

Abstract

Chemical compound space (CCS), the set of all theoretically conceivable combinations of chemical elements and (meta-)stable geometries that make up matter, is colossal. The first-principles based virtual sampling of this space, for example, in search of novel molecules or materials which exhibit desirable properties, is therefore prohibitive for all but the smallest subsets and simplest properties. We review studies aimed at tackling this challenge using modern machine learning techniques based on (i) synthetic data, typically generated using quantum mechanics based methods, and (ii) model architectures inspired by quantum mechanics. Such Quantum mechanics based Machine Learning (QML) approaches combine the numerical efficiency of statistical surrogate models with an ab initio view on matter. They rigorously reflect the underlying physics in order to reach universality and transferability across CCS. While state-of-the-art approximations to quantum problems impose severe computational bottlenecks, recent QML based developments indicate the possibility of substantial acceleration without sacrificing the predictive power of quantum mechanics.
1. Introduction
Promising applications of machine learning techniques have been rapidly gaining momentum throughout the chemical sciences. Apart from this present special issue in Chemical Reviews, a number of special issues in common theoretical chemistry community journals have appeared, including International Journal of Quantum Chemistry (2015),1Journal of Chemical Physics (2018),2Journal of Physical Chemistry (2018),3Journal of Physical Chemistry Letters (2020),4 and Nature Communications (2020).5 Books, essays, reviews, and opinion pieces have also been contributed by practitioners in the field.6−23 Such growth of interest prompted a general discussion in Angewandte Chemie within a trilogy of essays by Hoffmann and Malrieu on the seemingly conflicting nature of simulation and understanding in quantum chemistry.24−26 The overall enthusiasm in the hard sciences for machine learning has even led to the introduction of novel journals, such as Springer’s Nature Machine Intelligence, IOP’s Machine Learning: Science and Technology,27 or Wiley’s Applied Artificial Intelligence Letters.28
In this review, we attempt to provide a comprehensive overview on recent progress made regarding the problem of using machine learning models to train and predict quantum properties throughout chemical compound space (CCS) (Figure 1). In contrast to the current trend of machine learning in quantum computing, we here refer to the application of statistical learning to quantum properties as “quantum machine learning” (QML). This notation follows a common convention in atomistic simulation, where the quantum nature of the object to be studied corresponds to a prefix, while the actual algorithms are rather classical in nature. Examples include Quantum Monte Carlo or Quantum Molecular Dynamics (also known as ab initio or “first-principles” molecular dynamics).
Figure 1.
A cartoon of similarities among atoms across chemical compound space, not in conflict with quantum mechanics. The exemplary molecule aspirin is highlighted by bonds, and each of its atoms is superimposed with a similar atom in another molecule (hydrogens omitted for clarity). Green, yellow, gray, red, and blue refer to sulfur, phosphor, carbon, oxygen, and nitrogen, respectively. Reproduced with permission from ref (15). Copyright 2020 Springer Nature.
Within this introductory section, we will begin by first providing a qualitative description of chemical compound space (CCS) in terms of fundamental variables, which is consistent with the quantum mechanical picture within the Born–Oppenheimer approximation and neglecting nuclear quantum and relativistic effects. Thereafter, we briefly review related but complementary and system specific QML models which predominantly are not used throughout CCS but rather for training and predicting potential energies and forces in terms of conformational degrees of freedom, e.g., using molecular dynamics. Quantum mechanics based explorations for the purpose of materials design are mentioned subsequently, followed by a short subsection on studies which establish the quantitative and rigorous quantum chemistry based view on CCS.
1.1. Multiscale Nature of CCS
Figuratively speaking, CCS refers to the virtual set of all the theoretically (meta-)stable compounds one could possibly realize in this universe. To paraphrase Buckingham and Utting, a compound “...is a group of atoms...with a binding energy which is large in comparison with the thermal energy kT”.29 In other words, with respect to all its spatial degrees of freedom, it is that locally averaged atomic configuration, for which the free energy is in a local minimum surrounded by barriers sufficiently high to prevent spontaneous reactions within some observable lifetime. As such, CCS depends on external conditions. It loses all meaning, for example, when conditions are such that bonding spontaneously emerges and vanishes (e.g., aggregation state of plasma).
The mathematical number of compounds grows explosively with the number of constituting atoms due to the mutual enhancement of combinatorial scaling at three rather distinct but well established energetic scales: First, the number of possible stoichiometries for any given system size (in terms of electrons and total proton number) represents an integer partitioning problem which grows combinatorially, see ref (30), for example. The energetic variance among compounds that differ in stoichiometry is on the scale of chemical bonding due to having different number and different types of atoms. Second, the number of possible connectivity patterns, i.e., incomplete labeled undirected weighted graphs distinguishing constitutional isomers/allotropes (commonly drawn as Lewis structures) is mathematically known to grow combinatorially with number of atoms.31−33 The energetic variance among constitutional isomers is on the scale of differences in chemical bonding. Third, the number of possible conformational degrees of freedom grows combinatorially with number of atoms in a molecular graph (cf. Levinthal’s paradoxon for polymers), and one could even consider different atomic configurations of disconnected graphs, i.e., macromolecular or molecular condensed systems, to fall into this category of isomers. As such, the energetic variance among conformational isomers is on the scale of noncovalent intra- as well as intermolecular interactions. We note that stereoisomerism typically occurs among constitutional and conformational isomers. Its extension to compositional chirality has been proposed only recently.34 Given such size and diversity, highly universal, and efficient methods are in dire need in order to meaningfully explore CCS in search of deepened chemical insight and intuition and of new compounds and materials which exhibit desirable properties. While quantum mechanics and statistical mechanics offer the appropriate physical framework for dealing with CCS in an unbiased and universal manner, the computational complexity of the equations involved has hampered their widespread use.
We note that our ab initio definition of CCS implies that only those compounds are part of CCS that should, at least in principle, be experimentally accessible as long as sufficiently sophisticated synthetic chemical procedures and reservoirs of the necessary chemical elements are available. While any such synthetic procedure would have to follow the corresponding relevant free energy paths, by navigating the virtual analogue of CCS we do enjoy more design freedom and can, namely for any property that is a state function, also exploit unrealistic fictitious transformations in line with Hess’ law, i.e., without the need for direct correspondence to experimental realization (cf. “alchemical” transmutations).
We conclude this section by noting that our definition generalizes the more commonly made reference to CCS, which typically excludes conformational isomers, reactive intermediates, or minima in electronically excited states. For example, first steps toward an ab initio based representative exploration of the latter were also proposed in 2013 for drug-like compounds by Beratan and co-workers.35 However, for this review, we do not assume the most general view on CCS which would still be consistent with quantum mechanics, namely, that CCS comprises any chemical system, i.e., compounds with any chemical composition and any atomic configuration (being close to some state’s energy minimum or not). Such an encompassing definition would sacrifice the minimal free energy requirement mentioned above, and it would trivially correspond to the entire domain of CCS. Therefore, it would forego the useful link to observable lifetimes of systems as well as the appealing complementarity (not to be confused with orthogonality) to the well established problem of sampling potential energy hyper surfaces to study free energies or competing elementary reaction steps.
1.2. Machine Learning the Potential Energy Surface
While QM based studies of CCS are mostly concerned with (meta-)stable compounds, from inspection of the electronic Hamiltonian, it is quite clear that the effect of nuclear charges and nuclear coordinates are intimately linked. The well-known cusp condition due to Kato’s theorem36 explicitly links these two variables through the electron density observable. As such, ab initio studies of the PES aimed at calculating geometric distortion, transition states, or statistical mechanical averages are closely related to the topic of this review. More specifically, early attempts of QML have focused on the PES of homonuclear system (e.g., diamond37 or Sin cluster38) due to its relative simplicity (cf. compositional degree of freedom), for which many QML methods developed are also applicable to CCS. The distinction between CCS and the PES is somewhat arbitrary. For example, some molecular quantities of significant interest, such as libraries of ensemble properties of protein–ligand binding free energies, require accurate potentials as well as representative sampling of CCS. Also, instead of considering (meta-)stable constitutional or conformational isomers as distinct compounds, they can also equally well be viewed as local minima of the global PES hypersurface.
As mentioned above, within studies of the PES, the focus (at least currently) is typically placed on a single system and on computing energies and forces from scratch, i.e., ab initio. As such, one does not exploit correlations, constraints, and relationships, which only emerge through relationships observed among constitutional and compositional isomers, i.e., throughout all dimensions of CCS. The most common use-case of quantum methods for atomistic simulations deals with the problem of sampling the configurational degrees of freedom of the atoms of a given system. To develop a better informed understanding of the field, we now also briefly discuss relevant and select machine learning studies which touch upon the quantum based understanding of CCS but which primarily are concerned with the PES.
The question of how to best model a PES using some (physical or surrogate) function approximator and based on scarce and expensive potential energy surface data sets of specific systems, i.e., not through CCS, obtained from computationally demanding calculations, is long-standing. Potential energy hypersurfaces were traditionally studied for the purpose of molecular spectroscopy or for molecular dynamics applications of a given system. The development of empirical interatomic potentials, particularly the reactive force-field (ReaxFF) approach developed by van Duin and co-workers since 2001,39,40 amounts essentially to a traditional multidimensional regression problem for fixed functional basis functions and constitutes one of the mainstream efforts in this active field. Unlike traditional force field approaches, ReaxFF requires no predefined connectivity between atoms (topology) and casts the empirical interatomic potential within a formalism of bond order, which depends on the interatomic distances only. This improved adaptation of an atom to its environment allows for accurate descriptions of bond breaking and bond formation and has been applied extensively to model reactive chemistry at heterogeneous interfaces, involving typically very large systems,40 made up of millions of atoms.
The force-field approach, despite its efficiency, its chemical motivation, and its broad applicability and potential accuracy, suffers from the fixed functional forms imposed when relying on empirical interatomic potentials, implying that the model is hard to improve by adding more training data and could even fail catastrophically in certain regimes and classes. This limitation motivates interest in more flexible data-driven models. For example, already in 1994, Ischtwan and Collins improved the Shepard interpolation scheme for PES approximations.41 This paper illustrates the close relationship to QML: The authors utilized a formalism very similar to the modern kernel ridge regression, one of the workhorses of QML. The authors also already discussed one of the frequent challenges coming along with any new ML model project: How to best down-select optimal configurations for minimal data acquisition and training costs, and how to obtain systematic model improvements with increasing training set size.41 Early awareness of the trade-off between accuracy and training cost was also already addressed more than a decade earlier in the 1981 paper by Wagner, Schatz, and Bowman: Given a finite compute budget, data for which training instances should be acquired in order to obtain the most accurate model of the potential energy hypersurface?42 When facing the exploration of CCS with QML models, analogous questions must be addressed. For references to similar studies related to the problem of potential fitting and preceding 1989, we refer the reader to the comprehensive review by Schatz.43
Most of the data-driven models in the 1990s favored the neural network regressors for PES fitting. More specifically, in 1992, Sumpter and Noid published a neural network model for macromolecules.44 Additional neural network potentials were published by Blank et al. in 199545 for the CO/Ni(111) system, and Brown et al. in 199646 for the study of ground-state vibrational properties of two weakly bound molecular complexes: (FH)2 and FH–ClH. Neural networks were revisited for systems with increased size in the same year by Lorenz, Gross, and Scheffler47 for H2/K(2×2)/Pd(100) (with substrate fixed), followed by their application to represent high-dimensional potential energy surfaces for H2O2 by Manzhos and Carrington in 2006.48 Even larger systems, i.e., water clusters (up to 6 units), were dealt with by Handley and Popelier from 2009 onward,49 accounting for important electrostatic properties through learning of the atomic multipoles. These early developments used Cartesian/internal coordinates directly as the input of NN models, which is justified for modeling small to medium-sized systems; for large systems, however, this setup proves to be too inefficient. In 2007, Behler and Parinello published much improved deep neural network-based potentials,50 encoding molecular geometry effectively in terms of rotation, translation, and permutation invariant atom-centered symmetry function (ACSF), followed by molecular dynamics applications using metadynamics to identify Si phases under high pressure.51 A detailed overview of various neural network-based advances since 2010 was given in 2017.52 Starting in the same year, multiple, more universal, neural network models were introduced. In particular, Smith et al. advanced the idea of Behler’s symmetry functions in neural networks with the aforementioned normal mode displacements in order to generate a powerful neural network trained on millions of configurations of tens of thousands of organic molecules, called ANI.53 An accurate and transferable neural network exploiting an “on-the-fly” equilibration of atomic charges was introduced that same year by Faraji et al.,54 and soon thereafter, equally universal neural nets SchNet55 and PhysNet56 were published. An extensive review on neural network potentials for modeling the potential energy surfaces of small molecules and reaction is also part of the present issue in Chemical Reviews.57
Kernel models started to play a noticeable role for PES fitting in the late 1990s. In 1996, Ho and Rabitz presented kernel-based models for the fitting of potential energies58 for three small systems, He–He, He–CO, and H3+. Similar to early PES fitting works within NN framework, these early kernel-based models also utilized simple Cartesian/internal coordinates as input, and therefore applicability was limited. While the mathematics of kernel-based surrogate models was firmly established many decades ago, only from 2010 and onward, kernel-based models began to flourish, building on the seminal work contributed by Csanyi, Bartok, and co-workers within their “Gaussian-Approximated Potential” (GAP) method, relying on Gaussian process regression (GPR) and an atom index invariant bispectrum representation.59 In 2012, Henkelmann and co-workers introduced an interesting application of support vector machines (SVM) toward the identification of transition states.60 One year later, the first flavor of the smooth overlap of atomic positions (SOAP) representation for GPR based potentials was published.38 The SNAP61 method popularized the GAP idea using linear kernels in 2015, and other GPR applications with automatically improving forces were published the same year.62,63 Around the same time, a first stepping stone toward a universal force field, trained on atomic forces throughout the chemical space of molecules displaced along their normal modes, was established.64 Reproducing kernels were also shown in 2015 to be applicable toward dynamic processes in biomolecular simulations,65 and ever more accurate GPR based potentials were introduced in 201666 and in 2017.67,68 GPR was also applied to challenging processes in ferromagnetic iron69 and to the problem of the on-the-fly prediction of parameters in intermolecular force fields.70 Amorphous carbon was studied using SOAP based GPR/KRR models,71,72 and GDML, another series of highly robust and accurate GPR/KRR based molecular force fields, was introduced in refs (67 and 73−75) starting in 2017.
GPR and NN are currently the two most popular regressors for PES fitting, and each exhibits advantages and disadvantages. Seemingly very different in design, they do resemble each other to some extent in the sense that they take the role of basis functions (to be elaborated in section “Regressor”), although the similarity may be blurred within the framework of deep NN. Numerical comparison of the performance of these two methods is interesting. Most notably, such a comparison was made for modeling the potential energy surface of formaldehyde in 2018 by Manzhos and co-workers.76 A similar yet independent study on the same system was performed in 2020 by Meuwly and co-workers.77 Both studies confirm that kernel based QML models reach higher predictive power than neural network based models for same training set sizes. A highly related comparative study on modeling vibrations in formaldehyde was contributed by Käser et al.,77 also in 2020.
As for the active learning of interatomic potentials, most of the related studies relied on the kernel framework, some of them also detailed below in the section “Training Set Selection”. As early as in 2004, De Vita and co-workers proposed updating potential parameters to ab initio results during molecular dynamics runs (“learning-on-the-fly”),78 for a very large system, i.e., silicon systems composed of up to ∼200 000 atoms, although the reference level of theory is quite approximate. Podryabinkin and Shapeev proposed the so-called D-optimality criterion for selecting the most representative atomistic configurations for training on-the-fly as early as 2017.79 Using kernel ridge regression (KRR, a variant of GPR), Hammer and co-workers revisited the on-the-fly learning idea for structural relaxation in 2018,80 and investigated the exploration vs exploitation trade-off.81 In 2019, Weinan, Car, and co-workers contributed another active learning procedure for accurate potentials of Al–Mg alloys,82 and Westermayr et al. extended the use of neural networks for molecular dynamics in the electronic ground state toward photodynamics.83 Among the many purposes (also challenges) of QML for PES, one particular one is to scale to an extremely large (thus more realistic) system. Numerous efforts have pushed us closer to this goal, and most notably, Weinan, Car, and co-workers made full use of the Summit supercomputer to simulate 100 million atoms with ab initio accuracy using convolutional neural networks,84 for which they subsequently were awarded the Gordon Bell prize 2020 by the Association for Computing Machinery.
1.3. Navigating CCS from First Principles
The scientific research question of how properties trend across CCS lies at the core of the chemical sciences. Because of ever-improving hardware performance, improved approximations to Schrödinger’s equation, most notably within density functional theory and localized coupled cluster theory, QM data sets of considerable size have emerged, enabling the use of statistical learning to train surrogate QML models which can provide accurate and rapid quantum property estimates for new compounds within their applicability domain.
While quantum mechanics based computational materials design efforts had been undertaken as early as the 1990s85−88 with important progress made in the 1980s,89,90 the first-principles based computational high-throughput design has by now become an important success story.91 First attempts to employ machine learning and quantum predictions to discover new ternary materials databases date back to seminal work by Hautier and Ceder in 2010.92,93
As a promising alternative to ab initio high-throughput computations (or solving Schrodinger equation in general), one often assumes locality of atoms in molecules when constructing the mapping from molecular distance/similarity to difference in properties within QML, and the final predictive performance depends on how similar two local (and thus global) entities are, i.e., nuclear types covered by a test set are required to be retained in the training set. The capability of QML to treat species made up of elements not seen in training set is, however, very limited. There exists the so-called “alchemical” methods, being quite different in philosophy, allowing for effective and efficient treatment of the change of nuclear types, with or without the constraint that the number of electron number (Ne) being fixed (i.e., isoelectronic). We note in passing that alchemy is typically established within the density functional theory framework, as it would be tremendously simpler to expand molecular property (mostly energy) as a function of four variables (x, y, z, and Z) than 4NI ones in the case of wave function-based formulation.
Previous methodological works tackling chemical compound space from first-principles through variable (“alchemical”) nuclear charges were contributed by various pioneers, including Wilson’s formal four-dimensional density functional theory94 in 1962, which expresses the exact nonrelativistic ground-state energy of an electronic system as a functional of the electron density, which per se is a function of the spatial coordinates, and nuclear charges. Following Wilson’s idea, Politzer and Parr95 in 1972 made one step further toward practical computation by transforming Wilson’s formula into a functional of the total electrostatic potential V(r, Z) and derived some useful semiempirical formulas for the total energy of atoms and molecules, through the use of thermodynamic integration.95 Later in the 1980s, Mezey made some interesting discoveries96,97 about the global electronic energy bounds for a variety of isoelectronic polyatomic systems, which may be found useful for quantum-chemical synthesis planning, using multidimensional potential surfaces.
The theoretical alchemical research was resurrected in the new millennium. Among the numerous contributions, notable ones include a variational particle number (variable proton and electron number) approach for rational compound design90 proposed by one of the authors and collaborators, followed by a more detailed description of the underlying theories, in the name of molecular grand-canonical ensembles (GCE).98 In the same year, a reformulation of GCE in terms of linear combinations of atomic potentials (LCAP)99 (instead of Z and Ne as in GCE) was proposed by Wang et al., but for the optimization of molecular electronic polarizability and hyper-polarizability, with the optimal molecule determined analytically in the space of electron–nuclei attraction potentials. For the isoelectronic case, related works include the development of ab initio methods for the computation of higher-order alchemical derivatives100 by Lesiuk et al. in 2012, as well as the assessment of the predictability of alchemical derivatives101 by Munoz et al. in 2017. More recently, alchemical normal modes in CCS,102 alchemical perturbation density functional theory,103 and even a quantum computing algorithm for alchemical materials optimization104 were proposed, further enriching the field.
Starting in 1996 with stability of solid solutions,105 multiple promising applications, based on quantum alchemical changes, have been published over recent years, including thermodynamic integrations,106 mixtures in metal clusters,107,108 optimization of hyperpolarizabilities,109 reactivity estimates,110 chemical space exploration,111 covalent binding,112 water adsorption on BN-doped graphene,113 the nearsightedness of electronic matter,114 BN-doping in fullerenes,115 energy and density decompositioning,116 catalyst design,117−119 and protonation energy predictions120,121 Symmetry relations among perturbing Hamiltonians have also enabled the introduction of “alchemical chirality”.34
An extension of computational alchemy toward descriptions which go beyond the Born–Oppenheimer approximation has been introduced within path-integral molecular dynamics, enabling the calculation of kinetic isotope effects, already in 2011,122 and subsequently by Ceriotti and Markland.123
However, also varying the electron number is a long-standing concept within conceptual DFT.124,125 Actual variations have only more recently been considered, e.g., to estimate redox potentials,98,126 higher-order derivatives,100−102 or for the development of improved exchange-correlation potentials.127
2. Heuristic Approaches
Modern systematic attempts to establish quantitative structure–property relationships (QSPRs) have led to computationally advanced bio-, chem-, and materials-informatics methodologies. Unfortunately, conventional approaches in QSPR predominantly rely on heuristic assumptions about the nature of the forward problem, and are thus inherently limited to certain applicability domains. The implicit bias, often due to lacking basis in the underlying physics is known, as discussed, e.g. in a 2010 review by G. Schneider,128 and many improvements have been contributed more recently.20
While heuristic in nature, QSPR can still provide useful qualitative trends and insights for relevant applications, and sometimes yield accurate predictions for specific property subdomains and systems. Albeit not directly relying on the laws of quantum mechanics, these early developments are still valuable, in the sense that some just correspond to special variants of the more complicated models, for instance, a linear model can be mapped onto the framework of kernel method, by choosing a linear kernel, instead of say Gaussian kernel for Gaussian process regression (GPR). Other heuristic approaches, exhibiting more quantitative characteristics can be considered important precursors for modern QML. Such examples include Collin’s improved Shepard interpolation scheme41 for accurate representation of molecular potential energy surfaces, which resembles the form of kernel methods except that the weights are determined in a heuristic way, instead of being regressed as in GPR. One may also argue that Collins’ scheme could be recast into the kernel framework, except that a specific kernel is chosen such that the Shepard interpolation weights in Collins’ scheme are exactly reproduced (with the constraint that these weights sum up to 1). Another highly related concept is Ramon Carbo-Dorca’s quantum similarity (for a comprehensive review, see ref (129)), derived based on density matrix, or molecular orbitals, or other related quantum quantitites, it is also closely linked to kernel based methods and may be used directly as parameter-free kernel matrix elements (unlike in GPR, kernel matrix element characterizing similarity is typically hyper-parameter dependent).
In the sections below, we focus on relevant literature regarding three distinct perspectives which largely follow chronological order: (i) low-dimensional correlations or simple models from the early days of chemistry, (ii) coarse representations of molecules and derived quantities, mostly providing an overview of QSPR, and (iii) molecular representations based on properties.
2.1. Low-dimensional Correlations
Early practices of fundamental chemical research dealt with spotting correlations between inherent properties of the system and systematic changes of observed quantities. Possibly the most famous example for such work is Mendeleev’s discovery of the periodic table.130 Other important examples correspond to Pauling’s electronegativity concept and covalent bond postulate,131 or Pettifor’s Mendeleev numbering scheme.132,133 Work along such lines has been continued, and recent contributions include revisiting Pettifor scales,134,135 use of variational autoencoders to “rediscover” the ordering of elements in the periodic table,136 or the chemplitude model which extends Pauling’s concept,137 among many others. Free-energy relationships are the subject of yet another broad category of early research which is still active today. Relating logarithms of reaction constants (free energy difference) across CCS for related series of reactions138 has led to the famous Hammett equation, a 2D projection of all degrees freedom onto composition and reaction conditions.139−141 Similarly low-dimensional effective degrees of freedom have been identified within Hammond’s postulate,142 or Bell–Evans–Polanyi principle.143,144
Most of the aforementioned concepts were proposed to gain a better (or more useful) understanding of molecular behavior in the first place. For extended systems such as metallic surfaces, complexities arise and many of the simplified molecular models are no longer applicable. With the advent of density functional theory (DFT),145−149 alternative descriptors have been proposed during the past decades, playing an increasingly important role. Notable contributions include the d-band center model by Hammer et al.,150,151 the generalized coordination number,152,153 and the Fermi softness.154 Free-energy relationships are more robust against subtle changes in the electronic structure and are being widely applied in analyzing surface elementary reaction steps.155 Scaling relations between the energetics of adsorbed species on surfaces156 also enjoy extensive attention and have been proven useful for catalyst design regardless of the surface not being metallic.157−159 Many of the empirical chemical concepts such as electronegativity, softness/hardness, and electrophilicity/nucleophilicity can be rationalized and quantified within what is known as “conceptual” DFT.160,161 This specific field, as pioneered by Fukui or Parr and Yang,160 has been championed and furthered by many including Geerlings, De Proft, Ayers, Cardenas, and co-workers.161,162
We note that simple models, involving one or a few variables in general, represent effective coarse-grained schemes applicable to specific subdomains of chemistry. While they lack the desired transferability of quantum mechanics, they often do encode well tempered approximations and therefore are capable of capturing much of the essential physics. As such, they have much to offer, and they could, for example, serve the design of robust and general representations enabling the training and application of improved QML models (see below or refs (6 and 163)). Alas, this idea, to connect low-dimensional model, based on well established heuristics, with more recently developed generic ML models, is still largely unexplored, despite the fact that the latter often bear (magic) black box characteristics allowing for little qualitative insights. Unifying modern ML with low-dimensional model could therefore also help resolve open challenges in QML. For instance, how can we properly represent different electronic (spin-) states of molecules in the molecular representation or different oxidation states? Conceptual DFT derived linear or quadratic energy relationships suggest treating the number of electrons (Ne) and/or its powers as independent features might be a reasonable starting point. Another thus-inspired direction of research is to utilize conceptual DFT-based local indicators as properties of composing atoms/bonds/fragments of a target molecule as a starting point (much like the fundamental variables such as Z and R) for building representations. This might be necessary in order to address hard and outstanding problems such as building QML models of intensive properties or to account for multireference character in the electronic structure.
2.2. Stoichiometry
Given a fixed pattern of structure, stoichiometry alone can be used as a unique representation of the system under study. This idea has been demonstrated for an exhaustive QML based scan of the elpasolite (ABC2D6 stoichiometry) subspace of CCS, predicting cohesive energies of all the 2 million crystals made up from main-group elements.164 Elpasolites are the most abundant quaternary crystal form found in the Inorganic Crystal Structure Database. Comparison of the QML results to known competing ternary and binary phases enabled favorable stability predictions for nearly 90 crystals (convex hull) which subsequently have been added to the Materials Project database.165 A compact stoichiometry based representation in terms of period and group entry for elements A, B, C, and D was shown to reach the accuracy of explicit geometry based many-body potential representations at larger training set size, indicating the dominance of the former in large training data regimes.166 Similar work was subsequently done by Ye et al.167 as well as Marques et al. for perovskites on crystal stability,168 as well as by Legrain et al.,169 for predicting vibrational free energies and entropies for compounds, drawn from the Inorganic Crystal Structure Database.
A naive but useful derived concept is the so-called “dressed atom” concept,170 which characterizes the atom in a molecule of a fixed stoichiometry. When using this approach together with a linear regression model to approximate the total energy (or atomization energy), the accuracy turns out to be surprisingly reasonable,171 at least for common data sets of organic molecules with small variance among constitutional isomers. For instance, the corresponding mean absolute error (MAE) for QM7 data set is only 15.1 kcal/mol.170 Using bond counting, the MAE could be improved further to less than 10.0 kcal/mol, within reach of a conventional DFT GGA functionals.172 Therefore, it seems advisible to always use the dressed atom approach for centering the data for any fixed stoichiometry (i.e., averaging out constitutional and conformational isomers) before proceeding to the next level of QML training on the complete set of degrees of freedom. This normalization step can also be seen as data preprocessing, enabling the QML model to focus on “minor” deviations from the mean.164,173
2.3. Connectivity Graph
When the systems under study do not share some common structural skeleton, stoichiometry alone is not enough, and the covalent bonding connectivity between atoms, as well as conformations, may have to be also examined.
It is worth pointing out that chemists often assume a one-to-one relationship between the molecular graph and its associated global conformational minima (or the second lowest energy minima, or the third, etc.), and therefore it should be possible to build a QML model to predict relevant quantum properties of such ordered minima from graph-input only. In fact, the remarkable performance of extended Hückel theory for some systems could be explained in this way.
Because of the intuitive accessibility and applicability of (incomplete) graph based representations, such as Lewis structures and their extensions, for a wide range of molecular systems, associated ML methods have received broad attention and wide applications in many fields such as cheminformatics or bioinformatics. Examples of such representations include various fingerprint representations,174 such as the signature methodology.175−177 Another notable example corresponds to the so-called extended circular fingerprint (ECFP).178 ECFP and similar representations have been used for drug design179,180 and qualitative exploration of CCS.181,182 ECFP has also been used in KRR models for prediction of quantum properties of QM9 molecules. Numerical results for ECFP based QML models indicate a substantially worse performance compared to more complete, geometry derived representations.183
Modern molecular graphs, typically in SMILES format, based neural networks models, have gained considerable momentum during the past decade. A vast amount of related literature deal with chemical synthesis and retrosynthesis in such representation spaces (mainly in organic chemistry),184,185 typically favoring different deep learning architectures, chemical reaction network,186 as well as molecular design using variational autoencoder (VAE, which maps a molecule represented by SMILES string to some latent space187). The absence or presence of relationships between functional groups and binding affinity was also recently explored through use of random matrix theory in drug design.188 The incorporation of new and improved formats, such as SELFIES,189 might still lead to further improvements for such research.
In the context of a first-principles view on CCS, we note however that molecular graphs only encode a (biased) statistical average of the many conformational configurations for a molecule near some local minima in the potential energy surface. As such, they are naturally disposed for use of QML models of ensemble properties. Work along such lines still awaits being explored in the future. Albeit popular and justified for certain problems, graph-based approaches are inherently limited when it comes to noncovalent problems, such as supramolecular assembly processes governed by van der Waals interactions, metal cluster/bulk/surface adsorption involving “multivalent” (transition) metal elements controlled by weaker metallic bond (cf., covalent bond), or chemical reactions requiring the transformation of graphs from one into the other. In such situations, the intuitive concept of a graph is ill-defined, and the necessary corrections are not always obvious.
2.4. Coarse-grained
As the system size grows, the cost in training and prediction of QML models increases accordingly, although with more favorable scaling than typical quantum chemistry methods. Therefore, it may become very demanding or even impossible to deal with system sizes which cross certain thresholds. In such scenarios, one typically represents the systems in a coarse-grained fashion, meaning “superatoms” (groups of atoms in close proximity, or beads) in a molecule are being considered. Coarse-grained approaches can drastically reduce the number of degrees of freedom and are therefore the only feasible way to model systems at macroscopic scale. More importantly, they enable a significant collapse of the size of chemical space due to the transferability of beads by design.190
Current practices of coarse-grained ideas comprise mostly coarse-grained force fields (CGFF) for simulation of large systems such as macromolecular systems and soft matter. With the emerging need for systematic control of the accuracy of models of such systems, coarse-grained representation based QML models (CGQML) may be a rather promising alternative to CGFF, much the same as how QML models based on full-atom representation remedy the deficiencies of classical force field approaches for small to medium-sized molecules.19 Such comparison between QML and FF makes sense, as the most modern implementations of ML hold promise to approach the computational efficiency of FFs. Some of the first studies on coarse-grained representation used together with QML include John and Csányi’s free energy surface modeling of molecular liquids in 2017.191 Later, efforts to tackle complicated biosystems were reported by Bereau and co-workers in 2019,190 as well as by Clementi and co-workers.192 Compared to CGFF, CGQML could be significantly more accurate once the system information is properly encoded in the coarse-grained representation, as the QML part can recover what is missing in the CG part by careful selection of training data (vide infra).
2.5. Property Based
There exists another type of representation, typically referred to as descriptor and the least “ab initio” in spirit, in which the basic idea is to simply select a set of pertinent atomic/molecular properties as underlying degrees of freedom. The properties can stem from calculation and/or experiment and have to be relatively easy to obtain and are typically supposed to somehow “describe” the property of interest, and hence the name “descriptor”. This representation is often utilized in combination with some nonlinear regressor like a neural network, as the relationship between the chosen properties is commonly highly nonlinear. Although this approach could be universally applicable, no matter the size or composition of target systems, its predictive power is limited by construction due to its potential lack of uniqueness.30,163 Most of the studies following this direction can be traced back to the early applications of ML in chemistry and related fields, one example being Karthikeyan et al.’s work193 on melting and boiling points prediction of molecular crystals using the properties of standalone molecules as a feature vector. A more recent and systematic study of this idea has applied optimization algorithms toward the down-selection of descriptor candidates in order to build predictive ML models of formation energies of binary solids.194 From a first-principles point of view, however, such representations are questionable because relationships between different observables (or other arbitrary mathematical properties), obtained as expectation values of independent operators, are not necessarily well-defined.
3. QML Methodology
The fundamental idea to employ machine learning models in order to infer solutions to Schrödinger’s equation throughout CCS, rather than solving them numerically, was first introduced in 2011.195 The authors stated that ”....the external potential...uniquely determines the HamiltonianHof any system, and thereby the ground state’s potential energy by optimizing Ψ”, and they show that one can use QML instead (encoding the number of electrons implicitly by imposing charge neutrality). As such, the problem of predicting quantum properties throughout CCS belongs to what is commonly known as “supervised learning”. One typically distinguishes between unsupervised (compound data only) and supervised (data records including compounds and associated properties) learning. In this review, we focus on the latter, i.e., on the question how, given sufficient exemplary structure–property pairs, properties can be inferred for new, out-of-sample compounds.
The generic procedure for supervised learning requires first defining the model architecture, i.e., the mathematical expression for the statistical surrogate model f, which estimates some quantum property p as a function of any query compound M, pQML(M) ≈ f((M)|{Mi},{piref};{ci}), wherefcorresponds to the regressor, and regression coefficients and hyper parameters {ci} are obtained via minimization of training loss-function quantifying the deviation of pQML from {pi} for all training compounds {Mi}. In other words, f is parametric in regression coefficients and hyperparameter which, in return, are nonlinear functions in the training data. The origin (calculated or measured) as well as the actual existence (some properties, such as energies of atoms in molecules,116 are not observables but can still be inferred) of pref is secondary. Noise in the data (due to experimental or numerical uncertainty, or due to minor inconsistencies) can be accommodated to a certain degree through well-established regularization procedures. Converged cross-validation protocols help to avoid overfitting and to enable the optimization of hyper-parameters as well as meaningful estimates for any interpolative query. For introductory texts on kernel based regressors, the reader is referred to the book by Rasmussen et al.;197 as for representation and training sets, several reviews have recently been published.7,10,19,198
3.1. Regressor
When considering the problem of fitting a generic set of basis functions to precalculated data, some of the most commonly made choices in the field of atomistic simulation include support vector machines (kernel ridge regression), tantamount to Gaussian process regression in their specific model form, neural networks, random forests, or permutationally invariant polynomials (PIPs).197,199,200 While agnostic about the training labels by construction, the choice of these basis set expansions constitutes a crucial step. Most evidently for support vector machines, nonlinear kernel functions (based on feature representations vide infra) map any nonlinear high-dimensional regression problem into a low-dimensional kernel space within which the regression problem becomes linear and therefore straightforward to solve through a closed-form expression (“kernel-trick”). How kernel space relates to CCS is also quite intuitive to grasp when thinking about it as a graph of compounds. As displayed in Figure 2, each compound, being representable by a molecular graph (or derived matrix such as a Coulomb matrix or Cartesian coordinate and nuclear charge vector) is projected into higher-dimensional feature space (shown are only three principal dimensions from the infinite number of dimensions defined within the framework of KRR/GPR). The complete connection between all compounds in the new space form another type of graph, with each vertex corresponding to a compound and each edge corresponding to a similarity measure of compounds (edge length may indicates a metric distance between two compounds). Inferring the property of a new compound (labeled as pink “X” in Figure 2) may be conceptualized as summing up distance scaled property weights. Within this picture, it becomes intuitively obvious that the interpolating accuracy must improve with increasing compound density.
Figure 2.
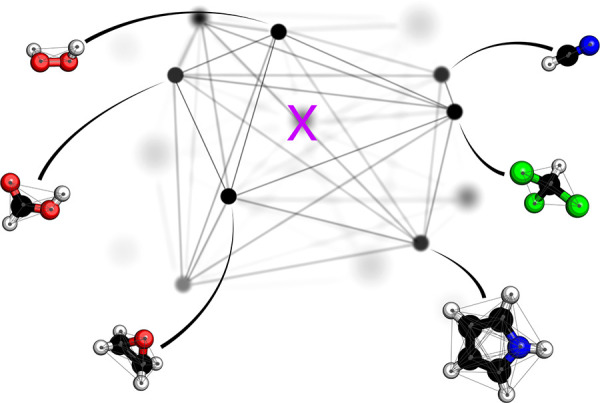
3D projection of high-dimensional kernel representation of chemical compound space. Within kernel ridge regression, chemical compound space corresponds to a complete graph where every compound is represented by a black vertex and black lines correspond to the edges which quantify similarities. Each compound, in return, can be represented by a molecular complete graph (e.g., the Coulomb matrix (CM)195) recording the elemental type of each atom and its distances to all other atoms. Given known training data for all compounds shown, a property prediction can be made for any query compound as illustrated by X. Choice of kernel-function, metric, and representation will strongly impact the specific shape of this space and thereby the learning efficiency of the resulting QML model.
While deep neural network models are very powerful and possess significant black-box character, their training requires data sets of very large size as well as a substantial calculation effort in order to optimize the regression coefficients and hyper-parameters (no closed-form solution is known). In this sense, kernel methods are rather lightweight and preferable in scarce data scenarios, as they enjoy the potential benefits of being more intuitive and faster to train. The specific architecture of the neural network will affect its performance and data efficiency dramatically. Deep, recurrent, convolutional, message passing, generative, adversarial, geometric neural networks, and other flavors, as well as choices of activation function, number of layers, and neurons, have all shown significant impact on the cost of training and on the predictive power in atomistic simulation.
In the case of GPR/KRR, the architecture is much simpler and hence of a lesser concern (GPR/KRR can be seen as a single-layer neural network model in the limit of infinite width201), but the specific kernel space does not only depend on the choice of kernel function but also on the choice of metric. While it is clear that one should avoid similarity measures which do not meet the mathematical criteria of how a metric is defined (identity, symmetry, triangle inequality), the impact of the specific metric choice has not yet been studied much in the field of atomistic simulation. Euclidean, Manhattan, or Frobenius norms are commonly used. Only most recently, the use of the Wasserstein norm has been proposed to gain permutational atom-index invariance while using index-dependent matrix representations.202 From inspection of Figure 2, it should be obvious that any nonlinear change in metric will strongly affect the shape of the kernel regression space and thereby the overall performance.
3.2. Learning Curves
Correct implementations of ML algorithms applied to noise-free data sets afford interpolating ML models, which avoid overfitting and enable statistically meaningful predictions of properties of out-of-sample compounds,199 after proper regularization and hyper-parametrization through converged cross-validation protocols, as discussed in great detail in the literature, for example, in refs (203 and 204).
On the basis of statistical learning theory, the leading order term of the out-of-sample prediction error (E) was shown to decay inversely with training set size N, i.e., E ∝ a/Nb, for GPR/KRR as well as for neural network models.205,206 This is not surprising, considering the great similarities shared between NN models and GPR/KRR model, as also mentioned in the preceding subsection. This asymptotic behavior for QML models has been confirmed numerically within numerous and independent studies, many of which are referenced herein. As illustrated in Figure 3, learning curves (LC), i.e., prediction error E vs training set size N, plotted on log–log scales assume linear form (log E = log a – b log N) and serve as a useful standard, facilitating systematic comparison and quality control of the efficiency of differing ML models. For maximal consistency, the QML models should be trained and tested on the exact same cross-validation splits stemming from the exact same data set. When the data contains noise, or when relevant degrees of freedom are neglected (e.g., through use of a nonunique representation, such as the bag of bond (BoB) representation,170 see section 4.1 for more details), the learning will cease eventually for some training-set size, manifesting itself visually through learning curves which level off, cf. solid-black line in Figure 3. For noise-free data and complete representations, however, a linear correlation between log(E) and log(N) is to be expected (see the dotted and dashed lines in Figure 3), with some slope b typically more or less a constant for different unique representations and related to the effective dimensionality of the problem, and some offset log a, which typically reflects the capability of the representation to capture the most relevant feature variations in kernel space. More specifically, the offset measures the degree to which the representation encodes the right physics. An illustrative example for this statement can be given by comparison of the learning curves obtained for the CM representation versus derived matrices with off-diagonal entries dependent on alternative interatomic power-laws.163 For interatomic off-diagonal elements approaching London’s R–6 law, the representation achieved lower offsets than for off-diagonal elements decaying according to Coulomb’s law. Correspondingly, representation matrices with off-diagonal elements linearly or quadratically growing with the interatomic distances resulted in LCs with dramatically increased offsets.163 At first glance, it might seem that the slope of LC (aka, “learning rate” of QML model) barely changes, when switching from one unique representation to another. It is therefore natural to ask if it is impossible to further speed up the learning process as indicated by the dotted-dashed learning curve in Figure 3, exhibiting a much steeper slope. Through an expert-informed reduction of effective dimensionality (through a priori removal of irrelevant information stored in randomly selected training data), it was shown that this is indeed possible. Such strategies for a more rational sampling of training data will be discussed in section 6. Note that in contrast to conventional curve fitting, training errors for properly trained machine learning models applied to synthetic data are typically orders of magnitude smaller than the variance of the signal. As such, they are negligible and carry little meaning because noise levels are typically close to zero or at least many orders of magnitude smaller than label variance. Consequences of model construction, i.e., choice of regressor, metric, optimizer, loss-function, representation, or computational efficiency, become immediately apparent in the characteristic shape of learning curves. When training a small parametric regressor, e.g., a shallow neural network with few neurons, to estimate a complex and high-dimensional target function, the learning curve will rapidly “saturate” and converge toward a finite optimal residual prediction error that can no longer be lowered by mere increase of training set size. As such, it should come as no surprise that learning curves have emerged as a crucial tool for development, validation, comparison, and demonstration purposes of QML models in the field.
Figure 3.
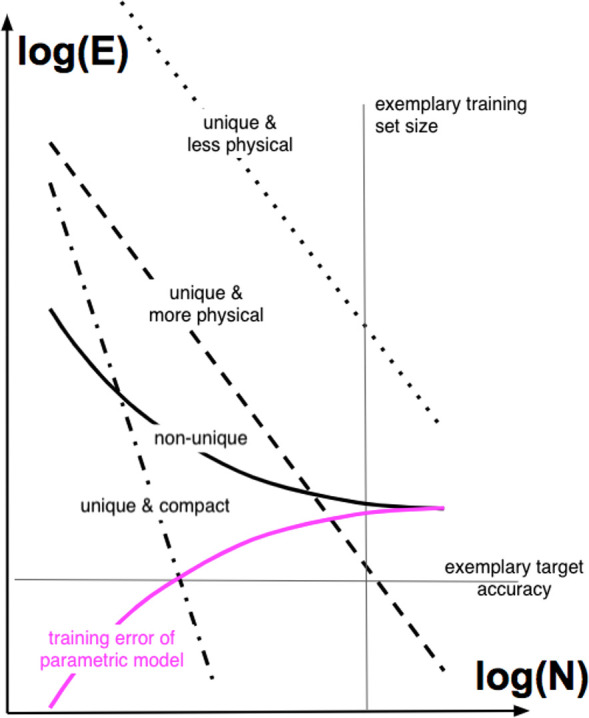
Illustration of learning curves: Errors (E) versus training set size (N). Horizontal and vertical thin lines illustrate exemplary target accuracy and available training set size, respectively. For functional ML models, training errors are close to zero (not shown), and prediction errors must decay linearly with N on log–log scales. Black-solid, dotted, dashed and dotted-dashed lines exemplify prediction errors of ML models with incomplete information (ceases to learn for large N due to being parametric, using nonunique representations, or training on noisy data), unique and less physical representation, unique and more physical representation, and explicit account of lowered effective dimensionality (i.e., “compact”), respectively. The solid-pink line corresponds to the training error for a parametric model. Training errors for ML models are negligible for noise-free data.
3.3. Loss Functions
Imposing differential relationships during training amounts to adaptation of the loss function to better reflect the problem at hand. In particular, inclusion of derivative information (gradients and Hessian) has led to dramatic improvements when tackling the problem of potential energy surface fitting.66,67,73,74 A generalization of this idea to adapt the loss function for response properties of any QM observable was established for KRR in 2018207−209 (exemplified for forces, Hessians, dipolemoments, and IR spectra) and for deep neural nets in 2020 (FieldSchNet exemplified moreover also for solvent effects and magnetic effects).210
While conventional machine learning assumes that train and test loss function are identical, for atomistic simulation (or other application domains for that matter), a mathematically, more “gready” alternative might exist. In particular, the role of gradients in loss functions differing for training and testing has been studied in ref (211), with results suggesting that for predicting atomization energies throughout a CCS of distorted structures, inclusion of gradients in training improves learning curves negligibly while surely inflating the number of necessary kernel basis functions. However, when it comes to predicting the potential energy surface of a given system, they do improve the energy predictions in the above referenced studies. Conversely, when predicting gradients throughout CCS, the use of energies alone in training offers no advantage over using forces, suggesting that the inclusion of forces (if computationally less demanding than energies) should always be beneficial.
4. Representations
One could consider the choice of functional form of the representation M to be part of the machine learning methodology. However, this is a much studied question which is at the heart of how one views CCS. More specifically, what are the truly defining aspects in a compound? And how does one measure similarity? These are old questions which have already been answered for an impressive array of applications and instruct much of the basic and fundamental textbook knowledge. For example, Hammett’s σ-parameter provides a low-dimensional quantitative data-driven measure of similarity between distinct functional groups in terms of their impact on reaction rates or yields.212,213 Within QML, physically more motivated representations are sought after for subsequent use within high-dimensional nonlinear interpolators which are more universal and transferable. As illustrated for KRR in Figure 2, also the specific form of the representation (as well as the metric used) can dramatically affect the way CCS is represented within the regressor. It should therefore not come as a surprise that the data efficiency of QML models was found to depend dramatically on the specifics of the representation used. Because the importance of the choice of the distance measures has already been mentioned above, in this section, we will focus on research that was done to find improved representations.
The choice of this particular compound representation, aka descriptor or feature, plays a particularly crucial role. Correspondingly, substantial research on the design of descriptors has already been made in the fields of chem-, bio-, or materials informatics where scarce data is typical.174,214 Often, a large set of prospective features is hypothesized and subsequently reduced within iterative procedures in order to distill the most relevant variables and low-dimensional projections pertinent to the problem at hand (see above). While it is certainly possible to also pursue this approach within a quantum mechanical description of CCS,194 its heuristic and speculative character remains as unsatisfactory as its lack of universality and transferability. Fortunately, the quantum nature of CCS allows us to follow more systematic and rigorous procedures in order to address this question.
For example, it is a necessary condition for any successful ML model to rely on uniqueness (or completeness) in the representation, as pointed out, proven and discussed several years ago in refs (215 and 216) and more recently in refs (217 and 218), Uniqueness is essential in order to avoid the introduction of spurious noise due to uncontrolled “coarsening” of that subset of degrees of freedom which is neglected. Molecular graphs based on covalent bond connectivity only, for example, do not account for conformational degrees of freedom. Consequently, their use as a representation will make it impossible to quench prediction errors below the variance of the target property’s conformational distribution, no matter how large the training set.
Other characteristics, desirable for representations to display, include compactness, computational efficiency, symmetries, invariances, and meaning. Representations, in conjunction with the regressor’s functional form, define the basis functions in which properties are being expanded and strongly affect the shape of the learning curves, e.g., accounting for a target property’s invariances through the representation typically leads to an immediate decrease of the learning curve’s offset.
While it is possible to model all QM properties using the same representation and kernel,219 as also demonstrated for neural nets with multiple outputs already in 2013,220 it should be stressed, however, that this is a distinct feature of QML which stands in stark contrast to conventional QSAR or QSPR, where the ML model is typically strongly dependent on the target property. If regressor, metric, and representation M are independent of the label, i.e., the quantum property, there is a strict analogy to quantum mechanics in the sense of the Hamiltonian (or the wave function) of a system not depending on the operator for which the expectation value of any given observable is calculated.8 This becomes obvious by considering the training of a KRR model where the regression coefficients are obtained through inversion of the kernel matrix, α = (K + λ I)−1pref, where for synthetic calculated data with signals being orders of magnitude smaller than noise, the regularization λ (also known as noise level) is typically close to zero. Using property independent representations, metrics, and kernel functions, it is therefore obvious that the regression coefficients adapt to each property only because of the reference property vector pref. In ref (219), this has been illustrated numerically by generating learning curves for various properties using always the same inverted kernel matrix for any fixed training set size.
The predictive accuracy for specific properties varies wildly as a function of representation and regressor choice.183 The historic development over years 2012–2018 for a selection of ever-improved machine learning models (due to improved representations and/or regressor architectures) can be exemplified for the prediction errors of atomization energies stored in the QM9 data set171 and has also recently been summarized in the context of the “QM9-IPAM-challenge” in refs (15, 16, and 18).
The inclusion of increasingly more (less) “physics” in the representation has been demonstrated to systematically improve (worsen) learning curves163 and has been followed by a series of developments which have all been benchmarked on the same set of atomization energies of small organic molecules in the QM9 data set171 and which demonstrate the progress made. While binding energies of “frozen” geometries still constitute an application somewhat remote from most real-world applications in chemistry, from a basic physics point of view they do represent a crucial intermediate step before tackling more complex properties. In other words, if machine learning models failed to predict binding energies, one should not expect them to work for free energies. But also from a practical point of view, the computational cost of single-point energy calculations typically dominates all quantum chemistry compute campaigns and therefore represents one of the most worthwhile targets for surrogate models used for the navigation of CCS.
We note that with the emergence of deep neural networks, the problem of also “learning” the representation can be mitigated to be incorporated in the overall learning problem.55,221 While many intriguing and sophisticated representations, such as Fourier-series expansions,216 wavelets,222 multitensors,223 or molecular orbitals224 have been proposed, most representations can be categorized to either correspond to discrete adjacency matrices or to continuous many-body expansions through distribution functions. We therefore limit ourselves to discuss in the following, both predominantly in the context of KRR based QML models. A comprehensive overview on representations for KRR based QML models has also recently been contributed by Rupp and co-workers.225
4.1. Discrete
Coordinate-free, bonding neighbors (covalently bonded atom pairs) based graphs, as well as their systematic extensions to arbitrary number of neighboring shells, have formed an important research direction in cheminformatics for many years.32,174,176,178,214 In 2011, supervised learning was proposed as an alternative to solving Schrödinger’s equation throughout a chemical compound space relying as a representation on a complete undirected labeled graph that encodes the simplex spanned by all atoms.195 More specifically, this graph was represented by the “Coulomb matrix” (CM), an atom by atom matrix with the nuclear Coulomb repulsion on off-diagonal elements and with approximate energy estimates of free atoms (EI ≈ 0.5ZI2.4226) as diagonal elements. Formal requirements such as uniqueness, translational and rotational invariance, as well as basic symmetry relations (symmetric atoms will share the same matrix elements in their respective rows or columns) are all met by the CM. Atom index invariance can be achieved through use of its eigenvalues (thereby sacrificing uniqueness215,227), sets of randomly permuted CMs,220 or sorting by norms of rows,203 thereby losing differentiability due to sudden switches in ranks.202 We reiterate once more that the atom indexing dependence can be mitigated through using more sophisticated distance measures such as the Wasserstein metric.202
Similarly encouraging findings of KRR based QML models applicable throughout CCS were quickly reproduced for other materials classes such as polymers228 or crystalline solids.229 While off-diagonal elements with a London dispersion power law (r–6) have subsequently been found to be preferable for QML models of atomization energies,163 other representations (vide infra) offer lower learning curve offsets. In particular, the bag of bonds representation (BoB) is worthwhile mentioning.170 Introduced in 2015, BoB groups the entries of the CM in separate sets for each combination of atomic element pairs within which all entries have been sorted. When calculating the similarity between two molecules, only Coulomb repulsions between atoms with the same nuclear charge are being compared, rendering thereby the similarity measurement more balanced and effectively lowering the learning curve offset. While even more compact than the CM, BoB lacks uniqueness due to being strictly a two-body representation which can not distinguish between homometric configurations.216 The generalization of BoB toward the explicit incorporation of covalent bond information, angles, as well as dihedrals in terms of a systematic expansion in Bond, Angle, and higher-order interactions (i.e., BAML representation) was accomplished in 2016163 by using functional forms and parameters from the universal force-field.230 A similar, but more elaborate, parameter-free, many-body dispersion (MBD) based representation involving two and three body terms231 was proposed later in 2018.
The CM has been essential as a baseline for the interpretation, analysis, and further development of subsequent QML models. It has also been adapted successfully to account for periodicity in the condensed phase, as evinced by learning curves for formation energies of solids.232 For other properties, such as forces, electronic eigenvalues, or excited states, the CM (or its inverse distance analogues for QML applications with fixed chemical composition) is still competitive with state of the art representations.67,73−75,83,233−236 Furthermore, because of its uniqueness, compactness, and obvious meaning, the CM (or its variants) are conveniently used to overcome frequent data analysis problems in atomistic simulations, such as removal of duplicates, quantification of noise levels, and simple learning tests.
Regarding the interatomic distance dependent decay rate of off-diagonal elements, it is also worthwhile to mention exponential functions, rather than 1/r. In particular, the overlap matrix between atomic basis functions of all atoms has been proposed237 and used with great success for QML models of basis-set effects238 and excited-state surfaces.239 The overlap matrix was also included within a recent sensitivity assessment of various state-of-the art representations and performed in impressive ways.218 A constant-size descriptor based on a combination of the CM with more common molecular graph fingerprints was also proposed in 2018.240
Viewing BoB and CM as first and second rank tensors, to the best of our knowledge, use of a third rank tensor (explicitly encoding the surface of all possible triangles in a compound) has not yet been tested.
4.2. Continuous
Aforementioned discrete and global representations such as BoB enjoy fast computation. One important requirement for this kind of representation to work, however, is to introduce atom indexing invariance by sorting atoms according to the magnitude of entries belonging to each bond or other many-body types. This is artificial and may introduce derivative discontinuities with unfavorable consequences in related applications such as force predictions.
The sorting and associated problems can be naturally overcome by selecting continuous or distribution based representations, which, in essence, integrate out atom index dependent terms such as distance (w/wo angle and dihedral angle) and/or nuclear charge (i.e., alchemically166) through use of smeared out projections (a Gaussian is commonly placed on each degree of freedom). Distribution based representations, also closely related to many-body or cluster expansion,241 have gained much popularity also within QML models building on Behler’s seminal work on atom-centered symmetry functions for training neural networks on potential energy surfaces,242 or through the subsequent introduction of smooth overlap of atomic potentials (SOAP) for use in GPR by Bartok et al. in 2013.38 The first variant of linearly independent distribution based representations for QML models, applicable throughout CCS, a Fourier series expansion of nuclear charge weighted radial distribution functions, was also contributed already in 2013,216 albeit published in its final version only in 2015. Radial distributions were also used for representing crystals in solids in 2014.229 The atomic spectrum of London Axilrod–Teller–Muto (aSLATM) terms was first presented in 2017 within the “AMON” approach by Huang et al.243 (vide infra), yielding unprecedentedly low offsets in learning curves for atomization energies in the QM9 data set.171 In that same year, SOAP based QML models were generalized and shown to be also applicable throughout CCS.244
The generic histogram of distances, angles, and dihedrals (HDAD),234 a continuous but simplified version of BAML, both including many-body terms up to torsions, was contributed in 2017. In the following year, Faber et al. conceived the idea of adding alchemical degrees of freedom in a structural distribution based many-body representation, dubbed FCHL18166 (FCHL indicating the first letters of the last names of the authors and 18 in the year 2018). The FCHL family of representations encodes a systematic interatomic many-body expansion in terms of Gaussians weighted by power laws due to the insights gained in ref (163). Power law exponents and Gaussian widths were optimized as hyper-parameters through nested cross-validation during training. FCHL18 consists of three parts: The one-body term corresponds to a two-dimensional Gaussian encoding the chemical identity of the atom in terms of groups and periods of the periodic table; the two-body term encodes the interatomic distance distribution scaled down by r–4, and the three-body term encodes all angular distributions and is scaled down by r–2. The impact of four-body terms has been tested on QM9 but was found to have negligible impact on learning curves.166 Most importantly within the context of CCS, FCHL18 based QML models have been demonstrated to be capable of accurately inferring property estimates of systems containing chemical elements which were not part of training. More specifically, consider the family of molecules of formula HnY ∼ X, where Y corresponds to an element from group IV (either C, Si, or Ge), where “∼” represents single, double, or triple bond depending on chemical element X being from groups VII, VI, or V, respectively, and where n is the number of H atoms that saturates the total valences. Semiquantitative covalent bond potential binding curves have been predicted for any X/Y/bond-order combination using QML models after training on corresponding DFT curves for all other molecules that neither contain X nor Y.(see the top- and left-most subplot in Figure 4 for an illustration). For example, the ML binding curve of HC#N was obtained after training on binding curves of all other molecules that neither contained N nor C, i.e., when predicting the blue curve in the upper left panel of Figure 4, the red and green curves of that panel were not part of training nor were any other blue curve from the other panels. FCHL19, a recent revision, has been shown to provide a substantial speed-up in training and testing while imposing only a small reduction in predictive accuracy.209
Figure 4.
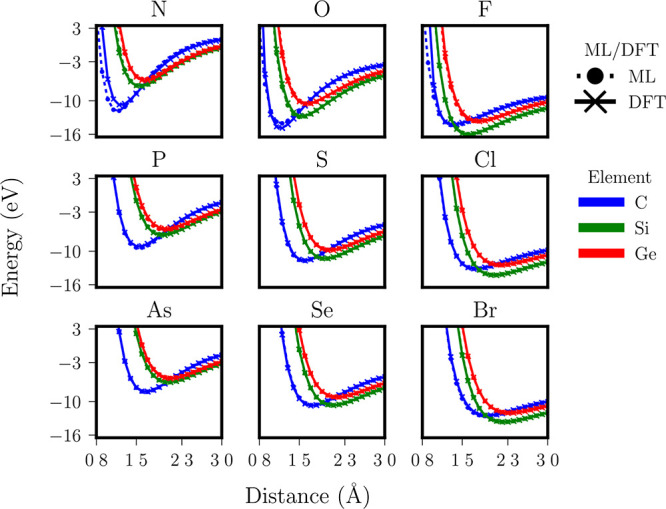
QML models infer properties for new chemical compositions. DFT and QML (FCHL+KRR) based predictions of covalent triple, double, and single bonding between groups IV and V (left column), VI (mid column), and VII (right column) elements, respectively. Open valencies in the group IV elements have been saturated with hydrogens. QML models were trained on the DFT results for all of those chemical elements that are not present in the query molecule. Reproduced with permission from ref (166). Copyright 2018 licensed under a Creative Commons Attribution (CC BY) license.
We note in passing also the related moment tensor model (MTM) by Shapeev and co-workers, introduced in 2018,245 as well as the unifying interpretation of many of the popular distribution based representations by Ceriotti and co-workers.246
5. Regressor
Depending on how regression parameters are being obtained, the incorporation of legacy methods in QML models applicable throughout CCS is typically done either within neural networks or within Gaussian process regression (GPR) (or kernel ridge regression, KRR for short). Here, we mostly focus on kernel methods, mentioning only shortly the idea of transfer learning in neural network models,247 which is also applicable to QML models as shown in 2018 by Smith et al.248
More specifically, five categories of QML models can easily be distinguished, each of which accounting for legacy information in its own way: QML models of parameters of existing models, QML models of corrections to existing models (Δ-ML), multifidelity ML (MF-ML), multilevel-grid-combination (MLGC), and transfer learning techniques. We briefly review each of these in the following.
5.1. ML Models of Parameters
Existing force-field models can capture nicely the essential physics of a wide range of chemical systems, the main drawback being that force-field parameters (e.g., atom charges, harmonic force constants, etc.) are often rigid and unable to adapt to different atomic environments. Therefore, it would be natural to make these parameters flexible and predicted by ML models. This idea dates back to the 1990s, and the first piece of related works was done by Hobday et al.,249 where they proposed a neural network model to predict parameters of the Tersoff potential for C–H systems. In 2009, Handley and Popelier proposed to use machine learning models for multipole moments.49 This idea was revisited in 2015, when learning curves for atomic QML models of electrostatic properties, such as atomic charges, dipole moments, or atomic polarizabilities were presented.250 Their use for the construction of universal noncovalent potentials was established in 2018.70 Neural-network based equilibrated atomic charges were also proposed in 2015 by Goedecker and co-workers54,251 and in 2018 by Roitberg, Tretiak, Isayev, and co-workers.252,253
Similar strategy could also be applied to semiempirical quantum chemistry methods relying on parameters typically fitted by computational/experimental data. In 2015, QML models of nuclear screening parameters were contributed by Pavlo and co-workers.254 In 2018, unsupervised learning for improved repulsion in tight-binding DFT was introduced by Elstner et al.,255 followed by substantial further improvements in 2020.256 Extended Hückel theory was revisited in 2019 by Tretiak and co-workers.257
5.2. Δ-ML
The idea to present QML models of label corrections applicable throughout CCS and which systematically improve with training data size was first established in 2015 in terms of Δ-machine learning. Numerical results provided overwhelming evidence for the success of this idea as demonstrated for modeling energy and geometry differences between various levels of theory, including PM7, PBE, BLYP, B3LYP, PBE0, G2MP4, HF, MP2, CCSD, and CCSD(T) for QM9171 and subsets thereof.258
Δ-ML also works for correcting complex and subtle properties, such as van der Waals interactions in extremely data-scarce limits, as illustrated for DFT corrections based on training sets with less than 100 training instances,259 or to model higher-order corrections to alchemical perturbation density functional theory based estimates of heterogeneous catalyst activity.260 Among many other applications, Δ-ML has enabled corrections to electron densities,261 electron correlation based on electronic structure representations within Hartree–Fock or MP2 level of theory,262 or DFT and CCSD(T) based potential energy surface estimates.263
For noise-free data and functional QML models (unique representations), numerical results for learning curves indicate a constant lowering of offset, no matter which training set size. Such nonvanishing improvement appears to turn into vanishing improvement when employing Δ-ML in order to correct low-quality or coarse-grained baselines, such as a semiempirical PM7258 or Hammett’s relation.213
5.3. Multifidelity
The success of Δ-ML is encouraging, enabling a significant reduction in high-accuracy reference quantum chemical data necessary for training, to reach the same level of predictive accuracy as traditional QML models. However, it still consumes a considerable amount of data calculated at some high level of theory, as its structure in design is too simple to fully exploit the underlying correlation between varied quality of properties. In fact, well-established quantum chemical methods abound in literature, exploiting effectively the underlying correlation, in the name of the so-called composite methods, for example, the famous Gn series.264−266 In essence, these methods approximate some specific part of correlation energy (e.g., energy lowering due to inclusion of diffuse orbital in basis set) from a high level of theory (for instance CCSD(T)) by the same quantity calculated from a relatively low level of theory (say MP2). Because of error cancellation, composite methods have been proven to be extremely effective toward reaching an accuracy of experimental quality and are widely used for calculations of high-quality thermochemical data.264−266
To do interpolation and meanwhile exploit error cancellation effectively, multifidelity ML (MF-ML) comes into play. The core idea of MF-ML is hereafter demonstrated by total energy (E) prediction. For brevity, we deal with two levels of theory (the low and high level are denoted by 0 and 1, respectively) and focus on one flavor of MF-ML, i.e., recursive KRR (r-KRR for short, or MF-KRR),267 which is similar to its counterpart, recursive GPR (r-GPR, or MF-GPR),268,269 and differs to MF-GPR to some extent, in analogy to the difference between KRR and GPR. Unlike Δ-ML, MF-ML comprises multiple machines with different labels to learn (two for our exemplified case). The first one is just a traditional QML model trained on a set of data (denoted as S0) associated with the low level of theory, i.e., Ej0 = Σi∈S0cik(i,j), where j ∈ S0 denotes the molecular representation vector and c0 is the regression coefficient associated with the low level of theory. This machine is also called the baseline model. Then we build a second machine with training set S1 satisfying S1 ⊂ S0 and energy delta, i.e., E1 – E0, as label, the same as a Δ-ML model. In math, ΔEn0→1 = En – En0 = Σm∈S1cmk(m,n), where n ∈ S1. Once trained separately for each machine, all cl’s are obtained and MF-KRR predicts the property of any query q out-of-sample at the high level of theory by Eq1= Eq + ΔEq0→1.
Extending r-KRR to more than two levels of theory is straightforward: except the baseline model for the lowest level, one needs to build one machine for every two adjacent levels of theory, and the final test energy is just the summation of the inferred energies by all machines, i.e., EqL = Eq + Σl = 0L–1 ΔEq, where l is the level indicator (starts from 0, the lowest level) and L corresponds to the largest l, or the target level. Bear in mind that S0 ⊂ S1 ⊂··· ⊂ SL.
We note in passing that MF-GPR has a rather different formulation compared to MF-KRR and benefits from the stochastic nature of GP, i.e., it is capable of providing the variance estimate of prediction. Like GPR, data at each level of theory in MF-GPR is modeled as a GP,268,269 and every two adjacent levels are connected by a linear transformation, i.e., El+1 = γEl + ϵ, where γ is a scaling factor and ϵ is a correction term respectively and both of which may depend on the two involved levels of theory (i.e., l and l + 1). Nevertheless, both MF-KRR and MF-GPR could end up with the same set of working equations under certain conditions. For detailed derivation of the equations of MF-GPR, the reader is referred to an early review on QML.10 Last but not least, one should note that MF-KRR converges toward the conventional KRR model associated with the highest level as the difference between training sets for each machine vanishes.
Albeit well-founded in mathematics decades ago, the power of MF-ML has not been harvested until recently. Applications include quantum collision for the Ar–C6H6 system by Cui et al.,270 bandgap prediction of solids done by Pilania et al.,271 dopant formation energy prediction in hafnia by Batra et al.,272 high-accuracy potential energy surface prediction for small molecules by Wiens et al.,273 and the recently performed molecular crystal structure prediction study by Egorova et al.274
5.4. Multilevel Grid Combination
In spite of the drastic improvement over Δ-ML, MF-ML has its own limitations. For one, the computational cost of the baseline evaluation for every query compound can still be considerable. Furthermore, it must be strictly satisfied that the increasingly more expensive training sets form a nested structure, implying that possible and beneficial correlations between non-nested reference data calculated at different level of theory are not being exploited. To overcome this drawback, Zaspel et al.267 proposed a multilevel model in 2018, combining successfully ML with sparse grid (SG),275 a numerical technique widely used to integrate/interpolate high dimensional functions.
The genuine SG approach assumes (quasi-)uniform grids along each dimension, which serves as basis functions (more precisely, centers of basis functions, such as triangular function) and based on tensor products of which any multidimensional function could be represented/expanded.275 The expansion weight for each tensor product is dependent on only the indices of associated grid and spacing along each dimension and determined by multivariant Boolean algorithm.276
Replacing such a grid by abstract variable (or combinations of which) such as electron correlation level (xC), basis set (xB) and expressing system property as a function of these abstract variables represents an appealingly rigorous alternative. For example, the total energy of a system could be expressed as E = E(xC, xB). Given some sparse grids comprising small xC combined with all xB’s, and small xB combined with all xC’s, and intermediate xB’s combined with intermediate xC’s, we are able to interpolate/extrapolate the E at some different combination of xC and xB. Of particular interest is extrapolation to regions unsampled, i.e., regions with large xC and xB. However, one major issue with such extension is the elusive nature of distance between two abstract variables, which is essential in determining the weight associated with each grid, as mentioned above. More specifically, it is unknown how to quantitatively characterize how distant HF and MP2 are along the dimension C, although qualitatively it is certain that HF lies closer to MP2 compared to CCSD(T). The B subspace, is understood much better, as the magnitude of xB could be at least characterized by the largest angular channel, or more straightforwardly, although less rigorously, by the number of basis functions. This ill definition of these abstract variables is absent in genuine SG, as grids there reside typically in Euclidean space and therefore distance is well-defined. To rise to this problem, a workaround is to assume uniformality of grids along each dimension (i.e., equidistant) and grids along each dimension is represented simply by indices starting from 0 (now weights depend solely on the indices of grids). This, however, should always be done with great care. In the original MLGC paper,267 electron correlation levels are reasonably chosen as HF, MP2, and CCSD(T), together with three basis sets, i.e., STO-3G, 6-31G, and cc-pVDZ (the number of basis functions increases by a factor of ∼2).
Note that the aforementioned SG approach deals with typically one system at a time. To incorporate it within ML framework, one extra variable has to be introduced, i.e., training set (xN), the size of which indicates the magnitude of xN.267 Accordingly, E = E(xC, xB, xN). Unlike subspace C or B, xN is well-defined with explicit value. Nevertheless, it has to be treated in a similar fashion as for xC/xB, i.e., given the minimal xN (aka. N0) and a ratio (s) between any two adjacent xN’s, training sets are to be assigned an array of indices starting from 0 (for N0) and an increment of 1. This assignment is necessary so as to comply to what has been done for subspaces C and B. Now each grid in this abstract space is a combination of three variables: (xC, xB, xN), with xI ∈{0, 1, . . ., Imax}, I ∈{C, B, N}. For each such combination, an associated ML model is trained (with N training instances of course). Given a query system, its energy is predicted as a weighted summation of test energies from all ML models, with weights derived from Boolean algorithm.267,276 Note that in practice, to reduce the cost of generation of reference quantum data, a large xN is associated with a low level of correlation and/or small basis set, while only few(er) labeled data are needed for high(er) correlation level and/or large(r) basis set.
With the above setting, Zaspel et al. were able to show267 that MLGC enables ∼10-fold reduction (cf. traditional single level ML model) in the costly highest level quantum data (i.e., CCSD(T)/cc-pVDZ) to reach chemical accuracy in predicting atomization energy of out-of-sample QM7b molecules. Last but not least, it is worth pointing out that MLGC reduces to MF-ML if only one dimension is being considered.
5.5. Transfer Learning
While multilevel methods are most naturally combined with kernel methods, they may have even more far-reaching effects for neural network (NN) models, as training a NN model, deep NN (DNN) in particular, is a nontrivial problem. Furthermore, current ad hoc DNN models are typically specialized, meaning a (D)NN model may need to be retrained for a slightly different task.
Transfer learning (TL)277 is one popular approach employing multiple levels in machine learning that can greatly alleviate the aforementioned problems, which reuses the knowledge gained through solving one task (base task) as a starting point for a second task (target task), different but highly related. For instance, knowledge obtained from learning to infer DFT energies could be applied to infer CCSD(T) energies.278 A successful transfer of knowledge can improve the performance of the target DNN model significantly. Speaking the language of learning curve, TL could offer277 (i) smaller offset as the transferred model per se provides a decent starting point and (ii) steeper learning curve due to the transferred model usually accounting for a parameter space rather close to the optimal one.
On the basis of the type of traditional ML algorithms involved, TL could be categorized into several variants. Here we focus on a variant named inductive transfer learning, in which the labeled source and target domains are the same, yet the source and target tasks are different from each other.
In TL, there are two essential ingredients: (i) a pretrained model, obtained by either training a network from scratch on some data set and a specific task, or simply from published models, and (ii) target network, to be trained on a target data set and task, but utilizing the learned features from (i). This process is likely to work only if the features are general (i.e., generic features) to both base and target tasks, as would be captured by the initial layers of NN models. When retraining the target model, one may choose to either freeze the initial layers in the base network to use them as feature extractors for the target model or fine-tune the last several layers further for improved performance. A rule of thumb is to freeze when target labels are scarce (to avoid overfitting), while fine-tune otherwise.
To develop a successful TL model, it is vital to choose the proper base model and associated training data set. However, it remains largely an open question how to make the choice. This may require profuse intuition developed via experience. Furthermore, there exists one major potential risk of using TL, i.e., negative transfer, which refers to scenarios where the reuse of base-task knowledge degrades the overall performance of the target task. To avoid negative transfer, one may have to resort to approaches that explicitly model relationships between tasks and include this information in the transfer method.279
Applications of TL covers mainly computer sciences, such as image recognition and natural language processing. For chemistry-related problems, TL is emerging as a promising approach. Examples include Smith et al.’s work on predicting CCSD(T)/CBS energies based on transferred knowledge gained through training on DFT energies278 for the ANI-1x data set (see section 8.1), Iovanac et al.’s work on property prediction of QM9 molecules,280 as well as Cai et al.’s recent work on drug discovery.281
6. Training Set Selection
Among all factors determining the performance of a QML model, training set selection plays another fundamentally important role in the sense that all knowledge essential for making confident predictions are implicitly encoded in the training data.
Several pertinent fundamental and distinct questions have remained open:
-
Q1
How to extract the most representative and least redundant general subset from a given data set?
-
Q2
How to quantitatively define the suitability of a given training set for a specific query at hand?
-
Q3
How to systematically select the most relevant training set for a specific system?
Because of their highly nonlinear impact of training instances on model parameters, these questions are challenging and have not been studied much. Of course, the problem of training set selection is not a problem unique to chemistry, and it is relevant to most supervised learning problems in other fields. Currently, the aforementioned issues are mostly addressed through random selection. Although universally applicable, random selection suffer inevitably from selection bias inherent in the data itself. More specifically, in the randomly selected training set, many instances can be ignored and their inclusion in training does not improve predictive performance of the QML model (due to redundancy) or could even degrade it (due to being irrelevant for a given query test or due to noise).
Bias could become a very serious issue as the systems under study are increasingly more complicated. The origins of the bias issue could be divided into two components: (i) Curse of dimensionality. This is mainly related to the size of the systems and plagued further by compositional diversity. More specifically, as the system size and/or the encompassing number of types of elements grow, the size of the thus-spanned CCS grows combinatorially (see above). (ii) the inhomogeneity of CCS. The energetics in chemistry typically favors one kind of bonding over another. For instance, hydrogen atom favors a single σ bond with other atoms, while carbon atom can exhibit several different bonding patterns such as sp3, sp2, and sp. Consequently, random sampling will introduce more subsampling of hydrogen environments, but proportionally fewer C–sp local environments leading to worse model estimates of properties for C than for H.
To tackle the bias issue, previous and ongoing research has been trying to almost exclusively tackle Q1, assuming a pre-existing data set (or a data set that is straightforward to generate, e.g., in molecular dynamics). Examples include the use of genetic algorithms (requiring labeled data to gradually expand the optimal training set),80,282 or “active learning” approaches,79,223 which selects the most representative subset “on-the-fly” from a given set of unlabeled configurations, i.e., no quantum chemical data is needed for making decisions about whether or not a query configuration is redundant. The AMONs concept proposed by the authors243 partially resolves question Q3 (cf. Q1 and Q2), at the same time allowing for significant dimension reduction of CCS as well as the effective removal of statistical redundancy of training sets (see below for details). Other related work shifts the attention to training set reduction instead, primarily in molecular dynamics simulations, for instance, Li et al.62 proposed a “learning-and-remembering” scheme, in which the decision to recompute QM data for a new configuration was taken every n steps. Another relevant contribution to active learning in CCS was made in 2018 by Smith et al.,283 relying on “query by committee”, i.e. ensemble information obtained through use of multiple neural networks (of the ANI kind53). Potentially promising alternative directions could possibly be inspired by recent developments in computer science, among many others notably the idea of artificial “soft” labels, curated through carefully blending features of training instances.284,285 In the original paper, this idea was tested on the MNIST data set (a database of handwritten digits) and similar performance was achieved with much fewer but soft labels, as compared to training on almost the entire data set. This idea should in principle also be applicable to CCS requiring the design of some fictitious averaged training molecules, interestingly probable to violate common rules of chemical bonding. In the following, we review the three most promising approaches toward training set selection: genetic algorithm, active learning, and the AMONs approach.
6.1. Genetic Algorithm
Genetic algorithms (GA) have been widely used in (global) optimization problems in quantum chemistry, such as first-principles based global structure optimization286 (for compounds with desired physio-chemical property), a key topic in the inverse-design problem.287 To the best of our knowledge, the first piece of work about using GA for training set selection within QML was done by Browning et al.282 for molecules, followed by Jacobsen’s work80 on SnO2(110) surface reconstruction.
In the following, we discuss the central idea of GA for the selection of the most representative set of QM9 molecules as done in ref (282). For applications to other properties and systems such as chemisorption systems,288 only technical details will differ. Given a set (S0) of N molecules, GA carries out three consecutive steps for optimization: (a) Generate M random sets of size N1. This forms a starting population of training sets (aka. the parent population), labeled as Ŝ(1) = {Si}, where i ∈ {1, 2, . . ., M}. Note that the initial size needs to be balanced against the diversity of the molecules for optimal performance. (b) Train a QML model on each set in Ŝ(1) and then test on some joint preselected out-of-sample molecules (i.e., not part of Ŝ(1)), the resulting test error ϵi (measured by for instance mean absolute error) serves as a “fitness” indicator, characterizing how fit si is as a training set (smaller ϵi means better fitness). (c) Evolution of Ŝ(1) takes place through three consecutive steps: selection, crossover, and mutation. In the selection step, decisions have to be made on which Si ∈ Ŝ(1) should be kept in the population to produce a temporary refined smaller set t̂(1), and a set with larger fitness value means higher probability to be kept in t̂(1). The crossover step involves the update on Ŝ(1) from t̂(1), and the resulting new population is relabeled as Ŝ(2), each of which is obtained by mixing molecules from two subsets of t̂(1). The last step mutation randomly modifies molecules in some subset of Ŝ(2) to promote diversity, e.g., replace −NH2 group by −CH3. To avoid introduction of chemical environments alien to the whole data set, the replacements in mutation have to be constrained locally in S0. (d) Go back to step (b) and repeat b–d until there is no improvement in the population and the fitness value has no significant improvements for over n iterations. The final converged set corresponds to a “optimal” training set and is labeled as Ŝ.
It is not a surprise that selected Ŝ should be able to represent all the typical atomic environments in S0, and therefore a QML model trained on Ŝ warrants significantly improved test results in comparison to randomly drawn training sets. As the fitness value decreases during the GA iterations, the QML models “tried” out the sensitivity with respect to inclusion of certain training instances, and this can serve their systematic inclusion/exclusion. The usefulness of the optimized set Ŝ has to be assessed by the generalizability of the QML model trained on Ŝ to new molecules absent in S0. Indeed, improved generalizability is observed for PubChem molecules compared to random sampling, as was reported in ref (282).
In spite of its power for solving hard optimization problems, such as finding the optimal training set composition, the drawback of most GA implementations is also obvious: It typically relies on the availability of labeled data to evaluate the fitness in each iteration. As such, it only offers computational cost savings in terms of QML model efficiency and not in terms of reducing the total need for available training data. Possible solution to circumvent this is to introduce heuristics in feature space, e.g., accounting for the fitness by some distance metric instead, meanwhile avoiding the costly training-test procedure in each iteration.289
6.2. Active Learning
Active learning (AL) is more interesting than GA for training set selection, as it can use directly unlabeled data, i.e., before the acquisition of costly labels. Intuitively, it makes sense that this should be possible as the quantum properties of any compound are implicit functions of its composition and geometry, which is the only input required for calculating rigorous representations. Among the many categories of AL algorithms used for determining which unlabeled data points should be labeled, below we mainly focus on the variance reduction query strategy, which labels only those points that would minimize output variance (uncertainty in prediction). Note that the task of variance estimation is fundamentally different from mean error estimation, and the variance based selection method differs significantly from the mean error based selection method (such as GA mentioned above) accordingly. Relevant works on active learning include the D-optimality approach79,223 and methods based on variance estimators using Gaussian process regression (GPR),290−292 as well as neural network (NN) models.
Rooted in linear algebra, the D-optimality approach79,223 takes advantage of (i) the dimension of features could in principle be significantly lower than the number of degrees of freedom spanned by the molecules (in particular for molecules that are in or close to their equilibrium states) and (ii) linearly parametrized local atomistic potential. Given a set of K molecules, the total energy of the qth molecule could be approximated as E(q) = Σi = 1NV(xi) = Σi = 1N Σj = 1θjbj(xi(q)) = Σj = 1θjBj(x(q)) (in matrix form, E = θB), where Bj(x(q)) = Σi = 1Nbj(xi) serves as the effective basis function of dimension m and bj(xi(q)) is some function dependent only on the local representation xi of the ith atom in q, N is the number of atoms of q. Then deriving the D-optimality criteria boils down to finding the “best” submatrix (of size m × m) from the overdetermined matrix AK×m (where K > m and Akl = Bk(x(l))) such that the absolute value of det A reaches its maximum. Well-established algorithms exist to achieve the D-optimality criteria, e.g., the maxvol algorithm.293 To obtain an optimal set, one typically has to iterate the procedure, one new query per time. If the corresponding magnitude of det A increases, it would be selected (query strategy) and discarded otherwise. Numerical results79 have shown much improved performance for long-time MD simulation compared to classical on-the-fly learning.62 However, the downside of D-optimality approach is also noticeable, that is, the model has to be updated at each iteration and application of the model could be prohibitive for a data set bearing a large feature space. Furthermore, the linear potential Bj depends on the proposed representation and the potential form, the latter of which in particular may suffer from lack of expressive power for some systems, i.e., the potential form may lack general applicability for a wide range of molecular systems. And last but not least, this approach relies on the choice of ratio (of det A values of two consecutive iterations) threshold manually chosen, which has to be tailored for a specific data set and may not be applicable to other data sets that only differ ever so slightly.
We note in passing that an alternative view79 of D-optimality criteria is to assume that the energy has a Gaussian random noise and the best submatrix A corresponds to the minimal variance in the solution of E = θB. Besides, consideration of other properties such as forces could be naturally incorporated into this framework by simply taking derivatives of Bj with respect to Cartesian coordinates, expanding the feature matrix B.79
Another variance-based approach relies on the GPR directly. That is, once trained, the model can estimate the variance directly, without referring to other criteria (as in the D-optimality approach). The estimated variance serves as a natural indicator, telling if any newly added data point would improve the model (if the variance is large with respect to a user-defined tolerance) or not (if the variance is very small). A small variance typically also means that the newly added data lie within or close to the current training space, distant otherwise. Methods like Gaussian process regression (GPR) are stochastic in nature and inherently capable of calculating the variance of prediction. More specifically, GPR aims to estimate the predictive distribution for any test data (unlike the kernel ridge regression model). Related works include that of Snyder et al.’s,290 in which the Bayesian predictive variance is shown to correlate with the actual error, and recently Reiher’s group291,292 used GPR to select optimal training sets in an automated fashion to explore chemical reaction network291 and subsequently adjust for systematic errors in D3-type dispersion corrections, with one (sequential scheme) or multiple systems (batchwise variance-based sampling, BVS) selected each time.
Neural network (NN) based methods also offer a quite distinct perspective on the confidence of predictions. The general finding is that for NN methods estimates tend to be overconfident,294 possibly due to the lack of principled uncertainty estimates295 (i.e., NN model typically produces one single value for an input instead of a preditive distribution like GPR) and/or that the tools for mean estimation perhaps do not generalize.295 In spite of the lack of native variance estimate, variance can still be modeled in practice through consideration of multiple parallel NN models. In analogy to GPR, uncertainty in NN models can be understood by taking a Bayesian view of the uncertainty of weight with some distribution assumed a priori and then updated by training data. There exists several variants of such NN models, including the ensemble neural network models,283,296−298 where NN models share the same architecture but varied parameters (typically, ensembles are generated by NN submodels training on distinct subsets of data), and the dropout regularized neural network,299 a lower cost framework for deriving uncertainty estimates (randomly dropout some nodes each time). These NN models are highly dependent on the training data, and therefore the predicted variance may not be reliable if the test data is distinct from training data, as is commonly expected for CCS exploration. Another type of NN model based uncertainty metrics, widely adopted, may alleviate this deficiency, which employs distances in feature space (or some latent space) of the test data point to the current training data to provide an estimate of similarity measure and thus model applicability.300 This kind of approach enjoys several other advantages, such as easy interpretation, model independence, as well as potentially fast computation, but suffer from high dependence on the representation.300
6.3. AMON Based QML
Having a closer look at all the selection methods presented above, one notices that there is always some footprint of random sampling, i.e., one prerequisite for all those methods is a pre-existing starting training data, usually randomly selected, and reaching convergence of training data through iterative addition of new feature inputs may be slow if the starting points barely represent the space spanned by test data. The AMONs approach243 attempts to mitigate these shortcomings through selection of the “optimal” training set on-the-fly, i.e., only after having been provided a given specific query test feature input. In essence, AMON based QML exploits the locality of an atom in molecule which allows to reconstruct extensive properties, such as the ground-state energy, in some analogy to the nearsightedness of electronic systems.301,302 For the sake of a succinct discussion, we turn our attention to valence saturated system only and we neglect hydrogens. However, extension to other systems (e.g., system involving radicals, charges, conformational changes, vibrations, reactions, or noncovalent interactions) are also possible207,243). Note that throughout the whole process, we are only concerned with heavy atoms.
The AMONs selection procedure243 can be divided into four major steps: (i) The connectivity graph G of a query molecule is constructed using its 3D geometry. (ii) Next, all subgraphs are enumerated (the ith subgraph is labeled as Gi) of G with increasing number of heavy atoms (denoted as NI). For a given Gi, one performs a series of checks to see if it is a representative subgraph: (a) Is it a connected subgraph? (b) does subgraph isomorphism hold true? (c) Are all atoms valency-saturated after rationalization of the subgraph? And (d), is ring structure retained when all associated nodes are present in the current subgraph? If all of these criteria are met, then Gi is ready for further filtering and discarded otherwise. Criteria b is concise yet informative: subgraph isomorphism ensures that hybridization states of all atoms in the subgraph are retained, implying that bonds in query graph with bond order larger or equal to 2 are not allowed to break for fragmentation. (iii) Perform geometry relaxation for the corresponding fragment (now with valencies saturated with hydrogen atoms) using, for example, Universal Force Field (UFF)303 or other force-field optimizer with dihedral angles fixed to match the local geometry of the query molecule (to avoid conformational changes in local environments). This step is followed by geometry relaxation using some quantum chemistry program. At this stage, it may happen that the subgraph candidate dissociates (turning into a disconnected graph) or is transformed into a molecule with different connectivity. In the former case, the fragment should be discarded, while in the latter case, the subgraph isomorphism has to be rechecked. (iv) One proceeds if the subgraph candidate has experienced no change in connectivity or if subgraph isomorphism is retained despite there being change in connectivity. The resulting fragment is selected for the AMON database.
As the number of atoms in the subgraph increases, one continues looping through {Gi} until the set has been exhausted. The resulting set of AMONs is considered the query-specific “optimal” set which is representative of all local chemistries in the query molecule.
Figure 5 shows all AMONs for an exemplified QM9 molecule named 2-(furan-2-yl)propan-2-ol with AMON size (NI) being at most 7 by applying the above algorithm. Not surprisingly, there exist only two molecules possessing NI = 1, i.e., CH4 and H2O. For NI = 2, a C=C double bond is allowed to be cleaved from the 5-membered ring, forming a valid AMON H2C=CH2, as the resulting AMON retains its original coordination number for C’s, meanwhile keeping their valence saturated (i.e., meeting octet rule). While a fragment such as H2C–OH, also extracted from the ring, is not a valid AMON as the valence of C atom is not saturated. Repeating similar arguments for increasingly larger NI’s, we end up with only 30 AMONs, but which as a whole represent the complete set of local atomic environments present in the target and has the potential to extrapolate accurately the properties of the exemplified target QM9 molecule, as well as infinitely many other molecules that share the same set of AMONs after fragmentation.
Figure 5.
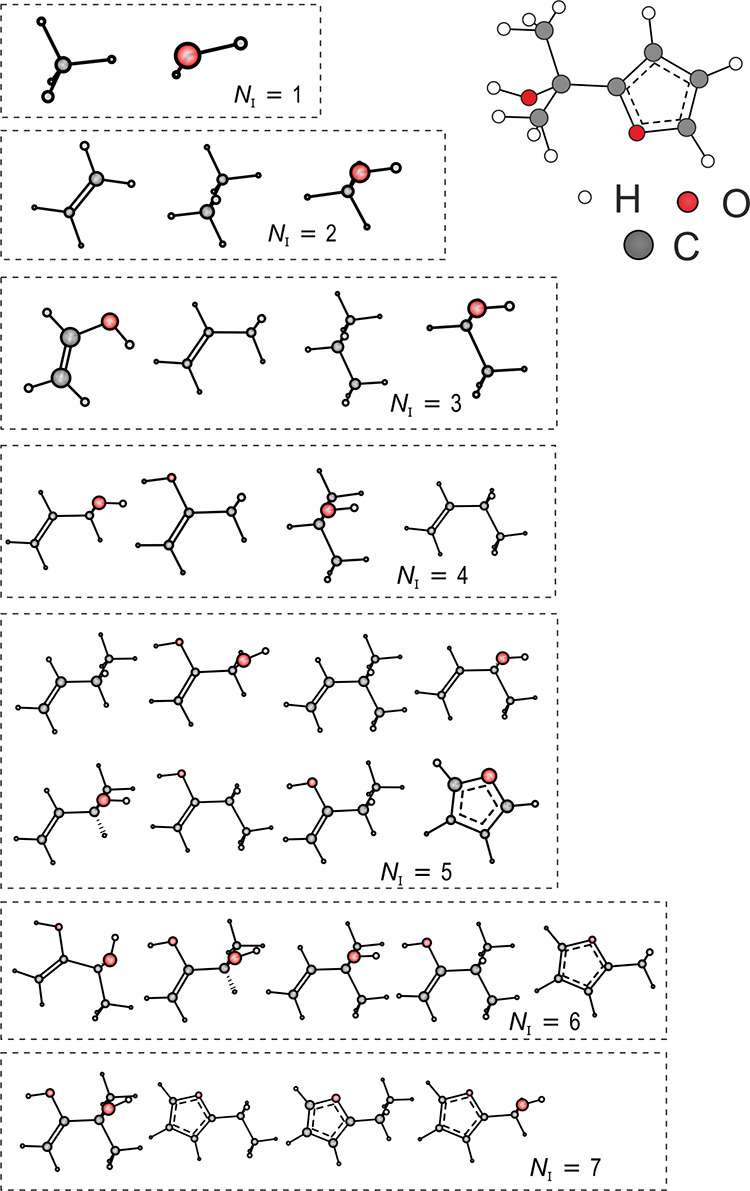
All AMONs sizes 1–7 for training system specific QML models of exemplary query molecule 2-(furan-2-yl)propan-2-ol (top right).
AMON based QML models exhibit improved slopes and offsets in learning curves, as evinced for thousands of molecules after reaching respective training set sizes of only ∼50 on average. By contrast, 20 times larger training set sizes are required using random sampling.243 One should note that graph based AMONs are not omnipotent. They are best suited for sampling chemical spaces of large systems. To extend AMONs to also handle configurational spaces is possible in principle, but not trivial as it faces challenges similar to modeling large systems without explicit graphs, such as metals, metal surfaces, or molecular crystals or liquids.
7. Properties
As we focus on supervised learning throughout this text, properties (or labels) of molecules have to be always paired with some molecular representation. Starting from regression of experimental properties, e.g., atomization energies, dipole moment, boiling point, in the early practises of machine learning, by now the scope of properties has been expanded significantly.
Because of its determining role for stability and dynamics, energy is among the most important properties, and it is also the primary target property of most studies. As early as in 2011,304 reorganization energies in a subspace of CCS consisting of polyaromatic hydrocarbons relevant to photovoltaic applications were already predicted using ML models. While the pioneering work on demonstrating the applicability of QML models for navigating CCS was published in 2012 for atomization energies only,195 a multiproperty neural network was published shortly after,220 covering not only atomization energies but also polarizabilities, molecular orbital eigenvalues, ionization potentials, electron affinities, and excited-states properties at various levels of theory. The correlations among these properties have confirmed some of the well-established physical principles as well as shown some interesting patterns. As illustrated in Figure 6, the ionization potential (IP) is well correlated with the HOMO energies, as expected from Koopman’s theorem; the polarizability is linked to the stability, as often implied by the hard–soft acid–base principle. Properties calculated at different levels of theory are strongly correlated, suggesting the possibility to exploit implicit correlations for the training of QML models with superior data efficiency. What is more interesting is that, for properties such as HOMO and atomization energies displaying little correlation, after training the neural network encodes some of the underlying and hidden correlations among these properties (box in Figure 6), indicating already in 2013 that neural network based QML models are amenable to “explainable AI”, as also illustrated subsequently in 2017 for effective atomic chemical potentials.305
Figure 6.

Property vs property matrix for ∼7k organic molecules at various levels of theory. A multiproperty neural net trained in CCS encodes underlying correlations as evinced by the first principal components of the last layer for 2k molecules not part of training. Reproduced with permission from ref (220). Copyritht 2013 licensed under a Creative Commons Attribution 3.0 license.
While QML commonly deals with properties which correspond to observables, other well-defined but more arbitrary labels can also be modeled. Examples include atomic charges or energies which do not have a unique definition. A more exotic application consists of successfully trained QML models of “time-to-solution” in terms of estimates of the number of iterations necessary to reach convergence for given initial conditions: In 2020, QML models of the computational cost of common quantum chemistry calculations have been demonstrated to enable optimal load-balancing and scheduling in ensemble calculations of high-throughput compute campaigns through CCS.306
To provide a more comprehensive perspective on the interesting subject of property, below we divide all properties into three main categories depending on the number of atoms/species involved: atomic property (atom/bond/functional group in a molecule), molecular property (the entire molecule), and intermolecular property (at least two molecular species). And within each section, we briefly review models of important properties in a rough chronological order. Note that the boundary between different categories is not clear-cut. For instance, the highest vibrational frequency of a molecule may be attributed to certain functional group but only in an approximate way, the exact value of which may still depend on all the other atoms in the molecule. For this and similar cases, we prefer to classify the relevant properties into atomic rather than molecular properties.
7.1. Atomic
Generally speaking, atomic properties are relatively easy to learn as they typically benefit the most from the general assumption of locality of an atom in a molecule. On the basis of the QM9 database, QML models were introduced for atomic properties, such as core level excitations, forces (see previous section), or NMR-shielding constants.64 Atomic QML models of electrostatic properties, such as atomic charges, dipole moments, or atomic polarizabilities, were introduced in 2015,250 and their use for the construction of universal noncovalent potentials was established in 2018.70 Deep neural networks for similar properties were also contributed in 2018 and 2019 by Unke and Meuwly.56,307 Information from topological atoms has also been used to build dynamic electron correlation QML models in 2017.308 In 2017 and 2018, atomic energies and potentials were also discussed in refs (166, 173, 309, and 310). QML models of polarizabilities based on tensorial learning were presented in 2020,311 and most recently, Gastegger and co-workers introduced external field effects within neural networks and demonstrated interesting performance for predictions of IR, Raman, and NMR spectra, as well as for continuum solvent effects on chemical reactions.210 Multiscale models of atomic properties have also been proposed.312
QML models of NMR shifts in molecules were first studied in 201564 and 2017,243 followed by shifts in solids in 2018,313,314 and NMR shifts in solvated proteins, coupling, a kaggle challenge, and an in-depth revisit of shifts in molecules were all contributed in 2020.315−318
In 2017, self-correcting KRR based models of potential energy surfaces and vibrational states were presented in ref (319) as well as neural network based molecular dynamics for the calculation of infrared spectra.320 Out of all QML models for properties studied in the 2017 overview study on the CCS of QM9,234 it was only for the highest vibrational (fundamental) frequency that random forests performed better than KRR or neural networks, the likely reason being that the QML model’s task consisted “only” of detecting if an O–H or N–H bond present on top of the C–H bonds, and to assign the typical corresponding bond-frequency, and that typically random forests work well for such classification tasks. Other 2018 studies dealing with infrared spectra include refs (207, 252, and 253).
7.2. Molecular
At the molecular level, properties are greatly diversified, ranging from properties for ground state to excited ones, from static to dynamic ones, as well as from single molecule in vacuum to condensed phase.
QML models of electronic properties, such as excited states, quantum transport, or correlation, have remained rather sparse over the years. Examples include QML models for electron transmission coefficients for transport across molecular bridges of varying composition,321 and Anderson impurity models322 in 2014, and dynamical mean field theory323 and excitation energies233 in 2015. Only recently QML has been extended to also study nonadiabatic excited states dynamics for given systems (conformational sampling) by Dral, Barbatti, and Thiel324 or Westermayr and Marquetand.83,325 And the recent introduction of SchNarc,326 a combination of the deep neural net architecture SchNet55 and the surface hopping ab initio molecular dynamics code SHARC,327 has led to promising first results for CCS studies involving small sets of small molecules.328 For more details we refer to the recently published reviews on this field.329−331
QML models of electron affinities and ionization potentials with deep neural networks have also recently been proposed.332 Symmetry conserving neural networks for efficient calculations of electronic and vibrational spectra have been presented in 2020.333
7.3. Intermolecular
As the system becomes more complicated, the associated properties also tend to show more interesting, and sometimes surprising patterns. Hereafter, we will focus on energetic properties, unless otherwise stated. Depending on whether or not the system has experienced significant reconstruction in the relative orientation between atoms, intermolecular energetics could be further divided into intermolecular binding energy or reaction energy/barriers. Below, we summarize relevant contributions for each of these two subcategories.
In terms of binding energies within assemblies of atoms, ever since the publication of ref (195) in 2012, a large variety of systems has been addressed, reaching from formation energy predictions of diverse inorganic materials,164,166,334 over models of chemical bonds in molecules,335 to models of electronic properties of transition metal complexes.336 GPR/KRR based QML models represent a unified approach, as demonstrated for applications to surface reconstructions, organic molecules, as well as protein ligands.244 Symmetry adapted learning of tensorial properties was introduced in 2018,337 as well as neural networks for atomic energies,310 on-the-fly learning for structural relaxation,80 crystal graph convolution networks for materials properties,338 solvation and acidity in complex mixtures,339 and a machine learning based understanding of the chemical diversity in metal–organic-frameworks.340 An extensive review of big data in metal–organic frameworks was also published in 2020.341
Accurate QML prediction of reaction related properties, the reaction barrier in particular, is a difficult task, as typically off-equilibrium configurations are involved, and the training space is undersampled.
The use of QML models to investigate properties relevant for catalysis represents another major domain of research. A GPR model was used in 2016 to estimate free energies of possible adsorbate coverage for surfaces in order to accelerate the construction of Pourbaix diagrams.342 In 2017, Ulissi et al. introduced a neural network based exhaustive search enabling the identification of active site motifs for CO2 reduction,343 as well as a GPR based estimator of adsorption energies for identifying the most import reaction step.344 QML models of reaction barriers of elementary reactions (using 236 dehydrogenation, 38 N2 dissociation, and 41 O2 dissociation examples) on surfaces were proposed by Singh et al. in 2019.345 Quantum machine learning based design of homogeneous catalyst candidates was presented in 2018.346 In 2020, QML models of competing reaction barriers and transition state geometries corresponding to SN2 and E2 reactions in the gas phase were successfully trained and applied throughout a CCS covering thousands of reactants,347 relying on the QMrxn data set.348 That same year, Bligaard and co-workers employed active learning to identify stable iridium oxide polymorphs and study their usefulness for the acidic oxygen evolution reaction,349 introduced a Bayesian framework for adsorption energies of bimetallic alloy catalyst candidates,350 and proposed a bond information based GPR as a means to speed up structural relaxation across different types of atomic systems.351 In 2020, neural networks have been proposed for the prediction of overpotentials relevant for heterogeneous catalyst candidates,352 as well as a higher-order correction scheme in alchemical perturbation density functional theory applications to catalytic activity.260 An overview on machine learning for computational heterogeneous catalysis was also contributed in 2019.353
8. Data Sets
As implied already in previous sections, the availability of training sets is vital for any machine learning. Admittedly, it would be ideal to generate training set only when necessary, i.e., to minimize the number of QM computations throughout CCS, or for converging the sampling using molecular dynamics. However, for general applications of QML, a pre-existing data set is indispensable, for instance, to tackle the inverse design problem to identify some compound with unknown composition and exhibiting specified and desirable ground-state physiochemical properties. Currently, this is only feasible with a given labeled data set being as representative as possible for the local chemistries that we know to affect the properties of interest.
Alongside the increasing popularity of QML in chemistry and related sciences, many data sets have emerged in recent years. By now, there are a multitude, built for various purposes. Here we detail all those data sets we know of that encode quantum information throughout CCS, with a coarsened and incomplete overview given in Table 1.
Table 1. Overview: Synthetic Quantum Data Sets in Three Data Families of Chemical Compound Space: Generated Data Base (GDB33,354,361), Transition Metal Complexes (TMC), and Periodic Systems (Crystalline Solids or Surfaces)a.
| family | data set | composition | size | method | properties | year | notes |
|---|---|---|---|---|---|---|---|
| GDB | QM7386 | C, O, N, S | 7165 | PBE0 | E | 2012 | |
| QM7b359 | C, O, N, S, Cl | 7211 | PBE0, ZINDO, GW | E, ε, α, E*, etc. | 2013 | ||
| QM9171 | C, O, N, F | 134k | B3LYP/6-31G(2df,p) | E, μ, α, ε, Pthermo, etc. | 2014 | ||
| QM8364 | C, H, O, N, F | 20k | TDDFT, CC2/def2-TZVP | E*, f1, f2 | 2015 | excited state | |
| ANI-1367 | C, O, N, F | 20M | w97x/6-31G(D) | E | 2017 | off-equilibrium | |
| QM7bMl267 | C, O, N, S, Cl | 7211 | {HF,MP2,CCSD(T)}/ {sto-3g, 6-31g, cc-pVDZ} | E | 2018 | multifidelity QML | |
| Alchemy363 | C, N, O, F, S, Cl | 119k | B3LYP/6-31G(2df,p) | E, μ, α, ε, Pthermo, etc. | 2019 | ||
| QM7-X360 | C, H, O, N, S, Cl | 4.2M | PBE0+MBD | E, f, ε, μ, α, qA, C6, etc. | 2020 | off-equilibrium | |
| ANI-1x368 | C, O, N, F | 5M | w97x/def2-TZVPP and CCSD(T)/CBS | E, f, μ, qA, etc. | 2020 | off-equilibrium | |
| AGZ7366 | B, C, N, O, F, Si, P, S, Cl, Br, Sn, I | 140k | B3LYP/cc-pVTZ | E, μ, α, ε, Pthermo, etc. | 2020 | ||
| TMC | tmQM383 | 3d, 4d and 5d transition metals, B, Si, N, P, As, O, S, Se, halogens | 86k | TPSSh-D3BJ/def2-SVP | E, μ, qA, ε, etc. | 2020 | GFN2-xTB geometry |
| (MIT)384,387 | Cr, Fe, Mn, Co, Ni, C, N, O, S, Cl | >2M | B3LYP/LANL2DZ (6-31g*) | E, ΔEH–L, redox potential | 2017, 2020 | ||
| periodic | Materials Project165 | across periodic table | >600k | PBE | E, electronic and response properties | 2011 | |
| AFlow388 | across periodic table | 3M | PBE | E, electronic and response properties | 2012 | ||
| OQMD389 | across periodic table | 300k | PBE | E, electronic and response properties | 2013 | ||
| OC20390 | across periodic table | >1M | RPBE | E, Eads | 2020 | ||
Properties covered include E (total energy (or atomization energy)), f (atomic forces), qA (atomic charges), μ (dipole moments), α (polarizability), ε (eigenvalues), E* (excitation energy), fi: oscillation strength for transition from ground state to the ith excited state (i = 1 or 2), ΔEH–L (high- and low-spin energy difference), C6 (London dispersion coefficients), Pthermo (thermochemical properties such as internal energies, enthalpy, free energy, and heat capacity); Eads (chemisorption energy).
8.1. GDB
The synthetic GDB (generated database) data sets created by Reymond and co-workers for the main purpose of exploring the CCS of organic drug-like molecules comprise the probably largest list of systematically generated molecular graphs (constitutional and compositional isomers only) of small to medium sized organic molecules of biochemical relevance.182,354−356 To date, GDB17182,356 represents the single largest set of molecules, which contains more than 166 billion molecules made up of H, C, N, O, S, and halogens (up to 17 non-hydrogen atoms), obeying certain chemical rules for stability and synthesizability. GDB17 has two main subsets, GDB11 (26M)354,357 and GDB13 (970M),355 together with a variety of smaller subsets featuring specificity of organic chemistry. Because of its systematic enumeration, interesting new structures have been identified and subsequently been synthesized, as exemplified by the synthesis of trinorbornane.358
Other than the implicit information that any compound listed corresponds to a stable constitutional isomer, the original GDB data sets are unlabeled in the sense that only molecular composition and connectivity information are detailed, without calculated quantum properties. The first extension of the GDB data set to also include quantum data, QM7195 consists of 7165 ground-state geometries and energies of molecules with up to 23 atoms (with up to 7 heavy atoms C, N, O, or S) calculated at the PBE0 level. QM7 is also the first quantum benchmark data set covering the organic subspace CCS for QML. Some extensions exists, such as QM7b,359 QM7b multilevel data set267 (QM7bMl for short), and QM7-X.360
QM7b359 extends QM7 by including chlorine-containing molecules (expanding the set size to 7211), and reporting 13 additional calculated electronic properties (e.g., polarizability, HOMO/LUMO energies, excitation energies). QM7bMl267 was designed for studying QML combinations with legacy quantum chemistry methods such as multilevel, multifidelity, or transfer learning. Starting from the original coordinates at PBE level, geometries of QM7b molecules were refined at the level of B3LYP/6-31G(D), and subsequently single-point energies were calculated at nine levels of theory, corresponding to all possible combinations of electron correlation treatment {HF, MP2, CCSD(T)} and basis sets {STO-3G, 6-31G, cc-pVDZ}). QM7-X, the largest extension of QM7, is a comprehensive data set comprising ∼4.2 M equilibrium and nonequilibrium structures of QM7b molecules, accompanied by 42 physicochemical properties computed at the PBE0+MBD level, covering global (molecular) and local (atom-in-a-molecule) properties ranging from ground-state quantities (such as atomization energies and dipole moments) to response quantities (such as polarizability tensors and dispersion coefficients).
Because of the limited molecular size, QM7 and its variants are scarcely scattered across CCS and barely begin to represent its full diversity and complexity. Targeting “big data” Ramakrishnan et al. released the QM9171 data set in 2014, derived from molecular graphs drawn from GDB17,361 totalling ∼134k organic molecules made up of C, H, O, N, or F, and up to nine non-hydrogen atoms. Except for equilibrium geometries and electronic ground-state properties, QM9 also records a series of thermochemical properties at 298 K and 1 atm pressure estimated based on harmonic frequencies, namely enthalpies, and free energies of atomization at the level of B3LYP/6-31G(2df,p). Alongside, additional QM data is reported for the subset of all of QM9’s 6k constitutional isomers with sum formula C7H10O2, i.e., thermochemical properties computed at the G4MP2 level. In 2020, QM9 was augmented by more accurate energies, calculated at multiple levels of theory, including M06-2X, wb97xd, and G4MP2.362 Another similar data set, dubbed alchemy363 (sized 119 487) expands the volume and diversity of QMx series and is made up of 9–14 C, N, O, F, S and Cl atoms, sampled from the GDB MedChem subset of GDB17.356
The only data set that deals with excited-state properties across CCS is QM8,364 totalling ∼20k structures subsampled from QM9 and comprising up to eight heavy atoms C, O, N, or F. Ground-state energies (S0) and the lowest two vertical electronic singlet–singlet excitation energies (S1 and S2) are included, calculated at two TDDFT levels employing the density functional theory/basis-set combination PBE0/def2-SVP or CAM-B3LYP/def2-TZVP, as well as post-Hartree–Fock level CC2/def2-TZVP. Corresponding oscillator strengths (f1) for each transition from S0 to S1 have also been recorded.
As also evinced for GDB17, when increasing the number of atoms per molecule, the data set quickly grows out of control, and it becomes prohibitive to conduct QM calculations for comprehensive subsets of CCS. The Amon based dictionary of building blocks designed to cover GDB361 and Zinc365 and containing no more than seven heavy atoms (AGZ7) has been introduced to alleviate this curse of dimensionality.366 It was obtained by systematically fragmenting all larger molecules (from GDB17 and zinc365) into smaller entities containing no more than seven non-hydrogen atoms (i.e., atom-in-molecule based fragements, aka, AMONs243). To date, AGZ7 is the most compact yet most diverse data set relevant for organic/biochemistry, totalling only 140k molecules but covering up to 13 elements (H, B, C, N, O, F, Si, P, S, Cl, Br, Sn, and I). It also includes a similar set of properties as in QM9 but relying on a slightly different level of theory (B3LYP/cc-pVTZ as well as pseudopotentials for Sn and I).
Apart from QM7-X,360 all data sets mentioned so far deal with equilibrium geometries only, representing the typical constraint for what defines a stable molecule. To enable the QML based study of dynamics and reactivity of nonequilibrium geometries throughout CCS, however, configurational sampling involving nonstationary geometries has to also be accounted for through the data sets. Similar to QM7-X, ANI-1367 also explores nonequilibrium geometries but for relatively larger systems drawn from GDB11.354,357 It consists of more than 20 M off-equilibrium structures (sampling both chemical and conformational degrees of freedom) and wB97x/6-31G(d) energies for 57 462 small organic molecules containing up to 11 CONF atoms. Two follow-up data sets expand ANI-1 considerably, i.e., ANI-1x and ANI-1ccx.368 The former contains multiple QM properties (density-derived properties and forces) from 5 M DFT calculations (wB97x/6-31G* and wB97x/def2-TZVPP), while the latter contains 500k CCSD(T) energies for estimated CBS limits.
For MD simulations, two main data sets are being frequently benchmarked. One is ISO17,55,309 containing MD trajectories of 129 molecules randomly drawn from the aforementioned 6k C7O2H10 isomers, each comprising 5000 conformational geometries with total energies and atomic forces calculated at PBE level plus van der Waals correction.369 The other is MD-17,73,370 which records energies and forces from ab initio molecular dynamics trajectories (133k to 993k frames) at the DFT/PBE+vdW-TS level of theory at 500 K for eight organic molecules: benzene, uracil, naphthalene, aspirin, salicylic acid, malonaldehyde, ethanol, and toluene. More accurate CCSD(T) energies and forces are also available but only for ethanol (with basis cc-pVTZ), toluene and malonaldehyde (cc-pVDZ), and CCSD/cc-pVDZ for aspirin. Recently, a revised MD-17 data set was published,211 with a lower noise floor in DFT forces thanks to tighter SCF convergence criteria and denser integration grids. In 2020, G4MP2 benchmarks of organic molecules with up to 14 non-hydrogen atoms were contributed by Dandu et al.,371 and resulting QML models were compared and discussed.
8.2. PubChem amd ZINC
While the GDB family currently dominates QML campaigns, GDB compounds resulted from virtual exhaustive graph enumeration campaigns and mostly correspond to molecules for which neither thermodynamics stability nor synthesizability has been established. Within practical applications, such aspects matter for the experimental design and fabrication of new chemical compounds. With respect to QML, some theoretically possible local chemical environments may not be viable within the entire molecular framework, and ruling out such possibilities when training could help to further improve data efficiency and transferability. PubChem372 is an ever-growing open chemistry database hosted at the National Institutes of Health (NIH). As of October 2020, there were over 111 million unique chemical structures records listed together with many a experimental property, as contributed by hundreds of data sources. To harvest the richness and popularity of this database, Maho Nakata, and co-workers lauched the so-called PubChemQC project,373 consisting of ground-state geometries and properties (at B3LYP/6-31G* level), as well as low-lying excited states of approximately four million molecules via time-dependent DFT at the level of B3LYP/6-31+G*. A PubChemQC derived subset, called pc9,374 covering over 99k molecules made up of CHONF was published afterward and encoded the same set of properties as QM9. The full potential of PubChemQC remains yet to be generally explored.
ZINC,365 yet another large database, focuses more on biochemistry, in particular drug design. Quantum calculations on this database per se have not taken place, except for its associated fragment set. That is, AZ7, a subset of AGZ7,366 contains all ZINC AMONs of up to seven non-hydrogen atoms (with optimized geometries and electronic properties, as for AGZ7 described above). AGZ7 could be considered as an effective set covering all local chemistries of ZINC and may serve as a scaffold for building larger drugs through a theoretical approach.
Beside PubChem and ZINC, there are several other public big databases being exploited within QML. One of them is the Cambridge Structural Database (CSD),375 on the basis of which Stuke et al.376 reported a diverse benchmark spectroscopy data set of 61 489 molecules, denoted OE62. Using geometries optimized by PBE plus vdW correction, OE62 provides total energies and orbital eigenvalues at PBE and PBE0 levels for all molecules in vacuum and at the PBE0 level for a subset of 30 876 molecules in (implicit) water. Also based on CSD, Schober et al.377 extracted 95 445 molecular crystals thereof and carried out computations on electronic couplings (at the level BLYP and fragment molecular orbital-based DFT) and intramolecular reorganization energies (by QM/MM with an ONIOM-scheme) as two main descriptors for charge mobility, hoping to facilitate the theoretical design and discovery of high mobility organic semiconductors.
8.3. Barriers and Spin
Quantum data sets on chemical reaction profiles are rather scarce. The QMrxn348 reports calculated quantum properties for SN2 and E2 reactions amounting to 4466 transition state and 143 200 reactant complex geometries and energies at MP2/6-311G(d) and single-point DF-LCCSD/cc-pVTZ level of theory, respectively. QMrxn covers the subset of CCS that is spanned by the substituents −NO2, −CN, −CH3, −NH2, and with −H,−F, −Cl, and −Br as nucleophiles and leaving groups. A different data set featuring elementary reactions comes from Grambow et al.,378 totalling 12k organic reactions that involve H, C, N, and O atoms, calculated at the wB97X-D3/def2-TZVP level, with optimized geometries and thermochemical properties for reactants, products, and transition states.
Going beyond mostly singlet-state chemistry, Schwilk et al. introduced QMspin,379 consisting of ∼5k (∼8k) singlet (triplet) state carbenes derived from 4k randomly selected QM9 molecules. QMspin also contains optimized geometries (B3LYP/def2-TZVP for triplet state and CASSCF(2e,2o)/cc-pVDZ-F12 for singlet state), as well as the singlet–triplet vertical spin gap computed at MRCISD+Q-F12/cc-pVDZ-F12 level of theory.
For the QML models of the computational cost of typical quantum chemistry computations (measured by the CPU wall time), Heinen et al. reported the QMt data set,380 consisting of timings of various tasks (single point energy, geometry optimization, and transition state search) for thousands of QM9 molecules at several levels of theory including B3LYP/def2-TZVP, MP2/6-311G(d), LCCSD(T)/VTZ-F12, CASSCF/VDZ-F12, and MRCISD+Q-F12/VDZ-F12.
Treating noncovalent interaction (NCI) within QML is an interesting and important research subject, with relevant large data sets emerging only as of recently. Most notably, several collections of NCI data sets have by now become publicly available,381 covering 3700 distinct types of interacting molecule pairs: (i) DES370 K, contains interaction energies for more than 370k dimer geometries with NCI energy calculated at the level of CCSD(T)/CBS (MP2(aVTZ, aVQZ) correlation energy is used for extrapolation, and (ii) DES5M, comprising NCI energies calculated using SNS-MP2, for nearly 5 M dimer geometries. The monomers involved include typical organic species, made up of common p-block elements as well as alkali metal ions, most of which containing no more than seven heavy atoms.
Data sets including artificial molecules which violate basic principles of chemical bonding may also of great interest for QML, i.e., they may serve the use of “soft” labels, where relatively few compounds might more effectively represent CCS than selected many. MB08-165,382 proposed by Grimme, exemplifies that idea, relying on systematic constraints rather than uncontrolled chemical biases. Originally, this data set was designed for benchmarking DFT methods. The potential of such “unbiased” artificial molecules as soft labels (training set) in QML has yet to be unraveled.
8.4. Transition Metals
Transition metal complexes (d-block atom/ion center plus ligands, TMC for short) are pervasive in chemistry and have been widely used and studied. Because of their complicated electronic structure and the resulting higher computational cost (in comparison to typical organic molecules), the effective exploration of the chemical space spanned by TMCs remains a challenge and current efforts into this subspace are constrained to relatively low level of theory, primarily DFTB or DFT method with some small basis. Examples include tmQM383 and the TMC data sets289,336,384 from Kulik and co-workers, as described below.
tmQM383 contains geometries and common electronic properties (as for QM9) of 86 665 mononuclear complexes extracted from the Cambridge Structural Database (CSD). tmQM includes Werner, bioinorganic and organometallic complexes based on a large variety of organic ligands and 30 transition metals. On the basis of the DFTB(GFN2-xTB) geometry, common quantum electronic properties (orbital energies, dipole meoment and atomic charges) were computed at the TPSSh-D3BJ/def2-SVP level.
The largest and most comprehensive TMC data sets are from Kulik’s group at MIT and have been contributed across multiple publications.289,336,384 Overall, they correspond to combinations of several metal centers (Cr, Mn, Fe, or Co, Ni) and a wide range of ligands, ranging from weak-field chloride (Cl–) to strong-field carbonyl (CO) along with representative intermediate-field ligands and connecting atoms, including S (SCN–), N (e.g., NH3), and O (e.g., acetylacetonate). Calculated properties are primarily energetic, including total energy, high and low spin-state energy difference (ΔEH–L), and redox potential and solubility in candidate M(II)/M(III) redox couples, at the level of theory B3LYP/LANL2DZ (6-31G* for ligands) with or without polarizable continuum model (PCM) for solvents. The total size could reach up to several millions.
Recently introduced metal–organic frameworks (MOF) data set by Rosen and co-workers,385 called Quantum MOF (QMOF), represent another broad category of metal complexes. QMOF consists of computed properties (energy, band gap, charge density, and density of states) at the PBE-D3(BJ) level of theory, for more than 14 000 experimentally synthesized MOFs, which are made up chemical elements that span nearly the entire periodic table.
8.5. Solid and Solid Surface
Compared to TMCs, solid and solid surfaces present a challenge on their own due to the diversity in composition and spatial arrangements, as well as the resulting complexity of electronic structure. Typically DFT based methods are used for generating large-scale (or high-throughput) data sets for these systems. The most frequently used method is GGA (PBE) or GGA+U with PAW (projected augmented wave) potentials. On the basis of relaxed geometry, associated calculated properties fall into either electronic properties, e.g., cohesive energy, band structure (and derived properties including density of states and band gap), or response properties such as elastic tensor, bulk modulus, and thermodynamic properties (vibrational spectra, free energy, specific heat, and entropy) within harmonic approximations.
Relevant well-known solid databases and compute platforms include (i) AFlow,388 an open data set of more than 3 M material compounds (including alloys, intermetallics, and inorganic compounds) with over 596 M calculated properties. (ii) The Open Quantum Materials database389 (OQMD), a high-throughput database currently consisting of nearly 300k total energy calculations of compounds from the Inorganic Crystal Structure Database (ICSD). (iii) The Materials Project165 (www.materialsproject.org) covers the properties of almost all known inorganic materials, currently containing over 131k inorganic compounds and more than 530k nonporous materials. (iv) The Materials Cloud (www.materialscloud.org),391 a platform designed to enable open and seamless sharing of resources for computational science, driven by applications in materials modeling. (v) The Novel Materials Discovery (NoMaD, http://nomad-repository.eu), led by Scheffler, Draxl et al. (vi) The Open Materials Database (http://openmaterialsdb.se, currently under development) spearheaded by Armiento. The latter both are public archives for hosting, sharing, and reusing material data in their raw form. Apart from comprehensive public repositories for solid data sets, there are also select contributions for select materials classes, including the aforementioned data set of ∼10k AB2C2 elpasolites covering all main-group elements up to Bi from Faber et al.164
Regarding solid surfaces, the new Open Catalyst Project390 aims to help discover and design new catalysts for renewable energy storage using ML (https://opencatalystproject.org), currently including mainly the OC20 data set,390 consisting of >1 M relaxations (over 26 M single point evaluations) at RPBE level for a wide range of adsorbates (C-, N- and O-containing species) and surfaces.
9. Software Packages
To perform and supplement the aforementioned studies with methods and data sets, numerous software packages have been developed over recent years. We briefly mention the available codes and categorize them into three main types, the first of which being those related to the acceleration of legacy quantum codes, such as ab initio molecular dynamics (MD) runs in VASP,392 Gaussian process based geometry optimization in ASE,393 machine learning adaptive basis sets within CP2K,238 as well as SNAP394 in LAMMPS, a machine-learning interatomic potential using bispectrum components to characterize the local neighborhood of each atom of the system.
Codes which fall into the second category are standalone packages, some of which having also been interfaced to other atomistic simulation software. QMLcode209,395 which is an open-source python-based package featuring the Coulomb-matrix,195 BoB,170 SLATM/aSLATM,243 FCHL18, and FCHL19166,209 and other representations. AQML code396 is a variant of QMLcode featuring the BAML163 representation, and on-the-fly selection of AMONs for training.243 PLUMED397 is an open-source, community-developed library that provides a wide range of methods including enhanced-sampling algorithms, free-energy methods, and MD data analysis capabilities. It also interfaces with some of the most popular MD engines. TensorMol398 is a package of neural networks for chemistry, capable of running many common tasks in quantum chemistry such as geometry optimizations, molecular dynamics, Monte Carlo, nudged elastic band calculations, etc. It can also take into account screened long-range electrostatic and van der Waals interactions. TorchANI399 is a PyTorch implementation of ANI. It can compute molecular energies, gradients, Hessian and derived properties from the 3D coordinates of molecules. It also include tools to work with ANI data sets (e.g., ANI-1, ANI-1x, etc.).
The third category of software packages deals predominantly with data set construction, management and analysis. In particular, specific platforms include AFlow388 which has been mentioned above in section 8.5, and AiiDA,400 an open-source infrastructure for automation, management, sharing and reproduction of the workflows associated with big data in computational sciences.
10. Compound Discovery
The computational design and discovery of new compounds can be generally conducted following either one of two distinct approaches. The Edisonian and more basic one is straightforward, within a brute-force high-throughput screening, through solving Schrödinger equations sequentially or in parallel for potential materials candidates one by one, followed by subsequent ranking and selection. Given sufficient coverage and having used the data for training, the ab initio solver could successively be replaced by QML models, capable of making faster and equally accurate predictions of target properties of interest. It is obvious that such an approach suffers from limited domains of compounds conceived in the first place, no matter what solver is used for computation of properties. Also, as the intended search domain expands in CCS, the number of possible potential candidates will grow combinatorially. Therefore, when adopting this strategy, one should refrain from generally expanding the search domain and rather focus on a constrained subdomain of compounds, sharing one or more common features, e.g., the same stoichiometry and space group, as was exemplified for the elpasolite family ABC2D6 by Faber et al.,164 where compounds with exotic atomic oxidation states were identified.
The second more sophisticated approach attempts to solve the problem in an inverse fashion; more specifically, given a specific (range of) value(s) for the target property, how to best locate the corresponding optimal (set of) compound(s) from CCS. One particularly promising variant is the gradient-based inverse design,14 which can be reformulated as a global optimization problem and has the potential to search chemical subspace for substantial domains, due to its analytical nature. Strictly speaking, almost all current ML-guided studies (mostly neural network based) on gradient-based inverse design (e.g., ref (187), for a review, see ref (287)) fall into the QSPR regime, as the input is seldomly 3D geometry, but rather SMILES or other molecular graph derived features (therefore, the mapping from representation to property is not unique). This strategy is however the only attainable way by now, as otherwise (i) the search subspace (when optimizing for the “optimal” compound) would become overwhelmingly large due to the explosion of conformational degrees of freedom (Levinthal’s paradox). (ii) There exists, to the best of our knowledge, no 3D geometry-based representation that is compact enough for decoding, i.e., restoring the original geometry from its representation vector/matrix/tensor (or simply x), even with the help of a neural network model like variational encoder (VAE), as the entries in x are highly intertwined (significantly more so than the SMILES string). The fact that many representations are still being haunted by the uniqueness issue, further plagues these efforts, as often only two- and three-body terms are included in distribution-based representation. While inclusion of four-body terms are mandatory for reconstructing geometry, as evinced by the Z-matrix representation of geometry, the resulting x would become very expensive for generation, and more importantly, this could further perplex the feature vector decoder. However, representing a molecule in its most native form in terms of nuclear charges and coordinates,{Z; R}, i.e., by the variables employed in the electronic Hamiltonian, or some transformed form, such as an external potential, one is free from such problems. This strategy would be consistent with the aforementioned GCE and LCAP approach detailed in section 1.3.
11. Outlook and Conclusion
While QML is still in its infancy, very encouraging progress has already been achieved. It is still a long way, however, before we will reach the goal of routinely designing and discovering novel molecules and materials on a computer. Some of the most fundamental problems, also among the most common tasks in quantum chemistry calculations, such as correctly predicting ground-state energy and forces of novel molecules or materials with high efficiency and accuracy, still remain unresolved at large. Such seemingly simple tasks are particularly challenging when it comes to systems that are highly distorted, charged, or multireference in nature or that involve long-range nonbonded interactions. Successful QML models could easily demonstrate their applicability by energy ranking of competing structures of real materials. We believe that such tasks will be crucial for subsequent more challenging QML applications.
Another interesting path to pursue might be the integration of alchemical perturbation theory into QML. Because the alchemical problem could be essentially reformulated as a ML problem that involves both energy and energy gradient with respect to nuclear charges. A corresponding extension would exploit similarities between alchemical interpolations in pseudopotential parameter space and compositional representations that explicitly account for group and period in the periodic table, on top of all the structural degrees of freedom. Within the FCHL representation,166 preliminary results for inferring properties of chemical elements absent in training have already been obtained (see Figure 4).
Besides the curse of dimensionality, imposed by the compositional, constitutional, and conformational diversity of CCS, the lack of a more theoretical underpinning of the genesis of data sets is maybe among the most severe shortcomings. Little, if nothing, is known about fundamental questions such as: (i) Are there any basic quantities characterizing the completenes of a molecular data set, for instance in terms of diversity and/or sparsity? (ii) On the basis of the inherent properties of the data set, representation and regressor, can we infer the performance of a model without actual training/test runs, as translated into the slope and offset of resulting learning curve. (iii) What, if any, characterizes the “correct” distribution in CCS. Answering such questions rigorously, i.e., based on the laws of physical chemistry, is not only of conceptual importance but would also benefit the practical design of more efficient/accurate QML models.
Other unresolved issues include (i) the lack of appropriate QML models that deal with intensive properties such as HOMO/LUMO energy, or dipole moments, which may require careful consideration of both local and long-ranged features of a molecule. (ii) The lack of high-accuracy data sets at the level of experimental quality (e.g., CCSD(T)-F12/CVQZ-F12 or multireference) for medium-sized molecules: published data sets of such quality are still limited to very few or small molecules, containing typically no more than three heavy atoms.
As the field has been growing massively and rapidly,16 we can unfortunately not guarantee completeness of our outlook. Furthermore, several related important new research directions, i.e., going beyond the mere supervised learning problem of the electronic Schrödinger equation, possibly being out of the scope of “conventional” QML, have not been mentioned. They include, for example, variational autoencoders which can be used to help solving the inverse design challenge in CCS (e.g., applied to the design of improved molecular electronics401), the reconstruction of quantum states,402 or the generation of molecular structures.403 Other intriguing efforts deal with tackling the problem of reaction planning,404−409 phase diagrams,410−415 studying the electronic structure in more depth and detail,261,416−419 or the systematic incorporation of experimental information in order to improve experimental design.420
To recap, we have provided succinct explanations and pointers to three major ingredients of QML: representation, regressor, and training set. We have briefly discussed select relevant studies which deal with the development and use of surrogate machine learning models of quantum properties throughout CCS. One of the primary goals of QML, i.e., rational computational discovery and design of compounds with desired properties, however, has not yet been achieved in general, and most of the relevant studies are either conducted in a high-throughput fashion, merely accelerated by QML, or rely on coarsening the problem through neglect of relevant degrees of freedom. We have pointed out several open questions and challenges that must be overcome to reach this general goal, as well as potential solutions, and suggestions about interesting new research directions. Given the overall rapid growth and the multiple success cases already achieved in this young field, we are optimistic about its future and strongly believe that QML will develop into a helpful component for solving some of the long-standing problems in the atomistic sciences.
Acknowledgments
O.A.v.L. acknowledges support from the Swiss National Science Foundation (407540_167186 NFP 75 Big Data) and from the European Research Council (ERC-CoG grant QML and H2020 projects BIG-MAP and TREX). This project has received funding from the European Union’s Horizon 2020 research and innovation programme under Grant Agreements no. 952165 and no. 957189. This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement no. 772834). This result only reflects the author’s view, and the EU is not responsible for any use that may be made of the information it contains. This work was partly supported by the NCCR MARVEL, funded by the Swiss National Science Foundation. We thank J. Wagner and F. A. Faber for helping with the design of Figures 1 and 2.
Biographies
Bing Huang (Hubei, China, 1987) was initially trained in physical chemistry under the supervision of Prof. Lin Zhuang in Wuhan University and completed his Ph.D. there in 2015, investigating and developing reactivity theory concerning solid surface. Afterwards, he moved to Basel, Switzerland, to work as a postdoc with Anatole von Lilienfeld at the Department of Chemistry, University of Basel, shifting research interests to the development of machine learning models and methods in quantum chemistry to explore chemical compound space. As of 2020, he has relocated to Vienna, Austria, to continue his postdoctoral research with Anatole von Lilienfeld at the Faculty of Physics, University of Vienna. His research interests include electronic structure theory, chemical reactivity theory, theoretical surface science and quantum machine learning.
O. Anatole von Lilienfeld (Rochester, Minnesota, USA, 1976) is a full university professor of computational materials discovery at the Faculty of Physics at the University of Vienna. Research in his laboratory deals with the development of improved methods for a first principles based understanding of chemical compound space using perturbation theory, machine learning, and high-performance computing. Previously, he was an associate and assistant professor at the University of Basel, Switzerland, and at the Free University of Brussels, Belgium. From 2007 to 2013, he worked for Argonne and Sandia National Laboratories after postdoctoral studies with Mark Tuckerman at New York University and at the Institute for Pure and Applied Mathematics at the University of California Los Angeles. In 2005, he was awarded a Ph.D. in computational chemistry from EPF Lausanne under the guidance of Ursula Röthlisberger. His diploma thesis work was done at ETH Zürich with Martin Quack and the University of Cambridge with Nicholas Handy. He studied chemistry at ETH Zürich, the Ecole de Chimie Polymers et Materiaux in Strasbourg, and the University of Leipzig. He serves as Editor-in-chief of the IOP journal Machine Learning: Science and Technology and on the editorial board of Science Advances. He has been on the editorial board of Nature’s Scientific Data from 2014 to 2019. He was the chair of the long IPAM UCLA program “Navigating Chemical Compound Space for Materials and Bio Design”, which took place in 2011. He is the recipient of multiple awards, including a Swiss National Science Foundation postdoctoral grant (2005), a Harry S. Truman postdoctoral fellowship (2007), a Thomas Kuhn Paradigm Shift award (2013), a Swiss National Science professor fellowship (2013), the Odysseus grant from the Flemish Science Foundation (2016), an ERC consolidator grant (2017), and the Feynman Prize in Nanotechnology (2018).
The authors declare no competing financial interest.
References
- Rupp M. Special issue on machine learning and quantum mechanics. Int. J. Quantum Chem. 2015, 115, 1003–1004. 10.1002/qua.24955. [DOI] [Google Scholar]
- Rupp M.; von Lilienfeld O. A.; Burke K. Guest Editorial: Special Topic on Data-Enabled Theoretical Chemistry. J. Chem. Phys. 2018, 148, 241401. 10.1063/1.5043213. [DOI] [PubMed] [Google Scholar]
- Schneider W. F.; Guo H. Machine Learning. J. Phys. Chem. A 2018, 122, 879. 10.1021/acs.jpca.8b00034. [DOI] [PubMed] [Google Scholar]
- Prezhdo O. V. Advancing Physical Chemistry with Machine Learning. J. Phys. Chem. Lett. 2020, 11, 9656–9658. 10.1021/acs.jpclett.0c03130. [DOI] [PubMed] [Google Scholar]
- Tkatchenko A. Machine learning for chemical discovery. Nat. Commun. 2020, 11, 4125. 10.1038/s41467-020-17844-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schütt K.; Chmiela S.; von Lilienfeld O.; Tkatchenko A.; Tsuda K.; Müller K.. Machine Learning Meets Quantum Physics; Lecture Notes in Physics; Springer International, 2020. [Google Scholar]
- Ramakrishnan R.; von Lilienfeld O. A.. Reviews in Computational Chemistry; John Wiley & Sons, 2017; Vol. 30; pp 225–256. [Google Scholar]
- von Lilienfeld O. A. Quantum machine learning in chemical compound space. Angew. Chem., Int. Ed. 2018, 57, 4164–4169. 10.1002/anie.201709686. [DOI] [PubMed] [Google Scholar]
- Kitchin J. R. Machine learning in catalysis. Nat. Catal. 2018, 1, 230–232. 10.1038/s41929-018-0056-y. [DOI] [Google Scholar]
- Huang B.; Symonds N. O.; Lilienfeld O. A. v. Quantum machine learning in chemistry and materials. Handbook of Materials Modeling: Methods: Theory and Modeling 2018, 1–27. 10.1007/978-3-319-42913-7_67-1. [DOI] [Google Scholar]
- Butler K. T.; Davies D. W.; Cartwright H.; Isayev O.; Walsh A. Machine learning for molecular and materials science. Nature 2018, 559, 547–555. 10.1038/s41586-018-0337-2. [DOI] [PubMed] [Google Scholar]
- Aspuru-Guzik A.; Lindh R.; Reiher M. The matter simulation (r) evolution. ACS Cent. Sci. 2018, 4, 144–152. 10.1021/acscentsci.7b00550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber F. A.; Anatole von Lilienfeld O. Modeling Materials Quantum Properties with Machine Learning. Materials Informatics: Methods, Tools and Applications 2019, 171–179. 10.1002/9783527802265.ch6. [DOI] [Google Scholar]
- Freeze J. G.; Kelly H. R.; Batista V. S. Search for Catalysts by Inverse Design: Artificial Intelligence, Mountain Climbers, and Alchemists. Chem. Rev. 2019, 119, 6595. 10.1021/acs.chemrev.8b00759. [DOI] [PubMed] [Google Scholar]
- von Lilienfeld O. A.; Müller K.-R.; Tkatchenko A. Exploring chemical compound space with quantum-based machine learning. Nat. Rev. Chem. 2020, 4, 347. 10.1038/s41570-020-0189-9. [DOI] [PubMed] [Google Scholar]
- von Lilienfeld O. A.; Burke K. Retrospective on a decade of machine learning for chemical discovery. Nat. Commun. 2020, 11, 4895. 10.1038/s41467-020-18556-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noé F.; Tkatchenko A.; Müller K.-R.; Clementi C. Machine learning for molecular simulation. Annu. Rev. Phys. Chem. 2020, 71, 361–390. 10.1146/annurev-physchem-042018-052331. [DOI] [PubMed] [Google Scholar]
- Faber F. A.; Christensen A. S.; von Lilienfeld O. A.. Machine Learning Meets Quantum Physics; Springer, 2020; pp 155–169. [Google Scholar]
- Unke O. T.; Chmiela S.; Sauceda H. E.; Gastegger M.; Poltavsky I.; Schütt K. T.; Tkatchenko A.; Müller K.-R.. Machine Learning Force Fields. arXiv 2020, arXiv:2010.07067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muratov E. N.; Bajorath J.; Sheridan R. P.; Tetko I. V.; Filimonov D.; Poroikov V.; Oprea T. I.; Baskin I. I.; Varnek A.; Roitberg A.; et al. QSAR without borders. Chem. Soc. Rev. 2020, 49, 3525. 10.1039/D0CS00098A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chibani S.; Coudert F.-X. Machine learning approaches for the prediction of materials properties. APL Mater. 2020, 8, 080701. 10.1063/5.0018384. [DOI] [Google Scholar]
- Dral P. O. Quantum chemistry in the age of machine learning. J. Phys. Chem. Lett. 2020, 11, 2336–2347. 10.1021/acs.jpclett.9b03664. [DOI] [PubMed] [Google Scholar]
- Haghighatlari M.; Li J.; Heidar-Zadeh F.; Liu Y.; Guan X.; Head-Gordon T. Learning to Make Chemical Predictions: The Interplay of Feature Representation. Data, and Machine Learning Methods 2020, 6, 1527–1542. 10.1016/j.chempr.2020.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann R.; Malrieu J.-P. Simulation vs. Understanding: A Tension, in Quantum Chemistry and Beyond. Part A. Stage Setting. Angew. Chem., Int. Ed. 2020, 59, 12590–12610. 10.1002/anie.201902527. [DOI] [PubMed] [Google Scholar]
- Hoffmann R.; Malrieu J.-P. Simulation vs. Understanding: A Tension, in Quantum Chemistry and Beyond. Part B. The March of Simulation, for Better or Worse. Angew. Chem., Int. Ed. 2020, 59, 13156–13178. 10.1002/anie.201910283. [DOI] [PubMed] [Google Scholar]
- Hoffmann R.; Malrieu J.-P. Simulation vs. Understanding: A Tension, in Quantum Chemistry and Beyond. Part C. Toward Consilience. Angew. Chem., Int. Ed. 2020, 59, 13694–13710. 10.1002/anie.201910285. [DOI] [PubMed] [Google Scholar]
- von Lilienfeld O. A. Introducing Machine Learning: Science and Technology. Mach. Learn.: Sci. Technol. 2020, 1, 010201. 10.1088/2632-2153/ab6d5d. [DOI] [Google Scholar]
- Pyzer-Knapp E. O.; Cuff J.; Patterson J.; Isayev O.; Maskell S. Welcome to the first issue of Applied AI Letters. Appl. AI Lett. 2020, 1, e8. 10.1002/ail2.8. [DOI] [Google Scholar]
- Buckingham A.; Utting B. Intermolecular forces. Annu. Rev. Phys. Chem. 1970, 21, 287–316. 10.1146/annurev.pc.21.100170.001443. [DOI] [Google Scholar]
- von Lilienfeld O. A. First principles view on chemical compound space: Gaining rigorous atomistic control of molecular properties. Int. J. Quantum Chem. 2013, 113, 1676–1689. 10.1002/qua.24375. [DOI] [Google Scholar]
- Faulon J. L. Stochastic generator of chemical structure: 1. Application to the structure elucidation of large molecules. J. Chem. Inf. Comp. Sci. 1994, 34, 1204–1218. 10.1021/ci00021a031. [DOI] [Google Scholar]
- Braun J.; Gugisch R.; Kerber A.; Laue R.; Meringer M.; Rücker C. MOLGEN-CID — A canonizer for molecules and graphs accessible through the internet. J. Chem. Inf. Comp. Sci. 2004, 44, 542–548. 10.1021/ci030404l. [DOI] [PubMed] [Google Scholar]
- Fink T.; Bruggesser H.; Reymond J.-L. Virtual exploration of the small-molecule chemical universe below 160 Da. Angew. Chem., Int. Ed. 2005, 44, 1504. 10.1002/anie.200462457. [DOI] [PubMed] [Google Scholar]
- von Rudorff G. F.; von Lilienfeld O. A. Simplifying inverse materials design problems for fixed lattices with alchemical chirality. Sci. Adv. 2021, 7, eabf1173 10.1126/sciadv.abf1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virshup A. M.; Contreras-García J.; Wipf P.; Yang W.; Beratan D. N. Stochastic Voyages into Uncharted Chemical Space Produce a Representative Library of All Possible Drug-Like Compounds. J. Am. Chem. Soc. 2013, 135, 7296–7303. 10.1021/ja401184g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato T. On the eigenfunctions of many-particle systems in quantum mechanics. Communications on Pure and Applied Mathematics 1957, 10, 151–177. 10.1002/cpa.3160100201. [DOI] [Google Scholar]
- Bartók A. P.; Payne M. C.; Kondor R.; Csányi G. Gaussian Approximation Potentials: The Accuracy of Quantum Mechanics, without the Electrons. Phys. Rev. Lett. 2010, 104, 136403. 10.1103/PhysRevLett.104.136403. [DOI] [PubMed] [Google Scholar]
- Bartók A. P.; Kondor R.; Csányi G. On representing chemical environments. Phys. Rev. B: Condens. Matter Mater. Phys. 2013, 87, 184115. 10.1103/PhysRevB.87.184115. [DOI] [Google Scholar]
- van Duin A. C. T.; Dasgupta S.; Lorant F.; Goddard W. A. III ReaxFF: A reactive force field for hydrocarbons. J. Phys. Chem. A 2001, 105, 9396–9409. 10.1021/jp004368u. [DOI] [Google Scholar]
- Senftle T. P.; Hong S.; Islam M. M.; Kylasa S. B.; Zheng Y.; Shin Y. K.; Junkermeier C.; Engel-Herbert R.; Janik M. J.; Aktulga H. M.; et al. The ReaxFF reactive force-field: development, applications and future directions. NPJ Comput. Mater. 2016, 2, 15011. 10.1038/npjcompumats.2015.11. [DOI] [Google Scholar]
- Ischtwan J.; Collins M. A. Molecular potential energy surfaces by interpolation. J. Chem. Phys. 1994, 100, 8080–8088. 10.1063/1.466801. [DOI] [Google Scholar]
- Wagner A. F.; Schatz G. C.; Bowman J. M. The evaluation of fitting functions for the representation of an O(3P)+H2 potential energy surface. I. J. Chem. Phys. 1981, 74, 4960–4983. 10.1063/1.441749. [DOI] [Google Scholar]
- Schatz G. C. The analytical representation of electronic potential-energy surfaces. Rev. Mod. Phys. 1989, 61, 669–688. 10.1103/RevModPhys.61.669. [DOI] [Google Scholar]
- Sumpter B. G.; Noid D. W. Potential energy surfaces for macromolecules. A neural network technique. Chem. Phys. Lett. 1992, 192, 455–462. 10.1016/0009-2614(92)85498-Y. [DOI] [Google Scholar]
- Blank T. B.; Brown S. D.; Calhoun A. W.; Doren D. J. Neural network models of potential energy surfaces. J. Chem. Phys. 1995, 103, 4129–4137. 10.1063/1.469597. [DOI] [Google Scholar]
- Brown D. F.; Gibbs M. N.; Clary D. C. Combining ab initio computations, neural networks, and diffusion Monte Carlo: An efficient method to treat weakly bound molecules. J. Chem. Phys. 1996, 105, 7597–7604. 10.1063/1.472596. [DOI] [Google Scholar]
- Lorenz S.; Gross A.; Scheffler M. Representing high-dimensional potential-energy surfaces for reactions at surfaces by neural networks. Chem. Phys. Lett. 2004, 395, 210. 10.1016/j.cplett.2004.07.076. [DOI] [Google Scholar]
- Manzhos S.; Carrington T. Jr. A random-sampling high dimensional model representation neural network for building potential energy surfaces. J. Chem. Phys. 2006, 125, 084109–084123. 10.1063/1.2336223. [DOI] [PubMed] [Google Scholar]
- Handley C. M.; Popelier P. L. A. Dynamically polarizable water potential based on multipole moments trained by machine learning. J. Chem. Theory Comput. 2009, 5, 1474. 10.1021/ct800468h. [DOI] [PubMed] [Google Scholar]
- Behler J.; Parrinello M. Generalized neural-network representation of high-dimensional potential-energy surfaces. Phys. Rev. Lett. 2007, 98, 146401. 10.1103/PhysRevLett.98.146401. [DOI] [PubMed] [Google Scholar]
- Behler J.; Martonak R.; Donadio D.; Parrinello M. Metadynamics simulations of the high-pressure phases of silicon employing a high-dimensional neural network potential. Phys. Rev. Lett. 2008, 100, 185501. 10.1103/PhysRevLett.100.185501. [DOI] [PubMed] [Google Scholar]
- Behler J. First principles neural network potentials for reactive simulations of large molecular and condensed systems. Angew. Chem., Int. Ed. 2017, 56, 12828–12840. 10.1002/anie.201703114. [DOI] [PubMed] [Google Scholar]
- Smith J. S.; Isayev O.; Roitberg A. E. ANI-1: An extensible neural network potential with DFT accuracy at force field computational cost. Chem. Sci. 2017, 8, 3192–3203. 10.1039/C6SC05720A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faraji S.; Ghasemi S. A.; Rostami S.; Rasoulkhani R.; Schaefer B.; Goedecker S.; Amsler M. High accuracy and transferability of a neural network potential through charge equilibration for calcium fluoride. Phys. Rev. B: Condens. Matter Mater. Phys. 2017, 95, 104105. 10.1103/PhysRevB.95.104105. [DOI] [Google Scholar]
- Schütt K. T.; Sauceda H. E.; Kindermans P.-J.; Tkatchenko A.; Müller K.-R. SchNet-A deep learning architecture for molecules and materials. J. Chem. Phys. 2018, 148, 241722. 10.1063/1.5019779. [DOI] [PubMed] [Google Scholar]
- Unke O. T.; Meuwly M. A reactive, scalable, and transferable model for molecular energies from a neural network approach based on local information. J. Chem. Phys. 2018, 148, 241708. 10.1063/1.5017898. [DOI] [PubMed] [Google Scholar]
- Manzhos S.; Carrington T. Jr. Neural network potential energy surfaces for small molecules and reactions. Chem. Rev. 2020, 10.1021/acs.chemrev.0c00665 [DOI] [PubMed] [Google Scholar]
- Ho T.; Rabitz H. A general method for constructing multidimensional molecular potential energy surfaces from abinitio calculations. J. Chem. Phys. 1996, 104, 2584–2597. 10.1063/1.470984. [DOI] [Google Scholar]
- Bartók A. P.; Payne M. C.; Kondor R.; Csányi G. Gaussian Approximation Potentials: The Accuracy of Quantum Mechanics, without the Electrons. Phys. Rev. Lett. 2010, 104, 136403. 10.1103/PhysRevLett.104.136403. [DOI] [PubMed] [Google Scholar]
- Pozun Z. D.; Hansen K.; Sheppard D.; Rupp M.; Müller K.-R.; Henkelman G. Optimizing transition states via kernel-based machine learning. J. Chem. Phys. 2012, 136, 174101–174109. 10.1063/1.4707167. [DOI] [PubMed] [Google Scholar]
- Thompson A.; Swiler L.; Trott C.; Foiles S.; Tucker G. Spectral neighbor analysis method for automated generation of quantum-accurate interatomic potentials. J. Comput. Phys. 2015, 285, 316–330. 10.1016/j.jcp.2014.12.018. [DOI] [Google Scholar]
- Li Z.; Kermode J. R.; De Vita A. Molecular Dynamics with On-the-Fly Machine Learning of Quantum-Mechanical Forces. Phys. Rev. Lett. 2015, 114, 096405. 10.1103/PhysRevLett.114.096405. [DOI] [PubMed] [Google Scholar]
- Botu V.; Ramprasad R. Adaptive machine learning framework to accelerate ab initio molecular dynamics. Int. J. Quantum Chem. 2015, 115, 1074–1083. 10.1002/qua.24836. [DOI] [Google Scholar]
- Rupp M.; Ramakrishnan R.; von Lilienfeld O. A. Machine Learning for Quantum Mechanical Properties of Atoms in Molecules. J. Phys. Chem. Lett. 2015, 6, 3309. 10.1021/acs.jpclett.5b01456. [DOI] [Google Scholar]
- Soloviov M.; Meuwly M. Reproducing kernel potential energy surfaces in biomolecular simulations: Nitric oxide binding to myoglobin. J. Chem. Phys. 2015, 143, 105103. 10.1063/1.4929527. [DOI] [PubMed] [Google Scholar]
- Glielmo A.; Sollich P.; De Vita A. Accurate interatomic force fields via machine learning with covariant kernels. Phys. Rev. B: Condens. Matter Mater. Phys. 2017, 95, 214302. 10.1103/PhysRevB.95.214302. [DOI] [Google Scholar]
- Chmiela S.; Tkatchenko A.; Sauceda H. E.; Poltavsky I.; Schütt K. T.; Müller K.-R. Machine learning of accurate energy-conserving molecular force fields. Sci. Adv. 2017, 3, e1603015 10.1126/sciadv.1603015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unke O. T.; Meuwly M. Toolkit for the construction of reproducing kernel-based representations of data: Application to multidimensional potential energy surfaces. J. Chem. Inf. Model. 2017, 57, 1923–1931. 10.1021/acs.jcim.7b00090. [DOI] [PubMed] [Google Scholar]
- Dragoni D.; Daff T. D.; Csányi G.; Marzari N. Achieving DFT accuracy with a machine-learning interatomic potential: Thermomechanics and defects in bcc ferromagnetic iron. Phys. Rev. Materials 2018, 2, 013808. 10.1103/PhysRevMaterials.2.013808. [DOI] [Google Scholar]
- Bereau T.; DiStasio Jr R. A.; Tkatchenko A.; Von Lilienfeld O. A. Non-covalent interactions across organic and biological subsets of chemical space: Physics-based potentials parametrized from machine learning. J. Chem. Phys. 2018, 148, 241706. 10.1063/1.5009502. [DOI] [PubMed] [Google Scholar]
- Deringer V. L.; Caro M. A.; Jana R.; Aarva A.; Elliott S. R.; Laurila T.; Csányi G.; Pastewka L. Computational Surface Chemistry of Tetrahedral Amorphous Carbon by Combining Machine Learning and Density Functional Theory. Chem. Mater. 2018, 30, 7438–7445. 10.1021/acs.chemmater.8b02410. [DOI] [Google Scholar]
- Caro M. A.; Aarva A.; Deringer V. L.; Csányi G.; Laurila T. Reactivity of amorphous carbon surfaces: rationalizing the role of structural motifs in functionalization using machine learning. Chem. Mater. 2018, 30, 7446–7455. 10.1021/acs.chemmater.8b03353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chmiela S.; Sauceda H. E.; Müller K.-R.; Tkatchenko A. Towards exact molecular dynamics simulations with machine-learned force fields. Nat. Commun. 2018, 9, 3887. 10.1038/s41467-018-06169-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chmiela S.; Sauceda H. E.; Poltavsky I.; Müller K.-R.; Tkatchenko A. sGDML: Constructing accurate and data efficient molecular force fields using machine learning. Comput. Phys. Commun. 2019, 240, 38. 10.1016/j.cpc.2019.02.007. [DOI] [Google Scholar]
- Sauceda H. E.; Gastegger M.; Chmiela S.; Müller K.-R.; Tkatchenko A. Molecular force fields with gradient-domain machine learning (GDML): Comparison and synergies with classical force fields. J. Chem. Phys. 2020, 153, 124109. 10.1063/5.0023005. [DOI] [PubMed] [Google Scholar]
- Kamath A.; Vargas-Hernández R. A.; Krems R. V.; Carrington Jr T.; Manzhos S. Neural networks vs Gaussian process regression for representing potential energy surfaces: A comparative study of fit quality and vibrational spectrum accuracy. J. Chem. Phys. 2018, 148, 241702. 10.1063/1.5003074. [DOI] [PubMed] [Google Scholar]
- Käser S.; Koner D.; Christensen A. S.; von Lilienfeld O. A.; Meuwly M. Machine Learning Models of Vibrating H2CO: Comparing Reproducing Kernels, FCHL, and PhysNet. J. Phys. Chem. A 2020, 124, 8853–8865. 10.1021/acs.jpca.0c05979. [DOI] [PubMed] [Google Scholar]
- Csányi G.; Albaret T.; Payne M. C.; De Vita A. D. Learn on the Fly”: A Hybrid Classical and Quantum-Mechanical Molecular Dynamics Simulation. Phys. Rev. Lett. 2004, 93, 175503. 10.1103/PhysRevLett.93.175503. [DOI] [PubMed] [Google Scholar]
- Podryabinkin E. V.; Shapeev A. V. Active learning of linearly parametrized interatomic potentials. Comput. Mater. Sci. 2017, 140, 171–180. 10.1016/j.commatsci.2017.08.031. [DOI] [Google Scholar]
- Jacobsen T.; Jørgensen M.; Hammer B. On-the-Fly Machine Learning of Atomic Potential in Density Functional Theory Structure Optimization. Phys. Rev. Lett. 2018, 120, 026102. 10.1103/PhysRevLett.120.026102. [DOI] [PubMed] [Google Scholar]
- Jørgensen M. S.; Larsen U. F.; Jacobsen K. W.; Hammer B. Exploration Versus Exploitation in Global Atomistic Structure Optimization. J. Phys. Chem. A 2018, 122, 1504–1509. 10.1021/acs.jpca.8b00160. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Lin D.-Y.; Wang H.; Car R.; E W. Active learning of uniformly accurate interatomic potentials for materials simulation. Phys. Rev. Mater. 2019, 3, 023804. 10.1103/PhysRevMaterials.3.023804. [DOI] [Google Scholar]
- Westermayr J.; Gastegger M.; Menger M. F.; Mai S.; González L.; Marquetand P. Machine learning enables long time scale molecular photodynamics simulations. Chem. Sci. 2019, 10, 8100–8107. 10.1039/C9SC01742A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia W.; Wang H.; Chen M.; Lu D.; Lin L.; Car R.; Weinan E; Zhang L., et al. Pushing the limit of molecular dynamics with ab initio accuracy to 100 million atoms with machine learning. SC20: International Conference for High Performance Computing, Networking, Storage and Analysis, Atlanta, GA 2020; p 1. 10.1109/SC41405.2020.00009 [DOI]
- Marder S. R.; Beratan D. N.; Cheng L.-T. Approaches for Optimizing the First Electronic Hyperpolarizability of Conjugated Organic Molecules. Science 1991, 252, 103–106. 10.1126/science.252.5002.103. [DOI] [PubMed] [Google Scholar]
- Kuhn C.; Beratan D. N. Inverse Strategies for Molecular Design. J. Phys. Chem. 1996, 100, 10595–10599. 10.1021/jp960518i. [DOI] [Google Scholar]
- Ceder G. Predicting properties from scratch. Science 1998, 280, 1099–1100. 10.1126/science.280.5366.1099. [DOI] [Google Scholar]
- Franceschetti A.; Zunger A. The inverse band-structure problem of finding an atomic configuration with given electronic properties. Nature 1999, 402, 60. 10.1038/46995. [DOI] [Google Scholar]
- Jóhannesson G. H.; Bligaard T.; Ruban A. V.; Skriver H. L.; Jacobsen K. W.; Nørskov J. K. Combined Electronic Structure and Evolutionary Search Approach to Materials Design. Phys. Rev. Lett. 2002, 88, 255506. 10.1103/PhysRevLett.88.255506. [DOI] [PubMed] [Google Scholar]
- von Lilienfeld O. A.; Lins R.; Rothlisberger U. Variational particle number approach for rational compound design. Phys. Rev. Lett. 2005, 95, 153002. 10.1103/PhysRevLett.95.153002. [DOI] [PubMed] [Google Scholar]
- Mounet N.; Gibertini M.; Schwaller P.; Campi D.; Merkys A.; Marrazzo A.; Sohier T.; Castelli I. E.; Cepellotti A.; Pizzi G.; et al. Two-dimensional materials from high-throughput computational exfoliation of experimentally known compounds. Nat. Nanotechnol. 2018, 13, 246–252. 10.1038/s41565-017-0035-5. [DOI] [PubMed] [Google Scholar]
- Hautier G.; Fischer C. C.; Jain A.; Mueller T.; Ceder G. Finding nature’s missing ternary oxide compounds using machine learning and density functional theory. Chem. Mater. 2010, 22, 3762. 10.1021/cm100795d. [DOI] [Google Scholar]
- George J.; Hautier G. Chemist versus Machine: Traditional Knowledge versus Machine Learning Techniques. Trends Chem. 2021, 3, 86. 10.1016/j.trechm.2020.10.007. [DOI] [Google Scholar]
- Wilson E. B. Jr. Four Dimensional Electron Density Function. J. Chem. Phys. 1962, 36, 2232. 10.1063/1.1732864. [DOI] [Google Scholar]
- Politzer P.; Parr R. G. Some new energy formulas for atoms and molecules. J. Chem. Phys. 1974, 61, 4258. 10.1063/1.1681726. [DOI] [Google Scholar]
- Mezey P. G. Electronic energy inequalities for isoelectronic molecular systems. Theor. Chim. Acta 1980, 59, 321–332. 10.1007/BF00553391. [DOI] [Google Scholar]
- Mezey P. G. New global constraints on electronic energy hypersurfaces. Int. J. Quantum Chem. 1986, 29, 85–99. 10.1002/qua.560290109. [DOI] [Google Scholar]
- von Lilienfeld O. A.; Tuckerman M. E. Molecular grand-canonical ensemble density functional theory and exploration of chemical space. J. Chem. Phys. 2006, 125, 154104. 10.1063/1.2338537. [DOI] [PubMed] [Google Scholar]
- Wang M.; Hu X.; Beratan D. N.; Yang W. Designing molecules by optimizing potentials. J. Am. Chem. Soc. 2006, 128, 3228. 10.1021/ja0572046. [DOI] [PubMed] [Google Scholar]
- Lesiuk M.; Balawender R.; Zachara J. Higher order alchemical derivatives from coupled perturbed self-consistent field theory. J. Chem. Phys. 2012, 136, 034104. 10.1063/1.3674163. [DOI] [PubMed] [Google Scholar]
- Munoz M.; Cardenas C. How predictive could alchemical derivatives be?. Phys. Chem. Chem. Phys. 2017, 19, 16003–16012. 10.1039/C7CP02755A. [DOI] [PubMed] [Google Scholar]
- Fias S.; Chang K. S.; von Lilienfeld O. A. Alchemical normal modes unify chemical space. J. Phys. Chem. Lett. 2019, 10, 30–39. 10.1021/acs.jpclett.8b02805. [DOI] [PubMed] [Google Scholar]
- von Rudorff G. F.; von Lilienfeld O. A. Alchemical perturbation density functional theory. Phys. Rev. Research 2020, 2, 023220. 10.1103/PhysRevResearch.2.023220. [DOI] [PubMed] [Google Scholar]
- Barkoutsos P. K.; Gkritsis F.; Ollitrault P. J.; Sokolov I. O.; Woerner S.; Tavernelli I.. Quantum algorithm for alchemical optimization in material design. arXiv 2020.arXiv:2008.06449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzari N.; de Gironcoli S.; Baroni S. Structure and Phase Stability of GaxIn1-xP solid solutions from computational Alchemy. Phys. Rev. Lett. 1994, 72, 4001. 10.1103/PhysRevLett.72.4001. [DOI] [PubMed] [Google Scholar]
- Beste A.; Harrison R. J.; Yanai T. Direct computation of general chemical energy differences: Application to ionization potentials, excitation, and bond energies. J. Chem. Phys. 2006, 125, 074101. 10.1063/1.2244559. [DOI] [PubMed] [Google Scholar]
- Weigend F.; Schrodt C.; Ahlrichs R. Atom distributions in binary atom clusters: A perturbational approach and its validation in a case study. J. Chem. Phys. 2004, 121, 10380. 10.1063/1.1811079. [DOI] [PubMed] [Google Scholar]
- Weigend F. Extending DFT-based genetic algorithms by atom-to-place re-assignment via perturbation theory: a systematic and unbiased approach to structures of mixed-metallic clusters. J. Chem. Phys. 2014, 141, 134103. 10.1063/1.4896658. [DOI] [PubMed] [Google Scholar]
- Rinderspacher B. C.; Andzelm J.; Rawlett A.; Dougherty J.; Beratan D. N.; Yang W. Discrete Optimization of Electronic Hyperpolarizabilities in a Chemical Subspace. J. Chem. Theory Comput. 2009, 5, 3321. 10.1021/ct900325p. [DOI] [PubMed] [Google Scholar]
- Sheppard D.; Henkelman G.; von Lilienfeld O. A. Alchemical derivatives of reaction energetics. J. Chem. Phys. 2010, 133, 084104. 10.1063/1.3474502. [DOI] [PubMed] [Google Scholar]
- Balawender R.; Welearegay M. A.; Lesiuk M.; De Proft F.; Geerlings P. Exploring Chemical Space with the Alchemical Derivatives. J. Chem. Theory Comput. 2013, 9, 5327–5340. 10.1021/ct400706g. [DOI] [PubMed] [Google Scholar]
- Chang K. Y. S.; Fias S.; Ramakrishnan R.; von Lilienfeld O. A. Fast and accurate predictions of covalent bonds in chemical space. J. Chem. Phys. 2016, 144, 174110. 10.1063/1.4947217. [DOI] [PubMed] [Google Scholar]
- Al-Hamdani Y. S.; Michaelides A.; von Lilienfeld O. A. Exploring dissociative water adsorption on isoelectronically BN doped graphene using alchemical derivatives. J. Chem. Phys. 2017, 147, 164113. 10.1063/1.4986314. [DOI] [PubMed] [Google Scholar]
- Fias S.; Heidar-Zadeh F.; Geerlings P.; Ayers P. W. Chemical transferability of functional groups follows from the nearsightedness of electronic matter. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 11633–11638. 10.1073/pnas.1615053114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balawender R.; Lesiuk M.; De Proft F.; Geerlings P. Exploring Chemical Space with Alchemical Derivatives: BN-Simultaneous Substitution Patterns in C60. J. Chem. Theory Comput. 2018, 14, 1154. 10.1021/acs.jctc.7b01114. [DOI] [PubMed] [Google Scholar]
- von Rudorff G. F.; von Lilienfeld O. A. Atoms in molecules from alchemical perturbation density functional theory. J. Phys. Chem. B 2019, 123, 10073–10082. 10.1021/acs.jpcb.9b07799. [DOI] [PubMed] [Google Scholar]
- Saravanan K.; Kitchin J. R.; von Lilienfeld O. A.; Keith J. A. Alchemical Predictions for Computational Catalysis: Potential and Limitations. J. Phys. Chem. Lett. 2017, 8, 5002–5007. 10.1021/acs.jpclett.7b01974. [DOI] [PubMed] [Google Scholar]
- Griego C. D.; Saravanan K.; Keith J. A. Benchmarking computational alchemy for carbide, nitride, and oxide catalysts. Adv. Theor. Simul. 2019, 2, 1800142. 10.1002/adts.201800142. [DOI] [Google Scholar]
- Griego C. D.; Kitchin J. R.; Keith J. A. Acceleration of catalyst discovery with easy, fast, and reproducible computational alchemy. Int. J. Quantum Chem. 2021, 121, e26380 10.1002/qua.26380. [DOI] [Google Scholar]
- von Rudorff G. F.; von Lilienfeld O. A. Rapid and accurate molecular deprotonation energies from quantum alchemy. Phys. Chem. Chem. Phys. 2020, 22, 10519–10525. 10.1039/C9CP06471K. [DOI] [PubMed] [Google Scholar]
- Muñoz M.; Robles-Navarro A.; Fuentealba P.; Cárdenas C. Predicting Deprotonation Sites Using Alchemical Derivatives. J. Phys. Chem. A 2020, 124, 3754–3760. 10.1021/acs.jpca.9b09472. [DOI] [PubMed] [Google Scholar]
- Pérez A.; von Lilienfeld O. A. Path integral computation of quantum free energy differences due to alchemical transformations involving mass and potential. J. Chem. Theory Comput. 2011, 7, 2358. 10.1021/ct2000556. [DOI] [PubMed] [Google Scholar]
- Ceriotti M.; Markland T. E. Efficient methods and practical guidelines for simulating isotope effects. J. Chem. Phys. 2013, 138, 014112. 10.1063/1.4772676. [DOI] [PubMed] [Google Scholar]
- Geerlings P.; De Proft F. D.; Langenaeker W. Conceptual Density Functional Theory. Chem. Rev. 2003, 103, 1793. 10.1021/cr990029p. [DOI] [PubMed] [Google Scholar]
- Yang W.; Zhang Y.; Ayers P. W. Degenerate ground states and fractional number of electrons in density and density reduced matrix functional theory. Phys. Rev. Lett. 2000, 84, 5172. 10.1103/PhysRevLett.84.5172. [DOI] [PubMed] [Google Scholar]
- Zeng X.; Hu H.; Hu X.; Cohen A. J.; Yang W. Ab initio quantum mechanical/molecular mechanical simulation of electron transfer process: Fractional electron approach. J. Chem. Phys. 2008, 128, 124510. 10.1063/1.2832946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori-Sánchez P.; Cohen A. J.; Yang W. Discontinuous nature of the exchange-correlation functional in strongly correlated systems. Phys. Rev. Lett. 2009, 102, 066403. 10.1103/PhysRevLett.102.066403. [DOI] [PubMed] [Google Scholar]
- Schneider G. Virtual screening: an endless staircase?. Nat. Rev. Drug Discovery 2010, 9, 273. 10.1038/nrd3139. [DOI] [PubMed] [Google Scholar]
- Bultinck P.; Gironés X.; Carbo-Dorcaz R. Molecular quantum similarity: theory and applications. Rev. Comput. Chem. 2005, 21, 127. 10.1002/0471720895.ch2. [DOI] [Google Scholar]
- Kaji M. Mendeleev’s Discovery of the Periodic Law: The Origin and the Reception. Found. Chem. 2003, 5, 189. 10.1023/A:1025673206850. [DOI] [Google Scholar]
- Pauling L.; Yost D. M. The additivity of the energies of normal covalent bonds. Proc. Natl. Acad. Sci. U. S. A. 1932, 18, 414. 10.1073/pnas.18.6.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettifor D. G. A chemical scale for crystal-structure maps. Solid State Commun. 1984, 51, 31–34. 10.1016/0038-1098(84)90765-8. [DOI] [Google Scholar]
- Glawe H.; Sanna A.; Gross E. K. U.; Marques M. A. L. The optimal one dimensional periodic table: a modified Pettifor chemical scale from data mining. New J. Phys. 2016, 18, 093011. 10.1088/1367-2630/18/9/093011. [DOI] [Google Scholar]
- Glawe H.; Sanna A.; Gross E.; Marques M. A. The optimal one dimensional periodic table: a modified Pettifor chemical scale from data mining. New J. Phys. 2016, 18, 093011. 10.1088/1367-2630/18/9/093011. [DOI] [Google Scholar]
- Allahyari Z.; Oganov A. R. Nonempirical definition of the Mendeleev numbers: Organizing the chemical space. J. Phys. Chem. C 2020, 124, 23867. 10.1021/acs.jpcc.0c07857. [DOI] [Google Scholar]
- Glushkovsky A.AI Discovering a Coordinate System of Chemical Elements: Dual Representation by Variational Autoencoders. arXiv 2020, arXiv:2011.12090 [Google Scholar]
- Huang B.; Zhuang L.; Xiao L.; Lu J. Bond-energy decoupling: principle and application to heterogeneous catalysis. Chem. Sci. 2013, 4, 606–611. 10.1039/C2SC21232C. [DOI] [Google Scholar]
- Wells P. R. Linear Free Energy Relationships. Chem. Rev. 1963, 63, 171–219. 10.1021/cr60222a005. [DOI] [Google Scholar]
- Hammett L. P. The Effect of Structure upon the Reactions of Organic Compounds. Benzene Derivatives. J. Am. Chem. Soc. 1937, 59, 96–103. 10.1021/ja01280a022. [DOI] [Google Scholar]
- Hansch C.; Leo A.; Taft R. W. A survey of Hammett substituent constants and resonance and field parameters. Chem. Rev. 1991, 91, 165–195. 10.1021/cr00002a004. [DOI] [Google Scholar]
- Hammett L. P. Some Relations between Reaction Rates and Equilibrium Constants. Chem. Rev. 1935, 17, 125–136. 10.1021/cr60056a010. [DOI] [Google Scholar]
- Hammond G. S. A Correlation of Reaction Rates. J. Am. Chem. Soc. 1955, 77, 334–338. 10.1021/ja01607a027. [DOI] [Google Scholar]
- Bell R. P. The theory of reactions involving proton transfers. Proc R. Soc. Lond., A 1936, 154, 414–429. 10.1098/rspa.1936.0060. [DOI] [Google Scholar]
- Evans M. G.; Polanyi M. Further considerations on the thermodynamics of chemical equilibria and reaction rates. Trans. Faraday Soc. 1936, 32, 1333–1360. 10.1039/tf9363201333. [DOI] [Google Scholar]
- Kohn W.; Sham L. J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133. 10.1103/PhysRev.140.A1133. [DOI] [Google Scholar]
- Hohenberg P.; Kohn W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864. 10.1103/PhysRev.136.B864. [DOI] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- Ernzerhof M.; Scuseria G. E. Assessment of the Perdew-Burke-Ernzerhof exchange-correlation functional. J. Chem. Phys. 1999, 110, 5029. 10.1063/1.478401. [DOI] [PubMed] [Google Scholar]
- Adamo C.; Barone V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. 10.1063/1.478522. [DOI] [Google Scholar]
- Hammer B.; Nørskov J. K. Electronic factors determining the reactivity of metal surfaces. Surf. Sci. 1995, 343, 211–220. 10.1016/0039-6028(96)80007-0. [DOI] [Google Scholar]
- Hammer B.; Norskov J. K. Why gold is the noblest of all the metals. Nature 1995, 376, 238–240. 10.1038/376238a0. [DOI] [Google Scholar]
- Calle-Vallejo F.; Martínez J. I.; García-Lastra J. M.; Sautet P.; Loffreda D. Fast Prediction of Adsorption Properties for Platinum Nanocatalysts with Generalized Coordination Numbers. Angew. Chem., Int. Ed. 2014, 53, 8316–8319. _eprint: 10.1002/anie.201402958. [DOI] [PubMed] [Google Scholar]
- Calle-Vallejo F.; Tymoczko J.; Colic V.; Vu Q. H.; Pohl M. D.; Morgenstern K.; Loffreda D.; Sautet P.; Schuhmann W.; Bandarenka A. S. Finding optimal surface sites on heterogeneous catalysts by counting nearest neighbors. Science 2015, 350, 185–189. 10.1126/science.aab3501. [DOI] [PubMed] [Google Scholar]
- Huang B.; Xiao L.; Lu J.; Zhuang L. Spatially Resolved Quantification of the Surface Reactivity of Solid Catalysts. Angew. Chem. 2016, 128, 6347–6351. _eprint: 10.1002/ange.201601824. [DOI] [PubMed] [Google Scholar]
- van Santen R. A.; Neurock M.; Shetty S. G. Reactivity Theory of Transition Metal Surfaces: A Bronsted-Evans-Polanyi Linear Activation Energy Free Energy Analysis. Chem. Rev. 2010, 110, 2005–2048. 10.1021/cr9001808. [DOI] [PubMed] [Google Scholar]
- Abild-Pedersen F.; Greeley J.; Studt F.; Rossmeisl J.; Munter T. R.; Moses P. G.; Skúlason E.; Bligaard T.; Nørskov J. K. Scaling Properties of Adsorption Energies for Hydrogen-Containing Molecules on Transition-Metal Surfaces. Phys. Rev. Lett. 2007, 99, 016105. 10.1103/PhysRevLett.99.016105. [DOI] [PubMed] [Google Scholar]
- Fernández E. M.; Moses P. G.; Toftelund A.; Hansen H. A.; Martínez J. I.; Abild-Pedersen F.; Kleis J.; Hinnemann B.; Rossmeisl J.; Bligaard T.; Nørskov J. K. Scaling Relationships for Adsorption Energies on Transition Metal Oxide, Sulfide, and Nitride Surfaces. Angew. Chem., Int. Ed. 2008, 47, 4683–4686. _eprint: 10.1002/anie.200705739. [DOI] [PubMed] [Google Scholar]
- Calle-Vallejo F.; Martínez J. I.; García-Lastra J. M.; Rossmeisl J.; Koper M. T. M. Physical and Chemical Nature of the Scaling Relations between Adsorption Energies of Atoms on Metal Surfaces. Phys. Rev. Lett. 2012, 108, 116103. 10.1103/PhysRevLett.108.116103. [DOI] [PubMed] [Google Scholar]
- Nørskov J. K.; Bligaard T.; Rossmeisl J.; Christensen C. H. Towards the computational design of solid catalysts. Nat. Chem. 2009, 1, 37. 10.1038/nchem.121. [DOI] [PubMed] [Google Scholar]
- Parr R. G.; Yang W.. Density Functional Theory of Atoms and Molecules; Oxford Science Publications, 1989. [Google Scholar]
- Geerlings P.; De Proft F.; Langenaeker W. Conceptual Density Functional Theory. Chem. Rev. 2003, 103, 1793–1874. 10.1021/cr990029p. [DOI] [PubMed] [Google Scholar]
- Geerlings P.; Fias S.; Boisdenghien Z.; DE Proft F. Conceptual DFT: chemistry from the linear response function. Chem. Soc. Rev. 2014, 43, 4989–5008. 10.1039/c3cs60456j. [DOI] [PubMed] [Google Scholar]
- Huang B.; von Lilienfeld O. A. Communication: Understanding molecular representations in machine learning: The role of uniqueness and target similarity. J. Chem. Phys. 2016, 145, 161102. 10.1063/1.4964627. [DOI] [PubMed] [Google Scholar]
- Faber F. A.; Lindmaa A.; von Lilienfeld O. A.; Armiento R. Machine Learning Energies of 2 Million Elpasolite (ABC2D6) Crystals. Phys. Rev. Lett. 2016, 117, 135502. 10.1103/PhysRevLett.117.135502. [DOI] [PubMed] [Google Scholar]
- Ong S. P.; Jain A.; Hautier G.; Kocher M.; Cholia S.; Gunter D.; Bailey D.; Skinner D.; Persson K. A.; Ceder G.. The Materials Project. 2011; http://materialsproject.org/.
- Faber F. A.; Christensen A. S.; Huang B.; von Lilienfeld O. A. Alchemical and structural distribution based representation for universal quantum machine learning. J. Chem. Phys. 2018, 148, 241717. 10.1063/1.5020710. [DOI] [PubMed] [Google Scholar]
- Ye W.; Chen C.; Wang Z.; Chu I.-H.; Ong S. P. Deep neural networks for accurate predictions of crystal stability. Nat. Commun. 2018, 9, 3800. 10.1038/s41467-018-06322-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt J.; Shi J.; Borlido P.; Chen L.; Botti S.; Marques M. A. Predicting the thermodynamic stability of solids combining density functional theory and machine learning. Chem. Mater. 2017, 29, 5090–5103. 10.1021/acs.chemmater.7b00156. [DOI] [Google Scholar]
- Legrain F.; Carrete J.; van Roekeghem A.; Curtarolo S.; Mingo N. How Chemical Composition Alone Can Predict Vibrational Free Energies and Entropies of Solids. Chem. Mater. 2017, 29, 6220–6227. 10.1021/acs.chemmater.7b00789. [DOI] [Google Scholar]
- Hansen K.; Biegler F.; von Lilienfeld O. A.; Müller K.-R.; Tkatchenko A.; et al. Interaction potentials in molecules and non-local information in chemical space. Phys. Rev. Lett 2012, 108, 058301. 10.1103/PhysRevLett.108.058301. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan R.; Dral P. D.; Rupp M.; von Lilienfeld O. A. Quantum chemistry structures and properties of 134 kilo molecules. Sci. Data 2014, 1, 140022. 10.1038/sdata.2014.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch W.; Holthausen M. C.. A Chemist’s Guide to Density Functional Theory; Wiley-VCH, 2002. [Google Scholar]
- Huang B.; von Lilienfeld O. A.. The “DNA” of chemistry: Scalable quantum machine learning with “amons". arXiv 2017, arXiv:1707.04146, submitted to Nature. [Google Scholar]
- Braun J.; Kerber A.; Meringer M.; Rücker C. Similarity of molecular descriptors: The equivalence of Zagreb indices and walk counts. MATCH Commun. Math. Comput. Chem. 2005, 54, 163–176. [Google Scholar]
- Visco J.; Pophale R. S.; Rintoul M. D.; Faulon J. L. Developing a methodology for an inverse quantitative structure activity relationship using the signature molecular descriptor. J. Mol. Graphics Modell. 2002, 20, 429–438. 10.1016/S1093-3263(01)00144-9. [DOI] [PubMed] [Google Scholar]
- Faulon J.-L.; Visco D. P. Jr.; Pophale R. S. The Signature Molecular Descriptor. 1. Using Extended Valence Sequences in QSAR and QSPR Studies. J. Chem. Inf. Comp. Sci. 2003, 43, 707. 10.1021/ci020345w. [DOI] [PubMed] [Google Scholar]
- Martin S.; Roe D.; Faulon J.-L. Predicting protein-protein interactions using signature products. Bioinformatics 2005, 21, 218–226. 10.1093/bioinformatics/bth483. [DOI] [PubMed] [Google Scholar]
- Rogers D.; Hahn M. Extended-Connectivity Fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. 10.1021/ci100050t. [DOI] [PubMed] [Google Scholar]
- Lounkine E.; Keiser M. J.; Whitebread S.; Mikhailov D.; Hamon J.; Jenkins J. L.; Lavan P.; Weber E.; Doak A. K.; Côté S.; Shoichet B. K.; Urban L. Large-scale prediction and testing of drug activity on side-effect targets. Nature 2012, 486, 361. 10.1038/nature11159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besnard J.; et al. Automated design of ligands to polypharmacological profiles. Nature 2012, 492, 215. 10.1038/nature11691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruddigkeit L.; van Deursen R.; Blum L. C.; Reymond J.-L. Enumeration of 166 Billion Organic Small Molecules in the Chemical Universe Database GDB-17. J. Chem. Inf. Model. 2012, 52, 2864–2875. 10.1021/ci300415d. [DOI] [PubMed] [Google Scholar]
- Fink T.; Bruggesser H.; Reymond J.-L. Virtual Exploration of the Small-Molecule Chemical Universe below 160 Da. Angew. Chem., Int. Ed. 2005, 44, 1504–1508. 10.1002/anie.200462457. [DOI] [PubMed] [Google Scholar]
- Faber F. A.; Hutchison L.; Huang B.; Gilmer J.; Schoenholz S. S.; Dahl G. E.; Vinyals O.; Kearnes S.; Riley P. F.; von Lilienfeld O. A. Prediction errors of molecular machine learning models lower than hybrid DFT error. J. Chem. Theory Comput. 2017, 13, 5255–5264. 10.1021/acs.jctc.7b00577. [DOI] [PubMed] [Google Scholar]
- Schwaller P.; Gaudin T.; Lányi D.; Bekas C.; Laino T. Found in Translation”: predicting outcomes of complex organic chemistry reactions using neural sequence-to-sequence models. Chem. Sci. 2018, 9, 6091–6098. 10.1039/C8SC02339E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwaller P.; Laino T.; Gaudin T.; Bolgar P.; Hunter C. A.; Bekas C.; Lee A. A. Molecular Transformer: A Model for Uncertainty-Calibrated Chemical Reaction Prediction. ACS Cent. Sci. 2019, 5, 1572–1583. 10.1021/acscentsci.9b00576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unsleber J. P.; Reiher M. The Exploration of Chemical Reaction Networks. Annu. Rev. Phys. Chem. 2020, 71, 121–142. _eprint: 10.1146/annurev-physchem-071119-040123. [DOI] [PubMed] [Google Scholar]
- Gómez-Bombarelli R.; Wei J. N.; Duvenaud D.; Hernández-Lobato J. M.; Sánchez-Lengeling B.; Sheberla D.; Aguilera-Iparraguirre J.; Hirzel T. D.; Adams R. P.; Aspuru-Guzik A. Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules. ACS Cent. Sci. 2018, 4, 268–276. 10.1021/acscentsci.7b00572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q.; Bassyouni A.; Butler C. R.; Hou X.; Jenkinson S.; Price D. A.; et al. Ligand biological activity predicted by cleaning positive and negative chemical correlations. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 3373–3378. 10.1073/pnas.1810847116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krenn M.; Häse F.; Nigam A.; Friederich P.; Aspuru-Guzik A. Self-Referencing Embedded Strings (SELFIES): A 100% robust molecular string representation. Mach. Learn.: Sci. Technol. 2020, 1, 045024. 10.1088/2632-2153/aba947. [DOI] [Google Scholar]
- Hoffmann C.; Menichetti R.; Kanekal K. H.; Bereau T. Controlled exploration of chemical space by machine learning of coarse-grained representations. Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top. 2019, 100, 033302. 10.1103/PhysRevE.100.033302. [DOI] [PubMed] [Google Scholar]
- John S. T.; Csányi G. Many-Body Coarse-Grained Interactions Using Gaussian Approximation Potentials. J. Phys. Chem. B 2017, 121, 10934–10949. 10.1021/acs.jpcb.7b09636. [DOI] [PubMed] [Google Scholar]
- Wang J.; Olsson S.; Wehmeyer C.; Pérez A.; Charron N. E.; de Fabritiis G.; Noé F.; Clementi C. Machine Learning of Coarse-Grained Molecular Dynamics Force Fields. ACS Cent. Sci. 2019, 5, 755–767. 10.1021/acscentsci.8b00913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karthikeyan M.; Glen R. C.; Bender A. General Melting Point Prediction Based on a Diverse Compound Data Set and Artificial Neural Networks. J. Chem. Inf. Model. 2005, 45, 581–590. 10.1021/ci0500132. [DOI] [PubMed] [Google Scholar]
- Ghiringhelli L. M.; Vybiral J.; Levchenko S. V.; Draxl C.; Scheffler M. Big data of materials science: Critical role of the descriptor. Phys. Rev. Lett. 2015, 114, 105503. 10.1103/PhysRevLett.114.105503. [DOI] [PubMed] [Google Scholar]
- Rupp M.; Tkatchenko A.; Müller K.-R.; von Lilienfeld O. A. Fast and Accurate Modeling of Molecular Atomization Energies with Machine Learning. Phys. Rev. Lett. 2012, 108, 058301. 10.1103/PhysRevLett.108.058301. [DOI] [PubMed] [Google Scholar]
- Rasmussen C. E.; Williams C. K. I.. Gaussian Processes for Machine Learning; Dietterich T., Ed.; MIT Press: Cambridge, 2006; www.GaussianProcess.org. [Google Scholar]
- Schmidt J.; Marques M. R. G.; Botti S.; Marques M. A. L. Recent advances and applications of machine learning in solid-state materials science. NPJ Comput. Mater. 2019, 5, 83. 10.1038/s41524-019-0221-0. [DOI] [Google Scholar]
- Vapnik V.The Nature of Statistical Learning Theory; Springer Science & Business Media, 2013. [Google Scholar]
- Brown A.; Braams B. J.; Christoffel K.; Jin Z.; Bowman J. M. Classical and quasiclassical spectral analysis of CH5+ using an ab initio potential energy surface. J. Chem. Phys. 2003, 119, 8790. 10.1063/1.1622379. [DOI] [Google Scholar]
- Neal R. M.Bayesian Learning for Neural Networks; Springer, 1996; pp 29–53. [Google Scholar]
- ćaylak O.; von Lilienfeld O. A.; Baumeier B. Wasserstein metric for improved quantum machine learning with adjacency matrix representations. Mach. Learn.: Sci. Technol. 2020, 1, 03LT01. 10.1088/2632-2153/aba048. [DOI] [Google Scholar]
- Hansen K.; Montavon G.; Biegler F.; Fazli S.; Rupp M.; Scheffler M.; von Lilienfeld O. A.; Tkatchenko A.; Müller K.-R. Assessment and Validation of Machine Learning Methods for Predicting Molecular Atomization Energies. J. Chem. Theory Comput. 2013, 9, 3404–3419. 10.1021/ct400195d. [DOI] [PubMed] [Google Scholar]
- Rupp M. Machine learning for quantum mechanics in a nutshell. Int. J. Quantum Chem. 2015, 115, 1058–1073. 10.1002/qua.24954. [DOI] [Google Scholar]
- Cortes C.; Jackel L. D.; Solla S. A.; Vapnik V.; Denker J. S. Learning curves: Asymptotic values and rate of convergence. Adv. Neur. Inform. Process. Syst. 1994, 6, 327–334. [Google Scholar]
- Müller K. R.; Finke M.; Murata N.; Schulten K.; Amari S. A numerical study on learning curves in stochastic multilayer feedforward networks. Neural Comp. 1996, 8, 1085. 10.1162/neco.1996.8.5.1085. [DOI] [PubMed] [Google Scholar]
- Christensen A. S.; Faber F. A.; von Lilienfeld O. A. Operators in quantum machine learning: Response properties in chemical space. J. Chem. Phys. 2019, 150, 064105. 10.1063/1.5053562. [DOI] [PubMed] [Google Scholar]
- Christensen A. S.; von Lilienfeld O. A. Operator quantum machine learning: Navigating the chemical space of response properties. Chimia 2019, 73, 1028–1031. 10.2533/chimia.2019.1028. [DOI] [PubMed] [Google Scholar]
- Christensen A. S.; Bratholm L.; Faber F. A.; von Lilienfeld O. A. FCHL revisited: Faster and more accurate quantum machine learning. J. Chem. Phys. 2020, 152, 044107. 10.1063/1.5126701. [DOI] [PubMed] [Google Scholar]
- Gastegger M.; Schütt K. T.; Müller K.-R.. Machine learning of solvent effects on molecular spectra and reactions. arXiv 2020, arXiv:2010.14942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen A. S.; von Lilienfeld O. A. On the role of gradients for machine learning of molecular energies and forces. Mach. Learn.: Sci. Technol. 2020, 1, 045018. 10.1088/2632-2153/abba6f. [DOI] [Google Scholar]
- Hammett L. P. The Effect of Structure upon the Reactions of Organic Compounds. Benzene Derivatives. J. Am. Chem. Soc. 1937, 59, 96. 10.1021/ja01280a022. [DOI] [Google Scholar]
- Bragato M.; von Rudorff G. F.; von Lilienfeld O. A. Data enhanced Hammett Equation: Reaction Barriers in Chemical Space. Chem. Sci. 2020, 11, 11859. 10.1039/D0SC04235H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todeschini R.; Consonni V.. Handbook of Molecular Descriptors; Wiley-VCH: Weinheim, 2009. [Google Scholar]
- Moussa J. E. Comment on ”Fast and Accurate Modeling of Molecular Energies with Machine Learning. Phys. Rev. Lett. 2012, 109, 059801. 10.1103/PhysRevLett.109.059801. [DOI] [PubMed] [Google Scholar]
- von Lilienfeld O. A.; Ramakrishnan R.; Rupp M.; Knoll A. Fourier series of atomic radial distribution functions: A molecular fingerprint for machine learning models of quantum chemical properties. Int. J. Quantum Chem. 2015, 115, 1084. 10.1002/qua.24912. [DOI] [Google Scholar]
- Pozdnyakov S. N.; Willatt M. J.; Bartók A. P.; Ortner C.; Csányi G.; Ceriotti M. Incompleteness of Atomic Structure Representations. Phys. Rev. Lett. 2020, 125, 166001. 10.1103/PhysRevLett.125.166001. [DOI] [PubMed] [Google Scholar]
- Parsaeifard B.; De D. S.; Christensen A. S.; Faber F. A.; Kocer E.; De S.; Behler J.; von Lilienfeld A.; Goedecker S. An assessment of the structural resolution of various fingerprints commonly used in machine learning. Mach. Learn.: Sci. Technol. 2021, 2, 015018. 10.1088/2632-2153/abb212. [DOI] [Google Scholar]
- Ramakrishnan R.; von Lilienfeld O. A. Many Molecular Properties from One Kernel in Chemical Space. Chimia 2015, 69, 182. 10.2533/chimia.2015.182. [DOI] [PubMed] [Google Scholar]
- Montavon G.; Rupp M.; Gobre V.; Vazquez-Mayagoitia A.; Hansen K.; Tkatchenko A.; Müller K.-R.; von Lilienfeld O. A. Machine learning of molecular electronic properties in chemical compound space. New J. Phys. 2013, 15, 095003. 10.1088/1367-2630/15/9/095003. [DOI] [Google Scholar]
- Gilmer J.; Schoenholz S. S.; Riley P. F.; Vinyals O.; Dahl G. E.. Neural Message Passing for Quantum Chemistry. Proceedings of the 34th International Conference on Machine Learning, ICML, 2017.
- Eickenberg M.; Exarchakis G.; Hirn M.; Mallat S.; Thiry L. Solid harmonic wavelet scattering for predictions of molecule properties. J. Chem. Phys. 2018, 148, 241732. 10.1063/1.5023798. [DOI] [PubMed] [Google Scholar]
- Gubaev K.; Podryabinkin E. V.; Shapeev A. V. Machine learning of molecular properties: Locality and active learning. J. Chem. Phys. 2018, 148, 241727. 10.1063/1.5005095. [DOI] [PubMed] [Google Scholar]
- Welborn M.; Cheng L.; Miller III T. F. Transferability in machine learning for electronic structure via the molecular orbital basis. J. Chem. Theory Comput. 2018, 14, 4772–4779. 10.1021/acs.jctc.8b00636. [DOI] [PubMed] [Google Scholar]
- Langer M. F.; Goeßmann A.; Rupp M.. Representations of molecules and materials for interpolation of quantum-mechanical simulations via machine learning. arXiv 2020, arXiv:2003.12081v2. [Google Scholar]
- Englert B.-G.Semiclassical Theory of Atoms; Springer, 1988. [Google Scholar]
- Rupp M.; Tkatchenko A.; Müller K.-R.; von Lilienfeld O. A. Reply to Comment on ”Fast and Accurate Modeling of Molecular Energies with Machine Learning. Phys. Rev. Lett. 2012, 109, 059802. 10.1103/PhysRevLett.109.059802. [DOI] [PubMed] [Google Scholar]
- Pilania G.; Wang C.; Jiang X.; Rajasekaran S.; Ramprasad R. Accelerating materials property predictions using machine learning. Sci. Rep. 2013, 3, 2810. 10.1038/srep02810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schütt K. T.; Glawe H.; Brockherde F.; Sanna A.; Müller K. R.; Gross E. K. U. How to represent crystal structures for machine learning: Towards fast prediction of electronic properties. Phys. Rev. B: Condens. Matter Mater. Phys. 2014, 89, 205118. 10.1103/PhysRevB.89.205118. [DOI] [Google Scholar]
- Rappé A. K.; Casewit C. J.; Colwell K. S.; Goddard W. A. III; Skiff W. M. UFF, a Full Periodic Table Force Field for Molecular Mechanics and Molecular Dynamics Simulations. J. Am. Chem. Soc. 1992, 114, 10024. 10.1021/ja00051a040. [DOI] [Google Scholar]
- Pronobis W.; Tkatchenko A.; Müller K.-R. Many-Body Descriptors for Predicting Molecular Properties with Machine Learning: Analysis of Pairwise and Three-Body Interactions in Molecules. J. Chem. Theory Comput. 2018, 14, 2991. 10.1021/acs.jctc.8b00110. [DOI] [PubMed] [Google Scholar]
- Faber F.; Lindmaa A.; von Lilienfeld O. A.; Armiento R. Crystal Structure Representations for Machine Learning Models of Formation Energies. Int. J. Quantum Chem. 2015, 115, 1094. 10.1002/qua.24917. [DOI] [Google Scholar]
- Ramakrishnan R.; Hartmann M.; Tapavicza E.; von Lilienfeld O. A. Electronic Spectra from TDDFT and Machine Learning in Chemical Space. J. Chem. Phys. 2015, 143, 084111. 10.1063/1.4928757. [DOI] [PubMed] [Google Scholar]
- Faber F. A.; Hutchison L.; Huang B.; Gilmer J.; Schoenholz S. S.; Dahl G. E.; Vinyals O.; Kearnes S.; Riley P. F.; von Lilienfeld O. A. Prediction errors of molecular machine learning models lower than hybrid DFT error. J. Chem. Theory Comput. 2017, 13, 5255–5264. 10.1021/acs.jctc.7b00577. [DOI] [PubMed] [Google Scholar]
- Stuke A.; Todorović M.; Rupp M.; Kunkel C.; Ghosh K.; Himanen L.; Rinke P. Chemical diversity in molecular orbital energy predictions with kernel ridge regression. J. Chem. Phys. 2019, 150, 204121. 10.1063/1.5086105. [DOI] [PubMed] [Google Scholar]
- Ghosh K.; Stuke A.; Todorović M.; Jørgensen P. B.; Schmidt M. N.; Vehtari A.; Rinke P. Deep learning spectroscopy: neural networks for molecular excitation spectra. Adv. Sci. 2019, 6, 1801367. 10.1002/advs.201801367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L.; Amsler M.; Fuhrer T.; Schaefer B.; Faraji S.; Rostami S.; Ghasemi S. A.; Sadeghi A.; Grauzinyte M.; Wolverton C.; et al. A fingerprint based metric for measuring similarities of crystalline structures. J. Chem. Phys. 2016, 144, 034203. 10.1063/1.4940026. [DOI] [PubMed] [Google Scholar]
- Schütt O.; VandeVondele J. Machine learning adaptive basis sets for efficient large scale density functional theory simulation. J. Chem. Theory Comput. 2018, 14, 4168–4175. 10.1021/acs.jctc.8b00378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babaei M.; Azar Y. T.; Sadeghi A. Locality meets machine learning: Excited and ground-state energy surfaces of large systems at the cost of small ones. Phys. Rev. B: Condens. Matter Mater. Phys. 2020, 101, 115132. 10.1103/PhysRevB.101.115132. [DOI] [Google Scholar]
- Collins C. R.; Gordon G. J.; von Lilienfeld O. A.; Yaron D. J. Constant size descriptors for accurate machine learning models of molecular properties. J. Chem. Phys. 2018, 148, 241718. 10.1063/1.5020441. [DOI] [PubMed] [Google Scholar]
- Drautz R. Atomic cluster expansion for accurate and transferable interatomic potentials. Phys. Rev. B: Condens. Matter Mater. Phys. 2019, 99, 014104. 10.1103/PhysRevB.99.014104. [DOI] [Google Scholar]
- Behler J. Atom-centered symmetry functions for constructing high-dimensional neural networks potentials. J. Chem. Phys. 2011, 134, 074106. 10.1063/1.3553717. [DOI] [PubMed] [Google Scholar]
- Huang B.; von Lilienfeld O. A. Quantum machine learning using atom-in-molecule-based fragments selected on the fly. Nat. Chem. 2020, 12, 945–951. 10.1038/s41557-020-0527-z. [DOI] [PubMed] [Google Scholar]
- Bartók A. P.; De S.; Poelking C.; Bernstein N.; Kermode J. R.; Csányi G.; Ceriotti M. Machine learning unifies the modeling of materials and molecules. Sci. Adv. 2017, 3, e1701816. 10.1126/sciadv.1701816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubaev K.; Podryabinkin E. V.; Shapeev A. V. Machine learning of molecular properties: Locality and active learning. J. Chem. Phys. 2018, 148, 241727. 10.1063/1.5005095. [DOI] [PubMed] [Google Scholar]
- Willatt M. J.; Musil F.; Ceriotti M. Atom-density representations for machine learning. J. Chem. Phys. 2019, 150, 154110. 10.1063/1.5090481. [DOI] [PubMed] [Google Scholar]
- Taylor M. E.; Stone P. Transfer learning for reinforcement learning domains: A survey. J. Mach. Learn. Res. 2009, 10, 1633. [Google Scholar]
- Smith J. S.; Nebgen B. T.; Zubatyuk R.; Lubbers N.; Devereux C.; Barros K.; Tretiak S.; Isayev O.; Roitberg A. E. Approaching coupled cluster accuracy with a general-purpose neural network potential through transfer learning. Nat. Commun. 2019, 10, 2903. 10.1038/s41467-019-10827-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobday S.; Smith R.; Belbruno J. Applications of neural networks to fitting interatomic potential functions. Modell. Simul. Mater. Sci. Eng. 1999, 7, 397. 10.1088/0965-0393/7/3/308. [DOI] [Google Scholar]
- Bereau T.; Andrienko D.; von Lilienfeld O. A. Transferable atomic multipole machine learning models for small organic molecules. J. Chem. Theory Comput. 2015, 11, 3225. 10.1021/acs.jctc.5b00301. [DOI] [PubMed] [Google Scholar]
- Ghasemi S. A.; Hofstetter A.; Saha S.; Goedecker S. Interatomic potentials for ionic systems with density functional accuracy based on charge densities obtained by a neural network. Phys. Rev. B: Condens. Matter Mater. Phys. 2015, 92, 045131. 10.1103/PhysRevB.92.045131. [DOI] [Google Scholar]
- Sifain A. E.; Lubbers N.; Nebgen B. T.; Smith J. S.; Lokhov A. Y.; Isayev O.; Roitberg A. E.; Barros K.; Tretiak S. Discovering a transferable charge assignment model using machine learning. J. Phys. Chem. Lett. 2018, 9, 4495–4501. 10.1021/acs.jpclett.8b01939. [DOI] [PubMed] [Google Scholar]
- Nebgen B.; Lubbers N.; Smith J. S.; Sifain A. E.; Lokhov A.; Isayev O.; Roitberg A. E.; Barros K.; Tretiak S. Transferable Dynamic Molecular Charge Assignment Using Deep Neural Networks. J. Chem. Theory Comput. 2018, 14, 4687–4698. 10.1021/acs.jctc.8b00524. [DOI] [PubMed] [Google Scholar]
- Dral P. O.; von Lilienfeld O. A.; Thiel W. Machine Learning of Parameters for Accurate Semiempirical Quantum Chemical Calculations. J. Chem. Theory Comput. 2015, 11, 2120–2125. PMID: 26146493 10.1021/acs.jctc.5b00141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranz J. J.; Kubillus M.; Ramakrishnan R.; von Lilienfeld O. A.; Elstner M. Generalized density-functional tight-binding repulsive potentials from unsupervised machine learning. J. Chem. Theory Comput. 2018, 14, 2341–2352. 10.1021/acs.jctc.7b00933. [DOI] [PubMed] [Google Scholar]
- Stöhr M.; Medrano Sandonas L.; Tkatchenko A. Accurate Many-Body Repulsive Potentials for Density-Functional Tight Binding from Deep Tensor Neural Networks. J. Phys. Chem. Lett. 2020, 11, 6835–6843. 10.1021/acs.jpclett.0c01307. [DOI] [PubMed] [Google Scholar]
- Zubatyuk T.; Nebgen B.; Lubbers N.; Smith J. S.; Zubatyuk R.; Zhou G.; Koh C.; Barros K.; Isayev O.; Tretiak S.. Machine Learned Hückel Theory: Interfacing Physics and Deep Neural Networks. arXiv 2019, arXiv:1909.12963. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan R.; Dral P.; Rupp M.; von Lilienfeld O. A. Big Data meets Quantum Chemistry Approximations: The A-Machine Learning Approach. J. Chem. Theory Comput. 2015, 11, 2087–2096. 10.1021/acs.jctc.5b00099. [DOI] [PubMed] [Google Scholar]
- Mezei P. D.; von Lilienfeld O. A. Noncovalent Quantum Machine Learning Corrections to Density Functionals. J. Chem. Theory Comput. 2020, 16, 2647–2653. 10.1021/acs.jctc.0c00181. [DOI] [PubMed] [Google Scholar]
- Griego C. D.; Zhao L.; Saravanan K.; Keith J. A. Machine Learning Corrected Alchemical Perturbation Density Functional Theory for Catalysis Applications. AIChE J. 2020, 66, e17041 10.1002/aic.17041. [DOI] [Google Scholar]
- Bogojeski M.; Vogt-Maranto L.; Tuckerman M. E.; Müller K.-R.; Burke K. Quantum chemical accuracy from density functional approximations via machine learning. Nat. Commun. 2020, 11, 5223. 10.1038/s41467-020-19093-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend J.; Vogiatzis K. D. Transferable MP2-Based Machine Learning for Accurate Coupled-Cluster Energies. J. Chem. Theory Comput. 2020, 16, 7453. 10.1021/acs.jctc.0c00927. [DOI] [PubMed] [Google Scholar]
- Nandi A.; Qu C.; Houston P.; Conte R.; Bowman J. M. Delta-Machine Learning for Potential Energy Surfaces: A PIP approach to bring a DFT-based PES to CCSD (T) Level of Theory. J. Chem. Phys. 2021, 154, 051102. 10.1063/5.0038301. [DOI] [PubMed] [Google Scholar]
- Curtiss L. A.; Raghavachari K.; Trucks G. W.; Pople J. A. Gaussian-2 theory for molecular energies of first-and second-row compounds. J. Chem. Phys. 1991, 94, 7221–7230. 10.1063/1.460205. [DOI] [Google Scholar]
- Curtiss L. A.; Redfern P. C.; Raghavachari K.; Rassolov V.; Pople J. A. Gaussian-3 theory using reduced Mo/ller-Plesset order. J. Chem. Phys. 1999, 110, 4703–4709. 10.1063/1.478385. [DOI] [Google Scholar]
- Curtiss L. A.; Redfern P. C.; Raghavachari K. Gaussian-4 theory. J. Chem. Phys. 2007, 126, 084108. 10.1063/1.2436888. [DOI] [PubMed] [Google Scholar]
- Zaspel P.; Huang B.; Harbrecht H.; von Lilienfeld O. A. Boosting quantum machine learning models with multi-level combination technique: Pople diagrams revisited. J. Chem. Theory Comput. 2019, 15, 1546. 10.1021/acs.jctc.8b00832. [DOI] [PubMed] [Google Scholar]
- Le Gratiet L.; Garnier J. Recursive co-kriging model for design of computer experiments with multiple levels of fidelity. Int. J. Uncertain. Quan. 2014, 4, 365. 10.1615/Int.J.UncertaintyQuantification.2014006914. [DOI] [Google Scholar]
- Kennedy M. C.; O’Hagan A. Predicting the output from a complex computer code when fast approximations are available. Biometrika 2000, 87, 1–13. 10.1093/biomet/87.1.1. [DOI] [Google Scholar]
- Cui J.; Krems R. V. Gaussian Process Model for Collision Dynamics of Complex Molecules. Phys. Rev. Lett. 2015, 115, 073202. 10.1103/PhysRevLett.115.073202. [DOI] [PubMed] [Google Scholar]
- Pilania G.; Gubernatis J. E.; Lookman T. Multi-fidelity machine learning models for accurate bandgap predictions of solids. Comput. Mater. Sci. 2017, 129, 156–163. 10.1016/j.commatsci.2016.12.004. [DOI] [Google Scholar]
- Batra R.; Pilania G.; Uberuaga B. P.; Ramprasad R. Multifidelity Information Fusion with Machine Learning: A Case Study of Dopant Formation Energies in Hafnia. ACS Appl. Mater. Interfaces 2019, 11, 24906. 10.1021/acsami.9b02174. [DOI] [PubMed] [Google Scholar]
- Wiens A. E.; Copan A. V.; Schaefer H. F. Multi-fidelity Gaussian process modeling for chemical energy surfaces. Chem. Phys. Lett.: X 2019, 3, 100022. 10.1016/j.cpletx.2019.100022. [DOI] [Google Scholar]
- Egorova O.; Hafizi R.; Woods D. C.; Day G. M. Multifidelity Statistical Machine Learning for Molecular Crystal Structure Prediction. J. Phys. Chem. A 2020, 124, 8065–8078. 10.1021/acs.jpca.0c05006. [DOI] [PubMed] [Google Scholar]
- Garcke J.; Griebel M.; Thess M. Data Mining with Sparse Grids. Computing 2001, 67, 225–253. 10.1007/s006070170007. [DOI] [Google Scholar]
- Delvos F. J. d-Variate Boolean interpolation. J. Approximation Theory 1982, 34, 99–114. 10.1016/0021-9045(82)90085-5. [DOI] [Google Scholar]
- Pan S. J.; Yang Q. A Survey on Transfer Learning. IEEE Trans. Knowl. Data Eng. 2010, 22, 1345–1359. Conference Name: IEEE Transactions on Knowledge and Data Engineering 10.1109/TKDE.2009.191. [DOI] [Google Scholar]
- Smith J. S.; Nebgen B. T.; Zubatyuk R.; Lubbers N.; Devereux C.; Barros K.; Tretiak S.; Isayev O.; Roitberg A. E. Approaching coupled cluster accuracy with a general-purpose neural network potential through transfer learning. Nat. Commun. 2019, 10, 2903. 10.1038/s41467-019-10827-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivas E. S.; Guerrero J. D. M.; Sober M. M.; Benedito J. R. M.; Lopez A. J. S.. Handbook Of Research On Machine Learning Applications and Trends: Algorithms, Methods and Techniques; IGI Publishing: Hershey, PA, 2009. [Google Scholar]
- Iovanac N. C.; Savoie B. M. Simpler is Better: How Linear Prediction Tasks Improve Transfer Learning in Chemical Autoencoders. J. Phys. Chem. A 2020, 124, 3679–3685. PMID: 32267698 10.1021/acs.jpca.0c00042. [DOI] [PubMed] [Google Scholar]
- Cai C.; Wang S.; Xu Y.; Zhang W.; Tang K.; Ouyang Q.; Lai L.; Pei J. Transfer Learning for Drug Discovery. J. Med. Chem. 2020, 63, 8683–8694. 10.1021/acs.jmedchem.9b02147. [DOI] [PubMed] [Google Scholar]
- Browning N. J.; Ramakrishnan R.; von Lilienfeld O. A.; Roethlisberger U. Genetic Optimization of Training Sets for Improved Machine Learning Models of Molecular Properties. J. Phys. Chem. Lett. 2017, 8, 1351. 10.1021/acs.jpclett.7b00038. [DOI] [PubMed] [Google Scholar]
- Smith J. S.; Nebgen B.; Lubbers N.; Isayev O.; Roitberg A. E. Less is more: Sampling chemical space with active learning. J. Chem. Phys. 2018, 148, 241733. 10.1063/1.5023802. [DOI] [PubMed] [Google Scholar]
- Wang T.; Zhu J.-Y.; Torralba A.; Efros A. A.. Dataset Distillation. arXiv 2020, arXiv:1811.10959. [Google Scholar]
- Sucholutsky I.; Schonlau M. ‘'Less Than One’-Shot Learning: Learning N Classes From M < N Samples. arXiv 2020, arXiv:2009.08449 [Google Scholar]
- Cerqueira T. F.; Sarmiento-Pérez R.; Amsler M.; Nogueira F.; Botti S.; Marques M. A. Materials design on-the-fly. J. Chem. Theory Comput. 2015, 11, 3955–3960. 10.1021/acs.jctc.5b00212. [DOI] [PubMed] [Google Scholar]
- Sanchez-Lengeling B.; Aspuru-Guzik A. Inverse molecular design using machine learning: Generative models for matter engineering. Science 2018, 361, 360–365. 10.1126/science.aat2663. [DOI] [PubMed] [Google Scholar]
- Vilhelmsen L. B.; Hammer B. Systematic Study of Au6 to Au12 Gold Clusters on MgO100. F Centers Using Density-Functional Theory. Phys. Rev. Lett. 2012, 108, 126101. 10.1103/PhysRevLett.108.126101. [DOI] [PubMed] [Google Scholar]
- Janet J. P.; Kulik H. J. Predicting electronic structure properties of transition metal complexes with neural networks. Chem. Sci. 2017, 8, 5137–5152. 10.1039/C7SC01247K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder J. C.; Rupp M.; Hansen K.; Müller K.-R.; Burke K. Finding Density Functionals with Machine Learning. Phys. Rev. Lett. 2012, 108, 253002. 10.1103/PhysRevLett.108.253002. [DOI] [PubMed] [Google Scholar]
- Simm G. N.; Reiher M. Error-Controlled Exploration of Chemical Reaction Networks with Gaussian Processes. J. Chem. Theory Comput. 2018, 14, 5238–5248. 10.1021/acs.jctc.8b00504. [DOI] [PubMed] [Google Scholar]
- Proppe J.; Gugler S.; Reiher M. Gaussian Process-Based Refinement of Dispersion Corrections. J. Chem. Theory Comput. 2019, 15, 6046–6060. 10.1021/acs.jctc.9b00627. [DOI] [PubMed] [Google Scholar]
- Goreinov S. A.; Oseledets I. V.; Savostyanov D. V.; Tyrtyshnikov E. E.; Zamarashkin N. L.. Matrix Methods: Theory, Algorithms and Applications: Dedicated to the Memory of Gene Golub; World Scientific, 2010; pp 247–256. [Google Scholar]
- Guo C.; Pleiss G.; Sun Y.; Weinberger K. Q.. On calibration of modern neural networks. Proceedings of the 34th International Conference on Machine Learning Sydney, NSW, Australia, 2017; Vol. 70, pp 1321–1330.
- Skafte N.; Jørgensen M.; Hauberg S. Reliable training and estimation of variance networks. Adv. Neur. Inform. Process. Syst. 2019, 6326–6336. [Google Scholar]
- Peterson A. A.; Christensen R.; Khorshidi A. Addressing uncertainty in atomistic machine learning. Phys. Chem. Chem. Phys. 2017, 19, 10978–10985. 10.1039/C7CP00375G. [DOI] [PubMed] [Google Scholar]
- Cortés-Ciriano I.; Bender A. Deep Confidence: A Computationally Efficient Framework for Calculating Reliable Prediction Errors for Deep Neural Networks. J. Chem. Inf. Model. 2019, 59, 1269–1281. 10.1021/acs.jcim.8b00542. [DOI] [PubMed] [Google Scholar]
- Musil F.; Willatt M. J.; Langovoy M. A.; Ceriotti M. Fast and Accurate Uncertainty Estimation in Chemical Machine Learning. J. Chem. Theory Comput. 2019, 15, 906–915. 10.1021/acs.jctc.8b00959. [DOI] [PubMed] [Google Scholar]
- Gal Y.; Ghahramani Z.. Dropout as a bayesian approximation: Representing model uncertainty in deep learning. International Conference on Machine Learning, 2016; pp 1050–1059.
- Janet J. P.; Duan C.; Yang T.; Nandy A.; Kulik H. J. A quantitative uncertainty metric controls error in neural network-driven chemical discovery. Chem. Sci. 2019, 10, 7913–7922. 10.1039/C9SC02298H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prodan E.; Kohn W. Nearsightedness of electronic matter. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 11635–11638. 10.1073/pnas.0505436102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fias S.; Heidar-Zadeh F.; Geerlings P.; Ayers P. W. Chemical transferability of functional groups follows from the nearsightedness of electronic matter. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 11633–11638. 10.1073/pnas.1615053114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappe A. K.; Casewit C. J.; Colwell K. S.; Goddard W. A. III; Skiff W. M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. 10.1021/ja00051a040. [DOI] [Google Scholar]
- Misra M.; Andrienko D.; Baumeier B.; Faulon J.-L.; von Lilienfeld O. A. Toward Quantitative Structure-Property Relationships for Charge Transfer Rates of Polycyclic Aromatic Hydrocarbons. J. Chem. Theory Comput. 2011, 7, 2549. 10.1021/ct200231z. [DOI] [PubMed] [Google Scholar]
- Schütt K. T.; Arbabzadah F.; Chmiela S.; Müller K. R.; Tkatchenko A. Quantum-chemical insights from deep tensor neural networks. Nat. Commun. 2017, 8, 13890. 10.1038/ncomms13890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinen S.; Schwilk M.; von Rudorff G. F.; von Lilienfeld O. A. Machine learning the computational cost of quantum chemistry. Mach. Learn.: Sci. Technol. 2020, 1, 025002. 10.1088/2632-2153/ab6ac4. [DOI] [Google Scholar]
- Unke O. T.; Meuwly M. PhysNet: A Neural Network for Predicting Energies, Forces, Dipole Moments, and Partial Charges. J. Chem. Theory Comput. 2019, 15, 3678–3693. PMID: 31042390 10.1021/acs.jctc.9b00181. [DOI] [PubMed] [Google Scholar]
- McDonagh J. L.; Silva A. F.; Vincent M. A.; Popelier P. L. Machine learning of dynamic electron correlation energies from topological atoms. J. Chem. Theory Comput. 2018, 14, 216–224. 10.1021/acs.jctc.7b01157. [DOI] [PubMed] [Google Scholar]
- Schütt K. T.; Arbabzadah F.; Chmiela S.; Müller K. R.; Tkatchenko A. Quantum-chemical insights from deep tensor neural networks. Nat. Commun. 2017, 8, 13890. 10.1038/ncomms13890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Jørgensen M. S.; Li J.; Hammer B. Atomic energies from a convolutional neural network. J. Chem. Theory Comput. 2018, 14, 3933. 10.1021/acs.jctc.8b00149. [DOI] [PubMed] [Google Scholar]
- Wilkins D. M.; Grisafi A.; Yang Y.; Lao K. U.; DiStasio R. A.; Ceriotti M. Accurate molecular polarizabilities with coupled cluster theory and machine learning. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 3401–3406. 10.1073/pnas.1816132116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grisafi A.; Nigam J.; Ceriotti M. Multi-scale approach for the prediction of atomic scale properties. arXiv 2021, 12, 2078. 10.1039/D0SC04934D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paruzzo F. M.; Hofstetter A.; Musil F.; De S.; Ceriotti M.; Emsley L. Chemical shifts in molecular solids by machine learning. Nat. Commun. 2018, 9, 4501. 10.1038/s41467-018-06972-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel E. A.; Anelli A.; Hofstetter A.; Paruzzo F.; Emsley L.; Ceriotti M. A Bayesian approach to NMR crystal structure determination. Phys. Chem. Chem. Phys. 2019, 21, 23385–23400. 10.1039/C9CP04489B. [DOI] [PubMed] [Google Scholar]
- Li J.; Bennett K. C.; Liu Y.; Martin M. V.; Head-Gordon T. Accurate prediction of chemical shifts for aqueous protein structure on “Real World” data. Chem. Sci. 2020, 11, 3180–3191. 10.1039/C9SC06561J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro-Vázquez A. A DFT/machine-learning hybrid method for the prediction of 3JHCCH couplings. Magn. Reson. Chem. 2021, 59, 414. 10.1002/mrc.5087. [DOI] [PubMed] [Google Scholar]
- Bratholm L. A.; Gerrard W.; Anderson B.; Bai S.; Choi S.; Dang L.; Hanchar P.; Howard A.; Huard G.; Kim S., et al. A community-powered search of machine learning strategy space to find NMR property prediction models. arXiv 2020, arXiv:2008.05994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A.; Chakraborty S.; Ramakrishnan R. Revving up 13C NMR shielding predictions across chemical space: Benchmarks for atoms-in-molecules kernel machine learning with new data for 134 kilo molecules. Mach. Learn.: Sci. Technol. 2021, 2, 035010. 10.1088/2632-2153/abe347. [DOI] [Google Scholar]
- Dral P. O.; Owens A.; Yurchenko S. N.; Thiel W. Structure-based sampling and self-correcting machine learning for accurate calculations of potential energy surfaces and vibrational levels. J. Chem. Phys. 2017, 146, 244108. 10.1063/1.4989536. [DOI] [PubMed] [Google Scholar]
- Gastegger M.; Behler J.; Marquetand P. Machine learning molecular dynamics for the simulation of infrared spectra. Chem. Sci. 2017, 8, 6924–6935. 10.1039/C7SC02267K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Bezanilla A.; von Lilienfeld O. A. Modeling electronic quantum transport with machine learning. Phys. Rev. B: Condens. Matter Mater. Phys. 2014, 89, 235411. 10.1103/PhysRevB.89.235411. [DOI] [Google Scholar]
- Arsenault L.-F.; Lopez-Bezanilla A.; von Lilienfeld O. A.; Millis A. J. Machine learning for Many-Body Physics: the case of the Anderson impurity model. Phys. Rev. B: Condens. Matter Mater. Phys. 2014, 90, 155136. 10.1103/PhysRevB.90.155136. [DOI] [Google Scholar]
- Arsenault L.-F.; von Lilienfeld O. A.; Millis A. J.. Machine learning for many-body physics: efficient solution of dynamical mean-field theory. arXiv 2015, http://arxiv.org/abs/1506.08858. [Google Scholar]
- Dral P. O.; Barbatti M.; Thiel W. Nonadiabatic excited-state dynamics with machine learning. J. Phys. Chem. Lett. 2018, 9, 5660–5663. 10.1021/acs.jpclett.8b02469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermayr J.; Faber F. A.; Christensen A. S.; von Lilienfeld O. A.; Marquetand P. Neural networks and kernel ridge regression for excited states dynamics of CH2NH: From single-state to multi-state representations and multi-property machine learning models. Mach. Learn.: Sci. Technol. 2020, 1, 025009. 10.1088/2632-2153/ab88d0. [DOI] [Google Scholar]
- Westermayr J.; Gastegger M.; Marquetand P. Combining SchNet and SHARC: The SchNarc machine learning approach for excited-state dynamics. J. Phys. Chem. Lett. 2020, 11, 3828–3834. 10.1021/acs.jpclett.0c00527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter M.; Marquetand P.; González-Vázquez J.; Sola I.; González L. SHARC: ab initio molecular dynamics with surface hopping in the adiabatic representation including arbitrary couplings. J. Chem. Theory Comput. 2011, 7, 1253–1258. 10.1021/ct1007394. [DOI] [PubMed] [Google Scholar]
- Westermayr J.; Marquetand P. Deep learning for UV absorption spectra with SchNarc: First steps toward transferability in chemical compound space. J. Chem. Phys. 2020, 153, 154112. 10.1063/5.0021915. [DOI] [PubMed] [Google Scholar]
- Westermayr J.; Marquetand P.. Machine learning for electronically excited states of molecules. Chem. Rev. 2020, 10.1021/acs.chemrev.0c00749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermayr J.; Marquetand P. Machine learning for nonadiabatic molecular dynamics. Machine Learning in Chemistry 2020, 17, 76. 10.1039/9781839160233-00076. [DOI] [Google Scholar]
- Westermayr J.; Marquetand P. Machine learning and excited-state molecular dynamics. Mach. Learn.: Sci. Technol. 2020, 1, 043001. 10.1088/2632-2153/ab9c3e. [DOI] [Google Scholar]
- Zubatyuk R.; Smith J.; Nebgen B. T.; Tretiak S.; Isayev O.. Teaching a neural network to attach and detach electrons from molecules. ChemRxiv 2020, 10.26434/chemrxiv.12725276.v2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Ye S.; Zhang J.; Hu C.; Jiang J.; Jiang B. Efficient and Accurate Simulations of Vibrational and Electronic Spectra with Symmetry-Preserving Neural Network Models for Tensorial Properties. J. Phys. Chem. B 2020, 124, 7284–7290. 10.1021/acs.jpcb.0c06926. [DOI] [PubMed] [Google Scholar]
- Ward L.; Liu R.; Krishna A.; Hegde V. I.; Agrawal A.; Choudhary A.; Wolverton C. Including crystal structure attributes in machine learning models of formation energies via Voronoi tessellations. Phys. Rev. B: Condens. Matter Mater. Phys. 2017, 96, 024104. 10.1103/PhysRevB.96.024104. [DOI] [Google Scholar]
- Yao K.; Herr J. E.; Brown S. N.; Parkhill J. Intrinsic Bond Energies from a Bonds-in-Molecules Neural Network. J. Phys. Chem. Lett. 2017, 8, 2689. 10.1021/acs.jpclett.7b01072. [DOI] [PubMed] [Google Scholar]
- Janet J. P.; Kulik H. J. Predicting electronic structure properties of transition metal complexes with neural networks. Chem. Sci. 2017, 8, 5137. 10.1039/C7SC01247K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grisafi A.; Wilkins D. M.; Csányi G.; Ceriotti M. Symmetry-Adapted Machine Learning for Tensorial Properties of Atomistic Systems. Phys. Rev. Lett. 2018, 120, 036002. 10.1103/PhysRevLett.120.036002. [DOI] [PubMed] [Google Scholar]
- Xie T.; Grossman J. C. Crystal Graph Convolutional Neural Networks for an Accurate and Interpretable Prediction of Material Properties. Phys. Rev. Lett. 2018, 120, 145301. 10.1103/PhysRevLett.120.145301. [DOI] [PubMed] [Google Scholar]
- Rossi K.; Juraskova V.; Wischert R.; Garel L.; Corminboeuf C.; Ceriotti M. Simulating Solvation and Acidity in Complex Mixtures with First-Principles Accuracy: The Case of CH3SO3H and H2O2 in Phenol. J. Chem. Theory Comput. 2020, 16, 5139. 10.1021/acs.jctc.0c00362. [DOI] [PubMed] [Google Scholar]
- Moosavi S. M.; Nandy A.; Jablonka K. M.; Ongari D.; Janet J. P.; Boyd P. G.; Lee Y.; Smit B.; Kulik H. J. Understanding the diversity of the metal-organic framework ecosystem. Nat. Commun. 2020, 11, 4068. 10.1038/s41467-020-17755-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jablonka K. M.; Ongari D.; Moosavi S. M.; Smit B. Big-Data Science in Porous Materials: Materials Genomics and Machine Learning. Chem. Rev. 2020, 120, 8066. 10.1021/acs.chemrev.0c00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulissi Z. W.; Singh A. R.; Tsai C.; Nørskov J. K. Automated Discovery and Construction of Surface Phase Diagrams Using Machine Learning. J. Phys. Chem. Lett. 2016, 7, 3931–3935. 10.1021/acs.jpclett.6b01254. [DOI] [PubMed] [Google Scholar]
- Ulissi Z. W.; Tang M. T.; Xiao J.; Liu X.; Torelli D. A.; Karamad M.; Cummins K.; Hahn C.; Lewis N. S.; Jaramillo T. F.; et al. Machine-learning methods enable exhaustive searches for active bimetallic facets and reveal active site motifs for CO2 reduction. ACS Catal. 2017, 7, 6600–6608. 10.1021/acscatal.7b01648. [DOI] [Google Scholar]
- Ulissi Z. W.; Medford A. J.; Bligaard T.; Nørskov J. K. To address surface reaction network complexity using scaling relations machine learning and DFT calculations. Nat. Commun. 2017, 8, 14621. 10.1038/ncomms14621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A. R.; Rohr B. A.; Gauthier J. A.; Nørskov J. K. Predicting chemical reaction barriers with a machine learning model. Catal. Lett. 2019, 149, 2347–2354. 10.1007/s10562-019-02705-x. [DOI] [Google Scholar]
- Meyer B.; Sawatlon B.; Heinen S.; von Lilienfeld O. A.; Corminboeuf C. Machine learning meets volcano plots: computational discovery of cross-coupling catalysts. Chem. Sci. 2018, 9, 7069–7077. 10.1039/C8SC01949E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinen S.; von Rudorff G. F.; von Lilienfeld O. A.. Quantum based machine learning of competing chemical reaction profiles. arXiv 2020, arXiv:2009.13429 [Google Scholar]
- von Rudorff G. F.; Heinen S. N.; Bragato M.; von Lilienfeld O. A. Thousands of reactants and transition states for competing E2 and S2 reactions. Mach. Learn.: Sci. Technol. 2020, 1, 045026. 10.1088/2632-2153/aba822. [DOI] [Google Scholar]
- Flores R. A.; Paolucci C.; Winther K. T.; Jain A.; Torres J. A. G.; Aykol M.; Montoya J.; Nørskov J. K.; Bajdich M.; Bligaard T. Active Learning Accelerated Discovery of Stable Iridium Oxide Polymorphs for the Oxygen Evolution Reaction. Chem. Mater. 2020, 32, 5854–5863. 10.1021/acs.chemmater.0c01894. [DOI] [Google Scholar]
- Mamun O.; Winther K. T.; Boes J. R.; Bligaard T. A Bayesian framework for adsorption energy prediction on bimetallic alloy catalysts. NPJ Comput. Mater. 2020, 6, 177. 10.1038/s41524-020-00447-8. [DOI] [Google Scholar]
- Garijo del Río E.; Kaappa S.; Garrido Torres J. A.; Bligaard T.; Jacobsen K. W. Machine Learning with bond information for local structure optimizations in surface science. J. Chem. Phys. 2020, 153, 234116. 10.1063/5.0033778. [DOI] [PubMed] [Google Scholar]
- Groenenboom M. C.; Anderson R. M.; Wollmershauser J. A.; Horton D. J.; Policastro S. A.; Keith J. A. Combined Neural Network Potential and Density Functional Theory Study of TiAl2O5 Surface Morphology and Oxygen Reduction Reaction Overpotentials. J. Phys. Chem. C 2020, 124, 15171–15179. 10.1021/acs.jpcc.0c02093. [DOI] [Google Scholar]
- Schlexer Lamoureux P.; Winther K. T.; Garrido Torres J. A.; Streibel V.; Zhao M.; Bajdich M.; Abild-Pedersen F.; Bligaard T. Machine learning for computational heterogeneous catalysis. ChemCatChem 2019, 11, 3581. 10.1002/cctc.201900595. [DOI] [Google Scholar]
- Fink T.; Reymond J.-L. Virtual Exploration of the Chemical Universe up to 11 Atoms of C, N, O, F: Assembly of 26.4 Million Structures (110.9 Million Stereoisomers) and Analysis for New Ring Systems, Stereochemistry, Physicochemical Properties, Compound Classes, and Drug Discovery. J. Chem. Inf. Model. 2007, 47, 342–353. 10.1021/ci600423u. [DOI] [PubMed] [Google Scholar]
- Blum L. C.; Reymond J.-L. 970 Million Druglike Small Molecules for Virtual Screening in the Chemical Universe Database GDB-13. J. Am. Chem. Soc. 2009, 131, 8732–8733. 10.1021/ja902302h. [DOI] [PubMed] [Google Scholar]
- Ruddigkeit L.; van Deursen R.; Blum L. C.; Reymond J.-L. Enumeration of 166 Billion Organic Small Molecules in the Chemical Universe Database GDB-17. J. Chem. Inf. Model. 2012, 52, 2864–2875. 10.1021/ci300415d. [DOI] [PubMed] [Google Scholar]
- Fink T.; Bruggesser H.; Reymond J.-L. Virtual Exploration of the Small-Molecule Chemical Universe below 160 Da. Angew. Chem., Int. Ed. 2005, 44, 1504–1508. _eprint: 10.1002/anie.200462457. [DOI] [PubMed] [Google Scholar]
- Delarue Bizzini L.; Müntener T.; Häussinger D.; Neuburger M.; Mayor M. Synthesis of trinorbornane. Chem. Commun. 2017, 53, 11399–11402. 10.1039/C7CC06273G. [DOI] [PubMed] [Google Scholar]
- Montavon G.; Rupp M.; Gobre V.; Vazquez-Mayagoitia A.; Hansen K.; Tkatchenko A.; Müller K.-R.; von Lilienfeld O. A. Machine learning of molecular electronic properties in chemical compound space. New J. Phys. 2013, 15, 095003. 10.1088/1367-2630/15/9/095003. [DOI] [Google Scholar]
- Hoja J.; Medrano Sandonas L.; Ernst B. G.; Vazquez-Mayagoitia A.; DiStasio R. A.; Tkatchenko A. QM7-X: A comprehensive dataset of quantum-mechanical properties spanning the chemical space of small organic molecules. Sci. Data 2021, 8, 43. 10.1038/s41597-021-00812-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum L. C.; Reymond J.-L. 970 Million Druglike Small Molecules for Virtual Screening in the Chemical Universe Database GDB-13. J. Am. Chem. Soc. 2009, 131, 8732. 10.1021/ja902302h. [DOI] [PubMed] [Google Scholar]
- Narayanan B.; Redfern P. C.; Assary R. S.; Curtiss L. A. Accurate quantum chemical energies for 133000 organic molecules. Chem. Sci. 2019, 10, 7449–7455. 10.1039/C9SC02834J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G.; Chen P.; Hsieh C.-Y.; Lee C.-K.; Liao B.; Liao R.; Liu W.; Qiu J.; Sun Q.; Tang J.; Zemel R.; Zhang S.. Alchemy: A Quantum Chemistry Dataset for Benchmarking AI Models. arXiv 2019, arXiv:1906.09427 [Google Scholar]
- Ramakrishnan R.; Hartmann M.; Tapavicza E.; von Lilienfeld O. A. Electronic spectra from TDDFT and machine learning in chemical space. J. Chem. Phys. 2015, 143, 084111. 10.1063/1.4928757. [DOI] [PubMed] [Google Scholar]
- Irwin J. J.; Shoichet B. K. ZINC - A Free Database of Commercially Available Compounds for Virtual Screening. J. Chem. Inf. Model. 2005, 45, 177–182. 10.1021/ci049714+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B.; von Lilienfeld O. A.. Dictionary of 140k GDB and ZINC derived AMONs. arXiv 2020, arXiv:2008.05260 [Google Scholar]
- Smith J. S.; Isayev O.; Roitberg A. E. ANI-1, A data set of 20 million calculated off-equilibrium conformations for organic molecules. Sci. Data 2017, 4, 170193. 10.1038/sdata.2017.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J. S.; Zubatyuk R.; Nebgen B.; Lubbers N.; Barros K.; Roitberg A. E.; Isayev O.; Tretiak S. The ANI-1ccx and ANI-1x data sets, coupled-cluster and density functional theory properties for molecules. Sci. Data 2020, 7, 134. 10.1038/s41597-020-0473-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkatchenko A.; Scheffler M. Accurate molecular van der Waals interactions from ground-state electron density and free-atom reference data. Phys. Rev. Lett. 2009, 102, 073005. 10.1103/PhysRevLett.102.073005. [DOI] [PubMed] [Google Scholar]
- Chmiela S.; Tkatchenko A.; Sauceda H. E.; Poltavsky I.; Schütt K. T.; Müller K.-R. Machine learning of accurate energy-conserving molecular force fields. Sci. Adv. 2017, 3, e1603015. 10.1126/sciadv.1603015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dandu N. K.; Ward L.; Assary R. S.; Redfern P. C.; Narayanan B.; Foster I. T.; Curtiss L. A. Quantum Chemically Informed Machine Learning: Prediction of Energies of Organic Molecules with 10 to 14 Non-Hydrogen Atoms. J. Phys. Chem. A 2020, 124, 5804. 10.1021/acs.jpca.0c01777. [DOI] [PubMed] [Google Scholar]
- Kim S.; Chen J.; Cheng T.; Gindulyte A.; He J.; He S.; Li Q.; Shoemaker B. A.; Thiessen P. A.; Yu B.; Zaslavsky L.; Zhang J.; Bolton E. E. PubChem 2019 update: improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. 10.1093/nar/gky1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakata M.; Shimazaki T. PubChemQC Project: A Large-Scale First-Principles Electronic Structure Database for Data-Driven Chemistry. J. Chem. Inf. Model. 2017, 57, 1300–1308. 10.1021/acs.jcim.7b00083. [DOI] [PubMed] [Google Scholar]
- Glavatskikh M.; Leguy J.; Hunault G.; Cauchy T.; Da Mota B. Dataset’s chemical diversity limits the generalizability of machine learning predictions. J. Cheminf. 2019, 11, 69. 10.1186/s13321-019-0391-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen F. H. The Cambridge Structural Database: a quarter of a million crystal structures and rising. Acta Crystallogr., Sect. B: Struct. Sci. 2002, 58, 380–388. 10.1107/S0108768102003890. [DOI] [PubMed] [Google Scholar]
- Stuke A.; Kunkel C.; Golze D.; Todorović M.; Margraf J. T.; Reuter K.; Rinke P.; Oberhofer H. Atomic structures and orbital energies of 61,489 crystal-forming organic molecules. Sci. Data 2020, 7, 58. 10.1038/s41597-020-0385-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schober C.; Reuter K.; Oberhofer H. Virtual Screening for High Carrier Mobility in Organic Semiconductors. J. Phys. Chem. Lett. 2016, 7, 3973–3977. 10.1021/acs.jpclett.6b01657. [DOI] [PubMed] [Google Scholar]
- Grambow C. A.; Pattanaik L.; Green W. H. Reactants, products, and transition states of elementary chemical reactions based on quantum chemistry. Sci. Data 2020, 7, 137. 10.1038/s41597-020-0460-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwilk M.; Tahchieva D. N.; von Lilienfeld O. A.. Large yet bounded: Spin gap ranges in carbenes. arXiv 2020, arXiv:2004.10600 [Google Scholar]
- Heinen S.; Schwilk M.; von Rudorff G. F.; von Lilienfeld O. A. Machine learning the computational cost of quantum chemistry. Mach. Learn.: Sci. Technol. 2020, 1, 025002. 10.1088/2632-2153/ab6ac4. [DOI] [Google Scholar]
- Donchev A. G.; Taube A. G.; Decolvenaere E.; Hargus C.; McGibbon R. T.; Law K.-H.; Gregersen B. A.; Li J.-L.; Palmo K.; Siva K.; Bergdorf M.; Klepeis J. L.; Shaw D. E. Quantum chemical benchmark databases of gold-standard dimer interaction energies. Sci. Data 2021, 8, 55. 10.1038/s41597-021-00833-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korth M.; Grimme S. Mindless” DFT Benchmarking. J. Chem. Theory Comput. 2009, 5, 993–1003. PMID: 26609608 10.1021/ct800511q. [DOI] [PubMed] [Google Scholar]
- Balcells D.; Skjelstad B. B. tmQM Dataset—Quantum Geometries and Properties of 86k Transition Metal Complexes. J. Chem. Inf. Model. 2020, 60, 6135. 10.1021/acs.jcim.0c01041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janet J. P.; Ramesh S.; Duan C.; Kulik H. J. Accurate Multiobjective Design in a Space of Millions of Transition Metal Complexes with Neural-Network-Driven Efficient Global Optimization. ACS Cent. Sci. 2020, 6, 513–524. 10.1021/acscentsci.0c00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen A. S.; Iyer S. M.; Ray D.; Yao Z.; Aspuru-Guzik A.; Gagliardi L.; Notestein J. M.; Snurr R. Q. Machine learning the quantum-chemical properties of metal-organic frameworks for accelerated materials discovery. Matter 2021, 4, 1578. 10.1016/j.matt.2021.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupp M.; Tkatchenko A.; Müller K.-R.; von Lilienfeld O. A. Fast and Accurate Modeling of Molecular Atomization Energies with Machine Learning. Phys. Rev. Lett. 2012, 108, 058301. 10.1103/PhysRevLett.108.058301. [DOI] [PubMed] [Google Scholar]
- Janet J. P.; Kulik H. J. Resolving Transition Metal Chemical Space: Feature Selection for Machine Learning and Structure-Property Relationships. J. Phys. Chem. A 2017, 121, 8939–8954. 10.1021/acs.jpca.7b08750. [DOI] [PubMed] [Google Scholar]
- Curtarolo S.; Setyawan W.; Hart G. L.; Jahnatek M.; Chepulskii R. V.; Taylor R. H.; Wang S.; Xue J.; Yang K.; Levy O.; Mehl M. J.; Stokes H. T.; Demchenko D. O.; Morgan D. AFLOW: An automatic framework for high-throughput materials discovery. Comput. Mater. Sci. 2012, 58, 218–226. 10.1016/j.commatsci.2012.02.005. [DOI] [Google Scholar]
- Saal J. E.; Kirklin S.; Aykol M.; Meredig B.; Wolverton C. Materials design and discovery with high-throughput density functional theory: the open quantum materials database (OQMD). JOM 2013, 65, 1501–1509. 10.1007/s11837-013-0755-4. [DOI] [Google Scholar]
- Chanussot L.; Das A.; Goyal S.; Lavril T.; Shuaibi M.; Riviere M.; Tran K.; Heras-Domingo J.; Ho C.; Hu W. The Open Catalyst 2020 (OC20) Dataset and Community Challenges. ACS Catal. 2021, 11, 6059. 10.1021/acscatal.0c04525. [DOI] [Google Scholar]
- Talirz L.; Kumbhar S.; Passaro E.; Yakutovich A. V.; Granata V.; Gargiulo F.; Borelli M.; Uhrin M.; Huber S. P.; Zoupanos S.; et al. Materials Cloud, a platform for open computational science. Sci. Data 2020, 7, 299. 10.1038/s41597-020-00637-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinnouchi R.; Miwa K.; Karsai F.; Kresse G.; Asahi R. On-the-Fly Active Learning of Interatomic Potentials for Large-Scale Atomistic Simulations. J. Phys. Chem. Lett. 2020, 11, 6946–6955. 10.1021/acs.jpclett.0c01061. [DOI] [PubMed] [Google Scholar]
- Garijo del Río E.; Mortensen J. J.; Jacobsen K. W. Local Bayesian optimizer for atomic structures. Phys. Rev. B: Condens. Matter Mater. Phys. 2019, 100, 104103. 10.1103/PhysRevB.100.104103. [DOI] [Google Scholar]
- Thompson A.; Swiler L.; Trott C.; Foiles S.; Tucker G. Spectral neighbor analysis method for automated generation of quantum-accurate interatomic potentials. J. Comput. Phys. 2015, 285, 316–330. 10.1016/j.jcp.2014.12.018. [DOI] [Google Scholar]
- Christensen A. S.; Faber F. A.; Huang B.; Bratholm L. A.; Tkatchenko A.; Müller K.-R.; von Lilienfeld O. A.. QML: A Python Toolkit for Quantum Machine Learning, 2017; https://github.com/qmlcode/qml.
- Huang B.; von Lilienfeld O. A.. AQML: Amons-based Quantum Machine Learning Code for Quantum Chemistry. 2020; https://github.com/binghuang2018/aqml. [DOI] [PubMed]
- Bonomi M. Promoting transparency and reproducibility in enhanced molecular simulations. Nat. Methods 2019, 16 (8), 670–673. 10.1038/s41592-019-0506-8. [DOI] [PubMed] [Google Scholar]
- Yao K.; Herr J. E.; Toth D. W.; Mckintyre R.; Parkhill J. The TensorMol-0.1 model chemistry: a neural network augmented with long-range physics. Chem. Sci. 2018, 9, 2261–2269. 10.1039/C7SC04934J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X.; Ramezanghorbani F.; Isayev O.; Smith J.; Roitberg A. TorchANI: A Free and Open Source PyTorch Based Deep Learning Implementation of the ANI Neural Network Potentials. J. Chem. Inf. Model. 2020, 60, 3408. 10.1021/acs.jcim.0c00451. [DOI] [PubMed] [Google Scholar]
- Huber S. P.; Zoupanos S.; Uhrin M.; Talirz L.; Kahle L.; Hauselmann R.; Gresch D.; Muller T.; Yakutovich A. V.; Andersen C. W.; et al. AiiDA 1.0, a scalable computational infrastructure for automated reproducible workflows and data provenance. Sci. Data 2020, 7 (1), 300. 10.1038/s41597-020-00638-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Bombarelli R.; Wei J. N.; Duvenaud D.; Hernández-Lobato J. M.; Sánchez-Lengeling B.; Sheberla D.; Aguilera-Iparraguirre J.; Hirzel T. D.; Adams R. P.; Aspuru-Guzik A. Automatic chemical design using a data-driven continuous representation of molecules. ACS Cent. Sci. 2018, 4, 268–276. 10.1021/acscentsci.7b00572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasquilla J.; Torlai G.; Melko R. G.; Aolita L. Reconstructing quantum states with generative models. Nat. Mach. Intell. 2019, 1, 155–161. 10.1038/s42256-019-0028-1. [DOI] [Google Scholar]
- Nesterov V.; Wieser M.; Roth V.. 3DMolNet: A Generative Network for Molecular Structures. arXiv 2020, arXiv:2010.06477 [Google Scholar]
- Segler M. H.; Preuss M.; Waller M. P. Planning chemical syntheses with deep neural networks and symbolic AI. Nature 2018, 555, 604. 10.1038/nature25978. [DOI] [PubMed] [Google Scholar]
- Schwaller P.; Gaudin T.; Lanyi D.; Bekas C.; Laino T. Found in Translation”: predicting outcomes of complex organic chemistry reactions using neural sequence-to-sequence models. Chem. Sci. 2018, 9, 6091–6098. 10.1039/C8SC02339E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair V. H.; Schwaller P.; Laino T. Data-driven Chemical Reaction Prediction and Retrosynthesis. Chimia 2019, 73, 997–1000. 10.2533/chimia.2019.997. [DOI] [PubMed] [Google Scholar]
- Schwaller P.; Laino T.; Gaudin T.; Bolgar P.; Hunter C. A.; Bekas C.; Lee A. A. Molecular transformer: A model for uncertainty-calibrated chemical reaction prediction. ACS Cent. Sci. 2019, 5, 1572–1583. 10.1021/acscentsci.9b00576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwaller P.; Petraglia R.; Zullo V.; Nair V. H.; Haeuselmann R. A.; Pisoni R.; Bekas C.; Iuliano A.; Laino T. Predicting retrosynthetic pathways using transformer-based models and a hyper-graph exploration strategy. Chem. Sci. 2020, 11, 3316–3325. 10.1039/C9SC05704H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesciullesi G.; Schwaller P.; Laino T.; Reymond J.-L. Transfer learning enables the molecular transformer to predict regio-and stereoselective reactions on carbohydrates. Nat. Commun. 2020, 11, 4874. 10.1038/s41467-020-18671-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasquilla J.; Melko R. G. Machine learning phases of matter. Nat. Phys. 2017, 13, 431–434. 10.1038/nphys4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ch’Ng K.; Carrasquilla J.; Melko R. G.; Khatami E. Machine learning phases of strongly correlated fermions. Phys. Rev. X 2017, 7, 031038. 10.1103/PhysRevX.7.031038. [DOI] [Google Scholar]
- Broecker P.; Carrasquilla J.; Melko R. G.; Trebst S. Machine learning quantum phases of matter beyond the fermion sign problem. Sci. Rep. 2017, 7, 8823. 10.1038/s41598-017-09098-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas-Hernández R. A.; Sous J.; Berciu M.; Krems R. V. Extrapolating quantum observables with machine learning: inferring multiple phase transitions from properties of a single phase. Phys. Rev. Lett. 2018, 121, 255702. 10.1103/PhysRevLett.121.255702. [DOI] [PubMed] [Google Scholar]
- Cheng B.; Engel E. A.; Behler J.; Dellago C.; Ceriotti M. Ab initio thermodynamics of liquid and solid water. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 1110–1115. 10.1073/pnas.1815117116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng B.; Mazzola G.; Pickard C. J.; Ceriotti M. Evidence for supercritical behaviour of high-pressure liquid hydrogen. Nature 2020, 585, 217–220. 10.1038/s41586-020-2677-y. [DOI] [PubMed] [Google Scholar]
- Carleo G.; Troyer M. Solving the quantum many-body problem with artificial neural networks. Science 2017, 355, 602–606. 10.1126/science.aag2302. [DOI] [PubMed] [Google Scholar]
- Schütt K.; Gastegger M.; Tkatchenko A.; Müller K.-R.; Maurer R. J. Unifying machine learning and quantum chemistry with a deep neural network for molecular wavefunctions. Nat. Commun. 2019, 10, 5024. 10.1038/s41467-019-12875-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann J.; Schätzle Z.; Noé F. Deep-neural-network solution of the electronic Schrödinger equation. Nat. Chem. 2020, 12, 891–897. 10.1038/s41557-020-0544-y. [DOI] [PubMed] [Google Scholar]
- Carrasquilla J. Machine learning for quantum matter. Adv. Phys. X 2020, 5, 1797528. 10.1080/23746149.2020.1797528. [DOI] [Google Scholar]
- Raccuglia P.; Elbert K. C.; Adler P. D.; Falk C.; Wenny M. B.; Mollo A.; Zeller M.; Friedler S. A.; Schrier J.; Norquist A. J. Machine-learning-assisted materials discovery using failed experiments. Nature 2016, 533, 73–76. 10.1038/nature17439. [DOI] [PubMed] [Google Scholar]