

Abstract
Urokinase-type plasminogen activator (uPA) is a serine proteinase that catalyzes the generation of plasmin on the cell surface and activates cell signaling pathways that promote remodeling and repair. Neuronal cadherin (NCAD) is a transmembrane protein that in the mature brain mediates the formation of synaptic contacts in the II/III and V cortical layers. Our studies show that uPA is preferentially found in the II/III and V cortical laminae of the gyrencephalic cortex of the non-human primate. Furthermore, we found that in murine cerebral cortical neurons and induced pluripotent stem cell (iPSC)-derived neurons prepared from healthy human donors, most of this uPA is associated with pre-synaptic vesicles. Our in vivo experiments revealed that in both, the gyrencephalic cortex of the non-human primate and the lissecephalic murine brain, cerebral ischemia decreases the number of intact synaptic contacts and the expression of uPA and NCAD in a band of tissue surrounding the necrotic core. Additionally, our in vitro data show that uPA induces the synthesis of NCAD in cerebral cortical neurons, and in line with these observations, intravenous treatment with recombinant uPA three hours after the onset of cerebral ischemia induces NCAD-mediated repair of synaptic contacts in the area surrounding the necrotic core.
Keywords: Cerebral ischemia, neuronal cadherin, neurorepair, synapse, urokinase-type plasminogen activator
Introduction
The mechanisms that underlie neurorepair after an ischemic lesion are largely similar to those involved in synaptic plasticity in the uninjured brain.1 Indeed, a large body of experimental work indicates that functional improvement following an ischemic stroke is driven by the recovery and remodeling of synaptic contacts in surviving neurons surrounding the necrotic core.2,3 More specifically, the seeds for repair after an ischemic stroke are surviving neurons in the peri-infarct cortex that undergo structural and functional remodeling during the recovery phase from the ischemic injury.4
Urokinase-type plasminogen activator (uPA) is a serine proteinase assembled by a epidermal growth factor domain that binds to its receptor (uPAR), a kringle domain that interacts with plasminogen activator inhibitor-1 (PAI-1), and a C-terminus domain that harbors the active site of the enzyme.5 A growing body of experimental evidence indicates that uPA binding to uPAR not only catalyzes the conversion of plasminogen into plasmin,6,7 but also activates cell signaling pathways via plasminogen-dependent and -independent mechanisms.8 In the developing nervous system (CNS), uPA is abundantly found in neurons and oligodendrocytes,9 and its binding to uPAR promotes neuritogenesis and neuronal migration via a plasminogen-independent mechanism.10 In contrast, the expression and function of uPA in the mature brain are less well understood. However, our recent work with cultured cerebral cortical neurons and an animal model of cerebral ischemia indicates that following an ischemic injury uPA promotes functional recovery and the repair of axons and dendritic spines damaged by the ischemic insult.11–15
Neuronal cadherin [N-cadherin (NCAD)] is a type I cadherin that in the developing CNS is abundantly found in most neurons. In contrast, in the mature brain NCAD is detected almost exclusively in glutamatergic synapses of neurons in the II/III and V cortical layers.16,17 Neuronal depolarization increases the synaptic abundance of NCAD,18 and the establishment of calcium-dependent homotypic interactions between NCAD monomers located in juxtaposed membranes leads to the development of adherent contacts between pre- and postsynaptic terminals.19 Importantly, NCAD inhibition has a harmful effect on synapse formation and function20–22 and interferes with the organization and stabilization of dendritic spines.23 The expression of NCAD mRNA increases after transient middle cerebral artery occlusion (tMCAO),24 but the mechanisms that regulate these changes and their functional consequences are unknown. Nevertheless, NCAD seems to play a central role in neurorepair, as denoted by its ability to promote the recovery of demyelinated lesions,25 axons that have suffered an ischemic lesion,13 and synapses exposed to soluble amyloid β.14
Here we show that uPA is abundantly found in the presynaptic terminal of murine cerebral cortical neurons and induced pluripotent stem cell (iPSC)-derived neurons prepared from healthy human donors. We show that, as described for NCAD, uPA is also abundantly found in the II/III and V cortical layers of the non-human primate cortex. We found that in both, the murine lissencephalic brain and the gyrencephalic cortex of the non-human primate, cerebral ischemia decreases the expression of uPA and NCAD in surviving neurons that surround the necrotic core. Importantly, our in vitro experiments reveal that uPA induces the synthesis of NCAD in cerebral cortical neurons and, in line with these observations, our in vivo work shows that treatment with recombinant uPA abrogates the harmful effects of cerebral ischemia on the abundance of NCAD, and promotes NCAD-mediated formation of synaptic contacts in surviving neurons in the peri-infarct cortex. These data reveal a new role for uPA in the ischemic brain with potential translational implication to promote neurorepair in patients that have suffered an acute ischemic stroke.
Materials and methods
Animals and reagents
Animals were 8–12 weeks-old male wild-type (Wt) mice on a C57BL/6J background, and 7–13 years/old female rhesus monkeys. Animal procedures were conducted following the guidelines of the Guide for the Care and Use of Laboratory Animals, and with the approval of the Institutional Animal Care & Use Committee (IACUC) of Emory University, Atlanta GA. Experiments were reported following ARRIVE guidelines for how to report animal experiments. Recombinant murine and human uPA were purchased from Molecular Innovations (Novi, MI; Cat. # MUPA and HUPA-HTC, respectively). Other materials were the N-cadherin inhibitor Exherin-1 (ADH1; AdooQ bioscience; Irvine, CA. Cat. # A13689), phalloidin Alexa 488, the nuclear staining Hoechst (ThermoFisher; Grand Island, NY; Cat. # A12389 and H3570, respectively); trypsin (Sigma-Aldrich; St. Louis, MO. Cat. # 25300-054), pepsin (GeneTex; Irvine, CA. Cat # GTX30935), puromycin (Calbiochem Millipore; Burlington, MA. Cat # 54022), mini-protean TGX and Criterion TGX Stain-Free precast gels (Bio-Rad; Hercules, CA; Cat # 456-8083 and 5671093, respectively), and antibodies against rabbit PSD-95 (Cell Signaling Technology; Danvers, MA. Cat # 2507S), mouse PSD-95 (Novus, Biologicals; Centennial, CO. Cat # NB300-556), mouse bassoon (Enzon; Farmingdale, NY. Cat # ENZ-ABS666-0200), rabbit N-cadherin and mouse synaptophysin (Abcam; Cambridge, MA. Cat. # ab76057 and ab8049, respectively), mouse uPA and rabbit uPA (Molecular Innovations; Novi, MI. Cat # H77A10 and ASMUP-GF, respectively), rabbit syntaxin I and NeuN (Millipore; Burlington, MA. Cat. # AB5820 and MAB377, respectively), and β-actin (Sigma-Aldrich; St. Louis, MO. Cat. # A1978). Secondary antibodies were IRDye 800CW donkey anti-rabbit, IRDye 800CW donkey anti-mouse, IRDye 680RD donkey anti-mouse, IRDye 680RD donkey anti-rabbit (LI-COR; Lincoln, NE. Cat. # 926-32213, 926-32212, 92668072, 926-68073, respectively); anti-rabbit Alexa 488, anti-mouse Alexa 488, anti-rabbit Alexa 594, and anti-mouse Alexa 594 (ThermoFisher; Grand Island, NY. Cat # A21203, A21207, A11017, A11008, respectively).
Murine neuronal cultures
Wt cerebral cortical neurons were cultured, as described elsewhere.26 Briefly, astrocytes cultured from 1-day old Wt mice as described elsewhere11 were plated on the bottom of a plastic well to which paraffin columns were previously attached. Then, the cerebral cortex from E16-18 mice was dissected, transferred into Hanks' balanced salt solution (Hyclone, GE Healthcare. Chicago, IL. Cat # SH30599.01) containing 100 units/ml penicillin/100 μg/ml streptomycin (Gibco TermoFisher, Grand Island, NY. Cat # 15140-122) and 10 mM HEPES (MilliPore. Burlington, MA. Cat # RDD002), and incubated in trypsin containing 0.02% DNase (Gibco TermoFisher. Grand Island, NY. Cat # 25300-054) at 37 °C for 15 min. Tissue was triturated and the supernatant was re-suspended in neurobasal medium (Gibco TermoFisher. Grand Island, NY. Cat # 21103-049) supplemented with B27 (Gibco TermoFisher. Grand Island, NY. Cat # 17504-044) containing 2 mM l-glutamine (Gibco TermoFisher. Grand Island, NY. Cat # A12860-01), and plated onto 0.1 mg/ml poly-l-lysine-coated coverslips (MilliPore. Burlington, MA. Cat # P1399), which were then placed on top of the paraffin columns attached to the bottom of the well with glial cells. Finally, cells were treated 72 hours later with 5 µM of cytosine arabinoside (Ara-C. USB. Cleveland, OH. Cat # 1241381). With this method, 2.95 +/− 1.16% of 281 cells from 4 different cultures are immunoreactive to GFAP (data not shown).
Generation of induced pluripotent stem cell (iPSC)-derived neurons
Induced pluripotent stem cells (iPSCs) were generated by reprograming human dermal fibroblasts from consenting deidentified healthy donors (Emory IRB approval 00,064,365) with the CytoTune Sendai Kit (ThermoFisher; Cat #. A16517). IPSCs were cultured in standard feeder free conditions using mTESR cell culture media (STEMCELL Technologies; Cat # N85850) in 5% CO2 at 37 °C. IPSCs were differentiated to neural progenitor cells (NPCs) by generating embryoid bodies and performing rosette selection and expansion as previously described.27 NPCs were maintained in media containing DMEM/F-12 (TermoFisher, Grand Island, NY. Cat # 11320033), B27 (GIBCO, ThermoFisher, Grand Island, NY. Cat # 17504-044), 1 M HEPES (Millipore; Burlington, MA. Cat # RDD002), N2 supplement (STEMCELL Technologies, Cambridge, MA. Cat # 7152), Glutamax (Gibco, TermoFisher. Grand Island, NY. Cat. # A12860-01), 20 ng/ml of fibroblast growth factor (FGF2; STEMCELL Technologies. Cambridge, MA. Cat # 78003), and penicillin/streptomycin. To generate neurons, NPCs were plated onto poly-ornithine/laminin -coated culture dishes, FGF2 was removed from the media and 5 µM of the rho kinase inhibitor Y-27632 2HCl (TOCRIS; Minneapolis, MN. Cat # 1254) was added for 4 days and then removed for 4 more days. Media was then switched to neurobasal A (Gibco TermoFisher. Grand Island, NY. Cat # 21103-049) with B27, Glutamax and penicillin/streptomycin. Cells were cultured for 2–4 weeks prior to performing experiments. Neurons were characterized by their immunoreactivity to the neuronal markers NeuN, bassoon and PSD-95 (data not shown).
Isolation of presynaptic vesicles and presynaptic cytoplasm
To isolate presynaptic vesicles and presynaptic cytoplasm we used a protocol described elsewhere.28 Briefly, cerebral cortices were homogenized and centrifuged at 2,000 g for 5 min in fractionation buffer containing 1 mM EGTA (MilliPore. Burlington, MA. Cat # E3889), 0.25 M sucrose (TermoFisher. Grand Island, NY. Cat # S6-12) and 25 mM HEPES pH 8.1. Then, supernatants were centrifuged at 32,000 g for 10 min. The pellet was resuspended and layered on top of a 5%–10% discontinuous Ficoll gradient (HE Healthcare. Chicago, IL. Cat # 17-0300-10) and centrifuged at 50,000 g for 20 min using a Optima TLX tabletop ultracentrifuge (Beckman. Irving, TX). Synaptoneurosomes (assembled by the sealed axonal bouton and the attached post-synaptic density) were collected from the 5%–10% interface and subjected to osmotic lysis with 10 mM HEPES and protease inhibitors, and centrifuged at 17,000 g during 30 minutes. Supernatant was centrifuged at 48,000 g during 25 min. The pellet was discarded, and the supernatant was centrifuged again at 133,000 g during 60 minutes. The pellet containing synaptic vesicles (identified by their immunoreactivity to syntaxin I and synaptophysin but not to PSD-95) and the concentrated supernatant containing the presynaptic cytoplasm were mixed with 1% SDS and loaded onto a gel for immunoblotting.
Murine model of cerebral ischemia
Transient occlusion of the middle cerebral artery (tMCAO) was induced in 8 – 12 weeks/old Wt mice with a 6-0 silk suture advanced from the external carotid artery (ECA) into the internal carotid artery until the origin of the middle cerebral artery (MCA) as described elsewhere.29 Briefly, a silicone-coated nylon monofilament (6-0, Ethicon; Issy Les Moulineaux, France) was introduced through the ECA and advanced up to the origin of the MCA. The suture was withdrawn after 60 minutes of cerebral ischemia, followed by different recovery times. Cerebral perfusion (CP) in the distribution of the MCA was monitored throughout the surgical procedure and after reperfusion with a laser Doppler (Perimed Inc., North Royalton, OH), and only animals with a > 70% decrease in CP after occlusion and complete recovery after suture withdrawn were included in this study. The rectal and masseter muscle temperatures were controlled at 37 °C with a homoeothermic blanket.
Non-human primate model of cerebral ischemia
7–13 years/old female rhesus macaques (Macaca mulatta) underwent right middle cerebral artery occlusion (MCAO) as described elsewhere.30 Briefly, animals were anesthetized with 3–5 mg/kg of telazol followed by intubation and induction of general anesthesia with 1.0–2.0% isoflurane mixed with 100% O2. Heart rate, temperature, respiratory rate, pulse oximetry, blood pressure, EKG, and end tidal CO2 were continuously monitored and maintained within physiological parameters. Animals were immobilized with a radiolucent head holder and placed in the supine position. The groin region was shaved and a #22-gauge vascular access needle was placed into the right common femoral artery under ultrasound guidance and exchanged over a 0.018 Cope Mandrel using a 4 French Cook microdilator. A 4 French 65 cm Terumo glide catheter was advanced under fluoroscopic guidance into the right internal carotid through which was placed an Excelsior 10 microcatheter into the distal segment of previously identified M3 branch of the right MCA. Multiple 5-mm segments of 4-0 silk suture were injected through the microcatheter to prevent collateral flow into the ischemic area and the Excelsior 10 microcatheter was then withdrawn into the M1-M2 bifurcation of the right MCA. A final 10 mm segment of suture was injected through the microcatheter which was then removed. A final 10 mm segment of suture was injected and the microcatheter was removed. Twenty-four hours later magnetic resonance imaging (MRI) scanning was performed on a 3 T Magnetom Trio MR imaging scanner (Siemens, Erlangen, German) with an 8-channel phased array knee coil (Invivo Inc. Gainesville, Florida). Then brains were harvested and 30 µM sections involving the necrotic core and perinecrotic area were cut and stained for the immunohistochemical studies described below.
Exposure to oxygen and glucose deprivation (OGD) conditions
Wt cerebral cortical neurons were kept either in glucose-free neurobasal medium (TermoFisher. Grand Island, NY. Cat # A24775-01) under physiological conditions, or maintained in an anaerobic chamber (Don Whitley Scientific) with <1% oxygen and glucose-free neurobasal medium. Five minutes later neurons were returned to physiological conditions with normal oxygen and glucose, and treated two and a half hours later with 5 nM of uPA or a comparable volume of vehicle (control). Thirty minutes later cells were fixed and immunostained as described below.
Immunohistochemistry
To study the expression of uPA in neuronal extensions, DIV-4, -6 and -12 Wt cerebral cortical neurons, and 2–4-weeks/old iPSC-derived neurons prepared from healthy human donors, were fixed in 4% paraformaldehyde (PFA. Electron Microscopy Sciences; Hattfield, PA. Cat # 15714-S). To study the effect of uPA on the formation of synaptic contacts in vitro, DIV-16 Wt cerebral cortical neurons and 4-weeks/old iPSC-derived neurons prepared from healthy human donors were incubated 30 minutes with vehicle (control) or 5 nM of murine (for experiments with murine neurons) or human uPA (for experiments with iPSC-derived neurons), alone or in combination with either 270 μM of puromycin (murine neurons) or 175 μM of ADH1 (iPSC-derived neurons). In all experimental groups, neurons were permeabilized with 0.1% triton (MilliPore, Burlington, MA. Cat # 9400), blocked with 3% BSA in TBS (TermoFisher. Grand Island, NY. Cat # J64655) during 20 min, and incubated overnight with antibodies against either murine uPA (1:100), or rabbit uPA (1:400) and murine bassoon (1:50), or rabbit uPA (1:400) and bassoon (1:100), or rabbit uPA and PSD-95 (1:100). Cells were then washed and incubated with a 1:500 dilution of the following secondary antibodies: anti-mouse Alexa 594, anti-rabbit Alexa 594, or anti-mouse Alexa 488. Confocal micrographs were taken at 60× (murine cells) or 156× magnification (human cells) and deconvoluted using 64 iterations of the CellSens dimension 1.17 software (Olympus. RRID: SCR_014551). Quantifications of uPA/phalloidin-, uPA/bassoon-, and uPA/PSD-95-positive puncta in individual neuronal extensions were performed using the ImageJ and the plugin puncta analyzer (NIH. RRID: SCR_003070).
To study the expression of uPA, NCAD, bassoon and PSD-95 in the cerebral cortex, 30 µm sections were obtained from the ischemic and non-ischemic hemispheres of Wt mice (n = 5) and rhesus monkeys (n = 6) 24 hours after the induction of tMCAO or MCAO, respectively, performed as described above. Before tMCAO, a sub-group of mice was injected into the third ventricle at bregma: –0.94 mm, medial: 0.0 mm and ventral: 2.5 mm 31 with 2 µl of a 87.6 mM solution of ADH1 or saline solution. Three hours after tMCAO animals were intravenously treated with 0.1 mg/Kg of recombinant uPA. Brain slices were permeabilized with 0.1% triton, blocked with 3% BSA (TermoFisher, Grand Island, NY. Cat # J64655) in TBS during 1 hour, and incubated overnight with antibodies against murine uPA (1:100), rabbit uPA (1:400), or N-cadherin (1:400), or with a combination of anti-bassoon and PSD-95 antibodies. Samples were then washed and incubated with Hoechst (1:5,000) and either anti-mouse Alexa 488 (1:500) or anti-rabbit Alexa 488 (1:500) secondary antibodies. Pictures were taken from the frontal cortex at a 40× magnification with a fluorescent microscope (Olympus IX83), and from each cortical layer at 282× magnification with a Fluoview FV10i automated confocal laser-scanning microscope (Olympus; Center Valley, PA). Quantification of uPA-positive puncta in the non-ischemic brain was performed in confocal micrographs taken from individual cortical layers. Values are given as percentage of the total number of uPA-positive puncta in the six cortical layers. Quantification of N-cadherin- and uPA/bassoon-positive puncta were performed in confocal micrographs obtained from the II/III cortical layers in the area surrounding the necrotic core and from a comparable area in the contralateral non-ischemic hemisphere. In both cases images were processed with ImageJ with the plugin puncta analyzer (NIH. RRID: SCR_003070). Values were normalized to the contralateral non-ischemic hemisphere.
To study the effect of uPA on N-cadherin expression, murine Wt and 4-weeks/old iPSC-derived neurons prepared from healthy human donors were treated during 30 minutes with 5 nM of uPA. To study the effect of uPA on synapse formation, Wt cerebral cortical neurons and 4-weeks/old iPSC-derived neurons prepared from healthy human donors were treated during 30 minutes with 5 nM of uPA, alone or in combination with 175 µM of the N-cadherin inhibitor ADH-1 diluted in DMSO or with DMSO alone. To investigate the effect of OGD on N-cadherin expression, murine Wt neurons were exposed to 5 minutes of OGD conditions, followed 2.5 hours later by treatment with 5 nM of uPA or vehicle (control), and fixation 10 minutes later with 4% of PFA. In both experiments, cells were permeabilized and blocked as described above and incubated with the following antibodies: rabbit N-cadherin (1:400), mouse β-catenin (1:100), mouse bassoon (1:100), rabbit bassoon (1:100), and rabbit PSD-95 (1:100). Cells were washed and incubated with the following secondary antibodies at a 1:500 dilution: anti-rabbit Alexa 488, and anti-mouse Alexa 594. Quantifications of N-cadherin/bassoon-, N-cadherin/β-catenin-, bassoon/PSD-95-, and N-cadherin-positive puncta in neuronal extensions were performed with the ImageJ and puncta analyzer plugin in confocal micrographs taken at 60× magnification and deconvoluted as described above. Besides the magnification used for each observation and noted in the corresponding figure legend, parameters for all confocal micrographs were: 1024 x 1024 pixels, 4.4 sec/frame; scanning device: 2 galvano-meter mirrors; XY resolution: 300 nm; lens: 60× UPLSAPO60X0 NA 1.35; immersion oil: IMMOIL F30CC; and pinhole: 50 µm.
Western blot analyses
To study the expression of uPA in the axonal bouton, extracts were prepared from the cytoplasm and synaptic vesicles isolated from the presynaptic terminal as described above. To study the expression of NCAD in the brain, extracts were prepared from the cerebral cortex of Wt mice 1, 3 and 24 hours after 60 minutes of tMCAO. A sub-group of mice was intravenously treated with 0.1 mg/Kg of recombinant uPA 2.5 hours after reperfusion and sacrificed 30 minutes later (3 hours after tMCAO). To study the expression of NCAD in neurons, extracts were prepared from DIV-16 Wt cerebral cortical neurons treated during 30 minutes with 5 nM of uPA or vehicle (control), alone or in combination with 270 μM of puromycin.
Brain tissue was lysed in 1000 µl of RIPA buffer (TEKNOVA, Hollister, CA. Cat # R3792) and the protease inhibitors pack EASYpack (Roche. Indianapolis, IN. Cat # 4693132001) using a Universal Laboratory Stirrer IKA EuroStar 60 Digital (MIlliPore. Burlington, MA. Cat # Z766976) and a 5 ml Potter-Elvehjem Tissue Homogenizer (Omni International, Kennesaw, GA. Cat # 07-358034) at 10 stroke – 900 rpm. Samples were then centrifuged at 21,000 g × 20 min 4 C. Cells were lysed in 150 µl of RIPA and centrifuged at 21,000 g × 20 minutes at 4 °C. In both cases (brains and cells) the supernatant was recovered and proteins were quantified using the BCA assay (TermoFisher. Grand Island. NY. Cat # 23225). Then, equal amounts of protein were loaded and separated onto a 4 -15% SDS-gel, transferred into a nitrocellulose membrane, blocked with the Intercept Blocking Buffer (LI-COR. Lincoln, NE. Cat # 927-60001), and incubated overnight at 4 °C with antibodies against uPA (1:100), syntaxin I (1:2,000), actin (1:50,000), synaptophysin (1:2,000), rabbit PSD-95 (1:1000), and rabbit N-cadherin (1:2000). Membranes were then washed and incubated during one hour with IRDye 800CW donkey anti-rabbit (1:10,000), IRDye 800CW donkey anti-mouse (1:10,000), IRDye 680RD donkey anti-mouse (1:10,000), and IRDye 680RD donkey anti-rabbit antibodies (1:10,000), and developed using a LI-COR Odyssey Fc reader (LI-COR). Quantifications were performed using the Image Studio software Version 5.2 (LI-COR. RRID: SCR_015795). Values are given as a ratio of intensity of the band/actin intensity and normalized to intensity in the control condition for each experiment.
Statistics
To confirm normal distribution of the data when the sample size was less than 10, we used visual inspection and the outliers test, as described elsewhere.32 Statistical analysis was performed with either two-tailed student’s t-test, or Mann-Whitney non-parametric test, or one- or two-way ANOVA with corrections, as deemed as appropriate and described in each figure legend. p-values ≤0.05 were considered as significant. The effect of the interaction between experimental groups was calculated using the Prism software (RRID: SCR_002798) with two-way ANOVA test.
Results
uPA in found in the presynaptic terminal of cerebral cortical neurons
First, we used confocal microscopy to study the expression of uPA in murine cerebral cortical neurons at different developmental stages. We found that at DIV-4 uPA is localized around the nucleus, whereas most of it is detected in growth cones at DIV-6, and in neuronal extensions at DIV-12 (Figure 1(a) and (b)). To determine whether uPA is localized in the presynaptic (axonal bouton) or postsynaptic (dendritic spine) compartment, we used confocal microscopy to quantify the number of uPA/bassoon- (detects uPA in the presynaptic terminal) and uPA/PSD-95 (denotes uPA in the postsynaptic compartment) -positive puncta in murine cerebral cortical neurons. Our data indicate that most of uPA is found in the presynaptic compartment (Figure 1(c) and (d); n = 14 extensions examined per culture from three different cultures; p = 0.04 when the number of uPA-positive puncta in the axonal bouton is compared to the number of puncta in the postsynaptic terminal). To further characterize these results, we used Western blot analysis to study the expression of uPA in extracts prepared from either presynaptic vesicles or the cytoplasm of the presynaptic terminal of murine cerebral cortical neurons, isolated as described in the Materials and methods section. The purity of our preparations was confirmed by their immunoreactivity to synaptophysin and syntaxin-I only in the fraction containing synaptic vesicles, and the lack of PSD-95 immunoreativity in both fractions. These studies revealed that in the presynaptic terminal uPA is associated with synaptic vesicles (Figure 1(e) and (f)). The translational relevance of these findings is underscored by our observation that in extensions of mature (4 weeks/old) iPSC-derived neurons prepared from healthy human donors as described in Materials and methods, uPA co-localizes preferentially with the presynaptic marker bassoon (Figure 1(g) to (i)).
Figure 1.

uPA is found the presynaptic terminal of cerebral cortical neurons. (a) Representative confocal micrographs at 60× magnification of DIV-4, -6 & -12 murine cerebral cortical neurons immunostained with anti-uPA antibodies (red) and phalloidin (green). (b) Percentage of uPA-positive puncta/20 μm in extensions of DIV 12 murine cerebral cortical neurons compared to uPA-positive puncta in DIV 6 neurons. Each experiment was repeated with cells from 5 different cultures. Each data point is the average of 12 (DIV 6) and 9 (DIV 12) observations per culture. Mann-Whitney non-parametric test. (c and d) Representative confocal micrographs at 60× magnification of uPA (red), bassoon (green in a and c) and PSD-95 (green in b and d) expression (c), and mean percentage of either uPA/bassoon- or uPA/PSD-95-positive puncta in relation to the total number of uPA-positive puncta/50 μm extensions (d). Arrow in panel d depicts an example of an uPA/PSD-95-positive puncta. Arrowhead points to an example of a PSD-95-positive puncta that does not colocalize with uPA. Each experiment was repeated with DIV-16 neurons from 3 different cultures. Each data point is the average of 14 observations per culture. Two-tailed student’s t-test. (e and f) Representative immunoblotting (e) and quantification of the mean intensity of the band (f) of uPA, synaptophysin (SYP), syntaxin I (STX I), PSD-95 and actin expression in synaptic vesicles (SV) and cytoplasm (Cyt) from DIV-16 murine cerebral cortical neurons. Each observation was repeated with cortices extracted from 4 different animals. Two-tailed student’s t-test. (g) Representative confocal micrographs at 156× magnification of 2- and 4-weeks old induced pluripotent stem cell (iPSC)-derived neurons from healthy human donors stained with phalloidin (green) and anti-uPA antibodies (red). (h and i) Mean percentage of uPA- (h) and uPA/bassoon-positive puncta (i) in 4-weeks/old iPSC-derived neurons compared to uPA- and uPA/bassoon-positive puncta in 2 weeks/old iPSC-derived neurons stained as described in G. Each experiment was repeated with cells from three different cultures. Each data point is the average of 6 observations per culture. Mann-Whitney non-parametric test (h and i).
Cerebral ischemia decreases the abundance of uPA and NCAD in surviving neurons surrounding the necrotic core
We then used confocal microscopy to study the expression of uPA in the gyrencephalic frontal cortex of the rhesus monkey. We found that although the six cortical layers harbor uPA-positive puncta, most of them are found in the II/III and V cortical layers (27.70% +/− 1.25 and 25.63% +/− 2.53 of the total of number of uPA-positive puncta throughout the six cortical layers, respectively. Figure 2(a) and (b); n = 6 rhesus monkeys). To study the effect of cerebral ischemia on the expression of uPA, we performed similar observations in the peri-necrotic cortex of the non-human primate 24 hours after MCAO. We chose this zone (Figure 2(c)) because our previous studies indicate that uPA-driven morphological and biochemical changes in surviving neurons in the peri-infarct cortex are linked to functional recovery following an ischemic stroke.12,15 Additionally, since our experiments reveal that the expression of uPA and NCAD in the cerebral cortex follow a similar pattern, we co-stained our samples with anti-NCAD antibodies. Our data indicate that the expression of uPA (Figure 2(e) and (f)) and NCAD (Figure 2(g) and (h)) decrease in extensions of surviving neurons surrounding the area irreversibly affected by the ischemic injury.
Figure 2.

The expression of uPA decreases in the area surrounding the necrotic core. (a) Representative confocal micrographs at 282× magnification of individual cortical layers of the frontal cortex of the rhesus monkey immunostained with anti-uPA antibodies (green) and Hoechst (blue). (b) Percentage of uPA-positive puncta in individual cortical layers from the frontal cortex of 6 rhesus monkeys in relation to the total number of puncta in the six cortical layers of each animal. Each value represents the average of 10 confocal micrographs per animal. One-way ANOVA with Holm-Sidak’s multiple comparisons test. (c) Representative diffusion-weighted imaging (DWI) of an axial section of the brain of a rhesus monkey 24 hours after MCAO. The white square depicts one of the regions of interest surrounding the necrotic core where observations were made. (d) Representative micrographs at 4× magnification from the frontal cortex of a rhesus monkey immunostained with antibodies against uPA and NCAD 24 hours after MCAO. The white square in a denotes an area surrounding the necrotic core where uPA and NCAD expression are noticeably decreased. Panel b corresponds to a comparable area in the contralateral non-ischemic hemisphere. (e) Representative micrographs at 20× magnification of uPA expression in the area surrounding the necrotic core (a) and a comparable area in the contralateral non-ischemic hemisphere (c) of a rhesus monkey 24 hours after tMCAO. b and d correspond to a 5-fold electronic magnification of the area demarcated by the white squares in a and c, correspondingly. (f) Mean percentage of uPA-positive puncta in the II/III cortical layers of the area surrounding the necrotic core of 3 rhesus monkeys 24 hours after MCAO, in relation to the number of uPA-positive puncta in a comparable area in the non-ischemic hemisphere. n = 3 animals. Each data point represents the average of 5 micrographs per animal. Mann-Whitney non-parametric test. (g) Representative micrographs at 20× magnification of NCAD expression in the area surrounding the necrotic core (a) and a comparable area in the contralateral non-ischemic hemisphere (c) of a rhesus monkey 24 hours after tMCAO. b and d correspond to a 5-fold electronic magnification of the area demarcated by the white squares in a and c, correspondingly. (h) Mean percentage of NCAD-positive puncta in the II/III cortical layers of the area surrounding the necrotic core of 3 rhesus monkeys 24 hours after MCAO, in relation to the number of NCAD-positive puncta in a comparable area in the non-ischemic hemisphere. n = 3 animals. Each data point represents the average of 5 micrographs per animal. Mann-Whitney non-parametric test.
Effect of uPA treatment on the abundance of NCAD in the peri-ischemic tissue
To further study the effect of cerebral ischemia on the expression of uPA and NCAD, we used confocal microscopy to quantify uPA- and NCAD-positive puncta in the II/III and V cortical layers of the frontal murine cortex 24 hours after tMCAO. Our results indicate that, as observed in the gyrencephalic cortex of the rhesus monkey, the expression of uPA and NCAD also decrease in a well-defined band of surviving neurons surrounding the necrotic core (Figure 3(a) to (c)). To better characterize these observations, we used immunoblotting to study the expression of NCAD in the murine cerebral cortex 1, 3 and 24 hours after 60 minutes of tMCAO. We found that the ischemic injury causes a rapid decrease in the abundance of NCAD in the cerebral cortex (Figure 3(d) and (e)), but that this effect is attenuated by intravenous treatment with recombinant uPA 2.5 hours after tMCAO (Figure 3(f) and (g)). Importantly, our previous work with FITC-labeled uPA indicates that it reaches the ischemic tissue following its intravenous administration after tMCAO (11,12,33 and data not shown).
Figure 3.

Effect of uPA treatment on NCAD expression in the area surrounding the necrotic core. (a) Representative 10× micrographs through the murine frontal cortex stained with antibodies against either uPA or NCAD, or with a combination of anti-uPA and -NCAD antibodies 24 hours after 60 minutes of transient middle cerebral artery occlusion (tMCAO). Asterisks indicate a band of tissue outside the necrotic area were observations were performed. (b and c) Mean percentage of uPA- (b) and NCAD-positive puncta (c) in the II/III cortical layers of the area surrounding the necrotic core, compared to the number of uPA- and NCAD-positive puncta in a comparable area in the contralateral non-ischemic hemisphere, of 6 Wt mice 24 hours after tMCAO. Each value represents the average of 10 micrographs per animal. Mann-Whitney non-parametric test. (d and e) Representative Western blot analysis (d) and quantification of mean the intensity of the band (e) of NCAD expression in the murine cerebral cortex of sham-operated mice (control: C), or 1, 3 and 24 hours after tMCAO. n = 4–5 mice/group. One-way ANOVA with Holm-Sidak’s multiple comparisons test. (f and g) Representative Western bot analysis (f) and quantification of the mean intensity of the band (g) of NCAD expression in the murine cerebral cortex 3 hours after either sham operation or tMCAO. A sub-group of animals was intravenously treated with uPA 30 minutes before harvesting the brains. n = 5 animals per group. Statistical analysis: one-way ANOVA with Tukey’s multiple comparisons test. (h) Representative confocal micrographs at 60× magnification of murine cerebral cortical neurons immunostained with anti-NCAD antibodies 3 hours after exposure to 5 minutes of oxygen and glucose deprivation (OGD). Neurons were treated 30 minutes before the end of the experiment (2.5 hours) with 5 nM of uPA or a comparable volume of vehicle (control). (i) Percentage of NCAD-positive puncta/50 µm in neurons exposed to the experimental conditions described in h, compared to the number of NCAD-positive puncta in cells kept under normoxic conditions. Each experiment was repeated with cells from 5 (c, OGD) and 4 (OGD + uPA) different cultures, and data point represents the average of quantifications obtained in 8 extensions. One-way ANOVA with Tukey’s multiple comparisons test.
Because a sub-population of astrocytes also express NCAD,34 then it is plausible that the observed effect of uPA-treatment on the abundance of NCAD is due to an increase in NCAD expression in glia but not in neurons. To assess this possibility, we used confocal microscopy to study the expression of NCAD in extensions from murine cerebral cortical neurons exposed to 5 minutes of oxygen and glucose deprivation (OGD; our early work indicates that this time of OGD does not trigger cell death but decreases the number of intact synaptic contacts, thus successfully modeling surviving neurons the perinecrotic cortex12) followed 2.5 hours later by treatment with 5 nM of uPA or vehicle (control). Thirty minutes after uPA-treatment cells were fixed and stained with anti-NCAD antibodies. We found that OGD decreases the number of NCAD-positive puncta and that this effect is attenuated by treatment with uPA (Figure 3(h) and (i)).
uPA induces the formation of NCAD-mediated synaptic contacts
To further investigate the observed effect of uPA on NCAD expression,14 we used immunoblotting to quantify its abundance in extracts prepared from cultured murine cerebral cortical neurons treated during 30 minutes with 5 nM of uPA or vehicle (control), alone or in the presence of 270 µM of puromycin. Our results indicate that uPA induces the synthesis of NCAD mRNA in cerebral cortical neurons (Figure 4(a) and (b)). Furthermore, our confocal microscopy studies with cultured murine cerebral cortical neurons exposed to similar experimental conditions show that uPA induces the formation of synaptic contacts, and that this effect is abrogated by co-treatment with puromycin (Figure 4(c) and (d)). The translational relevance of these observations is underscored by our findings that uPA also increases the presynaptic abundance of NCAD (Figure 4(e) and (f)) and induces the formation of synaptic contacts (Figure 4(g) and (h)) in iPSC-derived neurons prepared from healthy human donors. Importantly, the effect of uPA on the formation of synaptic contacts in iPSC-derived neurons was mediated by NCAD as it was abrogated by ADH1 (prevents the formation of NCAD dimers required for the establishment of homotypic interactions between the pre- and postsynaptic terminals).
Figure 4.

uPA induces N-Cadherin-mediated formation of synaptic contacts. (a and b). Representative Western blot analysis (a) and quantification of the mean intensity of the band (b) of N-Cadherin (NCAD) expression in extracts prepared from murine cerebral cortical neurons incubated during 30 minutes with vehicle (control) or 5 nM of uPA, alone or in the presence of 270 µM of puromycin. n = 4 observations per group. Two-way ANOVA with Holm-Sidak’s multiple comparisons test. uPA-puromycin interaction: 3.9%. (c) Representative confocal micrographs at 240× magnification of extensions of murine cerebral cortical neurons incubated during 30 minutes with 5 nM of uPA, alone or in the presence of 270 µM of puromycin and then stained with antibodies against bassoon (red) and NCAD (green). (d) Mean percentage of bassoon/NCAD puncta in Wt cerebral cortical neurons exposed to the experimental conditions described in C. n = 5. Each data point represent the average of 5 observations per culture, for a total of 25 observations. Two-way ANOVA with Holme-Sidak’s multiple comparisons test. Interaction uPA – puromycin: 12.52%. (e) Representative confocal micrographs at 156× magnification of 4-weeks/old iPSC-derived neurons immunostained with antibodies against NCAD (green) and bassoon (red) after 30 minutes of treatment with uPA or vehicle (control). Panels c and f correspond to a 5-fold electronic magnification of the area demarcated by the white squares in b and e. (f) Mean percentage of NCAD/bassoon-positive puncta in 4-weeks/old iPSC-derived neurons treated with uPA, in relation to NCAD/bassoon-positive puncta in cells treated with vehicle (control). Each observation was repeated with neurons from 3 different cultures, and each data point is the average of 21 (control) and 20 (uPA-treated) observations. Mann-Whitney non-parametric test. (g) Representative confocal micrographs at 240× magnification of extensions from 4-weeks/old iPSC-derived neurons immunostained with antibodies against bassoon (red) and PSD-95 (green) following 30 minutes of treatment with either vehicle (control; a and e), 5 nM of uPA (b and f), or a combination of 175 µM of the NCAD inhibitor ADH1 and either vehicle (control; c and g) or uPA (d and h). e, f, g and h correspond to a 5× magnification of the area indicated by the dashed squares in a, b, c and d. (h) Mean percentage of bassoon/PSD-95 puncta in 4-weeks/old iPSC-derived neurons exposed to the experimental conditions described in G. n = 3. Each data point represent the average of 12 observations per culture. Two-way ANOVA with Holme-Sidak’s multiple comparisons test. Interaction uPA – ADH: 23.13%.
Treatment with uPA induces NCAD-mediated formation of synaptic contacts in the area surrounding the necrotic core
To determine if the observed effect of uPA treatment on the abundance of NCAD in the ischemic brain leads to an increase in the number of synaptic contacts in the peri-ischemic cortex, mice were injected into the third ventricle before undergoing tMCAO with 2 µl of either a 87.6 mM solution of ADH1, or a comparable volume of saline solution. Three hours after tMCAO animals were intravenously treated with murine uPA or saline solution. Brains were harvested 24 hours after tMCAO and the number of intact synaptic contacts (bassoon/PSD-95-positive puncta) in the peri-necrotic cortex was quantified with confocal microscopy. These experiments revealed that cerebral ischemia reduces the number of intact synaptic contacts in the band of tissue surrounding the necrotic core, where we also detected a decrease in the expression of uPA and NCAD, and that this effect is attenuated by uPA treatment. Significantly, we found that NCAD mediates the effect of uPA, as it was abrogated in mice pre-treated with ADH1 (Figure 5(a) and (b); n = 6 mice per group, p = 0.04 when animals treated with saline solution are compared with uPA-treated mice, and p = 0.03 when animals treated with uPA are compared with mice treated with uPA and ADH1; one-way ANOVA with Holm-Sidak’s multiple comparisons test).
Figure 5.
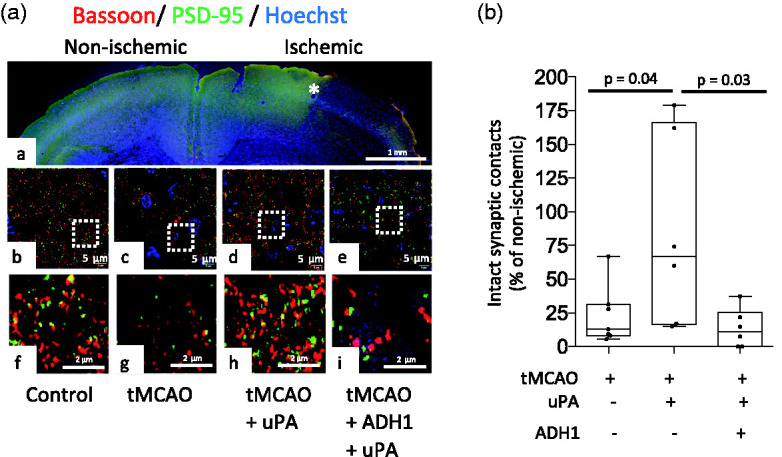
Treatment with uPA induces N-Cadherin-mediated formation of synaptic contacts in the area surrounding the necrotic core. (a) Panel a is a representative micrograph at 10× magnification of a coronal section through the frontal cortex of a murine brain stained with antibodies against bassoon and PSD-95, and the nuclear staining Hoechst, 24 hours after 60 minutes of tMCAO. The asterisk denotes the area outside the necrotic core where quantifications were made. Panels b–e correspond to representative confocal micrographs at 282× magnification in the II/III cortical later of Wt mice either sham operated (b) or 24 hours after tMCAO (c–e). Before the induction of surgical procedure a sub-group of animals were injected into the third ventricle with 2 µl of a 87.6 mM solution of ADH1 (e). Three hours after reperfusion animals were intravenously treated with 0.1 mg/Kg of murine uPA or saline solution (SS). Panels f, g, h and i correspond to a 5-fold electronic magnification of the areas depicted by white squares in b, c, d and e. (b) Percentage of bassoon/PSD-95-positive puncta in the II/III cortical layers of the area surrounding the necrotic core (denoted by an asterisk in A-panel e) in mice subjected to the experimental conditions described in A. Each point depicts the average of 10 micrographs taken from 6 animals per experimental group. Values are given as percentage compared to bassoon/PSD-95-positive puncta in comparable areas of the contralateral non-ischemic hemisphere. Statistical analysis: one-way ANOVA with Holm-Sidak’s multiple comparisons test.
Discussion
Since its initial discovery, it was believed that uPA’s only role was to catalyze the conversion of plasminogen into plasmin on the cell surface.35 However, it was soon evident that binding of uPA to uPAR also activates cell signaling pathways that promote survival, proliferation, motility and invasion, via mechanisms that not always require the conversion of plasminogen into plasmin.8 In the developing brain, uPA mRNA is abundantly detected in all neurons just before the onset of axogenesis, with the highest levels of message in large projection neurons.9 Furthermore, the expression of uPA mRNA has also been linked to tissue remodeling required for neural development.36 However, despite the relevance of these observations, the expression and function of uPA protein in cerebral cortical neurons are still unclear.
Our studies indicate that in immature cerebral cortical neurons uPA protein has a perinuclear distribution. In contrast, at DIV-6 uPA is detected in neurites and growth cones, and at DIV-12 its distribution is almost exclusively limited to neuronal extensions. Noticeably, this expression pattern resembles that of its receptor uPAR,13 and parallels the onset of synaptogenesis in neuronal cultures.37 Furthermore, our work indicates that these findings are not exclusive of murine neurons, as they are also observed in mature iPSC-derived neurons prepared from healthy human donors. Additionally, in both, murine neurons and iPSC-derived neurons prepared from healthy human donors, most of uPA is found associated with pre-synaptic vesicles in the axonal bouton, and to a significantly lesser extent is detected in the post-synaptic terminal.
A growing body of experimental evidence indicates that uPA is crucial for the remodeling and repair of several organs and tissues including skeletal muscle,38 lungs 39 and blood vessels.40 Likewise, the expression of its receptor uPAR increases in organs that require active remodeling, such as gestational tissue during placental development 41 and keratinocytes during epidermal wound healing.42 Our previous studies show that cerebral ischemia induces an early but transient increase in the expression of uPA in the ischemic tissue, and that genetic deficiency of uPA or inhibition of its binding to uPAR impairs neurological recovery after stroke.15 In contrast, the experimental design used in the present work does not allow us to make conclusions on the effect of endogenous uPA in the ischemic brain. Instead, here we show that 24 hours after an ischemic stroke the expression of uPA is decreased in the area surrounding the necrotic core, and that this effect and its harmful consequences on synaptic structure are antagonized by the administration of recombinant uPA.
Because most of the experimental observations published to this date have been performed in the lissencephalic murine brain, the expression of uPA protein in the gyrencephalic cerebral cortex has not been yet experimentally characterized. Our confocal microscopy studies reveal that uPA is abundantly found in the II/III and V cortical layers of both, the gyrencephalic cortex of the non-human primate and the lissencephalic murine brain. These findings are of particular importance because synapses from these cortical laminae receive the input from and are the main source of output to other cortical and subcortical areas of the brain.43 Therefore, these observations, together with our previous data indicating that uPA triggers the formation of axons and dendrites,13,15 and increases the amplitude and frequency of spontaneous postsynaptic excitatory currents (EPSCs) in cerebral cortical neurons,14 suggest that this plasminogen activator plays a role in the induction of plastic changes required for successful neurotransmission in the cerebral cortex. Together, our data indicate that the ability of uPA to induce plastic changes in the synapse underlies its reported capacity to trigger the recovery of synapses damaged by an ischemic injury.
The area surrounding the necrotic core plays a central role in the process of neurorepair following an ischemic stroke. Indeed, although neurons in this area survive the ischemic insult, their axonal boutons disappear and their post-synaptic terminals are engulfed within varicosities.44 However, upon successful reperfusion axonal boutons are newly formed and dendritic spines recover, following a sequence of events that require the release of uPA 11,45 and are associated with improved functional outcome.15 Here we report that uPA not only induces the remodeling and repair of pre-and post-synaptic terminals damaged by an ischemic insult, but also promotes the formation of synaptic contacts between them. However, despite the importance of these findings, to this date we do not know if this leads to the repair of tracts destroyed by the ischemic injury.
NCAD is crucial for the formation of synaptic contacts in the developing brain.19 Remarkably, we found that its expression pattern in synapses located in the II/III and V cortical layers of the mature brain is identical to that of uPA. Furthermore, our results reveal that uPA induces the synthesis of NCAD in neurons, and that NCAD mediates uPA-induced formation of synaptic contacts. The translational relevance of these findings is indicated by our observation that uPA also induces NCAD-mediated formation of synaptic contacts in mature non-ischemic iPSC-derived neurons prepared from healthy human donors. Importantly, although in the present work we focus on the synaptic effect of uPA-induced NCAD expression in cerebral cortical neurons, we are actively investigating if uPA also increases the expression of NCAD in astrocytes, and whether this effect contributes to the repair of synapses damaged by an ischemic injury.
The effect of cerebral ischemia on the expression and function of NCAD is poorly understood, and although the expression of its mRNA increases following an ischemic injury,24 the regulatory mechanisms and functional consequences of these changes remain unknown. We found that cerebral ischemia decreases the expression of NCAD in a band of tissue surrounding the necrotic core where the abundance of uPA is also decreased. Furthermore, we found that the expression of NCAD returns to baseline levels when animals are treated with recombinant uPA, and that this effect leads to the formation of new synaptic contacts in the area surrounding the necrotic core.
In summary, our data suggest a model in which uPA not only induces the structural and functional recovery of pre- and post-synaptic terminal in the area surrounding the necrotic core,13,15,45 but also prompts the formation of contacts between these repaired axonal boutons and dendritic spines via its ability to induce the synthesis of NCAD. This is a new role for uPA with translational implications for the development of therapeutic strategies to promote functional recovery following an ischemic stroke.
Supplemental Material
Supplemental material, sj-pdf-1-jcb-10.1177_0271678X211002297 for Urokinase-type plasminogen activator promotes N-cadherin-mediated synaptic recovery in the ischemic brain by Ariel Diaz, Paola Merino, Patrick McCann, Manuel A Yepes, Laura G Quiceno, Enrique Torre, Amelia Tomkins, Xiaodong Zhang, Chadwick M Hales, Frank C Tong and Manuel Yepes in Journal of Cerebral Blood Flow & Metabolism
Footnotes
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by National Institutes of Health Grant NS-NS091201 (to MY), VA MERIT Award IO1BX003441 (to MY), and American Heart Association Post-Doctoral Fellowship Grant 19POST34380009 (to AD).
Declaration of conflicting interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions: Ariel Diaz: designed and performed experiments, and analyzed data. Paola Merino: performed experiments. Patrick McCann: performed experiments. Manuel A Yepes: performed experiments. Laura Gutierrez Quiceno: performed experiments. Enrique Torre: performed experiments. Amelia Tompkins: performed experiments. Xiaodong Zhang: performed experiments. Chadwick Hales: performed experiments. Frank C Tong: designed and performed experiments. Manuel Yepes: designed experiments, analyzed data, and wrote paper.
ORCID iD: Manuel Yepes https://orcid.org/0000-0002-5224-9663
Supplementary material: Supplemental material for this article is available online.
References
- 1.Kleim JA, Jones TA.Principles of experience-dependent neural plasticity: implications for rehabilitation after brain damage. J Speech Lang Hear Res 2008; 51: S225–239. [DOI] [PubMed] [Google Scholar]
- 2.Hasbani MJ, Underhill SM, Erausquin G, et al. Synapse loss and regeneration: a mechanism for functional decline and recovery after cerebral ischemia. Neuroscientist 2000; 6: 110–119. [Google Scholar]
- 3.Hasbani MJ, Schlief ML, Fisher DA, et al. Dendritic spines lost during glutamate receptor activation reemerge at original sites of synaptic contact. J Neurosci 2001; 21: 2393–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murphy TH, Corbett D.Plasticity during stroke recovery: from synapse to behaviour. Nat Rev Neurosci 2009; 10: 861–872. [DOI] [PubMed] [Google Scholar]
- 5.Fazioli F, Resnati M, Sidenius N, et al. A urokinase-sensitive region of the human urokinase receptor is responsible for its chemotactic activity. EMBO J 1997; 16: 7279–7286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lijnen HR, Van Hoef B, Collen D.Activation with plasmin of tow-chain urokinase-type plasminogen activator derived from single-chain urokinase-type plasminogen activator by treatment with thrombin. Eur J Biochem 1987; 169: 359–364. [DOI] [PubMed] [Google Scholar]
- 7.Lijnen HR, Zamarron C, Blaber M, et al. Activation of plasminogen by pro-urokinase. I. Mechanism. J Biol Chem 1986; 261: 1253–1258. [PubMed] [Google Scholar]
- 8.Smith HW, Marshall CJ.Regulation of cell signalling by uPAR. Nat Rev Mol Cell Biol 2010; 11: 23–36. [DOI] [PubMed] [Google Scholar]
- 9.Dent MA, Sumi Y, Morris RJ, et al. Urokinase-type plasminogen activator expression by neurons and oligodendrocytes during process outgrowth in developing rat brain. Eur J Neurosci 1993; 5: 633–647. [DOI] [PubMed] [Google Scholar]
- 10.Lino N, Fiore L, Rapacioli M, et al. uPA-uPAR molecular complex is involved in cell signaling during neuronal migration and neuritogenesis. Dev Dyn 2014; 243: 676–689. [DOI] [PubMed] [Google Scholar]
- 11.Diaz A, Merino P, Manrique LG, et al. Urokinase-type plasminogen activator (uPA) protects the tripartite synapse in the ischemic brain via ezrin-mediated formation of peripheral astrocytic processes. J Cereb Blood Flow Metab 2019; 39: 2157--2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Diaz A, Merino P, Manrique LG, et al. A cross-talk between neuronal urokinase-type plasminogen activator (uPA) and astrocytic uPA receptor (uPAR) promotes astrocytic activation and synaptic recovery in the ischemic brain. J Neurosci 2017; 37: 10310–10322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Merino P, Diaz A, Jeanneret V, et al. Urokinase-type plasminogen activator (uPA) binding to the uPA receptor (uPAR) promotes axonal regeneration in the central nervous system. J Biol Chem 2017; 292: 2741–2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Diaz A, Merino P, Guo JD, et al. Urokinase-type plasminogen activator protects cerebral cortical neurons from soluble Abeta-induced synaptic damage. J Neurosci 2020; 40: 4251–4263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu F, Catano M, Echeverry R, et al. Urokinase-type plasminogen activator promotes dendritic spine recovery and improves neurological outcome following ischemic stroke. J Neurosci 2014; 34: 14219–14232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Benson DL, Tanaka H.N-cadherin redistribution during synaptogenesis in hippocampal neurons. J Neurosci 1998; 18: 6892–6904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Uchida N, Honjo Y, Johnson KR, et al. The catenin/cadherin adhesion system is localized in synaptic junctions bordering transmitter release zones. J Cell Biol 1996; 135: 767–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shan W, Yoshida M, Wu XR, et al. Neural (N-) cadherin, a synaptic adhesion molecule, is induced in hippocampal mossy fiber axonal sprouts by seizure. J Neurosci Res 2002; 69: 292–304. [DOI] [PubMed] [Google Scholar]
- 19.Tanaka H, Shan W, Phillips GR, et al. Molecular modification of N-cadherin in response to synaptic activity. Neuron 2000; 25: 93–107. [DOI] [PubMed] [Google Scholar]
- 20.Togashi H, Abe K, Mizoguchi A, et al. Cadherin regulates dendritic spine morphogenesis. Neuron 2002; 35: 77–89. [DOI] [PubMed] [Google Scholar]
- 21.Sanes JR, Yamagata M.Formation of lamina-specific synaptic connections. Curr Opin Neurobiol 1999; 9: 79–87. [DOI] [PubMed] [Google Scholar]
- 22.Honjo Y, Nakagawa S, Takeichi M.Blockade of cadherin-6B activity perturbs the distribution of PSD-95 family proteins in retinal neurones. Genes Cells 2000; 5: 309–318. [DOI] [PubMed] [Google Scholar]
- 23.Mendez P, De Roo M, Poglia L, et al. N-cadherin mediates plasticity-induced long-term spine stabilization. J Cell Biol 2010; 189: 589–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gertz K, Kronenberg G, Uhlemann R, et al. Partial loss of VE-cadherin improves long-term outcome and cerebral blood flow after transient brain ischemia in mice. BMC Neurol 2016; 16: 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klingener M, Chavali M, Singh J, et al. N-cadherin promotes recruitment and migration of neural progenitor cells from the SVZ neural stem cell niche into demyelinated lesions. J Neurosci 2014; 34: 9590–9606. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 26.Echeverry R, Wu J, Haile WB, et al. Tissue-type plasminogen activator is a neuroprotectant in the mouse hippocampus. J Clin Invest 2010; 120: 2194–2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Griesi-Oliveira K, Acab A, Gupta AR, et al. Modeling non-syndromic autism and the impact of TRPC6 disruption in human neurons. Mol Psychiatry 2015; 20: 1350–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ahmed S, Holt M, Riedel D, et al. Small-scale isolation of synaptic vesicles from mammalian brain. Nat Protoc 2013; 8: 998–1009. [DOI] [PubMed] [Google Scholar]
- 29.Wu F, Wu J, Nicholson AD, et al. Tissue-type plasminogen activator regulates the neuronal uptake of glucose in the ischemic brain. J Neurosci 2012; 32: 9848–9858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tong FC, Zhang X, Kempf DJ, et al. An enhanced model of Middle cerebral artery occlusion in nonhuman primates using an endovascular trapping technique. AJNR Am J Neuroradiol 2015. ; 36: 2354–2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Paxinos G, Franklin KBJ.The mouse brain in stereotaxic coordinates. San Diego, CA: Academic Press Inc., 2001, pp.1–93. [Google Scholar]
- 32.Aban IB, George B.Statistical considerations for preclinical studies. Exp Neurol 2015; 270: 82–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Merino P, Diaz A, Manrique LG, et al. Urokinase-type plasminogen activator (uPA) promotes ezrin-mediated reorganization of the synaptic cytoskeleton in the ischemic brain. J Biol Chem 2018; 293: 9234–9247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kanemaru K, Kubota J, Sekiya H, et al. Calcium-dependent N-cadherin up-regulation mediates reactive astrogliosis and neuroprotection after brain injury. Proc Natl Acad Sci U S A 2013; 110: 11612–11617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Macfarlane RG, Pilling J.Fibrinolytic activity of normal urine. Nature 1947; 159: 779. [DOI] [PubMed] [Google Scholar]
- 36.Sumi Y, Dent MA, Owen DE, et al. The expression of tissue and urokinase-type plasminogen activators in neural development suggests different modes of proteolytic involvement in neuronal growth. Development 1992; 116: 625–637. [DOI] [PubMed] [Google Scholar]
- 37.Cullen DK, Gilroy ME, Irons HR, et al. Synapse-to-neuron ratio is inversely related to neuronal density in mature neuronal cultures. Brain Res 2010; 1359: 44–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sisson TH, Nguyen MH, Yu B, et al. Urokinase-type plasminogen activator increases hepatocyte growth factor activity required for skeletal muscle regeneration. Blood 2009; 114: 5052–5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schuliga M, Jaffar J, Harris T, et al. The fibrogenic actions of lung fibroblast-derived urokinase: a potential drug target in IPF. Sci Rep 2017; 7: 41770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carmeliet P, Moons L, Herbert JM, et al. Urokinase but not tissue plasminogen activator mediates arterial neointima formation in mice. Circ Res 1997; 81: 829–839. [DOI] [PubMed] [Google Scholar]
- 41.Solberg H, Ploug M, Hoyer-Hansen G, et al. The murine receptor for urokinase-type plasminogen activator is primarily expressed in tissues actively undergoing remodeling. J Histochem Cytochem 2001; 49: 237–246. [DOI] [PubMed] [Google Scholar]
- 42.Romer J, Lund LR, Eriksen J, et al. The receptor for urokinase-type plasminogen activator is expressed by keratinocytes at the leading edge during re-epithelialization of mouse skin wounds. J Invest Dermatol 1994; 102: 519–522. [DOI] [PubMed] [Google Scholar]
- 43.Quiquempoix M, Fayad SL, Boutourlinsky K, et al. Layer 2/3 pyramidal neurons control the gain of cortical output. Cell Rep 2018; 24: 2799–. [DOI] [PubMed] [Google Scholar]
- 44.Zhang S, Boyd J, Delaney K, et al. Rapid reversible changes in dendritic spine structure in vivo gated by the degree of ischemia. J Neurosci 2005; 25: 5333–5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Merino P, Diaz A, Torre ER, et al. Urokinase-type plasminogen activator (uPA) regulates the expression and function of the growth- associated protein-43 (GAP-43) in the synapse. J Biol Chem 2020; 295: 619–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, sj-pdf-1-jcb-10.1177_0271678X211002297 for Urokinase-type plasminogen activator promotes N-cadherin-mediated synaptic recovery in the ischemic brain by Ariel Diaz, Paola Merino, Patrick McCann, Manuel A Yepes, Laura G Quiceno, Enrique Torre, Amelia Tomkins, Xiaodong Zhang, Chadwick M Hales, Frank C Tong and Manuel Yepes in Journal of Cerebral Blood Flow & Metabolism