

Emerging and re-emerging pathogens are great challenges to the public health (1). A cluster of pneumonia cases with an unknown cause occurred in Wuhan starting on December 21, 2019. As of January 20, 2020, a total of 201 cases of pneumonia in China have been confirmed. A team of professionals from the National Health Commission and China CDC conducted epidemiological and etiological investigations. On January 3, 2020, the first complete genome of the novel β genus coronaviruses (2019-nCoVs) was identified in samples of bronchoalveolar lavage fluid (BALF) from a patient from Wuhan by scientists of the National Institute of Viral Disease Control and Prevention (IVDC) through a combination of Sanger sequencing, Illumina sequencing, and nanopore sequencing. Three distinct strains have been identified, the virus has been designated as 2019-nCoV, and the disease has been subsequently named novel coronavirus-infected pneumonia (NCIP).
Phylogenetic analysis was conducted to determine the relationship between 2019-nCoVs and other sequences under the Orthocoronavirinae subfamily using MAFFT v7.455 (Figure 1) (2), and maximum likelihood inference was calculated using PhyML v3.3 (3), employing the GTR + I + Γ model of nucleotide substitution, and 1,000 bootstrap replicates. The 2019-nCoVs have features typical of the coronavirus family and were placed in the Betacoronavirus 2b lineage. Alignment of these strains’ full genomes and other available genomes of Betacoronavirus showed the closest relationship with Bat SARS-like coronavirus isolate bat-SL-CoVZC45 (Accession Number: MG772933.1) (Identity 87.99%). The typical crown-like particles of the 2019-nCoVs can be observed under transmission electron microscope (TEM) with negative staining ( www.gisaid.org). The origin of the 2019-nCoVs is still being investigated. However, all current evidence points to wild animals sold illegally in the Huanan Seafood Wholesale Market.
Figure 1.
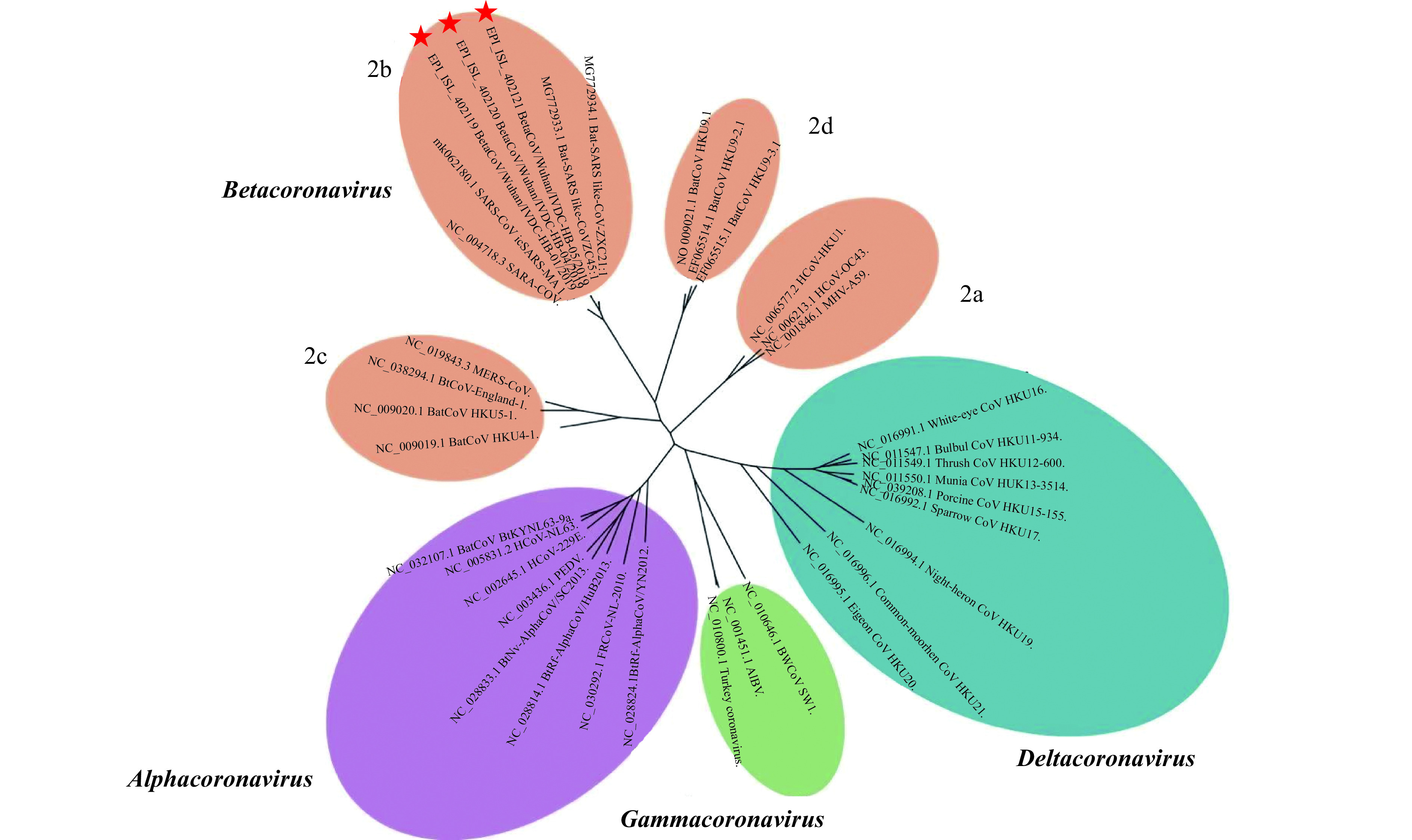
Phylogenetic relationships between the genomes of the new types of Betacoronavirus and other Orthocoronavirinae genomes. The viruses in the subfamily Orthocoronavirinae were classified into four genera (prototype or Refseq strains shown): Alphacoronavirus (purple), Betacoronavirus (orange), Gammacoronavirus (green), and Deltacoronavirus (blue). Classic subgroup clusters for the Betacoronavirus were labelled 2a–2d. The tree was based on complete genomes shown above using the maximum likelihood method under the GTR + I + Γ model of nucleotide substitution as implemented in PhyML. The new types of Betacoronavirus, labelled with red stars, were placed into the lineage of Betacoronavirus 2b, which contain the following: avian infectious bronchitis virus (AIBV), Middle East respiratory syndrome coronavirus (MERS-CoV), mouse hepatitis virus (MHV), porcine enteric diarrhea virus (PEDV), severe acute respiratory syndrome coronavirus (SARS-CoV), SARS-related coronavirus (SARSr-CoV), and Human coronavirus (HCoV).
Several complete genome sequences of 2019-nCoVs were successfully obtained and released recently via www.gisaid.org to provide a first look at the molecular characteristics of this emerging pathogen, and all related information has also been reported to the World Health Organization (WHO). Several rapid and sensitive detection tests have been developed by China CDC and will be applied to the prevention and control of this 2019-nCoV outbreak.
Data availability. The new Betacoronavirus genome sequence has been deposited in GISAID ( www.gisaid.org) under the accession number EPI_ISL_402119, EPI_ISL_ 402020 and EPI_ISL_402121.
Contributor Information
Wenjie Tan, Email: tanwj@ivdc.chinacdc.cn.
Guizhen Wu, Email: wugz@ivdc.chinacdc.cn.
References
- 1.George F. Gao From “A”IV to “Z”IKV: attacks from emerging and Re-emerging pathogens. Cell. 2018;172(6):1157–9. doi: 10.1016/j.cell.2018.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Katoh K, Standley DM MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30(4):772–80. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guindon S, Gascuel O A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52(5):696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]