

Abstract
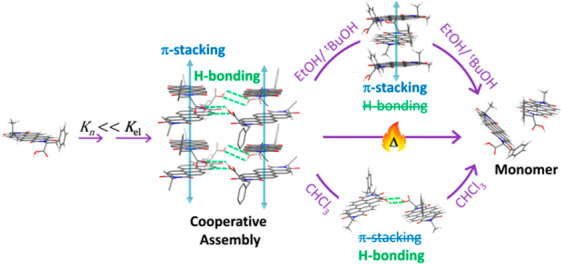
Cooperative interactions play a pivotal role in programmable supramolecular assembly. Emerging from a complex interplay of multiple noncovalent interactions, achieving cooperativity has largely relied on empirical knowledge. Its development as a rational design tool in molecular self-assembly requires a detailed characterization of the underlying interactions, which has hitherto been a challenge for assemblies that lack long-range order. We employ extensive one- and two-dimensional magic-angle-spinning (MAS) solid-state NMR spectroscopy to elucidate key structure-directing interactions in cooperatively bound aggregates of a perylene bisimide (PBI) chromophore. Analysis of 1H–13C cross-polarization heteronuclear correlation (CP-HETCOR) and 1H–1H double-quantum single-quantum (DQ-SQ) correlation spectra allow the identification of through-space 1H···13C and 1H···1H proximities in the assembled state and reveals the nature of molecular organization in the solid aggregates. Emergence of cooperativity from the synergistic interaction between a stronger π-stacking and a weaker interstack hydrogen-bonding is elucidated. Finally, using a combination of optical absorption, circular dichroism, and high-resolution MAS NMR spectroscopy based titration experiments, we investigate the anomalous solvent-induced disassembly of aggregates. Our results highlight the disparity between two well-established approaches of characterizing cooperativity, using thermal and good solvent-induced disassembly. The anomaly is explained by elucidating the difference between two disassembly pathways.
Short abstract
Strong π-stacking assisted by weak interstack H-bonding leads to cooperative self-assembly. Contrasting mechanisms of thermal vs solvent-induced disassembly complicate the analysis of cooperativity.
Introduction
Self-assembled one-dimensional organic nanostructures hold great promise in the area of systems chemistry and material science, and much of the success is owed to recent advances made in the area of programmable supramolecular polymerization.1,2 By virtue of reversibility and relaxed geometric restrictions, noncovalent interactions can drive a molecular self-assembly through multiple pathways and lead to a variety of assembled structures.3−6 However, in order to maximize performance, it is critical to achieve the desired assembly with high structural order and narrow polydispersity, while also maintaining a high degree of aggregation. In recent times, programmable supramolecular polymerization strategies have fulfilled this objective using a two-pronged approach: by steering the self-assembly toward a preferred pathway7−12 and ensuring that the assembled structure grows in a controlled manner.8−10,13−16 In terms of the underlying mechanism, both pathway selection and controlled growth are achieved through cooperative interactions.8−14
In the context of one-dimensional supramolecular polymerization, cooperative growth is characterized by a two-stage process: an initial, less-favored nucleation step that serves as a precursor to more spontaneous growth in the elongation stage. This deferred spontaneity, which results in the appearance of the well-known lag-phase in growth kinetics,8,14,17 is a consequence of additional noncovalent interactions that come into play after a well-defined nucleus is formed in solution.18−20 From a molecular design point of view, encoding such precise control over the sequence and chronology of noncovalent interactions is nontrivial. While empirical observations suggest a correlation between multiple noncovalent interactions and cooperativity,8−15,20−25 there are notable exceptions too.6,23,26 This underscores the need for a molecular-level characterization of noncovalent interactions that could elucidate how different interactions influence each other nonadditively and could lead to the development of a rational design principle to achieve cooperativity. Crystal structure can provide valuable information about the nature of molecular packing and the interactions involved, but its applicability is severely limited in most supramolecular assemblies that are inherently inhomogeneous, and/or lack long-range structural ordering. Small angle X-ray and/or neutron scattering techniques can look into the morphological heterogeneities of a molecular assembly but cannot comment on the nature of noncovalent interactions.27 Only in a few instances of hydrogen-bonded self-assemblies, a rationalization of cooperativity in terms of its molecular origin was done retrospectively, with the aid of computational methods and explicit calculation of stabilization energy per monomer.28−30
While investigating the molecular origin of cooperativity is challenging, its characterization and quantification are considered relatively straightforward. This typically involves monitoring the growth of a molecular self-assembly in solution, expressed in terms of the fraction of aggregated species or normalized degree of aggregation (αAgg), as a function of either solution temperature,31,32 monomer concentration,33 or solvent composition.34 Relevant thermodynamic parameters are extracted by analyzing these plots using appropriate mathematical models. All three approaches mentioned above, namely temperature, concentration, and solvent-composition dependence of αAgg, are considered equivalent in terms of the thermodynamic quantities one can obtain about the growth process. Therefore, preferring one approach over the other depends on the system under study, primarily governed by whether a particular variable can be suitably tuned to access the entire self-assembly equilibrium, from monomer (αAgg = 0) to the fully aggregated state (αAgg = 1).
The present work addresses both aspects of cooperativity discussed above. We employ an extensive magic-angle-spinning (MAS) solid-state NMR spectroscopy approach to investigate the origin of cooperativity in molecular self-assembly. MAS NMR does not require long-range structural order, allowing characterization of the local structure and dynamics of supramolecular assemblies.35−38 Notably, solid-state NMR spectroscopy has been previously used to investigate local ordering in disordered aggregates of molecular dyes.39 Further, one (1D) and two-dimensional (2D) MAS NMR techniques have been employed to understand key structure-directing interactions and phase behaviors in molecular self-assembly.40−45 With regards to the characterization of cooperative self-assembly, we show that the presumed equivalence of different experimental approaches is not strictly valid, thus highlighting the need to exercise caution in the analysis of solvent-composition dependent degree of aggregation, αAgg.
Results and Discussion
Cooperativity: Quantification and Molecular Origin
Figure 1a presents PBI-1, an unsymmetrically substituted perylene bisimide (PBI) with two prominent noncovalent interaction motifs: the aromatic PBI chromophore with strong π-stacking46 ability and the phenylalanine (Phe) side-group capable of hydrogen bonding. In methylcyclohexane (MCH), PBI-1 spontaneously aggregates with an H-type coupling between the neighboring chromophores. Figure 1b presents the evolution of optical absorption spectra as a solution of PBI-1 in MCH (15 μM) is heated under thermodynamic control (1 K/min). The room temperature spectrum of H-aggregated PBI-1 is characterized by an overall lowering of the absorption cross-section and a reduced 0–0 (515 nm) to 0–1 (479 nm) absorbance ratio (A0–0/A0–1) of 1.06. The broad, featureless absorption band in the 550–630 nm range is indicative of a rotationally twisted stacking of PBI chromophores, a feature commonly observed in PBI based H-aggregates.47 Disassembly at higher temperatures is evident from the overall increase in the absorbance and a monomer-like A0–0/A0–1 ratio of 1.52. A concomitant disappearance of the low-energy absorption band gives rise to a sharp isosbestic point at 532 nm. αAgg estimated from the absorbance at 515 nm exhibits a distinct nonsigmoidal profile (Figure 1c) that is characteristic of a cooperative self-assembly. Simulating the temperature-dependence of αAgg using the mass-balance model that takes into account an equilibrium between monomeric and aggregated PBI-1 in solution (Supporting Information, section 3)31 afforded a very good agreement with the experimental data. Thermodynamic parameters that characterize the nucleation and elongation stages of growth are presented in Table S1 (Supporting Information). The most important takeaway from the analysis of αAgg vs T is σ, the degree of cooperativity, expressed in terms of the ratio of nucleation to elongation stage equilibrium constants (σ = Kn/Kel). A low value of σ = 6.3 × 10–4 (at 293 K) translates to a 1600-fold increase in the elongation stage equilibrium constant, thus indicating a high degree of cooperativity in PBI-1 aggregates. Thermal disassembly of PBI-1 aggregates was also monitored using CD spectroscopy (Supporting Information, Figure S4a). PBI-1 aggregates in MCH exhibit a positive bisignated Cotton effect, suggesting a right-handed helical arrangement of PBI chromophores.48 Analysis of the temperature dependent αAgg, evaluated from the disappearance of CD signal upon heating (Figure S4b), agrees remarkably well with that of absorption spectroscopy: a cooperative assembly with σ = 6.06 × 10–4.
Figure 1.
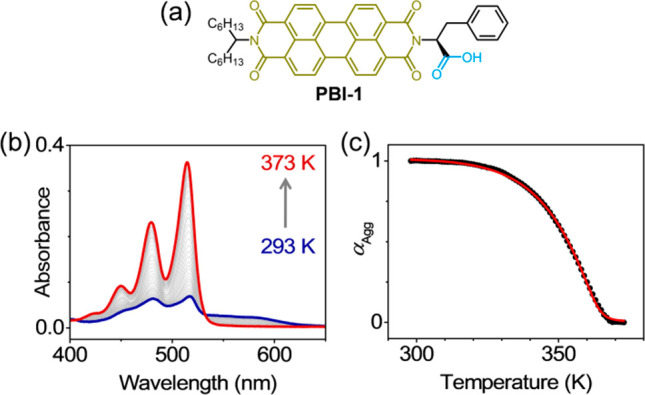
Cooperative self-assembly of PBI-1. (a) Molecular structure of PBI-1, showing the motifs responsible for π-stacking (olive) and hydrogen-bonding (blue). (b) Temperature-dependent optical absorption spectra of PBI-1 aggregates in methylcyclohexane (15 μM). (c) Variation of αAgg with temperature, derived from the 0–0 absorbance at 515 nm. The red line shows the simulated curve generated using mass-balance model for σ = 6.3 × 10–4, at 293 K.
In order to gain an insight into the molecular origin of cooperativity and the role of multiple interactions, we carried out a detailed MAS NMR investigation of solid PBI-1 aggregates grown from MCH solution. Figure 2a presents the 1H MAS NMR spectrum of PBI-1 aggregates. One can distinguish proton resonances corresponding to different functional groups of PBI-1 (assignments are color-coded as depicted in schematic structure and further validated by detailed 2D NMR analyses discussed subsequently): a broad signal from the branched aliphatic side-chain protons at 1.5 ppm, two benzylic protons at 3.2 and 4.2 ppm, two 1H–Cα next to the imide N at ∼5.6 ppm, and an intense signal at 7.3 ppm ascribed to the overlapped contributions from phenyl and PBI protons. We focus on the two functional motifs that are primarily responsible for PBI-1 self-assembly.
Figure 2.
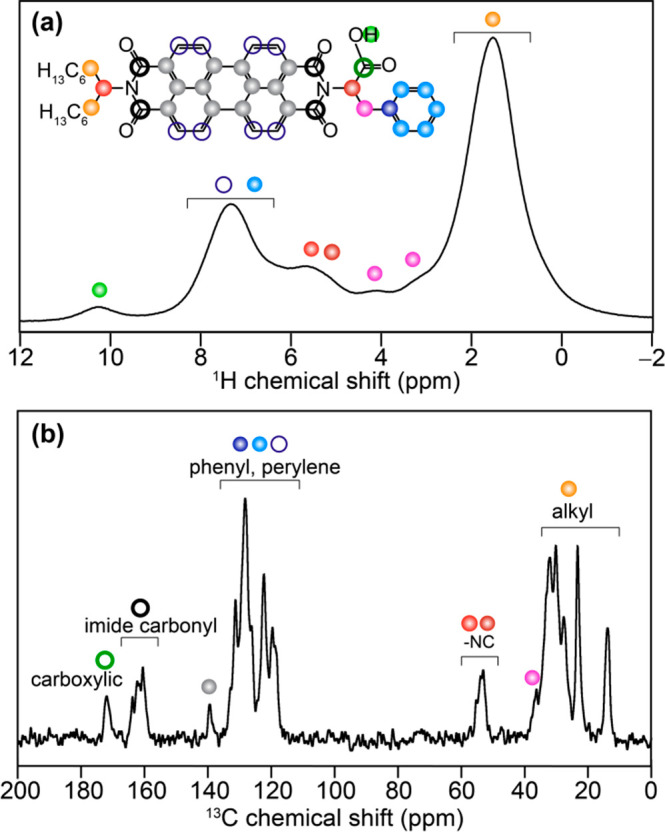
Solid-state 1D NMR spectra of solid PBI-1 aggregates acquired at 18.8 T (a) 1H MAS NMR (1H Larmor frequency = 800.1 MHz) recorded at 56 kHz MAS and (b) 13C CP-MAS NMR spectrum (13C Larmor frequency = 201.2 MHz) acquired at 50 kHz MAS using 2 ms CP contact time. 1H and 13C signals correspond to aliphatic and aromatic groups are colored as depicted in the inset to (a).
A comparison with the solution state 1H NMR spectrum of molecularly dissolved PBI-1 in CDCl3 (Supporting Information, Figure S1) reveals that the chemical shift difference (Δδ) between phenyl and PBI protons is greatly diminished in solid aggregates. In particular, the large shift toward lower frequency values (Δδ ∼ 1.2 ppm) experienced by the PBI protons is an indication of strong π-stacking between PBI chromophores.49−51 Such stacking can expose the protons of one PBI unit to the ring current of the neighboring PBI, resulting in a strong shielding. The spectrum also features a broad signal at 10.2 ppm, arising from weak hydrogen bonding interactions between carboxylic groups. This is corroborated by existing literature on the correlation between −COOH proton chemical shift and the strength of hydrogen-bond for a variety of carboxylic acids.52 It is possible that a weaker hydrogen-bonding between carboxyl acid side groups synergistically assists a stronger π-stacking interaction between the PBI units to give rise to cooperativity in self-assembled PBI-1.50 In the 13C cross-polarization (CP) MAS NMR spectrum of PBI-1 aggregates (Figure 2b), one can reliably assign signals corresponding to the terminal methyl (14.6 ppm) and intervening methylene (23.8 ppm) carbons of the alkyl side chain, perylene and phenyl group carbons (119–140 ppm), and the carboxylic acid carbon at 172 ppm. The solution-state 13C NMR spectrum (Supporting Information, Figure S2) agrees well with the 13C signals in the aliphatic and aromatic spectral regions. These 13C spectral assignments were further corroborated by DFT calculated chemical shifts (Supporting Information, Figure S5).
Next, we carried out an investigation into 1H···13C proximities using 2D 1H–13C CP heteronuclear correlation (CP-HETCOR) NMR spectroscopy. Analyses of 2D correlation peaks in HETCOR spectroscopy offer a unique way to identify different packing interactions, as well as reconcile that information with the emergence of cooperativity in molecular assembly. An advantage of 2D 1H–13C HETCOR NMR experiments lies in the resolution enhancement achieved by spreading 1H and 13C signals into two different frequency dimensions. This enables one to resolve correlation peaks associated with molecularly proximate and dipolar-coupled 1H···13C spin pairs with greater confidence. In order to characterize the intermolecular interactions responsible for PBI-1 self-assembly, it is important to first identify correlation signals that originate from the directly bonded C–H pairs and the ones at close intramolecular proximity. In doing so, we analyzed and compared the 2D 1H–13C HETCOR spectra acquired at different CP contact times. A preliminary analysis of 1D 1H → 13C CP signal intensity build-up as a function of the CP contact time revealed significant enhancements of 13C signals for a CP contact time of 4 ms and longer (Supporting Information, Figure S6). This was further corroborated by comparing 2D HETCOR spectra acquired with 2, 3, and 4 ms of CP contact time (Supporting Information, Figure S7 and Table S2), whereby the signals associated with short-range (covalently bonded) and long-range (through-space) 1H···13C proximities are distinguished. A detailed analysis of such 2D correlations associated with inter- and intramolecular interactions is presented in the Supporting Information (Figure S7c, Table S2).
In the 1H–13C HETCOR spectrum acquired with 4 ms of CP contact time (Figure 3a), 2D 1H–13C signals corresponding to the key structure-directing interactions that drive the assembly of PBI-1 molecules are indicated in solid and dashed ovals. The most notable of all are the 2D correlation signals between benzylic group (magenta) and the branched alkyl chain (orange dots). 1D 1H line-spectrum traced across the 13C dimension for the benzylic carbons at 37.2 ppm shows an intense peak at 1.8 ppm (Figure 3b), confirming dipolar coupling between the two groups. Similarly, 1H line-spectra corresponding to aliphatic C signals at 27.6 and 31.3 ppm also show sizable intensity at 3.2 ppm (benzylic protons). Interestingly, a similar correlation between terminal methyl carbons (14.5 ppm) and benzylic protons (3.2 ppm) is not apparent. Unlike all dipolar 1H···13C correlations discussed so far, these 2D correlation signals cannot result from intramolecular through-space interactions. Since the participating nuclei (benzylic and alkyl groups) are at opposite ends of the PBI-1 molecule and thus far separated, these 2D correlation signals present the first unambiguous evidence in favor of an antiparallel stacking. Such antiparallel stacking may bring Phe and branched alkyl groups on neighboring PBI-1 molecules in close proximity and also give rise to additional 2D correlation signals between phenyl ring carbons (blue) and alkyl protons (orange) at 123.4 (13C) and 1.8 ppm (1H). The 2D peak at 128.6 (13C) and 10.3 ppm (1H) is expected to originate from intermolecular close proximity between the carboxylic acid proton and the aromatic ring C of perylene and is consistent with the analogous 2D peak observed between the carboxylic 13C site (172.8 ppm) and perylene protons (7.8 ppm), as depicted in dashed ovals. Such proximity to the aromatic ring can shield the hydrogen-bonded carboxylic acid proton efficiently and be in part responsible for its low chemical shift of 10.3 ppm. In addition to these, 2D peaks involving the 13C signal of Cα (54.4 ppm, burgundy dot) and alkyl side chain protons (1.8 ppm) and between perylene carbons and alkyl protons at 128.6 (13C) and 1.6 ppm (1H) are attributed to intramolecular as well as intermolecular 1H···13C dipole–dipole interactions resulting from the antiparallel stacking of PBI-1 molecules. In addition, any intermolecular contacts originating from side-on interactions between adjacent π-stacked columns may interfere with the 2D correlation peaks but are likely to be of much weaker intensity. Consequently, disentangling the contribution of 1H–13C dipolar interactions along the π-stacking axis from those originating from side-on interactions is not straightforward.
Figure 3.
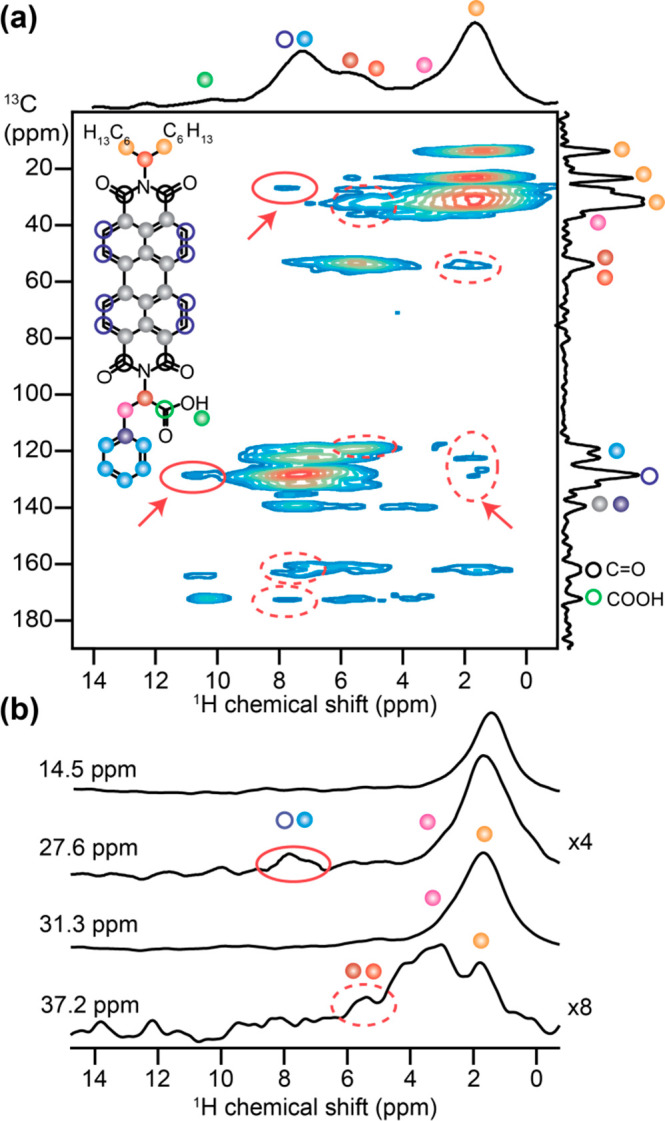
Solid-state (a) 2D 1H–13C HETCOR NMR spectrum of solid PBI-1 aggregates at 18.8 T, 298 K and 50 kHz MAS using 4 ms CP contact time, and (b) 1D 1H line-spectra traced across the 13C dimension for the aliphatic signals at 14.5, 27.6, 31.3, and 37.2 ppm. Solid and dashed ovals denote the 2D signals originating from purely intermolecular and from a combination of inter- and intramolecular C···H proximities, respectively. 1H and 13C signals associated with aliphatic and aromatic groups are colored as shown in the inset.
Information on local 1H···1H proximities at subnanometer (<0.5 nm) distances can be obtained from 1H double-quantum chemical shifts.40,45 In a 2D 1H double-quantum (DQ) single-quantum (SQ) correlation spectrum (Figure 4), the DQ coherences appear at the sum of two dipole-coupled SQ chemical shifts: the on- and off-diagonal peaks represent chemically equivalent and chemically distinct 1H···1H proximities, respectively. A detailed analysis of 2D correlation intensities arising from intramolecular1H···1H interactions in PBI-1 aggregates is presented in the Supporting Information (Figure S8, Table S3). Here we focus on the correlations arising out of the intermolecular interactions. Notably, the off-diagonal peak at δDQ of 1.6 + 7.4 = 9.0 ppm suggests through-space proximity between alkyl and aromatic protons, which is also consistent with the appearance of δDQ, 1.7 + 6.0 = 7.7 ppm and δDQ, 1.7 + 5.3 = 7.0 ppm peaks that indicate through-space 1H···1H proximities between alkyl protons and 1H–Cα. The latter correlation peaks are better resolved in the 1H DQ spectrum acquired with 71.4 μs DQ excitation time (Supporting Information, Figure S8b and Table S3). Since alkyl and aromatic protons in PBI-1 are spatially far from each other (>5 Å), a dipolar interaction between them is likely to be intermolecular and is consistent with the antiparallel stacking suggested earlier. Antiparallel stacking of PBI-1 molecules is best consolidated by the appearance of a reasonably intense 1H DQ coherence at δDQ, 5.3 + 6.0 = 11.3 ppm (red arrow), which indicates close proximity between two 1H–Cα protons. Clearly, the two 1H–Cα protons of the same PBI-1 molecule that are at a separation of over 5 Å cannot support such a strong dipolar coupling. A higher intensity of this DQ peak compared to the one at δDQ= 9.0 ppm suggests that the separation between the pair of 1H–Cα protons across the PBI stack is shorter than that between alkyl and aromatic protons. One expects that to be true for a rotationally twisted stack of PBI-1 molecules, in which the separation between the coupled nuclei increases as one moves away from groups that are centrally located, such as 1H–Cα, to ones that more peripheral, like alkyl and Phe groups. The 1H DQ peak at 7.4 + 10.3 = 17.7 ppm presumably arises from a combination of intra- and intermolecular 1H···1H dipolar coupling between carboxylic acid and aromatic protons. Finally, a weak intensity peak at δDQ, 10.3 + 10.3 = 20.6 ppm (red arrow) indicates hydrogen-bonding between carboxylic acid groups of neighboring PBI-1 molecules. We bear in mind that an antiparallel stacking of PBI-1 molecules rules out the possibility of any hydrogen-bonding between −COOH groups along the stacking axis. Therefore, we conclude that the observed hydrogen bonding must be between PBI-1 molecules belonging to the adjacent stacks. The intensity ratio of DQ peaks at a specific SQ frequency can be used to quantify interatomic distances for isolated 1H–1H pairs.53 The intensity ratio of DQ signals at 17.7 and 20.6 ppm at δSQ = 10.3 ppm is 4:1. From the ratio, we estimate that the distance between carboxylic acid protons of PBI-1 molecules hydrogen-bonded to each other is about ∼1.26 times greater than the intramolecular 1H–1H distance between the carboxylic acid proton and the nearest aromatic proton. This once again indicates that the hydrogen bonding interaction in PBI-1 aggregates is weak. Thus, while π-stacking is largely responsible for the longitudinal growth of PBI-1 aggregate, a weaker interstack hydrogen-bonding between −COOH groups contributes to its lateral growth. It is conceivable that in the early stages of self-assembly, a stronger π-stacking between PBI-1 molecules is primarily responsible for molecular association leading to the formation of a nucleus. In the subsequent elongation phase, additional interstack hydrogen-bonding comes into play that complements π-stacking to drive a more spontaneous growth of PBI-1 aggregates. Overall, the 2D ssNMR experiments provide a rich source of atomic-level information on the cooperatively bound molecular self-assembly, enabled by the identification of key structure directing intermolecular interactions with site-specificity.
Figure 4.
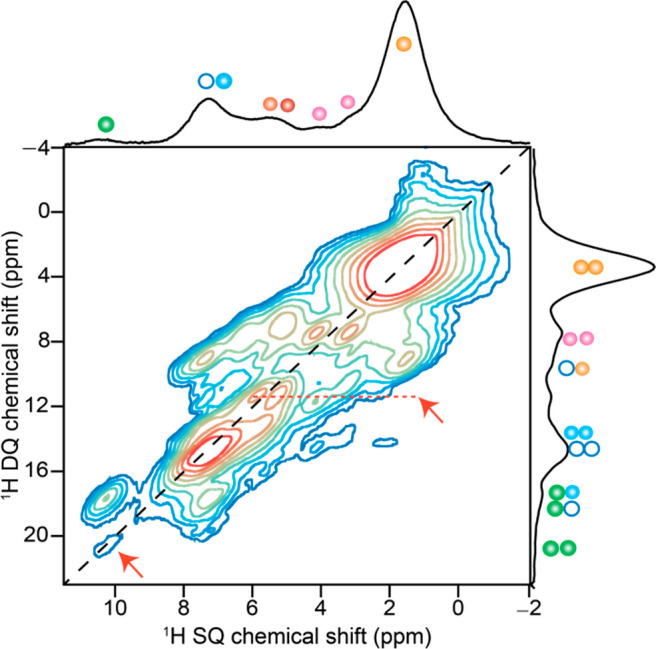
Solid-state 2D 1H–1H DQ-SQ correlation NMR spectrum of solid PBI-1 aggregates acquired at 800.1 MHz and 56 kHz MAS with 35.6 μs of DQ excitation time. 1H DQ correlation peaks that correspond to inter- and intramolecular H–H proximities (<5 Å) are colored as per the schematic of PBI-1 in Figures 2 and 3
Anomalous Solvent-Induced Disassembly
Having elucidated the role of multiple noncovalent interactions in the context of cooperative growth in PBI-1 aggregates, we investigate the effect of solvent composition on the aggregation process. It is generally accepted that analysis of solvent-composition dependence of αAgg affords very similar information about the self-assembly mechanism as temperature dependence. Analyzing cooperativity from solvent-induced disassembly is therefore a useful alternative for systems, where temperature cannot be suitably varied because of poor solubility and stability issues, or the possibility of a phase-transition.34 Investigation of solvent-composition dependence involves varying the ratio of a good-to-bad solvent. In our experiments, MCH was used as the bad solvent, whereas the good solvent was chosen by taking into account the specific nature of solvent–PBI-1 interactions. We carried out two sets of experiments, each with the aim to address a specific noncovalent interaction in PBI-1 aggregates. Figure 5a presents the effect of tert-butanol (tBuOH) on the optical absorption spectra of PBI-1 aggregates in MCH. Since tBuOH has strong hydrogen-bonding capabilities, its interaction with the −COOH group disrupts the intermolecular hydrogen bonding between adjacent PBI-1 molecules causing aggregate disassembly. A plot of αAgg versus the volume fraction of tBuOH (f), derived from the absorption spectra is presented in Figure 5b. A cursory examination of the plot quite surprisingly reveals the gradual nature of solvent induced disassembly. Absence of a critical volume fraction across which αAgg changes sharply is not consistent with the notion of a cooperatively bound system. We analyzed the solvent-composition dependence of αAgg within the framework of Goldstein and Stryer’s model (Supporting Information, section 8),33,54 which accounts for monomer association and cooperativity in terms of the elongation stage equilibrium constant (Kel) and σ (= Kn/Kel), respectively. A key assumption of this model is that the interaction between the aggregate and a good solvent is weak, which allows one to express the free energy change upon monomer binding (ΔG°′) as a linear function of f:
Here ΔG° is the free energy change in absence of a good solvent, and m quantifies the denaturing ability of the good solvent. In stark contrast to the temperature-dependence of αAgg presented earlier, the analysis of solvent-composition dependence indicates an isodesmic association in PBI-1 aggregates (σ = 1), where each monomer association step is equally spontaneous. We reconfirmed this anomalous disassembly behavior by analyzing αAgg vs ftBuOH plots for three different PBI-1 concentrations (Figure 5b). In each case, the best-fit to the experimental data afforded identical values for the parameters (ΔG°, m), thus validating the robustness of our analyses (see Table S4). In a separate set of experiments, CHCl3 was used as the good solvent to preferentially disrupt π-stacking between PBI-1 molecules (Figure 5c). Once again, analyses of αAgg vs fCHCl3 plots at two different PBI-1 concentrations (Figure 5d and Table S4) unambiguously suggest isodesmic aggregation. To further support our findings, we carried out solvent-composition dependent CD spectroscopy. Analyses of αAgg vs f obtained from CD spectra reaffirms the agreement with the isodesmic model (Supporting Information, Figure S9). It is worth noting that ΔG° values obtained from the study of solvent-composition dependence are consistently lower (−29 and −36.7 kJ/mol from tBuOH and CHCl3 induced disassembly, respectively; Table S4) than that obtained from temperature-dependence (−41 kJ/mol, Table S1) studies. These results clearly stress the need to reassess the applicability of these two approaches in elucidating the self-assembly mechanism.
Figure 5.
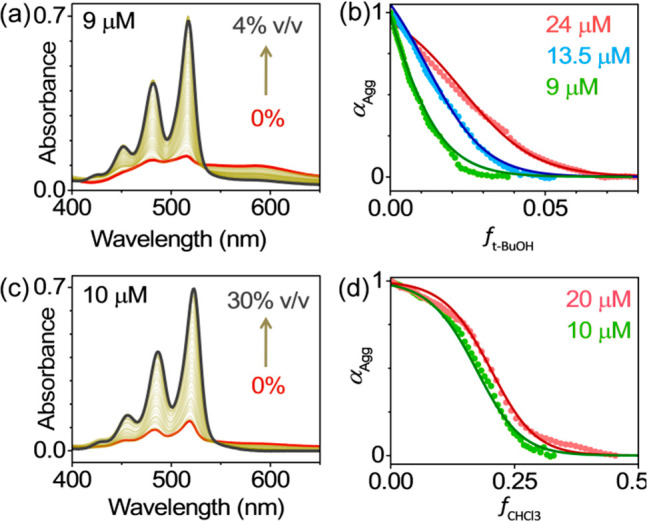
Good solvent induced disassembly. Absorption spectra of PBI-1 in MCH show progressive disassembly upon increasing the volume fraction of (a) tBuOH and (c) CHCl3. Analyses of corresponding αAggvs f plots in b and d show isodesmic behavior. The solid lines are fit to an isodesmic model.
Deducing the self-assembly mechanism from the disassembly process, be it thermal or solvent-induced, relies on one important assumption: that assembly and disassembly pathways are the same, and one process is just the reverse of the other. We envisage that a departure from this key assumption is responsible for the observed anomaly. One expects the effect of temperature to be straightforward: the monomer association constants (Kel, Kn) decrease with increasing temperature, in accordance with the van’t Hoff equation. Thus, in the absence of any competing pathways, thermal disassembly follows a course that is same as that of molecular assembly, only in reverse. This reversibility allows one to deduce the self-assembly mechanism from αAgg vs T behavior. The same, however, may not be true for disassembly caused by a good solvent. Since solvent-molecule interactions are very specific in nature, a good solvent can disproportionately affect different noncovalent interactions. Consequently, solvent-induced disassembly can follow a very different trajectory than the self-assembly process. This scenario is especially likely for aggregates, where interactions of very different natures are involved. In order to test this hypothesis, we carried out high-resolution MAS (HR-MAS) NMR titration experiments55 with variable spinning frequencies using two different good solvents (Figure 6). Our aim was to monitor the evolution of different noncovalent interactions in solid PBI-1 aggregates at an early stage of solvent-induced disassembly. Figure 6a presents a comparison of 1H HR-MAS NMR spectra of PBI-1 aggregates in the presence of different weight fractions of ethanol-d.56 A closer examination in the 6–12 ppm range reveals the changes brought about by the good solvent. A gradual loss of signal at 10.3 ppm suggests a progressive weakening of the intermolecular hydrogen-bonding between −COOH groups in the solid aggregate and eventually a complete disruption above 50 wt % of ethanol-d. Quite interestingly, the aromatic region of the NMR spectrum shows no change during this titration experiment. Clearly, ethanol causes a selective disruption of hydrogen-bonding, while retaining the π-stacking interactions in solid PBI-1 aggregates.57 In contrast, the evolution of 1H MAS NMR spectra upon the addition of CDCl3 to solid PBI-1 aggregates (Figure 6b) indicates a very different disassembly pathway. Initially, the changes are less distinct, as the spectrum becomes progressively narrower (see Supporting Information). This eventually leads to a significantly well-resolved 1H NMR spectrum at 78 wt % of CDCl3. While the initial line narrowing is presumably due to the breakdown of large aggregates into smaller aggregated domains, a large shift of the aromatic protons to higher frequencies (Δδ ∼ 0.9 ppm) unambiguously indicates a widespread disruption of PBI π-stacking. A largely unchanged signal at 10.3 ppm corroborates the retention of hydrogen-bonding interaction during titration with CDCl3.
Figure 6.
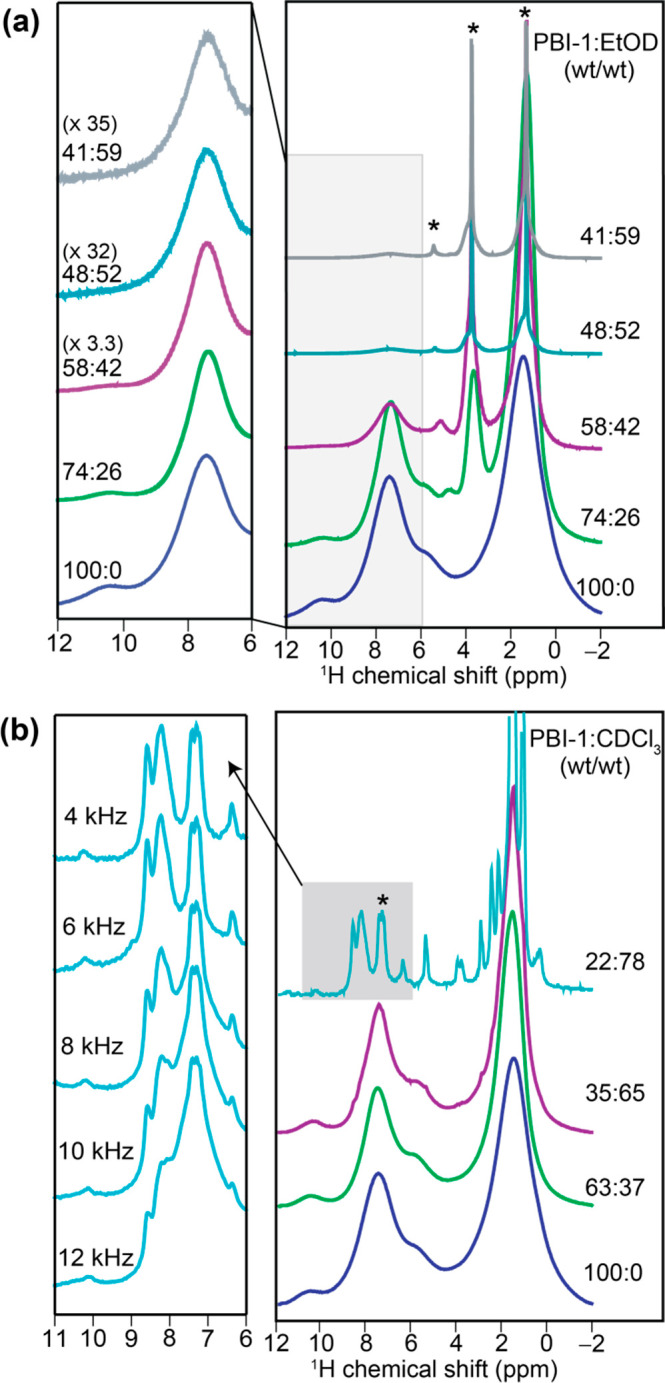
1D 1H HR-MAS NMR spectra of solid PBI-1 aggregates acquired at 800.1 MHz as a function of good solvent. (a) Bottom-up stack plot of PBI-1:ethanol-d, wt/wt, 100:0 (blue), 74:26 (green), 58:42 (violet), 48:52 (cyan), and 41:59 (gray) acquired at 20, 20, 18, 12, and 8 kHz of MAS, respectively. Expanded region (left) depicts normalized intensities of aromatic 1H signals. (b) Bottom-up stack plot of PBI-1:CDCl3, wt/wt 100:0 (blue), 63:37 (green), 35:65 (violet) acquired at 20 kHz MAS, and 22:78 (cyan) acquired at 4 kHz MAS. The expanded region (left) depicts aromatic regions of 1H spectra 22:78 (cyan) acquired at different MAS frequencies varying from 4 to 12 kHz (Supporting Information, section 9). * denotes solvent peaks associated with (a) ethanol-d (EtOD) and (b) chloroform-d (CDCl3).
Evidently, the manner in which solvent-induced disassembly is brought about in optical absorption vis-a-vis MAS NMR spectroscopy experiments has certain key differences. In the former, shorter soluble aggregates interact with the good-solvent and dissociate in a homogeneous solution-phase process. In contrast, the NMR experiments involved disassembly of much larger aggregates in an inhomogeneous solid-solvent phase. While this prohibits any quantitative comparison of the two disassembly processes, we are able to conclude that a good-solvent induced disassembly is achieved by disrupting one-interaction-at-a-time. Such interaction-specific aggregate dissociation is therefore driven primarily by a lowering of enthalpic gain (ΔHel) and is quite distinct from thermal disassembly, which is caused by an increase in the entropic loss (−TΔSel). This key distinction between the two disassembly processes can complicate analysis of self-assembly mechanism from the corresponding αAgg plots. We recall that our ability to distinguish between isodesmic and cooperative mechanisms relies on the behavior of αAgg vs f plots in the limit of very low αAgg.30,34 In this limit, isodesmic aggregates dissociate gradually, whereas cooperatively bound aggregates show a more drastic change in αAgg as the self-assembly process moves from nucleation to elongation regime. It is conceivable that a cooperative aggregate that dissociates one-interaction-at-a-time is initially reduced to a weakly bound aggregate state that is held together by a single noncovalent interaction. This change from a multi-interaction to a single-interaction aggregate, though significant from the standpoint of aggregate stability, may not affect the optical absorption spectra, as individual chromophores are still bound together, allowing excitonic interactions to operate. A subsequent disassembly of such monovalent aggregates to free monomers brings the most significant changes in the absorption spectra. Consequently, absorption spectroscopy does not probe the disassembly of the initial cooperatively bound aggregate, but that of an intermediate aggregated state. This intermediate aggregate, which is held together by a single noncovalent interaction, is more likely to exhibit the characteristics of an isodesmic association. Since thermal disassembly is free from such artifacts, temperature dependence of αAgg offers a more reliable method for characterizing self-assembly mechanism in small-molecule aggregates.
Conclusions
In conclusion, our work addresses two important questions pertaining to cooperative self-assembly of small molecules: molecular origin of cooperative interactions and its characterization. With regards to its origin, a correlation between multiple noncovalent interactions and cooperativity has been empirically observed in the past. However, the basis for such a correlation was not adequately understood in the absence of a detailed characterization of the local structure and the noncovalent interactions stabilizing it, particularly in assemblies that lack long-range order. To this end, we performed solid-state 1D and 2D MAS NMR spectroscopy on cooperatively bound aggregates of PBI-1. Our analyses of through-space dipole-coupling between nonbonded 1H···13C and 1H···1H spin pairs establish that PBI-1 molecules stack in a rotationally displaced, antiparallel fashion. This dominant π-stacking interaction is complemented by a weaker interstack hydrogen-bonding between the carboxylic acid side groups, resulting in a cooperative self-assembly. The ability to rationalize cooperativity at an atomistic level could offer a way to design molecular building blocks that can be made to assemble along a specific pathway to a desired supramolecular structure. In addition, we compared the conflicting conclusions about the extent of cooperativity in PBI-1 aggregates, derived from two well-established characterization methods, namely temperature and solvent-composition dependence of αAgg. HR-MAS NMR based titration experiments present a clear evidence that a good-solvent induced disassembly progresses in an interaction-specific manner. In aggregates that employ multiple noncovalent interactions of different kinds, this can lead to an anomalous characterization of cooperativity. Finally, our work underscores the importance of bridging the gap between solution-phase characterization techniques and solid-state structure elucidation using combined solid-state NMR and modeling approaches, the applicability of which extends beyond supramolecular chemistry to other disordered material systems.
Acknowledgments
The authors gratefully acknowledge EUSMI (Proposal: E191200366) for access to the 800 MHz solid-state NMR spectrometer. D.C. acknowledges IGSTC (Project: LABELONIK) and IISER Kolkata for financial support. G.N.M.R. acknowledges the University of Lille and region Hauts-de-France for financial support and Prof. Olivier Lafon and Dr. Julien Trébosc for insightful discussions.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.1c00604.
Experimental methods, synthesis and characterization, and additional figures and tables (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Yin P.; Choi H. M. T.; Calvert C. R.; Pierce N. A. Programming biomolecular self-assembly pathways. Nature 2008, 451, 318–322. 10.1038/nature06451. [DOI] [PubMed] [Google Scholar]
- Ong L. L.; Hanikel N.; Yaghi O. K.; Grun C.; Strauss M. T.; Bron P.; Lai-Kee-Him J.; Schueder F.; Wang B.; Wang P.; Kishi J. Y.; Myhrvold C.; Zhu A.; Jungmann R.; Bellot G.; Ke Y.; Yin P. Programmable self-assembly of three-dimensional nanostructures from 10,000 unique components. Nature 2017, 552, 72–77. 10.1038/nature24648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohr A.; Lysetska M.; Würthner F. Supramolecular stereomutation in kinetic and thermodynamic self- assembly of helical merocyanine dye nanorods. Angew. Chem., Int. Ed. 2005, 44, 5071–5074. 10.1002/anie.200500640. [DOI] [PubMed] [Google Scholar]
- Korevaar P. A.; George S. J.; Markvoort A. J.; Smulders M. M. J.; Hilbers P. A. J.; Schenning A. P. H. J.; De Greef T. F. A.; Meijer E. W. Pathway complexity in supramolecular polymerization. Nature 2012, 481, 492–496. 10.1038/nature10720. [DOI] [PubMed] [Google Scholar]
- Fennel F.; Wolter S.; Xie Z.; Plötz P.-A.; Kühn O.; Würthner F.; Lochbrunner S. Biphasic self- assembly pathways and size- dependent photophysical properties of perylene bisimide dye aggregates. J. Am. Chem. Soc. 2013, 135, 18722–18725. 10.1021/ja409597x. [DOI] [PubMed] [Google Scholar]
- Samanta S.; Chaudhuri D. Suppressing excimers in H-aggregates of perylene bisimide folda-dimer: role of dimer conformation and competing assembly pathways. J. Phys. Chem. Lett. 2017, 8, 3427–3432. 10.1021/acs.jpclett.7b01338. [DOI] [PubMed] [Google Scholar]
- Wagner W.; Wehner M.; Stepanenko V.; Ogi S.; Würthner F. Living supramolecular polymerization of a perylene bisimide dye into fluorescent J- aggregates. Angew. Chem., Int. Ed. 2017, 56, 16008–16012. 10.1002/anie.201709307. [DOI] [PubMed] [Google Scholar]
- Ogi S.; Sugiyasu K.; Manna S.; Samitsu S.; Takeuchi M. Living supramolecular polymerization realized through a biomimetic approach. Nat. Chem. 2014, 6, 188–195. 10.1038/nchem.1849. [DOI] [PubMed] [Google Scholar]
- Ogi S.; Fukui T.; Jue M. L.; Takeuchi M.; Sugiyasu K. Kinetic control over pathway complexity in supramolecular polymerization through modulating the energy landscape by rational molecular design. Angew. Chem., Int. Ed. 2014, 53, 14363–14367. 10.1002/anie.201407302. [DOI] [PubMed] [Google Scholar]
- Greciano E. E.; Matarranz B.; Sánchez L. Pathway complexity versus hierarchical self- assembly in N-annulated perylenes: structural effects in seeded supramolecular polymerization. Angew. Chem., Int. Ed. 2018, 57, 4697–4701. 10.1002/anie.201801575. [DOI] [PubMed] [Google Scholar]
- Mabesoone M. F. J.; Markvoort A. J.; Banno M.; Yamaguchi T.; Helmich F.; Naito Y.; Yashima E.; Palmans A. R. A.; Meijer E. W. Competing interactions in hierarchical porphyrin self-assembly introduce robustness in pathway complexity. J. Am. Chem. Soc. 2018, 140, 7810–7819. 10.1021/jacs.8b02388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner W.; Wehner M.; Stepanenko V.; Würthner F. Supramolecular block copolymers by seeded living polymerization of perylene bisimides. J. Am. Chem. Soc. 2019, 141, 12044–12054. 10.1021/jacs.9b04935. [DOI] [PubMed] [Google Scholar]
- Ogi S.; Stepanenko V.; Sugiyasu K.; Takeuchi M.; Würthner F. Mechanism of self-assembly process and seeded supramolecular polymerization of perylene bisimide organogelator. J. Am. Chem. Soc. 2015, 137, 3300–3307. 10.1021/ja511952c. [DOI] [PubMed] [Google Scholar]
- Endo M.; Fukui T.; Jung S. H.; Yagai S.; Takeuchi M.; Sugiyasu K. Photoregulated living supramolecular polymerization established by combining energy landscapes of photoisomerization and nucleation–elongation processes. J. Am. Chem. Soc. 2016, 138, 14347–14353. 10.1021/jacs.6b08145. [DOI] [PubMed] [Google Scholar]
- Ma X.; Zhang Y.; Zhang Y.; Liu Y.; Che Y.; Zhao J. Fabrication of chiral-selective nanotubular heterojunctions through living supramolecular polymerization. Angew. Chem., Int. Ed. 2016, 55, 9539–9543. 10.1002/anie.201602819. [DOI] [PubMed] [Google Scholar]
- Kang J.; Miyajima D.; Mori T.; Inoue Y.; Itoh Y.; Aida T. A rational strategy for the realization of chain growth supramolecular polymerization. Science 2015, 347, 646–651. 10.1126/science.aaa4249. [DOI] [PubMed] [Google Scholar]
- Baram J.; Weissman H.; Rybtchinski B. Supramolecular polymer transformation: a kinetic study. J. Phys. Chem. B 2014, 118, 12068–12073. 10.1021/jp507945t. [DOI] [PubMed] [Google Scholar]
- De Greef T. F. A.; Smulders M. M. J.; Wolffs M.; Schenning A. P. H. J.; Sijbesma R. P.; Meijer E. W. Supramolecular polymerization. Chem. Rev. 2009, 109, 5687–5854. 10.1021/cr900181u. [DOI] [PubMed] [Google Scholar]
- Hunter C. A.; Anderson H. L. What is cooperativity?. Angew. Chem., Int. Ed. 2009, 48, 7488–7499. 10.1002/anie.200902490. [DOI] [PubMed] [Google Scholar]
- Mahadevi A. S.; Sastry G. N. Cooperativity in noncovalent interactions. Chem. Rev. 2016, 116, 2775–2825. 10.1021/cr500344e. [DOI] [PubMed] [Google Scholar]
- Badjic J. D.; Nelson A.; Cantrill S. J.; Turnbull W. B.; Stoddart J. F. Multivalency and cooperativity in supramolecular chemistry. Acc. Chem. Res. 2005, 38, 723–732. 10.1021/ar040223k. [DOI] [PubMed] [Google Scholar]
- Chung M.-K.; Lee S. J.; Waters M. L.; Gagné M. R. Self-assembled multi-component catenanes: the effect of multivalency and cooperativity on structure and stability. J. Am. Chem. Soc. 2012, 134, 11430–11443. 10.1021/ja302347q. [DOI] [PubMed] [Google Scholar]
- Rest C.; Kandanelli R.; Fernandez G. Strategies to create hierarchical self-assembled structures via cooperative non-covalent interactions. Chem. Soc. Rev. 2015, 44, 2543–2572. 10.1039/C4CS00497C. [DOI] [PubMed] [Google Scholar]
- Kulkarni C.; Bejagam K. K.; Senanayak S. P.; Narayan K. S.; Balasubramanian S.; George S. J. Dipole moment driven cooperative supramolecular polymerization. J. Am. Chem. Soc. 2015, 137, 3924–3932. 10.1021/jacs.5b00504. [DOI] [PubMed] [Google Scholar]
- Li Y.; Wang Y.; Huang G.; Gao J. Cooperativity principles in self-assembled nanomedicine. Chem. Rev. 2018, 118, 5359–5391. 10.1021/acs.chemrev.8b00195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobko N.; Paraskevas L.; del Rio E.; Dannenberg J. J. Cooperativity in amide hydrogen bonding chains: implications for protein-folding models. J. Am. Chem. Soc. 2001, 123, 4348–4349. 10.1021/ja004271l. [DOI] [PubMed] [Google Scholar]
- Hollamby M. J.; Aratsu K.; Pauw B. R.; Rogers S. E.; Smith A. J.; Yamauchi M.; Lin X.; Yagai S. Simultaneous SAXS and SANS analysis for the detection of toroidal supramolecular polymers composed of noncovalent supermacrocycles in solution. Angew. Chem. 2016, 128, 10044–10047. 10.1002/ange.201603370. [DOI] [PubMed] [Google Scholar]
- Filot I. A. W.; Palmans A. R. A.; Hilbers P. A. J.; van Santen R. A.; Pidko E. A.; de Greef T. F. A. Understanding Cooperativity in Hydrogen-Bond-Induced Supramolecular Polymerization: A Density Functional Theory Study. J. Phys. Chem. B 2010, 114, 13667–13674. 10.1021/jp1072928. [DOI] [PubMed] [Google Scholar]
- Nakano Y.; Hirose T.; Stals P. J. M.; Meijer E. W.; Palmans A. R. A. Conformational analysis of supramolecular polymerization processes of disc-like molecules. Chem. Sci. 2012, 3, 148–155. 10.1039/C1SC00547B. [DOI] [Google Scholar]
- Kulkarni C.; Meijer E. W.; Palmans A. R. A. Cooperativity scale: a structure mechanism correlation in the self assembly of benzene-1,3,5-carbozamides. Acc. Chem. Res. 2017, 50, 1928–1936. 10.1021/acs.accounts.7b00176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smulders M. M. J.; Nieuwenhuizen M. M. L.; de Greef T. F. A.; van der Schoot P.; Schenning A. P. H. J.; Meijer E. W. How to distinguish isodesmic from cooperative supramolecular polymerisation. Chem. - Eur. J. 2010, 16, 362–367. 10.1002/chem.200902415. [DOI] [PubMed] [Google Scholar]
- ten Eikelder H. M. M.; Markvoort A. J.; de Greef T. F. A.; Hilbers P. A. J. An equilibrium model for chiral amplification in supramolecular polymers. J. Phys. Chem. B 2012, 116, 5291–5301. 10.1021/jp300622m. [DOI] [PubMed] [Google Scholar]
- Zhao D.; Moore J. S. Nucleation–elongation: a mechanism for cooperative supramolecular polymerization. Org. Biomol. Chem. 2003, 1, 3471–3491. 10.1039/B308788C. [DOI] [PubMed] [Google Scholar]
- Korevaar P. A.; Schaefer C.; de Greef T. F. A.; Meijer E. W. Controlling chemical self-assembly by solvent-dependent dynamics. J. Am. Chem. Soc. 2012, 134, 13482–13491. 10.1021/ja305512g. [DOI] [PubMed] [Google Scholar]
- Hansen M. R.; Graf R.; Spiess H. W. Interplay of structure and dynamics in functional macromolecular and supramolecular systems as revealed by magnetic resonance spectroscopy. Chem. Rev. 2016, 116, 1272–1308. 10.1021/acs.chemrev.5b00258. [DOI] [PubMed] [Google Scholar]
- Weingarth M.; Baldus M. Solid-state NMR-based approaches for supramolecular structure elucidation. Acc. Chem. Res. 2013, 46, 2037–2046. 10.1021/ar300316e. [DOI] [PubMed] [Google Scholar]
- Hansen M. R.; Graf R.; Spiess H. W. Solid-state NMR in macromolecular systems: insights on how molecular entities move. Acc. Chem. Res. 2013, 46, 1996–2007. 10.1021/ar300338b. [DOI] [PubMed] [Google Scholar]
- Tasios N.; Grigoriadis C.; Hansen M. R.; Wonneberger H.; Li C.; Spiess H. W.; Müllen K.; Floudas G. Self-assembly, dynamics, and phase transformation kinetics of donor–acceptor substituted perylene derivatives. J. Am. Chem. Soc. 2010, 132, 7478–7487. 10.1021/ja102150g. [DOI] [PubMed] [Google Scholar]
- Hansen M. R.; Graf R.; Sekharan S.; Sebastiani D. Columnar packing motifs of functionalized perylene derivatives: local molecular order despite long-range disorder. J. Am. Chem. Soc. 2009, 131, 5251–5256. 10.1021/ja8095703. [DOI] [PubMed] [Google Scholar]
- Seifrid M.; Reddy G. N. M.; Chmelka B. F.; Bazan G. C. Insight into the structures and dynamics of organic semiconductors through solid-state NMR spectroscopy. Nat. Rev. Mater. 2020, 5, 910–930. 10.1038/s41578-020-00232-5. [DOI] [Google Scholar]
- Reddy G. N. M.; Huqi A.; Iuga D.; Sakurai S.; Marsh A.; Davis J. T.; Masiero S.; Brown S. P. Co-existence of distinct supramolecular assemblies in solution and in the solid State. Chem. - Eur. J. 2017, 23, 2315–2322. 10.1002/chem.201604832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy G. N. M.; Marsh A.; Davis J. T.; Masiero S.; Brown S. P. Interplay of noncovalent interactions in ribbon-like guanosine self-assembly: an NMR crystallography study. Cryst. Growth Des. 2015, 15, 5945–5954. 10.1021/acs.cgd.5b01440. [DOI] [Google Scholar]
- Seifrid M. T.; Reddy G. N. M.; Zhou C.; Chmelka B. F.; Bazan G. C. Direct observation of the relationship between molecular topology and bulk morphology for a π-conjugated material. J. Am. Chem. Soc. 2019, 141, 5078–5082. 10.1021/jacs.8b13200. [DOI] [PubMed] [Google Scholar]
- Hansen M. R.; Schnitzler T.; Pisula W.; Graf R.; Müllen K.; Spiess H. W. Cooperative molecular motion within a self-assembled liquid-crystalline molecular wire: the case of a TEG-substituted perylenediimide disc. Angew. Chem., Int. Ed. 2009, 48, 4621–4624. 10.1002/anie.200900547. [DOI] [PubMed] [Google Scholar]
- Webber A. L.; Masiero S.; Pieraccini S.; Burley J. C.; Tatton A. S.; Iuga D.; Pham T. N.; Spada G. P.; Brown S. P. Identifying guanosine self assembly at natural isotopic abundance by high-resolution 1H and 13C solid-state NMR spectroscopy. J. Am. Chem. Soc. 2011, 133, 19777–19795. 10.1021/ja206516u. [DOI] [PubMed] [Google Scholar]
- Carter-Fenk K.; Herbert J. M. Electrostatics does not dictate the slip-stacked arrangement of aromatic π–π interactions. Chem. Sci. 2020, 11, 6758–6765. 10.1039/D0SC02667K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurthner F.; Saha-Moller C. R.; Fimmel B.; Ogi S.; Leowanawat P.; Schmidt D. Perylene bisimide dye assemblies as archetype functional supramolecular materials. Chem. Rev. 2016, 116, 962–1052. 10.1021/acs.chemrev.5b00188. [DOI] [PubMed] [Google Scholar]
- Smulders M. M. J.; Schenning A. P. H. J.; Meijer E. W. Insight into the mechanisms of cooperative self-assembly: the sergeants-and-soldiers principle of chiral and achiral C3-symmetric discotic triamides. J. Am. Chem. Soc. 2008, 130, 606–611. 10.1021/ja075987k. [DOI] [PubMed] [Google Scholar]
- Shao C.; Grüne M.; Stolte M.; Würthner F. Perylene bisimide dimer aggregates: fundamental insights into self-assembly by NMR and UV/vis Spectroscopy. Chem. - Eur. J. 2012, 18, 13665–13677. 10.1002/chem.201201661. [DOI] [PubMed] [Google Scholar]
- Sao S.; Naskar S.; Mukhopadhyay N.; Das M.; Chaudhuri D. Assisted π-stacking: a strong synergy between weak interactions. Chem. Commun. 2018, 54, 12186–12189. 10.1039/C8CC07207H. [DOI] [PubMed] [Google Scholar]
- Samanta S.; Mukhopadhyay N.; Chaudhuri D. Rapid and efficient electrochemical actuation in a flexible perylene bisimide dimer. Chem. Mater. 2019, 31, 899–903. 10.1021/acs.chemmater.8b04077. [DOI] [Google Scholar]
- Harris R. K.; Jackson P.; Merwin L. H.; Say B. J.; Hägele G. Perspectives in high-resolution solid-state nuclear magnetic resonance, with emphasis on combined rotation and multiple-pulse spectroscopy. J. Chem. Soc., Faraday Trans. 1 1988, 84, 3649–3672. 10.1039/f19888403649. [DOI] [Google Scholar]
- Bradley J. P.; Tripon C.; Filip C.; Brown S. P. Determining relative proton–proton proximities from the build-up of two-dimensional correlation peaks in 1H double-quantum MAS NMR: insight from multi-spin density-matrix simulations. Phys. Chem. Chem. Phys. 2009, 11, 6941–6952. 10.1039/b906400a. [DOI] [PubMed] [Google Scholar]
- Goldstein R. F.; Stryer L. Cooperative polymerization reactions: analytical approximations, numerical examples, and experimental strategy. Biophys. J. 1986, 50, 583–599. 10.1016/S0006-3495(86)83498-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manjunatha Reddy G. N.; Peters G. M.; Tatman B. P.; Rajan T. S.; Kock S. M.; Zhang J.; Frenguelli B. G.; Davis J. T.; Marsh A.; Brown S. P. Magic-angle spinning NMR spectroscopy provides insight into the impact of small molecule uptake by G-quartet hydrogels. Mater. Adv. 2020, 1, 2236–2247. 10.1039/D0MA00475H. [DOI] [Google Scholar]
- Disruption of hydrogen-bonding and a subsequent disassembly can be carried out using either tBuOH or ethanol. In absorption spectroscopy experiments, we used the former because of its higher boiling point that allowed us to accurately and reproducibly maintain a low volume fraction (f). In 1H-NMR titration experiments, ethanol-d was used
- Both ethanol and CHCl3 eventually disassemble the aggregate completely. The NMR titration experiment probes the early stages of aggregate disassembly, where only one interaction is preferentially disrupted
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.