

Abstract

Sialic acid-binding immunoglobulin-like lectins, also known as Siglecs, have recently been designated as glyco-immune checkpoints. Through their interactions with sialylated glycan ligands overexpressed on tumor cells, inhibitory Siglecs on innate and adaptive immune cells modulate signaling cascades to restrain anti-tumor immune responses. However, the elucidation of the mechanisms underlying these processes is just beginning. We find that when human natural killer (NK) cells attack tumor cells, glycan remodeling occurs on the target cells at the immunological synapse. This remodeling occurs through both the transfer of sialylated glycans from NK cells to target tumor cells and the accumulation of de novo synthesized sialosides on the tumor cells. The functionalization of NK cells with a high-affinity ligand of Siglec-7 leads to multifaceted consequences in modulating a Siglec-7-regulated NK-activation. At high levels of ligand, an enzymatically added Siglec-7 ligand suppresses NK cytotoxicity through the recruitment of Siglec-7 to an immune synapse, whereas at low levels of ligand an enzymatically added Siglec-7 ligand triggers the release of Siglec-7 from the cell surface into the culture medium, preventing a Siglec-7-mediated inhibition of NK cytotoxicity. These results suggest that a glycan engineering of NK cells may provide a means to boost NK effector functions for related applications.
Short abstract
With the formation of an immune synapse during an NK-based killing, a cancer cell upregulates its cell-surface sialylation level primarily via an accumulation of neosialylation, and its mediated immune inhibition can be blocked with a cis high-affinity ligand created chemoenzymatically, which resulted in a better tumor growth control.
Introduction
The development of immune checkpoint inhibitors for blocking the suppressive functions of cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed cell death protein 1 (PD-1) has offered curative hopes for many cancer patients.1−3 An effective checkpoint blockade results in a remarkable tumor regression, and clinically significant survival benefits for patients with a broad spectrum of advanced cancers have been observed clinically. However, more than 50% of cancer patients fail to respond to such treatments, and some initial responders eventually develop a resistance to these therapies with a relapsed disease.2 The mechanisms leading to such a resistance are varied, but they include the possibility of other immune checkpoints.4,5 Recently, the sialic acid-binding immunoglobulin-like lectins (Siglecs) family of sialic acid-binding proteins have been designated as glyco-immune checkpoints.5−10 Through their interaction with sialylated glycan ligands aberrantly expressed on tumor cells, inhibitory Siglecs such as Siglec-7 and -9 are found on immune cells, for example, natural killer (NK) and T cells, inhibiting immune signaling pathways to restrain immune responses.7,8,11−13 Likewise, an upregulation of Siglec-15 on cancer cells interacts with yet-to-be-identified T-cell membrane glycoproteins to suppress T-cell anti-tumor functions.5 Despite these exciting discoveries that correlate a Siglec-mediated immune suppression to cancer progression, we have a poor understanding of the prevalence of the Siglec-ligand expression. Moreover, the mechanisms that govern the Siglec-ligand interaction-mediated immune suppression are only partially elucidated.8,14,15
When Siglec-7 or -9 expressing NK cells encounter their target tumor cells, binding to trans ligands expressed by tumor cells recruits these Siglecs to the immune synapse, where their immunoreceptor tyrosine-based inhibition motifs (ITIMs) are phosphorylated by Src kinase, thereby creating a binding site for the tyrosine phosphatases SHP-1 and SHP-2.9,11,16 The binding of SHP-1/2 leads to a dephosphorylation of signaling components downstream of activation receptors to suppress NK cell activation and effector function. Therefore, it seems reasonable to postulate that tumor cells express higher levels of Siglec ligands to counteract NK-induced killing. However, here, when surveying tumor specimens and matched normal tissues, we observed that a high Siglec-7 ligand (Siglec-7L) expression was found in both normal and malignant tissues. Nevertheless, when live cancer cells encountered NK cells, a significant upregulation of Siglec-7 ligands was detected within 1 h, suggesting that this inhibitory pathway may serve as a negative feedback mechanism that follows NK cell infiltration. With sialyltransferase-mediated chemoenzymatic glycan editing, a high-affinity Siglec-7 ligand could be installed onto NK cells as a high-affinity cis ligand, which we found to induce multifaceted effects on Siglec-7 signaling and NK effector function.
Results
Siglec-7 Ligands Are Not Specifically Found on Cancer Cells and Malignant Tissues
To understand how the interactions between Siglec-7 and its sialoside ligands on cancer cells are involved in modulating NK cell-mediated tumor cell killing, we first assessed the expression of Siglec-7 ligands on a panel of cancer cell lines and primary human immune cells using a recombinant Siglec-7-Fc chimera17 and analyzed the binding by flow cytometry. Among the cancer cell lines that we screened, Raji B lymphoma and MDA-MB-435 melanoma cells were found to possess the highest level of Siglec-7 ligands (Figure 1a), whereas the prostate cancer cell line LNCaP and several ovarian and colorectal cancer cell lines only showed low levels. Although only basal levels of Siglec-7 ligands were found on the vascular endothelial cell line HUVEC, freshly isolated NK cells, peripheral blood mononuclear cells (PBMCs), and activated T cells expressed abundant Siglec-7 ligands. To assess insitu expression of Siglec-7 ligands, we analyzed a paraffin-embedded human tumor and adjacent tissue specimens from a number of malignancies. Significant Siglec-7-Fc staining was detected in almost all specimens analyzed (Figure 1b–d, Supporting Information Figures S1 and S2); both the primary tumor and the matched adjacent tissue specimens exhibited varying levels of Siglec-7-Fc staining, which was abolished upon neuraminidase treatment (Figure 1b,c). In particular, we examined 177 specimens from lung cancer patients and 20 specimens from normal lung tissues (Figure 1d,e, and Supporting Information Figure S2) and found that ∼20–40% of lung cancer tissues and 85% of normal lung tissues expressed high levels of Siglec-7 ligands. These observations demonstrate that Siglec-7 ligands are not specifically found on cancer tissues and, therefore, that their presence may not serve as a reliable marker for cancer cells.
Figure 1.

Profile of Siglec-7 ligand (Sigle-7L) expression on the cell surface of live cells and human tissue specimens. (a) Cell lines and primary immune cells from healthy human donors were stained with recombinant Siglec-7-Fc precomplexed with an anti-human Fc-APC antibody, and anti-human Fc-APC only was used to assess the ligand-binding specificity (Fc control). Error bar represents the standard deviation of three biological samples. (b) Probing the Siglec-7L expression in human breast cancer tissues and the normal adjacent tissues that were pretreated with neuraminidase to remove sialic acids or not. (c) Probing the Siglec-7L expression in different human malignant tissues. (d, e) Probing the Siglec-7L expression in lung cancer tissues and adjacent normal tissues from different donors (total of 197 samples). The lung malignant tumor samples (n = 2), stage I lung malignant (n = 75), stage II lung malignant (n = 38), stage III lung malignant (n = 58), stage IV lung malignant (n = 4), and normal lung tissue (n = 20) were used for Siglec-7L screening. Three representative Siglec-7L-high samples were shown (d). Siglec-7L levels were classified based on Siglec-7-Fc staining mean fluorescence intensity (MFI) that was determined via ImageJ with a cutoff value of MFI = 27.00. The sample size in each group was plotted using a pie chart, and the areas in each circle are equal to the sizes of each group (e), Siglec-7 low (gray) and high (red). The percentages of Siglec-7L high specimens were shown in the figure.
Desialylation of Siglec-7L-Expressing Target Cancer Cells Enhances NK-Cell Effector Functions
Because of the high expression of Siglec-7 ligands on Raji cells, we used these cells as the target (T) cells to determine the impact of the interaction between Siglec-7 on NK cells and cancer cell-expressed Siglec-7 ligands on NK cell effector functions. Primary NK cells18,19 isolated from the peripheral blood of healthy donors and NK-92MI cells20,21 were used as sources of NK effector (E) cells. NK cells isolated from healthy donors were expanded in a culture with recombinant human interleukin 2 (IL-2) and IL-15 to generate a large number of cells for an in vitro functional evaluation (these NK cells were defined as peripheral NK cells). The freshly isolated peripheral NK cells contained ∼70%–100% of Siglec-7 positive cells, and the frequency of this subset decreased slightly after a 10 d in vitro culture (Supporting Information Figure S3). Consistent with previous reports,22,23 the culture with IL-2 and IL-15 significantly improved the cytotoxicity of peripheral NK cells toward Raji cells (Supporting Information Figure S3c). To assess the NK-induced target cell killing, we incubated target Raji cells with peripheral NK cells for 4 h and determined the target cell killing using a lactate dehydrogenase (LDH) release assay. As shown in Figure 2a, NK-induced target-cell killing was enhanced along with the increased effector cell-to-target cell ratio (E/T ratio). Predesialylation of Raji cells by neuraminidase significantly enhanced their susceptibility to an NK-induced killing. Similarly, enhanced Raji cell killing was observed when a Siglec-7 functional blocking antibody9 was used.
Figure 2.
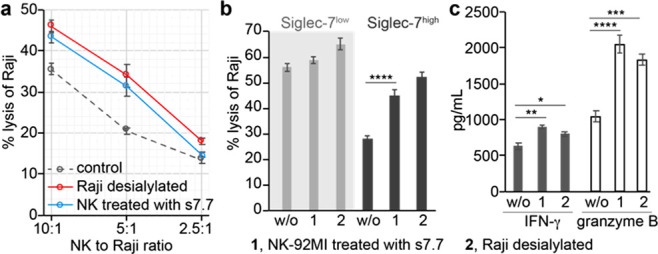
Cytotoxicity of NK cells was impaired by “Sialic acid-Siglec-7” interactions. (a, b) LDH release assay for quantifying the cytotoxicity of the peripheral NK cells (a) and NK-92MI (b) against Raji cells. The predesialylation of Raji cells was conducted by a neuraminidase treatment. The anti-Siglec-7 antibody (clone s7.7) blocking was performed before the coincubation of NK with Raji cells. (c) ELISA quantification of IFN-γ and granzyme B produced by NK-92MI-S7high cells after an incubation with Raji cells for 1h at an E/T ratio of 5:1. The bars represent the standard error of three biological replicates. The significance was analyzed with the two-sided Student’s t test. Note, *, p < 0.05; **, p < 0.01; ***, p < 0.005; ****, p < 0.001.
Next, we assessed target cell killing using NK-92MI cells, a constantly cytotoxic NK cell line currently undergoing clinical trials as an “off-the-shelf therapeutic” for treating both hematological and solid malignancies.20,21 NK-92MI cells were originally derived from a CD56bright-NK population,24 only a small subset (∼10%) of which expressed Siglec-7 (we define this status as NK-92MI-S7low). With increased passages in media supplemented with nonheat-inactivated horse serum, the Siglec-7 positive subset became the dominant subset. After ∼120 d in culture, ∼90% NK-92MI became Siglec-7 positive (we define this status as NK-92MI-S7high) (Supporting Information Figure S4). As expected, compared to NK-92MI-S7low cells, NK-92MI-S7high cells induced ∼55% reduced killing of Raji cells (E/T ratio = 5:1) (Figure 2b). The NK-92MI-S7high-associated cytotoxicity could be enhanced by the Siglec-7 blocking antibody (s7.7) that blocks the ability of Siglec-7 to bind its glycan ligands or by a predesialylation of Raji cells. Increased IFN-γ and granzyme B secretions were also observed in response to abrogated Siglec-7 ligand interactions (Figure 2c). The same treatments also increased the cytotoxicity of NK-92MI-S7low cells but to a much lower degree (Figure 2b). Similar results were observed using Daudi cells expressing moderate levels of Siglec-7 ligands as the target cells (Supporting Information Figure S5). Together, these results are consistent with what was reported previously by the Démoulins and Bertozzi groups7,8 and strongly indicate that Siglec-7 ligands on cancer cells protect them from NK cytotoxicity via an interaction with Siglec7 on NK cells.
Interactions between NK and Tumor Cells Lead to an Upregulation of Siglec-7 Ligands on Tumor Cells
During the course of monitoring interactions between Raji and NK-92MI or peripheral NK cells, we observed a rapid increase of sialylated glycans on Raji cell surface within 2 h, as revealed by a lectin staining with Sambucus nigra lectin (SNA, specific for α2–6-sialosides) and Maackia Amurensis lectin (MAA, specific for α2–3-sialosides) (Figure 3a–c and Supporting Information Figure S6). Notable increases (ca. four-fold) of the SNA staining were also observed on JIMT-1 human breast carcinoma and H1975 human nonsmall cell lung carcinoma cells after an incubation with NK-92MI cells under the same condition (Supporting Information Figure S7). We considered several possibilities through which cancer cells could elevate their cell-surface sialylated glycans: (1) transfer of sialylated glycoconjugates from NK cells, (2) an increase in de novo sialic acid synthesis, and/or (3) a reduced endocytosis on Raji cell. To examine the first possibility and determine if sialosides on Raji cells were transferred from NK cells, NK cells were cultured in the presence of peracetylated N-(4-pentynoyl)mannosamine (Ac4ManNAl)25 to metabolically incorporate an alkyne-containing Neu5Ac (SiaNAl) onto cell surface glycoconjugates. After 48 h, NK cells with newly synthesized sialosides labeled by SiaNAl were incubated with Raji cells for 1.5 h, and the cell mixture was reacted with biotin-azide via the ligand 3-[4-({bis[(1-tert-butyl-1H-1,2,3-triazol-4-yl)methyl]amino}methyl)-1H-1,2,3-triazol-1-yl]propyl hydrogen sulfate(BTTPS)-assisted copper-catalyzed azide–alkyne [3 + 2] cycloaddition reaction (CuAAC),26 followed by a flow cytometry analysis. A robust biotin signal was detected on NK cells that were cultured with Ac4ManNAl. When Ac4ManNA1 was incubated with Raji cells, a significant decrease in the NK-associated biotin signal was observed, along with the appearance of the biotin signal on Raji cells, indicating that alkyne-tagged sialic acids were transferred from NK to Raji cells upon immune interactions (Figure 3d,e).
Figure 3.
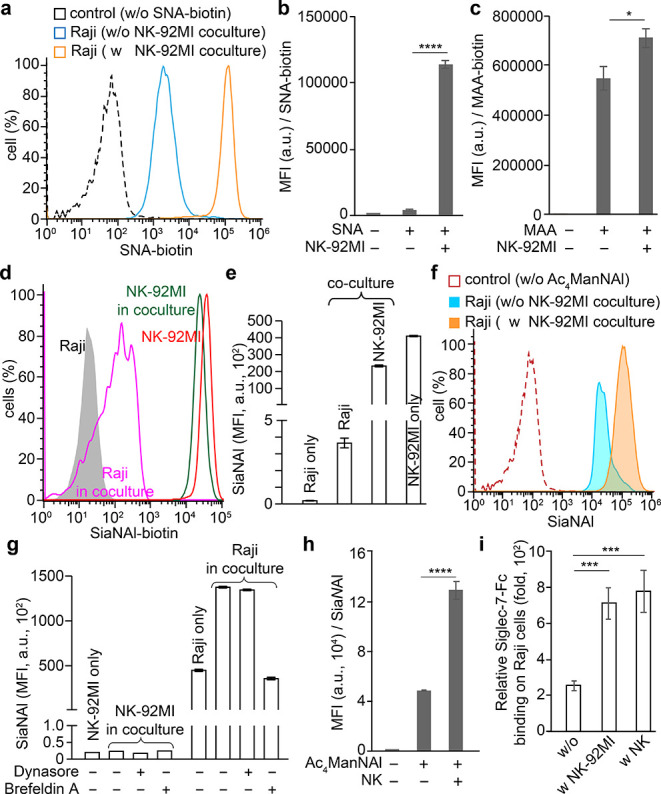
Interactions between NK and Raji cells lead to an upregulation of Siglec-7 ligands on Raji cells. (a) SNA staining of α2–6-sialic acids on Raji cells. (b, c) MFI of SNA (b) and MAA (c) lectin staining of Raji cells. (d, e) Transfer of SiaNAl-labeled sialosides from NK-92MI cells to Raji cells upon the coculture of both cells. (f, g) Ac4ManNAl-based metabolic glycan labeling of Raji cells in the coculture with NK-92MI-S7 cells, in the presence of dynasore, brefeldin A, or not. (h) Coculture with peripheral NK cells also significantly induced Raji cells increase sialylation levels, as revealed by Aci4ManNAl labeling. (i) NK-induced upregulation of sialylation on Raji cells further significantly increased the binding of Siglec-7-Fc. The relative Siglec-7 binding was determined via a division to the corresponding Fc controls stained with anti-human Fc-APC. (b, c, e, g-I) Bars represent the standard error of the MFI of three biological repeats of samples. The significance was analyzed with the two-sided Student’s t test. (*) p < 0.05; (***) p < 0.005; (****) p < 0.001.
We next examined the second and third possibilities, whether there was an increased de novo sialic acid synthesis and/or a reduced endocytosis on Raji cells upon NK cell encountering, respectively. Raji cells were metabolically labeled with Ac4ManNAl for 48 h and subjected to the CuAAC-mediated biotin conjugation before and after an incubation with NK-92MI cells. In comparison to Raji cells without a prior NK incubation, Raji cells that were incubated with NK cells exhibited a twofold higher biotinylation signal (Figure 3f,g). Notably, the endogenously increased sialylation was at least 200-fold higher than that transferred from NK cells (Figure 3e vs 3g). We observed that the increased SiaNAl incorporation on the cell surface was largely blocked by Brefeldin A,27 which inhibits the protein transport from the endoplasmic reticulum to the Golgi complex, but not by the endocytosis inhibitor Dynasore28 (Figure 3g and Supporting Information Figure S8). Importantly, the Brefeldin A treatment also exacerbated the susceptibility of Raji cells to the NK-induced killing (Supporting Information Figure S8b).
Because endocytosis would bring cell-surface biotinylated glycoconjugates into the cytosol, which prevents their detection by membrane-impermeable streptavidin-fluorophore conjugates, we then prelabeled Raji cell-surface glycans with biotinylated Neu5Ac by in situ ST6Gal1-mediated chemoenzymatic glycan editing,29−32 added the labeled cells to NK cells, and stained the cell surface using streptavidin at various time points. Whereas untreated Raji cells exhibited a rapid reduction of streptavidin staining, no significant changes of streptavidin signal were detected on Raji cells cocultured with NK-92MI cells over 2 h (Supporting Information Figure S9), providing strong evidence that the endocytosis of target cells was slowed. Furthermore, within the 2 h time window biotinylated Raji cells cultured alone in the fresh or the conditioned NK-92MI culture medium exhibited levels of streptavidin staining that were comparable to those of their counterparts that were cultured in a contactless, transwell-based coculture system together with NK-92MI cells (Supporting Information Figure S10), indicating that a direct NK contact is required to suppress endocytosis. Similarly, the coincubation of Raji cells with peripheral NK cells also triggered a quick accumulation of newly synthesized sialosides, which in turn significantly augmented the sialylation on the Raji cell surface (Figure 3h). Notably, no obvious transfer of sialic acids from Raji to NK cells was observed. As a consequence of the increased Raji cell-surface sialylation, a notable elevation of Siglec-7-Fc binding was detected (Figure 3i). In addition, the Siglec-7-specific blocking antibody did not prevent the increase of Raji cell-surface sialylation, suggesting that Siglec-7 may not be involved in triggering this process (Supporting Information Figure S11). Consistent with a recent study by Shao and co-workers,33 we found that HEK293T cells (an immortalized human embryonic kidney cell line) with Siglec-7 ligands on the surface (Figure 1a), unlike malignant cells, were not attacked by NK cells (Supporting Information Figure S12). When NK cells were encountered, only a slight upregulation of cell surface sialylation was detected, and the upregulated sialic acid was derived primarily from the NK cell surface rather than de novo synthesized (Supporting Information Figure S12e). Together, the above observations verified that interactions with NK cells lead to a sialoside transfer from NK cells to the encountered cells. However, the accumulation of de novo synthesized sialosides on the cell surface cannot be triggered unless the encountered cells are prone to NK-induced killing.
Last, we sought to determine if there is any correlation between an NK cell infiltration and the in situ expression of Siglec-7 ligands by analyzing both freshly frozen and paraffin-embedded human tumor and adjacent healthy tissue specimens. The NK density as quantified by CD56 staining was found to have a modest negative correlation with the Siglec-ligand expression in healthy lung tissue samples, whereas a weak positive correlation existed between an NK infiltration and the Siglec-ligand expression in malignant tissues (Supporting Information Figures S13 and S14). Importantly, cancer tissues with elevated levels of NK infiltration appeared to express much higher amounts of Siglec-7 ligands. By contrast, no such correlations were found between Siglec-7 ligand levels and the infiltration of cytolytic, preeminent IFN-γ secreting CD8+ T cells34 in frozen tissue arrays of consecutive samples (Supporting Information Figure S15).
NKG2D Signaling-Triggered Granule Secretion Upregulates Siglec-7 Ligands on Target Cells
Subsequently, we sought to explore how NK contact with target cancer cells is involved in upregulating target cell sialylation. At the immunological synapse, the engagement of the NK activation receptor NKG2D and NKG2D ligands, such as major histocompatibility complex class I-related chains A and B (MIC A/B), elicits cytolytic responses and is sufficient to trigger target cell cytolysis.34−36 We found that NK-92MI cells that were pretreated with an NKG2D-blocking antibody resulted in ∼50% reduction of NK-contact-induced upregulation of target cell sialosides (Supporting Information Figure S16c,e). NKG2D signaling triggers the secretion of “lytic granules” (perforin/granzyme) and IFN-γ (Supporting Information Figure S16a,b). Accordingly, we assessed whether these molecules were involved in regulating target cell sialylation. We used egtazic acid (EGTA)36 to chelate calcium that is required for activating the perforin/granzyme secretion and found that it reduced the sialylation increase on Raji cells by 35% (Supporting Information Figure S16d,e). We also pretreated NK cells with the vacuolar-type H+-ATPase inhibitor concanamycin A (CMA)37 to block perforin maturation and observed that the increase of Raji cell-surface sialosides and Siglec-7-Fc binding was almost completely blocked (Supporting Information Figure S16f,g). Finally, to assess the contribution of interferon γ (IFN-γ),38 which is known to trigger the upregulation of the inhibitory receptor PD-L1 on target cells, we cultured Raji cells with elevated concentrations of IFN-γ. Interestingly, an IFN-γ treatment only slightly augmented a de novo sialylation on Raji cells but had no impact on the Siglec-7-Fc binding (Supporting Information Figure S16h–k).
Engineering Glycocalyx of NK Cells Via Chemoenzymatic Glycan Editing
The above observations provided strong evidence that NK contact triggers the upregulation of Siglec-7 ligands on target cells. As a self-defense mechanism, the upregulated ligands engage with the NK-associated Siglec-7 to suppress NK-induced killing. We then sought to engineer NK cells that are resistant to this negative feedback mechanism. Previous studies by Crocker and co-workers revealed that Siglec-7 on the NK cell surface is masked by cis ligands, such as Neu5Acα2–8Neu5Acα2–3 disialic acids displayed by the ganglioside GD3, which partially block its interactions with trans ligands found on target cells.39,40 The unmasking of Siglec-7 induces a strong suppression of NK-induced tumor cell killing due to the binding of Siglec-7 with its trans ligands. On the basis of this precedent, we thus hypothesized that functionalizing NK cells with a high-affinity ligand of Siglec-7 may induce even stronger cis interactions to boost the NK-associated cytotoxicity by preventing Siglec-7-trans ligand interactions. Developed by Paulson and co-workers, FTMCNeu5Ac41 (2), a chemically functionalized version of Neu5Ac (1) whose C9 is substituted with 9-N-(1-(5-fluorescein)-1H-1,2,3-triazol-4-yl)methylcarbamate, when α2–6 or α2–3 linked to N-acetyllactosamine (type 2 LacNAc, Galβ1–4GlcNAc), serves as a high-affinity ligand of Siglec-7. Using sialyltransferase-mediated chemoenzymatic glycan editing, we may introduce FTMCNeu5Ac onto NK cells to directly create high-affinity Siglec-7 ligands in a cis configuration.
To assess this design, we first synthesized cytidine-5'-monophospho(CMP)-FTMCNeu5Ac (5) as the donor substrate. A commonly used CMP-Sia synthetase from Neisseria meningitides (NmCSS)42 was not capable of directly converting FTMCNeu5Ac to the corresponding CMP-Neu5Ac analogue. Alternatively, we prepared C9-N-propargyloxycarbonyl Neu5Ac (3, C9-CPgNeu5Ac)43 and converted it to CMP-C9-CPgNeu5Ac. The resulting CMP-C9-CPgNeu5Ac (4) was then reacted with 5-fluorescein azide41 to produce CMP-FTMCNeu5Ac (5) with an overall yield of ∼26% (Figure 4a and Supporting Information Figure S27).
Figure 4.
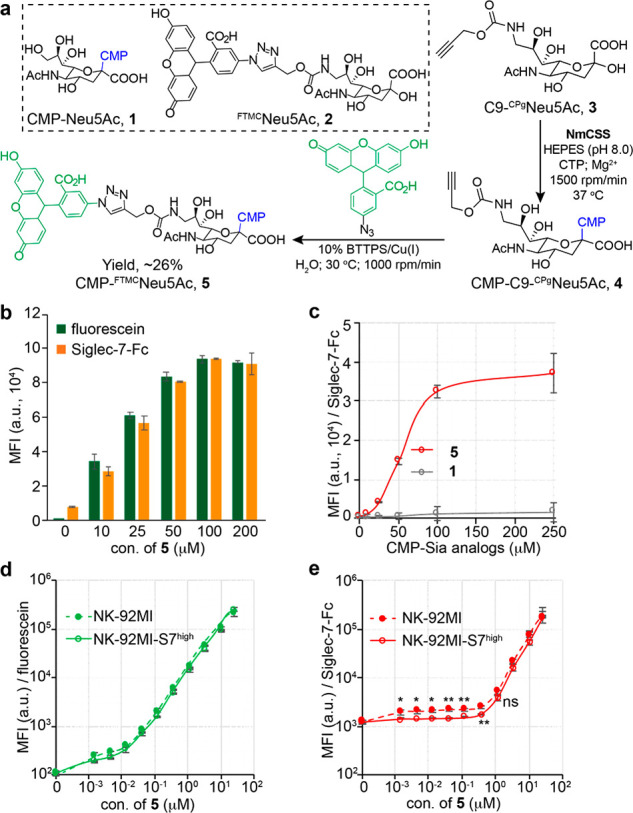
ST-assisted in situ creation of Siglec-7 high-affinity ligands on live cells. (a) One-pot synthesis of CMP-FTMCNeu5Ac (5). (b) ST6Gal1-assisted incorporation of FTMCNeu5Ac onto the peripheral NK cells was probed with the resultant fluorescein signals and Siglec-7-Fc binding. (c) ST6Gal1-assisted incorporation of FTMCNeu5Ac or natural Neu5Ac onto NK-92MI cells was probed by Siglec-7-Fc. (d, e) ST6Gal1-assisted incorporation of FTMCNeu5Ac onto the NK-92MI or NK-92MI-S7high cells was probed with the resultant fluorescein signals (d) and Siglec-7-Fc binding (e). (b–e) Bars represent the standard error of three biological repeats of samples. The significance was analyzed with the two-sided Student’s t test. (ns) not significant; (*) p < 0.05; (**) p < 0.01.
With the donor substrate in hand, we then assessed the feasibility of an ST6Gal1-mediated creation of high-affinity Siglec-7 ligands on peripheral NK and NK-92MI cells. Because FTMCNeu5Ac contains fluorescein, the cell-associated fluorescein fluorescence can be measured and used to quantify the number of FTMCNeu5Ac installed. A dose- and time-dependent increase of fluorescein signals was detected upon treating NK cells with ST6Gal1 and 5 (Figure 4b,d and Supporting Information Figure S17). When 100 μM of 5 was used for cell-surface glycan editing, Siglec-7-Fc binding reached the saturation level (Figure 4b,c). Notably, when less than 1 μM of 5 was used for the NK-92MI cell modification, we observed an interesting phenomenon: although comparable levels of FTMC-fluorescence could be detected on both NK-92MI Siglec-7low and Siglec-7high cells and fluorescence intensity increased along with an increasing dose of 5, staining these two modified cells with Siglec-7-Fc chimera gave distinct results (Figure 4e). The Siglec-7-Fc staining could be detected on Siglec-7low cells when as low as 10 nM of 5 was used, whereas the staining above background signals on Siglec-7high cells was only detectable when the concentration of 5 went beyond 100 nM. This observation suggests that the newly installed low-concentration FTMCNeu5Ac may be engaged in strong cis interactions with Siglec-7 on the NK cell surface, preventing its trans interactions with Siglec-7-Fc.
Modulate NK-Induced Tumor-Cell Killing Via a Newly Created cis High-Affinity Siglec-7L
After confirming that FTMCNeu5Ac could be installed directly onto NK cells to form high-affinity Siglec-7 ligands, we explored the possibility of using this approach to modulate Siglec-7 immunoinhibitory signaling. To our delight, the killing of Raji cells was significantly inhibited when NK-92MI-S7high cells were functionalized with FTMCNeu5Ac when a high concentration of 5 (i.e., >100 μM) was used as the donor (Supporting Information Figure S18). Similar inhibition effects were also observed when desialylated target Raji and Daudi cells were used (Figure 5a,b). The decreased cytotoxicity of modified NK-92MI cells toward desialylated Raji and Daudi cells was gradually rescued when the concentration of 5 used for glycan editing dropped below 1 μM. The lower the CMP-FTMCNeu5Ac concentration became the higher the NK-induced target cell killing. The NK cytotoxicity was fully rescued when the concentration of 5 dropped below 10 nM. Remarkably, compared to nontreated NK-92MI cells, NK-92MI-S7high cells modified with 10 nM of 5 exhibited a significantly enhanced killing of untrimmed Raji and Daudi cells with their natural sialylation intact (Figure 5c). For example, at the effector-to-target ratio of 5:1, the NK-induced target-cell killing was almost doubled upon FTMCNeu5Ac installation.
Figure 5.
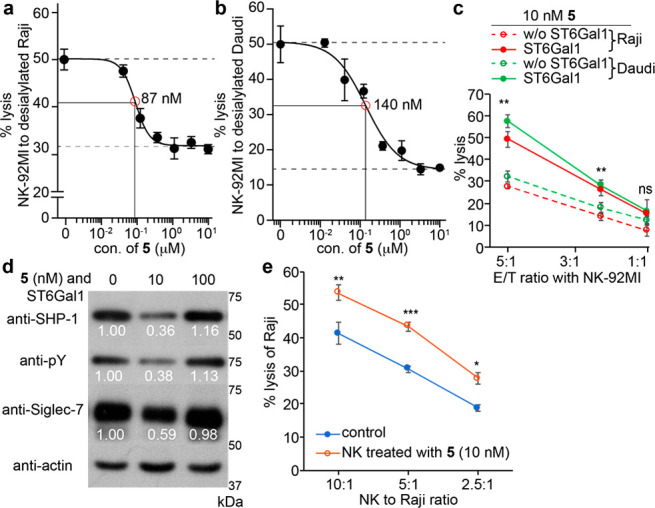
NK cell surface newly created cis high-affinity Siglec-7 ligands modulated NK-cytotoxicity against target cells. (a–c) LDH release assay for quantifying the cytotoxicity of the NK-92MI-S7high cells against Raji (a, c) and Daudi cells (b, c). The NK-92MI-S7high cells with or without FTMCNeu5Ac incorporation were used (d) Western blot analysis of Siglec-7 activation. NK-92MI-S7high cells with or without FTMCNeu5Ac modification were incubated with Raji cells and lysed, followed by an anti-Siglec-7 immunoprecipitation. A decrease in SHP-1 recruitment and Siglec-7 phosphorylation was seen in NK cells after a treatment with 10 nM of 5 but not 100 nM of 5. The numbers indicate the relative quantification of band intensity to that of actin by ImageJ. (e) The specific lysis of Raji using the sorted Siglec-7 positive peripheral NK cells with or without FTMCNeu5Ac modification. (a–c, e) Bars represent the standard error of three biological repeats of samples. The significance was analyzed with the two-sided Student’s t test. (ns) not significant; (*) p < 0.05; (**) p < 0.01; (***) p < 0.005.
Consistently, the cytotoxicity of peripheral NK cells and the fluorescence-activated cell sorting (FACS)-isolated Siglec-7 positive peripheral NK cells was also significantly improved upon an ST6Gal1-mediated glycan editing with 10 nM of 5 (Figure 5e and Supporting Information Figure S19).
When Siglec-expressing immune cells encounter their target cells, binding to the trans ligands that are expressed on target cells recruits Siglecs to the immune synapse, where their ITIMs are phosphorylated by Src kinases that are activated due to an activation receptor–ligand ligation, thereby creating a binding site for the tyrosine phosphatases SHP-1 and SHP-2.11 The binding of SHP-1/2 leads to the dephosphorylation of signaling components of the activatory receptors to suppress immune cell activation and effector function. Consistent with the decreased NK cytotoxicity triggered by the functionalization with high concentrations of 5, an enhanced phosphorylation of Siglec-7 was detected, which was accompanied by an increased SHP-1 recruitment (Figure 5d). However, when NK cells were functionalized with a low concentration of 5 (i.e., 10 nM), a significantly reduced phosphorylation of Siglec-7 and SHP-1 recruitment was observed. Simultaneously, a dramatically decreased Siglec-7 expression level was unexpectedly detected in NK cells (Figure 5d).
Newly Created Ligands Modulate the Siglec-7 Stability on NK Cells
To investigate the mechanism through which the high-affinity ligands installed in cis on NK cells modulate the Siglec-7 inhibitory signaling, we first imaged the Siglec-7 distribution on the cell surface of NK-92MI-S7high cells using fluorescence microscopy. As is known for Siglec-2 (CD22) on B cells,44 in the absence of trans ligands, Siglec-7 formed abundant clusters on NK-92MI-S7high cells (Supporting Information Figures S20–S23). After the removal of cell-surface sialic acid by neuraminidase, Siglec-7 clusters broke up (Supporting Information Figure S21). As expected, when NK-92MI-S7high cells encountered target Raji cells, Siglec-7 was recruited to the E/T interface (Supporting Information Figure S20a, first panel).
The installation of FTMCNeu5Ac onto the cell surface of NK-92MI-S7high also induced a Siglec-7 cluster formation in a dose-dependent manner (Supporting Information Figure S23). These clusters could be observed not only on the modified NK cells that were cultured by themselves but also on the modified NK cells cultured with Raji cells but without direct contact with Raji. Interestingly, when Raji cells were encountered, a dramatic decrease of cell-surface Siglec-7 was detected on NK-92MI-S7high cells that had been modified with 10 nM of 5, whereas no significant changes of Siglec-7 were observed on NK-92MI-S7high cells that had been modified with more than 100 nM CMP-FTMCNeu5Ac (shown in the second and third panels of Supporting Information Figure S20a).
Consistent with what was observed by imaging, a flow cytometry analysis revealed a dramatic decrease (∼20–30%) of the Siglec-7 positive NK cell population following the incubation of target Raji cells with NK-92MI-S7high cells that had been modified with 10 nM of 5 (Figure 6b). Similarly, when target cells were encountered, the decrease of the Siglec-7 positive subset was observed in both peripheral (Figure 6c) and the sorted Siglec-7 positive peripheral NK cells (Figure 6d,e) that were functionalized by 10 nM of 5. Interestingly, no notable increase of Siglec-7 endocytosis was detected, as revealed by the weak intracellular Siglec-7 signal (Supporting Information Figure S20a). The downregulation of Siglec-7 expression levels in NK cells (Figure 5d and Supporting Information Figure S20a), coupled with negligible changes in Siglec-7 endocytosis, prompted us to explore the possibility of a Siglec-7 release from the plasma membrane. A Western blot analysis of Siglec-7 in the culture supernatant revealed that, compared to the coculture supernatant of Raji and unmodified NK cells, a dramatic increase of Siglec-7 was detected in the coculture supernatant of Raji and NK-92MI (or peripheral NK) modified with 10 nM of 5 (Figure 6f and Supporting Information Figure S24), indicating that Siglec-7 was released from the NK plasma membrane upon target cell encountering, a process significantly exacerbated by the installation of superlow levels of FTMCNeu5Ac.
Figure 6.

ST6Gal1-assisted incorporation of FTMCNeu5Ac on NK-92MI-S7high or peripheral NK cells modulates Siglec-7 inhibitory signaling. (a, b) Siglec-7 positive populations of NK-92MI-S7high cells (a) or peripheral NK cells raised from different healthy donors (b) treated with ST6Gal1 under 10 nM of 5 were counted via flow cytometry. (c, d) The sorted Siglec-7 positive peripheral NK cells were treated with10 nM of 5 and then were cocultured with Raji cells at an E/T ratio of 10:1. (e) Western blot analysis of Siglec-7 released into the supernatant of the coculture of NK-92MI-S7high cells with Raji cells at an E/T ratio of 5:1. The significance was analyzed with the two-sided Students t test. (ns) not significant; (*) p < 0.05; (***) p < 0.005. (f) A schematic model of the ligand-mediated modulation of Siglec-7 inhibitory signaling on NK cells upon target cell encountering. The enzymatic creation of cis high-affinity Siglec-7 ligands at a low level mediates the release of Siglec-7 inhibitory receptors to the medium during an immune activation by target cancer cells.
Conclusion and Discussion
Tumor escape from an immune-mediated destruction has been associated with mechanisms that suppress immune system effector functions and trigger immune cell exhaustion.1−3,45,46 The Siglec-sialylated glycan interaction is a newly added member of tumor immune escape pathways.5−9,13−16 However, the mechanism of this interaction-dependent suppression of antitumor immunity is not well understood. Analogous to what has been confirmed for the upregulated expression of PD-L1 in the tumor microenvironment, which is driven by IFN-γ produced by tumor-infiltrating CD8+ T cells,38 here, we found that an encounter with NK cells triggered the accumulation of Siglec-7 ligands on tumor cells. This accumulation of Siglec-7 ligands is partially due to the secreion of lytic granules rather than IFN-γ by NK cells. With the formation of the immunological synapse, target cells slowed the endocytosis of cell-surface glycoconjugates, while the newly synthesized Siglec-7 ligands continued to build upon the cell surface to activate the Siglec-7 inhibitory signaling (Figure 6f and Supporting Information Figure S25). The decrease of sialoside endocytosis might result from the prominent actin accumulation in the synaptic contact, a mechanism exploited47 by certain cancer cells to restrict their membrane dynamic endocytosis for antagonizing lymphocytes-induced killing. Nevertheless, in contrast to initial preconceptions of a tumor-intrinsic high expression of Siglec-7 ligands, our data argue that tumor cells upregulate sialylated glycans, which counteract NK-induced killing via the Siglec-sialylated glycan interaction, a mechanism that is intrinsic to the immune system, and likely represent a physiological negative feedback loop.
Via an ST6Gal1-mediated cell-surface glycan editing, a high-affinity and specific ligand of Siglec-7 can be created on NK cells to modulate Siglec-7 signaling and NK effector function. Although many Siglecs are known to be masked by natural cis-ligands found on immune cells,39,40,49−51 unnatural, high-affinity ligands added in cis behave differently. On the one hand, with high levels of ligands added onto the NK cell surface, an enhanced phosphorylation of Siglec-7 and a recruitment of SHP-1 was observed, which in turn suppressed the NK-induced target tumor cell killing. On the other hand, at low levels, the same ligand induced the release of Siglec-7 from the NK cell surface to the culture medium, which completely restored the NK cytotoxicity. We confirmed that the ligand-triggered Siglec-7 secretion only took place after the target cell encounter, but not by the ligand installation itself. However, the detailed mechanism remains to be explored. One possibility is that the installation of low levels of high-affinity ligands in cis destroy the natural clusters of Siglec-7, which in turn increases the release of Siglec-7 via the secretion pathway that is triggered by the immune killing process (e.g., degranulation20,33). Although a chronic HCV infection is known to increase plasma levels of a soluble form of Siglec-7,52 to our knowledge, what was observed here is the first case that a high-affinity ligand induces the release of Siglec-7 from NK cell surface. It would be interesting to explore whether it is a common phenomenon that functionalizing immune cells with low levels of high-affinity Siglec ligands could release Siglecs from the cell surface. If so, this strategy may serve as a general approach to downregulate Siglecs’ inhibitory functions. In this endeavor, we have already demonstrated that high-affinity and specific ligands for other Siglecs, such as Siglec-2 (CD22)53 and Siglec-9,9,41,54 can be introduced onto live cells by employing similar strategies (Supporting Information Figures S26 and S28). A transplantation of expanded allogeneic NK cells has emerged as a promising strategy for cancer treatment;55−57 we envisage that, by modifying these NK cells with the high-affinity ligands prior to adoptive transfer, the constitutively expressed Siglec-7 could be released to enhance the NK effector functions for a better antitumor immunity.
Acknowledgments
This work was supported by the NIH (R01AI154138 to P.W., P41GM103390, P01GM107012, and R01GM130915 to K.W.M., R01AI050143, P01HL107151, and U19AI136443 to J.C.P.). Prof. M.S.M. thanks NSERC, Canada Research Chairs, and the Canadian Glycomics Network for funding. We thank Prof. M. Boyce (Duke University), Prof. Q. Xia (Peking University), and Prof. X. Chen (Peking University) for the insightful discussions.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.1c00064.
Experimental section and supplementary figures (PDF)
Author Contributions
The manuscript was written through the contributions of all authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Miller J. F.; Sadelain M. The journey from discoveries in fundamental immunology to cancer immunotherapy. Cancer Cell 2015, 27, 439–449. 10.1016/j.ccell.2015.03.007. [DOI] [PubMed] [Google Scholar]
- Ribas A.; Wolchok J. D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. 10.1126/science.aar4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P.; Allison J. P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- Hegde P. S.; Chen D. S. Top 10 challenges in cancer immunotherapy. Immunity 2020, 52, 17–35. 10.1016/j.immuni.2019.12.011. [DOI] [PubMed] [Google Scholar]
- Barkal A. A.; et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature 2019, 572, 392–396. 10.1038/s41586-019-1456-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; et al. Siglec-15 as an immune suppressor and potential target for normalization cancer immunotherapy. Nat. Med. 2019, 25, 656–666. 10.1038/s41591-019-0374-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jandus C.; et al. Interactions between Siglec-7/9 receptors and ligands influence NK cell–dependent tumor immunosurveillance. J. Clin. Invest. 2014, 124, 1810–1820. 10.1172/JCI65899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudak J. E.; Canham S. M.; Bertozzi C. R. Glycocalyx engineering reveals a Siglec-based mechanism for NK cell immunoevasion. Nat. Chem. Biol. 2014, 10, 69–75. 10.1038/nchembio.1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macauley M. S.; Crocker P. R.; Paulson J. C. Siglec-mediated regulation of immune cell function in disease. Nat. Rev. Immunol. 2014, 14, 653–666. 10.1038/nri3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RodrIguez E.; Schetters S. T. T.; van Kooyk Y. The tumour glyco-code as a novel immune checkpoint for immunotherapy. Nat. Rev. Immunol. 2018, 18, 204–211. 10.1038/nri.2018.3. [DOI] [PubMed] [Google Scholar]
- Ikehara Y.; Ikehara S. K.; Paulson J. C. Negative regulation of T cell receptor signaling by Siglec-7 (p70/AIRM) and Siglec-9. J. Biol. Chem. 2004, 279, 43117–43125. 10.1074/jbc.M403538200. [DOI] [PubMed] [Google Scholar]
- Kawasaki Y.; et al. Ganglioside DSGb5, preferred ligand for Siglec-7, inhibits NK cell cytotoxicity against renal cell carcinoma cells. Glycobiology 2010, 20, 1373–1379. 10.1093/glycob/cwq116. [DOI] [PubMed] [Google Scholar]
- Tao L.; Wang S.; Yang L.; Jiang L.; Li J.; Wang X. Reduced Siglec-7 expression on NK cells predicts NK cell dysfunction in primary hepatocellular carcinoma. Clin. Exp. Immunol. 2020, 201, 161–170. 10.1111/cei.13444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laubli H.; et al. Engagement of myelomonocytic Siglecs by tumor-associated ligands modulates the innate immune response to cancer. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 14211–14216. 10.1073/pnas.1409580111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams O. J.; Stanczak M. A.; von Gunten S.; Läubli H. Targeting sialic acid–Siglec interactions to reverse immune suppression in cancer. Glycobiology 2017, 28, 640–647. 10.1093/glycob/cwx108. [DOI] [PubMed] [Google Scholar]
- Bornhofft K. F.; Goldammer T.; Rebl A.; Galuska S. P. Siglecs: A journey through the evolution of sialic acid-binding immunoglobulin-type lectins. Dev. Comp. Immunol. 2018, 86, 219–231. 10.1016/j.dci.2018.05.008. [DOI] [PubMed] [Google Scholar]
- Rodrigues E.; et al. A versatile soluble siglec scaffold for sensitive and quantitative detection of glycan ligands. Nat. Commun. 2020, 11, 5091. 10.1038/s41467-020-18907-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivier E.; et al. Innate or adaptive immunity? The example of natural killer cells. Science 2011, 331, 44–49. 10.1126/science.1198687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veluchamy J. P.; et al. The rise of allogeneic natural killer cells as a platform for cancer immunotherapy: Recent innovations and future developments. Front. Immunol. 2017, 8, 631. 10.3389/fimmu.2017.00631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suck G.; et al. NK-92: an ‘off-the-shelf therapeutic’ for adoptive natural killer cell-based cancer immunotherapy. Cancer Immunol. Immunother. 2016, 65, 485–492. 10.1007/s00262-015-1761-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L.; et al. Natural Killer Cell (NK-92MI)-based therapy for pulmonary metastasis of anaplastic thyroid cancer in a nude mouse model. Front. Immunol. 2017, 8, 816. 10.3389/fimmu.2017.00816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hydes T.; et al. IL-12 and IL-15 induce the expression of CXCR6 and CD49a on peripheral natural killer cells. Immun., Inflammation Dis. 2018, 6, 34–46. 10.1002/iid3.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson W. E.; et al. Interleukin (IL) 15 is a novel cytokine that activates human natural killer cells via components of the IL-2 receptor. J. Exp. Med. 1994, 180, 1395–1403. 10.1084/jem.180.4.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong J. H.; Maki G.; Hlingemann H. G. Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia 1994, 8, 652–658. [PubMed] [Google Scholar]
- Wratil P. R.; et al. Metabolic glycoengineering with N-acyl side chain modified mannosamines. Angew. Chem., Int. Ed. 2016, 55, 9482–9512. 10.1002/anie.201601123. [DOI] [PubMed] [Google Scholar]
- Wang W.; et al. Sulfated ligands for the copper(I)-catalyzed azide-alkyne cycloaddition. Chem. - Asian J. 2011, 6, 2796–2802. 10.1002/asia.201100385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson J. G.; Finazzi D.; Klausner R. D. Brefeldin A inhibits Golgi membrane-catalysed exchange of guanine nucleotide onto ARF protein. Nature 1992, 360, 350–352. 10.1038/360350a0. [DOI] [PubMed] [Google Scholar]
- Macia E.; Ehrlich M.; Massol R.; Boucrot E.; Brunner C.; Kirchhausen T. Dynasore, a cell-permeable inhibitor of dynamin. Dev. Cell 2006, 10, 839–850. 10.1016/j.devcel.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Hong S.; et al. Bacterial glycosyltransferase-mediated cell-surface chemoenzymatic glycan modification. Nat. Commun. 2019, 10, 1799. 10.1038/s41467-019-09608-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moremen K. W.; et al. Expression system for structural and functional studies of human glycosylation enzymes. Nat. Chem. Biol. 2018, 14, 156–162. 10.1038/nchembio.2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbua N. E.; et al. Selective exo-enzymatic labeling of N-glycans on the surface of living cells by recombinant ST6GalI. Angew. Chem., Int. Ed. 2013, 52, 13012–13015. 10.1002/anie.201307095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez Aguilar A.; Briard J. G.; Yang L.; Ovryn B.; Macauley M. S.; Wu P. Tools for studying glycans: recent advances in chemoenzymatic glycan labeling. ACS Chem. Biol. 2017, 12, 611–621. 10.1021/acschembio.6b01089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z.; et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science 2020, 368, eaaz7548. 10.1126/science.aaz7548. [DOI] [PubMed] [Google Scholar]
- Mandelboim O.; et al. Nature killer activating receptors trigger interferon γ secretion from T cells and natural killer cells. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 3798–3803. 10.1073/pnas.95.7.3798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C.; et al. Chimeric antigen receptor-engineered NK-92 cells: An off-the-shelf cellular therapeutic for targeted elimination of cancer cells and induction of protective antitumor immunity. Front. Immunol. 2017, 8, 533. 10.3389/fimmu.2017.00533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esser M. T.; Haverstick D. M.; Fuller C. L.; Gullo C. A.; Braciale V. L. Ca2+ signaling modulates cytolytic T lymphocyte effector functions. J. Exp. Med. 1998, 187, 1057–1067. 10.1084/jem.187.7.1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kataoka T.; et al. Concanamycin A, a powerful tool for characterization and estimation of contribution of perforin- and Fas-based lytic pathways in cell- mediated cytotoxicity. J. Immunol. 1996, 156, 3678–3686. [PubMed] [Google Scholar]
- Spranger S.; et al. Up-regulation of PD-L1, IDO, and Tregs in the melanoma tumor microenvironment is driven by CD8+ T cells. Sci. Transl. Med. 2013, 5, 200ra116. 10.1126/scitranslmed.3006504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avril T.; North S. J.; Haslam S. M.; Willison H. J.; Crocker P. R. Probing the cis interactions of the inhibitory receptor Siglec-7 with alpha2,8-disialylated ligands on natural killer cells and other leukocytes using glycan-specific antibodies and by analysis of alpha2,8-sialyltransferase gene expression. J. Leukocyte Biol. 2006, 80, 787–796. 10.1189/jlb.1005559. [DOI] [PubMed] [Google Scholar]
- Nicoll G.; Avril T.; Lock K.; Furukawa K.; Bovin N.; Crocker P. R. Ganglioside GD3 expression on target cells can modulate NK cell cytotoxicity via siglec-7-dependent and -independent mechanisms. Eur. J. Immunol. 2003, 33, 1642–1648. 10.1002/eji.200323693. [DOI] [PubMed] [Google Scholar]
- Rillahan C. D.; et al. On-chip synthesis and screening of a sialoside library yields a high affinity ligand for Siglec-7. ACS Chem. Biol. 2013, 8, 1417–1422. 10.1021/cb400125w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiarto G.; et al. A sialyltransferase mutant with decreased donor hydrolysis and reduced sialidase activities for directly sialylating LewisX. ACS Chem. Biol. 2012, 7, 1232–1240. 10.1021/cb300125k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briard J. G.; Jiang H.; Moremen K. W.; Macauley M. S.; Wu P. Cell-based glycan arrays for probing glycan-glycan binding protein interactions. Nat. Commun. 2018, 9, 880–890. 10.1038/s41467-018-03245-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins B. E.; Smith B. A.; Bengtson P.; Paulson J. C. Ablation of CD22 in ligand-deficient mice restores B cell receptor signaling. Nat. Immunol. 2006, 7, 199–206. 10.1038/ni1283. [DOI] [PubMed] [Google Scholar]
- Wherry E. J.; Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. 10.1038/nri3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blank C. U.; et al. Defining ‘T cell exhaustion’. Nat. Rev. Immunol. 2019, 19, 665–674. 10.1038/s41577-019-0221-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abouzahr-Rifai S.; et al. Resistance of tumor cells to cytolytic T lymphocytes involves Rho-GTPases and focal adhesion kinase activation. J. Biol. Chem. 2008, 283, 31665–31672. 10.1074/jbc.M800078200. [DOI] [PubMed] [Google Scholar]
- Razi N.; Varki A. Masking and unmasking of the sialic acid-binding lectin activity of CD22 (Siglec-2) on B lymphocytes. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 7469–7474. 10.1073/pnas.95.13.7469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins B. E.; Blixt O.; DeSieno A. R.; Bovin N.; Marth J. D.; Paulson J. C. Masking of CD22 by cis ligands does not prevent redistribution of CD22 to sites of cell contact. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 6104–6109. 10.1073/pnas.0400851101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crocker P. R.; Paulson J. C.; Varki A. Siglecs and their roles in the immune system. Nat. Rev. Immunol. 2007, 7, 255–266. 10.1038/nri2056. [DOI] [PubMed] [Google Scholar]
- Varchetta S.; et al. Lack of Siglec-7 expression identifies a dysfunctional natural killer cell subset associated with liver inflammation and fibrosis in chronic HCV infection. Gut 2016, 65, 1–9. 10.1136/gutjnl-2015-310327. [DOI] [PubMed] [Google Scholar]
- Hong S.; et al. Glycoengineering of NK Cells with Glycan Ligands of CD22 and Selectins for B-Cell Lymphoma Therapy. Angew. Chem., Int. Ed. 2021, 60, 3603–3610. 10.1002/anie.202005934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rillahan C. D.; Schwartz E.; McBride R.; Fokin V. V.; Paulson J. C. Click and pick: identification of sialoside analogues for siglec-based cell targeting. Angew. Chem., Int. Ed. 2012, 51, 11014–11018. 10.1002/anie.201205831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veluchamy J. P.; Kok N.; van der Vliet H. J.; Verheul H. M. W.; de Gruijl T. D.; Spanholtz J. The rise of allogeneic natural killer cells as a platform for cancer immunotherapy: recent innovations and future developments. Front. Immunol. 2017, 8, 631. 10.3389/fimmu.2017.00631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberschmidt O.; Kloess S.; Koehl U. Redirected primary human chimeric antigen receptor natural killer cells as an ″off-the-shelf immunotherapy″ for improvement in cancer treatment. Front. Immunol. 2017, 8, 654. 10.3389/fimmu.2017.00654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alter G.; Malenfant J. M.; Altfeld M. CD107a as a functional marker for the identification of natural killer cell activity. J. Immunol. Methods 2004, 294, 15–22. 10.1016/j.jim.2004.08.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.