

Abstract
C1q/TNF-related protein 9 (CTRP9) acts as an adipokine and has been reported to exert numerous biological functions, such as anti-inflammatory and anti-oxidative stress effects, in ischemic heart disease. In the present study, the role of CTRP9 in neonatal rat cardiomyocytes (NRCMs) following hypoxia/reoxygenation (H/R) and the underlying mechanism was investigated. Adenoviral vectors containing CTRP9 or green fluorescent protein were transfected into NRCMs. A H/R model was constructed 2 days after transfection by 2 h incubation under hypoxia followed by 4 h of reoxygenation. Lactate dehydrogenase (LDH), creatine kinase (CK) and CK-myocardial band (CK-MB) levels were detected by a biochemical analyzer using biochemical kits. In addition, cell viability was detected using trypan blue staining to determine the extent of cell injury. Inflammatory cytokines TNF-α, IL-6 and IL-10 were measured by ELISA. Western blotting and reverse transcription-quantitative PCR were used to evaluate the expression levels of CTRP9, toll-like receptor 4 (TLR4), myeloid differentiation primary response (MyD88) and NF-κB. The DNA binding activity of NF-κB was also detected using an electrophoretic mobility shift assay. The results indicated that transfection with adenoviral vectors containing CTRP9 could markedly enhance CTRP9 expression. CTRP9 overexpression increased cell viability and decreased the release of LDH, CK and CK-MB. In addition, CTRP9 overexpression reduced TNF-α and IL-6 levels whilst increasing IL-10 levels, but decreased the expression of TLR4, MyD88 and NF-κB. Furthermore, the DNA binding activity of NF-κB under H/R was also decreased by CTRP9 overexpression. In conclusion, the results of the present study suggested that CTRP9 could protect cardiomyocytes from H/R injury, which was at least partially due to the inhibition of the TLR4/MyD88/NF-κB signaling pathway to reduce the release of inflammatory cytokines.
Keywords: C1q/TNF-related protein 9, cardiomyocytes, hypoxia-reoxygenation, inflammation
Introduction
Ischemic heart disease (IHD) is a major cause of disability and mortality worldwide (1). According to the World Health Organization, the number of acute myocardial infarction cases is ~32.4 million per year worldwide as of 2020(2). At present, timely revascularization is the most effective approach for reducing cardiomyocyte cell death (3). Nevertheless, reperfusion treatment can cause myocardial ischemia/reperfusion (I/R) injury (MIRI), which is a complex pathophysiological process that can cause additional myocardial damage (4). In recent years, evidence has shown that inflammation serves a crucial role in the pathogenesis of MIRI (5). Therefore, identification of effective approaches for reducing inflammation to attenuate MIRI remain in demand.
As a fat-derived plasma protein, C1q/TNF-related protein 9 (CTRP9) has reported beneficial effects on glucose metabolism and vascular function (6,7). Previous studies have indicated that CTRP9 can regulate the inflammatory response in the setting of various cardiovascular diseases, including atrial fibrillation (8), myocardial infarction (9), atherosclerosis (10) and heart failure (11). It was also demonstrated that CTRP9 is a potential protective factor against MIRI (12,13). Kambara et al (12) found that CTRP9 can protect the myocardium from I/R injury through an AMP-activated protein kinase (AMPK)-dependent mechanism. In another study, Zhao et al (13) revealed that cardiac-derived CTRP9 can protect against MIRI through calreticulin-dependent inhibition of apoptosis. However, to the best of our knowledge, the function of CTRP9 in MIRI remains unclear due to the complex molecular mechanism involved.
Previous studies have documented that I/R injury can induce the release of proinflammatory cytokines in cardiomyocytes by triggering toll-like receptor 4 (TLR4)/myeloid differentiation primary response 88 (MyD88)/NF-κB signaling (14,15). Inhibition of TLR4/MyD88/NF-κB-related signaling has been shown to decrease I/R injury-induced proinflammatory cytokine release and ameliorate cardiac dysfunction (16,17). Of note, TLR4/MyD88/NF-κB-related signaling transduction has also been found to be suppressed by CTRP9 during IHD (8,9). However, the effects of CTRP9 on inflammation and TLR4/MyD88/NF-κB activation during MIRI remain unclear.
In this experiment, adenoviral vectors containing CTRP9 were used to increase the expression of CTRP9 in neonatal rat cardiomyocytes (NRCMs), and then a hypoxia/reoxygenation (H/R) injury model was established to explore the role and mechanism of CTRP9 during MIRI.
Materials and methods
Chemicals and reagents
Adenoviruses encoding CTRP9 (Ad-CTRP9) or green fluorescent protein (GFP; Ad-GFP) were constructed and amplified by Gene Company, Ltd.. Lactate dehydrogenase (LDH; cat. no. A020-1-2), creatine kinase (CK; cat. no. A032-1-1) and CK-myocardial band (CK-MB; cat. no. E006-1-1) biochemical kits were obtained from Nanjing Jiancheng Bioengineering Institute. The following primary antibodies were purchased from Abcam: TLR4 (cat. no. ab13867; 1:1,000 dilution), MyD88 (cat. no. ab219413; 1:800 dilution), NF-κB (cat. no. ab220803; 1:1,000 dilution) and GAPDH (cat. no. ab8245; 1:1,000 dilution). HRP-conjugated anti-rabbit (cat. no. bs-0295G; 1:2,000 dilution) or anti-mouse secondary antibodies (cat. no. bs-0296G; 1:2,000 dilution) were obtained from BIOSS. A LightShift Chemiluminescent electrophoretic mobility shift assay (EMSA) kit (cat. no. SIDET201) was purchased from Viagene Biotech, Inc..
NRCM culture
The NRCMs were isolated from ~2-day old Sprague Dawley rats (n=5; weight, 8±2 g; sex, unknown), which were provided by the Experimental Animal Center of Southern Medical University (Shenzhen, China). The Sprague Dawley rats (housed at 23±2˚C with 50% relative humidity, 12-h light/dark cycles and free access to water) were anesthetized with 3% sodium pentobarbital (30 mg/kg) and euthanized via decapitation (18). Their hearts were then quickly removed and their large blood vessels carefully excised. The obtained heart tissues were rinsed in ice-cold PBS to remove any residual blood. Next, 0.08% collagenase type II and 0.125% trypsin were used to digest the tissues at 37˚C for 7 min. Finally, the NRCMs (fusiform or polygonal, determined with spontaneous pulsation) were centrifuged (1,000 x g at 4˚C for 10 min) and re-suspended in DMEM (Gibco; Thermo Fisher Scientific, Inc.) with 10% FBS (Gibco; Thermo Fisher Scientific, Inc.) and 1% penicillin/streptomycin at 37˚C with 5% CO2 and 95% O2. All experimental procedures were approved by the Ethics Committee of Experimental Animals of the Southern Medical University (approval no. 2020-01-03A).
Adenoviral transfection and establishment of the H/R injury model
NRCMs were transfected with Ad-CTRP9 or Ad-GFP (both containing GFP) at a MOI of 50, and the H/R injury model was established 2 days after transfection. Cells were observed using fluorescence microscopy after 48 h to visualize GFP expression. Cells no older than passage 5 were used for these experiments. Briefly, the cultured NRCMs were preserved in serum-free DMEM at 37˚C for 12 h. Next, NRCMs were incubated in an anerobic chamber (95% N2-5% CO2) at 37˚C for 2 h. NRCMs were then moved into a normal incubator (37˚C) for an additional 4 h to induce reoxygenation. The primary cardiomyocytes were randomly separated into the following four groups: Control, H/R, Ad-CTRP9 and Ad-GFP groups (both of which also underwent H/R). Each experiment was repeated ≥5 times.
Cell viability assay
Cell viability assay was performed to assess the cytotoxicity of adenovirus on NRCMs. Briefly, after the NRCMs (5x105/ml) were transfected with adenoviruses at various multiplicities of infection (MOI=5, 20, 50, 100 and 200) at 37˚C for 48 h, they were stained with 0.4% trypan blue 37˚C for 3 min and observed under a light microscope (magnification, x200). The ratio of unstained cells to total cell number was calculated to estimate cell viability.
Determination of markers of myocardial injury
In the present study, the supernatant of cultured NRCMs was collected and an automatic biochemical analyzer (Jinan Tianheng Technology Co., Ltd.) was used with the aforementioned biochemical kits, according to the manufacturers' protocols, to determine the LDH, CK and CK-MB levels.
Measurement of TNF-α, IL-6 and IL-10 levels
TNF-α (cat. no. SRTA00), IL-6 (cat. no. SR6000B) and IL-10 (cat. no. SR1000) ELISA kits (R&D Systems, Inc.) were used according to the manufacturer's protocol to determine the TNF-α, IL-6 and IL-10 levels in supernatants. Samples was centrifuged 500 x g at 4˚C for 10 min to obtain supernatants.
Western blotting
Western blotting was performed to analyze protein expression (19). Briefly, NRCMs were first homogenized and lysed in RIPA buffer (cat. no. R0278; MilliporeSigma). Next, the protein was extracted and the concentration was determined using a BCA assay (Beyotime Institute of Biotechnology). Subsequently, 10% SDS-PAGE was used to separate the extracted proteins (40 µg), which were then electrophoretically transferred onto PVDF membranes. The membranes were then blocked with 5% non-fat dry milk in PBS with 0.05% Tween-20 for 2 h at room temperature. Next, the membranes were incubated with antibodies against TLR4, MyD88 and NF-κB overnight at 4˚C. The next day, the membranes were incubated with HRP-conjugated anti-rabbit or anti-mouse secondary antibodies for another 2 h at room temperature. Finally, a Pierce™ ECL Western Blotting Substrate kit (cat. no. 32109; Pierce; Thermo Fisher Scientific, Inc.) was used to detect protein expression. An Odyssey Infrared Imaging system (model 9120; LI-COR Biosciences) was used to capture images of the membranes and Quantity One 1-D software (version 4.6.9; Bio-Rad Laboratories, Inc.) was used to quantify the protein bands.
Reverse transcription-quantitative PCR (RT-qPCR)
RT-qPCR was performed to detect mRNA levels. Briefly, TRIzol® reagent (Invitrogen; Thermo Fisher Scientific, Inc.) was used to extract total RNA from NRCMs. Obtained RNA (~4.0 µg) was then reverse transcribed into cDNA using SuperScript IV Reverse Transcriptase (Thermo Fisher Scientific, Inc.) at 37˚C for 60 min. Next, qPCR was performed using a SYBR green Master Mix kit (Thermo Fisher Scientific, Inc.) on a 7500 ABI PRISM system (Applied Biosystems; Thermo Fisher Scientific, Inc.). The qPCR thermocycling conditions were as follows: 45˚C for 2 min; 95˚C for 10 min, immediately followed by 45 cycles of 95˚C for 30 sec and 60˚C for 30 sec. The mRNA expression of CTRP9, TLR4, MyD88 and NF-κB were normalized to that of GAPDH. The 2-ΔΔCq method was used to calculate changes in mRNA expression (20). The following primers were used: CTRP9 forward, 5'-GGCTTCTACTGGTTATGGACGC-3' and reverse, 5'-GGAGCCTGGATCACCTTTGAT-3'; TLR4 forward, 5'-TGCTCAGACATGGCAGTTTC-3' and reverse, 5'-CTGGATTCAAGGCTTTTCCA-3'; MyD88 forward, 5'-GAGATCCGCGAGTTTGAGAC-3' and reverse, 5'-CTGTTTCTGCTGGTTGCGTA-3'; NF-κB forward, 5'-GGCAGCACTCCTTATCAACC-3' and reverse, 5'-GAGGTGTCGTCCCATCGTAG-3' and GAPDH forward, 5'-CGCTAACATCAAATGGGGTG-3' and reverse, 5'-TTGCTGACAATCTTGAGGGAG-3'.
EMSA
The binding activity of NF-κB was detected using an electrophoretic mobility shift assay. Briefly, nuclear extracts were prepared from the NRCMs and stored at -80˚C for the EMSA assay. The protein concentration was measured using a Bio-Rad protein assay reagent (Bio-Rad Laboratories, Inc.). Equal amounts (5 µg) of nuclear protein were incubated with poly (2'-deoxyinosinic-2'-deoxycytidylic acid) and synthesized NF-κB binding consensus oligonucleotides (sense, 5'-AGTTGAGGGGACTTTCCCAGGC-3'; antisense, 5'-GCCTGGGAAAGTCCCCTCAACT-3') for 20 min at room temperature using a LightShift EMSA Optimization and Control kit. Subsequently, protein-DNA complexes were separated via electrophoresis on a 6.5% non-denaturing polyacrylamide gel, transferred to a nylon membrane and cross-linked by UV light. The membrane was incubated with streptavidin-horseradish peroxidase and detected via enhanced chemiluminescence (Pierce; Thermo Fisher Scientific, Inc.).
Statistical analysis
SPSS 22.0 software (IBM Corp.) was used for data analysis. Data are presented as the mean ± SD (n=5). Student's unpaired t-test and one-way ANOVA were used for comparisons between groups. If interactions were significant, a Tukey post hoc test was used for multiple comparisons. P<0.05 was considered to indicate a statistically significant difference.
Results
NRCM viability
As shown in Fig. 1A, Ad-CTRP9 exerted no toxic effects on NRCM viability at MOI of <100. However, cell viability was 86.8% at an MOI of 200. After 48 h of transfection, GFP expression was assessed using a fluorescence microscope at an MOI of 50 (Fig. 1B). In the present study, the transfection efficiency at MOI of 20 was only ~84.7%, whereas that at MOI of 50 and 100 were ~92.6 and ~93.8% respectively, with no clear differences between MOI of 50 and 100 (data not shown). Considering these aforementioned results, MOI of 50 was used due to its higher transfection efficiency but minimal effects on NRCM viability.
Figure 1.

Transfection efficiency and cell viability in NRCMs after transfection with Ad-CTRP9 at different MOIs. (A) Cell viability following Ad-CTRP9 transfection at different MOIs (0-200). (B) NRCMs were transfected with Ad-CTRP9 (MOI=50) for 48 h and observed using fluorescence microscopy. Magnification, x100. Data are expressed as the mean ± SD (n=5). *P<0.05 vs. 0 MOI. Ad-CTRP9, adenovirus encoding CTRP9; MOI, multiplicity of infection; NRCM, neonatal rat cardiomyocytes.
Ad-CTRP9 induces CTRP9 upregulation in cardiomyocytes after H/R
Following adenoviral infection and the establishment of the H/R injury model, CTRP9 expression was examined by RT-qPCR. As shown in Fig. 2, the mRNA expression levels of CTRP9 were significantly reduced in the H/R and Ad-GFP groups compared with those in the control group. However, the mRNA expression of CTRP9 was significantly increased in the Ad-CTRP9 group compared with that in the Ad-GFP or H/R groups.
Figure 2.
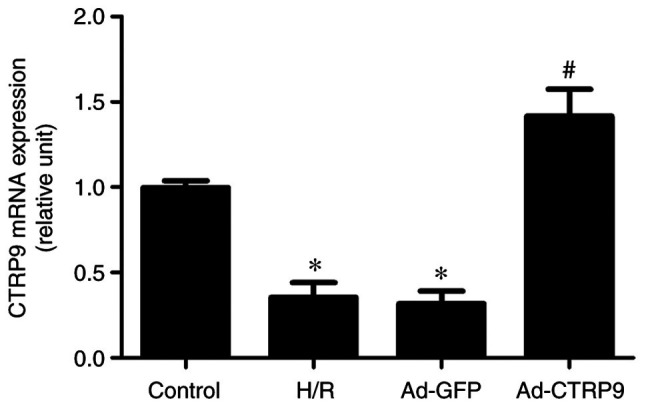
Ad-CTRP9 transfection induces CTRP9 upregulation in cardiomyocytes after H/R. CTRP9 mRNA expression was measured using reverse transcription-quantitative PCR. Data are presented as the mean ± SD (n=6). *P<0.05 vs. control. #P<0.05 vs. H/R or Ad-GFP. CTRP9, C1q/TNF-related protein 9; Ad-CTRP9, adenovirus encoding CTRP9; H/R, hypoxia/reoxygenation.
CTRP9 attenuates H/R-induced NRCM injury
LDH, CK and CK-MB are enzymes that are released by cardiomyocytes following severe injury, and their levels are used to estimate the severity of myocardial damage (17). To assess the effects of CTRP9 on H/R-induced cellular damage, cell viability, as well as LDH, CK and CK-MB activity in the cell culture supernatant, were evaluated. Cell viability was significantly suppressed by H/R compared with that in cells that underwent normoxic treatment in all three of the transfection groups (Fig. 3A). However, after H/R, cell viability was significantly increased in the Ad-CTRP9 group compared with that in the control group (Fig. 3A). LDH (Fig. 3B), CK (Fig. 3C) and CK-MB (Fig. 3D) activities were also significantly increased in the H/R group compared with those in the control group. However, the H/R-induced enzyme release was significantly reversed by CTRP9 overexpression (Fig. 3B-D).
Figure 3.

Overexpression of CTRP9 reduces H/R-induced damage in NRCMs. (A) Transfection with Ad-CTRP9 could markedly increase cell viability. (B) LDH, (C) CK and (D) CK-MB levels were markedly reduced following CTRP9 overexpression after H/R. Data are presented as the mean ± SD (n=6). *P<0.05 vs. the control group. #P<0.05 vs. H/R or Ad-GFP. CTRP9, C1q/TNF-related protein 9; Ad-CTRP9, adenovirus encoding CTRP9; H/R, hypoxia/reoxygenation; NRCM, neonatal rat cardiomyocyte; LDH, lactate dehydrogenase; CK, creatine kinase; CK-MB, CK-myocardial band.
CTRP9 alleviates inflammation after H/R injury
MIRI is closely associated with an excessive inflammatory response (5). In addition, CTRP9 has been shown to be involved in the progression of inflammation in the heart (9). ELISA was therefore used to measure TNF-α, IL-6 and IL-10 expression in cell culture supernatant. The levels of TNF-α and IL-6 were found to be significantly increased following H/R compared with those in the control group, but the levels of the anti-inflammatory cytokine IL-10 was significantly downregulated (Fig. 4). However, CTRP9 overexpression significantly decreased the levels of TNF-α and IL-6 whilst increasing the levels of IL-10 compared with those in the Ad-GFP or H/R groups (Fig. 4).
Figure 4.

CTRP9 overexpression alleviates inflammation after H/R damage. ELISA was performed to measure (A) TNF-α, (B) IL-6 and (C) IL-10 levels. TNF-α and IL-6 levels were markedly increased after H/R damage, but IL-10 levels decreased. CTRP9 overexpression reduced TNF-α and IL-6 levels but increased IL-10 levels. Data are presented as the mean ± SD (n=6). *P<0.05 vs. control. #P<0.05 vs. H/R or Ad-GFP. CTRP9, C1q/TNF-related protein 9; H/R, hypoxia/reoxygenation; Ad-GFP, adenovirus encoding green fluorescent protein.
CTRP9 inhibits TLR4/MyD88/NF-κB signaling
To further understand the possible mechanism underlying the CTRP9-mediated mitigation of H/R damage, the expression of components of TLR4/MyD88/NF-κB signaling was determined by western blotting and RT-qPCR. As shown in Fig. 5, H/R significantly upregulated the protein (Fig. 5A) and mRNA (Fig. 5B) expression levels of TLR4, MyD88 and NF-κB compared with those in the control group. However, CTRP9 overexpression after the onset of H/R significantly reversed these aforementioned increases. Similarly, the binding activity of NF-κB to DNA was markedly increased after H/R, but decreased following Ad-CTRP9 transfection (Fig. 5B). Therefore, this suggests that H/R injury may be ameliorated by CTRP9 overexpression, possibly through suppression of TLR4/MyD88/NF-κB signaling.
Figure 5.

CTRP9 overexpression inhibits TLR4/MyD88/NF-κB signaling. (A) Protein expression levels were determined using western blotting. (B) Quantification of western blotting from (A). (C) mRNA expression levels were determined using reverse transcription-quantitative PCR. (D) NF-κB binding activity was determined using an electrophoretic mobility shift assay. The arrow indicates the direction of sample travel during electrophoresis. Data are presented as the mean ± SD (n=5). *P<0.05 vs. control. #P<0.05 vs. H/R or Ad-GFP. CTRP9, C1q/TNF-related protein 9; TLR4, toll-like receptor 4; MyD88, myeloid differentiation primary response; Ad-GFP, adenovirus encoding green fluorescent protein; NSB, non-specific binding; F, free probe.
Discussion
MIRI leads to a range of pathological changes, including activation of the inflammatory response, which can lead to cell injury (21). Effectively reducing inflammation can improve the outcomes of MIRI in animal and cell models (22). Previous studies have found that CTRP9 exerts protective effects against ischemic heart injury through a variety of signaling pathways, such as the protein kinase A and ERK1/2 signaling pathways (23-25). In particular, cardiac-derived CTRP9 has been found to protect against MIRI through the inhibition of apoptosis and endoplasmic reticulum stress (13,26). However, to the best of our knowledge, the role of CTRP9 and possible underlying mechanism in MIRI and has not been completely elucidated. In the present study, a H/R model was established to simulate MIRI and CTRP9 was found to alleviate MIRI by significantly reducing myocardial inflammation, which was characterized by the upregulation of cell viability and reducing the release of CK, CK-MB and LDH. In addition, it was observed that CTRP9 overexpression markedly inhibited the TLR4/MyD88/NF-κB signaling pathway. These aforementioned results suggest that CTRP9 may possess the ability to ameliorate H/R-induced inflammation by regulating the TLR4/MyD88/NF-κB signaling pathway.
Previous studies have found that CTRP9 can regulate the inflammatory response in various pathological processes, such as myocardial infarction and endothelial dysfunction (27-29). In db/db mice, Li et al (30) found that overexpression of CTRP9 may reduce retinal inflammation and protect the blood-retinal barrier. Liu et al (9) revealed that overexpression of CTRP9 restored cardiac function following myocardial infarction by regulating TLR4/MD2/MyD88 and AMPK/NF-κB signaling in a rat model of myocardial infarction. In another study, Zhang et al (31) demonstrated that overexpression of CTRP9 attenuated a mouse model of atherosclerosis by inhibiting AMPK/NLR family pyrin domain containing 3 signaling. Qian et al (32) showed that overexpression of CTRP9 alleviated airway inflammation in a mouse model of asthma. Zhao et al (33) demonstrated that Ad-CTRP9 transfection weakened neuro-inflammation by activating adiponectin receptor 1 following intracerebral hemorrhage in mice. Therefore these previously reported biological activities of CTRP9 aforementioned attracted the interest of the research community. The present study demonstrated that CTRP9 overexpression may reverse the H/R-induced upregulated expression of TNF-α and IL-6 in addition to reversing the H/R-induced reduction in the levels of the anti-inflammatory cytokine IL-10.
H/R can directly decrease cardiomyocyte contractility and induce inflammation in cardiomyocytes by activating the TLR4/MyD88-dependent signaling pathway (16). The TLR4 signaling pathway has been extensively studied, where exerts its effects through the MyD88-mediated activation of NF-κB (34,35). As a transcription factor, NF-κB has been confirmed to be closely associated with inflammation activation (36). TLR4 signaling-related inflammatory activation is closely linked to myocardial injury during MIRI (37). A number of endogenous factors that can negatively regulate TLR4 signal transduction directly have been found (9,38). Among them, CTRP9 has particularly garnered interest (9). MyD88 and NF-κB are downstream molecules of TLR4, and the activation of MyD88 and NF-κB are partly dependent on TLR4; Liu et al (9) found that CTRP9 could directly bind to TLR4 to regulate the downstream molecules of MyD88 and NF-κB. In addition, ample evidence suggests that CTRP9 exerts pleiotropic effects, such as anti-inflammation, on a variety of pathological conditions by suppressing NF-κB, such as cardiac hypertrophy (39) and osteoarthritis (40). In terms of the possible involvement of CTRP9 in the suppression of TLR4/MyD88/NF-κB-related inflammatory signaling, the present study suggest that CTRP9 may exert protective effects on cardiomyocytes following H/R insult by conferring anti-inflammatory effects through suppressing TLR4/MyD88/NF-κB signaling downstream. However, it should be noted that the cause-effect relationship between TLR4 signaling and inflammation after overexpression of CTRP9 require further study. TLR4-knockout mice need to be established for further study, which will contribute to the understanding of the relationship between TLR4 signaling and inflammation after overexpression of CTRP9.
In conclusion, the present study indicated that the overexpression of CTRP9 could alleviate H/R by attenuating inflammation in a TLR4/MyD88/NF-κB-dependent manner. However, the pathophysiological process of MIRI is complex, such that the possibility of other signaling pathways being involved in the protective effects of CTRP9 in MIRI cannot be ruled out. Nevertheless, results from the present study suggest that CTRP9 can represent a novel therapeutic target for MIRI.
Acknowledgements
Not applicable.
Funding Statement
Funding: The present study was supported by the Health and Family Planning Commission of Wuhan Municipality (grant no. WX18A07).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
ZXZ, FA and ZYH were responsible for the conception and design of the study. DZ, FA and YJW provided administrative support. XLL, JH and JQL provided the study materials. ZYH, DZ and YJW conducted NRCM culture, adenoviral transfection and establishment of the H/R injury model; XLL and ZXZ conducted RT-qPCR and western blotting assays; JH and LSJ performed ELISA; and JQL and FA performed ELISA and cell viability assays. ZXZ and DZ were responsible for the analysis and interpretation of data. ZYH, DZ, FA and ZXZ confirmed the authenticity of all the raw data. All authors contributed to the writing of the manuscript and read and approved the final manuscript.
Ethics approval and consent to participate
All experimental procedures were approved by the Ethics Committee of Experimental Animals of the Southern Medical University (approval no. 2020-01-03A; Shenzhen, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Peng H, Abdel-Latif A. Cellular therapy for ischemic heart disease: An update. Adv Exp Med Biol. 2019;1201:195–213. doi: 10.1007/978-3-030-31206-0_10. [DOI] [PubMed] [Google Scholar]
- 2.Anderson JL, Morrow DA. Acute myocardial infarction. N Engl J Med. 2017;376:2053–2064. doi: 10.1056/NEJMra1606915. [DOI] [PubMed] [Google Scholar]
- 3.Godoy LC, Lawler PR, Farkouh ME, Hersen B, Nicolau JC, Rao V. Urgent revascularization strategies in patients with diabetes mellitus and acute coronary syndrome. Can J Cardiol. 2019;35:993–1001. doi: 10.1016/j.cjca.2019.03.010. [DOI] [PubMed] [Google Scholar]
- 4.Guan BF, Dai XF, Huang QB, Zhao D, Shi JL, Chen C, Zhu Y, Ai F. Icariside II ameliorates myocardial ischemia and reperfusion injury by attenuating inflammation and apoptosis through the regulation of the PI3K/AKT signaling pathway. Mol Med Rep. 2020;22:3151–3160. doi: 10.3892/mmr.2020.11396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Qian X, Zhu M, Qian W, Song J. Vitamin D attenuates myocardial ischemia-reperfusion injury by inhibiting inflammation via suppressing the RhoA/ROCK/NF-ĸB pathway. Biotechnol Appl Biochem. 2019;66:850–857. doi: 10.1002/bab.1797. [DOI] [PubMed] [Google Scholar]
- 6.Fujita T, Watanabe H, Murata Y, Hemmi S, Yabuki M, Fuke Y, Satomura A, Soma M. Plasma C1q/TNF-related protein 9: A promising biomarker for diabetic renal vascular injury. Minerva Urol Nefrol. 2017;69:195–200. doi: 10.23736/S0393-2249.16.02500-5. [DOI] [PubMed] [Google Scholar]
- 7.Liu Q, Zhang H, Lin J, Zhang R, Chen S, Liu W, Sun M, Du W, Hou J, Yu B. C1q/TNF-related protein 9 inhibits the cholesterol-induced Vascular smooth muscle cell phenotype switch and cell dysfunction by activating AMP-dependent kinase. J Cell Mol Med. 2017;21:2823–2836. doi: 10.1111/jcmm.13196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu M, Li W, Wang H, Yin L, Ye B, Tang Y, Huang C. CTRP9 ameliorates atrial inflammation, fibrosis, and vulnerability to atrial fibrillation in post-myocardial infarction rats. J Am Heart Assoc. 2019;8(e013133) doi: 10.1161/JAHA.119.013133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu M, Yin L, Li W, Hu J, Wang H, Ye B, Tang Y, Huang C. C1q/TNF-related protein-9 promotes macrophage polarization and improves cardiac dysfunction after myocardial infarction. J Cell Physiol. 2019;234:18731–18747. doi: 10.1002/jcp.28513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang J, Hang T, Cheng XM, Li DM, Zhang QG, Wang LJ, Peng YP, Gong JB. Associations of C1q/TNF-related protein-9 levels in serum and epicardial adipose tissue with coronary atherosclerosis in humans. Biomed Res Int. 2015;2015(971683) doi: 10.1155/2015/971683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao C, Zhao S, Lian K, Mi B, Si R, Tan Z, Fu F, Wang S, Wang R, Ma X, Tao L. C1q/TNF-related protein 3 (CTRP3) and 9 (CTRP9) concentrations are decreased in patients with heart failure and are associated with increased morbidity and mortality. BMC Cardiovasc Disord. 2019;19(139) doi: 10.1186/s12872-019-1117-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kambara T, Ohashi K, Shibata R, Ogura Y, Maruyama S, Enomoto T, Uemura Y, Shimizu Y, Yuasa D, Matsuo K, et al. CTRP9 protein protects against myocardial injury following ischemia-reperfusion through AMP-activated protein kinase (AMPK)-dependent mechanism. J Biol Chem. 2012;287:18965–18973. doi: 10.1074/jbc.M112.357939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao D, Feng P, Sun Y, Qin Z, Zhang Z, Tan Y, Gao E, Lau WB, Ma X, Yang J, et al. Cardiac-derived CTRP9 protects against myocardial ischemia/reperfusion injury via calreticulin-dependent inhibition of apoptosis. Cell Death Dis. 2018;9(723) doi: 10.1038/s41419-018-0726-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang J, Zhang J, Yu P, Chen M, Peng Q, Wang Z, Dong N. Remote ischaemic preconditioning and sevoflurane postconditioning synergistically protect rats from myocardial injury induced by ischemia and reperfusion partly via inhibition TLR4/MyD88/NF-κB signaling pathway. Cell Physiol Biochem. 2017;41:22–32. doi: 10.1159/000455815. [DOI] [PubMed] [Google Scholar]
- 15.Ye B, Chen X, Dai S, Han J, Liang X, Lin S, Cai X, Huang Z, Huang W. Emodin alleviates myocardial ischemia/reperfusion injury by inhibiting gasdermin D-mediated pyroptosis in cardiomyocytes. Drug Des Devel Ther. 2019;13:975–990. doi: 10.2147/DDDT.S195412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xue J, Ge H, Lin Z, Wang H, Lin W, Liu Y, Wu G, Xia J, Zhao Q. The role of dendritic cells regulated by HMGB1/TLR4 signalling pathway in myocardial ischaemia reperfusion injury. J Cell Mol Med. 2019;23:2849–2862. doi: 10.1111/jcmm.14192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li X, Yang J, Yang J, Dong W, Li S, Wu H, Li L. RP105 protects against myocardial ischemia-reperfusion injury via suppressing TLR4 signaling pathways in rat model. Exp Mol Pathol. 2016;100:281–286. doi: 10.1016/j.yexmp.2015.12.016. [DOI] [PubMed] [Google Scholar]
- 18.Simpson P, McGrath A, Savion S. Myocyte hypertrophy in neonatal rat heart cultures and its regulation by serum and by catecholamines. Circ Res. 1982;51:787–801. doi: 10.1161/01.res.51.6.787. [DOI] [PubMed] [Google Scholar]
- 19.Hnasko TS, Hnasko RM. The western blot. Methods Mol Biol. 2015;1318:87–96. doi: 10.1007/978-1-4939-2742-5_9. [DOI] [PubMed] [Google Scholar]
- 20.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 21.Xu W, Zhang K, Zhang Y, Ma S, Jin D. Downregulation of DEC1 by RNA interference attenuates ischemia/reperfusion-induced myocardial inflammation by inhibiting the TLR4/NF-κB signaling pathway. Exp Ther Med. 2020;20:343–350. doi: 10.3892/etm.2020.8706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang R, Xu L, Zhang D, Hu B, Luo Q, Han D, Li J, Shen C. Cardioprotection of ginkgolide B on myocardial ischemia/reperfusion-induced inflammatory injury via regulation of A20-NF-κB pathway. Front Immunol. 2018;9(2844) doi: 10.3389/fimmu.2018.02844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yan W, Guo Y, Tao L, Lau WB, Gan L, Yan Z, Guo R, Gao E, Wong GW, Koch WL, et al. C1q/tumor necrosis factor-related protein-9 regulates the fate of implanted mesenchymal stem cells and mobilizes their protective effects against ischemic heart injury via multiple novel signaling pathways. Circulation. 2017;136:2162–2177. doi: 10.1161/CIRCULATIONAHA.117.029557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun Y, Yi W, Yuan Y, Lau WB, Yi D, Wang X, Wang Y, Su H, Wang X, Gao E, et al. C1q/tumor necrosis factor-related protein-9, a novel adipocyte-derived cytokine, attenuates adverse remodeling in the ischemic mouse heart via protein kinase A activation. Circulation. 2013;128 (11 Suppl 1):S113–S120. doi: 10.1161/CIRCULATIONAHA.112.000010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weng CF, Wu CF, Kao SH, Chen JC, Lin HH. Down-regulation of miR-34a-5p potentiates protective effect of adipose-derived mesenchymal stem cells against ischemic myocardial infarction by stimulating the expression of C1q/tumor necrosis factor-related protein-9. Front Physiol. 2019;10(1445) doi: 10.3389/fphys.2019.01445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bai S, Cheng L, Yang Y, Fan C, Zhao D, Qin Z, Feng X, Zhao L, Ma J, Wang X, et al. C1q/TNF-related protein 9 protects diabetic rat heart against ischemia reperfusion injury: Role of endoplasmic reticulum stress. Oxid Med Cell Longev. 2016;2016(1902025) doi: 10.1155/2016/1902025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Y, Geng X, Wang H, Cheng G, Xu S. CTRP9 ameliorates pulmonary arterial hypertension through attenuating inflammation and improving endothelial cell survival and function. J Cardiovasc Pharmacol. 2016;67:394–401. doi: 10.1097/FJC.0000000000000364. [DOI] [PubMed] [Google Scholar]
- 28.Liu T, Xu B, Liu Z. CTRP9 alleviates inflammation to ameliorate myocardial infarction in rats by activating Nrf2. Minerva Endocrinol. 2020;45:268–270. doi: 10.23736/S0391-1977.19.03081-5. [DOI] [PubMed] [Google Scholar]
- 29.Jung CH, Lee MJ, Kang YM, Lee YL, Seol SM, Yoon HK, Kang SW, Lee WJ, Park JY. C1q/TNF-related protein-9 inhibits cytokine-induced vascular inflammation and leukocyte adhesiveness via AMP-activated protein kinase activation in endothelial cells. Mol Cell Endocrinol. 2016;419:235–243. doi: 10.1016/j.mce.2015.10.023. [DOI] [PubMed] [Google Scholar]
- 30.Li W, Ma N, Liu MX, Ye BJ, Li YJ, Hu HY, Tang YH. C1q/TNF-related protein-9 attenuates retinal inflammation and protects blood-retinal barrier in db/db mice. Eur J Pharmacol. 2019;853:289–298. doi: 10.1016/j.ejphar.2019.04.012. [DOI] [PubMed] [Google Scholar]
- 31.Zhang H, Gong X, Ni S, Wang Y, Zhu L, Ji N. C1q/TNF-related protein-9 attenuates atherosclerosis through AMPK-NLRP3 inflammasome singling pathway. Int Immunopharmacol. 2019;77(105934) doi: 10.1016/j.intimp.2019.105934. [DOI] [PubMed] [Google Scholar]
- 32.Qian M, Yang Q, Li J, Zhao B, Zhang Y, Zhao Y. C1q/TNF-related protein-9 alleviates airway inflammation in asthma. Int Immunopharmacol. 2020;81(106238) doi: 10.1016/j.intimp.2020.106238. [DOI] [PubMed] [Google Scholar]
- 33.Zhao L, Chen S, Sherchan P, Ding Y, Zhao W, Guo Z, Yu J, Tang J, Zhang JH. Recombinant CTRP9 administration attenuates neuroinflammation via activating adiponectin receptor 1 after intracerebral hemorrhage in mice. J Neuroinflammation. 2018;15(215) doi: 10.1186/s12974-018-1256-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Azam S, Jakaria M, Kim IS, Kim J, Haque ME, Choi DK. Regulation of toll-like receptor (TLR) signaling pathway by polyphenols in the treatment of age-linked neurodegenerative diseases: Focus on TLR4 signaling. Front Immunol. 2019;10(1000) doi: 10.3389/fimmu.2019.01000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ju M, Liu B, He H, Gu Z, Liu Y, Su Y, Zhu D, Cang J, Luo Z. MicroRNA-27a alleviates LPS-induced acute lung injury in mice via inhibiting inflammation and apoptosis through modulating TLR4/MyD88/NF-κB pathway. Cell Cycle. 2018;17:2001–2018. doi: 10.1080/15384101.2018.1509635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zheng Z, Zeng YZ, Ren K, Zhu X, Tan Y, Li Y, Li Q, Yi GH. S1P promotes inflammation-induced tube formation by HLECs via the S1PR1/NF-κB pathway. Int Immunopharmacol. 2019;66:224–235. doi: 10.1016/j.intimp.2018.11.032. [DOI] [PubMed] [Google Scholar]
- 37.Yuan X, Juan Z, Zhang R, Sun X, Yan R, Yue F, Huang Y, Yu J, Xia X. Clemastine fumarate protects against myocardial ischemia reperfusion injury by activating the TLR4/PI3K/Akt signaling pathway. Front Pharmacol. 2020;11(28) doi: 10.3389/fphar.2020.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guo X, Jiang H, Yang J, Chen J, Yang J, Ding JW, Li S, Wu H, Ding HS. Radioprotective 105 kDa protein attenuates ischemia/reperfusion-induced myocardial apoptosis and autophagy by inhibiting the activation of the TLR4/NF-κB signaling pathway in rats. Int J Mol Med. 2016;38:885–893. doi: 10.3892/ijmm.2016.2686. [DOI] [PubMed] [Google Scholar]
- 39.Appari M, Breitbart A, Brandes F, Szaroszyk M, Froese N, Korf-Klingebiel M, Mohammadi MM, Grund A, Scharf GM, Wang H, et al. C1q-TNF-related protein-9 promotes cardiac hypertrophy and failure. Circ Res. 2017;120:66–77. doi: 10.1161/CIRCRESAHA.116.309398. [DOI] [PubMed] [Google Scholar]
- 40.Zheng S, Ren J, Gong S, Qiao F, He J. CTRP9 protects against MIA-induced inflammation and knee cartilage damage by deactivating the MAPK/NF-κB pathway in rats with osteoarthritis. Open Life Sci. 2020;15:971–980. doi: 10.1515/biol-2020-0105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.