

Abstract
Responses to vaccination and to diseases vary widely across individuals, which may be partly due to baseline immune variations. Identifying such baseline predictors of immune responses and their biological basis are of broad interest given their potential importance for cancer immunotherapy, disease outcomes, vaccination and infection responses. Here we uncover baseline blood transcriptional signatures predictive of antibody responses to both influenza and yellow fever vaccinations in healthy subjects. These same signatures evaluated at clinical quiescence are correlated with disease activity in systemic lupus erythematosus patients with plasmablast-associated flares. CITE-seq profiling of 82 surface proteins and transcriptomes of 53,201 single cells from healthy high and low influenza-vaccination responders revealed that our signatures reflect the extent of activation in a plasmacytoid dendritic cell—Type I IFN—T/B lymphocyte network. Our findings raise the prospect that modulating such immune baseline states may improve vaccine responsiveness and mitigate undesirable autoimmune disease activities.
The immune system maintains health but also contributes to diverse pathologies1. The extent of immune responses to a perturbation or disease vary across individuals in the population and thus, there is a pressing need to uncover predictors and determinants of immune responsiveness in humans2–4. Human immune responses are shaped not only by genetics but are also markedly influenced by the environment3,5,6, e.g., antibody responses to vaccination show little heritability after infancy/early childhood7,8. Increasing evidence supports the hypothesis that the immune state of an individual prior to a perturbation can predict and determine immune response outcomes3. However, the molecular and cellular basis for the few existing baseline peripheral blood cell frequency or transcriptional predictors in humans9–12 remains largely unknown.
Some immunologic mechanisms that contribute to protective immune responses in vaccination and infection can mediate undesirable disease activities (DA) in patients suffering from an autoimmune disease. Notably, responses to influenza, yellow fever, and many other vaccines and infections are characterized by a plasmablast increase detectable in blood3. A plasmablast increase has been shown to coincide with DA in some autoimmune patients, such as a subset of the patients with systemic lupus erythematosus (SLE)13, a chronic, heterogeneous autoimmune disease that often presents clinically with episodic disease flares affecting multiple organs14. We thus hypothesized that there exist common baseline determinants that contribute to the responsiveness to both vaccination/infection and autoimmunity in the form of undesirable disease activities (DA). The determinants of DA, such as the intensity of flares in SLE, are poorly understood and predictors of DA remain elusive15. Given the availability of longitudinal blood transcriptomic data in SLE13 and the relevance of plasmablasts in both vaccination/infection and SLE, here we use SLE as a model for exploring common baseline signatures associated with vaccine responses in healthy individuals and autoimmune disease activities in patients.
We show that a baseline, peripheral blood signature predictive of antibody responses to influenza vaccination12 is also predictive of responses to the yellow fever vaccine in individuals naïve to the virus. The same signature evaluated at clinical quiescence (baseline-like) is associated with disease activity in SLE patients with flares characterized by elevation in plasmablast signature scores (Fig. 1a). Conversely, a biologically related baseline indicator of disease flares derived solely from the same subset of lupus patients is correlated with antibody responses to influenza vaccination in healthy subjects. We thus provide robust evidence for baseline “set point”2 signatures shared among vaccination and SLE. Simultaneous protein and transcriptome analysis16 (CITE-seq) of single peripheral blood mononuclear cells (PBMCs) from high and low healthy responders of influenza vaccination revealed that our signatures reflect the extent of activation in multiple immune cell populations at baseline, including plasmacytoid dendritic cells (pDC) and lymphocytes. These findings suggest that future responsiveness potential can be stably encoded by the activation status of a cellular network before perturbation and provide interventional targets that can potentially be modulated—at baseline and under homeostatic conditions—to improve human health.
Figure 1: Study questions and the derivation of a baseline, pre-vaccination signature predictive of response using an influenza fever vaccination cohort.
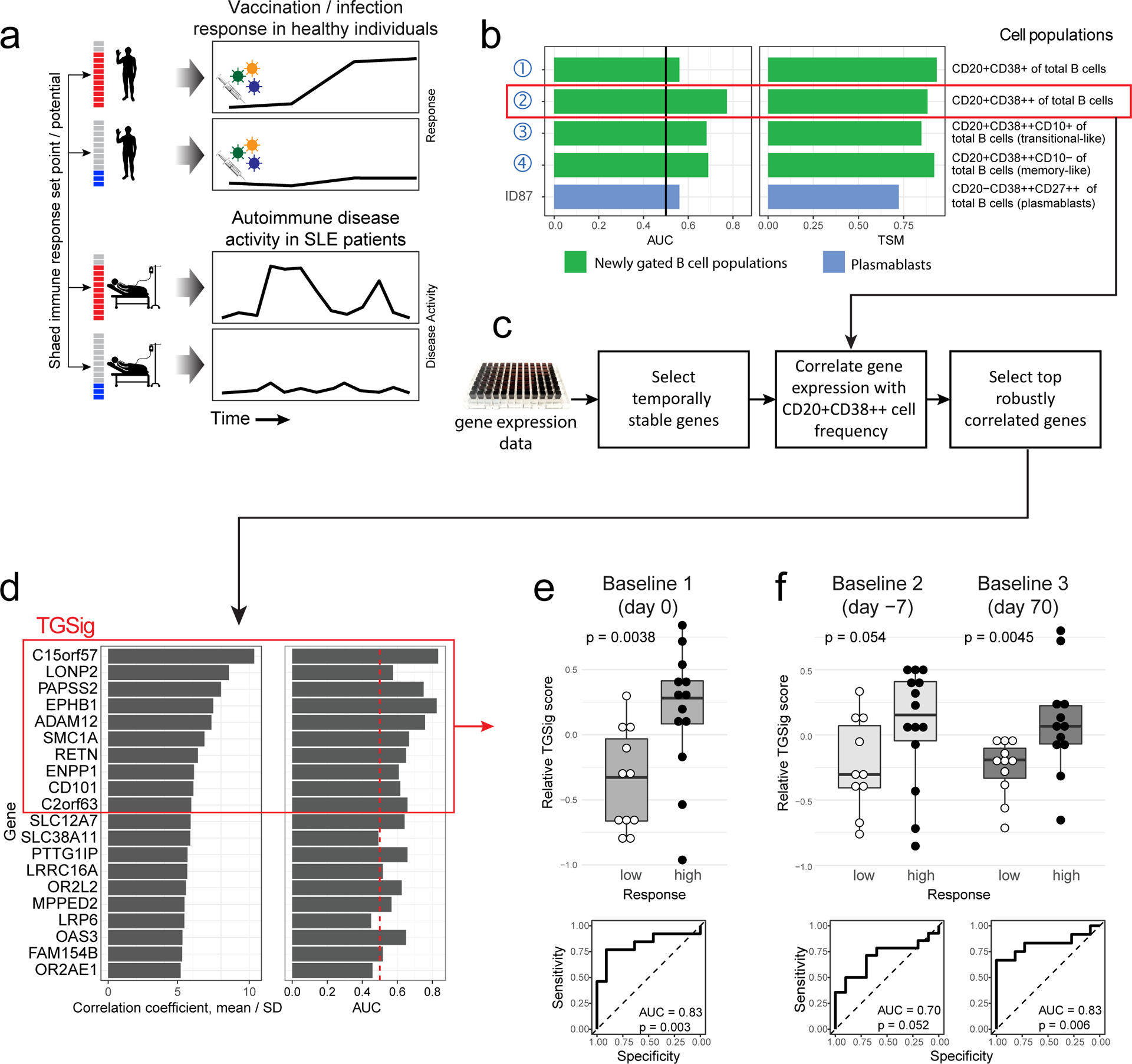
a, Overview of the study and research questions. b, Prediction performance for antibody response in the NIH influenza study12 using the frequency of several B cell subsets (y-axis) (see Methods and gating strategy outlined in Extended Data Fig. 1a). The left panel shows the AUC (area under receiver operator curve; x-axis) for predicting high and low responders (n = 23 with flow cytometry data) to the seasonal and pandemic H1N1 influenza vaccines in ref.12. The right panel shows the temporal stability metric (TSM) (x-axis); higher TSM indicates greater temporal stability over the three baseline time points (days −7 and 0 prior to vaccination and day 70 after vaccination) using 136 samples from 51 subjects. Population 2 (red box) is the CD19+CD20+CD38++ B cell population. c, Flow chart showing the steps to derive the gene expression-based surrogate signature (TGSig). d, Top temporally stable genes correlated with the frequency of CD19+CD20+CD38++ B cell and the selected genes in TGSig (red box). 22 high and low responders (those with both gene expression and flow cytometry data) are used to assess correlations and rank genes. Genes are ranked based on the average Spearman correlation divided by the standard deviation obtained from 231 iterations (as a safeguard against noise we iterated over all sub-cohorts containing 20 subjects by taking out 2 random subjects at a time [i.e., excluding 2 out of 22 subjects] to assess the correlation). See Extended Data Fig. 2a–d and Methods for further details about temporal stability, gene selection, and signature score calculation. e, Top: box plots comparing the TGSig score (y-axis) at day 0 (pre-vaccination) between low (n=11) and high (n=13) responders (x-axis) (Wilcoxon one-tailed p value shown); bottom: receiver operating curve (ROC) for assessing predictive capacity (area under the curve (AUC) and one-tailed permutation test p value shown). Boxplots’ center line corresponds to the median value, lower and upper hinges correspond to the first and third quartiles (the 25th and 75th percentiles); lower and upper whiskers extend from the box to the smallest or largest value correspondingly, but no further than 1.5x inter-quantile range. f, Similar to (e) but for the other two baseline time points: days −7 (10 low and 14 high responders) and day −70 (11 low and 12 high responders).
Results
Development of a temporally stable transcriptional baseline signature predictive of antibody responses to influenza vaccination
We previously identified a baseline (pre-vaccination), temporally stable signature predictive of antibody responses to influenza vaccination independent of age, gender, and pre-existing antibody levels12. The signature consisted of several B and T cell subpopulations without antigen-specific information. We subsequently noticed that all the predictive B cell populations expressed CD20 and high levels of CD38 (CD19+CD20+CD38++), suggesting that these markers together defined a core predictive population (Extended Data Fig. 1a). We quantified this and related populations in the three baseline time-points in our original NIH influenza vaccination study (days −7 and 0 prior to vaccination and day 70 when the parameters altered by vaccination had returned to their original values)12,17(Fig. 1b). As expected, high and low antibody responders (defined using the “adjMFC” metric that does not depend on pre-existing antibody levels against influenza12) could be distinguished by the frequency of the CD20+CD38++ cells at all three time points (Extended Data Fig. 1b,c). As observed previously12, this signature was independent of plasmablasts (CD20-CD38++), which alone could not predict the response (AUC=0.55; Fig. 1b).
To test whether our CD20+CD38++ signature could predict immune responsiveness in other scenarios using independent datasets, we developed a blood-based transcriptional surrogate because available public datasets either do not have flow cytometry data or, if present, do not measure the same cell subsets. We identified temporally stable genes robustly correlated with the frequency of CD20+CD38++ cells across subjects on day 0 in the NIH cohort to build a 10-gene signature (TGSig, see Glossary in Extended Data Fig. 1d; Figs. 1c–e and Extended Data Figs. 2a–f, Supplementary Table 1; see Methods). TGSig has AUCs comparable to the frequency of CD20+CD38++ B cells when evaluated in the other two baseline time-points (Fig. 1f) and it was robust against addition or removal of genes (Extended Data Fig. 2g). Similar and statistically significant trends were observed when middle responders were included (Extended Data Fig. 3a).
Figure 2: Assessing TGSig in independent influenza vaccination, yellow fever vaccination, and SLE datasets.
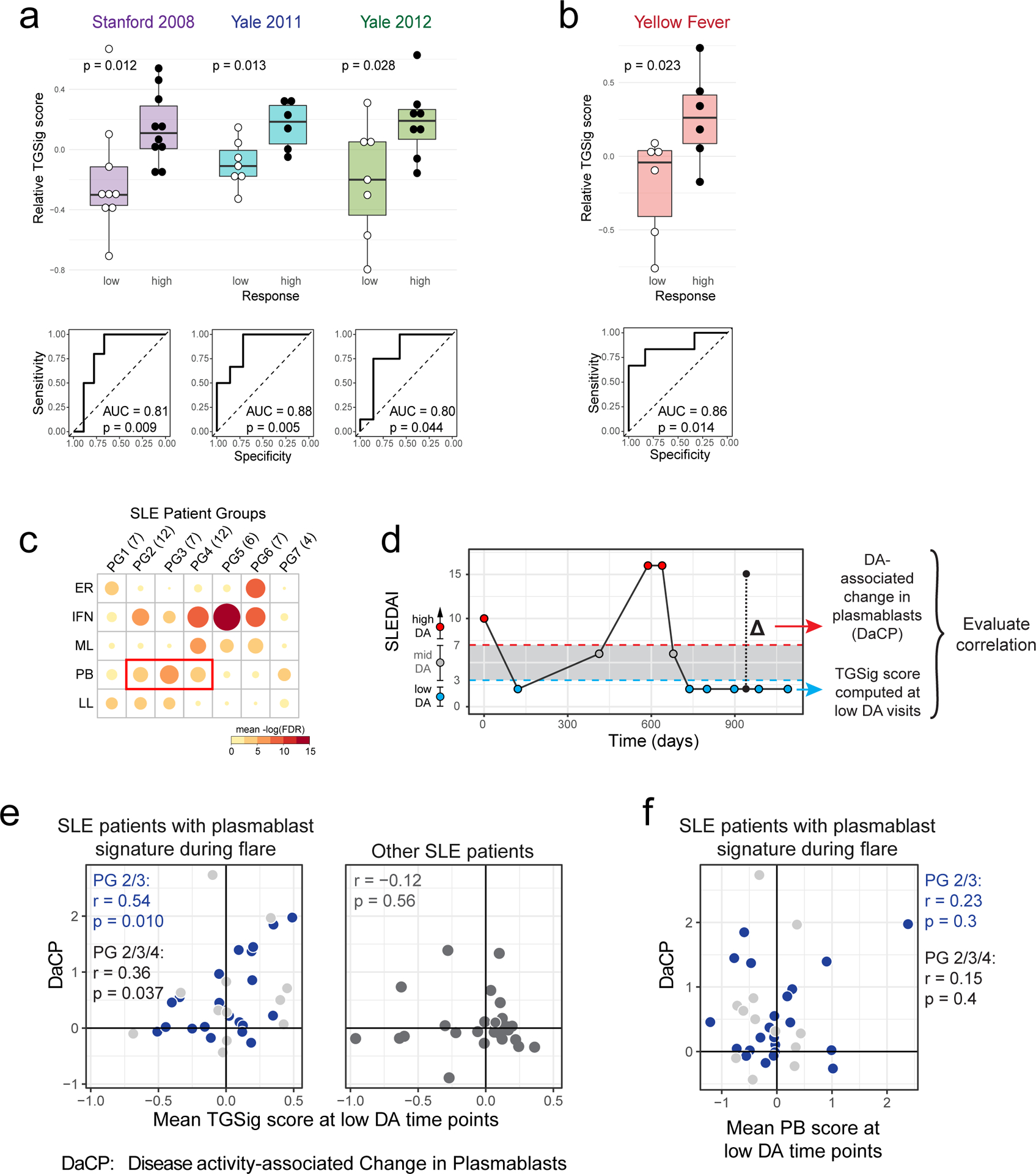
a, Same as Fig. 1e in terms of statistical tests and plot types but here showing the predictive performance of TGSig (evaluated at baseline/pre-vaccination) in the indicated independent datasets: Stanford 2008 (purple boxes; 10 high vs. 8 low responders), Yale 2011 (turquoise boxes; 6 high vs. 7 low responders) and Yale 2012 (green boxes; 8 high vs. 7 low responders) with the corresponding ROC shown at the bottom. b, Similar to (a) but for yellow fever vaccination. This cohort (trial #1) included 6 high and 5 low responders; see Extended Data Fig. 4a for results on a second, smaller cohort (trial #2) with 10 subjects (4 high and 3 low responders) from the same publication19). See also Extended Data Fig. 3 for boxplots that include middle responders. c, SLE patient groups determined based on the blood gene expression signatures associated with disease activity/flares (see also Extended Data Fig. 5a and Methods.) The color and size of the circle denote the average statistical significance (−log10 of BH-adjusted p-values from the CERNO test39) of the association across patients in the group. The patient group IDs (columns) are listed with the number of patients in each group in parentheses. The same phenotypic annotations from ref13 are used: ER = erthryopoiesis; IFN = IFN response/neutrophils; ML = myeloid linage/neutrophils; PB = plasmablasts; LL = Lymphoid lineage. The three groups with prominent PB signatures are boxed in red. d, Overview of the analysis approach. The dynamics of disease activity as measured by the SLEDAI score of an actual patient is shown. Individual visits are shown as dots (low DA (blue dots) = SLEDAI < 3, medium DA (grey dots) = SLEDAI: 3–7, high DA (red dots) = SLEDAI ≥ 8)). The TGSig score was computed from the low DA time points and the DaCP was computed from all time-points using a mixed-effect model accounting for treatment effects (see Methods); the correlation between TGSig (averaged across low DA time-points) and DaCP was then evaluated. e, Scatterplot showing the relationship between the DaCP (y-axis) and the mean TGSig score at low DA time points (x-axis) in (left panel) patient groups 2, 3, and 4 (34 subjects) and (right panel) subjects whose DA tended not to be associated with a plasmablast signature (patient groups 1, 5, 6, and 7; 27 subjects). Pearson correlation coefficient and two-tailed p values are shown, and for the left panel separately for patient groups 2 and 3 only (blue dots, n=22) or groups 2, 3, 4 (blue and grey dots; n=34). As a robustness check we also computed the Spearman correlation: rho=0.47 (p=0.029 two-tailed test) for groups 2 and 3; rho=0.36 (p=0.038 two-tailed test) for groups 2, 3 and 4; rho=−0.12 (p=0.56 two-tailed test) for the rest of the patient groups. f, Same as in (e) but the plasmablast (PB) signature score was computed from the low DA time-points instead of TGSig. Groups 2,3: Spearman rho=0.014 (p=0.95 two-tailed test); groups 2,3,4: rho=−0.006 (p=0.97 two-tailed test).
Evaluating TGSig in independent influenza and yellow fever vaccination datasets
We next assessed TGSig in independent influenza vaccination cohorts from multiple institutions. These data together with our NIH cohort span three geographic locations within the United States (US) and vaccination years (2008, 2009, 2011, and 2012)10 (Supplementary Table 2). Application of TGSig to the pre-vaccination, baseline data of multiple datasets as-is without any further model training showed good prediction performance (AUCs > 0.8; p<0.05 permutation test) (Fig. 2a; see also Extended Data Fig. 3b that includes middle responders). However, it lacked predictive power when applied to influenza vaccination data from another US institution covering four consecutive vaccination years (2008–2011)18, which became available after our initial analyses (Extended Data Fig. 3c). While many technical or biological factors could be responsible for this result, we have ruled out differences in low-level data processing or high/low-responder definition (see Methods).
We next investigated whether TGSig could predict response to a live viral infection by using yellow fever (YF) vaccination (YF-17D) given to healthy adults naïve to YF-17D as a model19. Unlike responses to the influenza (dead/inactivated) vaccine, YF-17D is a live, attenuated virus that would be expected to generate an antibody response through partially distinct immunological mechanisms. TGSig applied to pre-vaccination PBMC expression data was again able to separate high and low responders in the larger of two independent trials (Fig. 2b; AUC=0.86, p=0.014 permutation test; see also Extended Data Fig. 3d that includes middle responders). A similar, but not statistically significant trend was observed in a smaller second independent trial that had fewer subjects (4 high and 3 low responders; Extended Data Fig. 4a) (AUC=0.75; p=0.11 permutation test). A meta-analysis of the influenza vaccination datasets for which TGSig exhibited predictive capacity at the individual cohort level (Stanford 2008, NIH 2009, Yale 2011, Yale 2012) indicated that genes in TGSig had positive “meta” effect sizes and were thus positively associated with antibody responses (Extended Data Fig. 4b); the effect sizes were also qualitatively consistent with those in the yellow fever dataset (Extended Data Fig. 4c). Together, our data revealed that TGSig could predict the antibody response to a live attenuated virus in subjects naïve to the virus and to the inactivated seasonal influenza vaccine in several, but not all, healthy cohorts tested.
Evaluating TGSig in SLE
We next assessed the hypothesis that TGSig evaluated at clinically quiescent periods of SLE patients (i.e., low or no DA resembling “baseline”) might also be associated with the severity of flares (Fig. 1a), which is dynamic and conceptually similar to responses following vaccination or infection. We analyzed a longitudinal pediatric SLE cohort13, in which the SLE Disease Activity Index (SLEDAI), a clinical assessment score that combines DA in several organs and clinical categories,13 was used to quantify DA over time. Previous analysis of this cohort identified seven patient groups in which DA (i.e., SLEDAI score) correlated with distinct combinations of blood transcriptomic signatures, including a module enriched for plasma cells/plasmablasts13 (Fig. 2c). We hypothesized that for patients whose DA was correlated with a plasma cell/plasmablast signature (Fig. 2c and Extended Data Fig. 5a: PG2 and PG3 , and to a lesser extent, PG4), the DA-associated change in plasmablast score (DaCP, see Extended Data Fig. 1d) between periods of low and high DA may be correlated with the TGSig evaluated at clinically quiescent (low/no DA) periods (Fig. 2d).
We created a mixed-effects model to estimate the DaCP for PG2, PG3, and PG4 after accounting for treatments (see Methods). As expected, the DaCP values estimated by our procedure were correlated with the change in the average plasmablast score between low and high DA periods (Extended Data Fig. 5b). We detected a mild but significant correlation between the mean of TGSig across periods of low DA (SLEDAI<3) and DaCP (Pearson r=0.359, p=0.037, Fig. 2e). This correlation was stronger in PG2 and PG3 only (Pearson r=0.535, p=0.001) because PG4 often had weaker plasmablast signals associated with DA (Extended Data Fig. 5a). To further evaluate this hypothesis, we tested the correlation when we removed patients whose DaCP was lower than a set threshold. We observed that the correlation tended to increase as the threshold increased (Extended Data Fig. 5c,d), confirming that the correlation between TGSig and DaCP was specific to those patients with plasmablast-associated flares. As a control, we performed the same analyses for patients who did not show a plasmablast signature (PG1, PG5, PG6, and PG7, Fig. 2c) and found no correlation (Pearson r=−0.116, p=0.56, Fig. 2e right panel). We also confirmed that there was no correlation between the DaCP and the mean plasmablast score at low DA time points in PG2, PG3, and PG4 (Fig. 2f). Thus, the plasmablast signature evaluated at periods of low disease activity was not predictive and TGSig did not reflect plasmablast activity. These results suggest that for SLE patients whose DA was associated with a plasmablast signature, TGSig evaluated during clinical quiescence can inform the magnitude of disease flares. These findings also expand the predictive value of TGSig from response to vaccines and infections to that associated with flares in a specific subtype of SLE.
An independently derived baseline signature from SLE is associated with influenza vaccination responses
Our results suggest a biological parallel between vaccination/infection responses and lupus DA. We therefore examined whether an independently derived correlate of DaCP in SLE patients alone may conversely associate with the magnitude of antibody responses to vaccination. We focused again on patient groups 2, 3, 4 (Fig. 2c) and used Weighted Gene Co-expression Network Analysis (WGCNA)20 to ask whether there exist temporally stable (across low DA time-points) gene expression modules associate with DaCP (Fig. 3a and Supplementary Fig. 1a, Supplementary Table 3a; see Methods). One module (the “brown”) was mildly correlated with the DaCP (Fig. 3b and Supplementary Fig. 1b; Pearson r=0.31, p=0.04 permutation test; full results in Supplementary Table 3b), but it was intriguingly enriched for Type I IFN related blood transcriptomic modules (BTMs)21 (Fig. 3c and Supplementary Fig. 1c), consistent with Type I IFN signatures seen in many SLE patients22. Our observation suggests that the magnitude of a Type I IFN signature during clinical quiescence may be prognostic of disease activity for patients exhibiting plasmablast associated flares.
Figure 3: A transcriptional correlate of plasmablast-associated disease activity in SLE is also associated with influenza vaccination responses and functionally related to TGSig.
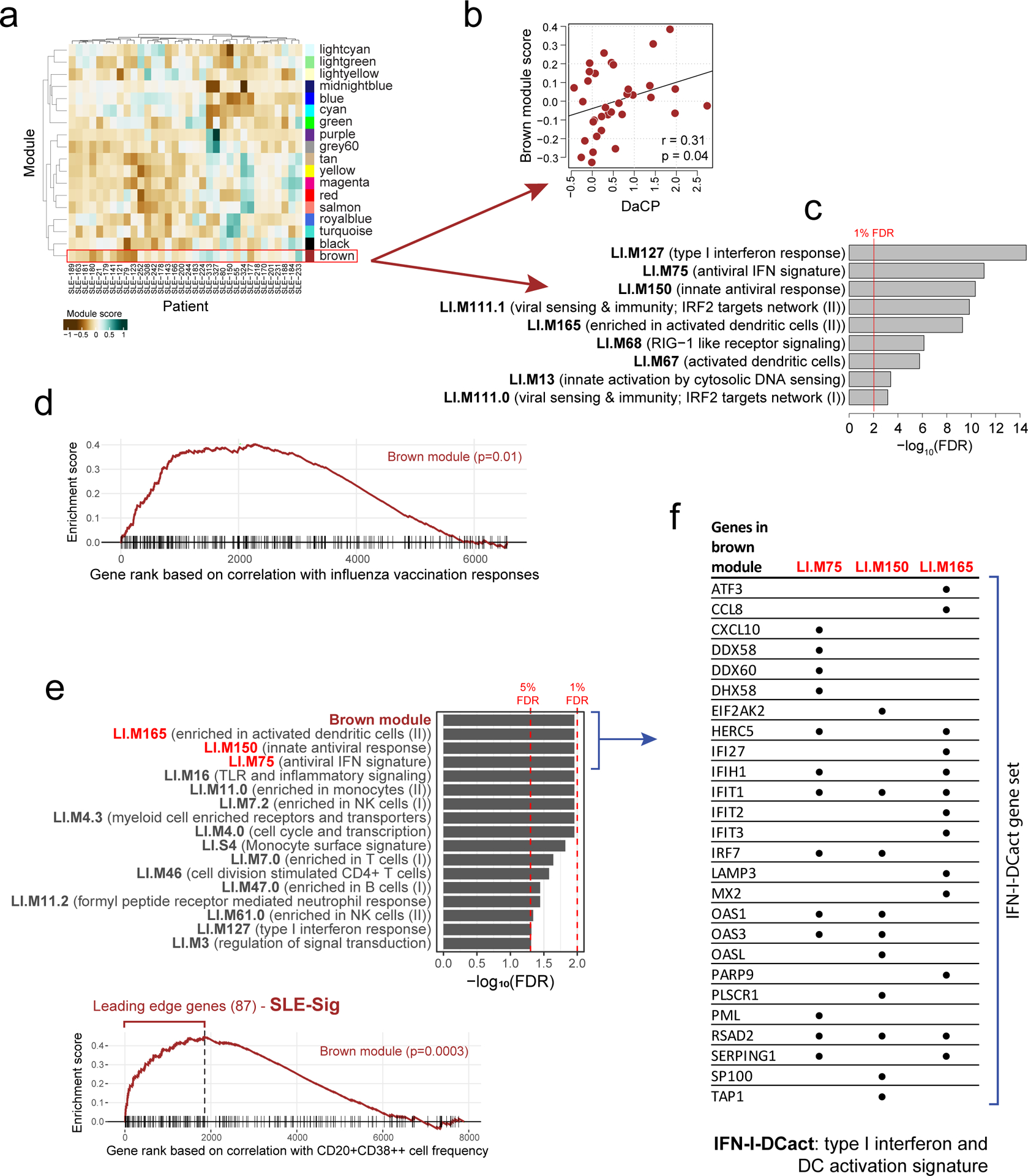
a, Identification of 18 transcriptomic modules from genes whose expression was temporally stable across low DA time-points in SLE patients from patient groups 2, 3, and 4. The heatmap shows the eigengene of each module averaged across low-DA time-points of each patient. Rows and columns correspond to modules and SLE patients, respectively. The number of genes in each module is shown in Supplementary Fig. 1a. b, Scatterplot showing the relationship between the brown module score and the DaCP for patients in groups 2, 3, and 4 as above (n=34; Pearson correlation=0.31, p=0.04 based on a one-tailed permutation test (Supplementary Fig. 1b)). Spearman rho=0.29 (p=0.05 one-tailed permutation test) c, Top enriched blood transcriptome modules21 (BTMs) of the brown module (370 genes) based on the hypergeometric test with BH-adjusted p values (FDR) shown; the red line corresponds to 1% FDR (no additional BTMs were identified at 5% FDR cutoff). Complete results of the enrichment analysis can be found in Supplementary Fig. 1c. d, Gene set enrichment analysis (GSEA) of the brown module genes (370 genes). 6563 genes were ranked by their magnitude of association with antibody responses based on a meta-analysis of four influenza vaccination datasets (the enrichment P value shown was computed from the GSEA test40). The tick marks denote the location of the genes in the brown module. e, (top) Enrichment analysis of blood transcriptome modules and the brown module in the temporally stable genes from the NIH influenza study as ranked by their correlation with the frequency of CD20+CD38++ B cells. A temporal stability score (see Methods) cutoff of 0.5 is used here to define 7889 stable genes; the enrichment results are robust to the threshold used. 5% and 1% FDR are indicated by the red dashed lines (BH-adjusted p values shown were computed from the GSEA test). (bottom) GSEA enrichment plot for the brown module. The top 87 genes in the brown module (based on the gene rank on the x axis) were identified by GSEA as the “leading edge genes” (i.e., the main driver of the enrichment signal) (we called this gene set SLE-Sig). The P value shown was computed from the GSEA test. The full list of leading-edge genes can be found in Supplementary Table 5. f, Genes in the brown module that are also in at least one of the three top Type I IFN /antiviral/dendritic cell activation BTMs from (e); black dots indicate that the gene is present in the indicated gene set.
To assess whether the brown module was correlated with vaccination responses, we used the meta-analysis results of influenza cohorts above to maximize statistical power (Supplementary Table 4a) and found that the brown module was significantly enriched for genes associated with antibody responses (p=0.01, GSEA test, Fig. 3d; Supplementary Table 4b). However, this was not the case for the yellow fever dataset (data not shown), perhaps due to the small cohort size and thus insufficient statistical power. Together, these observations provide independent support to our TGSig finding above that shared baseline signatures can exist for influenza vaccination responses and SLE disease activities.
Both baseline signatures are associated with Type I IFN responses and activation of dendritic cells
Given their qualitatively similar predictive profiles, TGSig and the brown module might reflect overlapping biology, even though they shared only one gene (EPHB1, out of the 370 genes in the brown module). Since TGSig was derived from the temporally-stable genes most robustly correlated with the frequency of CD20+CD38++ B cells (Fig. 1d), we ranked the temporally stable genes by their correlation with the frequency of these B cells and found that the most correlated genes were enriched for the brown module as well as Type I IFN and dendritic cell (DC) activation related BTMs (Fig. 3e; see Methods). These results established functional links between the brown module and TGSig, and suggest that they reflect aspects of the Type I IFN response/DC activation pathway. To capture the biology of this overlap for further analysis below we created the IFN-I-DCact (Type I IFN and DC activation; see Extended Data Fig. 1d) gene signature comprising genes in the brown module that are shared with at least one of the IFN-I/antiviral/DC activation BTMs (Fig. 3f).
We also evaluated the overlap among TGSig, the brown module, and IFN-I-Dcact from a predictive standpoint by using data pooled from the meta-analysis above (see Methods). As expected, each signature on its own contained predictive information (Extended Data Figs. 6a–c), and most of the predictive capacity of the brown module came from the 87 “leading edge” genes (SLE-Sig, see Extended Data Fig. 1d; Fig. 3e, Supplementary Table 5, Extended Data Fig. 6d). When TGSig was used together with SLE-Sig or IFN-I-DCact for prediction, TGSig was the dominant predictor (Extended Data Figs. 6e,f). The same was true for predicting DaCP in the SLE cohort (data not shown). Thus, TGSig, as a baseline signature, contains most of the predictive information in SLE-Sig and IFN-I-DCact for both influenza vaccination and SLE disease activity.
Dissecting the cellular origin of the baseline signatures: CITE-seq analysis of high and low influenza vaccination responders
To dissect the cellular origin of our signatures, we sorted CD19+CD20+CD38++ B cells from six healthy donors followed by RNA-seq (Extended Data Figs. 7a,b). However, genes differentially expressed in these cells compared to CD19+CD20+ B cells were not enriched for TGSig or SLE-Sig (Extended Data Figs. 7b–e), suggesting that both did not originate from these cells.
Given the large number of possible cellular origins for TGSig and SLE-Sig, we pursued an unbiased approach by adopting CITE-seq16 to simultaneously profile 82 surface proteins (covering lineage and phenotypic markers of diverse immune cells) and the transcriptomes of 53,201 single cells from the baseline (day 0/pre-vaccination) PBMC of 10 high and 10 low responders from the NIH cohort (Fig. 4a; average 2660 (SD=753) cells per donor). A major goal was to assess whether our baseline signatures reflect transcriptional state differences between the high and low responders in certain cell subsets.
Figure 4: CITE-seq (simultaneous protein and transcriptome expression profiling in single cells) analysis of high and low influenza vaccine responders.
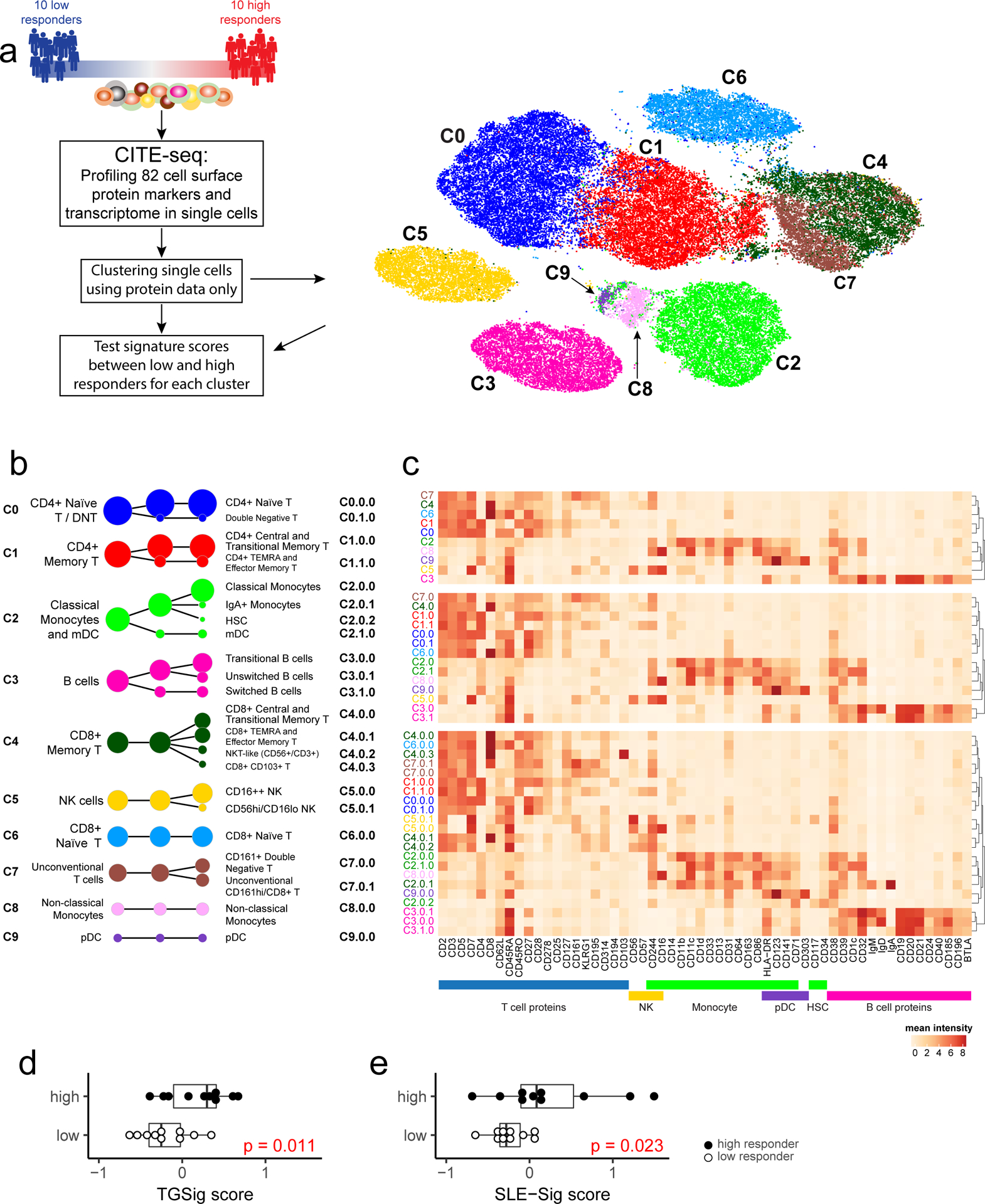
a, Experiment and analysis overview: single PBMCs from 10 high (red) and 10 low (blue) responders (as defined by adjMFC from ref.12) were profiled by CITE-seq (measuring 82 cell surface proteins and transcriptome). Cells from all subjects were clustered together using only surface protein expression profile at three increasingly detailed clustering resolutions (referred herein as levels 1–3, denoting the lowest to the highest resolutions; see Methods). 10 cell clusters (C0-C9) were identified at level 1 and shown in different colors in the tSNE plot. b, Cell clusters from levels 1–3 are shown in three columns and depicted as circles (size is proportional to the number of cells in the cluster). The edges denote containment relationship between the clusters at neighboring resolutions: an edge connecting one cluster to another cluster indicates that some fraction (or all) of the cells in the former are found in the latter. Annotations are provided for levels 1 (1st column) and 3 (3rd column) clusters. The clusters/circles are colored, matching those in the tSNE visualization. c, A heatmap showing the average expression of selected protein markers (columns) in each of the cell clusters (rows) derived from the three different clustering resolutions. The cell cluster names are color matched with those in (b). See Extended Data Fig. 8a–c and Supplementary Fig. 2 for additional details. d, Boxplot comparing the TGSig score between high (solid dot; n=10) and low (empty dot; n=10) responders using “pseudo bulk” data (average across all single cells within each subject; see Methods); p value from Wilcoxon one-tailed test. Boxplots’ center line corresponds to the median value, lower and upper hinges correspond to the first and third quartiles (the 25th and 75th percentiles); lower and upper whiskers extend from the box to the smallest or largest value correspondingly, but no further than 1.5x inter-quantile range. e, Same as (d) but for SLE-Sig.
We clustered the cells23 at several resolutions using their surface protein expression profile to reveal major cell types and subsets (Figs. 4a–c; Extended Data Figs. 8a–c; see Methods). The frequency of most cell clusters was not significantly associated with vaccination responses, except, e.g., negative associations involving effector CD4+ memory (cluster C1.1.0) (data not shown), which is consistent with the original study (ID36 in Figure 6C in ref12). TGSig computed using the “pseudo bulk” data (averaged across all single cells for each subject; see Methods) was significantly higher in the high than the low responders (Fig. 4d). The same holds for SLE-Sig (Fig. 4e) and the frequency of manually gated CD20+CD38++ B cells using CITE-seq data (Extended Data Figs. 8d,e; p=0.032 Wilcoxon one-tailed test). Thus, CITE-seq data reproduced our earlier findings from microarray and flow cytometric measurements.
Figure 6:
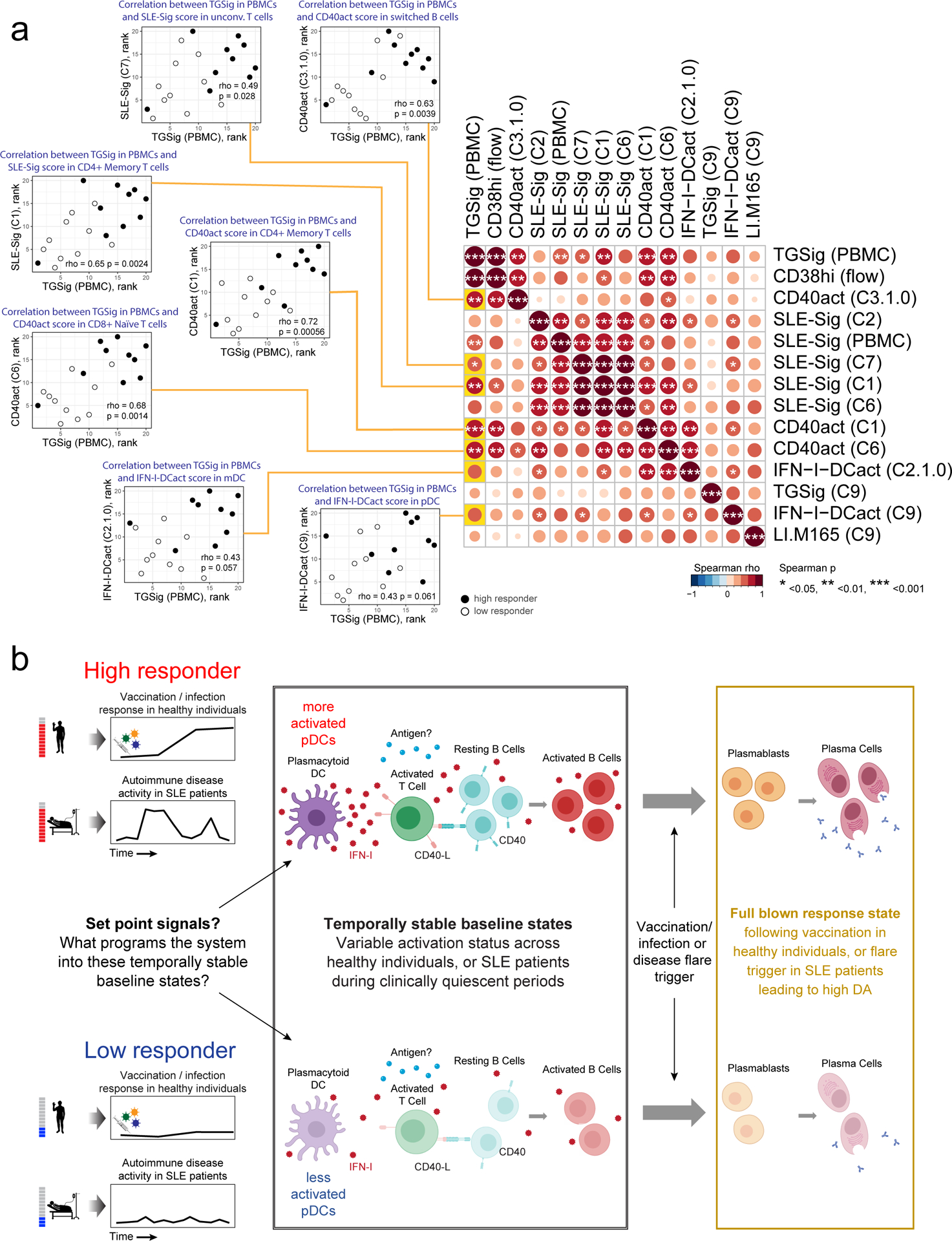
a, Matrix heatmap showing the pairwise Spearman correlation among the select signature scores across subjects. Example scatterplots similar to the one in Fig. 5f are shown for assessing the correlation between the original TGSig (computed using microarray data generated from PBMCs – see Figs. 1c–e) and the cell cluster based signature scores found to be significantly different between high (n=10) and low (n=10) responders. Spearman correlation and two-tailed p value are shown. The name of the cell cluster (see Fig. 4a,b) for which the indicated signature score was computed is in parentheses. Each example scatterplot corresponds to a highlighted (yellow) entry in the matrix on the right. The matrix is symmetrical: row and column profiles are identical. The size and shade of the circle indicate the correlation strength (Spearman rho) and asterisks denote significance level (two-tailed test) as shown in legend below. Note that SLE-Sig (PBMC) was computed using the original bulk microarray data (the same as TGSig (PBMC)). b, Model describing the molecular/cellular underpinnings and differences between high versus low responders. Activation of this entire circuit (including the components in the plasmablast/plasma cell box on the right) typically follows infection, vaccination, or during autoimmune disease flares. Here we propose that the high responders tend to have more activated pDCs and thus more Type I IFNs and activated B and T cells at baseline, but only upon additional antigenic and/or inflammatory co-stimulation (and flare trigger in the case of SLE patients) does the system mount a full-blown plasmablast/plasma cell response cumulating in the generation of antibodies. Open questions include: 1) What sets the system into such temporally stable “activated” states in pDCs, lymphocytes, and other myeloid cells?; 2) What constrains the activated immune baselines from mounting full-blown plasmablast/plasma cell responses?; 3) What is the antigen specificity repertoire of the activated lymphocytes at baseline?
We next evaluated TGSig and SLE-Sig differences between high and low responders within cell clusters (Supplementary Tables 6 and 7). Significant differences in the average expression of TGSig genes were found in plasmacytoid DCs (pDC – cluster C9), a major producer of Type I IFNs and other cytokines24 (Fig. 5a). SLE-Sig (Fig. 5b) was elevated broadly across cells clusters, including CD4+ central memory and CD8+ naïve T cells (C1 and C6), classical monocytes and myeloid DCs (mDCs) (C2), transitional B cells (C3.0.0), and unconventional T cells (C7) (Fig. 5b). While the other clusters were not statistically significant, SLE-Sig trended higher in the high responders, suggesting that most peripheral immune cells were broadly exposed to higher levels of IFNs in high responders at baseline. Similar results were obtained using an independently derived Type I IFN response gene set (Extended Data Fig. 8f; Supplementary Tables 6 and 7; see Methods).
Figure 5: Dissecting the cellular origin of baseline signatures.
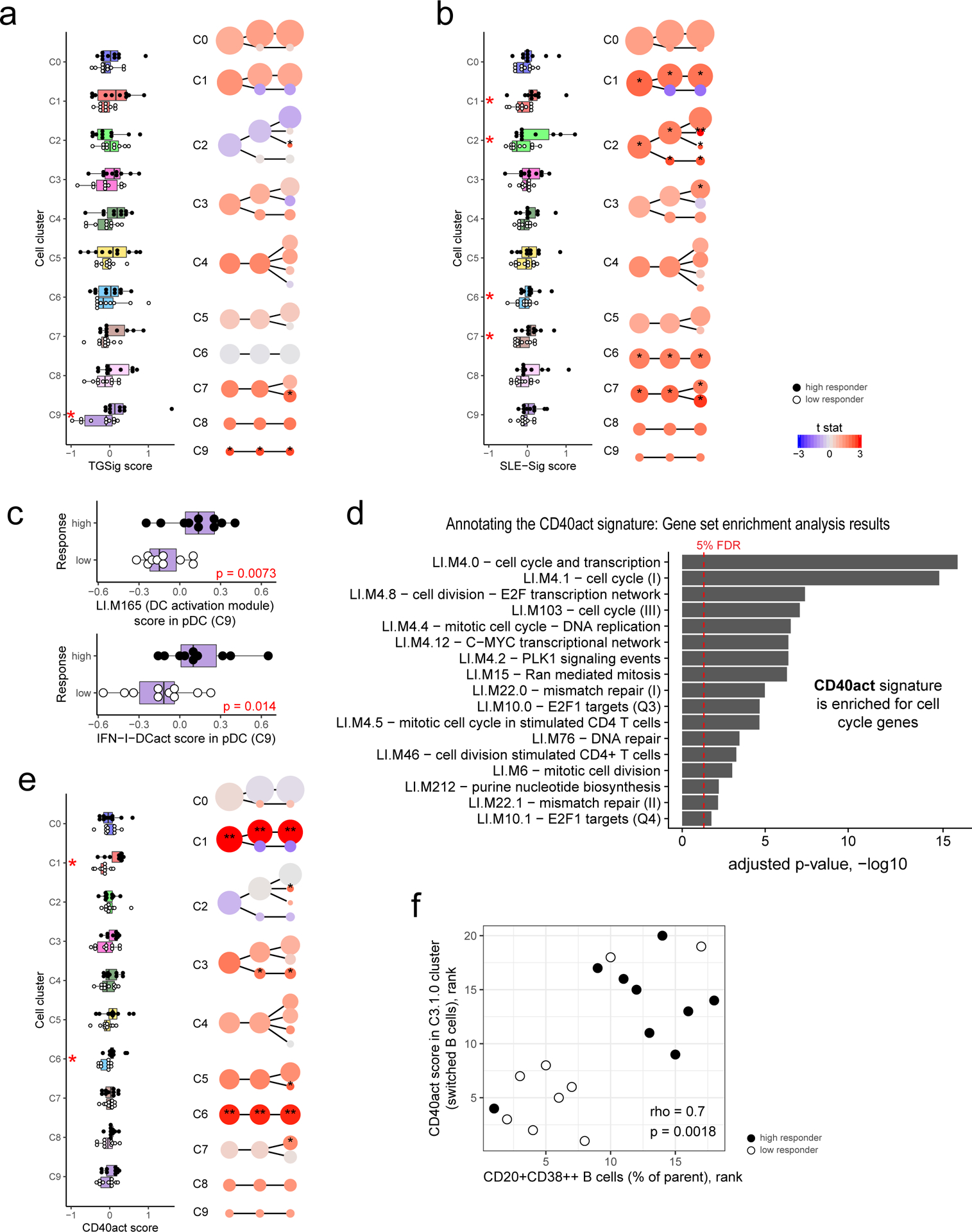
a, Evaluating the difference in TGSig score between high (n=10) and low (n=10) responders in each cell cluster from Fig. 4b (see Methods). Left panel: boxplot comparing high (solid dot) and low (empty dot) responders in each of the level 1 (1st column) clusters; each dot corresponds to the signature score of a subject. Red asterisks denote significance with p<0.05 (Wilcoxon one-tailed test; see also Supplementary Table 7). Right panel uses the same visualization as in Fig. 4b but here the color reflects the average normalized difference in TGSig signature score between the high and low responders (shown here as a t statistic). One or two asterisks denote significance with p<0.05 or p<0.01, respectively (Wilcoxon one-tailed test because we are interested in assessing whether the high responders are higher than the low responders; see also Supplementary Table 7). For all boxplots the center line corresponds to the median value, lower and upper hinges correspond to the first and third quartiles (the 25th and 75th percentiles); lower and upper whiskers extend from the box to the smallest or largest value correspondingly, but no further than 1.5x inter-quantile range. b, Same as (a) but for SLE-Sig. c, Similar to the boxplots in the left panel of (a) (10 high versus 10 low responders) but for the signature score of the LI.M165 BTM (top panel: enriched for dendritic cell activation) and the IFN-I-DCact (bottom panel; see Fig. 3f) gene sets evaluated for cells in the pDC cluster only (cluster C9). d, Enrichment analysis result of the CD40act gene set (Supplementary Table 6; 49 genes) using the hyper-geometric test against the BTMs from ref21. All 32,738 detected genes were used as a background. BTMs with an adjusted one-tailed p-value (FDR computed using the BH method) of 0.05 (red line) or lower are shown. e, Same as (a) but for CD40act. f, Scatterplot (based on ranks since Spearman correlation is being evaluated) assessing the correlation between the frequency of CD20+CD38++ B cells (see Fig. 1b) and the CD40 activation signature score in the switched B cell cluster (C3.1.0 – see Fig. 4b,c). Spearman correlation and two-tailed p value are shown (based on 9 high and 9 low responders because not all 20 subjects assessed by CITE-seq have corresponding flow cytometry data). Detailed test statistics for data shown in (a), (b), and (e) can be found in Supplementary Table 7.
Given that pDCs are major producers of Type I IFN24, the elevated Type I IFN status in high responder cells from diverse lineages may be due to the presence of more activated pDCs in these individuals; we indeed found that the average expression of the genes in LI.M165 and IFN-I-DCact—both reflective of DC activation and linked to TGSig and SLE-Sig (Figs. 3e,f)—was significantly elevated in pDCs of high responders (Fig. 5c).
Activated pDCs are known to activate T cells that then stimulate B cells via CD40L24,25. To test whether we can detect the activation status of this circuit even at baseline, we derived a CD40 activation gene signature (CD40act, see Extended Data Fig. 1d) (Supplementary Table 6) from two independent studies of human B-cells stimulated with CD40L26,27 (see Methods). CD40act was interestingly highly enriched for cell cycle processes (Fig. 5d), likely because CD40L-activated B cells are highly proliferative28. As hypothesized, CD40act was significantly increased in switched B cells (C3.1.0) in high responders (Fig. 5e). It was also elevated significantly in several lymphocyte clusters (see Figs. 4b,c and Extended Data Figs. 8a,c for cell cluster annotations), including CD4+ memory (C1.0.0), CD8+ naïve (C6), and unconventional CD161+ T cells (C7.0.0) (Fig. 5e). Together, these observations suggest that in high responders, the increased activation of pDCs and elevation of Type I IFNs led to the activation and proliferation of not only switched B cells, but also T lymphocytes. Furthermore, we hypothesized and indeed observed that CD40 activation status in switched B cells (C3.1.0) was correlated with the frequency of CD20+CD38++ B cells measured by flow cytometry (Fig. 5f). This suggests that the CD20+CD38++ cells, our original baseline signature (Fig. 1b, Extended Data Fig. 1a,b), overlapped with activated switched B cells.
TGSig captures the activation/cell cycle and Type I IFN response statuses of lymphocytes and myeloid cells
We next analyzed the correlation among the signatures, including TGSig assessed in “bulk”/PBMCs (Figs. 1c–e), the frequency of CD20+CD38++ B cells, and the significant cell cluster-based signatures emerged from CITE-seq analysis (Fig. 6a). TGSig correlated with and thus captured the status of several signatures at the cell cluster level that delineated high versus low responders as determined by CITE-seq (left scatterplots in Fig. 6a). These signatures include both the activation/proliferation (as reflected by CD40act) and the Type I IFN response statuses (as indicated by SLE-Sig) of switched B cell and several T cell clusters, and albeit more mildly, those of pDCs and mDCs. SLE-Sig (in PBMCs as measured by microarrays) also captured the Type I IFN response status of both lymphocytes and myeloid cells. However, SLE-Sig captured less well the activation/cell cycle status of switched B cells (C3.1.0) and T cell subsets (C1.0.0 and C6) (Fig. 6a), which were some of the most significant correlates of the response as revealed by CITE-seq (Fig. 5e).
Our single cell data gave us an opportunity to drop specific combinations of cell clusters from the data, and then evaluate whether the signature scores computed from the remaining cells are still associated with the response (Extended Data Fig. 8g). Consistent with the observations above, this analysis revealed that TGSig became uninformative of the response when both the CD4+ memory and CD8 naïve T cell clusters (C1 and C6) were dropped; for SLE-Sig the most important predictive information originated from the monocyte/mDC (C2) and CD4+ memory clusters (C1). Together, these observations suggest that even though TGSig only contains a few genes that were originally derived based on correlations with the frequency of the CD20+CD38++ B cell population, it captures the responsiveness-predicting states of multiple cell subsets in peripheral blood, particularly those of two subpopulations of T lymphocytes that represent a substantial fraction of cells in blood.
Discussion
To our knowledge, predictors of autoimmune disease activity, particularly from clinically quiescent periods, are rare29,30, and baseline set point2 signatures shared among vaccination, infection, and an autoimmune disease have not been reported. Our simultaneous deep immunophenotyping and transcriptome analysis of single cells suggests that these predictive signatures reflect the extent of cell cycle/activation and Type I IFN response statuses in a circuit comprising pDCs, switched B cells, and T lymphocytes (Fig. 6b), thus pointing to a shared origin for our two independently-derived, but biologically related set point signatures. The sustained activation of these and related circuits have been implicated in the pathogenesis of SLE and other autoimmune diseases31, however, here we provide fresh evidence that the elevated activation status of cells in this circuit during clinically quiescent periods in a subset of SLE patients may indicate higher plasmablast-associated disease activity. Intriguingly, this circuit was also more activated in certain healthy subjects stably over the course of months and these individuals tended to mount higher antibody responses to influenza or yellow fever vaccination. Thus, the future responsiveness potential to a perturbation can be encoded by the sustained activation status of a circuit that is typically activated full-blown only after an immune challenge (e.g., an infection). The mechanisms that restrain full-blown systemic immune activation before antigenic and inflammatory stimulation (particularly in healthy individuals) and the antigen-specificity repertoire of the activated lymphocytes at baseline remained to be dissected (Fig. 6b).
Through CITE-seq analysis we found that the CD20+CD38++ B cells we originally identified likely overlapped with activated switched B cells, some of which could also be precursors of plasmablasts32. It is unlikely that all of these cells were influenza specific or persistently activated by influenza at baseline and remained temporally stable over months. They could be enriched with influenza specific cells in some individuals, but the fact that TGSig could predict responses in YF naïve subjects (Fig. 2b) suggests that TGSig does not simply reflect the frequencies of memory lymphocytes specific for particular vaccine antigens, unless this was all due to cross-reactivity, which is also unlikely. Furthermore, previous analysis of antigen-specific B cells12 in the NIH cohort showed that baseline vaccine reactivity did not correlate with antibody responses, which was consistent with independent observations33 that the frequency of pre-existing influenza-specific B cells in peripheral blood does not correlate with antibody responses following vaccination. Similarly, higher frequencies of activated, influenza-specific CD4+ T cells in peripheral blood at baseline were not positively associated with T cell responses following influenza vaccination34.
pDCs were likely a major source of Type I IFNs that led to the elevated IFN response status across multiple cell lineages in the high responders (Fig. 5b and Extended Data Fig. 8f). Since they represent only a small fraction of circulating immune cells, TGSig and SLE-Sig likely reflect less on the activation status of the pDCs but instead capture pDCs’ downstream effects on lymphocytes and monocytes, which are much more abundant in blood. Together, these results also illustrate how deep single cell analysis in human subjects with distinct responsiveness phenotypes can help unmask cellular origins and provide mechanistic hypotheses on bulk blood transcriptomic biomarkers.
It remains to be determined why pDCs in healthy high responders were more activated—persistent triggers of TLR7/9 could be involved24. The microbiome could be responsible but seemed to largely affect naïve than recall responses based on a recent study in influenza vaccination6. CMV status is possible but less likely because a known correlate, the frequency of TEMRA cells35, was not significantly associated with responder class in our cohort (Extended Data Fig. 9a,b). While genetics could play a role, twin studies have indicated that antibody responses to influenza vaccination exhibit little heritability in adults7. Two relevant pDC phenotypes (HLA-DR and CD86 expression on pDCs (cluster C9))35 with strong genetic drivers were not significantly different between high and low responders (Extended Data Fig. 9c). Similarly, none of the genes in TGSig was associated with any trans expression quantitative trait loci based on a large-scale study of the genetics of blood gene expression in healthy subjects36. Age was not associated with TGSig in the NIH cohort (data not shown), but females tended to have higher TGSig scores than males (Extended Data Fig. 10a–c), which is consistent with earlier observation that females tend to mount higher responses to some vaccines but are more prone to autoimmunity37. Intriguingly, however, even within each sex, TGSig could predict responsiveness (Extended Data Figs. 10d,e), again suggesting that TGSig is a general baseline predictor.
While TGSig’s applicability in influenza (across three out of four US locations and four different years), yellow fever, and SLE supports the notion that it reflects common baseline determinants shared by these immune response scenarios, it was not predictive in influenza datasets from a US location over four consecutive years. It also remains to be determined whether it is applicable to other situations such as cancer immunotherapy and additional autoimmune diseases with flares associated with plasmablasts. TGSig was specific to a defined subtype of lupus as we had hypothesized (Fig. 2e) and all of the vaccination cohorts tested largely consisted of younger adults (age<45). In general, baseline set point signatures are likely not universally applicable but depend on factors such as age, ethnic background, and geographic location10,38. Rigorous clinical trials are ultimately needed to assess the applicability of these baseline signatures for different immune perturbations and diseases.
Given the centrality of the immune system in health and disease, our findings point to the prospect for longitudinal immune health assessment and monitoring as well as predicting future responses to vaccination, infection, and other perturbations using just a few markers in blood. Set point signatures could also be used to stratify populations in clinical trials and for properly accounting for baseline heterogeneity when analyzing trial outcomes. Our results motivate the search for additional shared baseline determinants and predictive signatures among vaccination, infection, cancer, and autoimmune and inflammatory diseases, which may reveal new biology including common mechanisms underpinning different types of immune responses.
Methods
Datasets
A summary of the datasets used in our study can be found in Supplementary Table 2. See also the “Life Sciences Reporting Summary” accompanying this paper and the Data Availability section.
Software
A summary of the software packages used and their versions can be found in Supplementary Table 8. See also the “Life Sciences Reporting Summary” accompanying this paper and the Code Availability section.
Predictive B cell populations
PBMC sample collection and processing are described in ref.12. The predictive baseline B cell populations identified in ref.12 (ID103, ID96, ID91, ID108; Figure 6, Table S2 in ref.12) had two important features: 1) high temporal stability (low within-subject variation (WSV)) with high inter-subject variation (ISV) (Fig. 2 in ref.12), and 2) a positive correlation between the baseline/pre-vaccination cell frequencies and vaccine antibody response as quantified by the adjusted maximum fold change (adjMFC) (Fig. 6C in ref.12). The adjMFC is a measure of antibody fold-change following vaccination after removing the nonlinear correlation between the maximum (log) fold change in antibody response and the baseline antibody titer, thus allowing for evaluation of predictive factors that are independent of the baseline antibody titer (see Methods in ref.12). As in ref.12, for all the influenza vaccination datasets we assessed in this study, we used the adjMFC metric to reflect the maximum titer to any of the vaccine components administered to a given subject, especially because the above B cell populations of interest, and therefore the associated transcriptional surrogate signatures we explored here, were not measured in an antigen specific manner.
Flow cytometry data processing
See also the “Life Sciences Reporting Summary” accompanying this paper. All flow cytometric quality control (e.g., sample exclusion) and gating was performed before the analyses described below, e.g., predictive modeling and surrogate transcriptional signature (TGSig) derivation. We gated four new subpopulations (based on CD38 and CD10 markers) inside the CD45+CD19+CD20+ live B cell gate (FlowJo ver.9.9.3, TreeStar/Becton Dickinson Co., Ashland, OR, on Mac OS X; Fig. 1b, Extended Data Fig. 1a), including Gate 1: CD38+ cells with CD38 (G610-A; PE-Texas Red) fluorescent intensity greater than 1000; Gate 2: CD38++ cells with fluorescent intensity greater than 10000; Gate 3: CD38++CD10+ cells with CD10 (R780-A; APC Cy7) fluorescent intensity greater than 1000; Gate 4: CD38++CD10- cells with CD10 fluorescent intensity less than 1000. We exported cell frequencies, expressed as the percentage of the parent population. The above gating strategy was designed based on the following considerations: 1) high CD38 expression was a common feature among the predictive CD20+ B cell populations from ref.12; 2) CD10 based gates were included because CD10 was expressed in one of the original predictive populations (ID91; Figure 6, Table S2 in ref.12) and these gates would also allow us to evaluate prediction performance with or without the inclusion of CD10 gates (Fig. 1b, Extended Data Fig. 1a). Note that delineating CD10+ vs. CD10- cells in healthy individuals can be challenging42, thus we were cautious in our interpretation of the CD10+/CD10- fractions and focused solely on evaluating relative prediction performance with or without the inclusion of our CD10 gates in CD20+CD38++ cells (Fig. 1b, Extended Data Fig. 1a).
Quality control:
Based on visual evaluation during gating, specifically taking into account: 1) low cell viability (<70%); 2) extremely small number of cells expressing high levels of CD38 (less than 5 cells in CD38++ gate); 3) non-discernable B cell populations (visually indistinct CD19+ population in CD45+ cells), we excluded 8 baseline samples (out of 78 baseline samples that: 1) had titer data published in ref.12 and 2) were either high or low responders based on adjMFC – only high/low responders were used in the subsequent analyses described below, although middle responders were also used to assess robustness (Extended Data Fig. 3); here again, baseline was defined as days 0, −7 and 70. Note that subject 262 (all three baseline time-points) was not used in subsequent analysis because it was erroneously labeled as an intermediate responder in the beginning, but during final quality check when preparing the manuscript we realized it was a high responder. In general, we followed the above cutoffs to remove samples, except that we also removed the day 70 sample of subject 250 (viability 78.1% and 20 counts of CD38++ cells) because the percent of CD38++ cells was exceedingly lower than day 0 (0.63% vs. 3.38%). After QC, we have the following number of samples for subsequent analyses:
| low | high | |
|---|---|---|
| Baseline 1 (day 0) | 11 | 12 |
| Baseline 2 (day −7) | 11 | 12 |
| Baseline 3 (day 70) | 10 | 11 |
Selection of CD19+CD20+CD38++ population (CBSig)
The predictive power of each of the four newly gated cell subsets (Fig. 1b, Extended Data Fig. 1a) were evaluated by AUC (see Performance assessment of model predictions). Of the four cell populations, population #2 (CD38++ of CD20+ B cells) had the highest AUC (Fig. 1b) and was selected as the population of interest (CBSig, red box). Note that the addition of CD10 in the gating scheme (CD10+CD38++ vs. CD10-CD38++) led to lower AUCs compared to CBSig, but the CD10+ and the CD10- subsets had similar predictive performance, indicating that CD10 status was not important for prediction (Fig. 1b). Variations of this gating scheme, such as lowering the CD38 expression level requirement or using CD10 to restrict to transitional or non-transitional subsets, did not result in better prediction performance (Fig. 1b), suggesting that CD19+CD20+CD38++ cells were indeed a core predictive population.
Construction of 10-gene signature (TGSig)
Microarray data processing:
To develop a gene-based surrogate predictive signature of CBSig, we used the same influenza vaccination dataset from our previous study (the NIH Center for Human Immunology (NIH/CHI) dataset) in which gene expression data was also generated from the same PBMC samples assessed by flow cytometry12. The raw gene expression data are available in GEO (GSE47353); the processed data are available on our data portal (https://chi.niaid.nih.gov). The expression data was RMA-normalized and batch-corrected as described in the “Extended Experimental Procedures” (“Low‐level microarray data processing” section) in ref.12. Briefly we found that the hybridization date (batch) of array was significantly correlated with most probe sets, and we removed this effect using linear regression for each probe set individually. For this purpose, for the i-th probe set we fitted the model (i-th probe set intensity) ~ (hybridization date) and only retained the residuals from this fit.
Assessing temporal stability:
To derive a surrogate gene signature, we looked for genes that: 1) correlated with the frequency of CD20+CD38++ B cells prior to vaccination (day 0), 2) have low within-subject variation across the three baseline time points (so that it is temporally stable), and 3) high inter-subject variation (ISV) to help potentially delineate differences in responsiveness.
Here we have a data vector X={xij} (individual: i, time-point: j), where each xij is associated with a subject and each subject has three measurements obtained from each of the baseline time-points. We then fitted a one-way ANOVA model (in R notation: X ~ Subject) to evaluate the total variance (total sum of squares) and partitioned the total sum of squares into components attributable to subject-to-subject variation and the rest as the fitted residual.
SSsubject is the sum of squares of differences between the subject mean and the overall mean. SSresidual is the remaining sum of squares. Here we assumed that the variance explained by SSsubject provides an estimate of subject-to-subject differences while the variance explained by the residual of this fit (SSresidual) provides an estimate of the sum of within-subject variation and other sources of noise, including technical noise. We then used this relationship:
to evaluate the fraction of variance explained by subject (SSsubject/SStotal; ISV) relative to the fraction explained by the residual of the fit (SSresidual/SStotal). We defined (SSsubject/SStotal; ISV) as the “stability score”, or the temporal stability metric (TSM), insofar as high ISV would indicate low residual variance and thus higher temporal stability within subjects over time. QCed expression data from all three baseline time-points were used in this analysis (see workflow below and ref.12 for QC procedures.)
Selection and evaluation of signature genes:
We selected a total of 726 genes with TSM ≥ 0.75 for correlation analysis. Using only data from the high- and low-responders with both day 0 flow cytometry (see above) and gene expression data passing QC in the NIH/CHI dataset (22 day 0 samples total), we correlated these 726 genes against the frequency of CD20+CD38++ cells using day 0 data. To mitigate influence from outlier subjects and account for sampling noise, we used multiple iterations of subsampling of the cohort to derive a “robust” measure of correlation as follows: in each iteration, we excluded two subjects and then calculated the Spearman correlation between each gene and the cell frequency. We performed a total of 231 iterations (the number of subject combinations to exclude two out of the 22 subjects). For each gene, the “robust correlation” is defined as the average correlation divided by its standard deviation across all iterations. Genes were ranked according to this robust correlation statistic, which considers both the magnitude of the correlation and sampling noise. Top genes from the list can be found in Supplementary Table 1 together with additional information, including the mean, standard deviation, the percent of iterations in which the gene was ranked among the top 20.
To determine the number of top genes to construct the surrogate signature, we compared AUCs generated from top k genes (k=1, 2, 3, …., 30) (see “Gene-based signature score calculation” below) for the three baseline time points (Extended Data Fig. 2c). We empirically chose k (k = 10, TGSig) so that that AUC was maximized in all three time-points while including a sufficient number of genes so that when we evaluate the signature in other datasets with different profiling platforms we can retain a reasonable number of these genes. In addition, we checked how exclusion of any single gene from the signatures affects prediction performance (Extended Data Fig. 2g).
To estimate the null-distribution of prediction performance we generated a set of 500 “random signatures” – top 10 genes based on the same ranking and evaluation scheme described above, except that the gene-cell population correlations were calculated using subject-label permuted data without applying the temporal stability filters. We used these “random signatures” generated for each dataset to evaluate whether the prediction performance of our signature is significantly higher than that expected by chance (Extended Data Fig. 2e).
Finally, we also checked that our signature genes retained their relative rank even if we relaxed the stability (TSM) threshold. Because the total number of “temporally stable” genes is dependent on the stability threshold, instead of using absolute ranks we normalized the ranks of signature genes by the total number of genes passing the stability threshold (Extended Data Fig. 2f). The ranking of the top genes is relatively stable across TSM thresholds ≥ 0.75, although additional random noise dominates when the TSM threshold exceeds 0.75 because the number of genes became small (Extended Data Fig. 2f).
Gene set-based signature score calculation
Given a set of signature genes (e.g., TGSig) and a dataset (e.g., Yale 2011), we calculated the signature scores for each dataset independently. Due to profiling platform differences, some genes in a signature were not present in a given dataset and those genes were not used in the signature score computation. We used a z-score transformation to standardize the expression of each gene to have mean 0 and standard deviation 1 across all the samples in the dataset. This step was carried out to ensure that expression values of different genes were on the same scale and thus comparable. The signature score of a sample was then calculated by averaging the standardized expression value of the signature genes (Extended Data Fig. 2d).
Evaluation of TGSig in non-CHI vaccination datasets
Dataset selection and data preparation:
We obtained influenza vaccination datasets from a recent meta-analysis of transcriptomic signatures of influenza vaccination conducted by the Human Immunology Project Consortium and CHI10. These data were derived from PBMC (SDY400 and SDY404) or whole blood (SDY212) (Supplementary Table 2). We downloaded the pre-processed expression data from ImmuneSpace (http://immunespace.org) using the ImmuneSpaceR Bioconductor package43. These studies are also available on ImmPort (http://immport.org44), under study IDs SDY212 (Stanford University, season 2008–2009), SDY400 (Yale University, season 2012–2013) and SDY404 (Yale University, season 2011–2012). See also the “Life Sciences Reporting Summary” accompanying this paper.
The influenza vaccination dataset from Nakaya et al18 (Emory; seasons 2008–2011) was downloaded from GEO (GSE29619 and GSE74817). The demographics and HAI titer data were received via private communication. Season 2007 was excluded from the analysis due to the small number (n=9) of subjects. The downloaded data (matrix data (probeset × sample) from GEO) was already pre-processed (including normalization and log-transformation), and we thus used it as-is in our analysis. We also conducted the same analyses by starting from the raw CEL files (using the same procedure as described below for yellow fever) and the results were similar to Extended Data Fig. 3c (data not shown.) We also tested defining high and low responders using the same method as reported in the original publication18 and the results were again similar in that the high and low responders were not separable statistically (data not shown).
The yellow fever vaccination dataset19 was downloaded from GEO (GSE13486). Since the pre-processed data was not available, we downloaded the CEL files and performed background correction and normalization of the expression data with RMA algorithm implemented in Affymetrix Power Tools (APT).
The systemic lupus erythematosus (SLE) dataset13 was downloaded from the webSLE data portal (http://websle.com) as an RData file, which contained ExpressionSet objects with pre-processed and pre-filtered expression data together with probe annotation and sample information.
For expression datasets containing only probe level information, we summarized probe level expression into gene level data as follows: 1) we retained only probes that were unambiguously mapped to a single gene; 2) in the case where multiple probes were mapped to the same gene, we performed principal component analysis of the probes using all samples in the dataset, and then selected the probe maximally correlated with the first principal component as the reporter for the expression of the gene.
All expression data were RMA normalized and log2-tranformed prior to analysis, except for the SLE data where we used the pre-processed data directly (which included log2-transformation.)
Note that for the NIH/CHI dataset, TGSig was computed only for subjects with both QCed gene expression and titer data (see workflow below.)
Identification of high and low responders:
High and low responder determination was performed before and independent of any predictive signature assessment. We used the adjMFC metric to quantify the antibody response to influenza vaccination independent of the initial/baseline titer (refs.12 and10). While there were differences in how antibody titer data were generated across different datasets/studies (e.g., the NIH/CHI dataset used microneutralization assays, while others used the hemagglutination inhibition (HAI) assay), adjMFC only reflects the normalized, relative response within a dataset/cohort, but not the absolute magnitude (see ref.12) and is thus applicable to all datasets to quantify the relative response (high vs. low responders) within each dataset. The detailed methodology can be found in the “Extended Experimental Procedures”/“Titer definitions” in ref.12).
For each of the influenza vaccination datasets, we classified subjects into high and low responders according to their adjMFC values and use these response class labels in subsequent analyses (see refs.12 and10 for the rationale behind only focusing on high and low responders). Briefly, subjects who were below the 30th percentile or above the 70th percentile adjMFC values in a dataset were classified as low and high responders, respectively. Following previous HIPC-CHI analyses, these thresholds were selected to allow enough high and low responders for analysis. This cutoff differs from that used in the original NIH/CHI influenza study, in which the subjects were discretized to low and high responders using the 20th and 80th percentile adjMFC values as cutoffs, respectively (see Methods in ref.12). To be consistent with the original NIH/CHI influenza study, we kept the same 20/80 cutoffs for defining high and low responders when analyzing the NIH/CHI dataset here. These cutoffs were all pre-determined before analyses started.
For the yellow fever vaccination dataset, neutralization titers against the yellow fever virus (YFV) measured on day 60 were used as the antibody response to vaccination because all subjects were naïve to the YFV at baseline. We discretized the original titer values within each of two reported trials. The thresholds to identify high and low responders were selected based on the distribution of titer values within each trial. Because the neutralizing titers were measured using different techniques (by cytopathic effect (CPE) for trial 1 and by plaque reduction neutralization test (PRNT) for trial 2), the exact thresholds we used were different. For trial 1, six subjects were classified as low responders (titer < 160), and six as high responders (titer > 160), while for trial 2 there are three low responders (titer < 640) and four high responders (titer > 640). Titer values of 160 and 640 were considered “middle responders” in each trial, respectively, and were excluded from analysis.
Predictive performance assessment:
To assess whether TGSig was predictive of high vs. low responders, we computed the AUC (area under the empirical Receiver Operating Characteristic (ROC) curve). A permutation procedure was used for estimating one-tailed p values – see Statistical Procedure below for further details. We also used the one-tailed Wilcoxon Rank Sum test to assess whether the signature score was significantly higher in high compared to low responders, which also provided another measure of statistical significance in addition to that computed for the AUC. We used a one-tailed test because we knew a priori that CBSig (and the original predictive populations from ref.12 from which CBSig was derived) had higher frequencies in high than low responders. Thus, we specifically test whether TGSig (a surrogate of CBSig) was significantly higher in high than low responders. All statistical analyses were performed using R/Bioconductor. ROC curves were computed with the pROC package45.
Assessing TGSig in independent vaccination datasets:
After selecting signature genes for TGSig using only the day 0 data of the CHI influenza dataset, we computed the TGSig signatures scores for each subject prior to vaccination in each of the influenza vaccination datasets (Stanford, Yale, and Emory) and the yellow fever vaccination dataset and evaluated the predictive performance of delineating high vs. low responders. The influenza datasets all had bi-modal age distributions (young: age<35 and older: age > 60) because they were originally designed to assess responses in older vs. younger individuals10; we thus focused on testing TGSig in young subjects only because the CHI dataset consisted of largely younger individuals. We did evaluate TGSig in older subjects and found that it was not predictive of high vs. low responders in those older than 60. For the Emory dataset, a higher cut-off for “young” (age < 60 years) was employed because using a cutoff of 35 would result in a very small dataset, but we also tested using 35 as the cutoff and the results were similar. The yellow fever dataset contains two trials with the first having a larger sample size than the second, and we tested the signature in each trial independently.
Additional statistics for assessing prediction of high versus low responders can be found in Supplementary Table 2b.
Application of TGSig to a pediatric systemic lupus erythematosus (SLE) cohort
See also the “Life Sciences Reporting Summary” accompanying this paper. In the SLE study, each patient had multiple samples (visits) collected over time, each of which may be collected when the patient had different levels of disease activity as reflected by the SLE Disease Activity Index (SLEDAI) score. The original publication13 defined seven groups of patients based on SLEDAI-associated gene expression patterns (correlation computed over time/multiple visits), and showed that for patient groups 2, 3 and 4, the SLEDAI score was correlated with genes associated with a plasma cell/plasmablast signature.
For reasons discussed in the main text, we focused on patients from groups 2, 3, and 4 and evaluated whether TGSig evaluated at low DA time-points was correlated with the changes in plasmablast signature score associated with DA (e.g., the extent of plasmablast score increase between low-DA and high-DA time-points). Note that we used a mixed effect model – see below for details – to estimate this latter quantity. For each patient in groups 2, 3, and 4, we calculated a plasmablast signature score for each visit (using the gene module DC.M4.11 as described below in “Plasmablast gene-based signature”). Because each patient had multiple visits and patients often had treatment (e.g., corticosteroids) that, in turn, may have an effect on the changes in plasmablast score as a function of DA, we used a linear mixed-effect model to integrate the data46 (in R/lme4 notation): PB ~ ∑Ti + SLEDAI|SUBJECT, where PB is the signature score, Ti is an indicator of treatment group (as defined in the original paper), SLEDAI is treated as a continuous variable, and subject is treated as the random effect. Here the subject-dependent, “random” coefficient for the SLEDAI term (the “slope”) would give us an estimate on the quantitative extent by which the plasmablast signature score would change as DA varies. Note that the plasmablast signature score is presumably reflective of plasmablast frequencies, as shown previously12. We fitted the mixed effect model and extracted the aforementioned personal “slope” by using the ranef function; hereon we will refer to this patient-specific quantity as the “disease activity-associated change in plasmablasts (DaCP)”. We also tested treating SLEDAI as a discrete/categorical variable (high and low DA only, or high, middle, and low DA – see below on definition) and the resulting estimate for the personal DaCP was similar to the that obtained by treating SLEDAI as a continuous variable (data not shown.) We further assessed the robustness of DaCP by comparing it to the simple difference between the average plasmablast score of high DA time-points and that of low DA time-points (Extended Data Fig. 5b), and as expected they were highly correlated.
To compute TGSig from clinically quiescent/“baseline”, low-DA time-points, we adopted the original SLE study’s classification of samples into low-, middle-, and high-DA groups based on the SLEDAI score (low DA: SLEDAI < 3, medium DA: SLEDAI: 3–7, high DA: SLEDAI ≥ 8). We then calculated the TGSig score at low DA time points. Because each subject may have more than one sample from a low DA time-point, subject-specific scores were computed by averaging the score from all low DA time points. We then tested whether the TGSig signature score calculated at low DA time points was correlated with DaCP. As a control, we performed the same analysis described above in patient groups 1, 5, 6, and 7 to assess if such a correlation exists for patients whose DA did not correlate with plasmablast scores.
Plasmablast gene signature
We chose the gene set DC.M4.11 “Plasma cells” from the Chaussabel blood transcriptomic modules to compute the plasmablast signature score47 because: 1) this was one of the plasma cell/plasmablast gene signatures that the original SLE study found to be correlated with DA (i.e., which helped define patient groups 2, 3, and 4); 2) we examined the within-module gene-gene correlation in this and additional plasmablast related gene modules (see below) and found that this gene set had the best coherency and exhibited the highest overall gene-gene correlations; 3) the score of DC.M4.11 was significantly correlated with the score computed from a gene set we derived earlier based on correlation with changes in plasmablast frequency on day 7 following influenza vaccination (see Fig. 7B in ref.12) (data not shown), thus suggesting that the signature score computed from DC.M4.11 was directly reflective of plasmablast frequencies.
| From ref.47 | |
| DC.M4.11 | Plasma Cells |
| DC.M7.7 | Undetermined |
| DC.M7.32 | Undetermined |
| From ref.21 | |
| LI.M156.0 | plasma cells & B cells, immunoglobulins |
| LI.M156.1 | plasma cells, immunoglobulins |
Computing gene set enrichment scores for transcriptional differences between high and low disease activity periods (Fig. 2c and Extended Data Fig. 5a)
For each gene and patient, we first computed the average gene expression for samples with high DA (or middle DA if no samples with high DA were available) and separately for those with low DA. Then for each subject the genes were ranked by the magnitude of the difference between these two average values (high DA – low DA), followed by gene set enrichment analysis using the CERNO test from tmod48 R package to generate the enrichment statistics for each subject and gene set. The enrichment for each gene set in each subject is shown as −log10 of the FDR-adjusted p-value in the heatmap of Extended Data Fig. 5a. To summarize the information in this heatmap and show patient group dependent enrichment, we averaged the enrichment values across each patient group and gene sets with similar phenotypic annotations (as determined previously in the original publication13) and generated the compressed heatmap of Fig. 2c.
Identification and characterization of coexpression modules at low disease-activity time points
To identify coexpression modules in SLE patients, we first calculated the average expression profile for each patient using samples from low DA time-points. We focused also on temporally stable genes (across low-DA time-points) because those are more likely to reflect stable, personal immune states. We estimated the TSM score for each gene across low DA time-points using a similar approach as described above for the development of TGSig in the NIH/CHI influenza data set, except that stable genes were selected using a more relaxed cutoff, i.e., using a FDR cutoff of 0.05 (calculated from the P values of the F statistics of the ANOVA model above), which resulted in 9601 “stable” genes, because we wanted to retain more genes for module analysis. We next used the WGCNA algorithm to determine modules20. We determined the algorithm’s hyperparameters following the authors’ recommended procedure, setting soft power to 4 (the lowest power for which the scale-free topology fit index curve flattens out upon reaching a high value, in this case, roughly 0.80), and network type to “signed hybrid”. We were able to assign ~2800 genes to 18 coexpression modules (Supplementary Table 3), with module size ranging from 25 to 719 genes (Fig. 3a and Supplementary Fig. 1a).
To assess the extent by which these modules were associated with the DaCP, we calculated Pearson correlation between the eigengene of each module (i.e., PC1) and the DaCP. To evaluate statistical significance, we generated an empirical null distribution for each module by randomly shuffling subject labels and recalculating correlations on the shuffled data (Supplementary Fig. 1b). Empirical p-values were determined by comparing the actual correlations with the corresponding null distributions.
For functional characterization, we evaluated whether the modules are enriched for known gene sets (such as the Blood Transcriptome Modules21, Supplementary Fig. 1c) with the hypergeometric test. The false discovery rate was controlled using the Benjamini-Hochberg procedure49.
Meta-analysis to identify genes associated with vaccine responses
We used random effect meta-analysis models50 to estimate the “meta” effect size of each gene by combining expression and antibody response data from multiple vaccination datasets. This method models the observed effect size yi (association between expression and vaccine response class as reflected by adjMFC high vs. low responders) as a draw from a distribution with study-specific mean θi and variance (i.e. intra-study sampling error). Furthermore, each θi is assumed to be a draw from a distribution with overall mean μ and variance τ2 (i.e. interstudy variability):
We identified influenza response genes by applying the method to the four influenza vaccination studies (Supplementary Table 4a). We only evaluated the temporally stable genes as defined above based on the low DA time-points because those were the genes we used to define co-expression modules. In addition, we applied the same method to the two trials of yellow fever vaccine. We found that the meta-analysis results are largely consistent with the first trial, but much less so with the second trial (data not shown). The relatively poor coherence between the two trials may be partly due to the very small sample size of the second trial (only 7 subjects). Our observation suggested that inclusion of data from trial two is more likely to introduce noise than signal, thus we decided to only focus on trial one.
Gene set enrichment analysis
Gene Set Enrichment Analysis (GSEA) is a widely-used approach to test if a particular gene set is enriched at the top of a ranked gene list51. Because the datasets we used are from peripheral blood samples, we decided to use known gene sets from the same tissue: the Blood Transcription Modules (BTMs)21. These gene modules were identified from network analysis of gene-gene correlations in multiple blood transcriptomic datasets. We used an efficient implementation of GSEA analysis from the fgsea R package40 with the BTM modules from the tmod R package48 (fgsea was used also to extract leading edge genes.)
We applied GSEA to assess whether the genes in the “brown” coexpression module identified in the SLE cohort are enriched at the top of the genes ranked by their meta-effect/association with antibody response to influenza vaccination (Fig. 3d); we also performed the same analysis for yellow fever trial #1 (see above) but the result was statistically insignificant.
To test for enrichment of BTMs in genes from the NIH/CHI influenza dataset ranked by correlation with CD20+CD38++ B cell frequency, we selected temporally stable genes with TSM ≥ 0.5 from the NIH/CHI influenza study dataset, and ranked them by their robust correlation metric (see “Baseline gene-based predictive signature” above). Here a more relaxed temporal stability cutoff was used to increase the number of genes and statistical power for enrichment analysis. P-values from the enrichment test were corrected for multiple‐testing using the Benjamini-Hochberg method49.
GSEA was also applied to identify “leading edge” genes, a subset of the query set that maximizes the enrichment score. Applying leading edge analysis to brown module and the genes from the NIH influenza study ranked by correlation with CD20+CD38++ B cell frequency, we identified a set (87 genes) of leading edge genes in the brown module (and called this gene set SLE-Sig) (Fig. 3e and Supplementary Table 5). To better understand the predictive property of genes in the brown module, we divided it into two subsets – the leading-edge set (87 genes – SLE-Sig), and the remaining genes. For each subset, we calculated the eigengene and used it in regression analysis (Extended Data Fig. 6).
Assessing the relative contribution of TGSig and SLE-Sig to predicting vaccination responses
We used logistic regression to model association with vaccine response (high vs. low responder based on adjMFC) by pooling all four influenza vaccination datasets (Extended Data Fig. 6). We fitted three models, all having the following form (high responder: response=1; low responder: response=0):
where X is a matrix of covariates, and β is a vector of coefficients estimated from the data. Model 1 includes both TGSig and SLE-Sig (represented by the eigengene or PC1 of genes in the respective set) as covariates, while Models 2 and 3 only include one of the two covariates. In addition, all three models include dataset ID (encoded by dummy variables) as a covariate. To assess statistical significance, we performed the likelihood ratio test comparing two nested models (e.g., Model 1 and Model 2).
The same approach was used to evaluate the relative contribution of TGSig and 1) genes outside the leading edge in brown module; 2) IFN-I-DCact genes. Similarly, instead of evaluating vaccine responses, we applied this same approach to evaluate the relative contribution of TGSig and these other gene signatures in predicting DaCP in SLE.
Sorting of B cell populations from healthy donors
Peripheral blood mononuclear cells (PBMCs) were obtained from peripheral blood of 6 healthy individuals (demographics shown in table below) by density-gradient centrifugation and cryopreserved until day of B cell analysis and sorting. Multicolor flow cytometry was performed to identify PBMC B cell populations (Extended Data Fig. 7a) using the following mAbs: CD19-PerCP-Cy5.5, CD27-PE-Cy7 (ThermoFisher); CD20-APC-H7, CD38-BV421 (BD Biosciences); CD3-BV510 (Biolegend). For transcriptional analysis, total CD19+CD20+ and CD19+CD20+CD38hi B cells (5000 each) were sorted into Trizol LS (Sigma) or ATACseq buffer using a FACSAria II (BD Biosciences). Flow data analyses were performed using FlowJo software (TreeStar/Becton Dickinson Co., Ashland, OR). Purity of B cell sorts was ~99%.
Donor demographic information is available in Supplementary Table 9. See also the “Life Sciences Reporting Summary”. Healthy donors were recruited under NIH IRB approved protocol NCT00001281. Informed consent was obtained from all study participants.
RNAseq library preparation and analysis
RNA was isolated from cells sorted into Trizol LS using the Zymo Direct-zol Micro-prep columns (Zymo Research, Irvine, California). Total RNA sequencing libraries were prepared using the NuGEN SoLo RNAseq library kit (NuGEN Technologies, San Carlos, California) according to the manufacturer’s instructions, using 1 ng of RNA as input. Libraries were sequenced using the Illumina NextSeq with version 2 sequencing reagents to an average of 20 million paired-end 37 base pair length reads per sample. Fastq files were generated from raw sequencing data using bcl2fastq version 2.17.1.14 (Illumina). Reads were mapped using STAR52 version 2.5.2 aligner and the UCSC hg19 genome annotation and the --quantMode GeneCounts option. Counts-per-feature data output from STAR was imported into R 3.4.1. The average number of exonic-feature mapped counts was 3.14 million per sample. Differential gene expression was estimated using the DESeq2 package53 version 1.16.1. MAplot shown in Extended Data Fig. 7b was produced using ggplot254 version 2.2.1(https://cran.r-project.org/web/packages/ggplot2/index.html).
Statistical evaluations
All statistical analyses were performed in R version 3.4.1, except for the low-level processing of single-cell data and some of the analyses in CITE-seq and RNA-seq (e.g., Extended Data Figs. 7–9), which were performed using R 3.5.1. Effect sizes and p-values of Pearson and Spearman correlations were computed using the cor.test function. P-values for Wilcoxon Rank Sum Test were computed using the wilcox.test function. Unless noted, all Wilcoxon tests were two-tailed. Hedges’ g effect sizes were computed using the cohen.d function in the effsize55 package. Meta-analysis was performed with the MetaDE56 package. Enrichment analysis was performed using the fgsea40 package in Bioconductor (when we want to extract leading edge genes; the similar CERNO test was used when leading edge genes are not needed, e.g., Fig. 2c and Extended Data Fig. 5a.) Jonckheere-Terpstra trend test for ordered differences among multiple classes was performed using jonckheere.test function in the clinfun package57. Area under ROC curve (AUC) and 95% confidential interval were computed using the pROC package45. The p value for AUCs was estimated by a permutation procedure: we shuffled the subject labels to create a mismatch between the responder class and the gene expression data and generated 1000 permutations test the null hypothesis that the observed AUC was drawn from this null distribution. Metrics to further assess prediction performance (Supplementary Table 2b) were computed using the MLmetrics package58. Some metrics required a cutoff for calling high vs. low responders: 1) for gene expression based predictors we used zero since we used scaled (z-score based) gene expression values to compute the score of a gene set for each subject; 2) for cell frequency based predictors the cutoff was set to the median of cell frequencies.
Simultaneous protein and transcriptomic single cell profiling via CITE-seq
Combined surface target protein and mRNA expression single cell analysis was performed using the CITE-seq methodology according to refs59–61 with the following modifications: n=20 high and low responders were split between two experimental batches with n=5 high and n=5 low responders in each batch. See also the “Life Sciences Reporting Summary”. The high and low responder groups were not significantly different in age and sex (p=0.16 for age and p=0.37 for sex, Wilcoxon two-tailed test). In each batch, samples were stained using cell “hashing”59 antibodies such that samples could be demultiplexed using a combination cell hashing identifiers and SNP genotype demultiplexing62. After cell hashing antibody staining, cells from each batch were pooled into one tube and stained with a mixture of oligo-labeled antibodies against target surface proteins. The two experimental batches were performed on consecutive days, using aliquots of the same pool of antibodies for each batch. Final antibody concentrations used for staining were determined using titration experiments; the concentration of antibody that appeared to saturate ligand was utilized, or for cases in which apparent saturation was not reached, the manufacturer’s recommended amount was used. Oligo-labelled antibodies for cell hashing and surface target protein detection were obtained from Biolegend (see Supplementary Table 10 for list of reagents). The same pool of donor cells from each batch were distributed evenly across 6 lanes of 10x Genomics Single Cell 3’ expression reagents (version 2). Cell hashing (HTO) and surface target protein (ADT) libraries were prepared according to59–61. cDNA tag libraries were prepared using the 10x Genomics v2 kit according to manufacturer’s instructions. Libraries were sequenced using the Illumina HiSeq 2500 and v4 reagents. Additional statistics: mean number of gene detected per cell was 746, mean number of UMIs per cell was 2139. The mean read depth per cell was approximately 43,000–84,000 as reported by CellRanger. The fraction of reads mapped to the genome was above 90% for all lanes; sequencing saturation was typically around 90%.
Computational low-level processing of CITE-seq data
Bcl2fastq version 2.20 (Illumina) was used to demultiplex the sequencing data. CellRanger version 3.0.1 (10x Genomics) was used for alignment (using the Hg19 annotation file provided by 10x Genomics) and counting unique molecular identifiers (UMI). CITE-seq ADT and HTO tag alignment and UMI counting was done with CITE-seqCount63 version 1.4.2. The following computational analysis of CITE-seq data was performed using the Seurat 2.3.4 R package23 and the SingleCellExperiment Bioconductor class64. Hashing antibodies were used to identify the response class (high vs. low adjMFC) in each batch. Samples demultiplexed with Seurat’s HTODemux k-means function corresponded to either one or two possible sample identities Sample labels were then assigned to each cell using demuxlet62; only cells that met the following conditions were retained: 1) the cell must be defined as a “singlet” by both hash demultiplexing and Demuxlet; 2) the identified donor from Demuxlet must match either of the two expected donors based on HTO hashing.
Surface protein data normalization:
For each protein, ADT data was normalized by taking the log + 1 of raw counts, then subtracting the mean and divided by the standard deviation of the corresponding antibody count from the same batch measured in negative droplets. In addition, to account for droplet-to-droplet differences in the ADT capture rate as well as background noise from unbound antibody, we denoised each cell by removing a covariate corresponding to the background counts for each cell using the removeBatchEffect function in limma65 package; this covariate was defined for each cell as the eigenvector of: 1) the mean of non-staining antibodies in that cell; 2) counts from 4 isotype control antibodies. mRNA data was normalized with scran66 after removing cells with greater or less than 3 median absolute deviations from the median library size.
Cell clustering and annotation
The cell clusters were identified by a shared nearest neighbor (SNN) modularity optimization based clustering algorithm67 implemented in the Seurat R package23. Only ADT data (surface protein expression) were used to generate the Euclidean distance matrix computed for all single cells. The matrix was then used to build the SNN graph followed by k-nearest neighbors clustering (k = 50). Three clustering resolutions were applied, 0.1, 0.3 and 1, to generate three sets of cell clusters; the relationship between these cluster sets were visualized using the clustree68 and ggraph69 R packages (Fig. 4b). The clusters were labeled accordingly to reflect this hierarchical relationship (e.g. C1→C1.0→C1.0.0). We annotated the protein-based clusters post hoc, linking them to canonical immune cell populations by visualizing both the average and distribution of all proteins in each cluster at multiple resolutions (Fig. 4c).
Single cell tSNE visualization
tSNE (t-distributed Stochastic Neighbor Embedding) analysis was performed using ADT (surface protein expression) data only with the RunTSNE function from the Seurat package. Note that tSNE was used only for visualization. We used PCA for dimensionality reduction: the number of dimensions (7) used for tSNE was determined with the “elbow” plot (not shown). In the tSNE plot (Fig. 4a), the 10 clusters resulted from the resolution=0.1 clustering above (with the perplexity parameter equal to 130) were shown.
Manual gating of CITE-seq data
To determine frequency of the CD19+CD20+CD38++ B cell population for each subject, we first gated the total B cells in the dataset as CD3-CD56-CD14-CD19+. The gates were determined based on the density plot of each marker. We then applied the CD20+CD38++ gate on total B cells. After we determined the cells in the gate, we used subject labels to compute the number of total B cells and CD20+CD38++ B cells for each subject, and then the frequency of CD20+CD38++ B cells as a fraction of the total number of cells.
Computing and evaluating subject-level gene set signature scores for cell clusters using CITE-seq data
Our goal was to assess, using only cells in a given cell cluster, whether the average (across single cells) relative expression of genes in a signature gene set is higher in the high responders compared to low responders. The procedure we devised below was used in two related situations: 1) given a bulk predictive signature (e.g., TGSig), determine whether average relative expression of genes in the signature is significantly higher within a specific cluster of cells in high than low responders—this was used to uncover the cellular contributors to a bulk predictive signature such as TGSig; 2) given a gene signature that reflects a cell state (e.g., CD40act), assess whether a particular cluster of cells tends to have higher average relative expression of genes in the signature in high than low responders—when interpreting results from this analysis we are often making the assumption that higher average relative expression corresponds to a greater strength of signal that drove the cells to or kept the cells in that state. Thus, for situation (2) we focused on signatures containing genes with evidence of increased expression when exposed to the “signal” (e.g., see CD40act below.) This assumption is sound because here we focus on genes that respond to signals such as CD40L and Type I interferons, which lead to activation of transcriptional programs that shape the mRNA profile within specific types of cells.
There were two levels of averaging in our procedure: 1) averaging across single cells – single cell mRNA expression data can be noisy and since our goal here is to evaluate cell clusters, we took advantage of the power of statistical averaging to increase robustness and derive conclusions at the cell cluster level; 2) averaging across genes—information obtained from individual genes can also be noisy and often does not reflect cell states, which are typically more robustly captured by the level of multiple genes as a set of genes, for example, can be shaped by common upstream regulators. As also demonstrated by the success of methods such as GSEA and eigengene analysis, gene set level information can be more robust and can better reflect cell state shifts that affect multiple genes. Our single cell normalization procedure (see above) and simple z-score scaling (see below and “Gene set-based signature score calculation” section above) helped to enable such averaging.
To compute the subject-level signature score for a gene set in a given cell cluster, we first created a cell-averaged gene expression data set for the cell cluster: for a given cell cluster and subject, we averaged the normalized single cell expression counts for each gene across single cells in that cell cluster for the subject. For each cell cluster (e.g., cluster C0—naïve CD4+ T cells; see Fig. 4b–c), this procedure generated a gene-by-subject expression matrix. We then applied the same z-score based procedure as we used for microarray gene expression data (see “Gene set-based signature score calculation” section above) to compute the gene set signature score for each subject. We then used the non-parametric one-tailed Wilcoxon test to evaluate whether the signature scores are significantly higher in the high responders compared to the low responders.
As a robustness check for results involving the SLE-Sig signature (Fig. 5b), which is enriched for Type I IFN responses and related processes (Fig. 3c,f), we also created another Type I IFN gene signature (IFN, see Supplementary Table 6) using modules from an independent collection (see ref.47). This signature was created by combining (i.e., the union) genes from the following three blood transcription modules47: DC.M1.2, DC.M3.4, and DC.M5.12.
Gene signature for CD40 activated B cells (CD40act)
Our goal was to construct a transcriptional signature that reflects CD40 activation in human B cells. We focused on genes with evidence of increased expression following CD40L stimulation because we wanted to evaluate whether these genes tended to be elevated in B cells in high responders compared to low responders using CITE-seq data. The CD40act gene signatures (49 genes) was generated as the intersection between two gene sets (Supplementary Table 6). The first gene set (163 genes) was obtained by reading off the heatmap in Supplemental Figure 1 in ref.26, which corresponded to genes/probe sets with higher expression in CD40 activated B cells in comparison to mock-activated B cells (i.e., those that appeared red and blue on the right and blue sides of the heatmap, respectively). Since the expression data was not available, we transcribed the probe set labels directly from the figure. The labels were then converted to probe set IDs with Affymetrix annotation and then converted to gene symbols by using the hgu95av2.db Bioconductor package70. The second gene set (295 genes) were generated by using publicly available expression data from ref.27. We downloaded the data from GEO (GSE54017 series) and selected genes more highly expressed in CD40-activated B cells than in resting B cells. The statistical cutoff was determined using a one-sided paired t-test: we selected genes with an FDR-adjusted p-value lower than 0.05 and a log2-fold-change greater than 1.5.
Code availability:
The source code and software pipeline to reproduce our analyses can be assessed at https://github.com/kotliary/baseline.
Data availability:
All data used in this study, including CITE-seq and flow data, are available at figshare (https://doi.org/10.35092/yhjc.c.4753772). The original/raw public gene expression data are available in the National Center for Biotechnology Information Gene Expression Omnibus (GEO) under accession numbers: GSE47353, GSE41080, GSE59654, GSE59743, GSE29619, GSE74817, GSE13486, and GSE65391 (see also Supplementary Table 2).
Extended Data
Extended Data Fig. 1. Identification and characterization of the CD19+CD20+CD38++ B cell population, a baseline, pre-vaccination cell frequency-based signature (CBSig) of antibody responses to influenza vaccination.
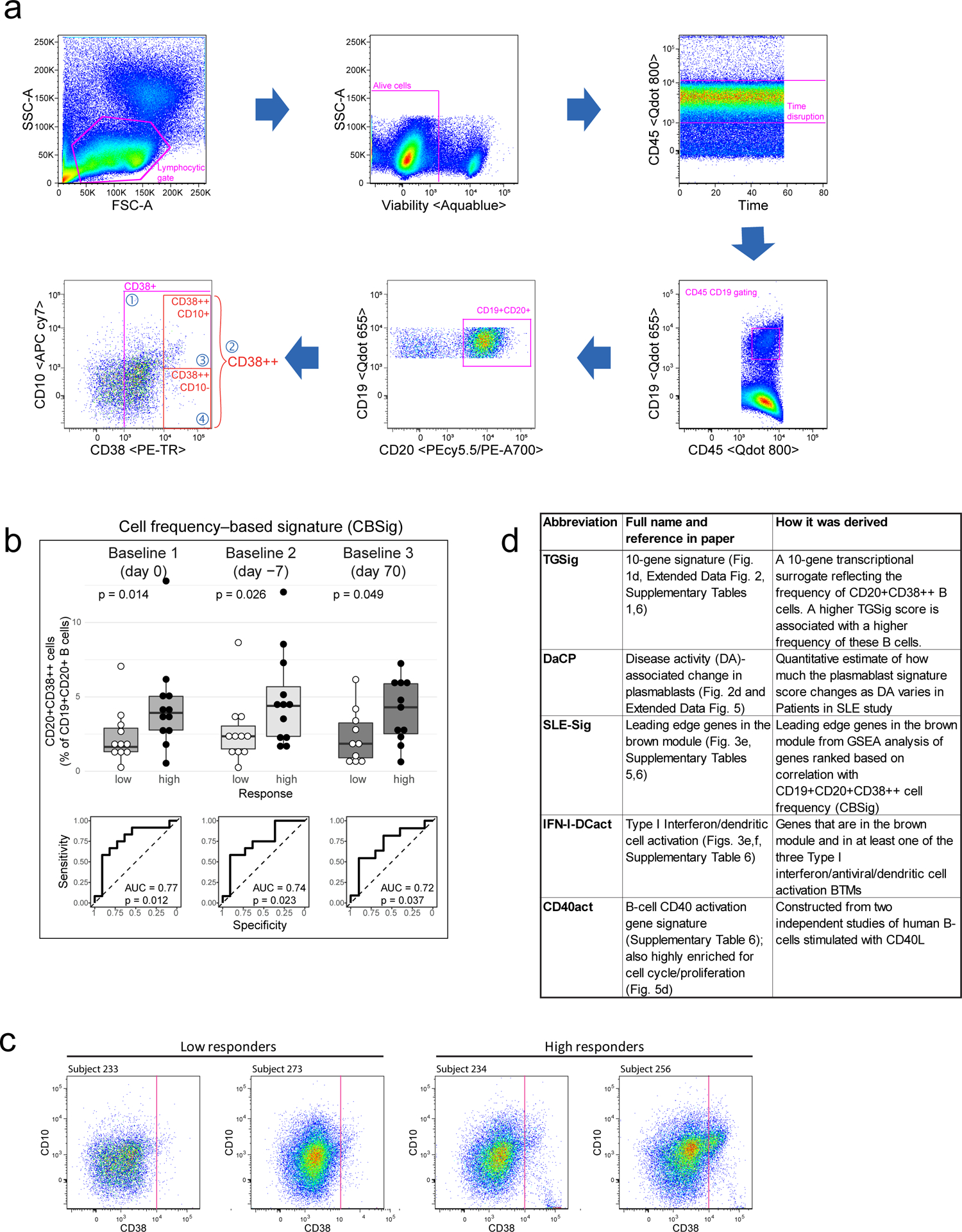
a, Flow cytometric gating strategy for the CD19+CD20+CD38++ B cell population. Populations 1–4 are further described in (Fig. 1b). b, Box plots (top) showing the frequency of CD19+CD20+CD38++ cells (CBSig; y-axis) at the three baseline time points from ref.12 (days −7 and 0 are prior to vaccination and day 70 is after vaccination) in low and high responders (x-axis) to the seasonal and pandemic H1N1 influenza vaccines as defined by the Adjusted Maximum Fold Change (adjMFC) metric (see ref.12). There are 11 low and 12 high responders for day −7 and 0, and 10 low and 11 high responders for day 70. P values from the Wilcoxon one-tailed test results are shown on the boxplots (based on results from ref.12 our hypothesis was that the high responders have higher frequencies of these cells than low responders). Boxplots’ center line corresponds to the median value, lower and upper hinges correspond to the first and third quartiles (the 25th and 75th percentiles); lower and upper whiskers extend from the box to the smallest or largest value correspondingly, but no further than 1.5x inter-quantile range. (Bottom) Corresponding receiver operator curves (ROC) for vaccine response at each of the above baseline time points and their AUC (area under the curve) and corresponding permutation based one-tailed p value are shown. c, Dot plots (CD38 vs. CD10 of CD19+CD20+ B cells) for example high and low responders.
Extended Data Fig. 2. Derivation of TGSig, a transcriptional surrogate signature for CBSig.
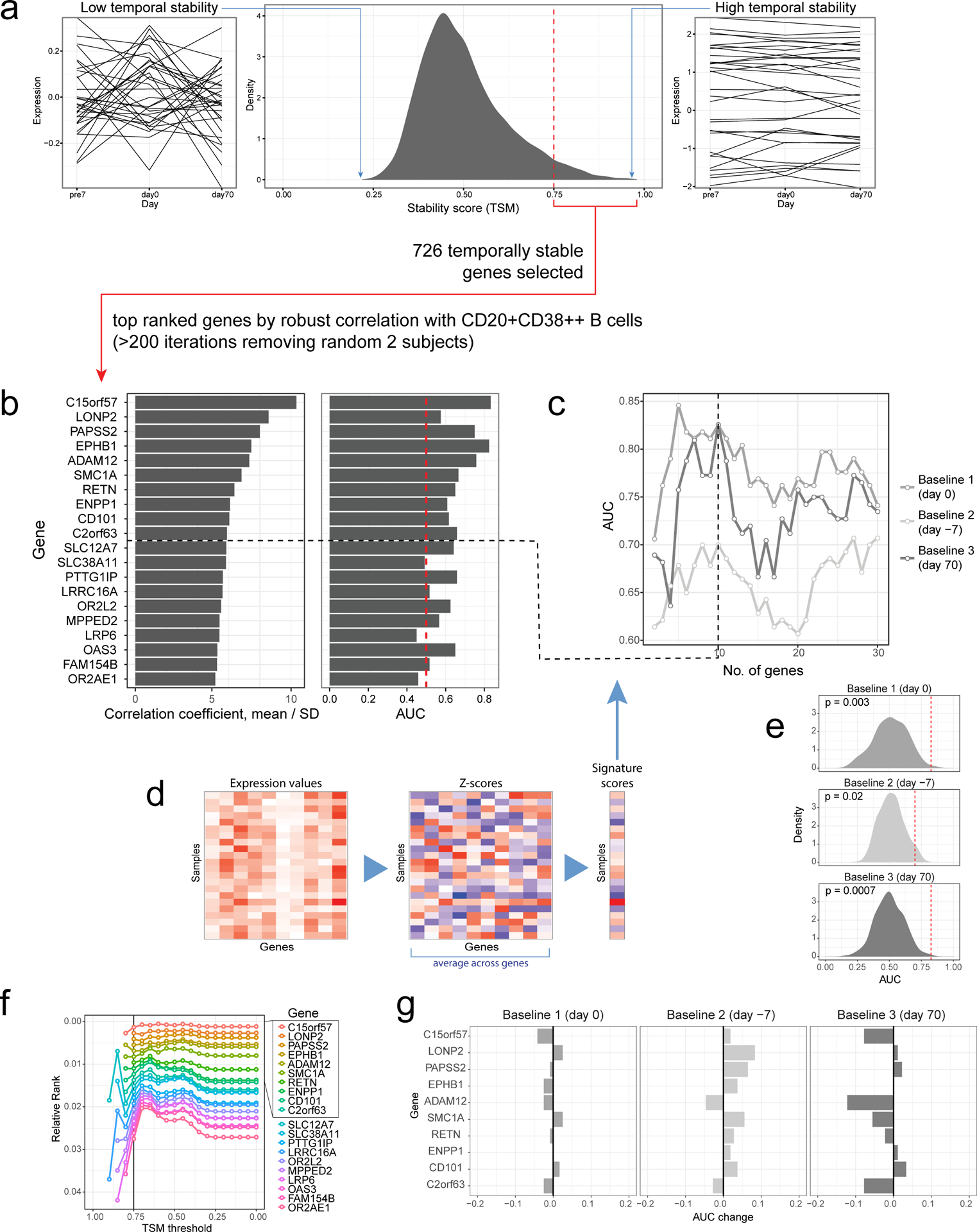
a, Step 1: Identification of genes with high temporal stability across the three baseline time points (days −7 and 0 are prior to vaccination and day 70 is after vaccination) in the NIH influenza study12. The middle box shows the distribution of the temporal stability metric (TSM) across all the genes. The boxes on the right and left show examples of genes with high and low temporal stability, respectively; each line corresponds to an individual. Genes with high temporal stability (≥0.75) across the three baseline time points (depicted to the right of the red dashed line) were subsequently evaluated for correlation with CD19+CD20+CD38++ B cell frequency. b, Step 2: 726 temporally stable genes were ranked by their “robust” correlation with CD19+CD20+CD38++ B cell frequency. Robustness is evaluated using all 231 random samplings of 20 subjects out of the cohort of 22 subjects (i.e., two random subjects were dropped out from each sampling); the mean Pearson correlation coefficient divided by the standard deviation across the samplings (x-axis left panel) was used to rank the genes. Top genes are shown together with the predictive performance of each gene evaluated at day 0 (AUC; right panel). The red dashed line in the right panel corresponds to AUC = 0.50 (prediction performance as expected by chance); the top 10 genes were selected in TGSig (the black dashed line; see Supplementary Table 1 for full list of ranked genes) based on (c). c, Performance (AUC; y-axis) of the gene signature by baseline time point (different lines) and number of top genes included in computing the signature score (x-axis). The vertical dashed line corresponds to a gene signature (TGSig) containing the top 10 genes achieving the best AUC across all three time-points. d, Schema for gene signature score calculation. Gene expression data is standardized through calculation of Z-scores for each gene (i.e., each gene would have mean 0 and standard deviation 1). The Z-scores for each gene in the signature are then averaged to generate the gene signature score. e, Distribution of predictive performance (AUC; x-axis) of 500 random top-10 gene signatures generated from subject-label shuffled gene expression data using the robust correlation metric approach described above. The dashed red line indicates the observed AUC of TGSig at each of the baseline time points. One-tailed empirical p-values are shown. f, Relative rank (rank position divided by the total number of genes) of the top 20 genes from (b) and Supplementary Table 1 at different TSM thresholds. The black line shows TSM cutoff = 0.75, the value used in selecting the top 10 genes for inclusion in TGSig (boxed). g, Change in AUC (x-axis) of TGSig score following removal of the indicated gene in the signature (y-axis) at each of the three baseline time points in the NIH influenza study.
Extended Data Fig. 3. Comparison among response classes by including middle responders and evaluating TGSig in the influenza datasets from Emory18.
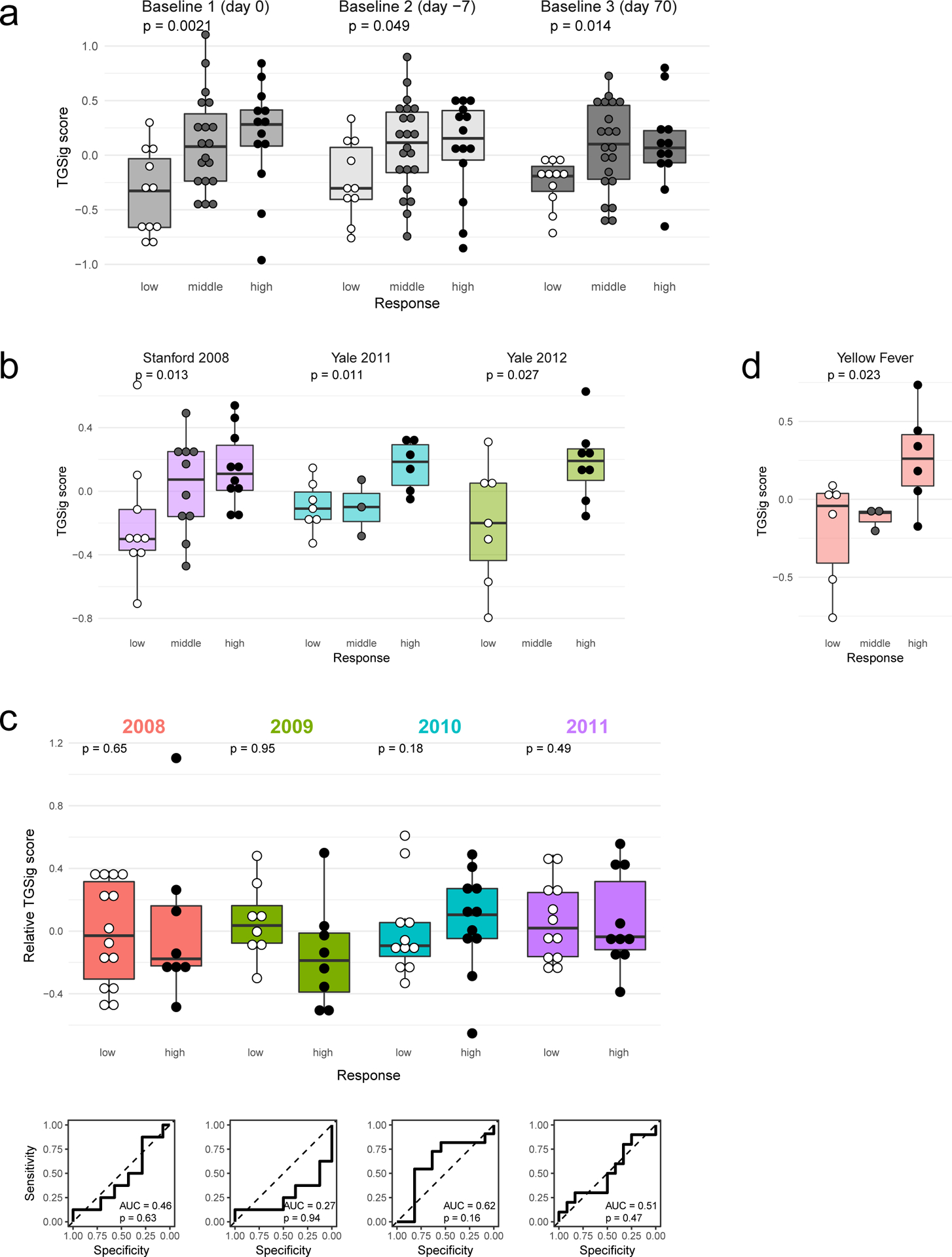
a, similar to Figs. 1e and 1f but including middle responders (for day 0 n=11/19/13 for low/middle/high responders, respectively; for day −7 n=10/22/14 and for day 70 n=11/21/12); p values from the Jonckheere trend test (with an a priori alternative hypothesis that the high responders >= middle responders >= low responders.) For all boxplots the center line corresponds to the median value, lower and upper hinges correspond to the first and third quartiles (the 25th and 75th percentiles); lower and upper whiskers extend from the box to the smallest or largest value correspondingly, but no further than 1.5x inter-quantile range. b, Similar to (a) but related to Fig. 2a; note that Yale 2012 does not have middle responders based on data retrieved from ImmuneSpace (n=9/10/10 (Stanford 2008), 7/3/6 (Yale 2010), 7/0/8 (Yale 2012)). c, Similar to Fig. 2a but testing TGSig using influenza datasets from Emory University over four years (n=14/8 low/high responders for year 2008, 8/8 for 2009, 11/11 for 2010, 12/10 for 2011). Box plots (top) showing the TGSig score (y-axis) in low and high responders (x-axis) as defined by adjMFC in the indicated season. P values shown on the boxplots were obtained from the Wilcoxon one-tailed test. (bottom) Corresponding receiver operator curves (ROC) for vaccine response and the AUC (area under the curve) and corresponding permutation-based one-tailed p values are shown. d, Similar to (a) but for yellow fever and related to Fig. 2b.
Extended Data Fig. 4. Further evaluation of TGSig in yellow fever and influenza datasets.
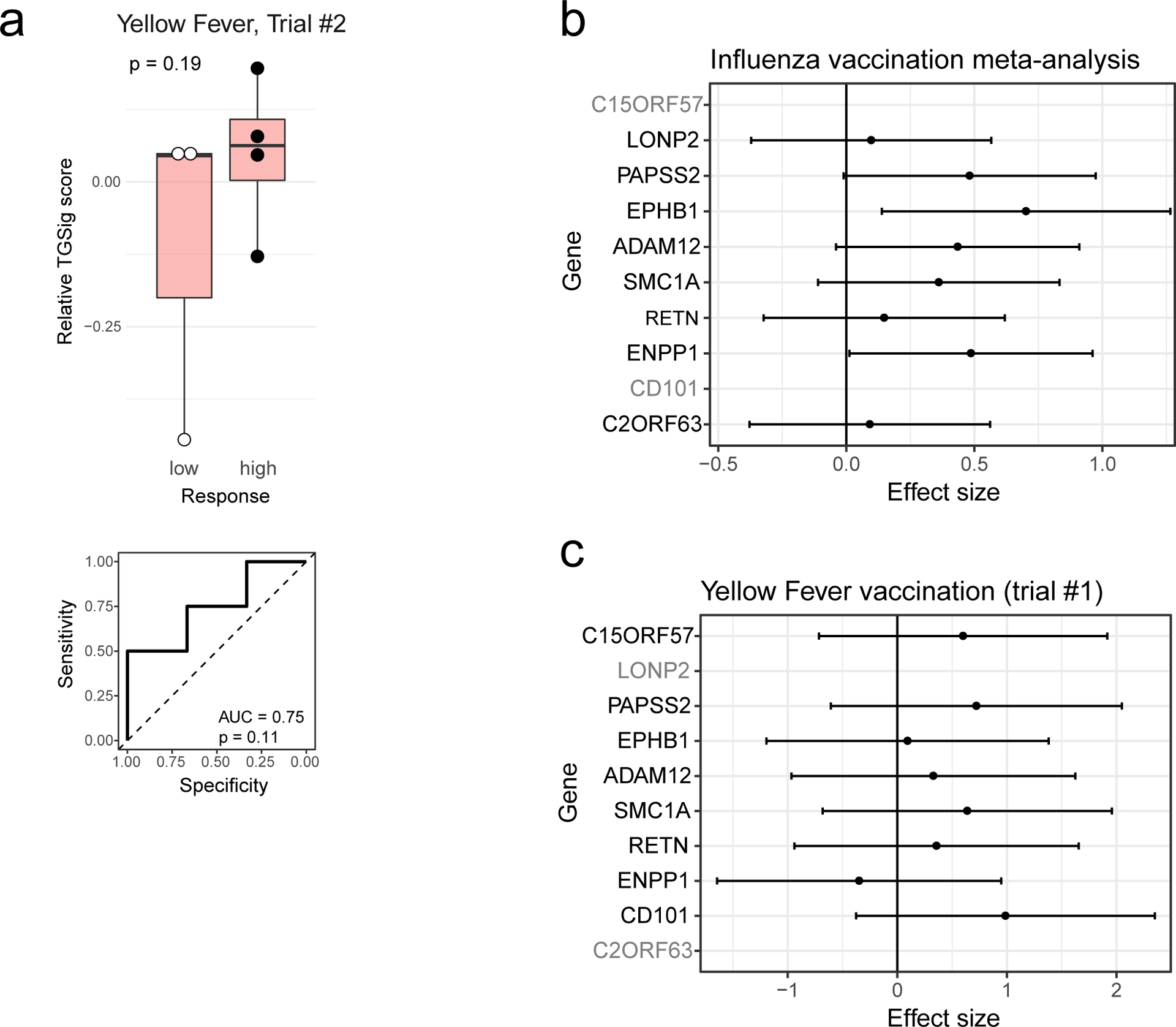
a, (top) Box plots (top) showing the TGSig score applied to pre-vaccination PBMC expression data (y-axis) between low and high responders (x-axis) to yellow fever vaccine in trial #2 (x-axis; 4 high vs. 3 low responders). (bottom) Corresponding receiver operator curves (ROC) for vaccine response and the AUC (area under the curve) and corresponding one-tailed permutation based p value are shown. This vaccination cohort included 10 subjects (see Fig. 2b for results on a first, larger trial with 15 subjects). Boxplots’ center line corresponds to the median value, lower and upper hinges correspond to the first and third quartiles (the 25th and 75th percentiles); lower and upper whiskers extend from the box to the smallest or largest value correspondingly, but no further than 1.5x inter-quantile range. P-value is from Wilcoxon one-tailed test. b, Forest plot showing the meta effect sizes (Hedge’s g reflecting correlation strength with adjMFC) of the TGSig genes from a meta-analysis of four influenza datasets (Stanford 2008, NIH 2009, Yale 2011, Yale 2012; genes in gray were not present in all datasets); the bars represent the 95% CI. c, Similar to (b) but for yellow fever vaccination (computed from trial #1).
Extended Data Fig. 5. SLE patient phenotypes based on DA-associated transcriptional signatures and assessing the association between TGSig (at low DA) and DaCP after the removal of patients with lower DA-associated plasmablast signature scores.
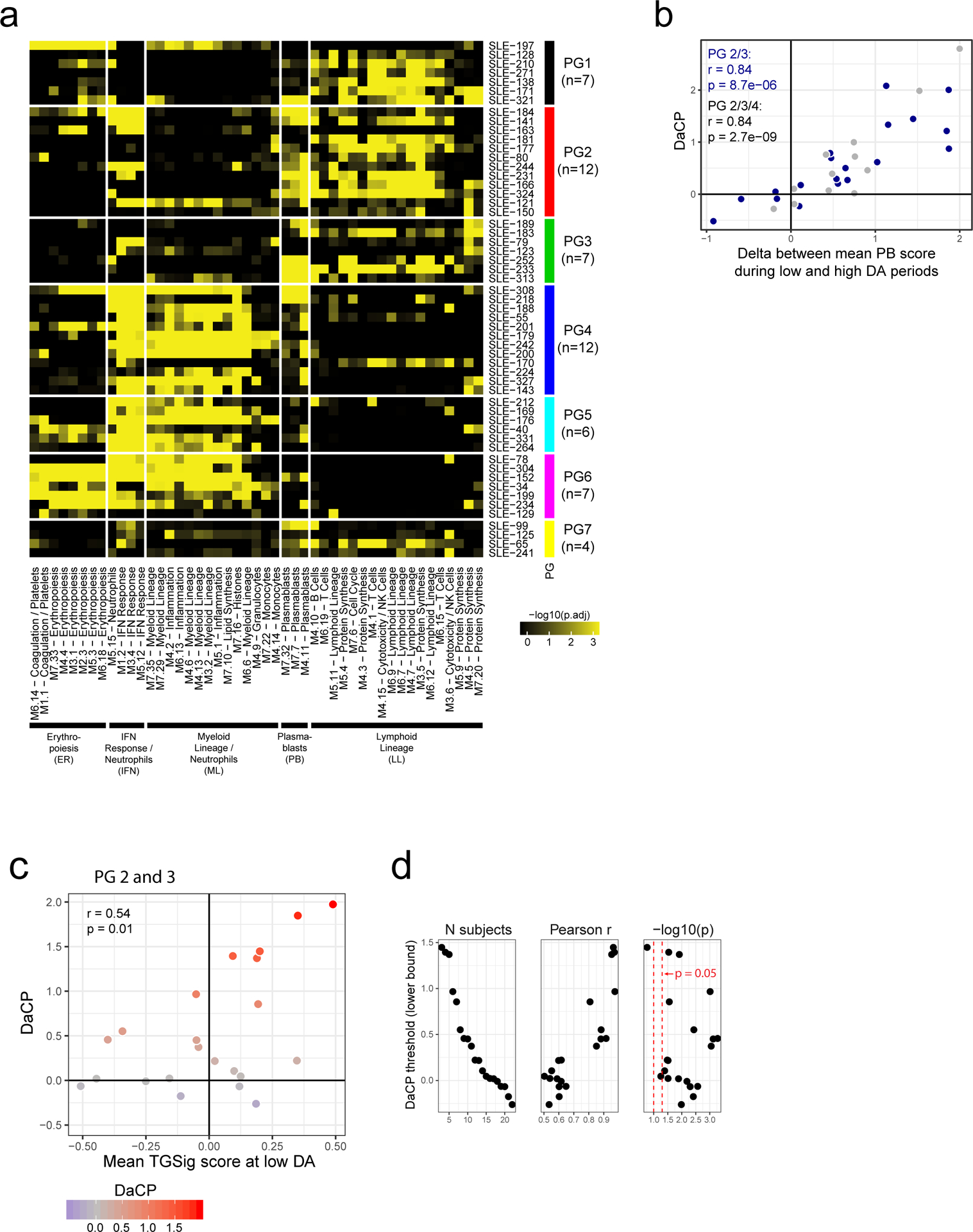
a, Recreation of Figure 6a from ref.13 but as a robustness check here we used a different method: for each patient we ranked genes based on the difference between their average expression at high/middle and low DA time points and then performed GSEA analysis using the blood transcriptomic modules (columns) to fingerprint the patient. The colors on the heatmap denote the statistical significance (−log10 of BH-adjusted p-values from CERNO39) of the gene set enrichment test. Here we kept the order of patients (rows) and modules (columns) the same as in the original heatmap, although some patients from the original heatmap were removed due to the absence of low DA time points. The original patient groups were used to annotate the patients (rows). The overall fingerprinting pattern is visually highly consistent with the original heatmap. This heatmap/matrix was then used to construct Fig. 2c by averaging individual patient values within each patient and phenotype group combination. b, DaCP versus average plasmablast score difference between high (or middle if no high was available) and low DA time points in SLE patients from patient groups 2, 3 (in blue, n=19) or groups 2, 3, and 4 (group 4 in grey, n=12) from ref.13 (see patient groups in Fig. 2c). Spearman rho for groups 2 and 3 is 0.87 (two-tailed p=2.2×10−16); and for groups 2, 3 and 4 is 0.85 (two-tailed p=4.9×10−7). c, Similar to Fig. 2e, but here patients are from patient groups 2 and 3 only (n=22) and are shaded based on their DaCP. Note that there are more patients here for analysis from groups 2 and 3 than those shown in (a) and (b) because there we required that every patient has at least one low DA and one high/mid DA sample, while here (and in Fig. 2) the DaCP was estimated using all patients with at least one sample including those without high/mid DA time-points (see Methods). Pearson correlation coefficient and two-tailed p values are shown. Spearman correlation coefficient is 0.47 (p=0.029 two-tailed). d, Evaluating the correlation between DaCP and mean TGSig score at low DA time-points (as in (c)) by removing patients with DaCP below the indicated cutoff (y-axis). The first panel shows the number of patients in the evaluation given the threshold; the second and third panels show the corresponding Pearson r and two-tailed p value (shown as −log10(p)).
Extended Data Fig. 6. Evaluating the predictive capacity and information overlap among the TGSig, SLE-Sig, and IFN-I-DCact signatures.
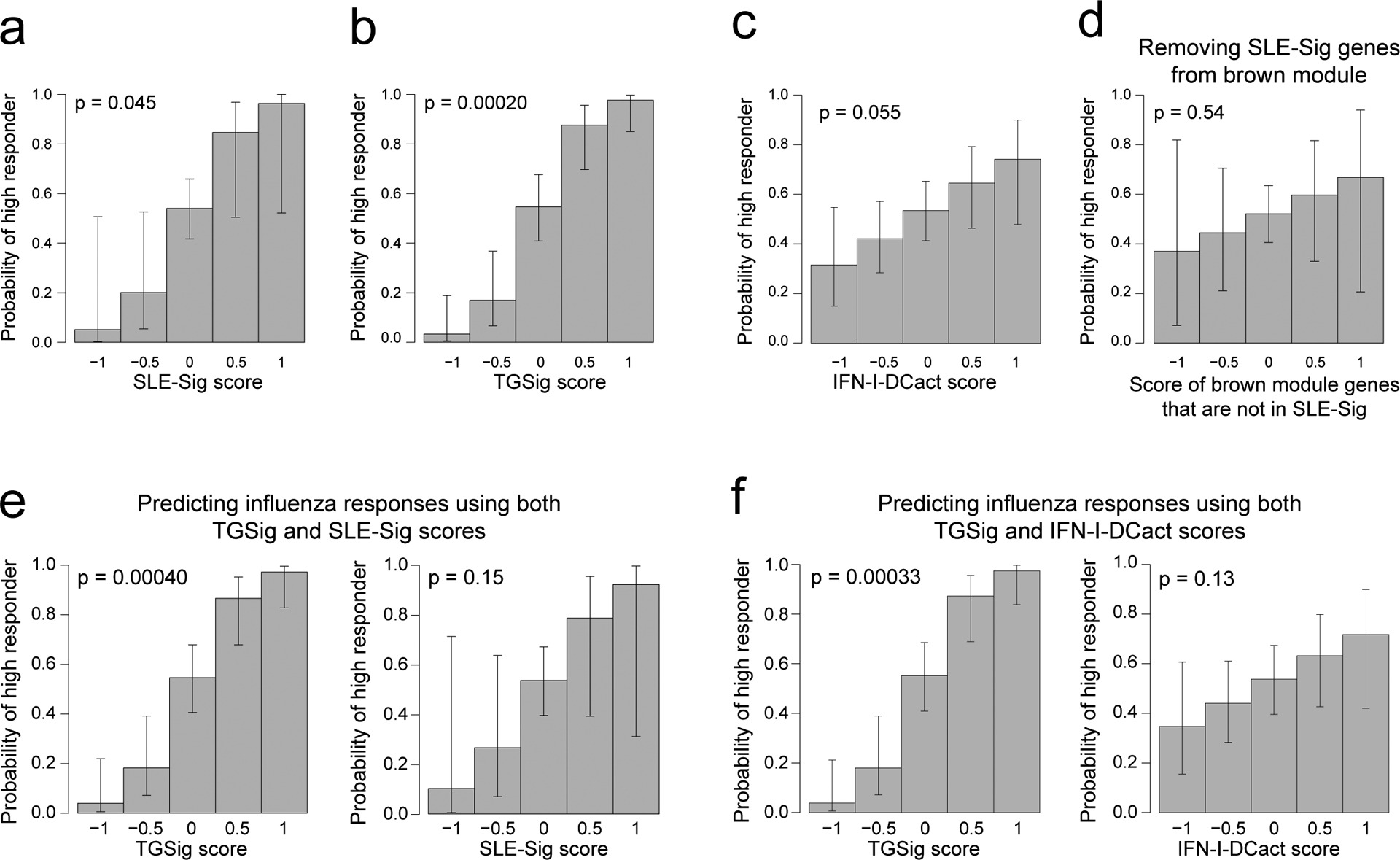
a-d, The predictive profile of SLE-Sig (Fig. 3e) (a), TGSig (b), IFN-I-DCact (Fig. 3f) (c), and the non-leading edge genes from the brown module (Fig. 3e) (d) used as the sole predictor in logistic regression models of high versus low influenza vaccination responder status. Influenza vaccination data pooled from four datasets (Stanford 2008, NIH 2009, Yale 2011, Yale 2012) were used (n = 71 high and low responders). Note that for the brown module (Fig. 3a–d), most of the predictive information come from the leading-edge genes since the signature score of genes outside of the leading edge is not predictive (shown in (d)). e, f when both TGSig and SLE-Sig were used as predictors in the logistic regression (e) or when both TGSig and IFN-I-DCact score were used as predictors (f). In these graphs, the predictor scores are shown on the x axis and the probability that a high responder falls within the predictor score bin is shown on the y axis. The error bars correspond to 95% CIs. The two-tailed p value indicates the probability that the coefficient (“effect”) of the term (e.g., TGSig score) in the logistic regression is 0.
Extended Data Fig. 7. RNA-seq analysis of CD19+CD20+CD38++ B cells sorted from healthy individuals.
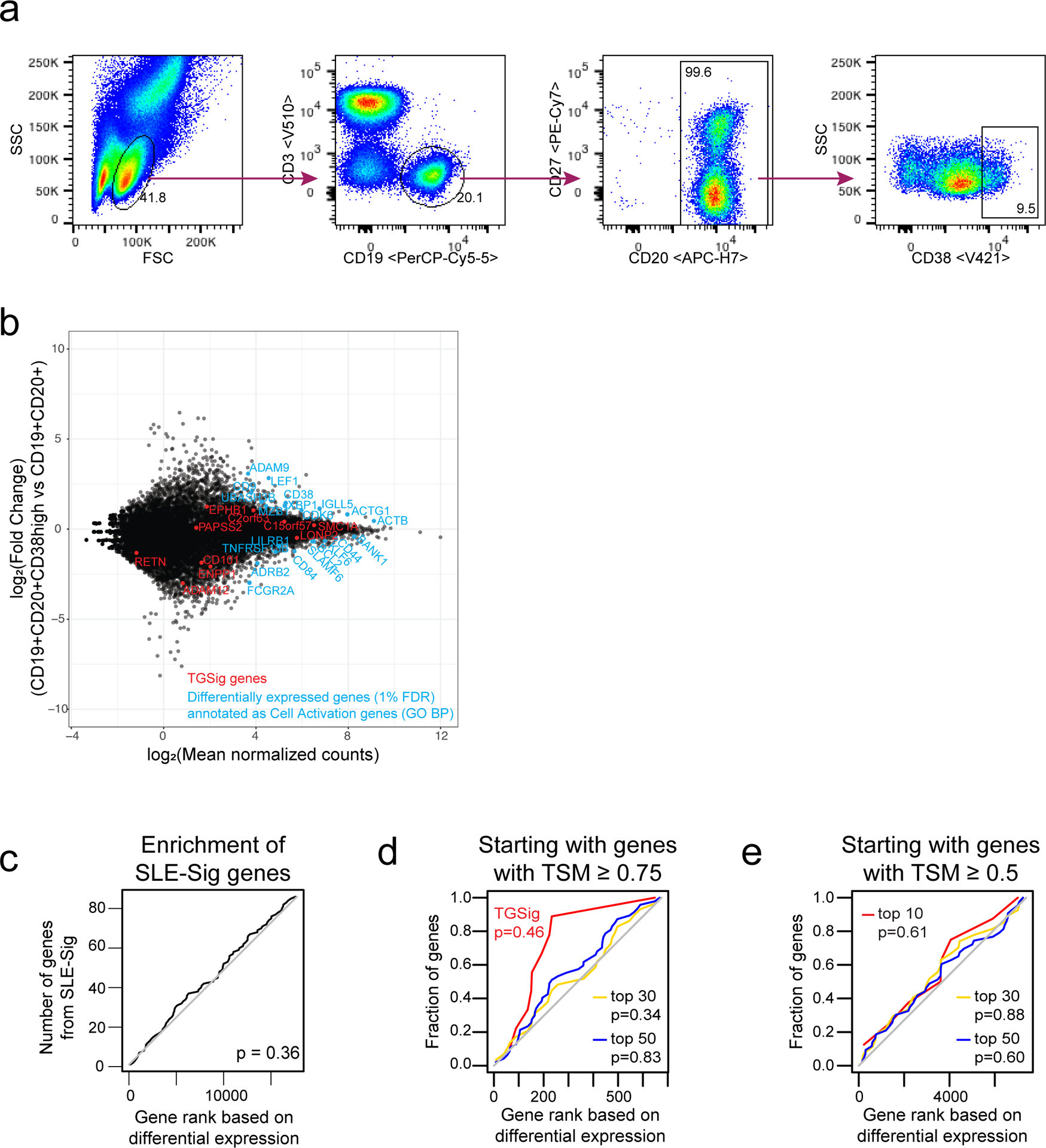
a, Sorting strategy and approach: CD19+CD20+ and CD20+CD38++ B cell populations were isolated by FACS from peripheral blood samples of six healthy donors. RNA-seq libraries were prepared for each isolated sample using the Nugen Ovation SoLo low-input RNAseq library preparation kit. b, Differential gene expression between the CD20+CD38++ B cells and parental CD19+CD20+ B cells from six paired samples was compared using DESeq2. Plot shows the log2 fold change versus the log2 of the mean normalized counts across all samples for each gene. TGSig genes are shown in red; genes from the differentially expressed gene set (BH-adjusted two-tailed p-value < 1%, log of mean normalized counts > 1; total genes in set: 105) that fall into the top enriched Gene Ontology Biological Processes category “Cell Activation” (enrichment analysis done using ToppGene41) are shown in cyan (21 genes). c, Enrichment of the 87 SLE-Sig genes in genes ranked by differential expression between CD19+CD20+CD38++ versus CD19+CD20+ cells. The p value shown was computed from the GSEA test. d, Similar to (c) but instead of the SLE-Sig genes here the top k (k=10 (TGSig), 30, 50) genes correlated with the frequency of CD20+CD38++ B cells is assessed (only 713 temporally stable genes with TSM≥ 0.75 were included in the analysis); also see Extended Data Fig. 2. e, Similar to (d) but using 7731 genes with TSM ≥ 0.5. A lower/more relaxed TSM cutoff was used to evaluate whether by starting with more genes (therefore potentially more statistical power for enrichment analysis) an enrichment signal can be detected.
Extended Data Fig. 8. Supporting data for CITE-seq single cell analysis to dissect the cellular origin of baseline signatures.
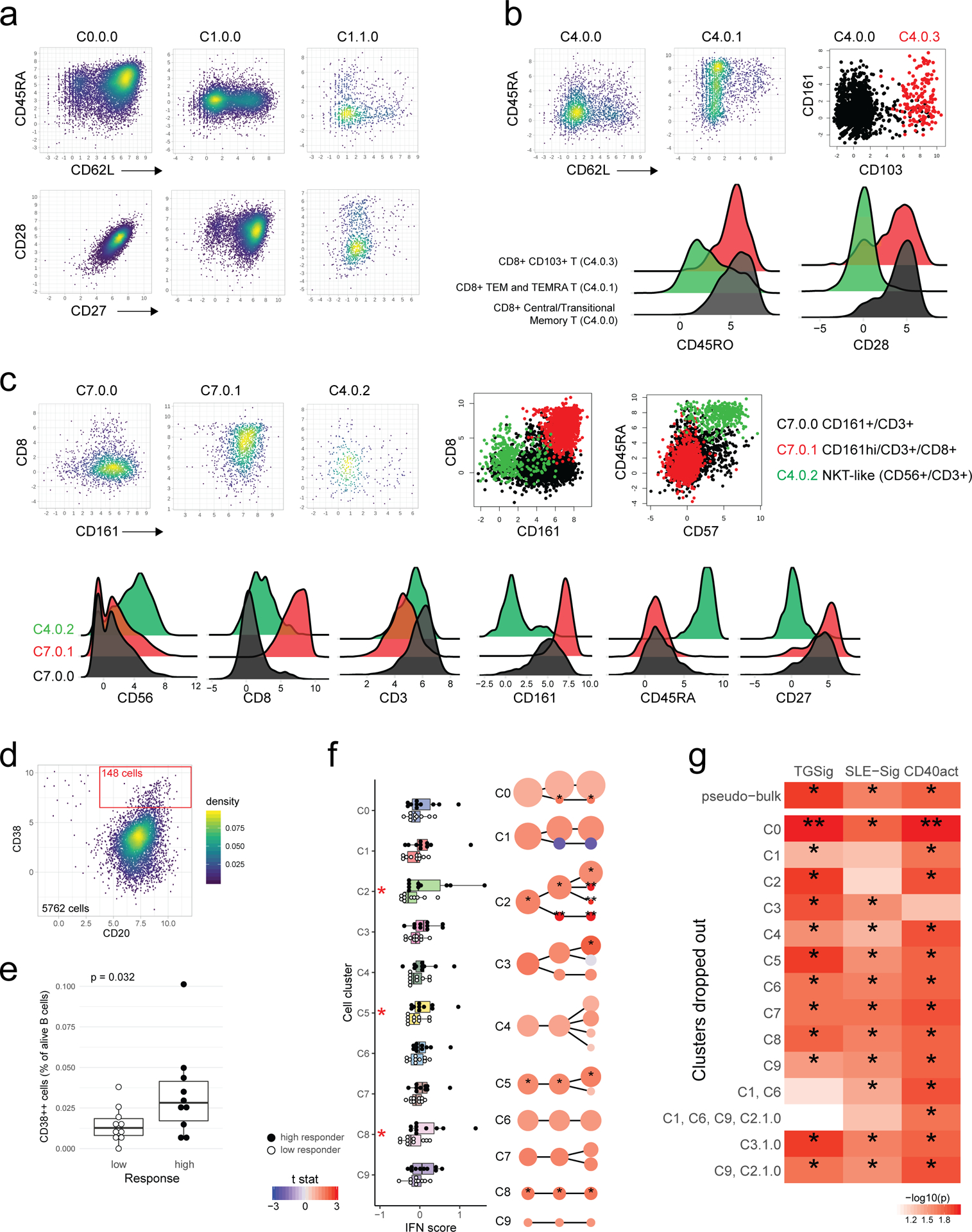
a, Single cell scatterplots of key markers in the CD4+ T cell clusters. C0.0.0 (Naïve), C1.0.0 (Central/Transitional Memory), and C1.1.0 (TEMRA/Effector memory) clusters show different distributions of CD62L, CD45RA, CD27, and CD28. b, Distributions of key markers in the CD8+ Memory T cell clusters. CD45RA vs. CD62L expression are shown in the top panels, ridge plots of CD45RO and CD28 are shown in lower panels. Similar to CD4+ cells, these CD8+ clusters show differences in CD62L and CD45RA distributions; C4.0.1 (TEMRA/Effector Memory) are mainly CD62L negative and CD28 negative, with CD45RA+ (TEMRA) and CD45RO+ (Effector Memory) subsets within this cluster. C4.0.0 and C4.0.3 show highly similar protein expression, with C4.0.3 being defined by high CD103 expression (upper right panel). c, Unconventional T cells (C7 clusters) show variable CD161, CD8, and CD56 expression. C7.0.0 and C7.0.1 are both CD3+/CD161+, with C7.0.1 being CD8 positive while C7.0.0 is CD8 negative. C4.0.2 (NKT-like) are also CD3 positive, but express CD57/56 and are CD8/CD161 negative or low. The C4.0.2 cluster also showed distinctly low CD27 and high in CD45RA compared to the C7 clusters, making it more similar to the C4.0.1 TEMRA/Effector memory CD8+ subset, except expressing CD56/CD57; this could also be consistent with a Terminal Effector phenotype. d, Hand gating strategy for CD20+CD38++ B cells using CITE-seq data (number of cells in the gate shown in red). e, Boxplot comparing the hand gated frequency of CD20+CD38++ B cells between 10 high and 10 low responders; p value from Wilcoxon one-tailed test. Boxplots’ center line corresponds to the median value, lower and upper hinges correspond to the first and third quartiles (the 25th and 75th percentiles); lower and upper whiskers extend from the box to the smallest or largest value correspondingly, but no further than 1.5x inter-quantile range. f, Similar to Fig. 5b (10 high versus 10 low responders) but for an independently obtained Type I IFN signature gene set (Supplementary Table 6: IFN gene set; see Methods). One or two asterisks denote significance with p<0.05 or p<0.01, respectively (Wilcoxon one-tailed test because we are interested in assessing whether the high responders are higher than the low responders; see also Supplementary Table 7). g, Results of the “drop out” analysis using CITE-seq data. The goal was to assess which cell clusters (or combination of clusters) that were individually significant delineators of high versus low responders were essential for prediction using the baseline signatures in bulk (i.e., simulating transcriptional data from PBMCs). The “pseudo bulk” results (average across all single cells for every subject in 10 low and 10 high responders) of the three signatures tested are shown on the first row. For subsequent rows cells from the indicated cell cluster(s) were dropped before repeating the pseudo-bulk analysis as in row 1. P values obtained from one-tailed Wilcoxon test: *: p<0.05; **: p<0.01.
Extended Data Fig. 9. Evaluating CMV correlates and pDC surface expression phenotypes strongly influenced by genetics from ref.35 in high versus low responders.
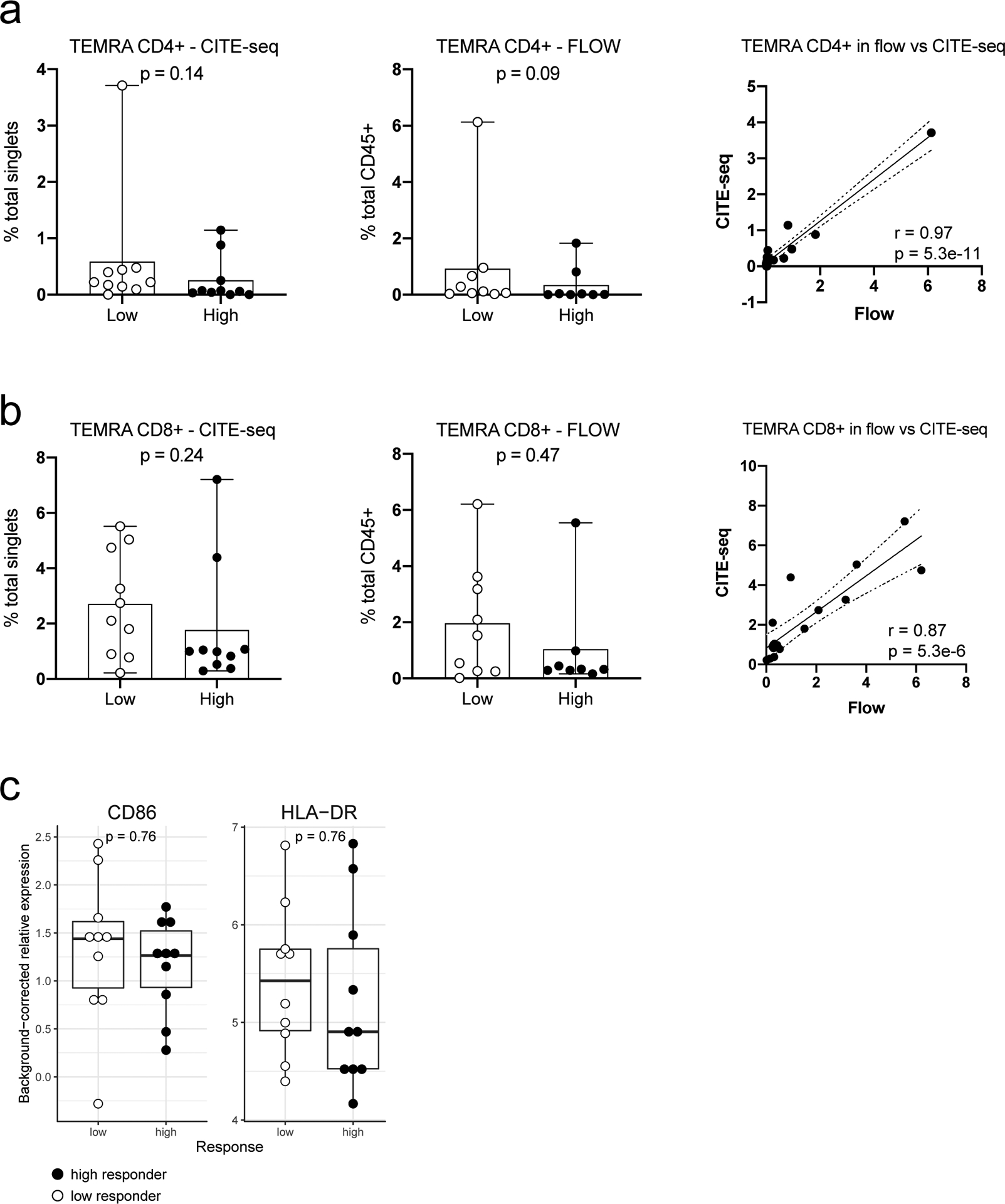
Since CMV status is not available for the cohorts we evaluated, we evaluated whether CMV correlates are significantly different between high and low responders in the NIH influenza vaccination cohort. a, Boxplots comparing the frequency of CD4+ TEMRA cells between 10 low and 10 high responders using CITE-seq (left panel) and between 9 low and 8 high responders using flow cytometry (center panel) data. Wilcoxon two-tailed p values are shown. The third panel is a scatter plot of CITE-seq versus the flow cytometry cell frequencies (n=17). Pearson correlation and two-tailed p values are shown. b, Same as (a) but for CD8+ TEMRA cells. c, Boxplots comparing the relative surface protein expression of CD86 and HLA-DR in pDCs (cluster C9) between 10 low and 10 high responders using CITE-seq data. Wilcoxon two-tailed p values are shown. For all boxplots the center line corresponds to the median value, lower and upper hinges correspond to the first and third quartiles (the 25th and 75th percentiles); lower and upper whiskers extend from the box to the smallest or largest value correspondingly, but no further than 1.5x inter-quantile range.
Extended Data Fig. 10. Relationship between sex and baseline signatures.
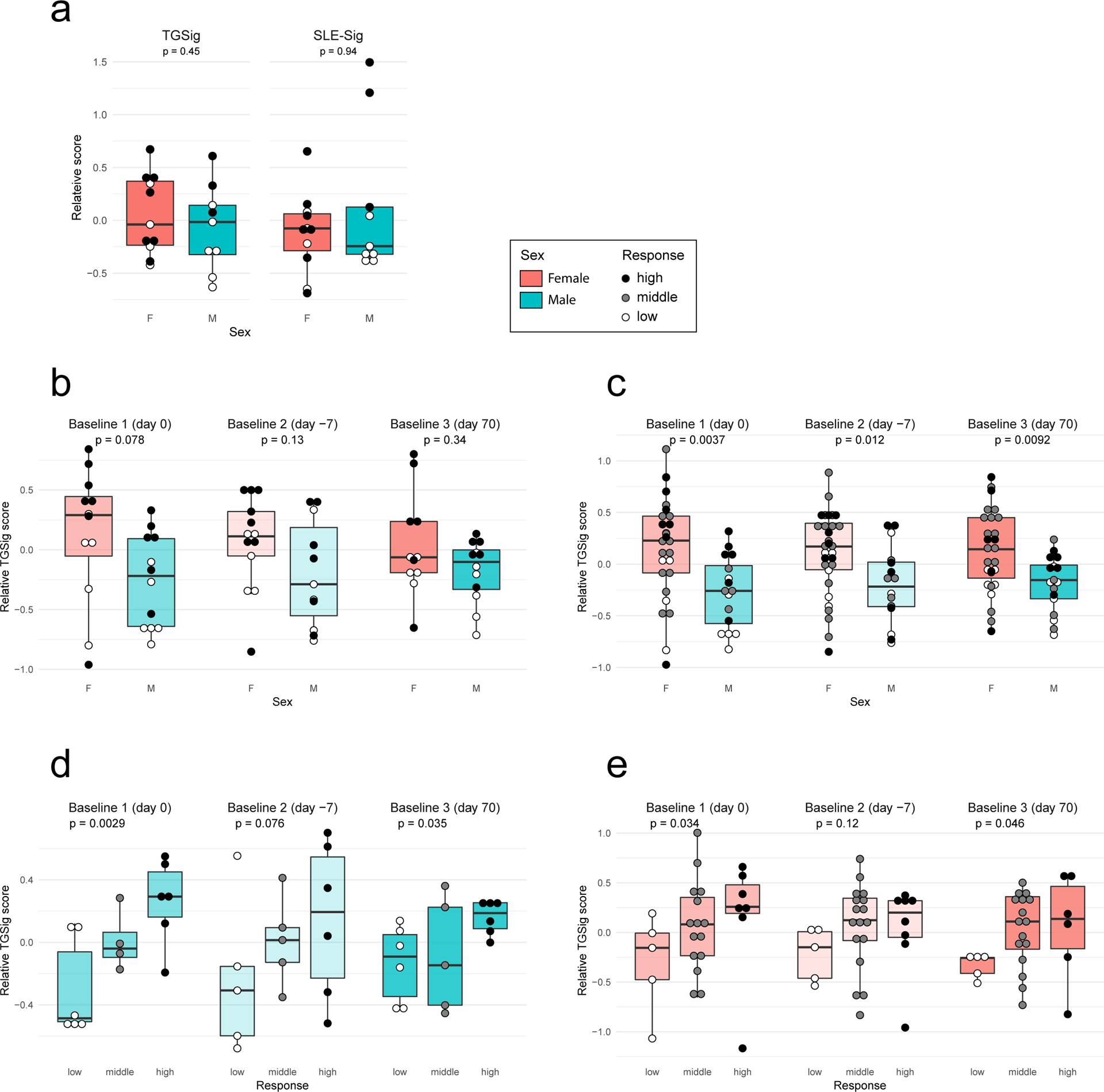
a, Box plots comparing the TGSig and SLE-Sig scores in females versus males; here only subjects with CITE-seq data are included to indicate that sex was not a driver of the differences between 10 high and 10 low responders emerged from CITE-seq data analysis (see Fig. 4). Wilcoxon two-tailed p values comparing 11 females and 9 males are shown. For all boxplots the center line corresponds to the median value, lower and upper hinges correspond to the first and third quartiles (the 25th and 75th percentiles); lower and upper whiskers extend from the box to the smallest or largest value correspondingly, but no further than 1.5x inter-quantile range. b, Same as (a) but including all high and low responders from original NIH study12. (day 0: 12 females and 12 males; day −7: 13 females and 11 males; day 70: 11 females and 12 males) c, same as (b) but including middle responders (i.e., all subjects in the study: day 0: 27 females and 16 males; day −7: 30 females and 16 males; day 70: 27 females and 17 males). d, Box plots comparing TGSig scores among low, middle, and high responders in males only (all subjects in the original NIH study12 used: day 0: 6/4/6 for low/middle/high responders, respectively; day −7: 5/5/6; day 70: 6/5/6). e, same as (d) but in females only (day 0: 5/15/17 for low/middle/high responders, respectively; day −7: 5/17/8; day 70: 5/16/6). All p values shown for two-group comparison were from the Wilcoxon two-tailed test; one-tailed p values shown for three-group comparison were from the Jonckheere trend test (with an a priori alternative hypothesis that the high responders >= middle responders >= low responders).
Supplementary Material
Acknowledgments:
We thank members of the Tsang Lab and the NIH Center for Human Immunology (CHI) for inputs and discussions; L. Failla for help with figures; D. Hirsch for testing the R code; R. Germain and R. Apps for comments on the manuscript; this research was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases (NIAID) and Intramural Programs of the NIH Institutes supporting the Center for Human Immunology. Figure 6 created with BioRender.com.
Footnotes
Competing Interests: all authors declare that there are no competing interests.
References
- 1.Kotas ME & Medzhitov R Homeostasis, inflammation, and disease susceptibility. Cell 160, 816–827 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen DS & Mellman I Elements of cancer immunity and the cancer-immune set point. Nature 541, 321–330 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Tsang JS Utilizing population variation, vaccination, and systems biology to study human immunology. Trends Immunol. 36, 479–493 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Willis JCD & Lord GM Immune biomarkers: the promises and pitfalls of personalized medicine. Nat. Rev. Immunol 15, 323 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Brodin P & Davis MM Human immune system variation. Nat. Rev. Immunol 17, 21–29 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hagan T et al. Antibiotics-Driven Gut Microbiome Perturbation Alters Immunity to Vaccines in Humans. Cell 178, 1313–1328.e13 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brodin P et al. Variation in the human immune system is largely driven by non-heritable influences. Cell 160, 37–47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marchant A et al. Predominant influence of environmental determinants on the persistence and avidity maturation of antibody responses to vaccines in infants. J. Infect. Dis 193, 1598–1605 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Krieg C et al. High-dimensional single-cell analysis predicts response to anti-PD-1 immunotherapy. Nat. Med 24, 144 (2018). [DOI] [PubMed] [Google Scholar]
- 10.HIPC-CHI Signatures Project Team & HIPC-I Consortium. Multicohort analysis reveals baseline transcriptional predictors of influenza vaccination responses. Sci. Immunol 2, eaal4656 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fourati S et al. Pre-vaccination inflammation and B-cell signalling predict age-related hyporesponse to hepatitis B vaccination. Nat. Commun 7, 1–12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsang JS et al. Global analyses of human immune variation reveal baseline predictors of postvaccination responses. Cell 157, 499–513 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Banchereau R et al. Personalized Immunomonitoring Uncovers Molecular Networks that Stratify Lupus Patients. Cell 165, 1548–1550 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Tsokos GC, Lo MS, Reis PC & Sullivan KE New insights into the immunopathogenesis of systemic lupus erythematosus. Nat Rev Rheumatol 12, 716–730 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Lee JC & Smith KGC Prognosis in autoimmune and infectious disease: new insights from genetics. Clin. Transl. Immunol 3, e15 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stoeckius M et al. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 14, 865–868 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu Y et al. Systematic Analysis of Cell-to-Cell Expression Variation of T Lymphocytes in a Human Cohort Identifies Aging and Genetic Associations. Immunity 45, 1162–1175 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakaya HI et al. Systems Analysis of Immunity to Influenza Vaccination across Multiple Years and in Diverse Populations Reveals Shared Molecular Signatures. Immunity 43, 1186–1198 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Querec TD et al. Systems biology approach predicts immunogenicity of the yellow fever vaccine in humans. Nat. Immunol 10, 116–125 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Langfelder P & Horvath S WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li S et al. Molecular signatures of antibody responses derived from a systems biology study of five human vaccines. Nat Immunol 15, 195–204 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muskardin TLW & Niewold TB Type I interferon in rheumatic diseases. Nat. Rev. Rheumatol 14, 214–228 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Butler A, Hoffman P, Smibert P, Papalexi E & Satija R Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Swiecki M & Colonna M The multifaceted biology of plasmacytoid dendritic cells. Nat. Rev. Immunol 15, 471–485 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jego G et al. Plasmacytoid dendritic cells induce plasma cell differentiation through type I interferon and interleukin 6. Immunity 19, 225–234 (2003). [DOI] [PubMed] [Google Scholar]
- 26.Gricks CS et al. Differential regulation of gene expression following CD40 activation of leukemic compared to healthy B cells. Blood 104, 4002–4009 (2004). [DOI] [PubMed] [Google Scholar]
- 27.Shimabukuro-Vornhagen A et al. Inhibition of protein geranylgeranylation specifically interferes with CD40-dependent B cell activation, resulting in a reduced capacity to induce T cell immunity. J. Immunol. Baltim. Md 1950193, 5294–5305 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Elgueta R et al. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol. Rev 229, 152–172 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arriens C, Wren JD, Munroe ME & Mohan C Systemic lupus erythematosus biomarkers: the challenging quest. Rheumatol. Oxf. Engl 56, i32–i45 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Verheul MK, Fearon U, Trouw LA & Veale DJ Biomarkers for rheumatoid and psoriatic arthritis. Clin. Immunol. Orlando Fla 161, 2–10 (2015). [DOI] [PubMed] [Google Scholar]
- 31.Banchereau J & Pascual V Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity 25, 383–392 (2006). [DOI] [PubMed] [Google Scholar]
- 32.Quách TD et al. Distinctions among Circulating Antibody-Secreting Cell Populations, Including B-1 Cells, in Human Adult Peripheral Blood. J. Immunol. Baltim. Md 1950 196, 1060–1069 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Andrews SF et al. High Preexisting Serological Antibody Levels Correlate with Diversification of the Influenza Vaccine Response. J. Virol 89, 3308–3317 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.He X-S et al. Baseline levels of influenza-specific CD4 memory T-cells affect T-cell responses to influenza vaccines. PloS One 3, e2574 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Patin E et al. Natural variation in the parameters of innate immune cells is preferentially driven by genetic factors. Nat. Immunol 19, 302–314 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Westra HJ et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat Genet 45, 1238–43 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Klein SL & Flanagan KL Sex differences in immune responses. Nat. Rev. Immunol 16, 626–638 (2016). [DOI] [PubMed] [Google Scholar]
- 38.Rees F, Doherty M, Grainge MJ, Lanyon P & Zhang W The worldwide incidence and prevalence of systemic lupus erythematosus: a systematic review of epidemiological studies. Rheumatol. Oxf. Engl 56, 1945–1961 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Zyla J et al. Gene set enrichment for reproducible science: comparison of CERNO and eight other algorithms. Bioinformatics doi: 10.1093/bioinformatics/btz447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sergushichev A An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation. bioRxiv (2016) doi: 10.1101/060012. [DOI] [Google Scholar]
- 41.Chen J, Bardes EE, Aronow BJ & Jegga AG ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res 37, W305–11 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van Lochem EG et al. Immunophenotypic differentiation patterns of normal hematopoiesis in human bone marrow: reference patterns for age-related changes and disease-induced shifts. Cytometry B Clin. Cytom 60, 1–13 (2004). [DOI] [PubMed] [Google Scholar]
- 43.Finak G et al. ImmuneSpaceR: A Thin Wrapper around the ImmuneSpace Database. (2014).
- 44.Bhattacharya S et al. ImmPort: disseminating data to the public for the future of immunology. Immunol Res 58, 234–9 (2014). [DOI] [PubMed] [Google Scholar]
- 45.Robin X et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics 12, 77 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bates D, Bolker BM, Maechler M & Walker SC Fitting Linear Mixed-Effects Models Using lme4. J. Stat. Softw 67, 1–48 (2015). [Google Scholar]
- 47.Chaussabel D et al. A modular analysis framework for blood genomics studies: application to systemic lupus erythematosus. Immunity 29, 150–64 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weiner J tmod: Feature Set Enrichment Analysis for Metabolomics and Transcriptomics. R package version 0.40. https://CRAN.R-project.org/package=tmod. (2018).
- 49.Benjamini Y & Hochberg Y Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 57, 289–300 (1995). [Google Scholar]
- 50.Choi JK, Yu U, Kim S & Yoo OJ Combining multiple microarray studies and modeling interstudy variation. Bioinforma. Oxf. Engl 19 Suppl 1, i84–90 (2003). [DOI] [PubMed] [Google Scholar]
- 51.Subramanian A et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U A 102, 15545–50 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinforma. Oxf. Engl 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wickham H ggplot2: Elegant Graphics for Data Analysis. (Springer-Verla, 2016). [Google Scholar]
- 55.Torchiano M effsize: Efficient Effect Size Computation. doi: 10.5281/zenodo.1480624, R package version 0.7.6, https://CRAN.R-project.org/package=effsize. (2019). [DOI] [Google Scholar]
- 56.Wang X et al. An R package suite for microarray meta-analysis in quality control, differentially expressed gene analysis and pathway enrichment detection. Bioinformatics 28, 2534–6 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Seshan V clinfun: Clinical Trial Design and Data Analysis Functions. R package version 1.0.15. https://CRAN.R-project.org/package=clinfun. (2018). [Google Scholar]
- 58.Yan Y MLmetrics: Machine Learning Evaluation Metrics. R package version 1.1.1. https://CRAN.R-project.org/package=MLmetrics. (2016). [Google Scholar]
- 59.Stoeckius M et al. Cell Hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol. 19, 224 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stoeckius M et al. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 14, 865–868 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.CITE-seq Protocols. CITE-seq https://cite-seq.com/protocol/ (2017).
- 62.Kang HM et al. Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nat. Biotechnol 36, 89–94 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Roelli Patrick, bbimber Bill Flynn, santiagorevale & Gege Gui. Hoohm/CITE-seq-Count: 1.4.2. (Zenodo, 2019). doi: 10.5281/zenodo.2590196. [DOI] [Google Scholar]
- 64.Lun ATL & Risso D SingleCellExperiment: S4 Classes for Single Cell Data. R package version 1.4.1. https://bioconductor.org/packages/release/bioc/html/SingleCellExperiment.html. (2019). [Google Scholar]
- 65.Ritchie ME et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lun L, A. T., Bach K & Marioni JC Pooling across cells to normalize single-cell RNA sequencing data with many zero counts. Genome Biol. 17, 75 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Waltman L & Eck N. J. van. A smart local moving algorithm for large-scale modularity-based community detection. Eur. Phys. J. B 86, 471 (2013). [Google Scholar]
- 68.Zappia L & Oshlack A Clustering trees: a visualization for evaluating clusterings at multiple resolutions. GigaScience 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pederson TL ggraph: An Implementation of Grammar of Graphics for Graphs and Networks. R package version 1.0.2. https://CRAN.R-project.org/package=ggraph. (2018). [Google Scholar]
- 70.Carlson M hgu95av2.db: Affymetrix Human Genome U95 Set annotation data (chip hgu95av2). R package version 3.2.3. (2016). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data used in this study, including CITE-seq and flow data, are available at figshare (https://doi.org/10.35092/yhjc.c.4753772). The original/raw public gene expression data are available in the National Center for Biotechnology Information Gene Expression Omnibus (GEO) under accession numbers: GSE47353, GSE41080, GSE59654, GSE59743, GSE29619, GSE74817, GSE13486, and GSE65391 (see also Supplementary Table 2).