

Abstract
Capsule:
Hyperandrogenemia in an obese PCOS mouse model results in altered glucose/insulin metabolism and mitochondrial structure and function in the oocytes, in part explaining adverse outcomes and inheritance patterns seen in PCOS.
Objective:
To study the oocyte quality by means of mitochondrial structure and function in a well-established classic PCOS mouse model.
Design:
Animal study using an obese PCOS mouse model compared with control.
Setting:
Animal research facility in a tertiary care university hospital setting
Animals:
C57/B6J mice
Intervention:
Three week old mice had subdermal implants of DHT controlled release pellet or placebo for 90 days.
Main Outcome Measures:
The mouse model was validated by performing glucose tolerance test, HbA1c levels, body weight and estrous cycle analyses. Oocytes were subsequently isolated and were used to investigate mitochondrial membrane potential, oxidative stress, lipid peroxidation, ATP production, mtDNA copy number, transcript abundance, histology and electron microscopy.
Results:
Results showed glucose intolerance and hyperinsulinemia along with dysregulated estrus cycle. Analysis of the oocytes demonstrated impaired inner mitochondrial membrane function, increased ATP production and mtDNA copy number, altered RNA transcript abundance and aberrant ovarian histology. Electron microscopy of the oocytes showed severely impaired mitochondrial ultrastructure.
Conclusion:
The obese PCOS mouse model shows a decreased oocyte quality related to impaired mitochondrial function.
Keywords: PCOS, Mitochondria, Oocyte, Androgen, Glucose intolerance
Introduction
Polycystic ovary syndrome (PCOS) is the most common ovulatory disorder in the world, affecting up to 10% of women, translating to more than 100 million women worldwide (1). PCOS may be described as the common endpoint of several metabolic disturbances, including altered hypothalamic-pituitary-ovarian signaling, insulin resistance and glucose intolerance, obesity and metabolic syndrome, hyperandrogenemia, and other environmental and genetic factors (1–4). Given this variability, the diagnosis is also complex, though the current accepted algorithm is the Rotterdam criteria: polycystic ovarian morphology, irregular menses and clinical or laboratory evidence of hyperandrogenemia (5). At least two of these three criteria will qualify a patient for the diagnosis of PCOS provided routine workup excludes other etiologies such as prolactin or thyroid disorders (5). The overall severity of the disease depends upon the combination of Rotterdam criteria a patient exhibits and hence this disease has a spectrum of phenotypes (6). Patients with all three criteria tend to fall on a more extreme end, and patients with only two of the three criteria will manifest a less pervasive phenotype (6).
Regardless of where a patient falls on this spectrum, PCOS has strong associations with many adverse outcomes. PCOS women have been shown to have increased obstetric risks including preeclampsia, gestational diabetes, preterm delivery and higher infant mortality (7, 8). Further, there are longstanding implications in chronic disease states such as hypertension, diabetes, depression, stroke and some cancers (7). In the realm of fertility, PCOS patients have higher miscarriage rates (9, 10), higher risk of ovarian hyperstimulation (OHSS), and lower APGAR scores in babies born after assisted reproduction technology (ART) (11). We recently showed that embryos from hyperandrogenic PCOS women grew faster until morula (12). Interestingly, PCOS patients also showed a higher miscarriage rate strongly indicating a hyperandrogenic ovarian microenvironment could alter fertility and obstetric outcomes (13).
As PCOS is more a compilation of subpopulations diagnosed under the varying combinations of the diagnostic criteria, this heterogeneity poses difficulty in its study. Indeed, the general PCOS population is abound with confounders, and this is particularly true in the case of obesity. Up to 80% of PCOS patients are obese, and given the epidemic of obesity in the 21st century, it is known that this well studied comorbidity is also similarly associated with a wide range of adverse events (14–16). Moreover, PCOS patients are more likely to have insulin resistance and glucose intolerance, commonly meeting criteria for metabolic syndrome as well (2). The degree to which hyperandrogenemia and hyperinsulinemia play a role in the downstream adverse outcomes of PCOS remains to be fully elucidated, but it appears that increasing obesity exacerbates the severity of the phenotype (17).
During folliculogenesis, an oocyte’s mitochondria increases in number from a handful to over 100,000 to provide the energy necessary for the embryo post fertilization (18, 19). Mitochondria are maternally inherited and essential for growth this process of rapid development for early embryo life is critical for survival. Prior studies in PCOS have shown associations of negative effects at the mitochondrial level with corresponding decreased oocyte quality, suggesting that mitochondrial competence in the oocyte warrants further study (20–22). Specifically, associations have been demonstrated regarding lower inner mitochondrial membrane potential and fertilization rates, increased reactive oxygen species and mitochondrial DNA damage with apoptosis, as well as lower ATP levels and lysis during early embryo development (19, 23). Furthermore, transgenerational inheritance of mitochondrial pathology has been reported, even with no further interventions in offspring (24–26). Despite the heritability of PCOS, research has not been able to identify any candidate genes to explain the inheritance pattern (4, 27–29). Given that offspring of patients with hyperandrogenic disorders such as congenital adrenal hyperplasia are at increased risk for PCOS, it is possible that androgen exposure may play a role at the level of the oocyte mitochondria (30).
Many labs have characterized the metabolic outcomes of PCOS in various mouse models noting increased lipids, glucose intolerance, insulin resistance, pancreatic dysfunction and cardiovascular disease (31, 32). Most of these PCOS models are created via administration of androgens, including testosterone, androstenedione, dehydroepiandrosterone (DHEA), or dihydrotestosterone (DHT), all of which may be given prenatally, pre-pubertally or post-pubertally (31). It is important to note that DHT is not aromatizable to more estrogenic products, and therefore may be considered a more “conserved” androgen. Studies using these models have also documented changes in reproductive function with decreased numbers of corpora lutea, increased follicular atresia and subfertility, though few studies have been performed on the oocyte or embryo (20, 22, 33). In a prior study, we investigated a lean PCOS mouse model with a normal body mass index (BMI) demonstrating adverse oocyte mitochondrial function and structure (34). Our objective in this study was to measure the mitochondrial function and to assess its structure in the oocytes of a classic obese PCOS phenotype using a DHT induced PCOS mouse model.
Materials and Methods
All experiments were approved through the Institutional Animal Care and Use Committee of Baylor College of Medicine (AN-7156). To create the obese PCOS mouse model, seventy-five 3 week old C57/B6J strain mice were obtained at weaning and randomly divided into treatment group (n=38) and control group (n=37). A 90 day controlled release pellet containing 2.5 mg DHT (27.5 μg/day, Innovative Research of America, Sarasota, Florida) or placebo was implanted under sterile surgical conditions. All mice were fed standard chow (PicoLab® Select Rodent 50IF/IR 5V5R from Lab Diet, Houston, TX, USA). In experiments involving oocytes, ‘n’ in each experiment denotes the number of mice used and not the number of oocytes. Varying numbers of oocytes from each animals were used for each experiments and the numbers used is mentioned in the appropriate experiments/legends. The methods for each of the experiments involving the oocytes have been previously published in detail, but are briefly summarized below (34).
Pups were weighed and measured from snout to anus in millimeters at 3, 4, 8, and 12 weeks up to euthanasia at 16 weeks to calculate their BMI. Testing for estrous cyclicity was performed at 3 months of age by observing the vaginal cytology using vaginal smears as previously described (35–37) for approximately 21 days, or 4 consecutive cycles. Glucose tolerance test (GTT) was performed 12 weeks old mice as in previously published mouse models (38, 39). Hemoglobin A1c levels were analyzed at the time of euthanasia at 16 weeks using the A1cNow+ test kit (PTS Diagnostics, Indianapolis, IN, USA) following the manufacturer’s instructions.
Plasma T was measured by using an ELISA kit (ADI-900-065, Enzo Life Sciences, Inc. NY. USA) following manufacturer’s instruction as reported earlier (40). Briefly, 100μl of plasma (diluted 1:40) and standards were added to each well pre-coated with antibody. Antibody solution (50 μl) was pipetted into each well, except the blank, total activity (TA) and non-specific wells and incubated for 1 hour at room temperature with constant shaking (~500 rpm). Conjugate solution was added into each well, except the TA and blank wells followed by an incubation for 1 hour at room temperature with constant shaking. The contents were emptied and washed thrice. Conjugate solution (5 μl) was added to the TA well followed with the addition of 200 μl of the substrate solution to every well. The plate was then incubated at 37°C for 1 hour without shaking. Stop solution (50 μl) was added to arrest the color development. The plate was then read at 405 nm using a plate reader (CLARIOstar™, BMG Labtech Inc. NC, USA).
Plasma DHT was measured using an ELISA kit from Abnova (Walnut, CA. USA) following manufacturer’s instruction. Briefly, 50μl of serum and standards were added to wells pre-coated with antibody. Following this, 100μl of antibody solution was then pipetted into each well, and the plate was incubated at 1 room temperature for 1 hour on a shaker. Next, contents were emptied and washed three times with diluted wash buffer. Following this, 150μl of TMB substrate was added into each well, the plate was shaken for 10 seconds then incubated for 10 minutes at room temperature. Finally, 50μl of stop solution was added into each well, and the plate was read on a plate reader at 405 nm (CLARIOstar™, BMG Labtech Inc. NC, USA). PCOS mice were sub-fertile and hence, it was necessary to perform superovulation to retrieve oocytes.
Mitochondrial function was evaluated on individual oocytes by single cell imaging experiments using fluorescent probes and the images were analyzed using Image J software (34). Inner mitochondrial membrane (IMM) potential was measured using JC-1 dye (Affymetrix, Santa Clara, CA., incubated for 30 minutes in 2μM concentration), reactive oxygen species (ROS) formation was measured using CellRox Green (ThermoFisher, Waltham, MA., incubated for 30 minutes in 5μM concentration), and lipid peroxidation was measured by using BODIPY® 581/591 (Life Technologies, Carlsbad, CA., incubated for 45 minutes in 1μg/ml concentration). Single cell imaging of oocytes for IMM, ROS and lipid peroxidation were performed using 2-5 oocytes/study/group from 5-8 mice.
ATP concentration was measured pooling 5-15 oocytes/mouse, using a luciferase assay kit (ThermoFisher, Waltham, MA) according to the manufacturer’s instruction. RNA transcript abundance was measured using qPCR. RNA was isolated and cDNA was amplified using 3-5 oocytes collected from each mouse. Previously validated genes known to be relevant to ovary and mitochondria were amplified using specific primers using cDNA library as templates. KEGG analysis was performed on all genes that showed altered abundance to predict involved pathways. Genomic DNA was isolated and amplified to measure mitochondrial DNA copy number. Mitochondrial gene cytochrome oxidase 1 (mtCo1) and genomic encoded tubulin was used as a reference gene, and delta delta Ct model was utilized for comparison between the groups (41). Details and primers were published earlier (34).
During euthanasia, ovaries from 4 month old unstimulated mice in diestrus were collected and processed for transmission electron microscopy (TEM) (n=4 in each group). The ‘n’ in each study corresponds to the number of mice used in each experiment. Student’s t test, chi square and ANOVA were used where appropriate for all above measurements, p< 0.05 was considered statistically significant. The data were analyzed using GraphPad Prism version 6.0 (San Francisco, CA, USA).
Results
DHT treated animals were obese with higher BMI
After the first 3 weeks, the experimental group weighed more than controls throughout the lifespan, Figure 1. The obese PCOS mice weighed more at week 4 (Control 14.94 ± 0.26 grams vs. obese PCOS 16.67 ± 0.22 grams, p<0.01), at week 8 (Control 19.56 ± 0.23 grams vs. obese PCOS 23.34 ± 0.31 grams, p<0.0001), at week 12 (Control 21.65 ± 0.34 grams vs. obese PCOS 26.41 ± 0.52 grams, p<0.0001), and at 16 weeks (Control 22.62 ± 0.38 grams vs. obese PCOS 27.56 ± 0.55 grams, p<0.0001). Further, the obese PCOS mice had an increased BMI at 12 weeks (Control 0.27 ± 0.007 g/cm2 vs. obese PCOS 0.34 ± 0.008 g/cm2, p<0.0001; Figure 1).
Figure 1.
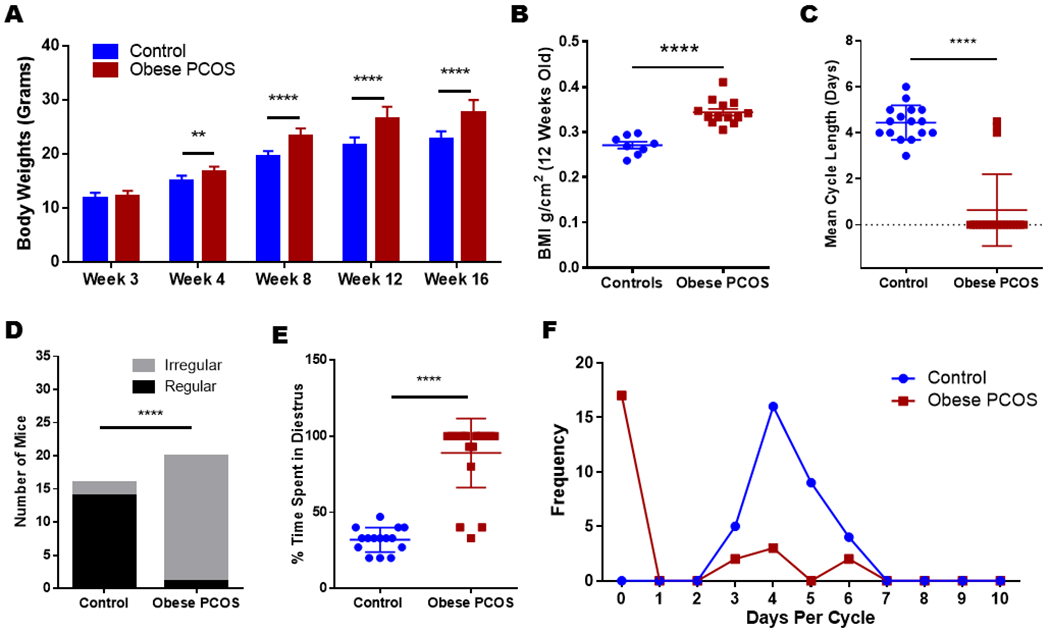
Body weights, BMI and estrous cycle: The obese PCOS mouse model displayed significantly increased body weights starting at week four (A), with a higher BMI (B). They also showed irregular cycles or acyclic when compared to controls. The mean cycle length (C) shows that most PCOS mice remained in diestrus (D&E), and were acyclic (E&F) ** p<0.01, **** p<0.0001, n=18 controls and 20 obese PCOS.
Obese PCOS mice had Irregular Cycles or Acyclic:
The obese PCOS mouse model displayed significantly irregular or no cycles when compared to controls; all but 3 of 20 animals remained in diestrus for the duration of monitoring period, indicating significant impairment in cyclicity (Control 4.44 ± 0.19 days vs. obese PCOS 0.64 ± 0.35 days, p<0.0001; Figure 1). Only 2 of the 18 control mice had irregular cycles; however, all but one of the obese PCOS mice had persistently irregular cycles during the monitoring period (Control 87.5% (n=16/18) with regular cyclicity vs. obese PCOS 5% (n=1/20) with regular cyclicity, p<0.0001). Finally, the percentage of time spent in the diestrus phase was significantly higher for the obese PCOS mouse group compared to controls (Control 31.93 ± 2.08% vs. Obese PCOS 89.0 ± 5.06%, p<0.0001; Figure 1).
Obese PCOS mice had elevated Androgen levels:
The obese model demonstrated higher testosterone (Control 2.23 ± 0.09 ng/ml vs. Obese PCOS 4.23 ± 0.6 ng/ml, p<0.01) with a two-fold increase and DHT (Control 61.91 ± 11.94 pg/ml vs. Obese PCOS 251.4 ± 33.95 pg/ml, p<0.001) levels with four-fold increase when compared to their respective controls, Figure 2).
Figure 2.
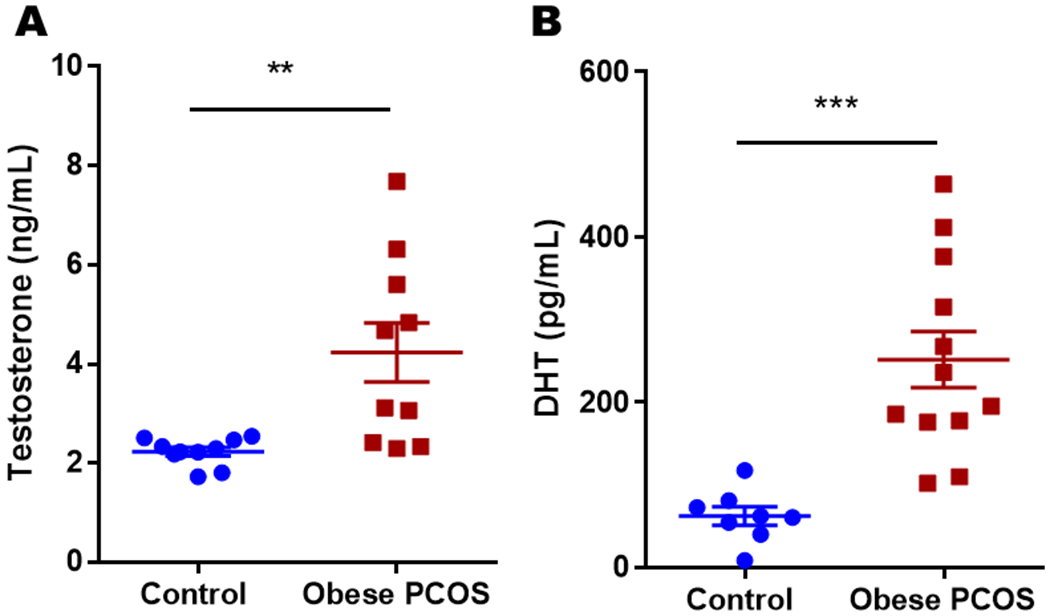
Testosterone and DHT levels: The obese PCOS mouse model showed significantly higher testosterone and DHT levels compared to controls. ** p<0.01, *** p<0.001, n=8-10 for controls and 10-12 for obese PCOS group.
Obese PCOS Mouse were Glucose Intolerant:
We performed GTT at 12 weeks of age. The obese PCOS model showed significant glucose intolerance and elevated insulin concentrations when compared to controls (Figure 3A and 3B). Obese PCOS mice had similar glucose values at 0 and 15 minutes (Control 7.43 ± 0.35 mmol/l vs. obese PCOS 9.36 ± 0.54 mmol/l at 0 minutes; Control 14.8 ± 0.89 mmol/l vs. obese PCOS 18.22 ± 0.71 mmol/l at 15 minutes, p>0.05). However, the obese PCOS mice had elevated glucose values at 30 minutes (Control 13.68 ± 0.69 mmol/l vs. obese PCOS 18.84 ± 1.2 mmol/l, p<0.05), at 60 minutes (Control 12.48 ± 0.81 mmol/l vs. obese PCOS 20.2 ± 1.88 mmol/l, p<0.0001), at 120 minutes (Control 9.93 ± 0.51 mmol/l vs. obese PCOS 15.81 ± 0.2.1 mmol/l, p<0.01), and at 180 minutes (Control 10.62 ± 0.89 mmol/l vs. obese PCOS 15.01 ± 1.6 mmol/l, p<0.05, Figure 3A). The overall area under the curve for the GTT showed higher glucose values for the obese PCOS model as well (Control 2062 ± 93.04 mmol/l*180 minutes vs. obese PCOS 3075 ± 281.9 mmol/l*180 minutes, p<0.01) (Figure 3C). Insulin levels at each time point were also assessed, and the obese PCOS model showed evidence of hyperinsulinemia at every time point: at 0 minutes (Control 44.06 ± 12.78 pmol/l vs. obese PCOS 120.36 ± 9.66 pmol/l, p<0.01); at 15 minutes (Control 86.62 ± 17.16 pmol/l vs. obese PCOS 214.38 ± 11.35 pmol/l, p<0.0001); at 30 minutes (Control 93.07 ± 17.14 pmol/l vs. obese PCOS 181.19 ± 23.25 pmol/l, p<0.001); at 60 minutes (Control 91.34 ± 12.53 pmol/l vs. obese PCOS 154.54 ± 19.66 pmol/l, p<0.05); at 120 minutes (Control 50.77 ± 14.9 pmol/l vs. obese PCOS 162.15 ± 9.54 pmol/l, p<0.0001); and at 180 minutes (Control 80.53 ± 11.82 pmol/l vs. obese PCOS 168.76 ± 11.9 pmol/l, p<0.001; Figure 3B). The overall area under the curve for insulin concentration was significantly higher in the obese PCOS model compared to controls (Control 13298 ± 1621 pmol/l*180 minutes vs. obese PCOS 29941 ± 1498 pmol/l*180 minutes, p<0.0001; Figure 3D). Moreover, there was a statistically significant difference in hemoglobin A1c between the obese PCOS mouse model and controls (Control 4.26 ± 0.05 vs. obese PCOS 4.83 ± 0.1, p<0.001; Figure 3E).
Figure 3.
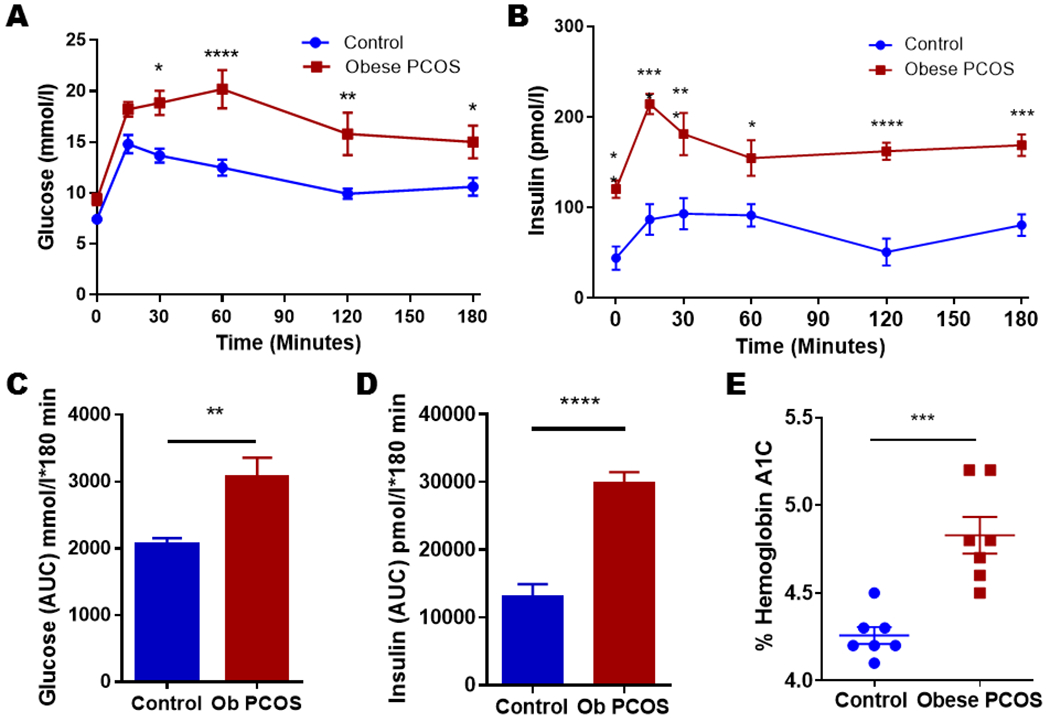
Glucose tolerance and Insulin levels: The obese PCOS mice displayed glucose intolerance at 30, 60, 120, and 180 minutes (A) with increased glucose AUC (C) as well as hyperinsulinemia (B) with increased insulin AUC (D) at all measured time points. The obese PCOS mouse model had increased hemoglobin A1c values (E) when compared to controls. * p<0.05, ** p<0.01, *** p<0.001 and **** p<0.0001, n=5-10 controls and 7-10 obese PCOS.
Oocytes from obese PCOS mouse had compromised IMM potential:
We measured IMM potential, ROS and lipid peroxidation in live oocytes obtained from obese PCOS mouse and controls. IMM potential was measured using JC-1. The obese PCOS mouse exhibited a lower red: green ratio, indicating a lower IMM potential (Control 1.66 ± 0.18 vs. obese PCOS 1.28 ± 0.07, p<0.05; Figure 4). Imaging for ROS using CellRox Green demonstrated similar concentrations of ROS between the obese PCOS model and controls (Control 12.65 ± 1.31 RFUs vs. obese PCOS 12.29 ± 1.72 RFUs, p>0.05; Supplementary Figure 1). There was no difference in relative fluorescence units in oocytes from obese PCOS mice exposed to BODIPY compared to controls (Control 19.32 ± 3 RFUs vs. PCOS 15.34 ± 2.1 RFUs, p>0.05; Supplementary Figure 2).
Figure 4.
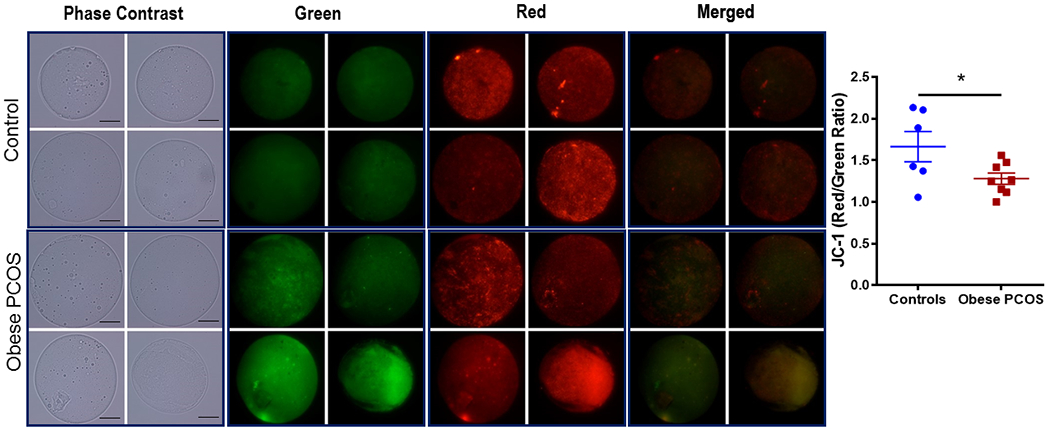
Inner Mitochondrial Membrane Potential: Pictorial representation of oocytes from control (above) and obese PCOS (below) mice. Brightfield microscopy, followed by green fluorescence, red fluorescence, and finally merged using JC-1 dye at 60x magnification. The obese PCOS mice show a lower red: green ratio, and therefore lower IMM potential compared to controls, 2-5 oocytes/mice. * p<0.05, n=6 controls and 8 obese PCOS, scale bar =20μm.
Oocytes from obese PCOS mice had higher mtDNA copy number and ATP levels:
The obese PCOS mice had a higher mtDNA copy number compared to controls (Control 61.23 ± 22.42 vs. obese PCOS 198 ± 6.35, p<0.05; Figure 5A). Further, the obese PCOS model had a higher concentration of ATP compared to controls (Control 23.13 ± 2.1 μM/oocyte vs. obese PCOS 34.86 ± 3.6 μM/oocyte, p=0.012; Figure 5B).
Figure 5.
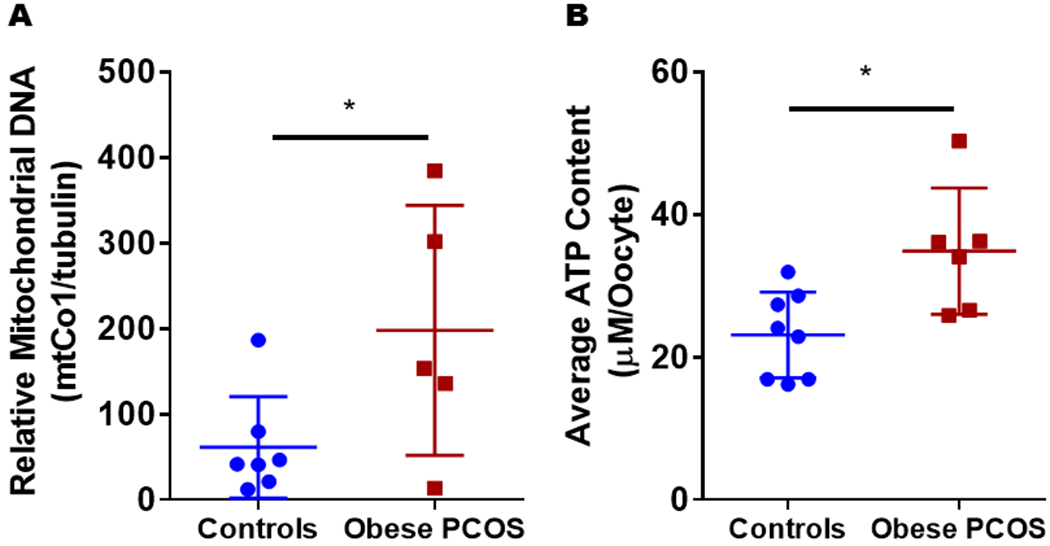
Mitochondrial Abundance and ATP Production: Obese PCOS mice demonstrated a higher mtDNA copy number (A) and ATP production (B) when compared to controls. * p<0.05, n=7-8 control and 5-6 obese PCOS.
RNA Transcript Abundance varied between control and obese PCOS oocytes:
Analysis of RNA transcript abundance that are known to play a role in PCOS were evaluated. Results demonstrated variable expression between the control and obese PCOS mouse model, Figure 6. Of the chromosomal genes that were investigated, there was a significant decrease in Ppp2ca, but an increase in Tuba1b, both related to meiosis, compared to controls, p<0.05. Similarly, regarding centrosome-related genes, there was a significant decrease in Tacc1, but an increase in Pcm1 in the obese PCOS mouse compared to controls, p<0.05. Mitochondrial genes related to complex I showed a decrease versus controls including mtNd1, mtNd4, and mtNd5, p<0.05. Mitochondrial genes including mtND2, mtND3, mtND4l, mtND6, mtCytb, mtCo1, mtCo2, mtCo3, mtATP6, mtATP8, mtRNr1, and mtRnr2 showed similar expression in both controls and obese PCOS groups (data not shown). Similarly, nuclear coded genes including Mfn1, Itga6, Dnm1, Ect2, Atrx, Cep70, Spast, Igf1, Igfr1, Gdf9, Nobox, Gja1, Bmp15, Diaph2, Zp3, Zmiz1, Kat2b, Xrcc1, Ppp2r1a, and Tuba1b showed similar expression in both controls and obese PCOS groups (data not shown).
Figure 6.
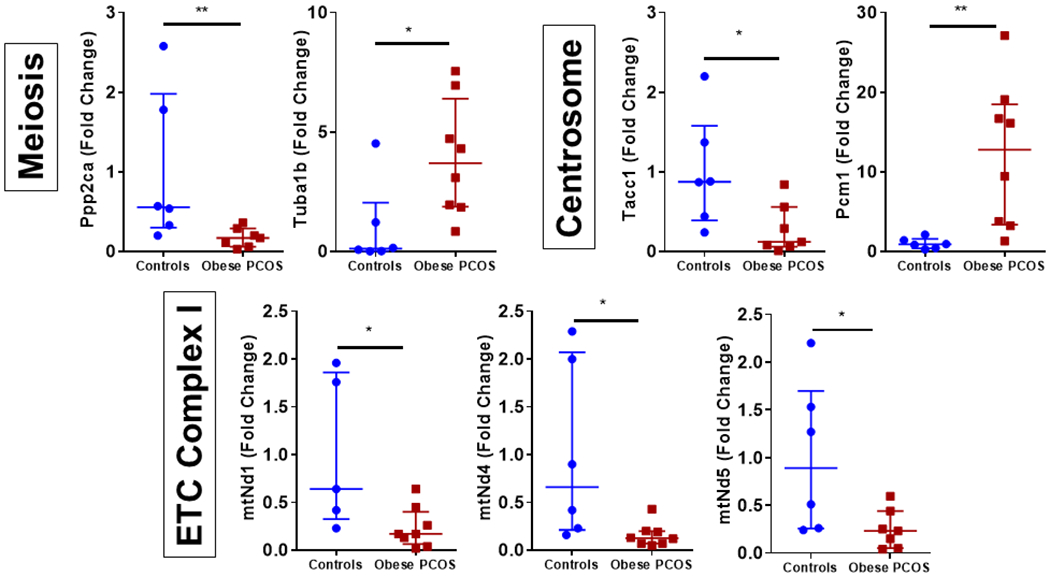
RNA Transcript Abundance: RNA transcript abundance in the obese PCOS mouse mice for genes pertaining to meiosis, centrosome and electron transport chain. * p<0.05, ** p<0.01, 3-5 oocytes/mice (n=5-8/group).
Oocytes from obese PCOS mice had altered mitochondrial ultrastructure:
Ultrastructural examination with electron microscopy showed that oocytes from obese PCOS mouse displayed structurally abnormal mitochondria when compared to the controls. We observed malformed cristae with concentric circles in some, or swollen cristae or a total loss of cristae in others in the oocytes from the obese PCOS mouse. The controls had well rounded mitochondria with normal looking cristae. Further, they also had normal looking electron dense materials inside the mitochondria (Figure 7).
Figure 7.
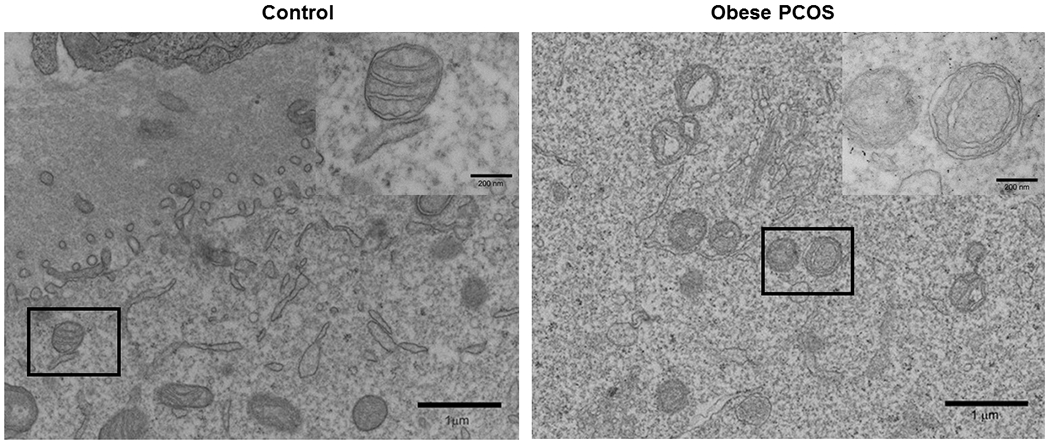
Transmission Electron Microscopy of Oocytes: Transmission electron microscopy image of control showing normal mitochondrial structure (A) and obese PCOS showing compromised structure with abnormally formed cristae (B). n=4/group.
Discussion
The obese PCOS mouse model demonstrated a phenotype consistent with classic PCOS with significantly altered cyclicity, elevated androgens, glucose intolerance, pervasive hyperinsulinemia, and elevated hemoglobin A1c (7, 42). Our results are consistent with prior studies of similar models using supra-physiologic androgens pre-pubertally that result in a severe form of PCOS and metabolic syndrome (34). In this study we show evidence for the first time that post-pubertal androgen administration affects oocyte mitochondrial structure and function; specifically, the obese PCOS mice displayed compromised IMM potential, increased ATP concentration as well as higher mitochondrial DNA copy number compared to controls. Qualitative studies using TEM showed abnormal mitochondrial ultrastructure. These findings are consistent with prior studies in other tissue types showing that exposure to DHT alters insulin and glucose metabolism and affects mitochondria (43). Taken together, these data present exciting implications for the relationship seen between hyperandrogenemia and metabolic syndrome, though further studies are needed to elucidate the tissue specific cellular pathways.
Initially, a primordial oocyte will only have a small number of mitochondria, though that number increases to well over 100,000 through folliculogenesis (44, 45). Once ovulated, there will be no further proliferation in mitochondria until blastulation (44, 45). Therefore, it is imperative to build up sufficient stores of healthy mitochondria to sustain the early processes of embryogenesis from fertilization through the first cell divisions (18). The mitochondria generate energy in the form of ATP, via the electron transport chain (ETC) (23). The ETC pumps protons across the IMM to generate a transmembrane gradient for the ATPase enzyme to utilize in creating ATP (46). If compromised, the ETC cannot produce adequate energy stores, and this has been shown to be a marker for poor mitochondrial function (46). Mitochondria are also integrally involved in ROS formation and scavenging which in part regulate apoptosis (47). Increased ROS is associated with adverse outcomes such as developmental arrest or even apoptosis, and physical DNA damage (19, 23, 48, 49). Thus, continued studies on mitochondrial competence in the oocyte or embryo may provide useful markers to the reproductive competence in the oocyte and embryo.
From the results of our study, hyperandrogenemia impacts both mitochondrial function and structure in the oocyte. Of interest in our findings was the lack of increased ROS despite altered IMM potential. Although we did not find increased ROS in the oocytes, it is likely that ROS could play a factor in the granulosa cells which could indirectly affect the oocytes. Lai et al showed that there is an increased ROS in the granulosa cells of PCOS patients leading to granulosa cell apoptosis (50). Taken in full, these results likely indicate the oocyte of the PCOS mice attempting compensation during folliculogenesis to increase mitochondrial abundance in an environment actively being exposed to elevated androgens. Indeed, the chronic exposure of the DHT could be directly affecting the oocyte as demonstrated in altered IMM potential; but the lack of ROS could be due to increased proliferation of mitochondria (as seen in increased ATP production and mtDNA copy number) to compensate for this impaired function. It is important to consider the role that mtDNA copy number may have played in the expression of RNA transcripts encoded in the mtDNA that was measured. However, as there was only variation in a few of the mitochondrial RNA transcripts, it is also possible other mechanisms are affecting the expression or persistence of these transcripts besides abundance of mtDNA copy number.
While there are limited studies of direct androgenic effects on the mitochondria of the oocyte, studies in other tissues have demonstrated similar findings as in the present study. Androgen effects in neuronal tissues have been described to have impacts in IMM potential, ATP production, as well as apoptotic pathways (51). While physiologic doses of androgens appear to be neuroprotective, higher levels of exposure of testosterone and DHT may induce mitochondrial dysfunction and apoptosis (52). Further, two studies using DHT in a pregnant rat model also demonstrated that hyperandrogenism and concurrent insulin resistance resulted in mitochondrial damage related to oxidative stress, ultimately affecting placentation and fetal survival (53, 54).
Importantly, the effects of androgens on mitochondria are dependent on several biological contexts such as age, sex, and tissue site (51). The above studies in the nervous system have shown both protective and toxic effects at different levels, other studies in skeletal and cardiac muscle have demonstrated protective effects of androgens in the apoptotic pathway (47). These data further highlight the importance of understanding the role of androgens and mitochondria in each tissue type.
Studies on mitochondrial structure have shown that increasing volume with decreasing cristae are the result of increased oxidative damage, and is commonly seen in routine aging (55). These prior findings would be consistent with our findings of swollen or even absent cristae and provides a foundation explaining in part the mechanism by which it may be occurring in our model.
PCR was performed to quantify genes involved in key functions in oocytes. In most cell lines, mRNA transcripts last a few hours (56), though an oocyte’s mRNA transcripts will last several days (56) to get through fertilization to blastulation and ultimately implantation (56). Therefore, studies of RNA expression are more appropriately considered to be measuring quantities of RNA transcripts created prior to ovulation (56, 57). In the present study, genes showed both up and downregulation in comparison to controls. KEGG analysis of the obese model found significance in oxidative phosphorylation, Parkinson’s, Alzheimer’s and Huntington’s disease, consistent with increasing findings of the role sex steroids play in chronic neuroprotection, though the correlation of these genes with the oocyte remains unclear at this time (51). Interestingly, there was also correlation with non-alcoholic fatty liver disease, which is known to have increased prevalence with obesity and PCOS (58). PPP2ca, encoding the catalytic unit of protein phosphatase 2, is a key regulator of cell cycle, was downregulated compared to controls, which is of great interest given the multi-pathway involvement of this gene. Additionally, the mitochondrial genome is known to have hormone response elements for both estrogens and androgens (57). Additionally, the mitochondrial genes mtNd1, mtNd4, and mtNd5 all showed decreased abundance compared to controls. These genes are related to complex I of the ETC, which has been shown to be impaired in neural tissue exposed to androgens (21). Our findings regarding the alterations in mRNA abundance may partly explain these earlier findings.
Finally, given that mitochondria are strictly maternally inherited, any alterations to the mitochondria of the oocyte are de facto transferred to the offspring. This may explain the inheritance pattern seen in PCOS offspring (59). While PCOS is hereditary, multiple studies have yet to identify strong gene candidates to explain this pattern (27–29). Moreover, it stands to reason that the propagation of these mitochondria from a fertilized egg to a live birth would result in abnormal mitochondrial function throughout the body. In fact, daughters of patients with PCOS have been shown to have lower birth weights and a higher prevalence of small for gestational age (SGA) as well as aberrations in follicle development with increased ovarian volumes and AMH concentrations (60, 61). In prior animal studies using a rat PCOS model, mitochondrial dysfunction in the pancreas (62) and kidney (63) have been noted, and human studies have now reported altered mitochondrial function in skeletal muscle (64) as well as in cumulus cells of PCOS patients (49).
We have previously published a similar study on a lean PCOS mouse model (34). Our present study on obese PCOS and our previous study in lean PCOS mouse model show that these two models have some common features but several distinct features. It appears that the timing of exposure determines the intensity of the phenotype. Obese PCOS mice have a severe phenotype with higher glucose intolerance, HbA1c levels, insulin resistance along with severely compromised estrous cycle. Further, mitochondrial copy number in the oocytes was affected in the obese PCOS mice and not in the lean model. Oocyte mitochondria from both lean and obese PCOS animal showed significant membrane depolarization, however only the lean model showed increased oxidative stress suggesting possible different mechanisms. It may be that the obese model, created via long term exposure of androgen, shows some compensation by increasing numbers of mitochondria as indicated by the elevated mtDNA copy number whereas the lean model, created with a brief early intervention, does not have the same compensation and thus does not have the mitochondrial numbers to control the balance of ROS. This is a plausible explanation for our findings, though requires more study. In addition, oocyte gene expression patterns were also distinct between the two models. Mitochondrial ultrastructure was compromised in both models. It is not clear how androgen inflicts mitochondrial damage to oocytes and warrants further future investigations.
Limitations in our study should be considered in interpretation of these results. Measurement of the competence or quality of an oocyte requires single cell study; however, there are an abundance of oocytes in each ovary. The conclusions made regarding the population of oocytes as a whole from a small proportion warrant caution. Further, we had to use superovulation to isolate oocytes. We do not know if the superovulation process affected the oocyte function and gene expression. Elevated androgens can lead to hyperinsulinemia as seen in the present study, and elevated insulin levels also lead to hyperandrogenemia; therefore, the relationship between these two is tightly linked and difficult to extricate. It may require studies at the cellular level, beyond animal models to isolate the role of the two and discover which, or perhaps both, are responsible for the downstream effects observed.
Our study shows that prepubertal exposure to androgens results in significant metabolic dysfunction with impact in oocyte mitochondrial function that results in compensatory efforts in response to the ongoing exposure through the reproductive lifespan. Of particular consequence, the changes in the oocyte can have significant ramifications in downstream pathology and potentially in adverse effects in offspring through inheritance of these compromised mitochondria.
Supplementary Material
Supplementary Figure 1. Reactive Oxygen Species: Pictorial representation of control (above) and obese PCOS (below) models. Brightfield microscopy, followed by green fluorescence using CellRox Green dye at 60x magnification. The obese PCOS mice have similar RFU values compared to controls, demonstrating no difference in ROS concentration, 2-5 oocytes/mice, scale bar =20μm (n=7/group).
Supplementary Figure 2. Lipid Peroxidation: Pictorial representation of control (above) and obese PCOS (below) models. Brightfield microscopy, followed by green fluorescence using BODIPY dye at 60x magnification. The obese PCOS mice have a similar RFU values, indicating no difference in lipid peroxidation compared to controls, 2-5 oocytes/mice, scale bar =20μm (n=7/group).
Acknowledgments:
The authors would like to acknowledge the Department of Obstetrics and Gynecology at the Baylor College of Medicine for their support in this project. The authors would also like to acknowledge Dr. Ignatia Van den Veyver and Dr. JoAnne Richards for their input as well as the Baylor College of Medicine Integrative Microscopy Core and the Animal Facility Core for their helpful assistance.
Funding:
This work was supported by training grants by the Department of Obstetrics and Gynecology, Baylor College of Medicine (N.C., B.Z., P.H. and A.S.) and R-01 research grant (Grant # DK114689) for C.S.B. from National Institutes of Health.
Footnotes
Conflicts of Interest: The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- 1.Goodarzi MO, Dumesic DA, Chazenbalk G, Azziz R. Polycystic ovary syndrome: etiology, pathogenesis and diagnosis. Nat Rev Endocrinol 2011;7:219–31. [DOI] [PubMed] [Google Scholar]
- 2.Azziz R Introduction: Determinants of polycystic ovary syndrome. Fertil Steril 2016;106:4–5. [DOI] [PubMed] [Google Scholar]
- 3.Baskind NE, Balen AH. Hypothalamic-pituitary, ovarian and adrenal contributions to polycystic ovary syndrome. Best Pract Res Clin Obstet Gynaecol 2016. [DOI] [PubMed] [Google Scholar]
- 4.Nisenblat V, Norman RJ. Androgens and polycystic ovary syndrome. Curr Opin Endocrinol Diabetes Obes 2009;16:224–31. [DOI] [PubMed] [Google Scholar]
- 5.Rotterdam EA-SPCWG. Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome. Fertil Steril 2004;81:19–25. [DOI] [PubMed] [Google Scholar]
- 6.Bozdag G, Mumusoglu S, Zengin D, Karabulut E, Yildiz BO. The prevalence and phenotypic features of polycystic ovary syndrome: a systematic review and meta-analysis. Hum Reprod 2016;31:2841–55. [DOI] [PubMed] [Google Scholar]
- 7.Palomba S, Santagni S, Falbo A, La Sala GB. Complications and challenges associated with polycystic ovary syndrome: current perspectives. Int J Womens Health 2015;7:745–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mills G, Badeghiesh A, Suarthana E, Baghlaf H, Dahan MH. Associations between polycystic ovary syndrome and adverse obstetric and neonatal outcomes: a population study of 9.1 million births. Hum Reprod 2020;35:1914–21. [DOI] [PubMed] [Google Scholar]
- 9.Liu L, Tong X, Jiang L, Li TC, Zhou F, Zhang S. A comparison of the miscarriage rate between women with and without polycystic ovarian syndrome undergoing IVF treatment. Eur J Obstet Gynecol Reprod Biol 2014;176:178–82. [DOI] [PubMed] [Google Scholar]
- 10.Luo L, Gu F, Jie H, Ding C, Zhao Q, Wang Q et al. Early miscarriage rate in lean polycystic ovary syndrome women after euploid embryo transfer - a matched-pair study. Reprod Biomed Online 2017. [DOI] [PubMed] [Google Scholar]
- 11.Grigorescu V, Zhang Y, Kissin DM, Sauber-Schatz E, Sunderam M, Kirby RS et al. Maternal characteristics and pregnancy outcomes after assisted reproductive technology by infertility diagnosis: ovulatory dysfunction versus tubal obstruction. Fertil Steril 2014;101:1019–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chappell NR, Barsky M, Shah J, Peavey M, Yang L, Sangi-Haghpeykar H et al. Embryos from polycystic ovary syndrome patients with hyperandrogenemia reach morula stage faster than controls. F&S Reports 2020;1:125–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chappell NR, Barsky M, Shah J, Peavey M, Yang L, Sangi-Haghpeykar H et al. Embryos from Polycystic Ovary Syndrome (PCOS) Patients with Hyperandrogenemia Reach Morula Stage Faster than Controls. Fertility and Sterility Reports 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Broughton DE, Moley KH. Obesity and female infertility: potential mediators of obesity’s impact. Fertil Steril 2017;107:840–7. [DOI] [PubMed] [Google Scholar]
- 15.Practice Committee of the American Society for Reproductive M. Obesity and reproduction: a committee opinion. Fertil Steril 2015;104:1116–26. [DOI] [PubMed] [Google Scholar]
- 16.Shah DK, Missmer SA, Berry KF, Racowsky C, Ginsburg ES. Effect of obesity on oocyte and embryo quality in women undergoing in vitro fertilization. Obstet Gynecol 2011;118:63–70. [DOI] [PubMed] [Google Scholar]
- 17.Bailey AP, Hawkins LK, Missmer SA, Correia KF, Yanushpolsky EH. Effect of body mass index on in vitro fertilization outcomes in women with polycystic ovary syndrome. Am J Obstet Gynecol 2014;211:163 e1–6. [DOI] [PubMed] [Google Scholar]
- 18.Babayev E, Seli E. Oocyte mitochondrial function and reproduction. Curr Opin Obstet Gynecol 2015;27:175–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van Blerkom J Mitochondrial function in the human oocyte and embryo and their role in developmental competence. Mitochondrion 2011;11:797–813. [DOI] [PubMed] [Google Scholar]
- 20.Huang Y, Yu Y, Gao J, Li R, Zhang C, Zhao H et al. Impaired oocyte quality induced by dehydroepiandrosterone is partially rescued by metformin treatment. PLoS One 2015;10:e0122370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Safiulina D, Peet N, Seppet E, Zharkovsky A, Kaasik A. Dehydroepiandrosterone inhibits complex I of the mitochondrial respiratory chain and is neurotoxic in vitro and in vivo at high concentrations. Toxicol Sci 2006;93:348–56. [DOI] [PubMed] [Google Scholar]
- 22.Tarumi W, Tsukamoto S, Okutsu Y, Takahashi N, Horiuchi T, Itoh MT et al. Androstenedione induces abnormalities in morphology and function of developing oocytes, which impairs oocyte meiotic competence. Fertil Steril 2012;97:469–76. [DOI] [PubMed] [Google Scholar]
- 23.Takahashi M Oxidative stress and redox regulation on in vitro development of mammalian embryos. J Reprod Dev 2012;58:1–9. [DOI] [PubMed] [Google Scholar]
- 24.Saben JL, Boudoures AL, Asghar Z, Thompson A, Drury A, Zhang W et al. Maternal Metabolic Syndrome Programs Mitochondrial Dysfunction via Germline Changes across Three Generations. Cell Rep 2016;16:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Turner N, Robker RL. Developmental programming of obesity and insulin resistance: does mitochondrial dysfunction in oocytes play a role? Mol Hum Reprod 2015;21:23–30. [DOI] [PubMed] [Google Scholar]
- 26.Abbott DH, Barnett DK, Bruns CM, Dumesic DA. Androgen excess fetal programming of female reproduction: a developmental aetiology for polycystic ovary syndrome? Hum Reprod Update 2005;11:357–74. [DOI] [PubMed] [Google Scholar]
- 27.Hayes MG, Urbanek M, Ehrmann DA, Armstrong LL, Lee JY, Sisk R et al. Genome-wide association of polycystic ovary syndrome implicates alterations in gonadotropin secretion in European ancestry populations. Nat Commun 2015;6:7502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu H, Zhao H, Chen ZJ. Genome-Wide Association Studies for Polycystic Ovary Syndrome. Semin Reprod Med 2016;34:224–9. [DOI] [PubMed] [Google Scholar]
- 29.Zhao H, Lv Y, Li L, Chen ZJ. Genetic Studies on Polycystic Ovary Syndrome. Best Pract Res Clin Obstet Gynaecol 2016. [DOI] [PubMed] [Google Scholar]
- 30.Dumesic DA, Abbott DH, Padmanabhan V. Polycystic ovary syndrome and its developmental origins. Rev Endocr Metab Disord 2007;8:127–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Caldwell AS, Middleton LJ, Jimenez M, Desai R, McMahon AC, Allan CM et al. Characterization of reproductive, metabolic, and endocrine features of polycystic ovary syndrome in female hyperandrogenic mouse models. Endocrinology 2014;155:3146–59. [DOI] [PubMed] [Google Scholar]
- 32.van Houten EL, Visser JA. Mouse models to study polycystic ovary syndrome: a possible link between metabolism and ovarian function? Reprod Biol 2014;14:32–43. [DOI] [PubMed] [Google Scholar]
- 33.Tarumi W, Itoh MT, Suzuki N. Effects of 5alpha-dihydrotestosterone and 17beta-estradiol on the mouse ovarian follicle development and oocyte maturation. PLoS One 2014;9:e99423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chappell NR, Zhou B, Schutt AK, Gibbons WE, Blesson CS. Prenatal androgen induced lean PCOS impairs mitochondria and mRNA profiles in oocytes. Endocr Connect 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Byers SL, Wiles MV, Dunn SL, Taft RA. Mouse estrous cycle identification tool and images. PLoS One 2012;7:e35538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nelson JF, Felicio LS, Randall PK, Sims C, Finch CE. A longitudinal study of estrous cyclicity in aging C57BL/6J mice: I. Cycle frequency, length and vaginal cytology. Biol Reprod 1982;27:327–39. [DOI] [PubMed] [Google Scholar]
- 37.Caligioni CS. Assessing reproductive status/stages in mice. Curr Protoc Neurosci 2009;Appendix 4:Appendix 4I. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Andrikopoulos S, Blair AR, Deluca N, Fam BC, Proietto J. Evaluating the glucose tolerance test in mice. Am J Physiol Endocrinol Metab 2008;295:E1323–32. [DOI] [PubMed] [Google Scholar]
- 39.Roland AV, Nunemaker CS, Keller SR, Moenter SM. Prenatal androgen exposure programs metabolic dysfunction in female mice. J Endocrinol 2010;207:213–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jones SL, Ismail N, King L, Pfaus JG. The effects of chronic administration of testosterone propionate with or without estradiol on the sexual behavior and plasma steroid levels of aged female rats. Endocrinology 2012;153:5928–39. [DOI] [PubMed] [Google Scholar]
- 41.Gonzalez-Hunt CP, Rooney JP, Ryde IT, Anbalagan C, Joglekar R, Meyer JN. PCR-Based Analysis of Mitochondrial DNA Copy Number, Mitochondrial DNA Damage, and Nuclear DNA Damage. Curr Protoc Toxicol 2016;67:20 11 1–20 11 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Norman RJ, Dewailly D, Legro RS, Hickey TE. Polycystic ovary syndrome. Lancet 2007;370:685–97. [DOI] [PubMed] [Google Scholar]
- 43.Navarro G, Allard C, Morford JJ, Xu W, Liu S, Molinas AJ et al. Androgen excess in pancreatic beta cells and neurons predisposes female mice to type 2 diabetes. JCI Insight 2018;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.St John JC, Facucho-Oliveira J, Jiang Y, Kelly R, Salah R. Mitochondrial DNA transmission, replication and inheritance: a journey from the gamete through the embryo and into offspring and embryonic stem cells. Hum Reprod Update 2010;16:488–509. [DOI] [PubMed] [Google Scholar]
- 45.Meldrum DR, Casper RF, Diez-Juan A, Simon C, Domar AD, Frydman R. Aging and the environment affect gamete and embryo potential: can we intervene? Fertil Steril 2016;105:548–59. [DOI] [PubMed] [Google Scholar]
- 46.Seidler EA, Moley KH. Metabolic Determinants of Mitochondrial Function in Oocytes. Semin Reprod Med 2015;33:396–400. [DOI] [PubMed] [Google Scholar]
- 47.Vasconsuelo A, Pronsato L, Ronda AC, Boland R, Milanesi L. Role of 17beta-estradiol and testosterone in apoptosis. Steroids 2011;76:1223–31. [DOI] [PubMed] [Google Scholar]
- 48.Treidel LA, Whitley BN, Benowitz-Fredericks ZM, Haussmann MF. Prenatal exposure to testosterone impairs oxidative damage repair efficiency in the domestic chicken (Gallus gallus). Biol Lett 2013;9:20130684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhao H, Zhao Y, Li T, Li M, Li J, Li R et al. Metabolism alteration in follicular niche: The nexus among intermediary metabolism, mitochondrial function, and classic polycystic ovary syndrome. Free Radic Biol Med 2015;86:295–307. [DOI] [PubMed] [Google Scholar]
- 50.Lai Q, Xiang W, Li Q, Zhang H, Li Y, Zhu G et al. Oxidative stress in granulosa cells contributes to poor oocyte quality and IVF-ET outcomes in women with polycystic ovary syndrome. Frontiers of Medicine 2018;12:518–24. [DOI] [PubMed] [Google Scholar]
- 51.Mohajeri M, Martin-Jimenez C, Barreto GE, Sahebkar A. Effects of estrogens and androgens on mitochondria under normal and pathological conditions. Prog Neurobiol 2019;176:54–72. [DOI] [PubMed] [Google Scholar]
- 52.Zup SL, Edwards NS, McCarthy MM. Sex- and age-dependent effects of androgens on glutamate-induced cell death and intracellular calcium regulation in the developing hippocampus. Neuroscience 2014;281:77–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hu M, Zhang Y, Guo X, Jia W, Liu G, Zhang J et al. Hyperandrogenism and insulin resistance induce gravid uterine defects in association with mitochondrial dysfunction and aberrant reactive oxygen species production. Am J Physiol Endocrinol Metab 2019;316:E794–E809. [DOI] [PubMed] [Google Scholar]
- 54.Zhang Y, Zhao W, Xu H, Hu M, Guo X, Jia W et al. Hyperandrogenism and insulin resistance-induced fetal loss: evidence for placental mitochondrial abnormalities and elevated reactive oxygen species production in pregnant rats that mimic the clinical features of polycystic ovary syndrome. J Physiol 2019;597:3927–50. [DOI] [PubMed] [Google Scholar]
- 55.Vasconsuelo A, Milanesi L, Boland R. Actions of 17beta-estradiol and testosterone in the mitochondria and their implications in aging. Ageing Res Rev 2013;12:907–17. [DOI] [PubMed] [Google Scholar]
- 56.Sirard MA. Factors affecting oocyte and embryo transcriptomes. Reprod Domest Anim 2012;47Suppl 4:148–55. [DOI] [PubMed] [Google Scholar]
- 57.Demonacos CV, Karayanni N, Hatzoglou E, Tsiriyiotis C, Spandidos DA, Sekeris CE. Mitochondrial genes as sites of primary action of steroid hormones. Steroids 1996;61:226–32. [DOI] [PubMed] [Google Scholar]
- 58.Perumpail BJ, Khan MA, Yoo ER, Cholankeril G, Kim D, Ahmed A. Clinical epidemiology and disease burden of nonalcoholic fatty liver disease. World J Gastroenterol 2017;23:8263–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pan JX, Zhang JY, Ke ZH, Wang FF, Barry JA, Hardiman PJ et al. Androgens as double-edged swords: Induction and suppression of follicular development. Hormones (Athens) 2015;14:190–200. [DOI] [PubMed] [Google Scholar]
- 60.Sir-Petermann T, Codner E, Maliqueo M, Echiburu B, Hitschfeld C, Crisosto N et al. Increased anti-Mullerian hormone serum concentrations in prepubertal daughters of women with polycystic ovary syndrome. J Clin Endocrinol Metab 2006;91:3105–9. [DOI] [PubMed] [Google Scholar]
- 61.Sir-Petermann T, Hitchsfeld C, Maliqueo M, Codner E, Echiburu B, Gazitua R et al. Birth weight in offspring of mothers with polycystic ovarian syndrome. Hum Reprod 2005;20:2122–6. [DOI] [PubMed] [Google Scholar]
- 62.Wang H, Wang X, Zhu Y, Chen F, Sun Y, Han X. Increased androgen levels in rats impair glucose-stimulated insulin secretion through disruption of pancreatic beta cell mitochondrial function. J Steroid Biochem Mol Biol 2015;154:254–66. [DOI] [PubMed] [Google Scholar]
- 63.Selen ES, Bolandnazar Z, Tonelli M, Butz DE, Haviland JA, Porter WP et al. NMR Metabolomics Show Evidence for Mitochondrial Oxidative Stress in a Mouse Model of Polycystic Ovary Syndrome. J Proteome Res 2015;14:3284–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cree-Green M, Rahat H, Newcomer BR, Bergman BC, Brown MS, Coe GV et al. Insulin Resistance, Hyperinsulinemia, and Mitochondria Dysfunction in Nonobese Girls With Polycystic Ovarian Syndrome. J Endocr Soc 2017;1:931–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Reactive Oxygen Species: Pictorial representation of control (above) and obese PCOS (below) models. Brightfield microscopy, followed by green fluorescence using CellRox Green dye at 60x magnification. The obese PCOS mice have similar RFU values compared to controls, demonstrating no difference in ROS concentration, 2-5 oocytes/mice, scale bar =20μm (n=7/group).
Supplementary Figure 2. Lipid Peroxidation: Pictorial representation of control (above) and obese PCOS (below) models. Brightfield microscopy, followed by green fluorescence using BODIPY dye at 60x magnification. The obese PCOS mice have a similar RFU values, indicating no difference in lipid peroxidation compared to controls, 2-5 oocytes/mice, scale bar =20μm (n=7/group).