

Abstract
Background
Human T-cell Leukemia Virus type-1 (HTLV-1) is a retrovirus that causes two diseases including Adult T-cell Leukemia/Lymphoma (ATLL cancer) and HTLV-1 Associated Myelopathy/Tropical Spastic Paraparesis (HAM/TSP, a neurodegenerative disease) after a long latency period as an asymptomatic carrier (AC). There are no obvious explanations about how each of the mentioned diseases develops in the AC carriers. Finding the discriminative molecular factors and pathways may clarify the destiny of the infection.
Methods
To shed light on the involved molecular players and activated pathways in each state, differentially co-expressed modules (DiffCoEx) algorithm was employed to identify the highly correlated genes which were co-expressed differently between normal and ACs, ACs and ATLL, as well as ACs and HAM/TSP samples. Through differential pathway analysis, the dysregulated pathways and the specific disease-genes-pathways were figured out. Moreover, the common genes between the member of DiffCoEx and differentially expressed genes were found and the specific genes in ATLL and HAM/TSP were introduced as possible biomarkers.
Results
The dysregulated genes in the ATLL were mostly enriched in immune and cancer-related pathways while the ones in the HAM/TSP were enriched in immune, inflammation, and neurological pathways. The differential pathway analysis clarified the differences between the gene players in the common activated pathways. Eventually, the final analysis revealed the involvement of specific dysregulated genes including KIRREL2, RAB36, and KANK1 in HAM/TSP as well as LTB4R2, HCN4, FZD9, GRIK5, CREB3L4, TACR2, FRMD1, LHB, FGF3, TEAD3, GRIN2D, GNRH2, PRLH, GPR156, and CRHR2 in ATLL.
Conclusion
The identified potential prognostic biomarkers and therapeutic targets are proposed as the most important platers in developing ATLL or HAM/TSP. Moreover, the proposed signaling network clarifies the differences between the functional players in the activated pathways in ACs, ATLL, and HAM/TSP.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12985-021-01643-8.
Keywords: HTLV-1, ACs, ATLL, HAM/TSP, Differentially co-expressed modules, Pathogenesis
Introduction
Human T-cell Leukemia Virus type-1 (HTLV-1) belongs to the family Retroviridae and subfamily Orthoretrovirinae [1]. The HTLV-1 infection may cause developing two major diseases including Adult T-cell Leukemia/Lymphoma (ATLL cancer) and Tropical Spastic Paraparesis/HTLV-1 Associated Myelopathy (TSP/HAM) after elapsing an asymptomatic carrier (AC) state. Although HTLV-1 is not a widespread virus all over the world, it is an endemic pathogen in sub-Saharan Africa, East north of Iran, the Caribbean region, Japan, and South America [2]. Approximately, 90% of the infected HTLV-1 are asymptomatic carriers (ACs) [3] with the capability of the silent transmission of the virus through blood contact, sexual intercourse, breastfeeding, etc.[4].
There are three main receptors on the cells for HTLV-1 including neuropilin, glucose transporter 1 (GLUT-1), and heperan sulfate proteoglycan [5]. The HTLV-1 proviral DNA is found in the immune cell types comprising dendritic cells, monocytes, CD8+ T-cells, B cells, and in higher extent in CD4+ T-cells [6, 7]. Two oncogenic proteins named as the HTLV-1 basic leucine zipper protein (HBZ) and the transactivator protein (Tax) are frequently expressed by the HTLV-1 genome. However, the tax gene expresses in only around 40% of ATLL patients [8]. Tax has a critical function in the viral pathogenesis of HAM/TSP patients through promoting the proliferation of infected cells by activating NFκB and AP-1 pathways, avoiding apoptosis, and activating cytotoxic T lymphocyte (CTL) response [3, 9]. On the other hand, HBZ prohibits NF-κB pathway, promotes tumor progression, and boosts T cell proliferation and lymphoma [10, 11].
One of the beneficial approaches to find the patterns of co-regulated genes is gene co-expression analysis [12]. Two common approaches are usually utilized to determine the mechanistic diversity between two conditions including identifying differential gene expression and differential gene co-expression. Through co-expression analysis, the highly correlated genes (a module) can be identified. Moreover, the differential co-expressed analysis determines the specific co-regulated genes in each condition. DiffCoEx is a sensitive and efficient method to find the gene co-expression differences which are grouped in various modules between multiple conditions [13]. The major advantage of this method is determining significant differential co-expressed gene groups even in the attendance of the within-group correlation across two conditions.
Since the pathogenesis mechanism of HTLV-1 as a virus-caused cancer and virus-caused neurologic disease have not been yet fully ascertained, the identification of differential co-regulated genes can determine new functional players in ACs, ATLL, and HAM/TSP. To this end, we integrated various gene expression datasets and then found the differential co-expressed genes between every two conditions employing DiffCoEx approach. The outcomes specified the gene players and activated pathways implicated in the development of each disease after infection.
Methods
Data collection, merging, and preprocessing
To find the relevant datasets correspond to our study purpose, Gene Expression Omnibus (GEO) repository database was searched. A total of four microarray datasets including GSE29312, GSE29332, GSE33615, and GSE55851 were selected to be involved in our analysis. In total, these datasets contain 58 normal samples, 43 ACs samples, 62 ATLL samples, and 20 HAM/TSP samples. Since datasets belong to different platforms, removeBatchEffect function in the R limma package was utilized to remove the batch effect across different datasets [14]. The expression data of samples related to each condition were merged, individually. A total of 15,565 common genes were finally used for further analysis. The merged data were quantile-normalized and log2-transformed. Moreover, to validate the identified modules, the ACs and HAM/TSP samples from GSE38537 dataset and ATLL samples from GSE43017 dataset were used. Table 1 contains more details related to each dataset. Moreover, Fig. 1 indicates the workflow of the utilized procedure in this study, which is explained in the following sections.
Table 1.
Characteristics of datasets involved in the analysis and validation
| Dataset | Platform | Number of Samples |
|---|---|---|
| Datasets for main analyses | ||
| GSE29312 |
Illumina HumanHT-12 V3.0 expression beadchip (GPL6947) |
Normal: 9 ACs: 20 HAM/TSP: 10 |
| GSE29332 |
Illumina HumanWG-6 v3.0 expression beadchip (GPL6884) |
Normal: 8 ACs: 17 HAM/TSP: 10 |
| GSE55851 |
Agilent-026652 Whole Human Genome Microarray 4 × 44 K v2 (GPL10332) |
Normal: 3 ACs: 6 ATLL: 12 |
| GSE33615 |
Agilent-014850 Whole Human Genome Microarray 4 × 44 K G4112F (GPL4133) |
Normal: 21 ATLL: 52 |
| Datasets for validation | ||
| GSE38537 |
Agilent-014850 Whole Human Genome Microarray 4 × 44 K G4112F (GPL6480) |
ACs: 4 HAM/TSP: 4 |
| GSE43017 |
Affymetrix Human Genome U133 Plus 2.0 Array (GPL570) |
ATLL: 7 |
Fig. 1.

Workflow of the proposed method
Determination of differential co-expressed modules
To find the differential co-expressed modules among two conditions, the DiffCoEx algorithm was employed [13]. DiffCoEx principally uses Weighted Gene Co-expression Analysis (WGCNA) [12] and contains five steps as follows: (1) An adjacency matrix is constructed for each condition in which their elements are the calculated correlations between each gene pairs. We used Pearson coefficient to measure correlations. (2) A matrix of adjacency difference (dij) is determined by calculating the signed squared correlation coefficients. The higher dij values are considered as significant co-expression changes between two genes. The soft threshold parameter β is determined so that the network follows a scale-free topology [15]. Then, dij values reach the power of β. (3) The topological overlap dissimilarity matrix (dissTOM) is constructed to detect the shared genes with common neighbors in the graph. It is constituted based on the adjacency matrix and contains the differential correlation network. (4) The hierarchical clustering is performed with flashClust [16]. Then, dynamicTreeCut function is used to extract gene groups (modules) from the obtained dendrogram and mergeCloseModules function is employed to merge close modules. Each module is specified with a color. (5) The statistical significance of differential co-expression is evaluated using the dispersion statistic to quantify the correlation alteration between two conditions. In this study, we identified differential co-expressed modules between normal and ACs, ACs and ATLL, ACs and HAM/TSP. Moreover, module-to-module co-expression changes were examined by evaluating the significance of the correlation alterations among the genes in each module pair. For this purpose, a similar “module-to-module” dispersion measure was determined and null distributions were produced from the same permutation approach.
Pathway enrichment analysis
The most connected genes in each module were enriched in the KEGG utilizing g:Profiler webtools (version: 1185_e69_eg16) [17] and Enrichr. The common gene expressions between ACs, ATLL, and HAM/TSP groups were considered as the background. The relevant terms with Benjamini–Hochberg FDR < 0.05 were employed as statistically significant.
Results
Identification of differential co-expressed modules
The DiffCoEx algorithm was employed to find the differential co-expressed gene modules between different sample groups. Through this algorithm, the co-regulated gene clusters that differentially co-expressed between two groups were determined. After computing Pearson correlation and then adjacency differences matrices according to Methods, the topological overlap matrices (TOMs) were constructed with β power of 2, 3, and 2 for Normal-ACs, ACs-ATLL, and ACs-HAM/TSP, respectively. These values are the lowest power for which the scale-free topology fit index reaches 0.80. Afterward, the differential co-expressed modules were identified in each comparative group utilizing hierarchical clustering and then merging adjacent clusters (Fig. 2a–c). Each module is specified with a distinctive color. The differences between the correlation pattern in each module are demonstrated in Fig. 3. A total of 8 differentially co-expressed modules were identified between Normal and ACs (DiffCoEx_NA) (Fig. 3a). Among them, the modules including brown, red, green, and blue are highly correlated in ACs samples and only pink module is highly correlated in Normal samples. Three of the five identified DiffCoEx modules between ACs and ATLL groups (DiffCoEx_AA) including orange, pink, and purple modules are mostly correlated in ATLL and the magenta module pursues the opposite pattern (Fig. 3b). All eight significant differential modules identified between ACs and HAM/TSP (DiffCoEx_AH) are highly correlated in HAM/TSP (Fig. 3c). The list of the identified genes in each module is mentioned in Additional file 1. Moreover, the module to module co-expression alterations related to each comparison group was determined by employing permutations. This matrix reveals the correlations between the differentially co-expressed modules. To this end, 1000 sample permutations across the two conditions were carried out. Afterward, the correlation alteration for each gene group (dispersion value) was calculated for each module pair. Figure 4 shows the module-to-module co-expression alterations across DiffCoEx_NA, DiffCoEx_AA, and DiffCoEx_AH, in which the within-module dispersion value for each module with permuted data than with original data is specified with p-value (the numbers in each cell divided to 1000) [13]. Figure 4a discloses that although there is no obvious differential correlation in black, magneta, and purple modules between ACs and normal groups, they were determined as a differentially co-expressed module due to their significant correlation with the genes in other differentially co-expressed modules (correlation of black and magenta with brown and red modules as well as correlation of purple with blue and green modules). Likewise, the black module in DiffCoEx_AA has a remarkable correlation with the genes in magenta (Fig. 4b) as well as pink and blue modules in DiffCoEx_AH with the green module (Fig. 4c). In order to find the most connected genes at the protein levels, modules were submitted to STRING and the modules with unconnected proteins were excluded. Therefore, modules brown, red, and blue in ACs, pink and purple in ATLL, and blue, turquoise, and pink in HAM/TSP were selected (Additional file 1).
Fig. 2.

Dendrogram of genes clustered based on (1-TOM) with assigned module colors of a ACs, b ATLL, and c HAM/TSP. The colored rows show the module membership acquired after merging modules by the dynamic tree cut method
Fig. 3.

Comparative correlation heatmap containing differentially co-expressed modules between a normal and ACs, b ACs and ATLL, and c ACs and HAM/TSP. The upper and lower diagonals of the main matrix represent a correlation between gene pairs among each studied group. Modules are specified in the heatmap by black squares
Fig. 4.

Module-to-module co-expression changes using permutations for a DiffCoEx_NA, b DiffCoEx_AA, and c DiffCoEx_AH. This figure is the result of the significance analyses. After 1000 permutations of the samples between every two conditions, the dispersion values were computed for each module and every possible module pair. The number in each cell shows the p-value (number/1000) identified for the within-module co-expression change. Black cells show significant p-value and light grey modules represent insignificant p value
Validation of modules in the external dataset
In order to evaluate the reliability of the identified differential co-expressed modules, their preservations in an independent dataset were surveyed. For this purpose, the function of “modulePreservation” was used to measure Zsummary and medianRank through a permutation test (200 times) [18]. Zsummary score is half the sum of the mean of Zdensity and Zconnectivity. MedianRank is a rank-based scale that depends on an observed preservation statistic and is not dependent on module size [19]. A lower medianRank score reveals the high preservation of the module. The module preservation analysis was employed to validate the ACs and HAM/TSP modules in GSE38537 and ATLL modules in GSE43017. Modules with a Zsummary > 4 and medianRank < 8 were regarded as moderate-high preserved modules [20, 21]. The results are mentioned in Additional file 2. Therefore, module brown from ACs and module purple from ATLL with Zsummary < 4 were excluded.
Pathway enrichment analysis
The further pathway enrichment analysis clarified that modules related to ACs were generally entered in pathways related to viral infection, Immune system, cancer, and inflammation like Human T-cell leukemia virus 1 infection, Toll-like receptor signaling pathway, Pathways in cancer, PI3K-Akt signaling pathway, Chemokine signaling pathway, Cell adhesion molecules (CAMs), NOD-like receptor signaling pathway, Th17 cell differentiation, Th1 and Th2 cell differentiation, JAK-STAT signaling pathway, and MAPK signaling pathway (Additional file 3, Sheet 1).
The identified differential modules in ATLL were mostly enriched in cancer-, viral- and immune-related pathways such as PI3K-Akt signaling pathway, Chemokine signaling pathway, Antigen processing and presentation, Viral carcinogenesis, Pathways in cancer, cAMP signaling pathway, Proteoglycans in cancer, Hippo signaling pathway, Th1 and Th2 cell differentiation, Th17 cell differentiation, ErbB signaling pathway, and VEGF signaling pathway (Additional file 3, Sheet 2). Moreover, the selected proteins in DiffCoEx_AH were enriched in viral infection, immune system, inflammation, and neurological pathways like Human T-cell leukemia virus 1 infection, PI3K-Akt signaling pathway, Th17 cell differentiation, Th1 and Th2 cell differentiation, MAPK signaling pathway, Apoptosis,, NF-kappa B signaling pathway, NOD-like receptor signaling pathway, Sphingolipid signaling pathway, JAK-STAT signaling pathway, Chemokine signaling pathway, Parkinson disease, and Neurotrophin signaling pathway (Additional file 3, Sheet 3). The results show that the immune and viral-related pathways are activated in ATLL and HAM/TSP, however, cancer pathways are mostly activated in ATLL and neurological pathways in HAM/TSP.
Specific genes-pathways
Although the identified modules were almost enriched in similar pathways, different genes were involved in the dysregulation of these pathways in each disease. To elucidate the specific genes, which may significantly dysregulate a biological pathway in each disease state, the unique pathways-genes were explored. Venn diagram depicted in Fig. 5 demonstrates the number of common and specific pathways-genes in each DiffCoEx group. The list of specific pathways-genes is also mentioned in Additional file 4. As the figure indicates, some pathways are activated after virus infection and also in two diseases. On the other hand, pathways including Proteoglycans in cancer and Rap1 signaling pathway are mostly dysregulated in ATLL as well as Sphingolipid signaling pathway, DNA replication, Parkinson disease, and Neurotrophin signaling pathway in HAM/TSP. These pathways considering their specific dysregulated genes can be furthermore studied to design proper treatment for ATLL or HAM/TSP.
Fig. 5.
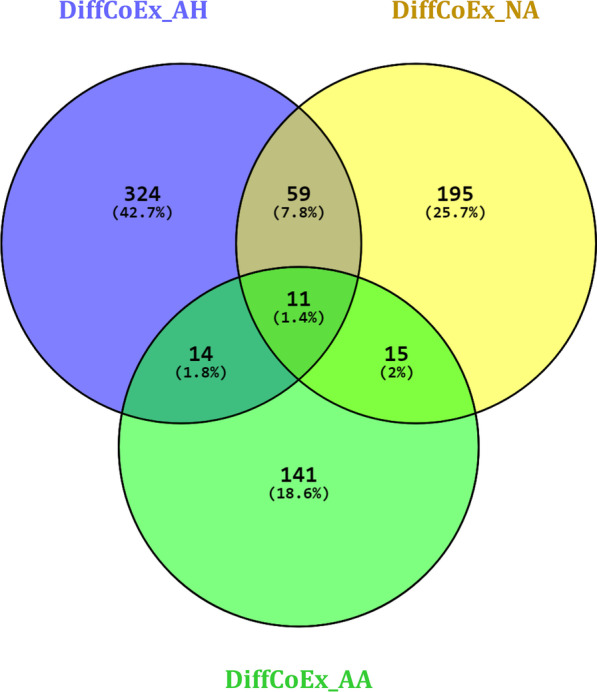
Venn diagram representing the number of common and specific pathways-genes in each DiffCoEx group
Finding potential biomarkers
In order to find potential biomarkers for each disease, the common genes between identified DiffCoEx and differentially expressed genes (DEGs) were found. To this end, DEGs between ACs and ATLL (DEGs_AA) as well as ACs and HAM/TSP (DEGs_HA) considering adj.p.val < 0.05 and |log FC|> 1 were found. Therefore, the shared genes between module pink in ATLL and DEGs_AA, as well as common genes between blue, pink, and turquoise modules in HAM/TSP and DEGs_HA, were identified. Eventually, the specific genes that were uncommon between ATLL and HAM/TSP were determined. As a result, KIRREL2, RAB36, and KANK1 were identified as specific genes and possible candidate biomarkers for HAM/TSP. Moreover, 77 specific genes were found for ATLL. To find the most important gene players in the progression of ATLL, pathway enrichment analysis was performed and the pathways enriched by at least 3 proteins were identified. Four pathways including Neuroactive ligand-receptor interaction, cAMP signaling pathway, Calcium signaling pathway, and Hippo signaling pathway were enriched by 15 genes including LTB4R2, HCN4, FZD9, GRIK5, CREB3L4, TACR2, FRMD1, LHB, FGF3, TEAD3, GRIN2D, GNRH2, PRLH, GPR156, and CRHR2. These genes may also be proposed as potential biomarkers for ATLL (Additional file 5).
Discussion
The functional cellular pathways implicated in the HTLV-1 infected ACs and two main HTLV-1 related diseases (including ATLL and HAM/TSP) have not been completely figured out. Herein, we tried to shed light partially on the differences between the involved genes and also activated pathways between these diseases and AC state through a differential co-expression analysis. To this end, we applied DiffCoEx algorithm to find differential co-expressed genes between normal and ACs, ACs and ATLL, ACs and HAM/TSP. The most important activated pathways in each condition are depicted as a schematic signaling network in Fig. 6.
Fig. 6.

The proposed signaling networks implicated AC state and pathogenesis of ATLL and HAM/TSP diseases
While the majority of the infected subjects by HTLV-1 remain as asymptomatic carriers, the most challenging question is the reason for progression from an asymptomatic viral infection toward ATLL or/and HAM/TSP. The identified differential co-regulated modules in ACs versus normal group revealed the activation of pathways related to the HTLV-1 infection which are expected.
HTLV-1 transfers between the infected and uninfected T cells through several mechanisms. One of the main mechanism is the formation of virological synapse (VS) [22]. This type of cell to cell transfer has notable advantages for viruses including more efficient transfer, elevating transfer speed, and confining the exposure to host immune defense mechanisms [23]. One of the major prerequisite steps to activate this transmission mechanism is the involvement of LFA-1 protein encoded by ITGLA. LFA-1 naturally binds to ICAM-1 that leads to changing both infected and uninfected cells. This provides the proper VS condition to transfer the virus. Tax and p12 implicate in the activation of LFA-1 and ICAM-1 expression [22, 24].
NF-kappa signaling is an important pathway in cell proliferation. It maintains cellular balance as well as Th1 and Th2 cell differentiation pathway through the expression alteration of the involved genes such as IKBKG and PLCG1. HTLV-1 can activate both Th1 and Th2 through interference in the NF-kappa signaling pathways based on the infection step. The downregulation of LCK was also observed in ACs which may be due to the expression of the viral regulatory tax gene [25]. Moreover, the obvious dysregulation of chemokine and cytokine-cytokine interaction pathways through down-regulation of CCR7 and CCR2 are also observed in this study. Our results showed that the virus in ACs keeps a balance in the successful infection by avoiding immune response. After latent infection of the virus, ATLL or HAM/TSP can be developed. Perhaps developing inflammation is the substantial difference between HAM-TSP and ACs.
HAM/TSP is known as an inflammatory disease in which several immune cells together have critical roles in the disease progression. The pathogenesis of HAM-TSP is explained in three levels: peripheral, transmit to the central nervous system, and damage to the nervous tissues. It appears that multiple genes and pathways are activated at each level which has been shown in this work.
Sphingolipid is a regulator of the cell signals through two secondary lipid messengers including ceramide (Cer) and sphingosine-1-phosphate (S1P). Cer stimulates apoptosis in the oligodendrocytes and neurons, and S1P speeds up the exit of lymphocytes from lymphoid tissues to blood [26]. Sphingolipid signaling pathway is affected in HAM/TSP that reveals the importance of theses metabolites and genes of MAPK8, PPP2R5E, GNAI3, S1PR1, MAPK1, RAF1, BID, NSMAF, TNFRSF1A, MAPK3 in the progression of disease.
Cer activates PP2A (PPP2R5E belongs to this protein family) and BID to stimulate apoptosis [27]. It also targets the kinase suppressor Ras which aids to regulate TNFα-mediated (may by the function of TNFRSF1A) actuation of MAPK3 and MAPK1 [28, 29], activation of RAF-1, and MAPK pathway [30]. Moreover, the overexpression of S1PR1 and interaction with GNAI3 helps to activation of MAPK pathway and promotes migration and cell survival [31]. Also, NSMAF is necessary for TNF-mediated activation of neutral sphingomyelinase and probably has a function in regulating responses of TNF-induced cellular such as inflammation [32].
DNA replication is another pathway that should be considered in the development of HAM/TSP. DNA replication is increased in the Tax-expressing cells causing progression of cell cycle, even in the presence of DNA damaging agents. Therefore, DNA damage can result in the accumulation of mutations in the Tax-expressing cell and also apoptosis [33]. In our previous studies, we showed the importance of apoptosis in the HAM/TSP pathogenesis [34, 35] which also confirms in this study.
Neurotrophin signaling pathway is mostly activated in HAM/TSP. This pathway is associated with the level of neurotrophin and downstream signaling cascades. The inhibitor factors in neurotrophin signaling pathway are strongly regulated by degradation and dephosphorylation. Neurotrophin signaling pathway implicates in several neurodegenerative disorders, such as Huntington's disease, Alzheimer's disease, and psychiatric disorders like depression and substance abuse [36, 37]. In this study, the function of co-regulated of genes including RPS6KA3, MAP3K3, PSEN1, SOS2, MAP2K5, RELA, ATF4 in the activation of Neurotrophin signaling pathway in HAM/TSP were clarified. TrkA is one of the main receptors that interact with neurotrophins. It leads to the activation of intracellular signaling cascades, actuation of Ras via GRB2 and SOS2, and further promotion of the ERK/MAPK pathway with the functional activity of MAP3K3 and MAP2K5. Moreover, RPS6KA3 phosphorylates ATF4 and has critical roles in cell survival, cellular differentiation, and axonal growth [38].
Three genes comprising KIRREL2, RAB36, and KANK1 were found as biomarkers for HAM/TSP. KIRREL2 (NEPH3) belongs to the immunoglobulin superfamily of cell adhesion molecules. NEPH3 is regulated by the cooperation of WT1 and NF-κB in podocytes. NEPH3 is a downstream target of Ptf1a in the progressing central nervous system. It is also expressed in early postmitotic neurons of the developing spinal cord. NEPH3 intercedes the adhesion between the Ptf1a-progenitors, which is essential for maturation, neuronal migration, and differentiation [39]. The dysregulation of NEPH3 may have a role in the progression of HAM/TSP and should be furthermore studied.
RAB36 is a member of the RAB family which is located on chromosome 22q11.2. It was reported that the depletion of RAB36 leads to G1 cell cycle arrest, disruption of mesenchymal-epithelial transition, and simplification of the malignant rhabdoid tumors dissemination [40, 41]. The information about the role of RAB36 in the development of diseases is limited, so further investigation is indispensable.
KANK1 belongs to the Kank family which is mainly involved in the cytoskeleton formation by regulating actin polymerization. The mutated KANK1 may develop the central nervous system disorder (cerebral palsy spastic quadriplegic type 2). It also inhibits the cell migration and formation of actin fiber. It can inhibit malignant peripheral nerve sheath tumors through the regulationof CXC5 as an apoptosis-related gene [42, 43].
ATLL is another disease caused by HTLV-1 infection. The overall identified DiffCoEx_AA and the activated pathways reveal that the viral infection provides a primary condition in the infected cells to malignancy in association with genetic susceptibility and other risk factors [44]. The differential co-expressed genes between ATLL and ACs implicates in cancer- and malignancy-related pathways. The final goals of these pathways are sustained angiogenesis, evading apoptosis, tissue invasion, block differentiation, proliferation, and genomic instability. Form this study, the dysregulation of pathways including Rap1 signaling pathway and Proteoglycans in cancer were identified in ATLL. Rap1 signaling pathway is activated with the involvement of several proteins. Rap1 upregulates ARAP3 which is a PI3K effector. ARAP3 selectively uses Rho as its substrate, and then Rho targets ACTB resulting in cell adhesion and migration. On the other hand, the expression of WNT3A has been reported along with the WNT signaling pathway components FZD9 and CTNNB1 [45]. Moreover, IQGAP1 is a significant scaffold in the EGF-stimulated MAPK cascade due to its direct interaction with MAPK1 and EGFR [46]. These events ultimately result in proliferation, cell migration and invasion, and also cytoskeleton activation as parts of the pathway of Proteoglycans in cancer.
Fifteen genes including LTB4R2, HCN4, FZD9, GRIK5, CREB3L4, TACR2, FRMD1, LHB, FGF3, TEAD3, GRIN2D, GNRH2, PRLH, GPR156, and CRHR2 were also determined as potential biomarkers for ATLL.
The importance of cAMP in developing hematological cancer has been reported previously [47]. The cAMP levels are regulated by the balance between the functions of two enzymes: adenylyl cyclase (AC) and cyclic nucleotide phosphodiesterase (PDE) [48]. cAMP binds to HCN4 and also mediates phosphorylation of proteins by PKA. PKA may increase the NMDAR currents (GRIN2D is a subunit of NMDA receptors). The overexpression of GRIN2D may cause an excessive influx of Ca2+ and as a result the regulation of CALM1 in the calcium signaling pathway [49]. On the other hand, regulation of transcription by PKA is obtained by direct phosphorylation of the CREB which finally may lead to cell survival/death [50].
TEAD3 is a transcription factor and FRMD1 is an activating transcription factor binding which have important functions in the Hippo signaling pathway, a pathway involved in tumor suppression by limiting proliferation and boosting apoptosis.
FGF3 belongs to the basic fibroblast growth factor (FGF) gene family which plays a major role in cell differentiation and proliferation and is involved in the calcium signaling pathway [51]. Furthermore, the contribution of GNRH2, PRLH, GPR156, GRIK5, LHB, and CRHR2 to the neuroactive ligand-receptor interaction pathway are specified in ATLL in consistent with some other cancers [52, 53].
LTB4R2 is one of the LTB4 receptors that is associated with invasion, survival, and metastasis. TACR2 is also a receptor for tachykinins that is mainly expressed in the periphery including inflammatory cells. Overexpression of TACR2 can develop the proliferation and migration of cancer cells by regulating the Wnt signaling pathway [54]. It seems that further in vivo and in vitro studies on the function and the involved proteins in this pathway may introduce a novel therapeutic way.
Our results reveal that more dysregulated genes and pathways are involved in the HAM/TSP pathogenesis in comparison to ATLL, which probably explains more prevalence of ATLL in the HTLV-1 infected patients [55]. Interestingly, some features which are implicated the HTLV-1 infection pathway and activated by differential co-expressed genes between HAM/TSP and ACs, do not observe in ATLL. Given these results, it seems that the host features and genetic susceptibility are the main players in ATLL. Moreover, the specific identified genes for each condition have of importance in the future proposed treatment for each disease.
Conclusion
The pathogenesis mechanism and development of diseases caused by HTLV-1 infection is obscure. Herein, through the identification of differential co-expressed module analysis and pathway enrichment, the major genes implicated the pathogenesis process of ACs toward ATLL and HAM/TSP were clarified. It seems that different dysregulated genes cause the activation of similar pathways in ATLL and HAM/TSP. They may be proposed as potential therapeutic targets. However, further studies should be designed and performed to evaluate the identified genes in each condition.
Supplementary Information
Additional file 1. List of the identified differential co-expressed modules in ACs, ATLL, and HAM/TSP.
Additional file 2. The preservation of modules in the external datasets (ACs, ATLL, and HAM/TSP).
Additional file 3. The enriched pathways by the connected proteins in each identified DiffCoEx_NA, DiffCoEx_AA, and DiffCoEx_AH.
Additional file 4. The list of specific pathways-genes in ACs, ATLL, and HAM/TSP.
Additional file 5. The list of DEGs, common between DEGs and DiffCoEx_AA, and specific genes for ATLL.
Acknowledgements
This research was supported by the Iran National Science Foundation (INSF) [Grant number 96012471]. The author would like to thank the University of Isfahan for supporting this research through Postdoctoral Fellowship Program.
Abbreviations
- HTLV-1
Human T-cell Leukemia Virus type-1
- ATLL
Adult T-cell Leukemia/Lymphoma
- HAM/TSP
HTLV-1 Associated Myelopathy/Tropical Spastic Paraparesis
- ACs
Asymptomatic carriers
- DiffCoEx
Differentially co-expressed modules
- TOMs
Topological overlap matrices
- DEGs
Differentially expressed genes
Authors' contributions
MZG performed bioinformatics and statistical analysis. MZG and MT-R interpreted the results and wrote the manuscript. S-HM revised the manuscript. RE supervised the study. All authors read and approved the final manuscript.
Availability of data and materials
All data generated or analyzed during this study are included in this published article [and its supplementary information files].
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors have no conflict of authors.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Quaresma JA, Yoshikawa GT, Koyama RV, Dias GA, Fujihara S, Fuzii HT. HTLV-1, immune response and autoimmunity. Viruses. 2016;8:5. doi: 10.3390/v8010005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Futsch N, Mahieux R, Dutartre H. HTLV-1, the other pathogenic yet neglected human retrovirus: from transmission to therapeutic treatment. Viruses. 2018;10:1. doi: 10.3390/v10010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gonçalves DU, Proietti FA, Ribas JGR, Araújo MG, Pinheiro SR, Guedes AC, Carneiro-Proietti ABF. Epidemiology, treatment, and prevention of human T-cell leukemia virus type 1-associated diseases. Clin Microbiol Rev. 2010;23:577–589. doi: 10.1128/CMR.00063-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mulherkar R, Karabudak A, Ginwala R, Huang X, Rowan A, Philip R, Murphy EL, Clements D, Ndhlovu LC, Khan ZK, Jain P. In vivo and in vitro immunogenicity of novel MHC class I presented epitopes to confer protective immunity against chronic HTLV-1 infection. Vaccine. 2018;36:5046–5057. doi: 10.1016/j.vaccine.2018.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matsuoka M, Mesnard J-M. HTLV-1 bZIP factor: the key viral gene for pathogenesis. Retrovirology. 2020;17:1–8. doi: 10.1186/s12977-020-0511-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koyanagi Y, Itoyama Y, Nakamura N, Takamatsu K, Kira J-I, Iwamasa T, Goto I, Yamamoto N. In vivo infection of human T-cell leukemia virus type I in non-T cells. Virology. 1993;196:25–33. doi: 10.1006/viro.1993.1451. [DOI] [PubMed] [Google Scholar]
- 7.Macatonia SE, Cruickshank J, Rudge P, Knight SC. Dendritic cells from patients with tropical spastic paraparesis are infected with HTLV-1 and stimulate autologous lymphocyte proliferation. AIDS Res Hum Retrovir. 1992;8:1699–1706. doi: 10.1089/aid.1992.8.1699. [DOI] [PubMed] [Google Scholar]
- 8.Takeda S, Maeda M, Morikawa S, Taniguchi Y, Ji Y, Nosaka K, Tanaka Y, Matsuoka M. Genetic and epigenetic inactivation of tax gene in adult T-cell leukemia cells. Int J Cancer. 2004;109:559–567. doi: 10.1002/ijc.20007. [DOI] [PubMed] [Google Scholar]
- 9.Jaworski E, Narayanan A, Van Duyne R, Shabbeer-Meyering S, Iordanskiy S, Saifuddin M, Das R, Afonso PV, Sampey GC, Chung M. Human T-lymphotropic virus type 1-infected cells secrete exosomes that contain Tax protein. J Biol Chem. 2014;289:22284–22305. doi: 10.1074/jbc.M114.549659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Satou Y, Yasunaga J-i, Zhao T, Yoshida M, Miyazato P, Takai K, Shimizu K, Ohshima K, Green PL, Ohkura N. HTLV-1 bZIP factor induces T-cell lymphoma and systemic inflammation in vivo. PLoS Pathog. 2011;7:e1001274. doi: 10.1371/journal.ppat.1001274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kataoka K, Nagata Y, Kitanaka A, Shiraishi Y, Shimamura T, Yasunaga J-i, Totoki Y, Chiba K, Sato-Otsubo A, Nagae G. Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat Genet. 2015;47:1304. doi: 10.1038/ng.3415. [DOI] [PubMed] [Google Scholar]
- 12.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tesson BM, Breitling R, Jansen RC. DiffCoEx: a simple and sensitive method to find differentially coexpressed gene modules. BMC Bioinform. 2010;11:497. doi: 10.1186/1471-2105-11-497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47–e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. 2005 doi: 10.2202/1544-6115.1128. [DOI] [PubMed] [Google Scholar]
- 16.Langfelder P, Horvath S. Fast R functions for robust correlations and hierarchical clustering. J Stat Softw. 2012 doi: 10.18637/jss.v046.i11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Raudvere U, Kolberg L, Kuzmin I, Arak T, Adler P, Peterson H, Vilo J. g: Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update) Nucleic Acids Res. 2019;47:W191–W198. doi: 10.1093/nar/gkz369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ghobadi MZ, Mozhgani S-H, Erfani Y. Identification of dysregulated pathways underlying HTLV-1-associated myelopathy/tropical spastic paraparesis through co-expression network analysis. J NeuroVirol. 2021;27:1–11. doi: 10.1007/s13365-020-00930-4. [DOI] [PubMed] [Google Scholar]
- 19.Ao ZX, Chen YC, Lu JM, Shen J, Peng LP, Lin X, Peng C, Zeng CP, Wang XF. Zhou RJOl: Identification of potential functional genes in papillary thyroid cancer by co-expression network analysis. Oncol Lett. 2018;16:4871–4878. doi: 10.3892/ol.2018.9306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bakhtiarizadeh MR, Mirzaei S, Norouzi M, Sheybani N, Vafaei Sadi MS. Identification of gene modules and hub genes involved in mastitis development using a systems biology approach. Front Genet. 2020;11:722. doi: 10.3389/fgene.2020.00722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen C, Meng Q, Xia Y, Ding C, Wang L, Dai R, Cheng L, Gunaratne P, Gibbs RA, Min S. POU3F2 is a regulator of a gene coexpression network in brain tissue from patients with neuropsychiatric disorders. Sci Transl Med. 2018;10:8eaat178. doi: 10.1126/scitranslmed.aat8178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nejmeddine M, Bangham CR. The HTLV-1 virological synapse. Viruses. 2010;2:1427–1447. doi: 10.3390/v2071427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mothes W, Sherer NM, Jin J, Zhong P. Virus cell-to-cell transmission. J Virol. 2010;84:8360–8368. doi: 10.1128/JVI.00443-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim S-j, Nair AM, Fernandez S, Mathes L, Lairmore MD. Enhancement of LFA-1-mediated T cell adhesion by human T lymphotropic virus type 1 p12I1. J Immunol. 2006;176:5463–5470. doi: 10.4049/jimmunol.176.9.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fraedrich K, Müller B, Grassmann R. The HTLV-1 Tax protein binding domain of cyclin-dependent kinase 4 (CDK4) includes the regulatory PSTAIRE helix. Retrovirology. 2005;2:54. doi: 10.1186/1742-4690-2-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jana A, Pahan K. Sphingolipids in multiple sclerosis. Neuromol Med. 2010;12:351–361. doi: 10.1007/s12017-010-8128-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marchesini N, Hannun YA. Acid and neutral sphingomyelinases: roles and mechanisms of regulation. Biochem Cell Biol. 2004;82:27–44. doi: 10.1139/o03-091. [DOI] [PubMed] [Google Scholar]
- 28.Chalfant CE, Kishikawa K, Mumby MC, Kamibayashi C, Bielawska A, Hannun YA. Long chain ceramides activate protein phosphatase-1 and protein phosphatase-2A. Activation is stereospecific and regulated by phosphatidic acid. J Biol Chem. 1999;274:20313–20317. doi: 10.1074/jbc.274.29.20313. [DOI] [PubMed] [Google Scholar]
- 29.Zhang Y, Yao B, Delikat S, Bayoumy S, Lin XH, Basu S, McGinley M, Chan-Hui PY, Lichenstein H, Kolesnick R. Kinase suppressor of Ras is ceramide-activated protein kinase. Cell. 1997;89:63–72. doi: 10.1016/S0092-8674(00)80183-X. [DOI] [PubMed] [Google Scholar]
- 30.Yan F, John SK, Polk DB. Kinase suppressor of Ras determines survival of intestinal epithelial cells exposed to tumor necrosis factor. Cancer Res. 2001;61:8668–8675. [PubMed] [Google Scholar]
- 31.Mahajan-Thakur S, Bien-Möller S, Marx S, Schroeder H, Rauch BH. Sphingosine 1-phosphate (S1P) signaling in glioblastoma multiforme—a systematic review. Int J Mol Sci. 2017;18:2448. doi: 10.3390/ijms18112448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Adam-Klages S, Adam D, Wiegmann K, Struve S, Kolanus W, Schneider-Mergener J, Krönke M. FAN, a novel WD-repeat protein, couples the p55 TNF-receptor to neutral sphingomyelinase. Cell. 1996;86:937–947. doi: 10.1016/S0092-8674(00)80169-5. [DOI] [PubMed] [Google Scholar]
- 33.Berneman Z. HTLV molecular biology and pathogenesis: Airlie Center, Warrenton, VA, USA, 17–19 March 2000. Leukemia. 2001;15:647–654. doi: 10.1038/sj.leu.2402032. [DOI] [PubMed] [Google Scholar]
- 34.Mozhgani S-H, Piran M, Zarei-Ghobadi M, Jafari M, Jazayeri S-M, Mokhtari-Azad T, Teymoori-Rad M, Valizadeh N, Farajifard H, Mirzaie M. An insight to HTLV-1-associated myelopathy/tropical spastic paraparesis (HAM/TSP) pathogenesis; evidence from high-throughput data integration and meta-analysis. Retrovirology. 2019;16:1–11. doi: 10.1186/s12977-019-0508-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mozhgani SH, Zarei-Ghobadi M, Teymoori-Rad M, Mokhtari-Azad T, Mirzaie M, Sheikhi M, Jazayeri SM, Shahbahrami R, Ghourchian H, Jafari M. Human T-lymphotropic virus 1 (HTLV-1) pathogenesis: a systems virology study. J Cell Biochem. 2018;119:3968–3979. doi: 10.1002/jcb.26546. [DOI] [PubMed] [Google Scholar]
- 36.Park H. Poo M-mJNRN: neurotrophin regulation of neural circuit development and function. Nat Rev Neurosci. 2013;14:7–23. doi: 10.1038/nrn3379. [DOI] [PubMed] [Google Scholar]
- 37.Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci. 2001;24:677–736. doi: 10.1146/annurev.neuro.24.1.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakagawa T, Ohta KJ. Quercetin regulates the integrated stress response to improve memory. Int J Mol Sci. 2019;20:2761. doi: 10.3390/ijms20112761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nishida K, Hoshino M, Kawaguchi Y, Murakami F. Ptf1a directly controls expression of immunoglobulin superfamily molecules Nephrin and Neph3 in the developing central nervous system. J Biol Chem. 2020;285:373–380. doi: 10.1074/jbc.M109.060657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mori T, Fukuda Y, Kuroda H, Matsumura T, Ota S, Sugimoto T, Nakamura Y, Inazawa J. Cloning and characterization of a NovelRab-family gene, Rab36, within the region at 22q11. 2 that is homozygously deleted in malignant rhabdoid tumors. Biochem Biophys Res Commun. 1999;254:594–600. doi: 10.1006/bbrc.1998.9968. [DOI] [PubMed] [Google Scholar]
- 41.Zhu Y, Liang S, Pan H, Cheng Z, Rui X. Inhibition of miR-1247 on cell proliferation and invasion in bladder cancer through its downstream target of RAB36. J Biosci. 2018;43:365–373. doi: 10.1007/s12038-018-9755-4. [DOI] [PubMed] [Google Scholar]
- 42.Vanzo RJ, Twede H, Ho KS, Prasad A, Martin MM, South ST. Wassman ER (2019) Clinical significance of copy number variants involving KANK1 in patients with neurodevelopmental disorders. Eur J Med Genet. 2019;62:15–20. doi: 10.1016/j.ejmg.2018.04.012. [DOI] [PubMed] [Google Scholar]
- 43.Cui Z, Shen Y, Chen KH, Mittal SK, Yang J-Y, Zhang GJ. KANK1 inhibits cell growth by inducing apoptosis through regulating CXXC5 in human malignant peripheral nerve sheath tumors. Sci Rep. 2017;7:1–13. doi: 10.1038/s41598-016-0028-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ghobadi MZ, Emamzadeh R, Mozhgani S-H. Deciphering microRNA-mRNA regulatory network in adult T-cell leukemia/lymphoma; the battle between oncogenes and anti-oncogenes. PLoS ONE. 2021;16:e0247713. doi: 10.1371/journal.pone.0247713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boyer A, Hermo L, Paquet M, Robaire B, Boerboom D. Seminiferous tubule degeneration and infertility in mice with sustained activation of WNT/CTNNB1 signaling in sertoli cells1. Biol Reprod. 2008;79:475–485. doi: 10.1095/biolreprod.108.068627. [DOI] [PubMed] [Google Scholar]
- 46.McNulty DE, Li Z, White CD, Sacks DB, Annan RS. MAPK scaffold IQGAP1 binds the EGF receptor and modulates its activation. J Biol Chem. 2011;286:15010–15021. doi: 10.1074/jbc.M111.227694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Caretta A, Mucignat-Caretta C. Protein kinase a in cancer. Cancer. 2011;3:913–926. doi: 10.3390/cancers3010913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sassone-Corsi P. The cyclic AMP pathway. Cold Spring Harbor Perspect Biol. 2012;4:011148. doi: 10.1101/cshperspect.a011148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Salter MW, Dong Y, Kalia LV, Liu XJ, Pitcher G. Regulation of NMDA receptors by kinases and phosphatases. Biol NMDA Recept. 2009;7:123–148. [PubMed] [Google Scholar]
- 50.Fenske S, Hennis K, Rötzer RD, Brox VF, Becirovic E, Scharr A, Gruner C, Ziegler T, Mehlfeld V, Brennan J. cAMP-dependent regulation of HCN4 controls the tonic entrainment process in sinoatrial node pacemaker cells. Nat Commun. 2020;11:1–22. doi: 10.1038/s41467-020-19304-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang X, Ibrahimi OA, Olsen SK, Umemori H, Mohammadi M, Ornitz DM. Receptor specificity of the fibroblast growth factor family: the complete mammalian FGF family. J Biol Chem. 2006;281:15694–15700. doi: 10.1074/jbc.M601252200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fang Z, Zang W, Chen R, Ye B, Wang X, Yi S, Chen W, He F, Ye G. Gene expression profile and enrichment pathways in different stages of bladder cancer. Genet Mol Res. 2013;12:1479–1489. doi: 10.4238/2013.May.6.1. [DOI] [PubMed] [Google Scholar]
- 53.Liu X, Wang J, Sun G. Identification of key genes and pathways in renal cell carcinoma through expression profiling data. Kidney Blood Press Res. 2015;40:288–297. doi: 10.1159/000368504. [DOI] [PubMed] [Google Scholar]
- 54.Jianfeng W, Yutao W, Jianbin B. TACR2 is associated with the immune microenvironment and inhibits migration and proliferation via the Wnt/β-catenin signaling pathway in prostate cancer. Cancer Cell Int. 2021;21:415. doi: 10.1186/s12935-021-02126-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Soltani A, Hashemy SI, Avval FZ, Soleimani A, Rafatpanah H, Rezaee SA, Griffith R, Mashkani B. Molecular targeting for treatment of human T-lymphotropic virus type 1 infection. Biomed Pharmacother. 2019;109:770–778. doi: 10.1016/j.biopha.2018.10.139. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. List of the identified differential co-expressed modules in ACs, ATLL, and HAM/TSP.
Additional file 2. The preservation of modules in the external datasets (ACs, ATLL, and HAM/TSP).
Additional file 3. The enriched pathways by the connected proteins in each identified DiffCoEx_NA, DiffCoEx_AA, and DiffCoEx_AH.
Additional file 4. The list of specific pathways-genes in ACs, ATLL, and HAM/TSP.
Additional file 5. The list of DEGs, common between DEGs and DiffCoEx_AA, and specific genes for ATLL.
Data Availability Statement
All data generated or analyzed during this study are included in this published article [and its supplementary information files].