

Abstract
The precise molecular effects that antiangiogenic drugs exert on the tumor vasculature remain poorly understood. We therefore set out to investigate the molecular and architectural changes that occur in the vasculature of two different tumor types that both respond to VEGFR2 inhibitor therapy.
Mice bearing Lewis Lung Carcinoma (LLC) or B16.F10 melanoma were treated with vandetanib (ZD6474), a VEGFR2/EGFR/RET kinase inhibitor, resulting in a significant 80% reduction of tumor outgrowth. While in LLC vascular density was not affected by vandetanib treatment, it was significantly decreased in B16.F10. In LLC, vandetanib treatment induced a shift in vascular gene expression toward stabilization, as demonstrated by upregulation of Tie2 and N-cadherin, and downregulation of Ang2 and integrin β3. In contrast, only eNOS and P-selectin responded to vandetanib treatment in B16.F10 vasculature. Strikingly, vandetanib reduced protein expression of VEGFR2 in both models, while mRNA remained unaffected. Analysis of miR-296 expression allowed us to exclude a role for the recently proposed microRNA-296 in VEGFR2 posttranslational control in LLC and B16.F10 in vivo.
Our data demonstrate that VEGFR2/EGFR inhibition through vandetanib slows down both LLC and B16.F10 tumor growth, yet the underlying molecular changes in the vasculature that orchestrate the antitumor effect differ between tumor types. Importantly, in both models vandetanib treatment induced loss of its pharmacological target, which was not directly related to miR-296 expression. Validation of our observations in tumor biopsies from VEGFR2 inhibitor-treated patients will be essential to unravel the effects of VEGFR2 inhibitor therapy on tumor vasculature in relation to therapeutic efficacy.
Keywords: tumor angiogenesis, VEGFR2 inhibition, vascular gene expression, vascular stabilization, receptor tyrosine kinase inhibitors
INTRODUCTION
For metastasis and growth, many tumors depend on angiogenesis, a tightly regulated process in which vascular endothelial growth factor (VEGF) plays a key regulatory role [1]. Indeed, high levels of VEGF alone are capable of initiating angiogenesis in a quiescent vasculature [2] and in most cancers VEGF overexpression is associated with disease progression [3], which makes VEGF an attractive target for antiangiogenic therapy. Currently three inhibitors of the VEGF pathway are approved by the FDA for use in cancer therapy [4]. Importantly, the cellular and molecular mechanisms that underlie efficient tumor growth inhibition through VEGF inhibitor therapy remain unknown. Several mechanisms of action have been proposed that contribute to the effect of VEGF-blocking agents, including inhibition of neovessel growth, blockade of incorporation of endothelial progenitor cells, induction of endothelial cell apoptosis, and vascular normalization [5, 6].
In many preclinical models, inhibition of tumor growth by inhibitors of VEGF signaling has been related to a decrease in microvascular density [7–9]. Yet, the changes in vascular gene expression upon VEGFR2 inhibition that orchestrate the tumor growth inhibitory effect in these models remain largely unknown. Studies in various in vitro and in vivo neovascularization models have shown that VEGF signaling upregulates the expression of many regulators of the angiogenic process, such as integrin αvβ3, the zinc finger transcription factor early growth response-3 (EGR3), the NR4A family of orphan nuclear receptors, matrix metalloproteinases, NO synthases, Robo1, Nur77, Tie2, and the Notch/Dll4/Jagged-1 signaling family, while downregulating expression of the vascular cell adhesion molecules ICAM and VCAM [10–13, 13–16]. The consequences of inhibition of VEGF signaling for endothelial gene expression patterns in vivo remain largely unknown.
In the present study, we aimed to identify the molecular changes that occur in the vasculature of Lewis Lung Carcinoma and B16.F10 melanoma upon antiangiogenic VEGFR2 inhibitor therapy. As a model antiangiogenic compound we used the small molecule VEGFR2 tyrosine kinase inhibitor vandetanib (ZD6474), that exerts its effect by preventing binding of ATP to the ATP-binding pocket of the receptor, thereby inhibiting autophosphorylation of the receptor dimers [17]. Vandetanib has additional affinity for epidermal growth factor receptor (EGFR) and REarranged during Transfection (RET) [18], and is currently in clinical trials for medullary thyroid cancer and a range of other tumor types [19]. We treated Lewis Lung Carcinoma and B16.F10 melanoma-bearing mice with vandetanib, monitored tumor growth during the course of therapy and analyzed the consequences for vascular architecture and density. Interestingly, we found that vandetanib-enforced tumor growth inhibition was accompanied by a significant decrease in vascular density in the B16.F10 melanoma model, while in Lewis Lung Carcinoma the overall vascular surface area remained unaffected. In order to unravel the molecular basis for this difference in vascular behavior, we set out to unravel the compartmentalized effects of vandetanib on gene expression in the vasculature of both models that was isolated by laser microdissection prior to transcriptional profiling. Based on the observed changes in vascular gene expression, we assessed the pattern of pericyte coverage in both models. Furthermore, we investigated the localization of expression of the pharmacological targets of vandetanib, and found that in both models, vandetanib decreased protein levels of VEGFR2. As the VEGFR2 mRNA level was not affected by therapy, we assessed whether the recently proposed VEGFR2-controlling microRNA-296 has a role in vandetanib-induced VEGFR2 protein loss.
MATERIALS & METHODS
Tumor cell culture and animal studies
B16.F10 murine melanoma cells were cultured in DMEM (Biowhittaker, Verviers, Belgium) supplemented with 10% fetal calf serum (FCS; Hyclone, Perbio Science, Etten-Leur, The Netherlands), 2 mM L-glutamine (Biowhittaker) and 1% gentamicin (Biowhittaker). Lewis Lung Carcinoma (LLC) cells (ATCC, Manassas VA, USA), were cultured in DMEM (Cellgro Mediatech, Manassas, VA, USA) supplemented with 5% FCS, 100 IU/ml penicillin and 100 μg/ml streptomycin (Cellgro Mediatech). Both cell lines were kept at 37°C and 5% CO2/95% air in a humidified incubator.
C57bl/6 male mice (Charles River, Frederick, MD, USA; and Harlan, Zeist, The Netherlands) were subcutaneously injected with 500,000 LLC cells or 100,000 B16.F10 cells in 100 μl PBS. Tumors were measured every other day by calipers, and the volume was calculated according to the formula: tumor volume = 0.52*length*width2, with ‘width’ being the shorter of the two diameters. When tumors appeared palpable, mice were randomly distributed into two groups and received i.p. injections with vandetanib (ZD6474; a kind gift from AstraZeneca, Macclesfield, UK) at 80 mg/kg in 5% arabic gum [20] or with vehicle alone, once a day. As for the Lewis Lung Carcinoma-bearing animals, injection with vandetanib was omitted at day 3 and 8 after start of treatment to prevent further weight loss during the course of therapy. After 10–12 days of treatment, the animals were sacrificed under anesthesia by inhalation of isoflurane/O2 or by CO2 euthanasia, and tumors were excised, immediately snap frozen in liquid N2, and stored at −80°C until further analysis.
All experiments were performed in accordance with the Animal Care and Use guidelines by the National Cancer Institute at Frederick and the University of Groningen.
Immunohistochemical and immunofluorescent staining of tumor tissues
Immunohistochemical detection of CD31, VEGFR2 and EGFR in acetone-fixed, 5 or 10 μm cryostat-cut tumor sections was performed using the DAKO Envision System-HRP kit (Dako Cytomation, Glostrup, Denmark) according to the supplier’s protocol. We used primary antibodies to CD31 (BD Pharmingen; clone MEC13.3), Flk1, and EGFR (both from R&D, Minneapolis MN, USA). Detection was performed with Rabbit-anti-Rat IgG (H+L, Vector Labs, Burlingame CA, USA) or Rabbit-anti-Goat-HRP (DAKO), and after incubation with an anti-rabbit HRP-conjugated polymer for 30 minutes, peroxidase activity was detected with 3-amino-9-ethylcarbazole (AEC) complex and sections were counterstained with Mayer’s hematoxylin (Merck, Darmstadt, Germany). Sections were embedded in Kaisers Glycerin (Merck) and examined using a Leica DMLB microscope (Leica, Wetzlar, Germany) equipped with Leica DFC 420C camera, and Leica Qwin V3.5.1 software. The percentage of CD31-positive surface area was quantified by morphometric analysis using the same software package.
To visualize pericytes, sections were double-stained with antibodies to either αSMA (Cy3-labeled; Sigma Aldrich, Steinheim, Germany) or desmin (Abcam, Cambridge, UK) together with antibodies against CD31. Subsequently, sections were incubated with Alexa fluor 568-conjugated Goat-anti-Rabbit IgG to detect desmin and Alexa fluor 488-conjugated Goat-anti-Rat IgG to detect CD31 (both H+L, Molecular Probes Invitrogen Detection Technologies, Eugene, Oregon), and nuclear counterstaining was performed using DAPI (F. Hoffmann-La Roche Ltd, Basel, Switzerland). Autofluorescence was reduced by incubation in 0.1% Sudan Black (Sigma-Aldrich) in 70% ethanol for 30 minutes, after which the sections were embedded in Citifluor (Citifluor Ltd., London, UK) and examined using a fluorescence microscope (DM RXA, Leica) and Leica Qwin V3 software.
mRNA extraction from whole tumor samples
Extraction of total RNA from cryostat-cut sections of Lewis Lung Carcinoma and B16.F10 tissue as a whole was carried out according to the protocol of RNeasy Mini Plus kit or microRNeasy Mini Kit (both from Qiagen, Leusden, The Netherlands; the latter was used to isolate total RNA including microRNAs). RNA was analyzed qualitatively by gel electrophoresis and quantitatively by Nanodrop ND-100 spectrophotometry (NanoDrop Technologies, Rockland, DE, USA) and consistently found to be intact and protein-free.
Laser microdissection of B16.F10 and LLC tumor vasculature
Nine-μm cryosections from B16.F10 tumors mounted on polyethylene-naphtalene membranes attached to normal glass slides (P.A.L.M. Microlaser Technology AG, Bernried, Germany) were fixed in acetone and stained with Mayer’s hematoxylin, washed with diethyl pyrocarbonate-treated water and air-dried. Tumor vascular segments (1–2×106 μm2 surface area, including vessel lumen area) were recognized based on a visible lumen and microdissected using the LMD6000 Laser Microdissection system (Leica). Blood vessels growing in Lewis Lung Carcinoma generally do not contain a visible lumen and are therefore not identifiable by hematoxylin staining. For this, we stained 9-μm acetone-fixed cryosections of LLC with a Goat antibody to Collagen IV (Southern Biotech, Birmingham, AL, USA) that was labeled with an Alexa488 conjugate using the Zenon Alexa fluor 488 Goat IgG labeling kit (Invitrogen). Sections were washed twice with PBS and air-dried prior to microdissection.
Total RNA was extracted according to the protocol of RNeasy Microkit or microRNeasy Mini Kit (Qiagen), and reverse transcribed for use in quantitative PCR as described below.
Gene expression analysis by quantitative RT-PCR (qRT-PCR)
Total RNA was reverse transcribed as described previously [21], using Superscript III Reverse Transcriptase (Invitrogen, Carlsbad, CA, USA) in a 20-μl final volume containing 250 ng of random hexamers (Promega, Madison, WI, USA) and 40 units of RNase OUT inhibitor (Invitrogen). mRNA expression analysis was performed in a real-time PCR-based, custom-designed, Low Density Array set-up (Applied Biosystems, Foster City, CA, USA). The Low Density Array was designed with exon-overlapping primers and minor groove-binding (MGB) probes of 45 genes selected for their involvement in angiogenesis, inflammation and basic influence on endothelial cell behavior, and of GAPDH as housekeeping gene. The Low Density Array card was processed according to the supplier’s protocol and analyzed in an ABI PRISM 7900HT Sequence Detector System (Applied Biosystems).
For a selection of genes, real-time PCR was performed in duplicate per sample with 1 μl cDNA per reaction in TaqMan PCR MasterMix in a total volume of 10 μl, with primer-probe sets (where appropriate corresponding to the ones pre-spotted on the Low Density Array cards) being purchased as Assay-on-Demand from Applied Biosystems (Nieuwekerk a/d IJssel, The Netherlands).
microRNA expression analysis
Isolated microRNA was transcribed using the Taqman MicroRNA Reverse Transcription Kit (Applied Biosystems) in a 15-μl final volume according to the suppliers’ protocol. MicroRNA-126 and microRNA-296 expression analysis was performed in a real-time PCR-based set up with 1 μl cDNA per reaction in TaqMan PCR MasterMix in a total volume of 10 μl, with primer-probe sets (MicroRNA assays) being purchased from Applied Biosystems (Nieuwekerk a/d IJssel, The Netherlands). Small nuclear RNA sno-202 (Applied Biosystems) was assayed as an internal control.
Statistics
Statistical significance of the observed differences was addressed by means of Student’s t-test or ANOVA with post hoc comparison using Bonferroni correction on the mean of duplicate qRT-PCR analyses per mouse. These statistical analyses were performed using GraphPad Prism V5.00 (GraphPad Software, San Diego, CA). Statistical significance of the differences in tumor volume at day 10 (LLC) or day 9 (B16.F10) after start of treatment were assessed by Mann-Whitney U test using Statistical Software Package SPSS. Differences were considered to be significant when p<0.05, unless stated otherwise.
RESULTS
Vandetanib treatment inhibited tumor growth in both LLC and B16.F10, but differentially affected vascular density
To investigate the molecular changes that occur in the tumor endothelium in vivo in a tumor which responds to antiangiogenic VEGFR2 inhibitor treatment, we treated LLC and B16.F10 tumor-bearing animals with the VEGFR2/EGFR tyrosine kinase inhibitor vandetanib at a dose of 80 mg/kg daily. Treatment significantly inhibited Lewis Lung Carcinoma tumor growth and B16.F10 tumor growth with 84% and 82% respectively (Fig 1). In both tumor models, vascular morphology shifted upon treatment from large, irregularly shaped vessels to smaller vessels that were characterized by a more round and regular shape (Fig 2A–D). A prominent difference between the two tumor models was however observed in the vascular density after vandetanib treatment. While in LLC vascular density remained unaffected, a significant decrease in vascular density had occurred in B16.F10 tumors (Fig 2E–F). Treatment significantly reduced the overall CD31-positive surface area of B16.F10 tumors ~2-fold, which was confirmed by a 3-fold reduction in mRNA levels of the endothelial marker molecules CD31 and VE-cadherin in the tumor (Fig 2G–H).
Figure 1: Vandetanib treatment significantly reduced outgrowth of Lewis Lung Carcinoma and B16.F10 melanoma.
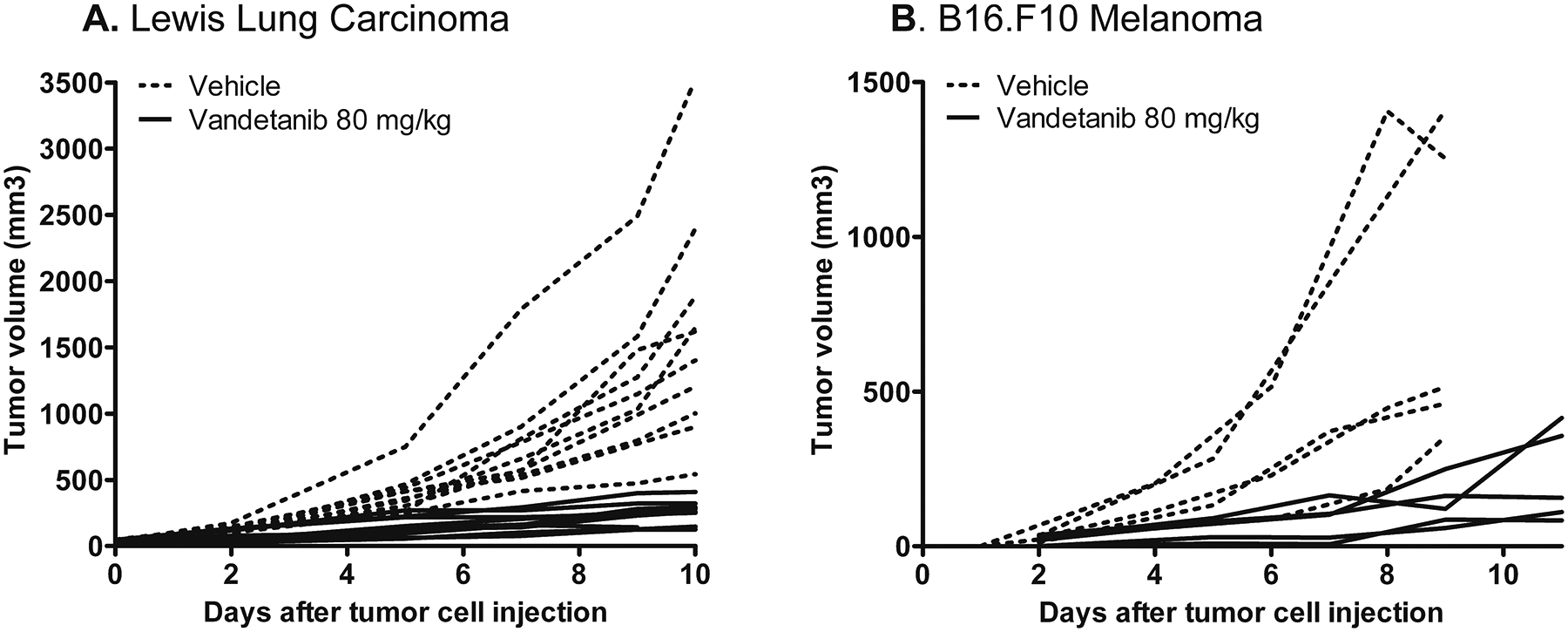
A+B: LLC and B16.F10-bearing mice were treated i.p. daily with 80 mg/kg of the VEGFR2 inhibitor vandetanib in 5% arabic gum or with vehicle alone. Treatment was started when tumors appeared palpable and continued for 10 (LLC vehicle and vandetanib group), 9 (B16 vehicle group) or 12 (B16 vandetanib group) days. In the LLC model, treatment was omitted at the 3rd and 8th day after start of therapy. Graph represents tumor growth curves of each mouse (tumor) separately. The average tumor volumes in vandetanib-treated mice at day 10 (LLC) or day 9 (B16.F10) after start of treatment were significantly smaller than in vehicle-treated mice (Mann-Whitney U test; p<0.05).
Figure 2: Vandetanib treatment reduced vascular surface area from B16.F10 melanoma, but not from Lewis Lung Carcinoma.
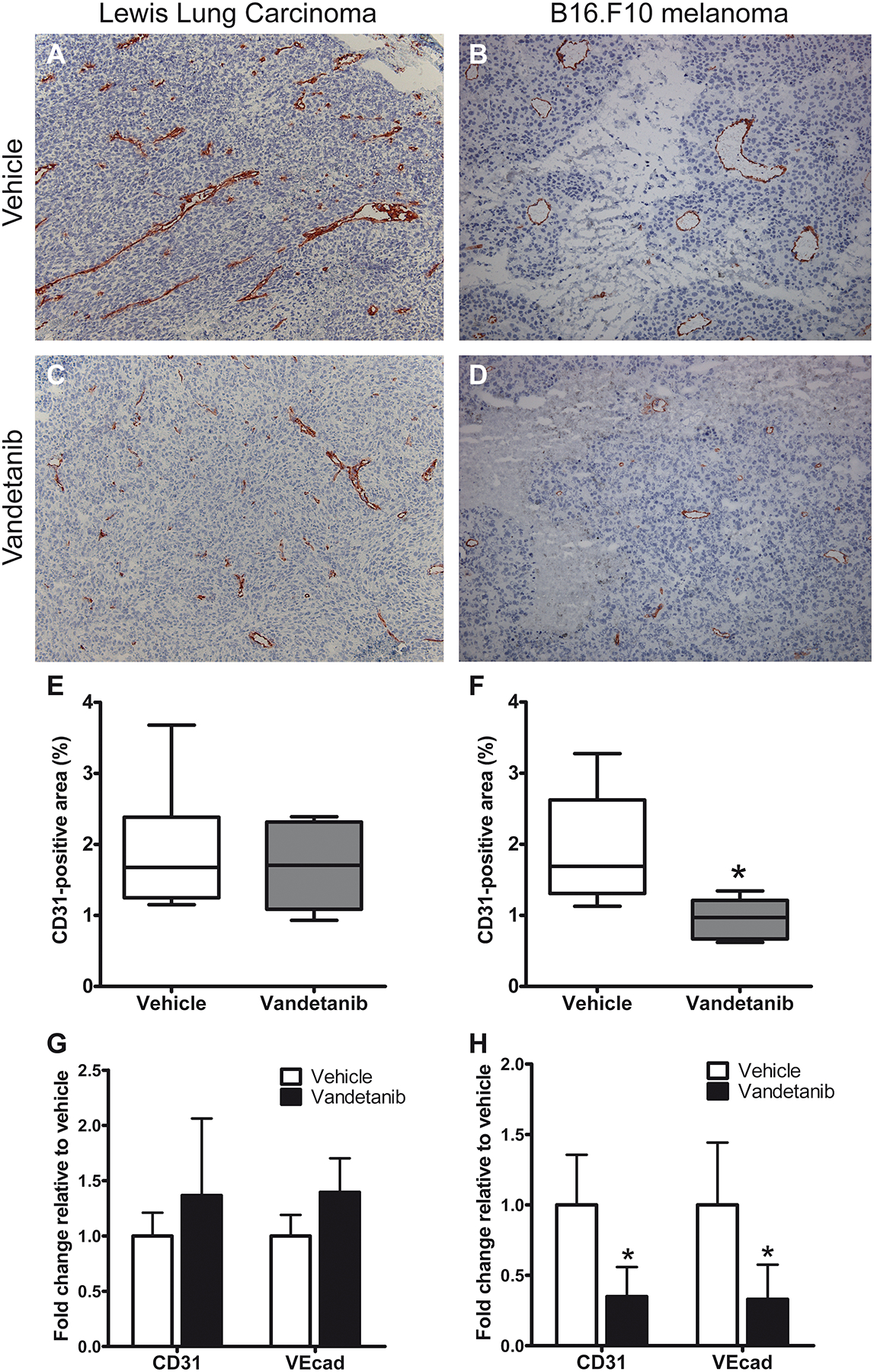
A–D: Vascular morphology of vandetanib and vehicle-treated LLC and B16.F10 tumors. Immunohistochemical staining for CD31, magnification 100x. E+F: Quantification of CD31-positive surface area in tumor sections using morphometric analysis. Graph represents median of 6 (LLC) or 5 (B16) tumors, minimum and maximum. G+H: mRNA levels of endothelial marker genes CD31 and VE-cadherin in B16.F10 and LLC tumors of vandetanib and vehicle-treated mice, normalized to GAPDH. Values represent mean of duplicate qRT-PCR analysis of 6 (LLC) or 5 (B16.F10) animals per group. *p<0.05 vs vehicle-treated control.
Vandetanib treatment differentially affected angio-gene expression in LLC and B16.F10
As vascular density in LLC and B16.F10 tumors was differentially affected by vandetanib treatment, we hypothesized that vandetanib has a different effect on the underlying molecular behavior of the tumor endothelium in these two models. We thus compared the changes in expression of genes that are known to be involved in angiogenesis and vascular stabilization upon vandetanib treatment in the vasculature of LLC to those in B16.F10 vasculature.
VEGFR2/EGFR inhibitor therapy significantly downregulated mRNA expression of the pro-angiogenic/vascular destabilization molecules integrin β3 and Ang2 3-fold and significantly upregulated mRNA levels of the vascular stability molecules Tie2 and N-cadherin approximately 3-fold in Lewis Lung Carcinoma (Fig 3). Furthermore, an approximately 1.5-fold decrease in PDGFRβ expression was observed in LLC tumor vasculature upon vandetanib, and a 9-fold upregulation of desmin, that however did not reach statistical significance (p=0.08) due to interindividual variation between the mice. In contrast, vandetanib treatment induced a 2.3-fold increase in eNOS mRNA in B16.F10 vasculature. In addition, PlGF showed a 3-fold downregulation upon vandetanib that did not reach statistical significance (p=0.09). Although expression of integrin β3, PDGFRβ and N-cadherin showed a similar, albeit non-significant, trend in B16.F10 compared to LLC, only P-selectin expression was similarly affected in both models, showing a significant >10-fold upregulation upon vandetanib. The mRNA levels from all genes under study, as measured in the vasculature of both the vehicle- and vandetanib-treated groups of LLC and B16.F10 tumors are depicted in Table 1.
Figure 3: Vandetanib-induced changes in gene expression in LLC and B16.F10 tumor vasculature.
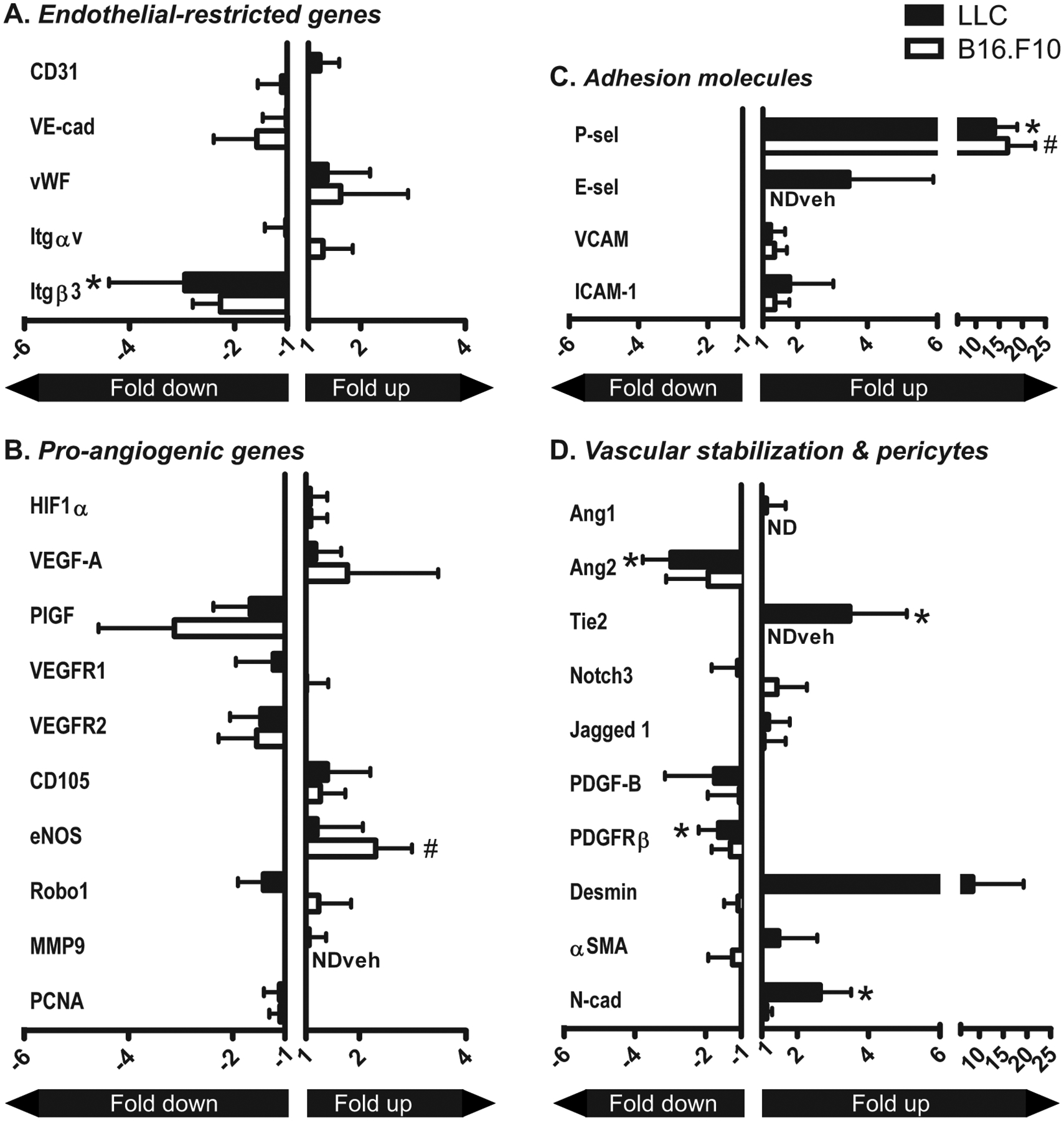
A–D: mRNA expression levels of various angiogenesis-associated and vascular behavior-determining genes in tumor vasculature that was isolated by laser microdissection from LLC and B16.F10 tumors treated with vandetanib or vehicle alone. Values represent fold increase or decrease in mRNA expression in the vasculature of vandetanib-treated tumors compared to vehicle-treated tumors, normalized to GAPDH. Mean + SD of 6 (LLC) or 5 (B16.F10) animals per group. NDveh = not detectable in the vehicle-treated mice, no ratio could be calculated. ND = not detectable in both vehicle- and vandetanib-treated group. *p<0.05 vs vehicle-treated control.
Table 1:
mRNA levels adjusted to GAPDH (x1000) of angiogenesis and vascular behavior-assoicated genes in tumor vasculature from vehicle and ZD6474-treated tumors.
| Gene | B16.F10 melanoma | Lewis Lung Carcinoma | ||
|---|---|---|---|---|
| Vehicle | Vandetanib | Vehicle | Vandetanib | |
| CD31 | 6.40 ± 3.26 | 5.79 ± 2.34 | 109.16 ± 16.22 | 133.07 ± 40.42 |
| VEcad | 5.33 ± 4.36 | 3.16 ± 2.13 | 165.95 ± 72.60 | 169.18 ± 53.22 |
| vWF | 1.65 ± 0.71 | 2.67 ± 2.12 | 66.33 ± 40.42 | 89.93 ± 54.54 |
| Itgαv | 1.06 ± 0.31 | 1.36 ± 0.60 | 22.95 ± 6.93 | 22.36 ± 8.69 |
| Itgβ3 | 0.96 ± 0.76 | 0.42 ± 0.10 | 46.88 ± 17.76 | 15.91 ± 7.76 |
| HIF1α | 3.48 ± 1.19 | 3.79 ± 1.09 | 32.66 ± 20.46 | 35.36 ± 10.43 |
| VEGF-A | 0.77 ± 0.24 | 1.37 ± 1.31 | 42.95 ± 10.31 | 50.59 ± 20.70 |
| PlGF | 1.61 ± 1.46 | 0.52 ± 0.24 | 8.11 ± 4.02 | 4.89 ± 2.09 |
| VEGFR1 | 0.66 ± 0.40 | 0.67 ± 0.27 | 27.03 ± 6.79 | 22.12 ± 12.99 |
| VEGFR2 | 1.17 ± 0.97 | 0.76 ± 0.36 | 72.96 ± 20.07 | 50.05 ± 20.32 |
| CD105 | 9.09 ± 2.75 | 11.52 ± 4.28 | 85.64 ± 58.69 | 120.25 ± 69.03 |
| eNOS | 0.26 ± 0.07 | 0.61 ± 0.18 | 10.60 ± 4.88 | 12.86 ± 9.12 |
| Robo1 | 0.18 ± 0.07 | 0.23 ± 0.11 | 1.00 ± 0.20 | 0.71 ± 0.25 |
| MMP9 | ND | 0.15 ± 0.03 | 22.67 ± 17.75 | 24.01 ± 7.28 |
| PCNA | 23.53 ± 4.85 | 21.62 ± 4.16 | 24.13 ± 10.28 | 21.85 ± 5.92 |
| Ang1 | ND | ND | 1.27 ± 0.83 | 1.43 ± 0.69 |
| Ang2 | 1.12 ± 0.57 | 0.58 ± 0.36 | 30.05 ± 5.81 | 10.09 ± 2.70 |
| Tie2 | ND | 0.57 ± 0.35 | 3.68 ± 1.60 | 12.78 ± 5.89 |
| Notch3 | 0.68 ± 0.39 | 0.98 ± 0.57 | 7.79 ± 3.83 | 7.05 ± 4.65 |
| Jag1 | 1.02 ± 0.59 | 1.08 ± 0.62 | 38.72 ± 7.32 | 46.13 ± 23.13 |
| PDGF-B | 3.67 ± 2.43 | 3.49 ± 2.95 | 62.32 ± 22.85 | 35.56 ± 28.45 |
| PDGF-Rβ | 1.40 ± 0.61 | 1.08 ± 0.44 | 43.41 ± 9.72 | 26.50 ± 9.00 |
| Desmin | 0.15 ± 0.08 | 0.14 ± 0.05 | 0.63 ± 0.22 | 4.61 ± 5.75 |
| αSMA | 16.50 ± 6.21 | 13.35 ± 7.49 | 28.61 ± 10.06 | 42.45 ± 31.00 |
| N-cad | 3.93 ± 1.13 | 4.48 ± 0.60 | 1.54 ± 0.69 | 4.50 ± 1.34 |
| P-sel | 0.21 ± 0.17 | 3.53 ± 1.25 | 1.27± 0.63 | 17.84 ± 6.22 |
| E-sel | ND | 0.65 ± 0.10 | 0.73 ± 0.01 | 2.53 ± 1.77 |
| VCAM-1 | 0.28 ± 0.07 | 0.37 ± 0.10 | 67.87 ± 22.36 | 82.95 ± 28.83 |
| ICAM-1 | 0.61 ± 0.38 | 0.83 ± 0.25 | 7.47 ± 2.02 | 13.20 ± 9.30 |
Vandetanib treatment did not affect the overall pattern of pericyte coverage
As the genes that were most extensively affected by vandetanib treatment, such as Ang2, Tie2 and N-cadherin, play an (indirect) role in endothelial-pericyte adhesion, we hypothesized that vandetanib treatment increased coverage of the tumor vessels with pericytes. We therefore set out to study tumor vessel pericyte coverage in both LLC and B16.F10 by immunofluorescent double-labeling for αSMA or desmin together with CD31. The pattern of desmin coverage of Lewis Lung Carcinoma and B16.F10 blood vessels did not change upon vandetanib treatment (Fig 4). Expression of αSMA was much more heterogeneously distributed throughout the tumors of both types than desmin expression. Not only was αSMA restricted to the merely lumen-containing large vessels, also the intensity of the staining largely varied between blood vessels. Quantification revealed that the percentage of αSMA-positive vessels was not changed by treatment in either of the two tumor models (data not shown). Furthermore, we found that in general B16.F10 tumor vessels were more intensely covered by αSMA-positive pericytes than Lewis Lung Carcinoma vessels (Fig 4 and data not shown).
Figure 4: Vandetanib does not affect the pattern of pericyte coverage in LLC and B16.F10.
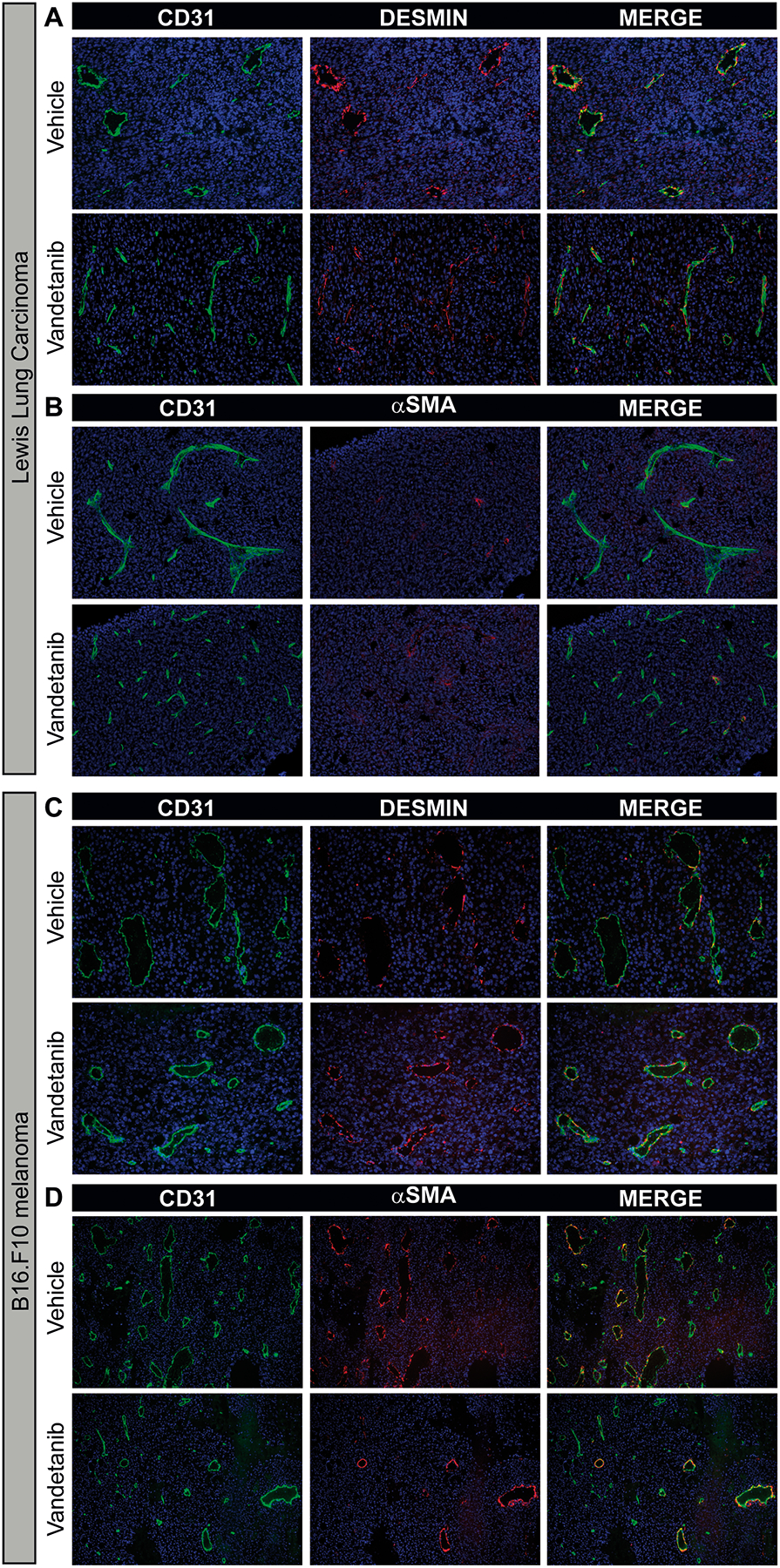
Immunofluorescent double staining for CD31 (green) and desmin (red) or αSMA (red) and nuclear counterstain (DAPI; blue) from a representative LLC tumor (A+B) and B16.F10 tumor (C+D). Magnification 200x for desmin staining and 100x for αSMA staining.
Vandetanib-induced loss of target protein expression is not posttranslationally controlled by miR-296
As vascular density was differentially affected by vandetanib in LLC and B16.F10 tumors, we hypothesized that the drug might target different compartments within the tumor in these two models. We therefore assessed the localization of the drug’s main target receptors. In both tumor models, expression of VEGFR2 was restricted to the endothelium and covered the full vasculature of the tumor prior to start of vandetanib treatment. Strikingly, VEGFR2 staining was strongly diminished after treatment, in both LLC and B16.F10 tumors (Fig 5A–L). Since part of vandetanib’s pharmacological activity can be attributed to EGFR inhibitory activity, we analyzed the localization of EGFR expression. As also EGFR expression is restricted to the vasculature (Fig 5A–L), this implies that in the two tumor models studied, vandetanib selectively targeted the tumor blood vessels. In contrast to VEGFR2, EGFR was expressed only on a subset of the vasculature, predominantly on lumen-containing vessels while smaller vascular structures were devoid of expression. Vandetanib treatment also reduced EGFR expression, although this reduction was less pronounced than for VEGFR2.
Figure 5: Pharmacological targeting of VEGFR2/EGFR with vandetanib resulted in downregulation of the target receptors at the protein level, while mRNA remained unaffected.
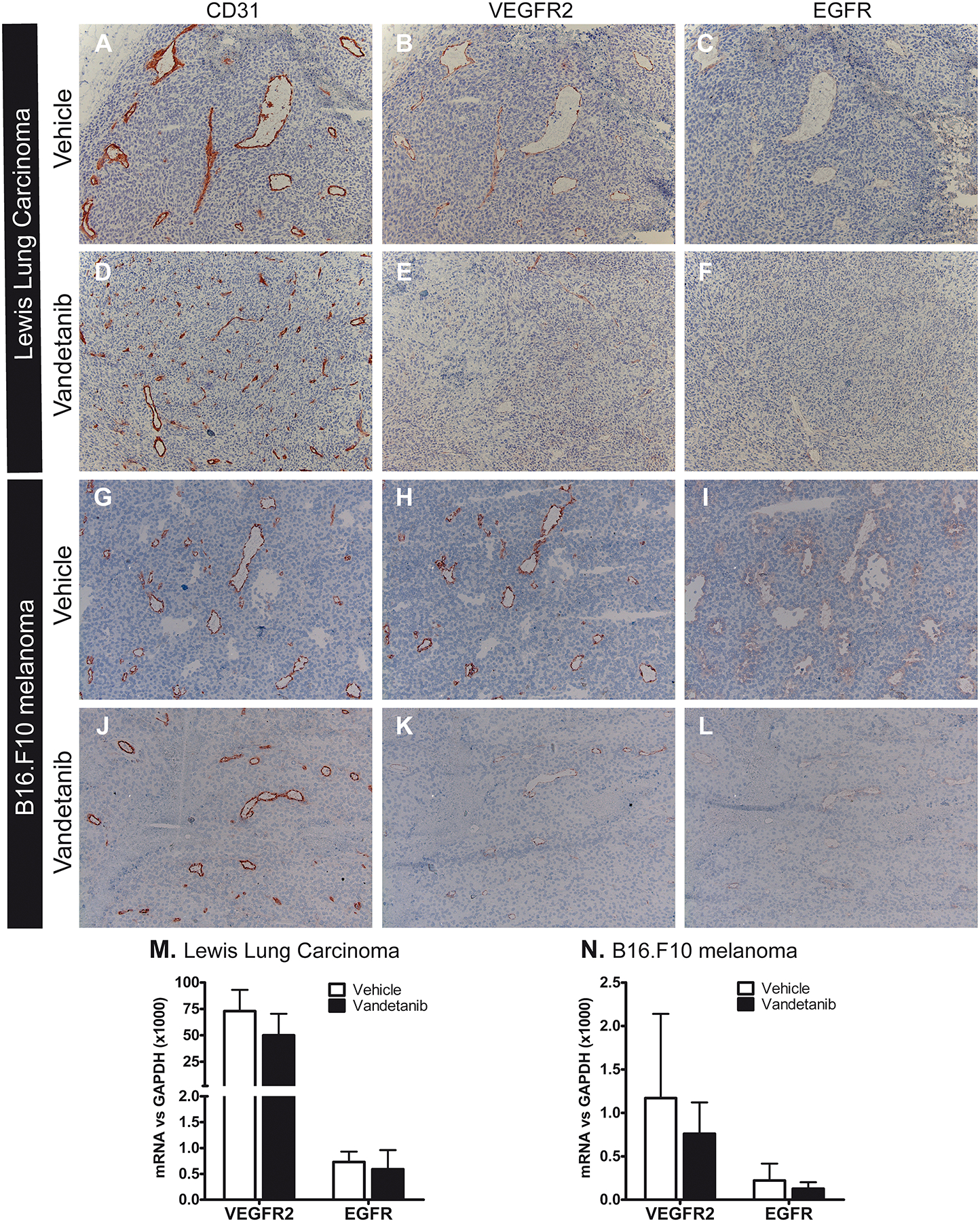
A–L: Protein expression of both VEGFR2 and EGFR was significantly reduced after treatment in LLC (A–F) and B16.F10 (G–L), as visualized by immunohistochemical staining of consecutive sections for VEGFR2, EGFR and CD31, magnification 100x. Interstitial dermal cells and some vessels in the skin surrounding the tumor strongly stained for EGFR, representing a good internal positive control (not shown). M+N: mRNA levels of VEGFR2 and EGFR in microdissected tumor vasculature from LLC (M) and B16.F10 (N) tumors. Values represent mean of duplicate qRT-PCR analysis + SD of 6 (LLC) or 5 (B16) animals per group. *p<0.05.
Interestingly, while protein levels of these target receptors decreased, their mRNA levels remained unaffected in LLC and B16.F10 tumor vasculature (Fig 5M+N). This difference between RNA and protein suggests that the observed downregulation of VEGFR2 upon therapy might be the result of posttranslational regulation. Recently, Würdinger et al. reported that the microRNA miR-296 can control VEGFR2 protein expression. Angiogenic growth factors have been demonstrated to increase the level of miR-296, that in turn increased protein expression of VEGFR2 [22]. To assess whether a change in miR-296 levels is associated with the vandetanib-induced loss of VEGFR2 protein expression observed in both our tumor models, we analyzed expression levels of miR-296 in whole tumor samples from LLC and B16.F10 tumors, and in microdissected B16.F10 tumor vasculature. We corrected for the amount of endothelium in our sample by including the endothelial-restricted miR-126. Vandetanib treatment did not change the expression levels of miR-296 in LLC and B16.F10 tumors, nor in microdissected B16.F10 tumor vasculature (Fig 6), suggesting that the vandetanib-induced downregulation of VEGFR2 protein is not posttranslationally controlled by miR-296.
Figure 6: The downregulation of VEGFR2 protein was not related to microRNA-296 expression.
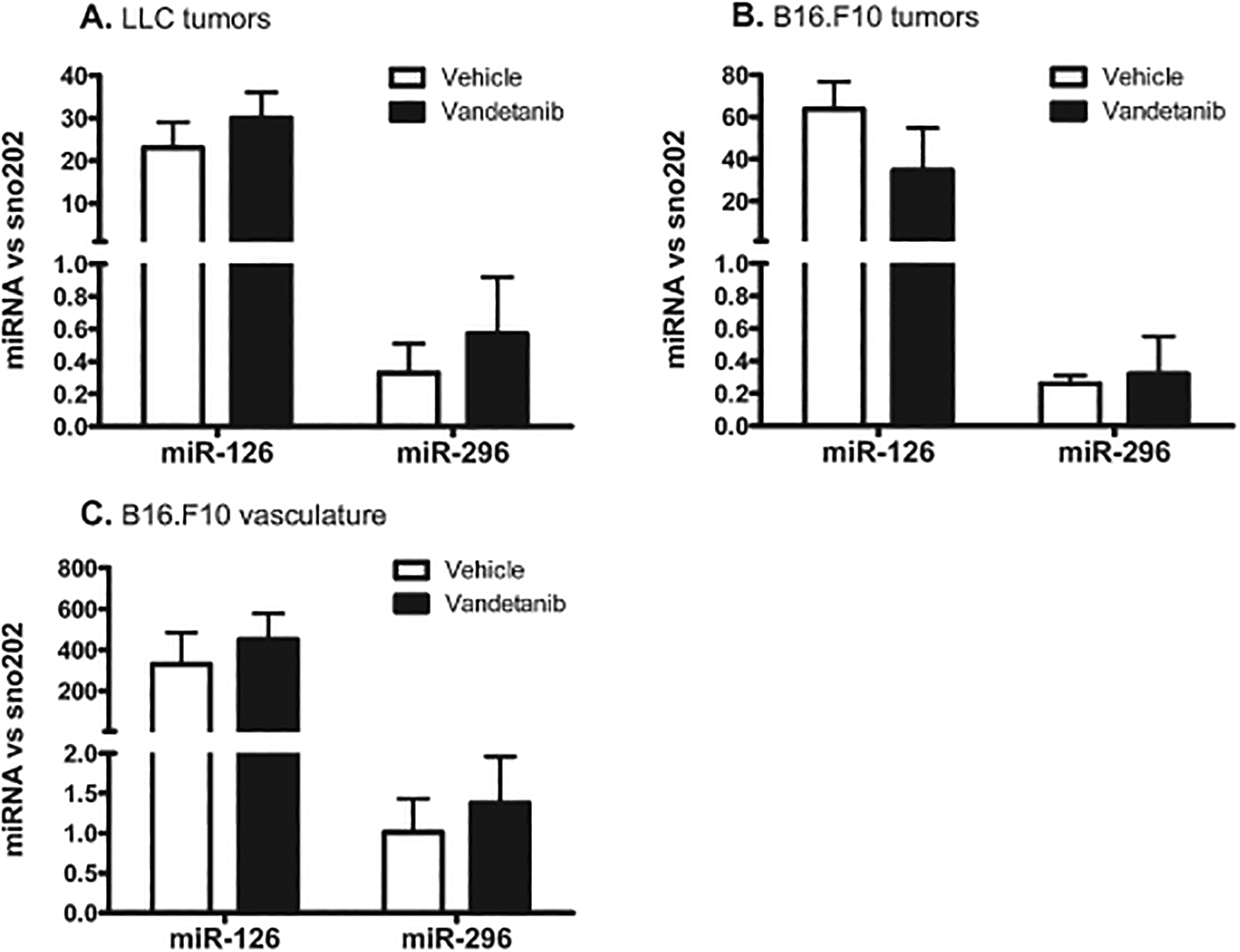
Vandetanib treatment did not affect miR-296 expression levels in the LLC (A) and B16.F10 (B) tumor as a whole, nor in B16.F10 tumor vasculature isolated by laser microdissection prior to qRT-PCR analysis (C). Values represent miR-296 expression levels normalized to the small nuclear RNA sno-202, mean of triplicate qRT-PCR analysis of 6 (LLC) or 5 (B16) animals per group. The endothelial-restricted miR-126 was included as control.
DISCUSSION
The first direct clinical evidence that anti-VEGF therapy exerts anti-vascular effects came from the landmark study by Willett et al, who demonstrated that bevacizumab treatment together with chemotherapy reduced tumor vascular density and blood flow, interstitial fluid pressure and the number of viable circulating endothelial progenitor cells in patients with advanced rectal cancer [23]. Despite a few clinical successes, VEGFR2-specific inhibitors have shown little activity in solid tumors when used as a monotherapy in the clinic [6]. Importantly, the molecular events that orchestrate the above-mentioned cellular effects of antiangiogenic therapy are until now poorly understood, and their contribution to successful antitumor effects in the pre-clinical setting remains unclear. We here demonstrated that efficient tumor growth inhibition induced by vandetanib treatment is accompanied by a significant decrease in microvascular density in B16.F10 tumors. In contrast, vandetanib treatment induced a similar degree of tumor growth inhibition in LLC, yet leaving vascular density unaffected. While vandetanib treatment downregulated expression of the pro-angiogenic genes integrin β3 and Ang2, and upregulated the expression of the vascular stability molecules Tie2 and N-cadherin in LLC vasculature, thereby inducing a shift toward vascular stabilization, none of these genes were significantly affected in B16.F10 vasculature. In contrast, B16.F10 vasculature exhibited increased expression of eNOS upon treatment. This suggests that vandetanib exerts its anti-tumor effect in Lewis Lung Carcinoma by inhibition of angiogenic sprouting and promoting a more stabilized vascular phenotype, while in B16.F10 other molecular changes play a role in creating the antitumor effect observed. Interestingly, in neither LLC nor B16.F10 the pattern of pericyte coverage was significantly altered by vandetanib treatment. As both in LLC and B16.F10, VEGFR2 and EGFR were restricted to the vasculature, our data indicate that pharmacologically targeting the vasculature alone can exert a potent antitumor effect. Moreover, the observation that VEGFR2 was more intensely expressed by the vasculature than EGFR, together with the fact that vandetanib has a 10-fold higher affinity for VEGFR2 than for EGFR [18], implies that the observed effects of vandetanib are mediated by VEGFR2 inhibition. The vandetanib-induced loss of VEGFR2 protein expression, which was not related to changes in miR-296 expression, requires further studies as it may severely hamper therapeutic efficacy when longer time periods of treatment are required.
The results of our study suggest that pharmacological targeting of the vasculature alone can induce a potent antitumor effect, and as such supports the observation previously published by Mavria and Porter [24]. They showed that selective transduction of proliferating endothelium with thymidine kinase followed by gangciclovir treatment inhibited tumor growth of KS Y-1 xenografts just as efficiently as selectively targeting the tumor cell compartment [24]. Our study demonstrated that effective tumor growth inhibition by targeting the vasculature alone does not per se coincide with a reduced overall vascular surface area, and that the effect of the drug on this parameter is highly dependent on the tumor type under study. Our data confirm previous reports that vessel density does not necessarily associate with antiangiogenic efficacy [25–27], although the majority of preclinical studies with small molecule VEGFR2 tyrosine kinase inhibitors have related efficient tumor growth inhibition by targeting the vasculature to a reduction in tumor vascular density [7, 9, 28].
Vandetanib-induced inhibition of LLC tumor growth was accompanied by a significant decrease in the expression of the integrin subunit β3. Two independent studies have demonstrated that angiogenic activation of endothelial cells is characterized by elevated expression of this molecule [14, 29], hence, its loss may be indicative of a reduction in angiogenic activity. Moreover, vandetanib treatment downregulated the expression of Ang2 and upregulated the expression of Tie2 in LLC endothelium, which points to a shift in Ang1/Ang2/Tie2 balance towards Ang1-Tie2 driven vascular stabilization [30]. This suggests that vandetanib exerts its antitumor effect in this specific tumor model by inhibiting angiogenic sprouting and promoting a more stabilized vascular phenotype. An initiating molecular mechanism to facilitate the occurrence of vascular stabilization under pharmacological pressure of a VEGFR2 inhibitor is that prevention of VEGFR2 signaling inhibits the assembly of a complex consisting of VEGFR2 and PDGFRβ on pericytes, thereby disabling VEGF/VEGFR2 induced inhibition of endothelial-pericyte attachment [31]. Hence VEGFR2 inhibition increased pericyte coverage and tumor vessel maturation [25, 32–34]. Once vessels are matured, they may loose their dependency on VEGF and thus their sensitivity for VEGFR2 inhibition during the course of therapy [33], as also suggested by the observed loss of VEGFR2 protein. Although our data did not reveal a change in the number of pericytes covering the vasculature per se, the enhanced degree of vascular stabilization strengthens the concept that more efficient antiangiogenic therapy should target both endothelial cells and pericytes [35]. In light of the loss of target protein, future studies should validate whether VEGFR2 inhibitor-induced loss of the target receptor occurs in human cancers, to evaluate the use of VEGFR2 as a target of choice. This emphasizes the need for tumor biopsies from patients treated with anti-VEGF therapy, ideally at different time points after treatment taken.
Our data in Lewis Lung Carcinoma indicate that VEGFR2/EGFR inhibition through vandetanib slows down tumor growth by inhibition of angiogenic sprouting and by promoting a more stabilized and quiescent vascular phenotype. These findings fit the current hypothesis that VEGFR2 inhibitor treatment restores the balance between pro- and antiangiogenic signaling by inactivating the surplus of VEGF activity. Normalization of this balance would result in a more regular vascular morphology, improved blood flow and reduced vascular permeability, thereby reducing the elevated interstitial pressure and facilitating uptake of e.g., chemotherapy into the tumor tissue [36, 37]. Importantly, several aspects of vascular normalization, such as a decrease in vascular diameter, require activation of Tie2 through Ang1 [38]. The observed upregulation of Tie2 mRNA, and downregulation of mRNA of its antagonist Ang2, suggest enhanced signaling of Tie2, which would thus contribute to vandetanib-induced vascular normalization.
While gene expression in the Lewis Lung Carcinoma showed a clear shift toward stabilization upon vandetanib treatment, B16.F10 vasculature responded to vandetanib only by upregulating eNOS and P-selectin. This demonstrates that, while the outcome of therapy can be the same, the underlying molecular changes that orchestrate the antitumor effect differ between tumor types. It is likely that human tumors will respond to therapy with an even larger degree of heterogeneity, emphasizing that successful evaluation of therapeutic efficacy of antiangiogenic treatment will require identification and validation of specific biomarkers for each type of tumor [39].
A striking observation in both tumor models was the loss of VEGFR2 protein expression upon vandetanib treatment, while VEGFR2 mRNA levels in the vasculature remained unaffected. This suggested a role for posttranslational modification of growth factor receptor expression. Recently, Würdinger et al. demonstrated that microRNA-296 indirectly regulates expression of VEGFR2 by inhibiting the translation of hepatocyte growth factor-regulated tyrosine kinase substrate (HGS), a protein that is involved in targeted degradation of VEGFR2. Importantly, in this system, expression of miR-296 was upregulated by VEGF signaling through VEGFR2 in human brain microvascular endothelial cells co-cultured with glioma cells. Administration of antagomirs to miR-296 to mice bearing U87 tumors reduced neovascularization, and in clinical glioblastoma specimens increased miR-296 expression was associated with elevated VEGFR2 protein levels in the vasculature [22]. We thus hypothesized that blockade of VEGF signaling upon vandetanib treatment would reduce the expression of miR-296, thereby leading to enhanced degradation of VEGFR2 protein. Our data, however, demonstrated that a role for miR-296 in the vandetanib-induced loss of VEGFR2 in these two tumor models could be excluded. Other explanations for the loss of VEGFR2 protein include inhibition of VEGF-activated recycling of VEGFR2 to the membrane by vandetanib [40] or the involvement of a hypoxia-induced decline in the VEGFR2 protein levels [41], as a result of inhibition of neovascularization by the drug. As this was not the aim of our study, we did not investigate these possibilities in more detail.
In summary, we demonstrated that vandetanib treatment inhibits tumor growth by interfering with different molecular processes in LLC compared to B16.F10, even though the pharmacological target is in both models present in the same compartment, i.e., the vasculature. Furthermore, we demonstrated that vandetanib treatment induced loss of VEGFR2 protein expression, which had no relation to the VEGFR2-controlling microRNA-296 recently reported in glioma vasculature. Future studies should focus on validation of our observations in human tumor biopsies, and where possible extension thereof by genome wide transcriptional analyses and kinome profiling [42–44], for which proper clinical specimens are an essential prerequisite. Importantly, in these studies each tumor type and each tumor in its own location should be appreciated by its own merits [45]. Such knowledge will contribute to a full understanding of the molecular effects of antiangiogenic therapy and their relation to creating a successful antitumor effect in the clinic.
Acknowledgements
We thank Mrs. Anita Meter-Arkema for excellent technical assistance and Mr. M. Schipper for excellent morphometrical analysis, and we thank AstraZeneca (Macclesfield, United Kingdom) for the generous gift of vandetanib.
Source of funding:
This research was funded by the University of Groningen.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest:
None declared.
REFERENCES
- 1.Dvorak HF. Vascular permeability factor/vascular endothelial growth factor: a critical cytokine in tumor angiogenesis and a potential target for diagnosis and therapy. J Clin Oncol. 2002; 20:4368–4380. [DOI] [PubMed] [Google Scholar]
- 2.Pettersson A, Nagy JA, Brown LF, Sundberg C, Morgan E, Jungles S, et al. Heterogeneity of the angiogenic response induced in different normal adult tissues by vascular permeability factor/vascular endothelial growth factor. Lab Invest. 2000; 80:99–115. [DOI] [PubMed] [Google Scholar]
- 3.Jain RK, Duda DG, Clark JW, Loeffler JS. Lessons from phase III clinical trials on anti-VEGF therapy for cancer. Nat Clin Pract Oncol. 2006; 3:24–40. [DOI] [PubMed] [Google Scholar]
- 4.Folkman J Angiogenesis: an organizing principle for drug discovery? Nat Rev Drug Discov. 2007; 6:273–286. [DOI] [PubMed] [Google Scholar]
- 5.Ellis LM, Hicklin DJ. VEGF-targeted therapy: mechanisms of anti-tumour activity. Nat Rev Cancer. 2008; 8:579–591. [DOI] [PubMed] [Google Scholar]
- 6.Duda DG, Batchelor TT, Willett CG, Jain RK. VEGF-targeted cancer therapy strategies: current progress, hurdles and future prospects. Trends Mol Med. 2007; 13:223–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wedge SR, Ogilvie DJ, Dukes M, Kendrew J, Chester R, Jackson JA, et al. ZD6474 inhibits vascular endothelial growth factor signaling, angiogenesis, and tumor growth following oral administration. Cancer Res. 2002; 62:4645–4655. [PubMed] [Google Scholar]
- 8.Laird AD, Vajkoczy P, Shawver LK, Thurnher A, Liang C, Mohammadi M, et al. SU6668 is a potent antiangiogenic and antitumor agent that induces regression of established tumors. Cancer Res. 2000; 60:4152–4160. [PubMed] [Google Scholar]
- 9.Sasaki T, Kitadai Y, Nakamura T, Kim JS, Tsan RZ, Kuwai T, et al. Inhibition of epidermal growth factor receptor and vascular endothelial growth factor receptor phosphorylation on tumor-associated endothelial cells leads to treatment of orthotopic human colon cancer in nude mice. Neoplasia. 2007; 9:1066–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Witmer AN, van Blijswijk BC, van Noorden CJ, Vrensen GF, Schlingemann RO. In vivo angiogenic phenotype of endothelial cells and pericytes induced by vascular endothelial growth factor-A. J Histochem Cytochem. 2004; 52:39–52. [DOI] [PubMed] [Google Scholar]
- 11.Hicklin DJ, Ellis LM. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J Clin Oncol. 2005; 23:1011–1027. [DOI] [PubMed] [Google Scholar]
- 12.Suehiro J, Hamakubo T, Kodama T, Aird WC, Minami T. Vascular endothelial growth factor activation of endothelial cells is mediated by early growth response-3. Blood 2010; 115:2520–2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zeng H, Qin L, Zhao D, Tan X, Manseau EJ, Van Hoang M, et al. Orphan nuclear receptor TR3/Nur77 regulates VEGF-A-induced angiogenesis through its transcriptional activity. J Exp Med. 2006; 203:719–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abdollahi A, Schwager C, Kleeff J, Esposito I, Domhan S, Peschke P, et al. Transcriptional network governing the angiogenic switch in human pancreatic cancer. Proc Natl Acad Sci U S A. 2007; 104:12890–12895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holderfield MT, Hughes CC. Crosstalk between vascular endothelial growth factor, notch, and transforming growth factor-beta in vascular morphogenesis. Circ Res. 2008; 102:637–652. [DOI] [PubMed] [Google Scholar]
- 16.Dirkx AE, Oude Egbrink MG, Kuijpers MJ, van der Niet ST, Heijnen VV, Bouma-ter Steege JC, et al. Tumor angiogenesis modulates leukocyte-vessel wall interactions in vivo by reducing endothelial adhesion molecule expression. Cancer Res. 2003; 63:2322–2329. [PubMed] [Google Scholar]
- 17.Hennequin LF, Stokes ES, Thomas AP, Johnstone C, Ple PA, Ogilvie DJ, et al. Novel 4-anilinoquinazolines with C-7 basic side chains: design and structure activity relationship of a series of potent, orally active, VEGF receptor tyrosine kinase inhibitors. J Med Chem. 2002; 45:1300–1312. [DOI] [PubMed] [Google Scholar]
- 18.Ryan AJ, Wedge SR. ZD6474--a novel inhibitor of VEGFR and EGFR tyrosine kinase activity. Br J Cancer. 2005; 92Suppl 1:S6–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morabito A, Piccirillo MC, Falasconi F, De FG, Del GA, Bryce J, et al. Vandetanib (ZD6474), a dual inhibitor of vascular endothelial growth factor receptor (VEGFR) and epidermal growth factor receptor (EGFR) tyrosine kinases: current status and future directions. Oncologist. 2009; 14:378–390. [DOI] [PubMed] [Google Scholar]
- 20.Wagemakers M, van der Wal GE, Cuberes R, Alvarez I, Andres EM, Buxens J, et al. COX-2 Inhibition Combined with Radiation Reduces Orthotopic Glioma Outgrowth by Targeting the Tumor Vasculature. Transl Oncol. 2009; 2:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuldo JM, Westra J, Asgeirsdottir SA, Kok RJ, Oosterhuis K, Rots MG, et al. Differential effects of NF-{kappa}B and p38 MAPK inhibitors and combinations thereof on TNF-{alpha}- and IL-1{beta}-induced proinflammatory status of endothelial cells in vitro. Am J Physiol Cell Physiol. 2005; 289:C1229–C1239. [DOI] [PubMed] [Google Scholar]
- 22.Wurdinger T, Tannous BA, Saydam O, Skog J, Grau S, Soutschek J, et al. miR-296 regulates growth factor receptor overexpression in angiogenic endothelial cells. Cancer Cell. 2008; 14:382–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Willett CG, Boucher Y, di TE, Duda DG, Munn LL, Tong RT, et al. Direct evidence that the VEGF-specific antibody bevacizumab has antivascular effects in human rectal cancer. Nat Med. 2004; 10:145–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mavria G, Porter CD. Reduced growth in response to ganciclovir treatment of subcutaneous xenografts expressing HSV-tk in the vascular compartment. Gene Ther. 2001; 8:913–920. [DOI] [PubMed] [Google Scholar]
- 25.Cesca M, Frapolli R, Berndt A, Scarlato V, Richter P, Kosmehl H, et al. The effects of vandetanib on paclitaxel tumor distribution and antitumor activity in a xenograft model of human ovarian carcinoma. Neoplasia. 2009; 11:1155–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hlatky L, Hahnfeldt P, Folkman J. Clinical application of antiangiogenic therapy: microvessel density, what it does and doesn’t tell us. J Natl Cancer Inst. 2002; 94:883–893. [DOI] [PubMed] [Google Scholar]
- 27.Sandstrom M, Johansson M, Andersson U, Bergh A, Bergenheim AT, Henriksson R. The tyrosine kinase inhibitor ZD6474 inhibits tumour growth in an intracerebral rat glioma model. Br J Cancer. 2004; 91:1174–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eichhorn ME, Strieth S, Luedemann S, Kleespies A, Noth U, Passon A, et al. Contrast enhanced MRI and intravital fluorescence microscopy indicate improved tumor microcirculation in highly vascularized melanomas upon short-term anti-VEGFR treatment. Cancer Biol Ther. 2008; 7:1006–1013. [DOI] [PubMed] [Google Scholar]
- 29.Palmowski M, Huppert J, Ladewig G, Hauff P, Reinhardt M, Mueller MM, et al. Molecular profiling of angiogenesis with targeted ultrasound imaging: early assessment of antiangiogenic therapy effects. Mol Cancer Ther. 2008; 7:101–109. [DOI] [PubMed] [Google Scholar]
- 30.Augustin HG, Koh GY, Thurston G, Alitalo K. Control of vascular morphogenesis and homeostasis through the angiopoietin-Tie system. Nat Rev Mol Cell Biol. 2009; 10:165–177. [DOI] [PubMed] [Google Scholar]
- 31.Greenberg JI, Shields DJ, Barillas SG, Acevedo LM, Murphy E, Huang J, et al. A role for VEGF as a negative regulator of pericyte function and vessel maturation. Nature. 2008; 456:809–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vecchiarelli-Federico LM, Cervi D, Haeri M, Li Y, Nagy A, Ben-David Y. Vascular endothelial growth factor--a positive and negative regulator of tumor growth. Cancer Res. 2010; 70:863–867. [DOI] [PubMed] [Google Scholar]
- 33.Helfrich I, Scheffrahn I, Bartling S, Weis J, von Felbert V, Middleton M, et al. Resistance to antiangiogenic therapy is directed by vascular phenotype, vessel stabilization, and maturation in malignant melanoma. J Exp Med. 2010; 207:491–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Palmowski M, Huppert J, Hauff P, Reinhardt M, Schreiner K, Socher MA, et al. Vessel fractions in tumor xenografts depicted by flow- or contrast-sensitive three-dimensional high-frequency Doppler ultrasound respond differently to antiangiogenic treatment. Cancer Res. 2008; 68:7042–7049. [DOI] [PubMed] [Google Scholar]
- 35.Bergers G, Song S, Meyer-Morse N, Bergsland E, Hanahan D. Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. J Clin Invest. 2003; 111:1287–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005; 307:58–62. [DOI] [PubMed] [Google Scholar]
- 37.Claes A, Leenders W. Vessel normalization by VEGF inhibition. A complex story. Cancer Biol Ther. 2008; 7:1014–1016. [DOI] [PubMed] [Google Scholar]
- 38.Winkler F, Kozin SV, Tong RT, Chae SS, Booth MF, Garkavtsev I, et al. Kinetics of vascular normalization by VEGFR2 blockade governs brain tumor response to radiation: role of oxygenation, angiopoietin-1, and matrix metalloproteinases. Cancer Cell. 2004; 6:553–563. [DOI] [PubMed] [Google Scholar]
- 39.Hanrahan EO, Lin HY, Kim ES, Yan S, Du DZ, McKee KS, et al. Distinct patterns of cytokine and angiogenic factor modulation and markers of benefit for vandetanib and/or chemotherapy in patients with non-small-cell lung cancer. J Clin Oncol. 2010; 28:193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gampel A, Moss L, Jones MC, Brunton V, Norman JC, Mellor H. VEGF regulates the mobilization of VEGFR2/KDR from an intracellular endothelial storage compartment. Blood. 2006; 108:2624–2631. [DOI] [PubMed] [Google Scholar]
- 41.Olszewska-Pazdrak B, Hein TW, Olszewska P, Carney DH. Chronic hypoxia attenuates VEGF signaling and angiogenic responses by downregulation of KDR in human endothelial cells. Am J Physiol Cell Physiol. 2009; 296:C1162–C1170. [DOI] [PubMed] [Google Scholar]
- 42.Stockwin LH, Vistica DT, Kenney S, Schrump DS, Butcher DO, Raffeld M, et al. Gene expression profiling of alveolar soft-part sarcoma (ASPS). BMC Cancer. 2009; 9:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Buckanovich RJ, Sasaroli D, O’Brien-Jenkins A, Botbyl J, Hammond R, Katsaros D, et al. Tumor vascular proteins as biomarkers in ovarian cancer. J Clin Oncol. 2007; 25:852–861. [DOI] [PubMed] [Google Scholar]
- 44.Sikkema AH, Diks SH, den Dunnen WF, ter Elst A, Scherpen FJ, Hoving EW, et al. Kinome profiling in pediatric brain tumors as a new approach for target discovery. Cancer Res. 2009; 69:5987–5995. [DOI] [PubMed] [Google Scholar]
- 45.Langenkamp E, Molema G. Microvascular endothelial cell heterogeneity: general concepts and pharmacological consequences for anti-angiogenic therapy of cancer. Cell Tissue Res. 2009; 335:205–222. [DOI] [PubMed] [Google Scholar]