

Abstract
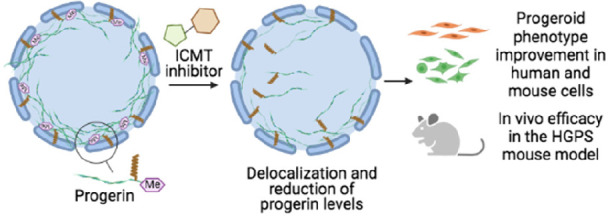
Hutchinson–Gilford progeria syndrome (HGPS, progeria) is a rare genetic disease characterized by premature aging and death in childhood for which there were no approved drugs for its treatment until last November, when lonafarnib obtained long-sought FDA approval. However, the benefits of lonafarnib in patients are limited, highlighting the need for new therapeutic strategies. Here, we validate the enzyme isoprenylcysteine carboxylmethyltransferase (ICMT) as a new therapeutic target for progeria with the development of a new series of potent inhibitors of this enzyme that exhibit an excellent antiprogeroid profile. Among them, compound UCM-13207 significantly improved the main hallmarks of progeria. Specifically, treatment of fibroblasts from progeroid mice with UCM-13207 delocalized progerin from the nuclear membrane, diminished its total protein levels, resulting in decreased DNA damage, and increased cellular viability. Importantly, these effects were also observed in patient-derived cells. Using the LmnaG609G/G609G progeroid mouse model, UCM-13207 showed an excellent in vivo efficacy by increasing body weight, enhancing grip strength, extending lifespan by 20%, and decreasing tissue senescence in multiple organs. Furthermore, UCM-13207 treatment led to an improvement of key cardiovascular hallmarks such as reduced progerin levels in aortic and endocardial tissue and increased number of vascular smooth muscle cells (VSMCs). The beneficial effects go well beyond the effects induced by other therapeutic strategies previously reported in the field, thus supporting the use of UCM-13207 as a new treatment for progeria.
Short abstract
Isoprenylcysteine carboxylmethyltransferase (ICMT) inhibitor induces progerin delocalization from the nuclear rim and decreases its levels, significantly improving the main hallmarks of progeria.
Introduction
Hutchinson–Gilford progeria syndrome (HGPS), or progeria, is an extremely rare pediatric disorder, characterized by premature aging and death at an average age of 14.6 years.1−3 The molecular cause of HGPS is an autosomal spontaneous dominant point mutation in the LMNA gene (encoding nuclear A-type lamins)4,5 that leads to the synthesis of progerin, a permanently farnesylated and methylated truncated version of prelamin A.6 This mutant protein accumulates abnormally in the inner nuclear membrane, causing the multiple and characteristic cellular and organismal alterations of the disease.1,2
In November 2020, lonafarnib (Zokinvy), an inhibitor of the enzyme farnesyltransferase aimed at decreasing progerin farnesylation, received the landmark approval by the US Food and Drug Administration (FDA), becoming the first (and only) approved drug for treating progeria. However, the outcome of the lonafarnib clinical trial showed limited benefits,7 possibly due to alternative prenylation of prelamin A and progerin by geranylgeranyl transferase upon farnesyltransferase inhibition.8 Yet, treatment of HGPS children with combination therapies inhibiting both prenylation pathways did not seem to improve the benefits achieved with lonafarnib monotherapy.9 In addition, it is possible that other targets of farnesyltransferase, apart from progerin, are affected by lonafarnib, eliciting adverse effects in progeroid patients. In line with this concern, since lonafarnib was originally developed for the treatment of Ras-dependent tumors,10 it is antiproliferative, a feature that can potentially limit its positive effects on progeroid cells, in which pro-proliferative effects are needed. Alternative approaches in HGPS animal models have explored the potential use of N-acetyltransferase 10 (NAT10) inhibitors,11 CRISPR/Cas9-based therapy,12,13 and microbiome-based interventions,14 but these strategies cannot as yet be easily translated to humans. Finally, a moderate extension of longevity (by 12%) has been reported in the progerin-expressing LmnaG609G/G609G mouse model after increasing the levels of ATP and pyrophosphate, but the relevance of this finding to human HGPS remains to be addressed.15 Hence, there is a clear and urgent need for better therapeutic strategies for treatment of HGPS. Building on the findings that genetic blockade of the enzyme isoprenylcysteine carboxylmethyltransferase (ICMT) ameliorated disease phenotype in the Zmpste24-null mouse model of progeria and in HGPS human cells,16 we hypothesized that pharmacological inhibitors of ICMT may be beneficial drugs for progeroid patients. Specifically, we considered that small molecule inhibitors of ICMT, an approach previously not explored in progeria, could avoid the carboxymethylation of progerin and consequently induce its delocalization from the nuclear membrane and as such decrease its accumulation, leading to an improvement of HGPS progression. However, the lack of noncytotoxic and bioavailable ICMT inhibitors have to date precluded the therapeutic validation of such a therapeutic strategy. Of note, while the present work was under review, the ability of a previously described ICMT inhibitor to delay senescence of HGPS cells was reported.17 However, the low bioavailability of this compound has hampered its in vivo assessment. Here, we describe the development of a new potent and selective ICMT inhibitor (compound 21, UCM-13207) and demonstrate its ability to significantly improve fundamental progeroid features in vitro and in vivo, including increased survival in LmnaG609G/G609G mice. Collectively, our findings indicate that small molecule inhibition of ICMT could be an effective therapy for progeria. Furthermore, as progeria can be considered as an accelerated model of physiological senescence, our findings could have implications for aging-related conditions, one of the major challenges nowadays.18
Results and Discussion
Design and Synthesis of New ICMT Inhibitors
We have previously carried out an extensive medicinal chemistry program aimed at the development of new ICMT inhibitors.19,20 Based on this work and using the key criteria of potency (80% inhibition at 50 μM) and low cellular toxicity (cellular viability >80% at 10 μM), we selected a new compound (1) as our initial hit. Our findings suggested that the 3-amino-N-phenylpropanamide moiety as well as the n-octyl chain of compound 1 were key elements for interaction with ICMT.20 Accordingly, we envisioned that the phenyl group could be further explored for optimizing cellular viability without significantly compromising ICMT inhibition (Figure 1a). This focused library was synthesized as detailed in the Supporting Information. In all cases, the obtained structural data were in agreement with the proposed structures (see the Supporting Information for details). All new compounds were screened for ICMT inhibition. In addition, those compounds that blocked >60% of ICMT activity were assayed for cellular viability in mouse wild-type and progerin-expressing fibroblasts (LmnaG609G/G609G) (Supporting Table S1). All compounds that showed the strongest capacity to enhance the viability of progeroid fibroblasts (values >70% at 2 μM) were selected for further biological assays with the exception of the furan-containing derivative 13. The reason for the latter was the narrow efficacy window observed for this compound when tested at a higher concentration (Supporting Figure S1).
Figure 1.

New ICMT inhibitors improve progeria hallmarks in mouse and human HGPS cells. (a) Design of new ICMT inhibitors. (b) Growth curves of Lmna+/+ and LmnaG609G/G609G mice fibroblasts incubated with 0.1% DMSO (vehicle, veh) or ICMT inhibitors at 10 μM over 14 days (n ≥ 4 independent experiments; two-tailed Student′s t test). (c) Growth curves of human wild type (WT) or HGPS fibroblasts incubated with vehicle or different ICMT inhibitors at 2 μM over 24 days (n ≥ 4 independent experiments; two-tailed Student′s t test). (d) Immunofluorescence images of human WT or HGPS fibroblasts treated with vehicle or different ICMT inhibitors at 2 μM for 17 days and stained with antiprogerin (green) and Hoechst (blue). Magnification shows subnuclear distribution of progerin, that was delocalized from the nuclear rim toward the nucleoplasm (1, quantification in middle plot) and/or decreased (2, quantification in right plot) after treatment with ICMT inhibitors (at least 47 cells quantified per condition from ≥2 independent experiments; two-tailed Student′s t test). Scale bar: 20 μm. (e) Immunoblot of progerin from Lmna+/+ or LmnaG609G/G609G mouse fibroblasts incubated with vehicle or ICMT inhibitors (2 μM) for 14 days. Protein levels were normalized against total GAPDH (n ≥ 3 independent experiments; plot shows mean ± sem, two-tailed Student′s t test). (f) Immunoblot and quantification of phosphorylated Akt (pAkt) and phosphorylated histone 2AX (pH2AX) from Lmna+/+ or LmnaG609G/G609G mouse fibroblasts incubated with vehicle or ICMT inhibitors (2 μM) for 14 days. Protein levels were normalized against total Akt or GAPDH, respectively (n ≥ 3 independent experiments; plots show mean ± sem, two-tailed Student′s t test).
New ICMT Inhibitors Improve the Cellular Hallmarks of Progeria in Fibroblasts from LmnaG609G/G609G Mice and HGPS Patients
Next, we sought to confirm whether compounds 14, 17, and 21 could counteract some of the most characteristic molecular and cellular defects found in both progeroid mouse and human cells. This included growth arrest, progerin accumulation in the perinuclear rim, Akt inhibition, and increased level of phosphorylated histone 2AX (pH2AX). We found that the three compounds augmented the proliferation rate of progeroid mouse cells (Figure 1b) and, in particular, incubation with compound 21 increased population doubling values almost to the level of Lmna+/+ cells. As the LmnaG609G/G609G mouse cells are SV40LT immortalized cells, they are p53 and Rb deficient, and therefore do not undergo senescence. Hence, to rule out the effects of cell immortalization in our results, we confirmed a significant positive proliferative effect of the compounds in primary HGPS human cells (Figure 1c). To further corroborate that the effects regarding the proliferation rate were due to the specific inhibition of ICMT activity, we knocked down its expression and studied the effects of compound 21. As expected, the effect of the inhibitor in the proliferation rate of progeroid mouse fibroblasts was absent in siRNA-induced ICMT knockout cells, indicating that the beneficial effects exerted by compound 21 are selectively elicited by ICMT inhibition (Supporting Figure S2). Under these conditions, we did not observe a significant effect of the siRNA alone on increasing cellular viability. It is possible that the time point (72 h) and the cells used (immortalized progeroid mouse fibroblasts) are not optimal for observing the direct siRNA effects, which may require longer times to become phenotypically evident, as siRNA and small molecule effect may differ in the extension of the observed effect or the optimal window time. In support of this, published experiments with short hairpin (sh) RNAs of ICMT conducted in human progeroid cells have shown that at least 20 days are needed to observe the effect of the ICMT shRNA alone on cellular viability.16
Consistent with their ICMT inhibitory activity, all three compounds induced a significant delocalization of progerin from the nuclear rim in human HGPS cells (Figure 1d, left plot). This effect was accompanied by a decrease in total levels of progerin, as shown by both immunofluorescence experiments using human HGPS cells (Figure 1d, right plot) and by Western blot analysis using mouse progeroid cells (Figure 1e and Supporting Figure S3).
Furthermore, and consistent with the increase in cellular proliferation, phospho-Akt levels were higher in treated progeroid cells (Figure 1f). In addition, the phosphorylated levels of histone H2AX, a marker of nuclear damage associated with aging,21 were also significantly reduced in the presence of compounds 14, 17, and 21. Finally, no significant effect was observed in the number of misshapen nuclei in cells treated with these compounds (Supporting Figure S4). This is in agreement with the results reported in the ICMT genetically deficient mouse model.16 Taken together, these results demonstrate that the new ICMT inhibitors described here significantly ameliorate the main cellular hallmarks of HGPS in progeroid fibroblasts from both LmnaG609G/G609G mice and HGPS patients, suggesting their potential for the treatment of the disease.
Selection of Compound 21 (UCM-13207) for In-Depth Studies
Among the three tested compounds, derivative 21 (UCM-13207) systematically induced the strongest effect in all the functional assays and showed a good IC50 value of 1.4 μM (Supporting Figure S5) in the ICMT in vitro assay. Compound 21 was also able to inhibit the ICMT activity in whole cells, as confirmed by prelamin accumulation in Lmna+/+ cells (Supporting Figure S6), accumulation of carboxymethylated acetylfarnesylcysteine (the minimal substrate recognized by ICMT), and Ras mislocalization in LmnaG609G/G609G cells (Supporting Figure S7). Hence, it was selected for further exploration of its toxicity-efficacy window in vitro and key pharmacokinetic parameters before extending its evaluation to in vivo efficacy assessment using a mouse model of progeria. UCM-13207 did not cause appreciable cellular toxicity at least up to 10 μM (Figure 2a), whereas its efficacy to preserve the proliferation of progeroid cells was dose-dependent, with 2 μM showing the maximal effect (Figure 2b). Consistent with these results, compound 21 at 2 and 10 μM induced a decrease in progerin level (Figures 2c and 1d and e) without significantly affecting levels of wild-type lamin A and lamin C (Supporting Figure S8). The decrease in progerin levels seemed to be mediated by the proteasome pathway since blocking its activity with the specific inhibitor MG-132 reversed the effects of compound 21, whereas treatment with the lysosome pathway inhibitor bafilomycin A did not significantly affect progerin downregulation induced by compound 21 (Figure 2d). Cycloheximide turnover studies indicated the ability of the compound to exert a reduction in half-life of progerin (Supporting Figure S9). The decrease in total levels of progerin was not initially expected, since the effect of an ICMT inhibitor should be mainly the mislocalization of progerin from the nuclear rim and increased progerin levels have been reported in HGPS cells treated with C75, another ICMT inhibitor.17 However, our results consistently showed decreased progerin levels both in human (Figure 1d) and mouse cells (Figures 1e, 2c, 2d, and Supporting Figure S3) treated with derivative 21. We hypothesize that the extent of progerin mislocalization produced by the compound (around 36% for derivative 21 compared to vehicle as shown in Figure 1d) may be an important factor that contributes to decrease stability of progerin and to make it more prone to proteasome degradation. This effect has been observed for other inhibitors of the interaction between progerin and lamin A/C, such as compounds JH4 and SLC-DO11.22,23 Both compounds induce a reduction in progerin levels that is reversed in the presence of a proteasome inhibitor, and a decrease in the half-life of progerin in experiments using cycloheximide. These results are in line with the findings reported here.
Figure 2.

Compound 21 ameliorates the progeria phenotype in mouse and human HGPS fibroblasts. (a) Lmna+/+ fibroblasts were incubated with compound 21 at different concentrations for 72 h, and viability was determined by an MTT assay (n ≥ 4 independent experiments; one-way ANOVA). (b) Lmna+/+ or LmnaG609G/G609G fibroblasts were incubated with compound 21 for 72 h at different concentrations, and viability was assessed by MTT assay (n ≥ 4 independent experiments; one-way ANOVA). (c) Cropped immunoblot of lamin A, progerin, and lamin C from Lmna+/+ or LmnaG609G/G609G fibroblasts incubated with 0.1% DMSO (vehicle, veh) or compound 21 at 2 μM for 14 days (n ≥ 3 independent experiments; plot shows progerin level ± sem, two-tailed Student’s t test). (d) Cropped immunoblot and quantification of lamin A, progerin, and lamin C from Lmna+/+ or LmnaG609G/G609G fibroblasts. Cells were incubated with vehicle or compound 21 at 2 μM for 14 days and then bafilomycin A (24 h at 25 nM) or MG-132 (5 h at 5 μM) was added when indicated (n = 3 independent experiments; plot shows mean ± sem, one-way ANOVA). (e) Cropped immunoblot and quantification of phosphorylated Akt (pAkt) and phosphorylated histone 2AX (pH2AX) from wild type (WT) or HGPS human fibroblasts incubated with vehicle or compound 21 at 2 μM for 10 days. Protein levels were normalized against total Akt or GAPDH (n ≥ 3 independent experiments; plot shows mean ± sem, one-way ANOVA). (f) Human WT or HGPS fibroblasts were incubated with vehicle or compound 21 at 2 μM for 15 days. The activity of senescence-associated (SA) β-galactosidase was determined by incubating the cells with fluorescein di-β-d-galactopyranoside (FDG) for 24 h and measuring the resultant fluorescein production (n ≥ 3 independent experiments in triplicates; plot shows mean ± sem, two-tailed Student′s t test). (g) Lmna+/+ C57BL/6 mice (n = 2 females; n = 2 males per time point) were treated with compound 21 (intraperitoneal injection, 40 mg/kg), blood was extracted at different time points, and the compound concentration in serum was determined by high-performance liquid chromatography coupled to mass spectrometry.
Moreover, human HGPS fibroblasts treated with compound 21 at 2 μM significantly increased phospho-Akt, reduced phospho-H2AX levels (Figure 2e) and diminished the levels of senescence-associated (SA) β-galactosidase activity (Figure 2f), a biomarker of senescent cells.24
With respect to the pharmacokinetic parameters, we first determined the cell culture and serum stability, the intrinsic clearance, and the serum free drug or fraction unbound (Fu) of the compound. Our in vitro stability results indicated excellent values for the half-life of compound 21 in both human and mouse cell culture media as well as in human serum (t1/2 > 24 h), although a more moderate value was obtained in the case of mouse serum (t1/2 = 29 ± 2 min). The in vitro microsomal intrinsic clearance was 40 ± 6 μL/min/mg protein in human samples and 144 ± 29 μL/min/mg protein in mouse samples. These values are predictive of a medium and high in vivo clearance, respectively, but since the Fu of compound 21 was 0.02 (with a KD value for human serum albumin of 7.2 μM), this might reduce the metabolic clearance by the liver and, in turn, increase its half-life. Indeed, the in vivo half-life curve after intraperitoneal (ip) administration showed a significant and sustained concentration of the compound up to 6 h postadministration, reaching a maximum concentration close to 40 μM after 5 h (Figure 2g). In addition, signs of acute toxicity, tissue, or pathological damage were not detected at doses up to 80 mg/kg of compound 21.
Compound 21 (UCM-13207) Improved the Overall Phenotype of Progeroid LmnaG609G/G609G Mice and Improved Cardiac Tissue Condition
Having demonstrated the in vitro efficacy of compound 21 in mouse and human progeroid cells, we evaluated its therapeutic potential in the progeroid LmnaG609G/G609G mice, a model that recapitulates the majority of the alterations observed in HGPS patients.25−28
Remarkably, progeroid mice treated with the ICMT inhibitor 21 showed significantly improved body weight at all ages tested (Figures 3a,b) and increased survival (Figure 3c). Thus, the mean survival of mice treated with the compound was extended to 173 days compared to 134 days of the vehicle treated mice (P = 0.0001, Figure 3c). Furthermore, the maximum survival increased from 164 to 194 days while the minimum survival from 110 to 158 days between untreated and treated animals, respectively. The beneficial effects of compound 21 were observed in both males and females (Supporting Figures S10–S12). Moreover, no signs of toxicity were observed in wild-type mice receiving the same dose of compound (Supporting Figure S13). Encouraged by these results, we explored other phenotypic features characteristic of the LmnaG609G/G609G progeria model such as reduced serum glucose levels, reduced grip strength, bone defects, and marked involutions of thymus and spleen.25 Treatment of progeroid mice with compound 21 increased serum glucose levels and grip strength close to the values observed in wild-type controls (Figures 3d, e). Additionally, administration of the compound slightly improved lordokyphosis (abnormal convexity in the curvature of the spine when viewed from the side), increased spleen size (Figures 3f, g), and significantly increased the size of the thymus (Figure 3g).
Figure 3.

Improvement of the progeroid phenotype in vivo upon treatment with compound 21. (a) Representative photograph of a 3-month-old Lmna+/+ mouse, a LmnaG609G/G609G mouse, and a LmnaG609G/G609G mouse treated with compound 21 (40 mg/kg). (b) Body weight versus age plot of LmnaG609G/G609G mice treated with vehicle (n = 6) or with compound 21 (n = 9) (plot shows mean ± sem, two-tailed Student′s t test, p-value * < 0.05, ** < 0.005, *** < 0.0005). (c) Kaplan–Meier survival plot showing a significant increase in life span in LmnaG609G/G609G mice treated with compound 21 (n = 9) compared with LmnaG609G/G609G mice treated with vehicle (n = 9) (P = 0.0001, log-rank/Mantel–Cox test). (d) Glycemia in Lmna+/+ and LmnaG609G/G609G mice treated with compound 21 or vehicle (n = 6 per condition). Data are represented by box plots, and whiskers are minimum to maximum values (one-way ANOVA followed by Bonferroni–Holm post hoc test, n.s. non significant P = 0.11). (e) Grip strenght in Lmna+/+ and LmnaG609G/G609G mice treated with compound 21 or vehicle (n = 5 per condition, one-way ANOVA followed by Bonferroni–Holm post hoc test, n.s. non significant P = 0.107). (f) Magnetic resonance imaging of a 3-month-old Lmna+/+ mouse, a vehicle-treated LmnaG609G/G609G mouse, and a compound 21-treated LmnaG609G/G609G mouse. Note that the marked curvature of the spine called lordokyphosis, a key feature of progeria, is reduced upon treatment with compound 21. The plot represents the average of inner column angle (yellow arrowhead) of ≥4 mice per condition (one-way ANOVA). (g) Representative photographs of thymus and spleen from a 3-month-old Lmna+/+ mouse, a vehicle-treated LmnaG609G/G609G mouse, and a compound 21-treated LmnaG609G/G609G mouse. Plots represent average of thymus or spleen weight from ≥3 mice per condition (one-way ANOVA followed by Bonferroni–Holm post hoc test, n.s. non significant P = 0.17).
Cardiovascular cells and tissues are major targets of progerin in animal models and in humans, being cardiovascular problems the main cause of death in progeria patients.2,27,29−31 As such, specific decrease of progerin accumulation in cardiovascular tissues could be critical in maintaining the cardiovascular wellbeing. In line with this notion, treatment of LmnaG609G/G609G mice with compound 21 substantially reduced progerin expression and increased the number of vascular smooth muscle cells (VSMCs) in the aortic arch (Figure 4a). Progerin levels were also decreased in endocardial tissue (Figure 4b), although the reduction was less evident in arterioles (Supporting Figure S15). Nonetheless, these results are relevant considering that progerin expression in VSMCs has been described as the main cause of vessel contraction impairment observed in HGPS32 and that VSMCs loss is an important hallmark of vascular disease in HGPS.27 Accordingly, specific decrease of progerin in aortic arch and endocardial tissue, together with an increase in the number of VSMCs, highlights that compound 21 is able to correct some of the critical primary causes of the cardiovascular complications that lead to the early death of HGPS patients. Importantly, compound 21 decreased fibrosis and microvascular cell loss in the heart of progeroid mice (Supporting Figures S14 and S15). In addition, treatment of mice with compound 21 led to an improvement in global tissue senescence in other organs such as liver and kidney (Supporting Figure S16) as assessed by quantification of the levels of SA β-galactosidase activity.
Figure 4.

Reduced progerin expression in progeric LmnaG609G/G609G mice treated with compound 21. Mice of the indicated genotypes were treated with compound 21 or vehicle, starting at 6 weeks of age, and were sacrificed at 12 weeks of age. Cross sections of aortic arch and heart were stained with antiprogerin antibody (white) and Hoechst 33342 to visualize nuclei (blue). Representative immunofluorescence images are shown. (a) Aortic arch. Scale bar: 100 μm (entire image) and 20 μm (magnified view). L, lumen; m, media; adv, adventitia. The left plot shows mean ± sem number of VSMCs in the medial layer of the aortic arch (n = 4 animal per condition, 2 sections per animal). The right plot shows mean ± desvest of progerin signal intensity in VSMC from the medial layer of the aortic arch (n = 646 nuclei for LmnaG609G/G609G treated with vehicle and 532 nuclei for LmnaG609G/G609G treated with compound 21, from 4 animals per condition, two-tailed Student′s t test). (b) Endocardial tissue. The bottom plot shows mean ± desvest of progerin signal intensity (n = 400 nuclei for LmnaG609G/G609G treated with vehicle and 412 nuclei for LmnaG609G/G609G treated with compound 21, from 5 animals per condition, two-tailed Student′s t test). Scale bar: 25 μm.
Conclusion
Here, we report a novel strategy to reduce the anomalous accumulation of progerin in the nuclear rim membrane, which is considered to be the main cause of the fatal phenotype developed by HGPS patients. Specifically, we show that compound 21 (UCM-13207), a new ICMT inhibitor, significantly induces the mislocalization of progerin and reduces its protein expression level, leading to a substantial overall amelioration of the cellular hallmarks of progeria in both mouse and human cells. Importantly, treatment with compound 21 significantly improved the phenotype of progeroid LmnaG609G/G609G mice, including lifespan extension in both males and females. Collectively, these findings have important clinical implications regarding the validation of the clinical relevance of ICMT as a promising therapeutic target for progeria treatment, providing a new pharmacological strategy in a field dramatically devoid of validated therapeutic targets beyond farnesyltransferase. Furthermore, our results pave the way for targeting ICMT as a new pharmacological strategy for succeeding in the long sought objective of making a meaningful difference to the lives of HGPS patients.
Materials and Methods
Safety statement
No unexpected or unusually high safety hazards were encountered.
Compound Syntheses
The synthesis and structural characterization of compound 21 (UCM-13207) is detailed below. Full details regarding the synthetic procedures and characterization data of all compounds are given in the Supporting Information.
Synthesis of N2-Octyl-N1-phenyl-N2-(pyridin-2-ylcarbonyl)-β-alaninamide (21, UCM-13207)
To a solution of picolinic acid (133 mg, 1.1 mmol) in anhydrous DCM (4 mL/mmol), EDC (207 mg, 1.1 mmol) and HOBt (146 mg, 1.1 mmol) were added. The reaction mixture was stirred at rt for 1 h. Then, a solution of N2-octyl-N1-phenyl-β-alaninamide (150 mg, 0.54 mmol) in anhydrous DCM (2.0 mL/mmol) was added and the reaction mixture was stirred at rt for 16 h. The reaction crude was washed with saturated aqueous solutions of NaHCO3 and NaCl, consecutively. The organic extracts were dried over Na2SO4, filtered, and the solvent was removed under reduced pressure. The residue was purified by column chromatography (hexane/EtOAc, 2:8) to yield compound 21 in 92% yield (189 mg).
Rf (hexane/EtOAc, 3:7): 0.30. IR (ATR, cm–1): 3309 (NH), 1685 (CON), 1615, 1547, 1495, 1443 (Ar). 1H NMR (CDCl3, δ): Amide rotamers A:B, 2:1; 0.85 (t, J = 6.7 Hz, 3H, CH3), 1.11–1.25 (m, 10H, (CH2)5CH3), 1.57 (m, 2H, CH2(CH2)5CH3), 2.81 (t, J = 6.4 Hz, 2H, CH2CO), 3.37 (t, J = 7.4 Hz, 2H, (CH2)6CH2N, rotamer A), 3.46–3.47 (m, 2H, (CH2)6CH2N, rotamer B), 3.69–3.75 (m, 2H, COCH2CH2N, rotamer B), 3.87 (t, J = 6.3 Hz, 2H, COCH2CH2N, rotamer A), 7.05 (t, J = 7.1 Hz, 1H, H4), 7.24–7.32 (m, 3H, H3, H5, H5′), 7.54–7.56 (m, 3H, H2, H6, H4′), 7.71–7.76 (m, 1H, H3′), 8.55 (d, J = 4.4 Hz, 1H, H6′), 8.64 (br s, 1H, NH, rotamer B), 8.92 (br s, 1H, NH, rotamer A). 13C NMR (CDCl3, δ): 14.0 (CH3), 22.6, 26.5, 28.8, 29.0 (2C), 31.7 ((CH2)6CH3), 36.5 (CH2CO), 43.2 (CH2N), 50.0 ((CH2)6CH2N), 119.9 (C2, C6), 123.1, 123.9, 124.4 (C4, C3′, C5′), 128.8 (C3, C5), 137.0 (C4′), 138.4 (C1), 148.5 (C6′), 154.5 (C2′), 169.5, 169.7 (CONH, CON). HRMS (ESI, m/z): Calculated for C23H31N3O2Na [M + Na]+: 404.2308. Found: 404.2276.
Determination of ICMT Activity
Synthesized compounds were tested for their ability to inhibit human ICMT activity using Sf9 membranes containing the recombinantly expressed enzyme. In this assay, a mixture of biotin-farnesyl-l-cysteine and tritiated S-adenosylmethionine in the presence or absence of the compound under study was prepared, and the reaction was initiated by the addition of the Sf9 membrane homogenates. The inhibitory capacity of the compounds was expressed as percentage of inhibition of the methyl esterification step, in which the tritiated methyl group of the methyl donor S-adenosylmethionine was transferred to the substrate biotin-farnesyl-l-cysteine as described previously,33 and the radioactivity incorporated was quantified.
Cell Lines and Culture
Progeroid mouse fibroblasts (LmnaG609/G609) and their wild-type counterparts were kindly donated by Prof. Carlos López Otín (Oviedo University, Spain). Cells were grown in Dulbecco’s modified eagle medium (DMEM, Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum (FBS, HyClone), 1% l-glutamine (Invitrogen), 1% sodium pyruvate (Invitrogen), 50 U/mL penicillin, and 50 μg/mL streptomycin (Invitrogen). Human progeroid or healthy fibroblasts (HGADFN167, HGADFN003, HGADFN143, and HGFDFN168) were obtained from The Progeria Research Foundation and cultured in 15% DMEM, 50 U/mL penicillin and 50 μg/mL streptomycin. All cells were incubated in a humidified atmosphere at 37 °C in the presence of 5% CO2.
Cell Viability and Proliferation Assays
The effect of the different compounds on the cellular viability was determined through standard MTT assays.34−36 Full details are given in the Supporting Information.
RNA Interference-Mediated Silencing of the ICMT Gene
Progeroid mouse fibroblasts (LmnaG609/G609) were transfected with an ICMT siRNA (m) or with a control siRNA commercially available from Santa Cruz Biotechnology (sc-146137 and sc-37007, respectively), using lipofectamine and following the manufacturer’s instructions. To determine cell viability, the MTT protocol indicated above was followed.
Immunoblot and Immunocytofluorescence Analysis
Western blot analysis was carried out as described previously.37−39 Full details are given in the Supporting Information.
Senescence-Associated (SA) β-Galactosidase Activity
SA β-galactosidase activity was determined by the FDG method.40 Full details are provided in the Supporting Information.
Stability Assays
Stability in cell culture medium, in mouse and human serum, and in mouse and human liver microsomes was assayed as detailed in the Supporting Information.
Human Serum Albumin (HSA) Binding Assay
Determination of the binding of the compound to HSA was performed by incubating a fixed concentration of the compound with different concentrations of immobilized HSA, using the TRANSILXL HSA Binding Kit (TMP-0210-2096, Sovicell) as described in the Supporting Information.
Animal Experiments
All scientific procedures with animals were conformed to EU Directive 2010/63 EU and Recommendation 2007/526/EC, enforced in Spanish law under Real Decreto 53/2013. Animal protocols were approved by the Committee of Animal Experimentation of Universidad Complutense de Madrid and the Animal Protection Area of the Comunidad Autónoma de Madrid (PROEX 159/18). Animal studies were carried out in LmnaG609G/G609G knock-in mice ubiquitously expressing progerin25 and control Lmna+/+ littermates. Mice were maintained in the animal facility of the Universidad Complutense de Madrid under specific pathogen free conditions. Full details about the in vivo characterization and the pathological and immunohistofluorescence analysis are provided in the Supporting Information.
Statistics
All data were analyzed using GraphPad Prism 6 software. Data were presented as mean ± standard error of the media (SEM) with at least three biologically independent experiments. Representative morphological images were taken from at least three biologically independent experiments with similar results. The Student t test or one-way ANOVA were used for comparison between groups. Survival analysis was performed using the Kaplan–Meier method. Differences between survival distributions were analyzed using the log rank test. Hazard ratio and confidence interval were obtained by Mantel–Haenszel analysis. Differences with P < 0.05 were considered significant.
Acknowledgments
This work was supported by grants from The Progeria Research Foundation (PRF 2016-65) and the Spanish MINECO (PID2019-106279RB-I00, PID2019-108489RB-I00). The authors thank Fundación La Caixa (A.G.), CEI Moncloa (N.I.M.-R.), MINECO (F.J.O.-N. and M.B.) and Ministerio de Ciencia, Innovación y Universidades (N.K.-F.) for predoctoral fellowships. The authors thank C. López-Otín for kindly donating LmnaG609G/G609G progeroid and their corresponding wild-type fibroblasts and UCM’s CAIs Cytometry and Fluorescence Microscopy, Genomics, NMR, and Mass Spectrometry, for their assistance. The CNIC is supported by the Ministerio de Ciencia e Innovación, the Instituto de Salud Carlos III, and the pro-CNIC Foundation, and is a Severo Ochoa Center of Excellence (grant SEV-2015-0505). The generation of the antiprogerin antibody was funded by the Wellcome Trust (098291/Z/12/Z to S.N.).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.0c01698.
Supporting Table S1, Supporting Figures showing the effect of compounds in the viability of Lmna+/+ or LmnaG609G/G609G mouse fibroblasts; lack of effect of compound 21 in the viability of siRNA-induced silencing of ICMT LmnaG609G/G609G mouse fibroblasts, on misshapen nuclei, and in lamin A or lamin C levels; concentration–response curve for ICMT inhibition of compound 21; effect of compound 21 in the total levels of progerin, in prelamin accumulation in Lmna+/+ cells, in Ras mislocalization, and in methylated acetylfarnesylcysteine accumulation in LmnaG609G/G609G cells; half-life of progerin in the presence of cycloheximide; size, body weight, and survival improvement in male and female progeroid mice after administration of compound 21; lack of in vivo toxicity of compound 21 in male and female mice; reduction of heart fibrosis and microvascular cell loss in progeroid mice treated with compound 21; SA β-galactosidase activity in kidney and liver of progeroid treated mice; detailed synthetic methods; full details for cell viability and proliferation assays, for immunoblot and immunocytofluorescence analysis, for SA β-galactosidase activity determination, for stability, and human serum albumin binding assay, for the in vivo studies, for the pathological and immunofluorescence analysis, and for the determination of the levels of methylated AFC in cells (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Gordon L. B.; Rothman F. G.; Lopez-Otin C.; Misteli T. Progeria: a paradigm for translational medicine. Cell 2014, 156, 400–407. 10.1016/j.cell.2013.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorado B.; Andres V. A-type lamins and cardiovascular disease in premature aging syndromes. Curr. Opin. Cell Biol. 2017, 46, 17–25. 10.1016/j.ceb.2016.12.005. [DOI] [PubMed] [Google Scholar]
- Merideth M. A.; Gordon L. B.; Clauss S.; Sachdev V.; Smith A. C.; Perry M. B.; Brewer C. C.; Zalewski C.; Kim H. J.; Solomon B.; Brooks B. P.; Gerber L. H.; Turner M. L.; Domingo D. L.; Hart T. C.; Graf J.; Reynolds J. C.; Gropman A.; Yanovski J. A.; Gerhard-Herman M.; Collins F. S.; Nabel E. G.; Cannon R. O. 3rd; Gahl W. A.; Introne W. J. Phenotype and course of Hutchinson-Gilford progeria syndrome. N. Engl. J. Med. 2008, 358, 592–604. 10.1056/NEJMoa0706898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson M.; Brown W. T.; Gordon L. B.; Glynn M. W.; Singer J.; Scott L.; Erdos M. R.; Robbins C. M.; Moses T. Y.; Berglund P.; Dutra A.; Pak E.; Durkin S.; Csoka A. B.; Boehnke M.; Glover T. W.; Collins F. S. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 2003, 423, 293–298. 10.1038/nature01629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Sandre-Giovannoli A.; Bernard R.; Cau P.; Navarro C.; Amiel J.; Boccaccio I.; Lyonnet S.; Stewart C. L.; Munnich A.; Le Merrer M.; Levy N. Lamin A truncation in Hutchinson-Gilford progeria. Science 2003, 300, 2055. 10.1126/science.1084125. [DOI] [PubMed] [Google Scholar]
- Davies B. S.; Fong L. G.; Yang S. H.; Coffinier C.; Young S. G. The posttranslational processing of prelamin A and disease. Annu. Rev. Genomics Hum. Genet. 2009, 10, 153–174. 10.1146/annurev-genom-082908-150150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon L. B.; Shappell H.; Massaro J.; D’Agostino R. B. Sr.; Brazier J.; Campbell S. E.; Kleinman M. E.; Kieran M. W. Association of lonafarnib treatment vs no treatment with mortality rate in patients with Hutchinson-Gilford progeria syndrome. Jama 2018, 319, 1687–1695. 10.1001/jama.2018.3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varela I.; Pereira S.; Ugalde A. P.; Navarro C. L.; Suarez M. F.; Cau P.; Cadinanos J.; Osorio F. G.; Foray N.; Cobo J.; de Carlos F.; Levy N.; Freije J. M.; Lopez-Otin C. Combined treatment with statins and aminobisphosphonates extends longevity in a mouse model of human premature aging. Nat. Med. 2008, 14, 767–772. 10.1038/nm1786. [DOI] [PubMed] [Google Scholar]
- Gordon L. B.; Kleinman M. E.; Massaro J.; D’Agostino R. B. Sr.; Shappell H.; Gerhard-Herman M.; Smoot L. B.; Gordon C. M.; Cleveland R. H.; Nazarian A.; Snyder B. D.; Ullrich N. J.; Silvera V. M.; Liang M. G.; Quinn N.; Miller D. T.; Huh S. Y.; Dowton A. A.; Littlefield K.; Greer M. M.; Kieran M. W. Clinical trial of the protein farnesylation inhibitors lonafarnib, pravastatin, and zoledronic acid in children with Hutchinson-Gilford progeria syndrome. Circulation 2016, 134, 114–125. 10.1161/CIRCULATIONAHA.116.022188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin-Ramos N. I.; Ortega-Gutierrez S.; Lopez-Rodriguez M. L. Blocking Ras inhibition as an antitumor strategy. Semin. Cancer Biol. 2019, 54, 91–100. 10.1016/j.semcancer.2018.01.017. [DOI] [PubMed] [Google Scholar]
- Balmus G.; Larrieu D.; Barros A. C.; Collins C.; Abrudan M.; Demir M.; Geisler N. J.; Lelliott C. J.; White J. K.; Karp N. A.; Atkinson J.; Kirton A.; Jacobsen M.; Clift D.; Rodriguez R.; Adams D. J.; Jackson S. P. Targeting of NAT10 enhances healthspan in a mouse model of human accelerated aging syndrome. Nat. Commun. 2018, 9, 1700. 10.1038/s41467-018-03770-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago-Fernandez O.; Osorio F. G.; Quesada V.; Rodriguez F.; Basso S.; Maeso D.; Rolas L.; Barkaway A.; Nourshargh S.; Folgueras A. R.; Freije J. M. P.; Lopez-Otin C. Development of a CRISPR/Cas9-based therapy for Hutchinson-Gilford progeria syndrome. Nat. Med. 2019, 25, 423–426. 10.1038/s41591-018-0338-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyret E.; Liao H. K.; Yamamoto M.; Hernandez-Benitez R.; Fu Y.; Erikson G.; Reddy P.; Izpisua-Belmonte J. C. Single-dose CRISPR-Cas9 therapy extends lifespan of mice with Hutchinson-Gilford progeria syndrome. Nat. Med. 2019, 25, 419–422. 10.1038/s41591-019-0343-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcena C.; Valdes-Mas R.; Mayoral P.; Garabaya C.; Durand S.; Rodriguez F.; Fernandez-Garcia M. T.; Salazar N.; Nogacka A. M.; Garatachea N.; Bossut N.; Aprahamian F.; Lucia A.; Kroemer G.; Freije J. M. P.; Quiros P. M.; Lopez-Otin C. Healthspan and lifespan extension by fecal microbiota transplantation into progeroid mice. Nat. Med. 2019, 25, 1234–1242. 10.1038/s41591-019-0504-5. [DOI] [PubMed] [Google Scholar]
- Villa-Bellosta R. ATP-based therapy prevents vascular calcification and extends longevity in a mouse model of Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 23698–23704. 10.1073/pnas.1910972116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim M. X.; Sayin V. I.; Akula M. K.; Liu M.; Fong L. G.; Young S. G.; Bergo M. O. Targeting isoprenylcysteine methylation ameliorates disease in a mouse model of progeria. Science 2013, 340, 1330–1333. 10.1126/science.1238880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Yao H.; Kashif M.; Revêchon G.; Eriksson M.; Hu J.; Wang T.; Liu Y.; Tüksammel E.; Strömblad S.; Ahearn I. M.; Philips M. R.; Wiel C.; Ibrahim M. X.; Bergo M. O. A small-molecule ICMT inhibitor delays senescence of Hutchinson-Gilford progeria syndrome cells. eLife 2021, 10.7554/eLife.63284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertozzi C. R.; Chang C. J.; Davis B. G.; Olvera de la Cruz M.; Tirrell D. A.; Zhao D. Grand challenges in chemistry for 2016 and beyond. ACS Cent. Sci. 2016, 2, 1–3. 10.1021/acscentsci.6b00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Rodríguez M. L.; Ortega-Gutiérrez S.; Martín-Fontecha M.; Balabasquer M.; Ortega-Nogales F. J.; Marín-Ramos N. I.. Novel Inhibitors of the Enzyme Isoprenylcysteine Carboxyl Methyltransferase (ICMT). WO2014118418A1, 2014.
- Marin-Ramos N. I.; Balabasquer M.; Ortega-Nogales F. J.; Torrecillas I. R.; Gil-Ordonez A.; Marcos-Ramiro B.; Aguilar-Garrido P.; Cushman I.; Romero A.; Medrano F. J.; Gajate C.; Mollinedo F.; Philips M. R.; Campillo M.; Gallardo M.; Martin-Fontecha M.; Lopez-Rodriguez M. L.; Ortega-Gutierrez S. A potent isoprenylcysteine carboxylmethyltransferase (ICMT) inhibitor improves survival in Ras-driven acute myeloid leukemia. J. Med. Chem. 2019, 62, 6035–6046. 10.1021/acs.jmedchem.9b00145. [DOI] [PubMed] [Google Scholar]
- Ocampo A.; Reddy P.; Martinez-Redondo P.; Platero-Luengo A.; Hatanaka F.; Hishida T.; Li M.; Lam D.; Kurita M.; Beyret E.; Araoka T.; Vazquez-Ferrer E.; Donoso D.; Roman J. L.; Xu J.; Rodriguez Esteban C.; Nunez G.; Nunez Delicado E.; Campistol J. M.; Guillen I.; Guillen P.; Izpisua Belmonte J. C. In vivo amelioration of age-associated hallmarks by partial reprogramming. Cell 2016, 167, 1719–1733. 10.1016/j.cell.2016.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. J.; Jung Y. S.; Yoon M. H.; Kang S. M.; Oh A. Y.; Lee J. H.; Jun S. Y.; Woo T. G.; Chun H. Y.; Kim S. K.; Chung K. J.; Lee H. Y.; Lee K.; Jin G.; Na M. K.; Ha N. C.; Barcena C.; Freije J. M.; Lopez-Otin C.; Song G. Y.; Park B. J. Interruption of progerin-lamin A/C binding ameliorates Hutchinson-Gilford progeria syndrome phenotype. J. Clin. Invest. 2016, 126, 3879–3893. 10.1172/JCI84164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang S. M.; Yoon M. H.; Ahn J.; Kim J. E.; Kim S. Y.; Kang S. Y.; Joo J.; Park S.; Cho J. H.; Woo T. G.; Oh A. Y.; Chung K. J.; An S. Y.; Hwang T. S.; Lee S. Y.; Kim J. S.; Ha N. C.; Song G. Y.; Park B. J. Progerinin, an optimized progerin-lamin A binding inhibitor, ameliorates premature senescence phenotypes of Hutchinson-Gilford progeria syndrome. Commun. Biol. 2021, 4, 1. 10.1038/s42003-020-01540-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debacq-Chainiaux F.; Erusalimsky J. D.; Campisi J.; Toussaint O. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat. Protoc. 2009, 4, 1798–1806. 10.1038/nprot.2009.191. [DOI] [PubMed] [Google Scholar]
- Osorio F. G.; Navarro C. L.; Cadinanos J.; Lopez-Mejia I. C.; Quiros P. M.; Bartoli C.; Rivera J.; Tazi J.; Guzman G.; Varela I.; Depetris D.; de Carlos F.; Cobo J.; Andres V.; De Sandre-Giovannoli A.; Freije J. M.; Levy N.; Lopez-Otin C. Splicing-directed therapy in a new mouse model of human accelerated aging. Sci. Transl. Med. 2011, 3, 106ra107. 10.1126/scitranslmed.3002847. [DOI] [PubMed] [Google Scholar]
- Villa-Bellosta R.; Rivera-Torres J.; Osorio F. G.; Acín-Pérez R.; Enriquez J. A.; López-Otín C.; Andrés V. Defective extracellular pyrophosphate metabolism promotes vascular calcification in a mouse model of Hutchinson-Gilford progeria syndrome that is ameliorated on pyrophosphate treatment. Circulation 2013, 127, 2442–2451. 10.1161/CIRCULATIONAHA.112.000571. [DOI] [PubMed] [Google Scholar]
- Hamczyk M. R.; Villa-Bellosta R.; Gonzalo P.; Andres-Manzano M. J.; Nogales P.; Bentzon J. F.; Lopez-Otin C.; Andres V. Vascular smooth muscle-specific progerin expression accelerates atherosclerosis and death in a mouse model of Hutchinson-Gilford progeria syndrome. Circulation 2018, 138, 266–282. 10.1161/CIRCULATIONAHA.117.030856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Campo L.; Sanchez-Lopez A.; Salaices M.; von Kleeck R. A.; Exposito E.; Gonzalez-Gomez C.; Cusso L.; Guzman-Martinez G.; Ruiz-Cabello J.; Desco M.; Assoian R. K.; Briones A. M.; Andres V. Vascular smooth muscle cell-specific progerin expression in a mouse model of Hutchinson-Gilford progeria syndrome promotes arterial stiffness: Therapeutic effect of dietary nitrite. Aging Cell 2019, 18, e12936. 10.1111/acel.12936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamczyk M. R.; Villa-Bellosta R.; Quesada V.; Gonzalo P.; Vidak S.; Nevado R. M.; Andrés-Manzano M. J.; Misteli T.; López-Otín C.; Andrés V. Progerin accelerates atherosclerosis by inducing endoplasmic reticulum stress in vascular smooth muscle cells. EMBO Mol. Med. 2019, 11, e9736. 10.15252/emmm.201809736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osmanagic-Myers S.; Kiss A.; Manakanatas C.; Hamza O.; Sedlmayer F.; Szabo P. L.; Fischer I.; Fichtinger P.; Podesser B. K.; Eriksson M.; Foisner R. Endothelial progerin expression causes cardiovascular pathology through an impaired mechanoresponse. J. Clin. Invest. 2019, 129, 531–545. 10.1172/JCI121297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorado B.; Pløen G. G.; Barettino A.; Macías A.; Gonzalo P.; Andrés-Manzano M. J.; González-Gómez C.; Galán-Arriola C.; Alfonso J. M.; Lobo M.; López-Martín G. J.; Molina A.; Sánchez-Sánchez R.; Gadea J.; Sánchez-González J.; Liu Y.; Callesen H.; Filgueiras-Rama D.; Ibáñez B.; Sørensen C. B.; Andrés V. Generation and characterization of a novel knockin minipig model of Hutchinson-Gilford progeria syndrome. Cell Discovery 2019, 5, 16. 10.1038/s41421-019-0084-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Campo L.; Sánchez-López A.; González-Gómez C.; Andrés-Manzano M. J.; Dorado B.; Andrés V. Vascular smooth muscle cell-specific progerin expression provokes contractile impairment in a mouse model of Hutchinson-Gilford progeria syndrome that is ameliorated by nitrite treatment. Cells 2020, 9, 656. 10.3390/cells9030656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter-Vann A. M.; Baron R. A.; Wong W.; dela Cruz J.; York J. D.; Gooden D. M.; Bergo M. O.; Young S. G.; Toone E. J.; Casey P. J. A small-molecule inhibitor of isoprenylcysteine carboxyl methyltransferase with antitumor activity in cancer cells. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 4336–4341. 10.1073/pnas.0408107102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamo A. M.; Gonzalez-Vera J. A.; Rueda-Zubiaurre A.; Alonso D.; Vazquez-Villa H.; Martin-Couce L.; Palomares O.; Lopez J. A.; Martin-Fontecha M.; Benhamu B.; Lopez-Rodriguez M. L.; Ortega-Gutierrez S. Chemoproteomic approach to explore the target profile of GPCR ligands: Application to 5-HT1A and 5-HT6 receptors. Chem. - Eur. J. 2016, 22, 1313–1321. 10.1002/chem.201503101. [DOI] [PubMed] [Google Scholar]
- Marin-Ramos N. I.; Alonso D.; Ortega-Gutierrez S.; Ortega-Nogales F. J.; Balabasquer M.; Vazquez-Villa H.; Andradas C.; Blasco-Benito S.; Perez-Gomez E.; Canales A.; Jimenez-Barbero J.; Marquina A.; Del Prado J. M.; Sanchez C.; Martin-Fontecha M.; Lopez-Rodriguez M. L. New inhibitors of angiogenesis with antitumor activity in vivo. J. Med. Chem. 2015, 58, 3757–3766. 10.1021/jm5019252. [DOI] [PubMed] [Google Scholar]
- Marín-Ramos N. I.; Piñar C.; Vázquez-Villa H.; Martín-Fontecha M.; González Á.; Canales Á.; Algar S.; Mayo P. P.; Jiménez-Barbero J.; Gajate C.; Mollinedo F.; Pardo L.; Ortega-Gutiérrez S.; Viso A.; López-Rodríguez M. L. Development of a nucleotide exchange inhibitor that impairs Ras oncogenic signaling. Chem. - Eur. J. 2017, 23, 1676–1685. 10.1002/chem.201604905. [DOI] [PubMed] [Google Scholar]
- Hernandez-Torres G.; Cipriano M.; Heden E.; Bjorklund E.; Canales A.; Zian D.; Feliu A.; Mecha M.; Guaza C.; Fowler C. J.; Ortega-Gutierrez S.; Lopez-Rodriguez M. L. A reversible and selective inhibitor of monoacylglycerol lipase ameliorates multiple sclerosis. Angew. Chem., Int. Ed. 2014, 53, 13765–13770. 10.1002/anie.201407807. [DOI] [PubMed] [Google Scholar]
- Hernandez-Torres G.; Enriquez-Palacios E.; Mecha M.; Feliu A.; Rueda-Zubiaurre A.; Angelina A.; Martin-Cruz L.; Martin-Fontecha M.; Palomares O.; Guaza C.; Pena-Cabrera E.; Lopez-Rodriguez M. L.; Ortega-Gutierrez S. Development of a fluorescent bodipy probe for visualization of the serotonin 5-HT1A receptor in native cells of the immune system. Bioconjugate Chem. 2018, 29, 2021–2027. 10.1021/acs.bioconjchem.8b00228. [DOI] [PubMed] [Google Scholar]
- Turrado C.; Puig T.; García-Cárceles J.; Artola M.; Benhamú B.; Ortega-Gutiérrez S.; Relat J.; Oliveras G.; Blancafort A.; Haro D.; Marrero P. F.; Colomer R.; López-Rodríguez M. L. New synthetic inhibitors of fatty acid synthase with anticancer activity. J. Med. Chem. 2012, 55, 5013–5023. 10.1021/jm2016045. [DOI] [PubMed] [Google Scholar]
- Yang N. C.; Hu M. L. A fluorimetric method using fluorescein di-beta-D-galactopyranoside for quantifying the senescence-associated beta-galactosidase activity in human foreskin fibroblast Hs68 cells. Anal. Biochem. 2004, 325, 337–343. 10.1016/j.ab.2003.11.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.