

Abstract
Effective treatment of pediatric solid tumors has been hampered by the predominance of currently ‘undruggable’ driver transcription factors. Improving outcomes while decreasing the toxicity of treatment necessitates the development of novel agents that can directly inhibit or degrade these elusive targets. MYCN in pediatric neural-derived tumors, including neuroblastoma and medulloblastoma, is a paradigmatic example of this problem. Attempts to directly and specifically target MYCN have failed due to its similarity to MYC, the unstructured nature of MYC family proteins in their monomeric form, the lack of an understanding of MYCN-interacting proteins and ability to test their relevance in vivo, the inability to obtain structural information on MYCN protein complexes, and the challenges of using traditional small molecules to inhibit protein-protein or protein-DNA interactions. However, there is now promise for directly targeting MYCN based on scientific and technological advances on all of these fronts. Here we discuss prior challenges and the reasons for renewed optimism in directly targeting this ‘undruggable’ transcription factor, which we hope will lead to improved outcomes for pediatric cancer patients and create a framework for targeting driver oncoproteins regulating gene transcription.
MYCN as an attractive drug target
Transcription factors in the MYC family are dysregulated in the majority of human tumors, including most pediatric malignancies1,2. This family is composed of three genes, MYC (c-MYC), MYCN (n-MYC), and MYCL, with conserved roles in central cellular processes including regulating transcription, metabolism, and cell division. Whereas MYC is altered across a wide range of cancers, MYCN has a more narrow role—primarily as a driver of pediatric malignancies derived from central and peripheral nervous system tissues, including neuroblastoma, medulloblastoma, retinoblastoma, astrocytoma, atypical teratoid rhabdoid tumors (ATRTs), and glioblastoma multiforme, among others, with emerging roles as a driver of therapy-resistant neuroendocrine variants of lung and prostate cancer2.
MYCN is in many ways an ideal therapeutic target. Unlike MYC, its physiologic expression is tightly lineage restricted during development, with limited expression in normal pediatric or adult tissues, suggesting a wide therapeutic index for MYCN-specific drugs. When present in tumors, MYCN amplification is generally thought to be a truncal initiating event that is required for ongoing tumor maintenance. It is rarely a subclonal finding and is not acquired or lost during tumor progression or relapse. In addition, transgenic expression of MYCN in the appropriate progenitor cells in mouse models can drive tumorigenesis that faithfully recapitulates human neuroblastoma and medulloblastoma, respectively3,4.
Despite intensive investigative efforts, indirectly targeting modulators of MYCN transcription and stability or the downstream mediators of MYCN function has failed to result in the identification of MYCN-specific therapeutics. For example, after initial identification of Bromodomain and Extra-Terminal (BET) protein inhibitors, there was optimism that they could serve as universal and specific MYC-targeting drugs5,6. However, it was clear early on that these compounds did not discriminate among MYC-family proteins, as they also impacted MYCN7 and MYCL8. Preclinical data in neuroblastoma models showed tumor growth delay in some models, but no anti-tumor activity in others, with likewise variable influence on MYCN protein levels9,10. In addition, while there has been some clinical efficacy in early adult trials, most notably in NUT midline carcinoma with a canonical BRD4-NUT fusion oncoprotein11, objective response rates have been low and mostly transient in other diseases12–14 and it has become clear that tumor cells can adapt in ways that restore MYC despite continued BET inhibition15–17.
Another illustrative example is the interaction between Aurora Kinase A (AURKA) and MYCN and the effects of AURKA inhibitors. Early preclinical testing showed excellent activity of the AURKA inhibitor alisertib (MLN8237) in pediatric solid tumors18, though this was independent of MYCN status and MYC-driven tumors are also sensitive to AURKA inhibition19. Subsequent clinical testing in neuroblastoma demonstrated substantial toxicity and largely disappointing responses20,21 However, AURKA binds to MYCN and sequesters it from degradation independent of its kinase activity.22 This raises the possibility that targeting this scaffolding function of AURKA may be more effective and more specific for MYCN, as requirement for a similar stabilizing interaction has not been reported between AURKA and MYC. Small molecules have been identified that bind to AURKA and alter its conformation in a way that prevents binding to MYCN and result in rapid MYCN degradation23. More recently, a chemical degrader approach has also been taken24. Together, this illustrates how targeting the synthetic lethal interaction between MYCN and the kinase activity of AURKA has largely failed to provide an efficacious and specific therapeutic, but it remains possible that targeting the MYCN stabilizing function of the AURKA-MYCN complex may prove more successful. These and other examples support the hypothesis that sustained and specific inhibition of MYCN activity will require direct targeting of the deregulated protein or the MYCN complex.
Barriers to direct and specific inhibition
Directly targeting MYCN poses substantial challenges that can be generalized to many transcription factors, but also some challenges that are unique to MYCN. Like many transcription factors, MYCN functions in the nucleus, has no known enzymatic function, and mediates its effects in the context of several multi-protein complexes that involve numerous protein-protein interactions and protein-DNA interactions25. The protein-protein interaction surfaces in particular tend to be large and lack the defined hydrophobic pockets typically targeted by drug-like small molecules26, and complex formation tends to involve cooperation of multiple low-affinity interactions that are individually difficult to target.
MYCN also poses some unique challenges as a drug target. The N-terminal transcription activating domains of MYC family proteins are intrinsically disordered in their monomeric forms27,28, and the C-terminal basic helix-loop-helix-leucine zipper (bHLH-LZ) domain lacks the deep hydrophobic pockets into which drug-like small molecules can be easily designed to bind29. Certain N-terminal domains become structured in complex with binding partners, enabling limited structural studies of these domains30,31, but interactome studies have identified hundreds of interacting proteins32,33. Lastly, MYCN is highly homologous to MYC and MYCL within the basic helix-loop-helix-leucine zipper (bHLH-LZ) domain and the 5 MYC boxes that have been shown to mediate much of MYC family protein function2,32,34. Although there are some clear functional differences between MYC and MYCN, particularly with regard to their respective interactions with MIZ135, it remains a conceptual challenge to inhibit the oncogenic function of MYCN while preserving the physiologic functions of MYC, including those in normal cell division and in wound healing.
Although these challenges remain formidable, a clearly appealing approach (outlined in Figure 1) would be to identify small molecules that bind to MYCN in complex with an essential and specific binding partner, using structural information to guide drug design and/or optimization, then link the small molecule to an E3 ligase binder to induce MYCN degradation. Here we discuss the prior difficulties with such an approach and the scientific and technological advances that may now make it feasible. These concepts were discussed at a meeting in November of 2019 that brought together experts in pediatric cancer, MYC and MYCN biology, structural biology, biochemistry, medicinal chemistry, and protein degradation technology to address the goal of directly targeting MYCN.
Figure 1: Highlighted challenges and technologic advances in identification and validation of a direct MYCN targeting compound.
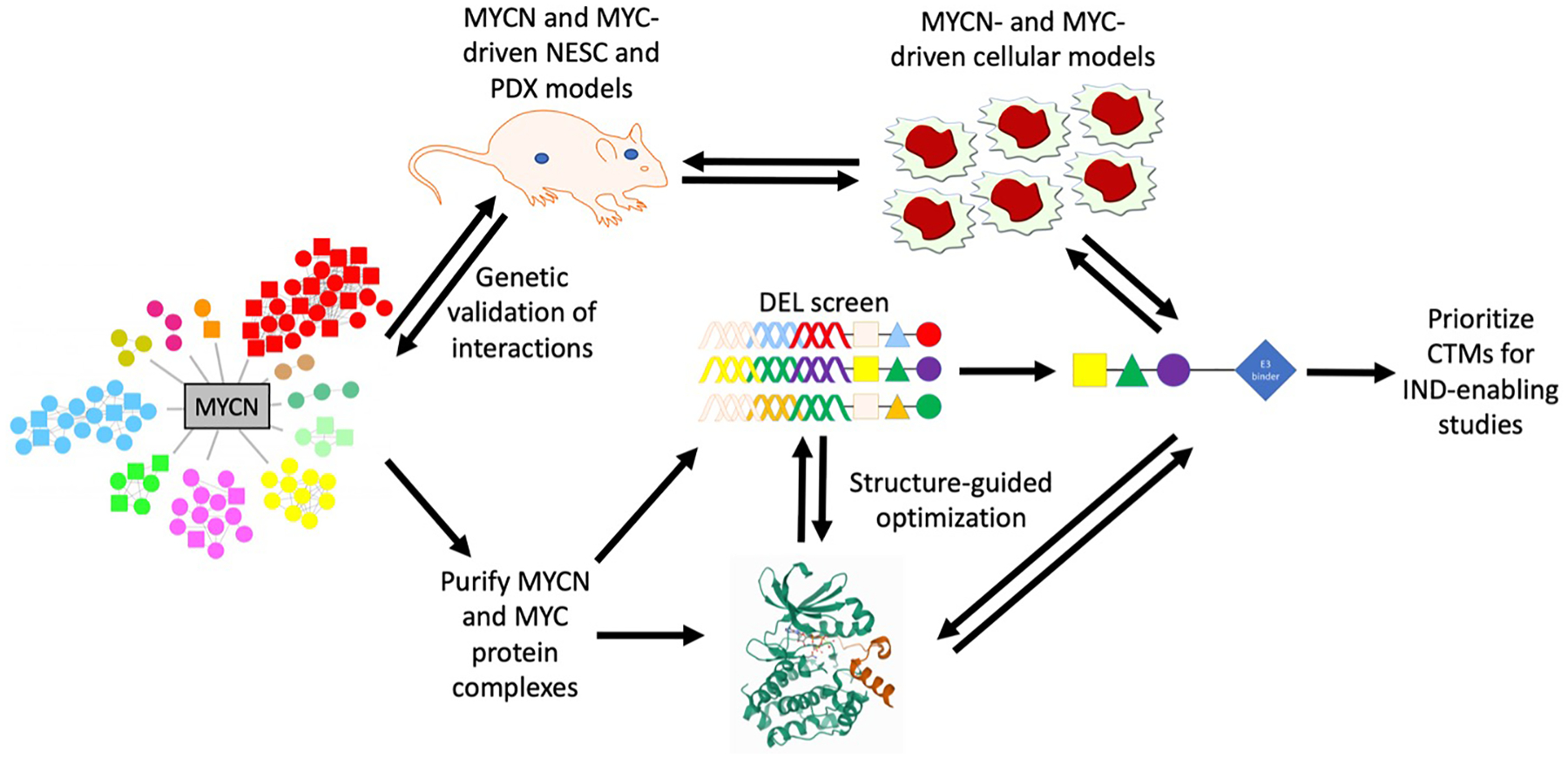
Biochemically-derived MYCN interactomes have recently revealed a large number of potential complexes to target and are contrasted to MYC interactomes (not shown). Which complexes are essential to tumor maintenance can now be identified using genetic loss-of-function testing in appropriate in vivo models (e.g. neural epithelial stem [NES] cell and patient-derived xenograft [PDX] models). Once oncogenic complexes are identified, these can be purified and used in structural studies, aided by advancements in cryo-EM technology. Challenges to the identification of small molecule inhibitors can be addressed by using a bind and degrade approach. DEL screens can facilitate the identification of binders unique to MYCN complexes as compared to MYC complexes. These compounds can then be linked to E3 ligase ligands to create proteolysis-targeting chimeras (PROTACs), optimized using structure as a guide, and tested in cell line and mouse models to confirm degradation, ensure anti-tumor activity, and determine selectivity. Once active and selective PROTACs are identified, they can be prioritized for IND-enabling studies and eventual clinical testing. Interactome modified from http://pennlab.ca/research/#, protein structure from the PDB (https://www.rcsb.org/structure/5G1X), and drawing tools from motifolio.com.
Targeting MYCN in its oncogenic context
Modern target-based drug discovery has relied on identifying the structure of fragments or lead molecules bound to their target protein, which then allows for the rational optimization of the molecule to improve potency while preserving and/or improving physico-chemical drug-like properties36. This process cannot be applied to intrinsically-disordered proteins such as MYC-family proteins in their monomeric form. Although there have been efforts – and some progress – in targeting the intrinsically disordered state of MYC27,37, the compounds developed to date have generally suffered from low potency and have not yet been turned successfully into credible lead drug compounds. However, it has long been appreciated that MYC-family proteins require binding partners to exert their tumorigenic function38, and individual domains of MYC-family proteins have been demonstrated to form stable structures when complexed with interacting proteins. The most prominent of these is the structure of the bHLH-LZ domain of MYC in complex with MAX29. Although the structure of MYCN in complex with MAX has not been solved itself, it is thought to be highly homologous to the MYC-MAX complex. Indeed, MYC-MAX disrupters have been identified and these also disrupt the MYCN-MAX interaction, further supporting the concept that MYC family proteins interact with MAX in a highly similar fashion39. The structure of other domains of MYC-family proteins have been solved in complex with other interacting proteins, including KPNA1 (importin-α), BIN1, WDR5, TBP, and AURKA28,30,31,40–43. These examples clearly demonstrate that MYC-family proteins can assume ordered states in the context of multi-protein complexes, providing a potential avenue for applying the tools of modern structure-based drug discovery.
Although targeting MYCN in a structured complex with an interacting protein has clear appeal, determining which interacting protein(s) to choose is challenging. The identity of the full complement of MYCN-interacting proteins has only recently been catalogued. This has not allowed for a substantial narrowing of focus, however, as interactome profiling has identified hundreds of proteins that can complex with MYC-family proteins32,33. This large number strongly suggests that there is not a single MYCN complex, but rather a number of different complexes that may differ in function and in their contribution to tumorigenesis and tumor maintenance and that change in composition throughout the cell cycle and across cell types or tumor types. Comparison between MYC and MYCN binding proteins has revealed large numbers of common interactors, but also many MYCN-unique interactors.
Prior to undertaking laborious structural biology and drug discovery campaigns against a given target, the MYCN-interacting protein complex should first be demonstrated to be essential for tumor maintenance in an appropriate in vivo model and not required for survival of normal cells. Such models and the appropriate genetic manipulation tools have only recently become available. For example, human neuroepithelial stem (NES) cell-based models provide one potentially appealing approach for this type of validation44. NES cells can be stably cultured and are amenable to gene editing technology. MYCN expression can be introduced, which results in medulloblastoma after orthotopic implantation45, and similar models of neuroblastoma are also under development. A moderate number of interacting proteins can now be screened via genetic loss-of-function approaches to prioritize those that (when lost) result in tumor regression in MYCN-driven tumors but do not impede the growth of analogous MYC-driven tumors or of untransformed NES cells. Although alternative models could be used for validation of the centrality of a given interacting protein to MYCN’s oncogenic function, it is clear that this type of genetic validation is essential prior to initiating further drug discovery efforts and that appropriate models are now available or will soon be available.
Limitations to obtaining structures of MYCN in complex
Although structures of individual domains of MYC proteins have been identified in complex with interacting proteins (see discussion above), limitations in structural biology have made it difficult to obtain more extensive structural information about MYC complexes. All of the three central techniques used in structural biology (X-ray crystallography, nuclear magnetic resonance [NMR], and cryo-electron microscopy [cryo-EM]) are likely to have value and play complementary roles in obtaining the structural information about MYCN complexes needed to aid drug development. However, recent advances in the ability to use cryo-EM to obtain structures of complexes of a variety of sizes is particularly important and is likely to make obtaining conformational data on oncogenic MYCN interactions more feasible. This information is not necessary to embark on a DNA-Encoded chemical Library (DEL) screen (see below) but will be critically important for understanding hits and developing drug candidates.
Two parallel approaches could be undertaken to obtain the structure of purified MYCN complexes. The first, similar to what has been done previously30 including with the AURKA43, is to map interaction domains using either NMR or cross-linking and mass spectroscopy, and then to use this information to pursue crystallographic determination of substructures of the MYCN complex. The advantages of this approach include identification of physiologically relevant MYCN interactions that might be of lower affinity, and a possibility of obtaining high resolution structures of such complexes. However, an interaction requiring a large portion of MYCN and/or requiring multiple interactors may be difficult to probe using crystallography. Cryo-EM offers an alternative that can provide Ångström-level resolution of multi-protein complexes that are unlikely to crystallize in their full-length form, such as transcriptional complexes46. These techniques can be used in a complementary fashion, with cryo-EM contributing an overall structure of the complex and X-ray crystallography or NMR focusing on smaller stable sub-complexes. Structural insights from these complementary techniques can then be incorporated to provide higher resolution views of individual side chains in domains that may be less well visualized by cryo-EM. Lower affinity interactions can be stabilized by cross-linking of nearby residues. These different modalities have been useful in the examination of complexes involving intrinsically disordered proteins like MYC, for example with the FACT complex (Facilitates Chromatin Transcription)47,48. In addition, cryo-EM has successfully identified structures of such proteins when they form ordered structures in complex49, though NMR has been applied more widely50.
In addition to playing a role in the determination of structures of known MYCN complexes, advances in cryo-EM may also offer an opportunity to identify novel complexes. Graphene oxide (GO) covered cryo-EM functionalized with affinity tags51 can be used to purify transient/lower affinity MYCN complexes from tumors or cell lysates directly on the cryo-EM grid. In addition to providing structural information simultaneously on multiple different MYCN complexes, computational advances in cryo-EM analysis can potentially allow for the determination of previously unidentified proteins in these complexes52.
While multiple approaches could be pursued in parallel, the ultimate goal should be both to solve atomic resolution structures of oncogenic MYCN complexes that will be suitable for structure-based drug development and to obtain purified protein complexes that can be used for inhibitor screens. Given the described technological advances, this is now a much more achievable goal.
Challenges in inhibitor screening and advances in degrader technology
Identifying small molecules that bind to transcription factors like MYC proteins is a substantial hurdle. Even if an effective binder is identified, the traditional approach to developing a therapeutic requires that the compound also interferes with protein function, a formidable challenge for a protein lacking enzymatic activity and functioning through protein-protein and protein-DNA interactions. However, the advent of proteolysis-targeting chimeras (PROTACs, also referred to as chimeric targeting molecules)53,54 over the past 5 years has rendered ‘undruggable’ targets such as MYCN potentially druggable. Rather than requiring a specific inhibitor of MYCN’s function, it may be sufficient to identify a MYCN-specific or MYCN complex-specific binder that can be linked to an E3 ubiquitin ligase ligand to drive the rapid ubiquitination and degradation of MYCN. In addition to requiring only a binder (and not an inhibitor), this approach also may allow for enhanced specificity through choice of the E3 ligase that is engaged by the PROTAC. For example, tissue-specific expression of E3 ligases has been described55, hence recruiting an E3 ligase that is only expressed in tumor cells or neural cells could enhance tumor-specific activity and therapeutic index. Alternatively, by engaging only an E3 ligase with expression limited to MYCN-high cells, a molecule that binds both MYC and MYCN could be made into a de facto MYCN-specific degrader. One potential drawback of a degrader approach is that downregulation of the E3 ligase provides an additional potential resistance mechanism. For this reason, it is important to choose an E3 that is essential to tumor maintenance or to simultaneously apply ligands for multiple different ligases.
Importantly, adopting a degradation approach shifts the challenge in MYCN targeting to the identification of a molecule that specifically binds to MYCN or the MYCN oncogenic complex, with little or no binding to MYC or MYC complexes. Advances in small molecule screening technology has also made this challenge easier to address. Fragment-based screening can identify low affinity binders that can then be evolved into high affinity lead compounds using structure-guided compound “growing” or by linking fragments together56. If high-quality structures are available, advances in computational docking can identify synthetically accessible potential binders57. Particularly promising is the advent of DELs that have allowed hundreds of millions to billions of drug-like compounds to be rapidly screened for affinity against proteins or protein complexes of interest58. Ideally, a systematic approach could be taken to use DELs to 1) identify binders to MYCN and several essential MYCN complexes validated in vivo as critical for tumorigenic functions of MYCN, followed by 2) hit re-synthesis and binding validation, then 3) linkage to a number of different E3 ligase ligands, and 4) analysis of the effect of the compounds on MYCN protein or complex stability. Such a comprehensive screening and follow up campaign may exceed the capacity of academic investigators, but there are companies that are well-tooled to perform such experiments and may be willing to participate in novel types of public-private partnerships.
Once validated binders and degraders are identified, structures incorporating the MYCN complex, the compound, and the appropriate E3 ligase can be solved in order to rationally optimize both binding moieties as well as the linker, in addition to making modifications to enhance predicted pharmacological properties.
Rigorous validation and characterization of compound activity and molecular diagnostic-linked early phase clinical trials
Even if putative inhibitors of MYCN or a MYCN oncogenic complex had been identified previously, properly modeling their efficacy and specificity would have been a substantial challenge. However, there are now available a range of genetically defined models, both in vitro and in vivo, to validate potential inhibitors or degraders. Cell lines that are functionally dependent upon MYCN or MYC can both be used to demonstrate MYCN-specific degradation and growth inhibition, with the caveat that changes in expression in tissue culture of the E3 and of competing E3 substrates may influence specificity. Ideally, compounds advanced to in vivo testing for efficacy should demonstrate nanomolar potency, several-fold MYCN selectivity, and undergo pharmacokinetic testing, including blood-brain barrier penetration analysis. The latter is essential to understand how the drug might be used in patients with brain tumors, as well as neuroblastomas that can metastasize to the central nervous system. Drugs may need to be modified to enhance blood-brain barrier penetration or may require combination with novel methods to transiently open the blood-brain barrier to enable drug delivery59,60.
For in vivo testing, both patient-derived xenograft models (PDXs) and genetic-engineered mouse models (GEMMs) are now widely available for the relevant diseases. Extensive PDX models of neuroblastoma, medulloblastoma, and ATRTs61–63 have the advantages of being human cells with human MYCN and MYCN interactors, of providing sufficient diversity to model genetic heterogeneity among MYCN-driven tumors, and of providing MYC-driven tumors that can be used as controls. While in autochthonous neuroblastoma and medulloblastoma GEMMs MYCN-interacting proteins are murine, non-germline GEMMs utilizing human cells enable models in which interacting proteins are human. The GEMMs also do not provide as much heterogeneity as PDXs. However, given the long-appreciated role of MYCN in suppressing antigen presentation and creating an immune-depleted tumor microenvironment64–66, it is essential to profile alterations in immune interactions upon MYCN depletion to understand the possible engagement of the adaptive immune system and how this might be enhanced. Orthogonal preclinical development of drug candidates will be essential for prioritizing the optimal drug(s) for early phase clinical trials.
Advances in molecular diagnostics should allow for more precise early phase clinical trials. Patients can be selected that have tumors with clear hyperactive MYCN signaling, both through copy number changes and transcriptional profiling. Response can be followed over time both through traditional imaging modalities as well as through detection of MYCN in cell-free circulating tumor DNA67. Non-invasive imaging modalities to detect the in vivo activity of transcription factors have been developed in some cases, such as for EWS-FLI1 in Ewing Sarcoma68, and comparable reagents could be pursued for MYCN. Lastly, patients on early phase clinical trial typically have suffered multiple relapses and received extensive immunosuppressive therapy. If pre-clinical testing demonstrates that drug efficacy depends on intact immunity, it may be necessary to incorporate immune function criteria into early phase trials.
Conclusions
Driver transcription factors such as MYCN historically represent ‘undruggable’ targets. For MYCN, this has been due to limitations in understanding the biochemistry and structural biology of MYCN complexes, the inability to model those complexes in vivo, and the difficulty in identifying small molecule inhibitors of non-enzymatic proteins like MYCN. No single advance, but rather progress on all of these fronts suggests that it is time to revisit a direct targeting approach, particularly in light of the continued failures of indirect approaches to produce an effective therapeutic. Here we describe how developments in MYCN biology, structural biology (especially cryo-EM), drug screening, modeling of pediatric cancers in mice, and PROTAC/degrader technology have made direct targeting of MYCN a practical and feasible goal, which we expect will produce an important new therapeutic for several devastating childhood tumors.
Acknowledgements
The meeting and this report were supported by a grant from the Alex’s Lemonade Stand Foundation and the meeting was held at their Childhood Cancer Data Lab in Philadelphia.
Footnotes
To develop a novel discovery pipeline for the development of MYCN-targeting therapeutics, Alex’s Lemonade Stand Foundation brought together a small group of multi-disciplinary researchers November 6–8 2019 to discuss the challenges and opportunities to directly target the MYCN oncoprotein, which is an essential driver of many childhood cancers.
The authors declare the following potential conflicts of interest:
C.V.D serves on the boards of Rafael Pharmaceuticals, Inc, Polaris Pharmaceuticals, Inc, and the Barer Institute, Inc. M.R. is a cofounder and consultant to Nurix Therapeutics, a public company in the ubiquitin space. G.M.H is an employee and shareholder of Nurix Therapeutics. W.W. is a cofounder of StemSynergy Therapeutics Inc. The other authors declare no potential conflicts of interest.
References
- 1.Dang CV MYC on the path to cancer. Cell 149, 22–35 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rickman DS, Schulte JH & Eilers M The Expanding World of N-MYC-Driven Tumors. Cancer Discov 8, 150–163 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Weiss WA, Aldape K, Mohapatra G, Feuerstein BG & Bishop JM Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO Journal 16, 2985–2995 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Swartling FJ et al. Pleiotropic role for MYCN in medulloblastoma. Genes Dev 24, 1059–72 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Delmore JE et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 146, 904–17 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Filippakopoulos P et al. Selective inhibition of BET bromodomains. Nature 468, 1067–73 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Puissant A et al. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Discov 3, 308–23 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kato F et al. MYCL is a target of a BET bromodomain inhibitor, JQ1, on growth suppression efficacy in small cell lung cancer cells. Oncotarget 7, 77378–77388 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iniguez AB et al. Resistance to Epigenetic-Targeted Therapy Engenders Tumor Cell Vulnerabilities Associated with Enhancer Remodeling. Cancer Cell 34, 922–938 e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Healy JR et al. Limited antitumor activity of combined BET and MEK inhibition in neuroblastoma. Pediatr Blood Cancer 67, e28267 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stathis A et al. Clinical Response of Carcinomas Harboring the BRD4-NUT Oncoprotein to the Targeted Bromodomain Inhibitor OTX015/MK-8628. Cancer Discov 6, 492–500 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amorim S et al. Bromodomain inhibitor OTX015 in patients with lymphoma or multiple myeloma: a dose-escalation, open-label, pharmacokinetic, phase 1 study. Lancet Haematol 3, e196–204 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Berthon C et al. Bromodomain inhibitor OTX015 in patients with acute leukaemia: a dose-escalation, phase 1 study. Lancet Haematol 3, e186–95 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Lewin J et al. Phase Ib Trial With Birabresib, a Small-Molecule Inhibitor of Bromodomain and Extraterminal Proteins, in Patients With Selected Advanced Solid Tumors. J Clin Oncol 36, 3007–3014 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Rathert P et al. Transcriptional plasticity promotes primary and acquired resistance to BET inhibition. Nature 525, 543–547 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fong CY et al. BET inhibitor resistance emerges from leukaemia stem cells. Nature 525, 538–42 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kurimchak AM et al. Resistance to BET Bromodomain Inhibitors Is Mediated by Kinome Reprogramming in Ovarian Cancer. Cell Rep 16, 1273–1286 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maris JM et al. Initial testing of the aurora kinase A inhibitor MLN8237 by the Pediatric Preclinical Testing Program (PPTP). Pediatr Blood Cancer 55, 26–34 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mollaoglu G et al. MYC Drives Progression of Small Cell Lung Cancer to a Variant Neuroendocrine Subtype with Vulnerability to Aurora Kinase Inhibition. Cancer Cell 31, 270–285 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mosse YP et al. Pediatric phase I trial and pharmacokinetic study of MLN8237, an investigational oral selective small-molecule inhibitor of Aurora kinase A: a Children’s Oncology Group Phase I Consortium study. Clin Cancer Res 18, 6058–64 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mosse YP et al. A Phase II Study of Alisertib in Children with Recurrent/Refractory Solid Tumors or Leukemia: Children’s Oncology Group Phase I and Pilot Consortium (ADVL0921). Clin Cancer Res 25, 3229–3238 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Otto T et al. Stabilization of N-Myc is a critical function of Aurora A in human neuroblastoma. Cancer Cell 15, 67–78 (2009). [DOI] [PubMed] [Google Scholar]
- 23.Gustafson WC et al. Drugging MYCN through an allosteric transition in Aurora kinase A. Cancer Cell 26, 414–427 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adhikari B et al. PROTAC-mediated degradation reveals a non-catalytic function of AURORA-A kinase. Nat Chem Biol 16, 1179–1188 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dang CV, Reddy EP, Shokat KM & Soucek L Drugging the ‘undruggable’ cancer targets. Nat Rev Cancer 17, 502–508 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bushweller JH Targeting transcription factors in cancer - from undruggable to reality. Nat Rev Cancer 19, 611–624 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Metallo SJ Intrinsically disordered proteins are potential drug targets. Curr Opin Chem Biol 14, 481–8 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Andresen C et al. Transient structure and dynamics in the disordered c-Myc transactivation domain affect Bin1 binding. Nucleic Acids Res 40, 6353–66 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nair SK & Burley SK X-ray structures of Myc-Max and Mad-Max recognizing DNA. Molecular bases of regulation by proto-oncogenic transcription factors. Cell 112, 193–205 (2003). [DOI] [PubMed] [Google Scholar]
- 30.Bayliss R, Burgess SG, Leen E & Richards MW A moving target: structure and disorder in pursuit of Myc inhibitors. Biochem Soc Trans 45, 709–717 (2017). [DOI] [PubMed] [Google Scholar]
- 31.Wei Y et al. Multiple direct interactions of TBP with the MYC oncoprotein. Nat Struct Mol Biol 26, 1035–1043 (2019). [DOI] [PubMed] [Google Scholar]
- 32.Kalkat M et al. MYC Protein Interactome Profiling Reveals Functionally Distinct Regions that Cooperate to Drive Tumorigenesis. Mol Cell 72, 836–848 e7 (2018). [DOI] [PubMed] [Google Scholar]
- 33.Buchel G et al. Association with Aurora-A Controls N-MYC-Dependent Promoter Escape and Pause Release of RNA Polymerase II during the Cell Cycle. Cell Rep 21, 3483–3497 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kohl NE et al. Human N-myc is closely related in organization and nucleotide sequence to c-myc. Nature 319, 73–7 (1986). [DOI] [PubMed] [Google Scholar]
- 35.Vo BT et al. The Interaction of Myc with Miz1 Defines Medulloblastoma Subgroup Identity. Cancer Cell 29, 5–16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van Montfort RLM & Workman P Structure-based drug design: aiming for a perfect fit. Essays Biochem 61, 431–437 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hammoudeh DI, Follis AV, Prochownik EV & Metallo SJ Multiple independent binding sites for small-molecule inhibitors on the oncoprotein c-Myc. J Am Chem Soc 131, 7390–401 (2009). [DOI] [PubMed] [Google Scholar]
- 38.Dang CV et al. Intracellular leucine zipper interactions suggest c-Myc hetero-oligomerization. Mol Cell Biol 11, 954–62 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Han H et al. Small-Molecule MYC Inhibitors Suppress Tumor Growth and Enhance Immunotherapy. Cancer Cell 36, 483–497 e15 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pineda-Lucena A et al. A structure-based model of the c-Myc/Bin1 protein interaction shows alternative splicing of Bin1 and c-Myc phosphorylation are key binding determinants. J Mol Biol 351, 182–94 (2005). [DOI] [PubMed] [Google Scholar]
- 41.Conti E & Kuriyan J Crystallographic analysis of the specific yet versatile recognition of distinct nuclear localization signals by karyopherin alpha. Structure 8, 329–38 (2000). [DOI] [PubMed] [Google Scholar]
- 42.Thomas LR et al. Interaction with WDR5 promotes target gene recognition and tumorigenesis by MYC. Mol Cell 58, 440–52 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Richards MW et al. Structural basis of N-Myc binding by Aurora-A and its destabilization by kinase inhibitors. Proc Natl Acad Sci U S A 113, 13726–13731 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McLaren D et al. Automated large-scale culture and medium-throughput chemical screen for modulators of proliferation and viability of human induced pluripotent stem cell-derived neuroepithelial-like stem cells. J Biomol Screen 18, 258–68 (2013). [DOI] [PubMed] [Google Scholar]
- 45.Huang M et al. Engineering Genetic Predisposition in Human Neuroepithelial Stem Cells Recapitulates Medulloblastoma Tumorigenesis. Cell Stem Cell 25, 433–446 e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vos SM et al. Structure of activated transcription complex Pol II-DSIF-PAF-SPT6. Nature 560, 607–612 (2018). [DOI] [PubMed] [Google Scholar]
- 47.Liu Y et al. FACT caught in the act of manipulating the nucleosome. Nature 577, 426–431 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tsunaka Y, Ohtomo H, Morikawa K & Nishimura Y Partial Replacement of Nucleosomal DNA with Human FACT Induces Dynamic Exposure and Acetylation of Histone H3 N-Terminal Tails. iScience 23, 101641 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gilman MSA et al. Structure of the Respiratory Syncytial Virus Polymerase Complex. Cell 179, 193–204 e14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schneider R, Blackledge M & Jensen MR Elucidating binding mechanisms and dynamics of intrinsically disordered protein complexes using NMR spectroscopy. Curr Opin Struct Biol 54, 10–18 (2019). [DOI] [PubMed] [Google Scholar]
- 51.Wang F et al. General and robust covalently linked graphene oxide affinity grids for high-resolution cryo-EM. bioRxiv, 657411 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ho CM et al. Bottom-up structural proteomics: cryoEM of protein complexes enriched from the cellular milieu. Nat Methods 17, 79–85 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Burslem GM & Crews CM Proteolysis-Targeting Chimeras as Therapeutics and Tools for Biological Discovery. Cell (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Winter GE et al. DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 348, 1376–81 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kaneko M et al. Genome-wide identification and gene expression profiling of ubiquitin ligases for endoplasmic reticulum protein degradation. Sci Rep 6, 30955 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Erlanson DA, Fesik SW, Hubbard RE, Jahnke W & Jhoti H Twenty years on: the impact of fragments on drug discovery. Nat Rev Drug Discov 15, 605–619 (2016). [DOI] [PubMed] [Google Scholar]
- 57.Lyu J et al. Ultra-large library docking for discovering new chemotypes. Nature 566, 224–229 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Neri D & Lerner RA DNA-Encoded Chemical Libraries: A Selection System Based on Endowing Organic Compounds with Amplifiable Information. Annu Rev Biochem 87, 479–502 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mainprize T et al. Blood-Brain Barrier Opening in Primary Brain Tumors with Non-invasive MR-Guided Focused Ultrasound: A Clinical Safety and Feasibility Study. Sci Rep 9, 321 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen EM et al. Biodegradable PEG-poly(omega-pentadecalactone-co-p-dioxanone) nanoparticles for enhanced and sustained drug delivery to treat brain tumors. Biomaterials 178, 193–203 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stewart E et al. Orthotopic patient-derived xenografts of paediatric solid tumours. Nature 549, 96–100 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rokita JL et al. Genomic Profiling of Childhood Tumor Patient-Derived Xenograft Models to Enable Rational Clinical Trial Design. Cell Rep 29, 1675–1689 e9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Smith KS et al. Patient-derived orthotopic xenografts of pediatric brain tumors: a St. Jude resource. Acta Neuropathol 140, 209–225 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bernards R, Dessain SK & Weinberg RA N-myc amplification causes down-modulation of MHC class I antigen expression in neuroblastoma. Cell 47, 667–74 (1986). [DOI] [PubMed] [Google Scholar]
- 65.Layer JP et al. Amplification of N-Myc is associated with a T-cell-poor microenvironment in metastatic neuroblastoma restraining interferon pathway activity and chemokine expression. Oncoimmunology 6, e1320626 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wei JS et al. Clinically Relevant Cytotoxic Immune Cell Signatures and Clonal Expansion of T-Cell Receptors in High-Risk MYCN-Not-Amplified Human Neuroblastoma. Clin Cancer Res 24, 5673–5684 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chicard M et al. Genomic Copy Number Profiling Using Circulating Free Tumor DNA Highlights Heterogeneity in Neuroblastoma. Clin Cancer Res 22, 5564–5573 (2016). [DOI] [PubMed] [Google Scholar]
- 68.Osgood CL et al. (18)F-FLT Positron Emission Tomography (PET) is a Pharmacodynamic Marker for EWS-FLI1 Activity and Ewing Sarcoma. Sci Rep 6, 33926 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]