

Abstract
What is already known on this topic?
Coronavirus disease 2019 (COVID-19), a disease caused by a novel human coronavirus named the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) or COVID-19 virus, was reported in December 2019. Complete genomes of the COVID-19 virus from clinical samples using next generation sequencing (NGS) have been reported.
What is added by this report?
Here we provide the technical data for sequencing complete genome of COVID-19 virus from clinical samples using the Sanger method. Two complete COVID-19 virus genome sequences (named WH19004-S and GX0002) were obtained from clinical samples of COVID-19 patients, and two single nucleotide polymorphisms (SNPs) in ORF7a (T/C, nt 27,493) and ORF8 (T/C, nt 28,253) of WH19004-S were identified by Sanger sequencing.
What are the implications for public health practice?
The COVID-19 virus genome sequencing by Sanger method reported here could be used to generate data of high enough quality without requirement for expensive NGS equipment, which support sequencing complete genomes from clinical samples and monitoring of viral genetic variations of COVID-19 infections.
In December 2019, a novel coronavirus from patients with pneumonia was identified and subsequently named the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (1-3). SARS-CoV-2 has caused a coronavirus disease 2019 (COVID-19) pandemic with high morbidity and mortality. Analyzing the genome of SARS-CoV-2 (also referred to as COVID-19 virus) from clinical samples is crucial for the understanding of viral spread and viral evolution as well as for vaccine development (4-6). Presently, whole genome sequencing of the COVID-19 virus was often generated by next generation sequencing (NGS) (7). Although NGS methods have many advantages in terms of speed and parallelism, the accuracy and read length of Sanger sequencing is still superior and has confined the use of NGS mainly to resequencing genomes (8).
Here we introduce a detailed method to rapidly obtain COVID-19 virus whole-genome sequence from clinical samples. This method is based on multiple nucleic acid amplified fragments for Sanger sequencing. We applied this method to obtain 2 complete genome sequences of COVID-19 virus from clinical samples of patients with COVID-19.
MATERIALS AND METHODS
Clinical Samples
In this study, bronco-alveolar lavage samples were collected from patients with COVID-19 in Hubei, China. COVID-19 virus RNA was identified as positive (Ct value: 28.78 and 31.86) by a real-time fluorescence-based reverse transcriptase polymerase chain reaction (rRT-PCR) assay as previously reported (7).
Nucleic Acid Extraction and Fragment Amplification
Viral RNA was extracted from 140 μL of sample using QIAamp Viral mini kits (Qiagen, Germany) according to the manufacturer’s instructions. RNA was eluted in 80 μL of elution buffer. A total of 38 sets of specific primers covering the whole COVID-19 virus genome were designed (Table 1) according to the reference sequence (WH19004, Accession ID: EPI_ISL_402120) obtained by NGS as previously reported (7). Overlapping fragments were obtained by RT-PCR conducted as follow: 5 μL of extracted RNA were amplified with the QIAGEN OneStep RT-PCR Kit (Qiagen, Germany) and RT-PCR programs were run as follows: 50 ℃ 30 min; 95 ℃ for 15 min; 95 ℃ for 30 s, 50/55 ℃ 30 s, 72 ℃1/2 min, 40 cycles; 72 ℃ 5 min. All PCR products were confirmed by gel electrophoresis analysis and sequenced using the Sanger method.
Table 1. Primers used for whole-genome sequencing of the COVID-19 virus.
| Set | Name | Start* | End* | Primer sequence (5’→3’) |
| *The location on the reference genome, accession ID: EPI_ISL_402120. | ||||
| 1 | 1F | 64 | 86 | CTCTAAACGAACTTTAAAATCTG |
| 1R | 1,048 | 1,068 | CCATTGAAGGTGTCAAATTTC | |
| 2 | 2F | 706 | 729 | CGAGCTTGGCACTGATCCTTA |
| 2R | 1,398 | 1,419 | GCAAGACTATGCTCAGGTCCTA | |
| 3 | 3F | 950 | 970 | TACTGCTGCCGTGAACATGAG |
| 3R | 2,183 | 2,203 | CCAACCGTCTCTAAGAAACTC | |
| 4 | 4F | 1,999 | 2,020 | GAGACTCATTGATGCTATGATG |
| 4R | 3,099 | 3,120 | TCAGTACCATACTCATATTGAG | |
| 5 | 5F | 2,352 | 2,374 | GTGGAGCTAAACTTAAAGCCTTG |
| 5R | 3,452 | 3,473 | CTCCTCCATGTTTAAGGTAAAC | |
| 6 | 6F | 2,846 | 2,865 | ACAGTTGAACTCGGTACAGA |
| 6R | 4,068 | 4,088 | CAATGTCACTAACAAGAGTGG | |
| 7 | 7F | 3,884 | 3,904 | CCTAAAGAGGAAGTTAAGCCA |
| 7R | 5,153 | 5,172 | TGGTAGTACTCAAAAGCCTC | |
| 8 | 8F | 4,787 | 4,807 | GCTGGTTCCTATAAAGATTGG |
| 8R | 6,146 | 6,165 | ACATCACCATTTAAGTCAGG | |
| 9 | 9F | 5,976 | 5,997 | ATTCTTATTTCACAGAGCAACC |
| 9R | 7,178 | 7,200 | GAAATGGTAATTTGTATAGTTTC | |
| 10 | 10F | 6,977 | 6,999 | GTTTGCCTAGGTTCTTTAATCTA |
| 10R | 8,183 | 8,204 | CTACATCTGAATCAACAAACCC | |
| 11 | 11F | 7,985 | 8,006 | CAGGCATTAGTGTCTGATGTTG |
| 11R | 9,167 | 9,188 | CTCTAACAGAACCTTCAAGGTA | |
| 12 | 12F | 8,966 | 8,986 | AAACTTATAGAGTACACTGAC |
| 12R | 10,166 | 10,185 | CAGATCACATGTCTTGGACA | |
| 13 | 13F | 9,900 | 9,921 | ATAAGTACAAGTATTTTAGTGG |
| 13R | 11,114 | 11,133 | GCAGACATAGCAATAATACC | |
| 14 | 14F | 10,901 | 10,922 | GGTAGTGCTTTATTAGAAGATG |
| 14R | 12,175 | 12,196 | AAGAACAACTTCAGAATCACCA | |
| 15 | 15F | 12,024 | 12,043 | CCATGCAGGGTGCTGTAGAC |
| 15R | 13,205 | 13,225 | GGATTCTTGATCCATATTGGC | |
| 16 | 16F | 12,970 | 12,991 | CAACCTAAATAGAGGTATGGTA |
| 16R | 14,290 | 14,312 | TCCCAATATTTAAAATAACGGTC | |
| 17 | 17F | 13,775 | 13,795 | CACATATATCACGTCAACGTC |
| 17R | 14,999 | 15,019 | GTGCATCTTGATCCTCATAAC | |
| 18 | 18F | 14,756 | 14,777 | ACTTCTTCTTTGCTCAGGATGG |
| 18R | 15,989 | 16,011 | TTCAATCATAAGTGTACCATCTG | |
| 19 | 19F | 15,929 | 15,850 | GCAAAATGTTGGACTGAGACTG |
| 19R | 17,014 | 17,036 | GCAACATTGCTAGAAAACTCATC | |
| 20 | 20F | 16,832 | 16,853 | CCTTTGAAAAAGGTGACTATGG |
| 20R | 17,956 | 17,977 | GGTCTCTATCAGACATTATGCA | |
| 21 | 21F | 17,530 | 17,549 | ATAGGTCCAGACATGTTCCT |
| 21R | 18,781 | 18,801 | TTGTAGGTTACCTGTAAAACC | |
| 22 | 22F | 18,487 | 18,506 | ATACCACTTATGTACAAAGG |
| 22R | 19,618 | 19,639 | AAGCCACATTTTCTAAACTCTG | |
| 23 | 23F | 19,438 | 19,459 | CCACTAAAGTCTGCTACGTGTA |
| 23R | 20,568 | 20,589 | GTCAATAGTCACTTTGACAACC | |
| 24 | 24F | 20,363 | 20,384 | TACATCTACTGATTGGACTAGC |
| 24R | 21,658 | 21,678 | GGGTAATAAACACCACGTGTG | |
| 25 | 25F | 19,828 | 19,850 | AAAATACTCAATAATTTGGGTGT |
| 25R | 21,019 | 21,041 | ATAATGAGATCCCATTTATTAGC | |
| 26 | 26F | 20,428 | 20,447 | CCTATGGACAGTACAGTTAA |
| 26R | 21,665 | 21,684 | TTGTCAGGGTAATAAACACC | |
| 27 | 27F | 21,332 | 21,354 | ATGCAAATTACATATTTTGGAGG |
| 27R | 22,539 | 22,560 | GTAATATTAGGAAATCTAACAA | |
| 28 | 28F | 22,433 | 22,449 | TGTGCACTTGACCCTCT |
| 28R | 23,345 | 23,364 | CCTGGTGTTATAACACTGAC | |
| 29 | 29F | 23,123 | 23,142 | CCAGCAACTGTTTGTGGACC |
| 29R | 24,095 | 24,116 | CACAAATGAGGTCTCTAGCAGC | |
| 30 | 30F | 23,339 | 23,360 | GGTGGTGTCAGTGTTATAACAC |
| 30R | 24,328 | 24,349 | ACTATTAAATTGGTTGGCAATC | |
| 31 | 31F | 23,948 | 23,971 | GATTTTGGTGGTTTTAATTTTTCA |
| 31R | 25,157 | 25,176 | TTTCCAAGTTCTTGGAGATC | |
| 32 | 32F | 24,960 | 24,981 | TCAACAACACAGTTTATGATCC |
| 32R | 26,171 | 26,192 | GGTTCATCATAAATTGGTTCCA | |
| 33 | 33F | 25,837 | 25,857 | TGGCATACTAATTGTTAYGAC |
| 33R | 27,033 | 27,052 | GAAAGCGTTCGTGATGTAGC | |
| 34 | 34F | 26,815 | 26,834 | CTTCTTTCAGACTGTTTGCG |
| 34R | 27,948 | 27,968 | ACATGACTGTAAACTACATTC | |
| 35 | 35F | 27,389 | 27,406 | CGAACATGAAAATTATTC |
| 35R | 28,550 | 28,568 | CGTCACCACCACGAATTCG | |
| 36 | 36F | 28,322 | 28,341 | TTTGGTGGACCCTCAGATTC |
| 36R | 29,543 | 29,561 | CCATCTGCCTTGTGTGGTC | |
| 37 | 37F | 29,149 | 29,170 | CAGACAAGGAACTGATTACAAA |
| 37R | 29,836 | 29,857 | GAAGCTATTAAAATCACATGGG | |
| 38 | 38F | 26,539 | 26,559 | GTACTATTACCGTTGAAGAGC |
| 38R | 27,102 | 27,122 | CCTGTAGCGACTGTATGCAGC | |
| 5′-RACE | 39R | 1,048 | 1,068 | CCATTGAAGGTGTCAAATTTC |
| 40R | 493 | 512 | GACCATGAGGTGCAGTTCGA | |
| 3′-RACE | 41F | 29,149 | 29,170 | CAGACAAGGAACTGATTACAAA |
| 42F | 29,438 | 29,459 | CAGCAAACTGTGACTCTTCTTC | |
5’ and 3’ Ends of Genome Sequencing
The 5’ and 3’ ends of the genome were determined by rapid amplification of cDNA ends (RACE) using the Invitrogen 5’ RACE System and 3’ RACE System (Invitrogen, USA) according to the manufacturer’s instructions. Gene-specific primers for 5’ and 3’ RACE PCR amplification were designed to obtain a fragment of approximately 400–500 bp for the two regions. Purified PCR products were cloned into the pMD18-T Simple Vector (TaKaRa, Takara Biotechnology, Dalian, China) and chemically competent Escherichia coli (DH5α cells, TaKaRa), according to the manufacturer’s instructions. PCR products were sequenced with use of M13 forward and reverse primers.
Genome Sequence Assembly
All sequencing fragments were assembled using DNAStar software. The open reading frames of the verified genome sequences were predicted using Geneious (version 11.1.5) and annotated using the Conserved Domain Database. Sequence alignment of the COVID-19 virus with reference sequences was done with Mafft software (version 7.450). The SNPs of each sequence were defined as the site’s variant from the reference sequence.
RESULTS
Primer Design
The primers were designed in entire genome regions to obtain overlapping amplicons of approximately 1,000–1,200 bp leading to a list of 38 primer pairs. Meanwhile, 5’ and 3’ terminal sequencing primers were designed to obtain amplicons of 400–500 bp for sequencing (Table. 1).
Genomic Characterization
Using DNAStar software all sequencing fragments were assembled, 2 complete sequences named WH19004-S and GX0002 were obtained from the clinical samples (Figure 1). Of which, WH19004-S(29896 nt) is consistent with WH19004-NGS (accession ID: EPI_ISL_402120), except that there are 2 variants in nucleotide (nt) 27,493 (T/C) and 28,253 (T/C) respectively and identified these positions as single nucleotide polymorphisms by alignment with a large number of COVID-19 genome sequences (Figure 2). Nt 27,493 located in ORF7a (amino acid position 34), T or C translated to different amino acid (Ser or Pro), while nt 28,253 in ORF8 (amino acid position 120), no changes in amino acid.
Figure 1.
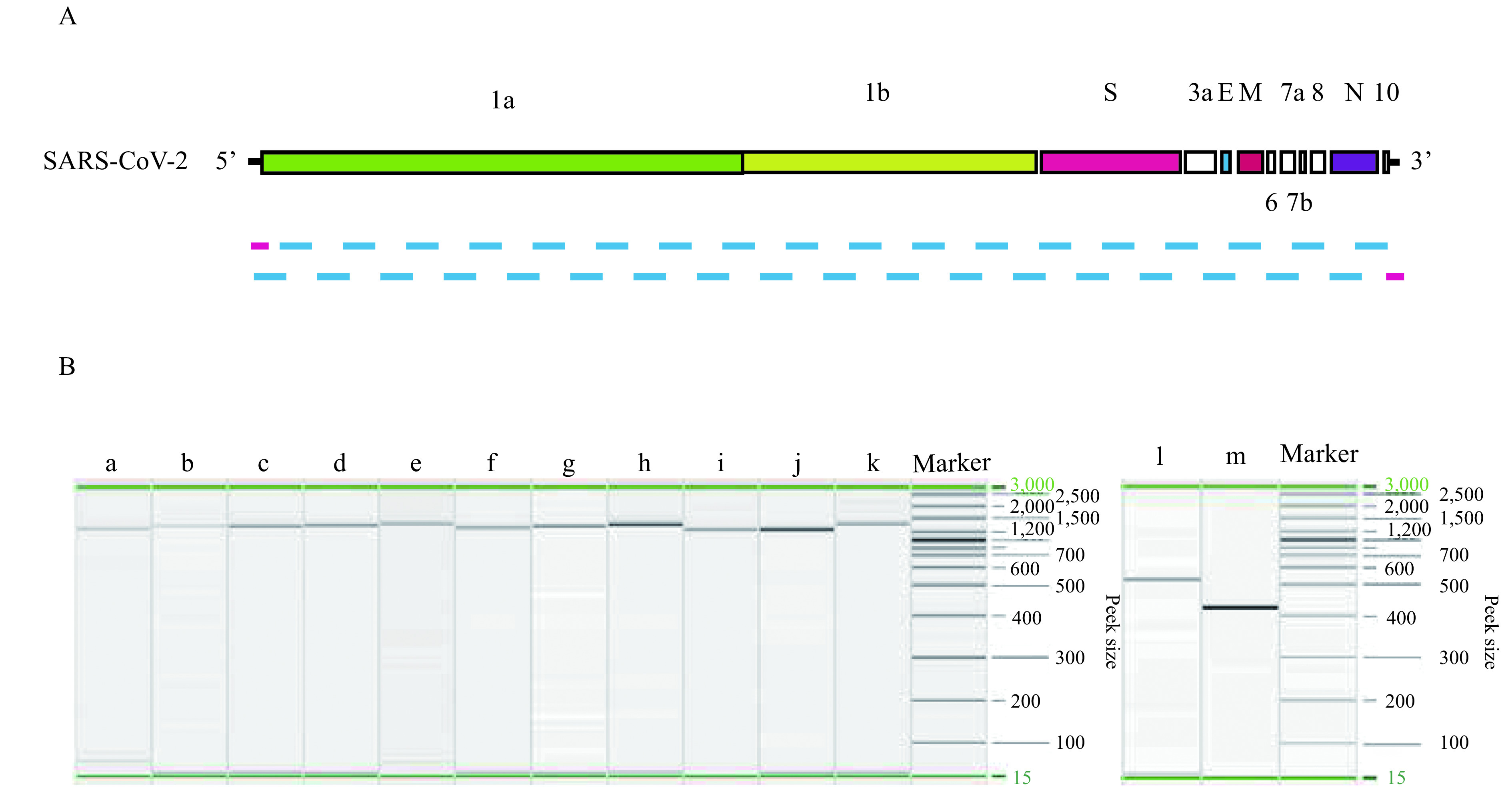
COVID-19 virus genome fragment amplification by RT-PCR. A: Schematic representation of the amplificatory fragments of the genome. B: Capillary electrophoresis profiles of RT-PCR products of the obtained partial fragment. a to k: RT-PCR products of the fragment 1–11; l to m: RT-PCR products of the end of 5’ and 3’ of the genome.
Figure 2.
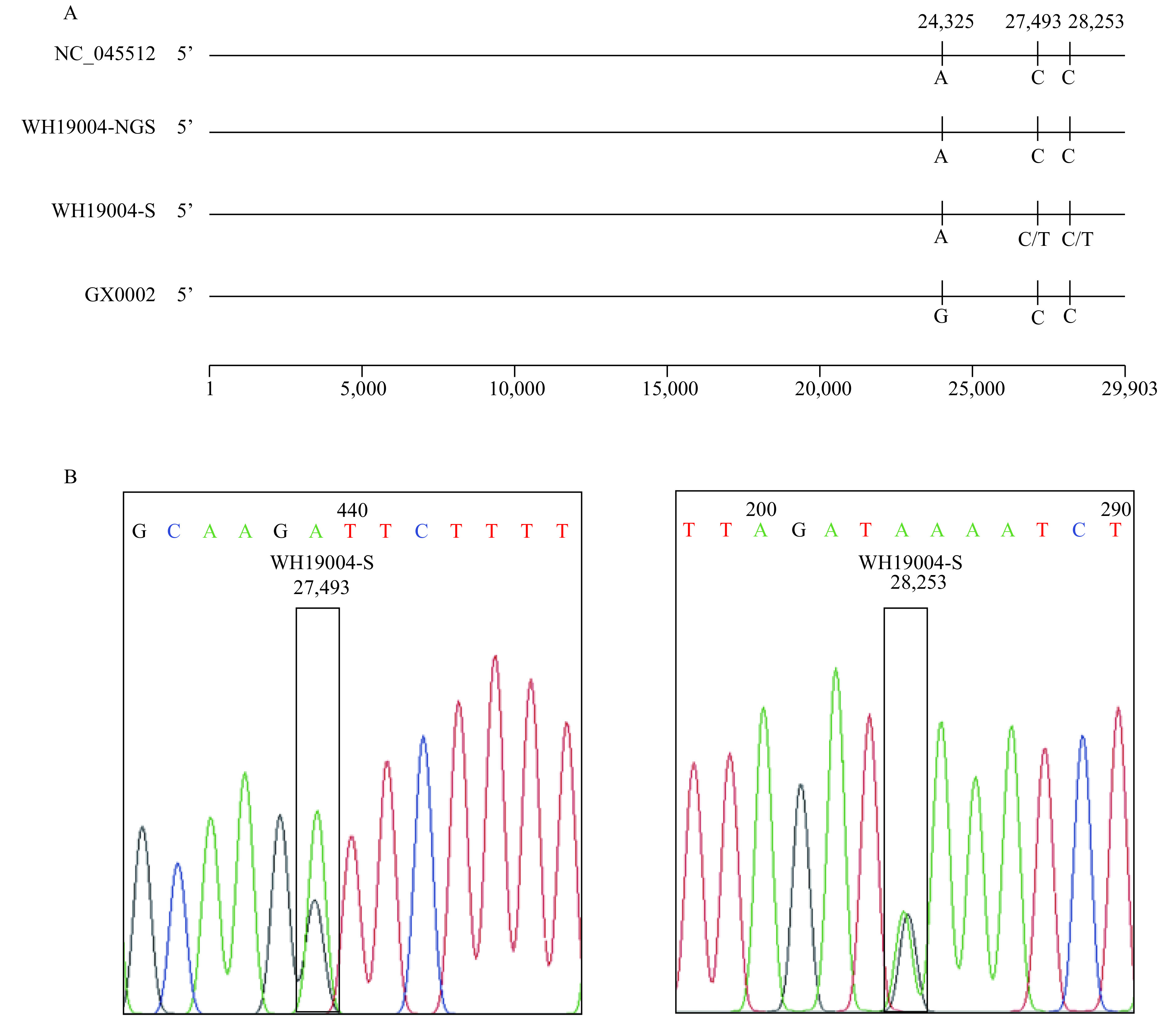
Sequence comparison and Sanger sequencing diagram of COVID-19 virus in this study. (A) Sequence alignment of 4 whole-length genomes of COVID-19 virus. (B) Sanger sequencing diagram of WH19004-S by reverse primers 29R and 30R, which shows the SNPs in ORF7 and ORF8 of WH19004-S.
The GX0002 strain (accession ID: EPI_ISL_434534) was 29,892 nt in length, including a 5’ untranslated region (UTR) (nt 1 to 265), replicase complex open reading frame 1ab [ORF1ab] (nt 266 to 21,555), S gene (nt 21,563 to 25,384), ORF3a (nt 25,393 to 26,220), E gene (nt 26,245 to 26,472), M gene (nt 26,523 to 27,191), ORF6 (nt 27,202 to 27,387), ORF7a (nt 27,394 to 27,759), ORF7b (nt 27,756 to 27,887), ORF8 gene (nt 27,894 to 28,259), N gene (nt 28,274 to 29,533), ORF10 gene (nt 29,558 to 29,674), and 3’ UTR (nt 29,675 to 29,892). Compared with the reference strain (GenBank no. NC_045512), the GX0002 strain only has a nucleotide variant in nt 24,325 position (G/A) in the S gene and no changes in amino acid (Figure 2).
DISCUSSION
To accelerate our investigation of this virus and the disease it causes, a practical protocol for viral genome research of clinical samples is urgently needed. In this study, we obtained 2 COVID-19 virus complete genome sequences WH19004-S and GX0002 from clinical samples using the Sanger sequencing method.
While NGS is the current mainstream sequencing method with the characteristics of high-throughput, rapidity, etc., it also has some drawbacks such as its relatively short reads. As a result, NGS lacks the capacity to link independent variations on the same nucleic molecule, so it is not well suited to discriminate and phase alleles to their respective parental homolog (9). In addition, the abundance of COVID-19 virus in clinical samples is often low, so the application of conventional NGS requires deeper sequencing of each sample in order to obtain sufficient coverage and depth of the whole viral genome, which increases the time and cost of sequencing. Nevertheless, as one of the earliest sequencing methods, the Sanger method has the characteristics of high accuracy, long reads, no requirement for expensive equipment, etc. Sanger sequencing has been used for analyzing genes where NGS fails to achieve sufficient depth of coverage or to generate data of high enough quality. Sanger sequencing is also used for confirming NGS variants before they are clinically reported (10). Especially when the general laboratory have common PCR machine and lack of expensive NGS platform, Sanger method is more prefer to be applied. In this study, we identified two SNPs in ORF7a (T/C, nt 27,493) and ORF8 (T/C, nt 28,253) of WH19004-S using Sanger sequencing compared with WH19004-NGS derived from NGS. The SNP in ORF7a of WH19004-S translated to two different amino acid (Ser or Pro). The roles of the SNPs in COVID-19 virus genetic evolution and whether it causes functional changes still need further investigation.
In summary, we reported here a rapid, versatile, and clinic-friendly approach for sequencing the complete genome of COVID-19 virus from clinical samples using the Sanger method, which will facilitate monitoring of viral genetic variations during outbreaks, both current and future.
Conflict of interest: No conflicts of interest were reported.
Funding Statement
This work was supported by the National Key Research and Development Program of China (2016YFD0500301).
References
- 1.Wang C, Horby PW, Hayden FG, Gao GF A novel coronavirus outbreak of global health concern. Lancet. 2020;3(10223):470–3. doi: 10.1016/S0140-6736(20)30185-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tan WJ, Zhao X, Ma XJ, Wang WL, Niu PH, XU WB, et al. Notes from the field: a novel coronavirus genome identified in a cluster of pneumonia cases-Wuhan, China 2019−2020. China CDC Weekly 2020; 2(4): 61-2. http://weekly.chinacdc.cn/en/article/id/a3907201-f64f-4154-a19e-4253b453d10c.
- 3.Zhu N, Zhang DY, Wang WL, Li XW, Yang B, Song JD, et al A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;3(8):727–33. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang CL, Wang YM, Li XW, Ren LL, Zhao JP, Hu Y, et al Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;3(10223):15–21. doi: 10.1016/s0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coronaviridae Study Group of the International Committee on Taxonomy of Viruses The species Severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2 . Nat Microbiol. 2020;3(4):536–44. doi: 10.1038/s41564-020-0695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Su S, Wong G, Shi WF, Liu J, Lai ACK, Zhou JY, et al Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiol. 2016;3(6):490–502. doi: 10.1016/j.tim.2016.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu RJ, Zhao X, Li J, Niu PH, Yang B, Wu HL, et al Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;3(10224):565–74. doi: 10.1016/s0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Quiñ ones-Mateu ME, Avila S, Reyes-Teran G, Martinez MA, et al Deep sequencing: becoming a critical tool in clinical virology. J Clin Virol. 2014;3(1):9–19. doi: 10.1016/j.jcv.2014.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tewhey R, Bansal V, Torkamani A, Topol EJ, Schork NJ, et al The importance of phase information for human genomics. Nat Rev Genet. 2011;3(3):215–23. doi: 10.1038/nrg2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mu WB, Lu HM, Chen J, Li SW, Elliott AM Sanger confirmation is required to achieve optimal sensitivity and specificity in next-generation sequencing panel testing. J Mol Diagn. 2016;3(6):923–32. doi: 10.1016/j.jmoldx.2016.07.006. [DOI] [PubMed] [Google Scholar]