for the TCR in regulatory T cell function. Nat. Immunol 15, 1070–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Germain RN, and Gerner MY (2017). Multiplex, quantitative cellular analysis in large tissue volumes with clearing-enhanced 3D microscopy (Ce3D). Proc. Natl. Acad. Sci. U.S.A 114, E7321–E7330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao W, Lin J-X, and Leonard WJ (2011). IL-2 family cytokines: new insights into the complex roles of IL-2 as a broad regulator of T helper cell differentiation. Curr. Opin. Immunol 23, 598–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim H-S, Cordoba S-P, Dushek O, Goyette J, Taylor A, Rudd CE, and van der Merwe PA (2015). Costimulation of IL-2 Production through CD28 Is Dependent on the Size of Its Ligand. J Immunol 195, 5432–5439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Gerner MY, Van Panhuys N, Levine AG, Rudensky AY, and Germain RN (2015). Immune homeostasis enforced by co-localized effector and regulatory T cells. Nature 528, 225–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livet J, Weissman TA, Kang H, Draft RW, Lu J, Bennis RA, Sanes JR, and Lichtman JW (2007). Transgenic strategies for combinatorial expression of fluorescent proteins in the nervous system. Nature 450, 56–62. [DOI] [PubMed] [Google Scholar]
- Malhotra D, Linehan JL, Dileepan T, Lee YJ, Purtha WE, Lu JV, Nelson RW, Fife BT, Orr HT, Anderson MS, et al. (2016). Tolerance is established in polyclonal CD4(+) T cells by distinct mechanisms, according to self-peptide expression patterns. Nat. Immunol 17, 187–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandl JN, Liou R, Klauschen F, Vrisekoop N, Monteiro JP, Yates AJ, Huang AY, and Germain RN (2012). Quantification of lymph node transit times reveals differences in antigen surveillance strategies of naive CD4+ and CD8+ T cells. Proc. Natl. Acad. Sci. U.S.A 109, 18036–18041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchingo JM, Kan A, Sutherland RM, Duffy KR, Wellard CJ, Belz GT, Lew AM, Dowling MR, Heinzel S, and Hodgkin PD (2014). T cell signaling. Antigen affinity, costimulation, and cytokine inputs sum linearly to amplify T cell expansion. Science 346, 1123–1127. [DOI] [PubMed] [Google Scholar]
- Marson A, Housley WJ, and Hafler DA (2015). Genetic basis of autoimmunity. J. Clin. Invest 125, 2234–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matloubian M, Lo CG, Cinamon G, Lesneski MJ, Xu Y, Brinkmann V, Allende ML, Proia RL, and Cyster JG (2004). Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature 427, 355–360. [DOI] [PubMed] [Google Scholar]
- McHugh RS, Shevach EM, Margulies DH, and Natarajan K (2001). A T cell receptor transgenic model of severe, spontaneous organ-specific autoimmunity. Eur. J. Immunol 31, 2094–2103. [DOI] [PubMed] [Google Scholar]
- van der Merwe PA, Bodian DL, Daenke S, Linsley P, and Davis SJ (1997). CD80 (B7-1) binds both CD28 and CTLA-4 with a low affinity and very fast kinetics. J. Exp. Med 185, 393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MJ, Hejazi AS, Wei SH, Cahalan MD, and Parker I (2004). T cell repertoire scanning is promoted by dynamic dendritic cell behavior and random T cell motility in the lymph node. Proc. Natl. Acad. Sci 101, 998–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle M, and Lazebnik YA (1995). Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature 376, 37–43. [DOI] [PubMed] [Google Scholar]
- Oyler-Yaniv A, Oyler-Yaniv J, Whitlock BM, Liu Z, Germain RN, Huse M, Altan-Bonnet G, and Krichevsky O (2017). A Tunable Diffusion-Consumption Mechanism of Cytokine Propagation Enables Plasticity in Cell-to-Cell Communication in the Immune System. Immunity 46, 609–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace L, Tempez A, Arnold-Schrauf C, Lemaitre F, Bousso P, Fetler L, Sparwasser T, and Amigorena S (2012). Regulatory T cells increase the avidity of primary CD8+ T cell responses and promote memory. Science 338, 532–536. [DOI] [PubMed] [Google Scholar]
- Pandiyan P, Zheng L, Ishihara S, Reed J, and Lenardo MJ (2007). CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat. Immunol 8, 1353–1362. [DOI] [PubMed] [Google Scholar]
- Park K, Prüstel T, Lu Y, and Tsang JS (2019). Machine learning of stochastic gene network phenotypes. BioRxiv 825943. [Google Scholar]
- Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, Baker J, Jeffery LE, Kaur S, Briggs Z, et al. (2011). Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science 332, 600–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapp M, Wintergerst MWM, Kunz WG, Vetter VK, Knott MML, Lisowski D, Haubner S, Moder S, Thaler R, Eiber S, et al. (2019). CCL22 controls immunity by promoting regulatory T cell communication with dendritic cells in lymph nodes. J. Exp. Med 216, 1170–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reay PA, Kantor RM, and Davis MM (1994). Use of global amino acid replacements to define the requirements for MHC binding and T cell recognition of moth cytochrome c (93-103). J. Immunol 152, 3946–3957. [PubMed] [Google Scholar]
- Ross AE, and Pompano RR (2018). Diffusion of cytokines in live lymph node tissue using microfluidic integrated optical imaging. Anal. Chim. Acta 1000, 205–213. [DOI] [PubMed] [Google Scholar]
- Rubtsov YP, Niec RE, Josefowicz S, Li L, Darce J, Mathis D, Benoist C, and Rudensky AY (2010). Stability of the regulatory T cell lineage in vivo. Science 329, 1667–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallusto F, Cella M, Danieli C, and Lanzavecchia A (1995). Dendritic cells use macropinocytosis and the mannose receptor to concentrate macromolecules in the major histocompatibility complex class II compartment: downregulation by cytokines and bacterial products. J. Exp. Med 182, 389–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheinecker C, McHugh R, Shevach EM, and Germain RN (2002). Constitutive Presentation of a Natural Tissue Autoantigen Exclusively by Dendritic Cells in the Draining Lymph Node. J Exp Med 196, 1079–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nature Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert D, Bode C, Kenefeck R, Hou TZ, Wing JB, Kennedy A, Bulashevska A, Petersen B-S, Schäffer AA, Grüning BA, et al. (2014). Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat. Med 20, 1410–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scrucca L, Fop M, Murphy TB, and Raftery AE (2016). mclust 5: Clustering, Classification and Density Estimation Using Gaussian Finite Mixture Models. R J 8, 289–317. [PMC free article] [PubMed] [Google Scholar]
- Setoguchi R, Hori S, Takahashi T, and Sakaguchi S (2005). Homeostatic maintenance of natural Foxp3(+) CD25(+) CD4(+) regulatory T cells by interleukin (IL)-2 and induction of autoimmune disease by IL-2 neutralization. J. Exp. Med 201, 723–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevach EM (2009). Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity 30, 636–645. [DOI] [PubMed] [Google Scholar]
- Shim J, Lim H, Yates JR, and Karin M (2002). Nuclear Export of NF90 Is Required for Interleukin-2 mRNA Stabilization. Mol. Cell 10, 1331–1344. [DOI] [PubMed] [Google Scholar]
- Shvartsman SY, Wiley HS, Deen WM, and Lauffenburger DA (2001). Spatial range of autocrine signaling: modeling and computational analysis. Biophys. J 81, 1854–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simeonov DR, Gowen BG, Boontanrart M, Roth TL, Gagnon JD, Mumbach MR, Satpathy AT, Lee Y, Bray NL, Chan AY, et al. (2017). Discovery of stimulation-responsive immune enhancers with CRISPR activation. Nature 549, 111–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simeonov DR, Wong HS, Cortez JT, Young A, Li Z, Nguyen V, Park K, Umhoefer J, Indart AC, Woo JM, et al. (2020). T cell subset-selective IL2RA enhancers shape autoimmune diabetes risk. BioRxiv 2020.07.22.216564. [Google Scholar]
- Smigiel KS, Richards E, Srivastava S, Thomas KR, Dudda JC, Klonowski KD, and Campbell DJ (2014). CCR7 provides localized access to IL-2 and defines homeostatically distinct regulatory T cell subsets. J. Exp. Med 211, 121–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snippert HJ, Flier LG van der, Sato T, van Es JH, van den Born M, Kroon-Veenboer C, Barker N, Klein AM, van Rheenen J, Simons BD, et al. (2010). Intestinal Crypt Homeostasis Results from Neutral Competition between Symmetrically Dividing Lgr5 Stem Cells. Cell 143, 134–144. [DOI] [PubMed] [Google Scholar]
- Spangler JB, Tomala J, Luca VC, Jude KM, Dong S, Ring AM, Votavova P, Pepper M, Kovar M, and Garcia KC (2015). Antibodies to Interleukin-2 Elicit Selective T Cell Subset Potentiation through Distinct Conformational Mechanisms. Immunity 42, 815–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugár IP, Das J, Jayaprakash C, and Sealfon SC (2017). Multiscale Modeling of Complex Formation and CD80 Depletion during Immune Synapse Development. Biophys. J 112, 997–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suri-Payer E, Amar AZ, Thornton AM, and Shevach EM (1998). CD4+CD25+ T cells inhibit both the induction and effector function of autoreactive T cells and represent a unique lineage of immunoregulatory cells. J. Immunol 160, 1212–1218. [PubMed] [Google Scholar]
- Tadokoro CE, Shakhar G, Shen S, Ding Y, Lino AC, Maraver A, Lafaille JJ, and Dustin ML (2006). Regulatory T cells inhibit stable contacts between CD4+ T cells and dendritic cells in vivo. J. Exp. Med 203, 505–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Q, Adams JY, Tooley AJ, Bi M, Fife BT, Serra P, Santamaria P, Locksley RM, Krummel MF, and Bluestone JA (2006). Visualizing regulatory T cell control of autoimmune responses in nonobese diabetic mice. Nat. Immunol 7, 83–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Q, Adams JY, Penaranda C, Melli K, Piaggio E, Sgouroudis E, Piccirillo CA, Salomon BL, and Bluestone JA (2008). Central role of defective interleukin-2 production in the triggering of islet autoimmune destruction. Immunity 28, 687–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Termeer C, Benedix F, Sleeman J, Fieber C, Voith U, Ahrens T, Miyake K, Freudenberg M, Galanos C, and Simon JC (2002). Oligosaccharides of Hyaluronan activate dendritic cells via toll-like receptor 4. J. Exp. Med 195, 99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thauland TJ, Koguchi Y, Dustin ML, and Parker DC (2014). CD28-CD80 interactions control regulatory T cell motility and immunological synapse formation. J. Immunol 193, 5894–5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurley K, Gerecht D, Friedmann E, and Höfer T (2015). Three-Dimensional Gradients of Cytokine Signaling between T Cells. PLoS Comput. Biol 11, e1004206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkach KE, Barik D, Voisinne G, Malandro N, Hathorn MM, Cotari JW, Vogel R, Merghoub T, Wolchok J, Krichevsky O, et al. (2014). T cells translate individual, quantal activation into collective, analog cytokine responses via time-integrated feedbacks. Elife 3, e01944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubo NJ, Pagán AJ, Taylor JJ, Nelson RW, Linehan JL, Ertelt JM, Huseby ES, Way SS, and Jenkins MK (2013). Single naive CD4+ T cells from a diverse repertoire produce different effector cell types during infection. Cell 153, 785–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vahl JC, Drees C, Heger K, Heink S, Fischer JC, Nedjic J, Ohkura N, Morikawa H, Poeck H, Schallenberg S, et al. (2014). Continuous T cell receptor signals maintain a functional regulatory T cell pool. Immunity 41, 722–736. [DOI] [PubMed] [Google Scholar]
- Voisinne G, Nixon BG, Melbinger A, Gasteiger G, Vergassola M, and Altan-Bonnet G (2015). T Cells Integrate Local and Global Cues to Discriminate between Structurally Similar Antigens. Cell Rep 11, 1208–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrisekoop N, Monteiro JP, Mandl JN, and Germain RN (2014). Revisiting thymic positive selection and the mature T cell repertoire for antigen. Immunity 41, 181–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HM, and Smith KA (1987). The interleukin 2 receptor. Functional consequences of its bimolecular structure. J. Exp. Med 166, 1055–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidemann T, Worch R, Kurgonaite K, Hintersteiner M, Bökel C, and Schwille P (2011). Single Cell Analysis of Ligand Binding and Complex Formation of Interleukin-4 Receptor Subunits. Biophys. J 101, 2360–2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P-H, Giri A, and Wirtz D (2015). Statistical analysis of cell migration in 3D using the anisotropic persistent random walk model. Nat. Protoc 10, 517–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing Y, and Hogquist KA (2012). T-cell tolerance: central and peripheral. Cold Spring Harb Perspect Biol 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Huynh A, Whitcher G, Chang J, Maltzman JS, and Turka LA (2013). An obligate cell-intrinsic function for CD28 in Tregs. J. Clin. Invest 123, 580–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Gothe F, Pennamen P, James JR, McDonald D, Mata CP, Modis Y, Alazami AM, Acres M, Haller W, et al. (2019). Human interleukin-2 receptor β mutations associated with defects in immunity and peripheral tolerance. J. Exp. Med 216, 1311–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1, related toFigures 1 and 2. Paracrine IL-2 signaling enhances local Treg functionality, forming a negative feedback circuit topology that constrains IL-2 production and sensing in self-activated T cells (A) Representative fixed bandwidth KDE of segmented T cell positional coordinates. The resulting probability density function (PDF) was used to define the boundaries of the LN paracortex. Scale bar = 200 μm (B) Discrete gating to distinguish CD4+ T cells from Tregs within the LN paracortex. (C) In situ quantification of PD-1 expression in paracortical CD4+ T cells. Measurements were made in popliteal LNs of WT or PD-1−/− animals. The PDFs were determined by KDE, pooling cells from LNs of n = 2 animals per condition. Dashed red line represents the top 2% of the distribution in WT animals. (D) In situ quantification of PD-1 expression in paracortical CD4+ T cells and Tregs 24h after injecting an I-Ab blocking Ab or isotype control. T cells were initially trapped in the LN using a combination of CD62L blocking Abs and FTY-720. The PDFs were determined by KDE. n = 4 animals per condition from 2 independent experiments. (E) Local empirical densities of Tregs surrounding PD-1+ CD4+ T cells or randomly sampled PD-1− CD4+ T cells from the same LN paracortex in GF mice. Measurements were made in the inguinal LNs. Solid lines represent local regressions determined by the LOESS method. Error bars represent 95% confidence intervals. Data are representative of LNs from n = 5 animals from 2 independent experiments. (F) Quantification of IL-2 secretion in vitro using flow cytometry. Transgenic 5C.C7 Rag2−/− (5C.C7) cells were stimulated by splenocytes pulsed with cognate K3 peptide. At various timepoint post-stimulation, 5C.C7 cells were incubated with an IL-2 capture reagent and secreted cytokine was captured for 45 minutes. Representative scatterplot shows single-cell IL-2 and PD-1 measurements at the 12h timepoint. (G) The fraction of IL-2+ and PD-1+ 5C.C7 cells was determined at 12, 24, 36, and 60h post-stimulation. Data points are mean ± SEM from 2 independent experiments. Solid lines represent local regressions determined by the LOESS method. Shaded error bars represent 95% confidence intervals. (H) Detection of endogenous IL-2 in situ. Animals were euthanized and rapidly perfused with paraformaldehyde (see STAR Methods). Representative confocal micro-graph highlights two IL-2+ (red puncta) CD4+ T cells (dashed white lines) surrounded by pSTAT5+ Tregs. Inset – magnified XY optical slice illustrating a CD4+ IL-2+ T cell enriched in PD-1. Scale bars = 10 μm and 5 μm (inset). (I) Single-cell quantification of PD-1 expression on IL-2 producers and non-producers in situ. IL-2 producers were pooled from the popliteal LNs of n = 3 animals. Data are from 2 independent experiments. The p value was determined using a two-tailed KS test. (J) Log2 fold-change in global Treg Foxp3, CTLA-4, and IL-2Rα expression 24h after injecting α-IL2 blocking antibodies. Dashed red line represents the isotype control condition. Individual cells were pooled from n=3 animals. Data are mean ± 95% confidence intervals derived using non-parametric bootstrapping. ***p < 0.0001 determined using a one-tailed Student’s T test. (K) Log2 fold-change in global Treg Foxp3, CTLA-4, and IL-2Rα expression 7h after injecting Foxp3DTR+/+ with DTX. Dashed red line represents the PBS control condition. Data are mean ± 95% confidence intervals derived using non-parametric bootstrapping. ***p < 0.0001 determined using a one-tailed Student’s T test. (L) In situ quantification of Tregs as a percentage of total CD4+ T cells within the LN. Foxp3DTR +/+ mice were injected as in K. Data are mean ± SEM. Each dot represents an individual mouse pooled from 2 independent experiments. (M) In situ quantification of pSTAT5 expression among paracortical CD4+ T cells. Representative distribution is shown from a Foxp3DTR+/+ mouse injected with DTX for 7h. Dashed red line represents a discrete threshold of pSTAT5 expression (pSTAT5+) set at 3 times above the population median. Inset = zoomed tail of the distribution.
Figure S2, related toFigure 3. TxA23 cells are activated in the presence of Tregs but fail to respond IL-2. (A) 1x105 TxA23 cells and WT cells were differentially labeled with fluorescent dyes and co-transferred into healthy BALB/c recipients. Top panel: representative confocal micrograph highlights an area of the gastric LN containing both transferred cell types at 48h post-transfer. Bottom panel: magnified ROI (white dashed boxes) from the top panel. Scale bars = 20 μm and 10 μm (insets). (B) Quantification of PD-1 expression over time. PD-1 fluorescence intensity values were normalized to the median of co-transferred WT cells (dashed red line) at each time point. (C) In situ single-cell quantification of pSTAT5 signal within TxA23 cells compared to Tregs over time. Data are normalized to the median of TxA23 cells at 12.5 post-transfer. Dashed red line represents a discrete cut off for pSTAT5 signal. For B and C, k = 17-70 cells were pooled from gastric LN sections of n = 6-7 animals. Data are from 2 independent experiments.
Figure S3, related toFigure 3. TxA23 cells proliferate despite local IL-2 constraints. (A) TxA23 cell proliferation in the gastric LN, 48h post-transfer. Left: TxA23 cells, WT cells, and Tregs were visualized in an entire gastric LN volume using Ce3D imaging. Scale bar = 200 μm. Right: Zoomed insets highlighting heterogeneous fluorescence intensities of TxA23 cells. Scale bar = 20 μm. (B) Quantification of A. A GMM with unequal variance was fit to the distribution of CellTracker DeepRed using the EM algorithm and Bayesian information criterion (BIC) (see Figure 3C). The graph depicts the fraction of TxA23 cells within each mixture component. (C) TxA23 cell proliferation in the gastric LN, 96h post-transfer. Plot of the BIC values determining the best model (i.e., equal vs. unequal variance) and optimal number of mixture components. (D) and (E) CellTrace Violet distributions of co-transferred WT cells in the gastric LN at 48h and 96h post-transfer. For each timepoint, the fluorescence intensity values were normalized the median of the distribution. Model fitting was performed as in B and C. Both histograms fit a single component, consistent with 0 rounds of cell division. (F) Experimental schematic illustrating the timing of anti-CD62L blocking antibodies, FTY720, and IL-2:S4B61 immunocomplex injections in G-J (G) Ce3D imaging of an entire gastric LN at 72h post-transfer with the IL-2:S4B61 immunocomplex. Insets: magnified ROIs (white dashed boxes) highlighting multiple TxA23 cell clusters. Scale bars = 100 μm and 20 μm (Inset). (H) The absolute number of TxA23 or WT cells per gastric LN was determined by Ce3D imaging at the indicated time points. Each small dot represents a LN from a single mouse. Large dots and error bars represent mean ± SEM. **p < 0.001 determined using a two-tailed Student’s T test. (I) Quantification of WT and TxA23 cell clusters in the gastric LN at 72h post-transfer +/− IL-2:S4B61 immunocomplex using the DBSCAN algorithm (see STAR Methods). (J) Related to I. Quantification of the number of TxA23 cells per cluster using the DBSCAN algorithm.
Figure S4, related toFigures 4 and 5 Paracrine IL-2 signaling enhances local Treg proliferation, promoting micro-domain formation and growth (A) In situ quantification of pSTAT5 signal in Tregs. Violin and dot plots illustrate single-cell distributions in C57BL/6 animals injected with IL-2 blocking Abs or isotype controls. Popliteal LNs were assessed 24h after Ab injections. Dashed red line represents a discrete threshold for pSTAT5 expression. (B) Quantification of the global (i.e., mean) Treg density within the gastric LN of BALB/c mice. LNs were assessed 24h after injecting IL-2 blocking Abs or isotype controls. Each dot represents a density measurement from 20 μm LN section. Sections were pooled from LNs of n = 4 animals. (C) Quantification of Foxp3 expression in cells expressing fluorescent proteins 14 days after injecting tamoxifen into Foxp3CreERT2+/−-Confettifl/− mice. Each dot represents an individual animal. (D) Representative confocal micrographs illustrating Treg clonality near a CD4+ PD-1+ T cell. Dashed white circle highlights a CD4+ PD-1+ T cell of interest. Dashed white traces outline individual Tregs of interest and highlight Foxp3 expression. XZ optical slices highlight CD4 and PD-1 expression (arrow). Scale bar = 10 μm.
Figure S5, related toFigure 6A computational model predicts that modest reductions in micro-domain size or functionality can enable self-activated T cells to respond to IL-2. (A) and (B) Scatter plots showing the concordance between the independently simulated pSTAT5CD4-max (x-axis) and those predicted by a RFML regression model (y-axis) using out-of-bag (OOB) data (i.e., data not used for training). Two RFML models are shown: A is based on the initial simulations (“initial data”) only while B is based on initial data plus additional simulations enriched for parameter configurations with large pSTAT5CD4-max values (“combined data”), thus increasing prediction performance. Each r value represents the Pearson correlation coefficient between the predicted and simulated pSTAT5CD4-max values. Each dot represents the pSTAT5CD4-max for a specific parameter configuration. (C) Averaged local variable importance (LVI) of the parameter configurations above the cut-off for the pSTAT5CD4-max shown in Figure 6B. The LVI was calculated using the combined data model described above. “Permutation importance” (x-axis) represents the increase in prediction errors for the OOB data after permuting the indicated variables. (D), (E), and (F) Dynamical trajectories of IL-2 secretion, IL-2Rα protein copy number, and pSTAT5 signal in a CD4+ T cell as Tregλ is varied. The control condition represents an inactive configuration that was picked at random from parameter space. (G), (H) and (I) Dynamical trajectories as in D-F, but this time CTLA-4 or IL-2Rβ expression was reduced by 50% in Tregs. (J) Prediction performance of the RFML classification model comparing susceptible and active configurations. Performance was assessed using the receiver operator characteristic (ROC) and precision-recall curves; area under the curve (AUC) was used as the quantitative metric. (K) and (L) Distributions of values for the top two parameters (i.e Tregλ and TregContact_Baseline) in main Figure 6I and the calculated Tregλ-effective (Tregλ * TregContact_Baseline). Distributions are shown for active, inactive, and susceptible configurations.
Figure S6, related toFigure 7Modest reductions in Treg micro-domain size or functionality promote non-linear breakdowns in control, enabling self-activated T cells to subvert pruning (A) Quantification of cell-surface TCR-β in Tregs harvested from SLOs of Foxp3CreERT2+/+-TCR-αfl/fl. Measurements were made by flow cytometry 5 days after the final injection of corn oil (vehicle control) or tamoxifen. Discrete gating was performed on CD3, CD4, and Foxp3. (B) PDFs of Treg spatial coordinates overlaid with the positions of individual PD-1+ CD4+ T cells (black dots) in Foxp3CreERT2+/+-TCR-αfl/fl animals. Representative inguinal LNs 5 days after the final injection of corn oil (vehicle control) or tamoxifen. Scale bar = 200 μm (C) Local, empirical densities of Tregs surrounding individual PD-1+ CD4+ T cells within inguinal LNs of Foxp3CreERT2+/+-TCR-αfl/fl injected with corn oil (vehicle) or tamoxifen. Solid lines represent local regressions determined by the LOESS method. Error bars represent 95% confidence intervals. For A-C, data are representative of LNs from n = 5 animals from 2 independent experiments. (D) Quantification of chimerism 10 weeks after irradiation and bone marrow reconstitution of C57BL/6 mice. Irradiated recipient animals were injected with different ratios of WT and Foxp3DTR+/+ bone marrow. The DTR is fused to an eGFP (DTR-eGFP) in the Foxp3DTR+/+ strain. Measurements were made by flow cytometry using peripheral blood. Discrete gating was performed on CD3, CD4, and Foxp3. Percentages represent the mean ± SEM from LNs of n = 5 animals. (E) Representative confocal micrograph illustrates a clustered micro-domain of WT and DTR expressing Tregs. Popliteal LN sections were prepared from a 60% WT + 40% Foxp3DTR+/+ chimera that was not injected with DTX. Scale bar = 20 μm. (F) Local empirical densities of Tregs surrounding individual PD-1+ CD4+ T cells within the popliteal LN paracortex of 60% WT + 40% Ubi-GFP+/+ or 60% WT + 40% Foxp3DTR+/+ chimeras, both injected with DTX. Solid lines represent local regressions determined by the LOESS method. Error bars represent 95% confidence intervals. Data are representative of LNs from n = 3-5 animals per condition (G) In situ quantification of CTLA-4 and IL-2Rβ in Tregs from the indicated animals. Analysis was performed 7 days after the final injection of corn oil (vehicle) or tamoxifen. Measurements were made in the popliteal LNs. Data are representative of LNs from n = 4 animals per condition from 2 independent experiments. (H) PDFs of IL-2Rα expression in IL-2 responders (pSTAT5+) vs. non-responders (pSTAT5−) in each indicated experimental condition. Single-cell distributions were pooled from LNs of n = 3-4 animals for each KDE.
Figure S7, related toFigure 7. Local Treg feedback gives rise to a sharp discriminatory boundary between self and non-self (A) In situ quantification of paracortical Tregs frequencies in the indicated animal strains. *p < 0.05 determined using a two-tailed, Student’s t test (B) In situ quantification of Tregs within 40 μm of pSTAT5+ CD4+ T cells (i.e., emergent active configurations). Each dot represents an individual active configuration pooled from n = 3 animals. ***p < 0.0001 was determined using a two-tailed Kolmogorov-Smirnov test. (C) Distributions of TCR off-rates predicted by MAPPA in inactive, susceptible, and active configurations. (D) 5C.C7 and WT cells were differentially labelled with fluorescent dyes and co-transferred into healthy B10.A recipients. 24h post-transfer, 1 μg of T102S peptide was injected subcutaneously. Quantification of PD-1 expression post peptide injection at the indicated time points in the draining popliteal LN. PD-1 fluorescence intensity values were normalized to the median of co-transferred WT cells (dashed red line) at each time point. (E) Maximum deviation in λTreg (Δ λTreg) or λpSTAT5+Treg (ΔλpSTAT5+Treg) between 5C.C7 and WT cells over time. Animals were injected subcutaneously with 1 μg of MCC88-103 or T102S and popliteal LNs were assessed at the indicated time points. Colored curves represent local regressions with 95% confidence intervals determined by the LOESS method. (F) Quantification of pSTAT5 signal within 5C.C7 cells responding to T102S or MCC(88-103). Dashed red line represents discrete cutoff for pSTAT5 expression. For D, E, and F k = 24-240 cells pooled from 3 animals per timepoint. Data are from 2 independent experiments. (G) A GMM was fit to the normalized CellTracker DeepRed distributions of 5C.C7s as in Figure 3C. Fluorescence measurements were made in draining popliteal LNs using confocal microscopy 48h post peptide injection. The illustrated histogram is decomposed into its individual mixture components. (H) Fraction of 5C.C7 cells belonging to each mixture component. Nonparametric bootstrapping was used to determine the robustness of the model. Data are Mean ± 95% confidence intervals derived from 999 bootstrap samples. For G and H, k = 83 cells for the T102S condition and k = 175 cells for the MCC88-103 condition. Data are from two independent experiments.
Movie S1, related toFigure 3Ce3D imaging enables accurate localization and enumeration of rare CD4+ T cells. The entire volume of the gastric LN is shown 24h after co-transferring 5x104 TxA23 (orange) and WT cells (cyan) into healthy BALB/c recipients. Endogenous Foxp3 (purple) depicts Tregs.
Movie S2, related toFigure 3TxA23 cells proliferate in the presence of functional Tregs. Ce3D imaging of the entire gastric LN 72h after co-transferring 5x104 TxA23 (orange) and WT cells (cyan) into healthy BALB/c recipients. See Figure 3A for experimental details. Endogenous Foxp3 (purple) depicts Tregs.
Movie S3, related toFigure 3TxA23 cells are rapidly pruned from the host following proliferation. Ce3D imaging of the entire gastric LN 192h after co-transferring 5x104 TxA23 (orange) and WT cells (cyan) into healthy BALB/c recipients. See Figure 3A for experimental details. Endogenous Foxp3 (purple) depicts Tregs.
Data Availability Statement
The code supporting the current study has been deposited on Github