

Abstract
Oncogenic protein tyrosine phosphatases have long been viewed as drug targets of interest, and recently developed allosteric inhibitors of SH2 domain-containing phosphatase-2 (SHP2) have entered clinical trials. However, the ability of phosphatases to regulate many targets directly or indirectly and to both promote and antagonize oncogenic signaling may make the efficacy of phosphatase inhibition challenging to predict. Here we explore the consequences of antagonizing SHP2 in glioblastoma, a recalcitrant cancer where SHP2 has been proposed as a useful drug target. Measuring protein phosphorylation and expression in glioblastoma cells across 40 signaling pathway nodes in response to different drugs and for different oxygen tensions revealed that SHP2 antagonism has network-level, context-dependent signaling consequences that affect cell phenotypes (e.g., cell death) in unanticipated ways. To map specific signaling consequences of SHP2 antagonism to phenotypes of interest, a data-driven computational model was constructed based on the paired signaling and phenotype data. Model predictions aided in identifying three signaling processes with implications for treating glioblastoma with SHP2 inhibitors. These included PTEN-dependent DNA damage repair in response to SHP2 inhibition, AKT-mediated bypass resistance in response to chronic SHP2 inhibition, and SHP2 control of hypoxia-inducible factor expression through multiple mitogen-activated protein kinases. Model-generated hypotheses were validated in multiple glioblastoma cell lines, in mouse tumor xenografts, and through analysis of TCGA data. Collectively, these results suggest that in glioblastoma, SHP2 inhibitors antagonize some signaling processes more effectively than existing kinase inhibitors but can also limit the efficacy of other drugs when used in combination.
Keywords: PTPN11, SHP099, temozolomide, hypoxia, kinase inhibitors, AP-1 complexes
Introduction
SH2 domain-containing phosphatase-2 (SHP2) is a proto-oncogene known for its ability to promote extracellular signal-regulated kinase (ERK) activity downstream of receptor tyrosine kinases (RTKs) (1,2). While SHP2 is an attractive therapeutic target in cancer (3), it has been difficult to target with specificity until recently (4,5). Allosteric SHP2 inhibitors such as SHP099 and TNO155 are now available and may help overcome adaptive resistance such as ERK reactivation after MEK inhibition in carcinomas (6). SHP2 targeting is also of longstanding interest for glioblastoma multiforme (GBM) (7–9) due to largely ineffective standard of care [including temozolomide chemotherapy (10)] and involvement of SHP2 in RTK signaling commonly dysregulated in GBM (11). SHP2’s ubiquitous expression and broad functions may also make it an attractive target for overcoming issues of tumor heterogeneity and bypass signaling in GBM (12,13). Although SHP2 inhibitors are in clinical trials, they remain untested in GBM.
While SHP2 antagonism may effectively target glioma stem cells (7) or PDGFR-driven GBM (14), the ultimate consequences of SHP2 inhibition may be challenging to foresee given that SHP2 can both promote and antagonize oncogenic signaling. In a previous study from our lab, SHP2 knockdown impeded GBM cell/tumor growth and hypoxia-inducible factor (HIF) expression but simultaneously promoted resistance to targeted therapy (15). These effects were attributed, in part, to the ability of SHP2 to drive ERK activity and concomitantly antagonize STAT3 phosphorylation. Other conflicting roles of SHP2 have been reported [e.g., (16,17)]. These complexities may be rooted in the ability of SHP2 to regulate a broader network of signaling pathways than has been characterized and in a way that depends on specific cell setting.
We hypothesized that SHP2 controls a network of GBM signaling processes that impact the effects of SHP2 inhibition in ways that depend on presence of other drugs, cell background, and tumor microenvironmental factors. Because we anticipated that the scope of SHP2-dependent signaling could be large, we adopted a data science approach wherein we broadly characterized signaling and phenotypic changes in GBM cells for a variety of perturbations and used the data to develop a quantitative multivariable regression model. Specifically, we constructed a partial least squares regression (PLSR) data-driven model based on dynamic measurements of 40 signaling nodes (protein phosphorylation or expression) for SHP2-depleted or -replete GBM cells treated with RTK inhibitors or temozolomide and cultured under normoxic or hypoxic conditions. Such data science approaches are increasingly used in cancer research because they can reduce the dimensionality of multivariate cell decision processes to a realistic number of actionable drug targets (18–20). The PLSR model we developed efficiently separated experimental conditions across three principal components, while exceeding accepted thresholds for model fit and predictive power. The model was used to develop three testable hypotheses, validation of which led to novel inferences about the ability of SHP2 inhibition to promote DNA damage repair or AKT bypass resistance to therapy and the ability of SHP2 to control HIF expression through multiple MAPKs. Validation in multiple GBM cell backgrounds and mouse tumor xenografts, and through analysis of publicly available patient data, suggests these phenomena may be relevant in the GBM clinical setting and lends support to biomedical data science models as useful tools for designing clinical trials.
Materials and Methods
Cell culture
U87MG cells expressing EGFRvIII (Dr. Frank Furnari), U118MG (ATCC), T98G (ATCC), LN229 (ATCC), LN18 (ATCC), and LentiX™ 293T (Takara Bio) cells were maintained in DMEM with 10% fetal bovine serum (VWR), 1 mM L-Glutamine, 100 units/mL penicillin, and 100 μg/mL streptomycin (Gibco). G88 (Dr. Jakub Godlewski), TS528 (Dr. Frank Furnari), T3691 (Dr. Celeste Simon), and T4302 (Dr. Celeste Simon) glioma stem cells were maintained in suspension culture in Neurobasal™ Medium with B27™ and N-2 supplements (Gibco), 0.25 mM L-Glutamine, 100 units/mL penicillin, and 100 μg/mL streptomycin (Gibco) with 50 ng/mL recombinant human EGF (Peprotech) and human basic FGF (Peprotech). G88 cells expressing click beetle red luciferase (CBLuc) were described previously (21). Cell characteristics are listed in Table S1. For hypoxic experiments, cells were cultured in 1% oxygen using a Forma Steri-cycle i160 tri-gas incubator. For chronic SHP2 inhibition, medium containing SHP099 was replenished every 2–3 days. U87MG and U118MG parental or transduced cell lines were tested for mycoplasma using the MycoAlert system (Cambrex) and used within 15 passages from testing. G88 cells were authenticated by short tandem repeat profiling within six months of use.
shRNA and expression vectors
Oligonucleotides for SHP2 shRNA (shRNA#1: 5’ – GTATTGTACCAGAGTATTA – 3’; shRNA#2: 5’ – GGACGTTCATTGTGATTGA – 3’) and control shRNA (5’ – ATCACAGAATCGTCGTATGCA – 3’) were cloned into the lentiviral pLKO.1-puro plasmid (Broad Institute, The RNAi Consortium), and vectors were used to generate U87MG, U118MG, T98G, and LN229 cells expressing shRNA. Identical oligonucleotides were cloned into TET-pLKO-puro (Dr. Dmitri Wiederschain, Addgene plasmid #21915) and used to generate U87MG cells expressing doxycycline-inducible shRNA. Identical oligonucleotides, or a control non-targeting sequence, were cloned into the lentiviral pSicoR-GFP plasmid (Dr. Tyler Jacks) and used to generate G88 and TS528 GSCs expressing shRNA. Unless indicated otherwise, SHP2 shRNA#1 was used. The lentiviral pLKO.1-puro vector expressing HIF-1α shRNA and pCDH-puro vector expressing HIF-1α cDNA were gifts from Dr. Celeste Simon. The pBABE-hygro retroviral vector expressing PTEN was cloned previously (22) using cDNA provided by Dr. Frank Furnari.
Virus production and cell line engineering
Lentivirus was produced by calcium phosphate-mediated transfection of LentiX™ 293T cells with pLKO.1-puro, pSicoR-GFP, or pCDH-puro and packaging vectors pCMV-VSVg, pMDL-gp-RRE, and pRSV-Rev. Retrovirus was produced by calcium phosphate-mediated transfection of LentiX™ 293T with a pBABE-hygro plasmid and packaging vectors pCMV-VSVg and pUMVC.
Luminex
MILLIPLEX Cell Signaling Multiplex Assay kits (Millipore Sigma) were used according to manufacturer recommendations. Catalog numbers and analyte lists are provided in Table S2.
Western blotting
Whole-cell lysates were prepared and protein concentrations determined as described for Luminex assays. ~20 μg of total protein was loaded per lane on 4–12% gradient polyacrylamide gels (Thermo Fisher Scientific) under denaturing and reducing conditions and transferred to 0.2 μm nitrocellulose membranes (Bio-Rad). After antibody probing, membranes were imaged on a LI-COR Odyssey CLx. Densitometry was performed using Image Studio (LI-COR). Antibody information is provided in Supplementary Materials and Methods.
Quantitative reverse transcription PCR (qRT-PCR) and qPCR arrays
RNA was extracted using the RNeasy kit (Qiagen), and equal amounts were reverse transcribed (High Capacity cDNA Reverse Transcription Kit, Applied Biosystems). qRT-PCR was performed using SYBR Green PCR Master Mix (Applied Biosystems) on a StepOnePlus or QuantStudio3 Real-Time PCR System (Applied Biosystems). All qRT-PCR primers are listed in Table S3.
Immunofluorescence microscopy
Cells on glass coverslips were maintained in 6-well dishes, fixed in 4% paraformaldehyde in PBS for 20 min, permeabilized with 0.25% Triton X-100 in PBS for 5 min, and stained overnight at 4°C with primary antibodies. The next day, samples were washed and stained with secondary antibodies conjugated to Alexa Fluor fluorochromes (Thermo Fisher Scientific) and Hoechst (Thermo Fisher Scientific).
Flow cytometry
Flow cytometry measurements for cell death assays and cell cycle analysis were performed on a BD Biosciences FACSCalibur at the UVA Flow Cytometry Core Facility.
AP-1 luciferase reporter assay
For AP-1 reporter assays, cells were transfected with expression vector 3xAP1pGL3 and RSVpGL3 (Dr. Alexander Dent; Addgene plasmids #40342, 40343).
Orthotopic tumor xenografts
2,000 G88 cells (parental or expressing control or SHP2 shRNA) were stereotactically injected into the right striatum of 4–8-week-old, female, BALB/c SCID mice (Charles River). After recovery, animals were randomly assigned to treatment groups (n ≥ 5 for all treatment groups; see figure legends for specific numbers for each experiment). Drugs preparations were: SHP099, 10 or 50 mg/kg in corn oil, oral administration; temozolomide, 15 mg/kg in sterile water, intraperitoneal injection. All experiments were approved by the UVA Institutional Animal Care and Use Committee and performed in accordance with NIH guidelines.
Immunohistochemistry
Charged slides with 4–5-micron thick sections from formalin-fixed, paraffin-embedded tissues were generated by the UVA Research Histology Core. Antigen-retrieved tissues, prepared by the UVA Biorepository and Tissue Research Facility, were permeabilized with 0.10% Triton X-100 in PBS for 15 min, blocked for 1 h at room temperature, and stained overnight at 4°C with antibodies at manufacturer-recommended dilutions. The next day, samples were washed and stained with Alexa Fluor-conjugated secondary antibodies and Hoechst (Thermo Fisher Scientific).
TCGA analysis
GBM patient data were downloaded from The Cancer Proteome Atlas portal (https://tcpaportal.org/tcpa/) and cBioPortal (https://www.cbioportal.org/) and analyzed as described in detail in Supplementary Materials and Methods.
Partial Least Squares Regression (PLSR)
Luminex data were normalized per signal to average baseline values (t = 0, DMSO, 21% oxygen) in control shRNA expressing cells for each replicate. Normalized Luminex and phenotype measurements were mean-centered and variance-scaled (Table S4). PLSR was performed using SIMCA-P (Umetrics). To improve the model, t = 0 time points, for which mean-centered and variance scaled values are identical for all analytes, and time points with qualitatively identical trends for a given analyte were removed from the X (signaling) matrix. A three-component model was chosen to maximize fit (R2X, R2Y) and predictive power (Q2) with the fewest principal components. The final three-component model met accepted criteria for model performance, with R2X and R2Y “goodness of fit” parameter values > 0.75 and a Q2 “goodness of prediction” parameter value > 0.55, as in other cancer-specific models (23,24). Table S5 provides loadings for the first three principal components. For “filtered” PLSR loadings plots, only SHP2-dependent signals (p-value < 0.05 and log2 fold changes > 0.5 or < −0.5 between control or SHP2 shRNA) were shown. Comparisons were made between control or SHP2 shRNA for (1) DMSO, 21% oxygen (2) G+P, 21% oxygen, (3) TMZ, 21% oxygen, or (4) DMSO, 1% oxygen. See Table S6 for p-value and fold change calculations for all comparisons. P-values were determined using a two-tailed Student’s t-test (Excel) followed by FDR adjustment by the Benjamini-Hochberg method (GraphPad).
Statistical analysis
Comparisons between two groups were made using a two-tailed Student’s t-test (Excel), with significance determined as p < 0.05. Multiple comparisons were made using one-, two-, or three-way ANOVA (GraphPad), with p-values calculated by Tukey’s post hoc testing and significance determined as p < 0.05. Unless stated otherwise, all data are representative of 3 independent replicates. Pearson’s correlation coefficients were calculated in Excel. P-values for log-rank comparison tests of survival probability curves were calculated in the R package ‘survival.’
Supplementary Materials and Methods
Additional details for the methods listed above, plus those for PTEN immunoprecipitation, subcellular fractionation, siRNA, and inhibitors/other reagents, are provided in Supplementary Information.
Results
SHP2 knockdown impedes GBM cell cycle and hypoxia response, but simultaneously promotes resistance to therapy.
Constructing a data-driven model of SHP2 signaling control of GBM phenotypes requires paired measurements of cell phenotypes and signaling for a common set of conditions. We began with phenotype measurements. To characterize relationships among SHP2 expression and responses to drugs and hypoxia, an adherent GBM cell line expressing control or SHP2 shRNA was treated with gefitinib and PHA665752 (EGFR and MET inhibitors, respectively), temozolomide, or DMSO, and cultured in 21% or 1% oxygen. Measurements were made of cell cycle distribution, cell death, and HIF-1α expression (Fig. 1A–1C, S1A). U87MG cells expressing epidermal growth factor variant III (EGFRvIII), which is expressed in ~25% of GBM, were chosen for this dataset after screening three cell lines for two expected effects of SHP2 knockdown, reduced ERK phosphorylation and cell cycle arrest (Fig. S1B–S1D). EGFRvIII-expressing U87MG cells are hereafter referred to as U87MG for simplicity. Specificity of SHP2 knockdown effects was confirmed using a second non-overlapping shRNA (Fig. S1E–S1G).
Figure 1. SHP2 knockdown promotes cell phenotypes expected to promote and antagonize GBM progression.
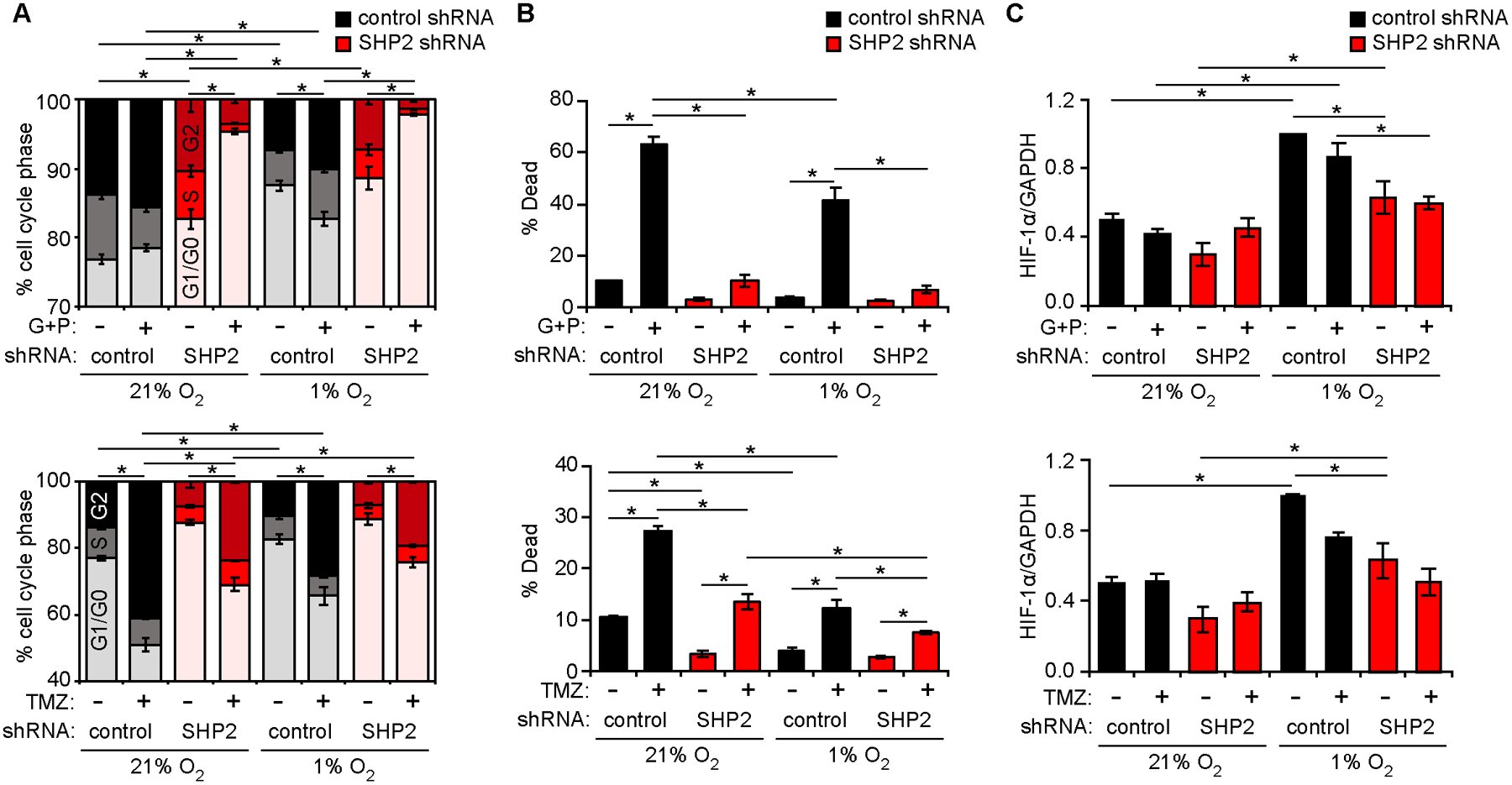
U87MG cells expressing control or SHP2 shRNA were cultured in 21% or 1% oxygen, and treated with 10 μM gefitinib (G) + 3 μM PHA665752 (P), 500 μM temozolomide (TMZ), or DMSO. 48 h after treatments, (A) cell cycle distribution or (B) cell death was measured by flow cytometry. (C) Lysates from cells treated for 6 h were analyzed by HIF-1α western blotting (Fig. S1A) with densitometry. The SHP2 shRNA used in Figure 1, and throughout main figures, corresponds to SHP2 shRNA#1, defined in Materials and Methods. See Fig. S1E–S1G for a comparison of SHP2 shRNA#1 against the non-overlapping shRNA#2. Throughout the panels, error bars indicate mean ± s.e.m. of three replicates; * p < 0.05 for indicated comparisons from Tukey’s post-hoc comparison following three-way ANOVA. For Fig. 1A, comparisons are made for percentage of cells in G1/G0.
Fig. 1A–1C demonstrate some expected trends based on prior work (15), including that SHP2 depletion promoted cell cycle arrest (Fig. 1A), increased resistance to EGFR and MET inhibitors (Fig. 1B), and impaired HIF-1α expression (Fig. 1C). At the same time, additional trends were revealed that motivate a data-driven model. For example, the ability of SHP2 knockdown to promote resistance to temozolomide (Fig. 1B) may have important implications for combining SHP2 inhibitors with standard of care GBM chemotherapy.
A partial least squares regression model identifies key SHP2-regulated signaling pathways that govern GBM cell response to therapy and hypoxia.
To identify signaling processes responsible for the phenotypes in Fig. 1, we generated lysates from U87MG cells expressing control or SHP2 shRNA (plated in parallel) using the same conditions as in Fig. 1 and for t ≤ 48 h after treatment (Fig. 2A). Lysates were analyzed using Luminex assay kits selected for coverage of pathways reported to be regulated by SHP2, including MAPK/SAPK, AKT/mTOR, STAT, RTK, apoptosis, and DNA damage signaling (25–30). Most Luminex antibodies targeted phosphorylated proteins. Signaling and phenotypic data were then pre-processed as described in Materials and Methods (Fig. 2B, Table S4) and PLSR was performed. PLSR efficiently separated experimental conditions (SHP2 expression, drug treatments, hypoxia/normoxia), with >75% of the data variance explained by three principal components (PCs) and sufficient predictive power [Q2 > 0.55; (31)] (Fig. 2C–2E, S2A, Table S5). See Materials & Methods for additional details on model goodness of fit and prediction.
Figure 2. A data-driven model predicts SHP2-regulated signaling governing GBM cell response to therapy.
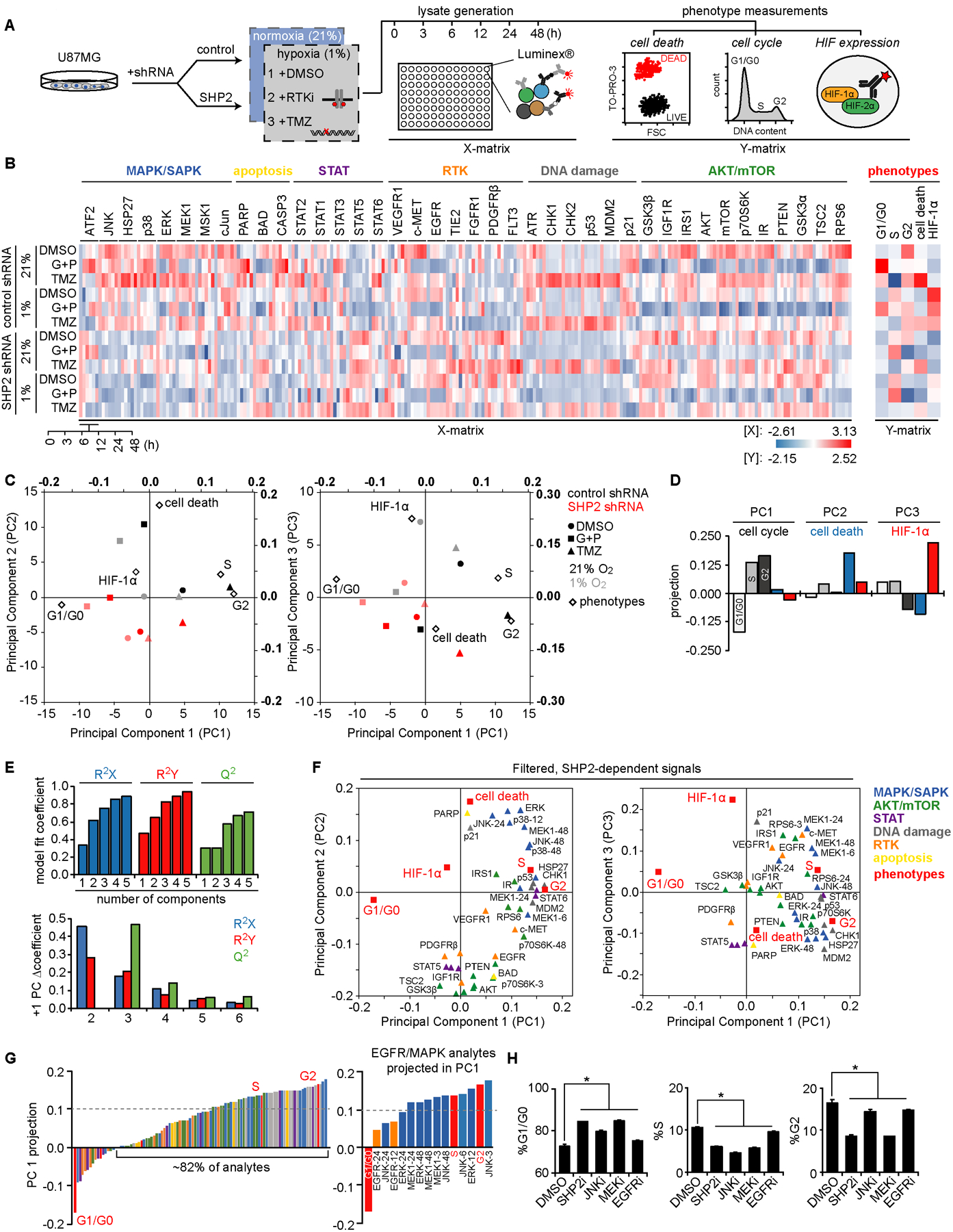
(A) U87MG cells expressing control or SHP2 shRNA were cultured in 21% or 1% oxygen and treated with gefitinib (G) and PHA665752 (P), temozolomide (TMZ), or DMSO. Cells were lysed 0, 3, 6, 12, 24, and 48 h after treatment, and lysates were analyzed via Luminex. (B) Mean-centered, variance-scaled signaling and phenotypic measurements are represented by a heat map, with Luminex kits indicated. Table S2 provides analyte post-translational modifications. (C) Bi-plots of scores (bold numbered axes; top/right) for conditions and loadings (bottom/left) of Y-matrix phenotypes for principal component 2 (PC2) or PC3 vs. PC1. (D) Phenotype projections into PCs 1–3 are plotted. (E) Model fit (R2X, R2Y) and predictive (Q2) coefficients are plotted versus PC. Coefficient changes (Δ) with additional components is shown. (F) Loadings are plotted of signals (triangles, color coded by Luminex kit) and phenotypes (red squares) for the three-component model, displaying only SHP2-regulated analytes. Times are indicated where two or more time points projected in opposite directions for an analyte. Loadings are listed in Table S5. (G) PC1 signal and phenotype projections are plotted, smallest to largest. Strongly projecting EGFR (orange) and MAPK (blue) analytes, and cell cycle phases (red), are highlighted. (H) U87MG cells were treated for 48 h with 10 μM SHP099 (SHP2i), 20 μM SP600125 (JNKi), 5 μM CI-1040 (MEKi), 10 μM gefitinib (EGFRi), or DMSO, and cell cycle distribution was analyzed. Error bars indicate mean ± s.e.m. of three replicates; * p < 0.05 for the indicated comparisons from Tukey’s post-hoc testing following one-way ANOVA.
To focus on likely signaling consequences of targeting SHP2, we filtered the model to retain analytes that displayed a significant difference (p < 0.05) and log2 fold change > 0.5 or < −0.5 between control and SHP2 shRNA conditions, with comparisons made for identical drug treatment, oxygen tension, and time (Fig. 2F, Table S6). One obvious trend was that >80% of analytes projected positively in principal component 1, along with S and G2 cell cycle phases (Fig. 2G). Many top analytes in principal component 1 (e.g., EGFR, MEK/ERK, and JNK) have established relationships with SHP2 and regulate cell cycle progression (25,28). The direction of their projection suggested that their phosphorylation promotes cell cycle progression. As expected, EGFR, MEK, and JNK inhibitors, as well as SHP2 inhibitor SHP099, antagonized cell cycle progression (Fig. 2H). Similar effects of SHP099 were observed in GSCs (Fig. S2B, S2C). In this broad sense, validated model predictions argue for the utility of SHP2 inhibitors in GBM, consistent with previous reports (7,8,14). The remainder of this study presents three specific and more nuanced inferences from the model relating to principal components 2 and 3.
Highlighted model inference (I): Adaptations in AKT signaling impact cell response to combined SHP2 and RTK inhibition.
In Fig. 2H, SHP2 inhibition produced cell cycle effects similar to SHP2 knockdown. Based on that and projection of SHP2 knockdown conditions antiparallel to cell death in principal component 2 (Fig. 2C), we anticipated that SHP2 inhibition would generally antagonize cell death. However, acute SHP2 inhibition surprisingly promoted cell death (Fig. 3A, S3A). We hypothesized that these differences could involve signaling adaptation with stable SHP2 knockdown. To pursue this, we generated U87MG cells expressing doxycycline-inducible control or SHP2 shRNA and, using them, observed time-dependent changes in cell death and ERK phosphorylation after doxycycline treatment (Fig. 3B, S3B, S3C). Moreover, in differentiated GBM and GSC lines, chronic SHP2 inhibition (> 4 days) promoted resistance to RTK inhibitors (Fig. 3C, S3D). [The marginally increased U87MG cell death in response EGFR and MET inhibition when combined with acute SHP099 (Fig. 3A) was lost with shorter treatment.] Furthermore, ERK inhibition in response to SHP2 inhibition was not durable, even with SHP099 replenishment (Fig. 3D, S3E), again suggesting adaptation. Importantly, acute and chronic SHP2 inhibition and doxycycline-induced SHP2 knockdown all led to cell cycle effects similar to constitutive SHP2 knockdown (Fig. S3F, S3G, S3H).
Figure 3. Prolonged SHP2 antagonism promotes AKT-mediated resistance to EGFR and MET inhibitors.
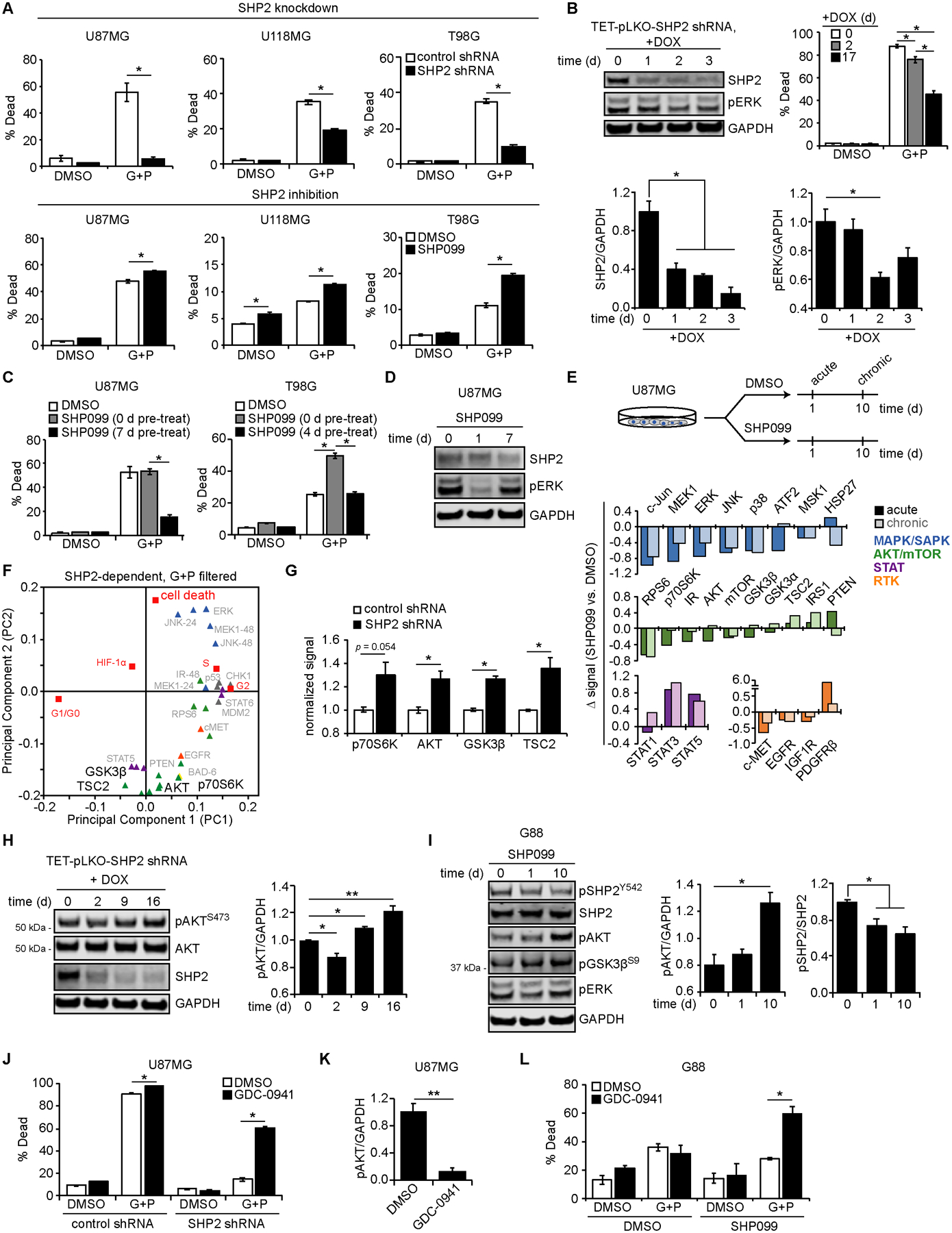
(A) Indicated cell lines expressing control or SHP2 shRNA, or parental cells treated with 10 μM SHP099 or DMSO, were treated for ≤ 72 h with 10 μM gefitinib (G) + 3 μM PHA665752 (P) or DMSO, and cell death was measured by flow cytometry. (B) U87MG cells expressing doxycycline (DOX)-inducible SHP2 shRNA were treated with 5 μg/mL DOX as indicated. Lysates were analyzed by western blotting using antibodies against indicated proteins, with densitometry normalized to values at t = 0 days (d). SHP2 shRNA-expressing cells treated with DOX for indicated times were treated for 72 h with 10 μM G + 3 μM P or DMSO, and cell death was measured by flow cytometry. Control shRNA results shown in Fig. S3B, S3C. (C) U87MG and T98G cells were pre-treated with 5 μM SHP099 or DMSO for indicated times, followed by 48 h treatment with 10 μM SHP099, 10 μM G + 3 μM P, 10 μM SHP099 + 10 μM G + 3 μM P, or DMSO, and cell death was measured by flow cytometry. (D) Lysates from parental U87MG cells treated with SHP099 for indicated times were analyzed by western blotting using antibodies against indicated proteins. pERK densitometry shown in Fig. S3E. (E) Lysates from parental U87MG cells treated with 10 μM SHP099 or DMSO for 1 (acute) or 10 (chronic) d were analyzed via Luminex. Differences (Δ) between average, normalized Luminex signals for acute or chronic SHP099 versus DMSO are shown. (F) Loadings of signals differentially regulated by SHP2 for G+P treatment are shown, highlighting AKT/mTOR analytes projected opposite cell death in PC2. (G) Luminex signals (normalized to control shRNA), for DMSO treatment and 21% oxygen, are plotted for phosphorylated p70S6K (T412; 48 h), AKT (S473; 0 h), GSK3β (S9; 0 h), and TSC2 (S939; 0 h). (H) Cells described in (B) were treated with 5 μg/mL DOX for indicated times. Lysates were analyzed by western blotting using antibodies against indicated proteins, with pAKT densitometry normalized to t = 0 d. Matched no-DOX lysates shown in Fig. S3K. (I) Lysates from G88 cells treated with 5 μM SHP099 for indicated times were analyzed by western blotting using antibodies against indicated proteins, with pAKT and pSHP2 (Y542) densitometry normalized to t = 0 days. (J) U87MG cells expressing control or SHP2 shRNA were treated for 72 h with 5 μM GDC-0941, 10 μM G + 3 μM P, 5 μM GDC-0941, or DMSO, and cell death was measured by flow cytometry. (K) pAKT blot densitometry for U87MG cells treated for 24 h with 1 μM GDC-0941 or DMSO is shown. (L) G88 cells treated for 19 d with 5 μM SHP099 or DMSO were treated for 48 h with 1 μM GDC-0941, 5 μM G + 1 μM P, 1 μM GDC-0941, or DMSO, and cell death was measured by flow cytometry. Throughout the panels, representative blot images are shown, and error bars indicate mean ± s.e.m. for three replicates. Statistical comparisons were made by Tukey’s post-hoc testing following two-way ANOVA (panels A, B cell death, C, J, L), Tukey’s post-hoc testing following one-way ANOVA (panels B western blotting, H, I), or Student’s t-test (panels G, K). * p < 0.05 and ** p < 0.01 for indicated comparisons.
While lack of durable ERK suppression could explain differences between acute and chronic SHP099 treatment, positive projection of ERK in principal component 2 (Fig. 2F) and evidence from our previous work (15) suggest that other SHP2-regulated pathways are relevant. To identify other pathways, lysates from cells treated for 1 (acute) or 10 (chronic) days with SHP099 were analyzed by Luminex (Fig. 3E). To further narrow candidates, we compared Fig. 3E with a version of the filtered PLSR model shown in Fig. 2F displaying only analytes that were significantly and substantially perturbed by SHP2 knockdown (p < 0.05 and log2 fold change > 0.5 or < −0.5 between control and SHP2 shRNA conditions) when EGFR and MET inhibitors were present and focusing on analytes projecting antiparallel to cell death in principal component 2 (Fig. 3F). Although STAT5 meets the criteria, STAT5 and STAT3 phosphorylation increased in response to both acute and chronic SHP2 inhibition, suggesting they are unlikely responsible for the adaption. It is worth recalling that STAT3 was previously identified as promoting resistance to RTK inhibitors with stable SHP2 knockdown (15), and that relationship was observed here (Fig. S3I, S3J).
The remaining analytes that met the criteria were predominantly from the AKT/mTOR pathway. As expected, based on projections of control and knockdown conditions in Fig. 2C, constitutive SHP2 knockdown promoted phosphorylation of AKT pathway analytes (Fig. 3G). Moreover, in response to doxycycline-induced SHP2 knockdown, AKT phosphorylation briefly decreased (2 days) and then increased above baseline (9 and 16 days) (Fig. 3H, S3K). Similar increases in AKT and GSK3β phosphorylation (without acute decrease) were observed in G88 GSCs treated with SHP099 (Fig. 3I). Interestingly, decreased AKT phosphorylation with acute SHP2 inhibition was only observed in a subset of differentiated GBM lines (Fig. S3L), perhaps consistent with context-dependent SHP2 regulation of AKT (26). PI3K inhibition (GDC-0941) sensitized cells with SHP2 knockdown to EGFR and MET inhibition (Fig. 3J, 3K), confirming AKT’s importance. Moreover, while PI3K inhibition did not augment response to gefitinib and PHA665752 in SHP099-naive G88 cells, it promoted response to gefitinib and PHA665752 in cells chronically treated with SHP099 (Fig. 3L). Thus, PI3K and AKT inhibitors could be utilized to overcome resistance mechanisms induced by prolonged SHP2 inhibition.
Highlighted model inference (II): SHP2 controls of AP-1 transcriptional activity and HIF expression via multiple MAPK pathways.
We previously reported that HIF-1α and HIF-2α expression in GBM cells was impaired by SHP2 knockdown (15). This is important because hypoxia in the GBM microenvironment promotes tumor cell stemness and tumor progression (32,33). While we previously attributed this effect to SHP2 control of ERK, the PLSR model implicated JNK, p38, and ERK MAPKs together, based on their projection in principal component 3 (Fig. 4A). We note that HIF-2α expression mirrored HIF-1α trends (Fig. S4A), that ectopic HIF-1α expression promoted cell cycle arrest and resistance to RTK inhibition (Fig. S4B, S4C, S4D), and that HIF-1α antagonized resistance to RTK inhibitors in hypoxia (Fig. S4E, S4F). Thus, HIF-1α measurements captured more general HIF trends, and HIF-1α may be directly responsible for SHP2-dependent phenotypes.
Figure 4. SHP2 antagonism impairs HIF expression via decreased, MAPK-dependent AP-1 transcriptional activity.
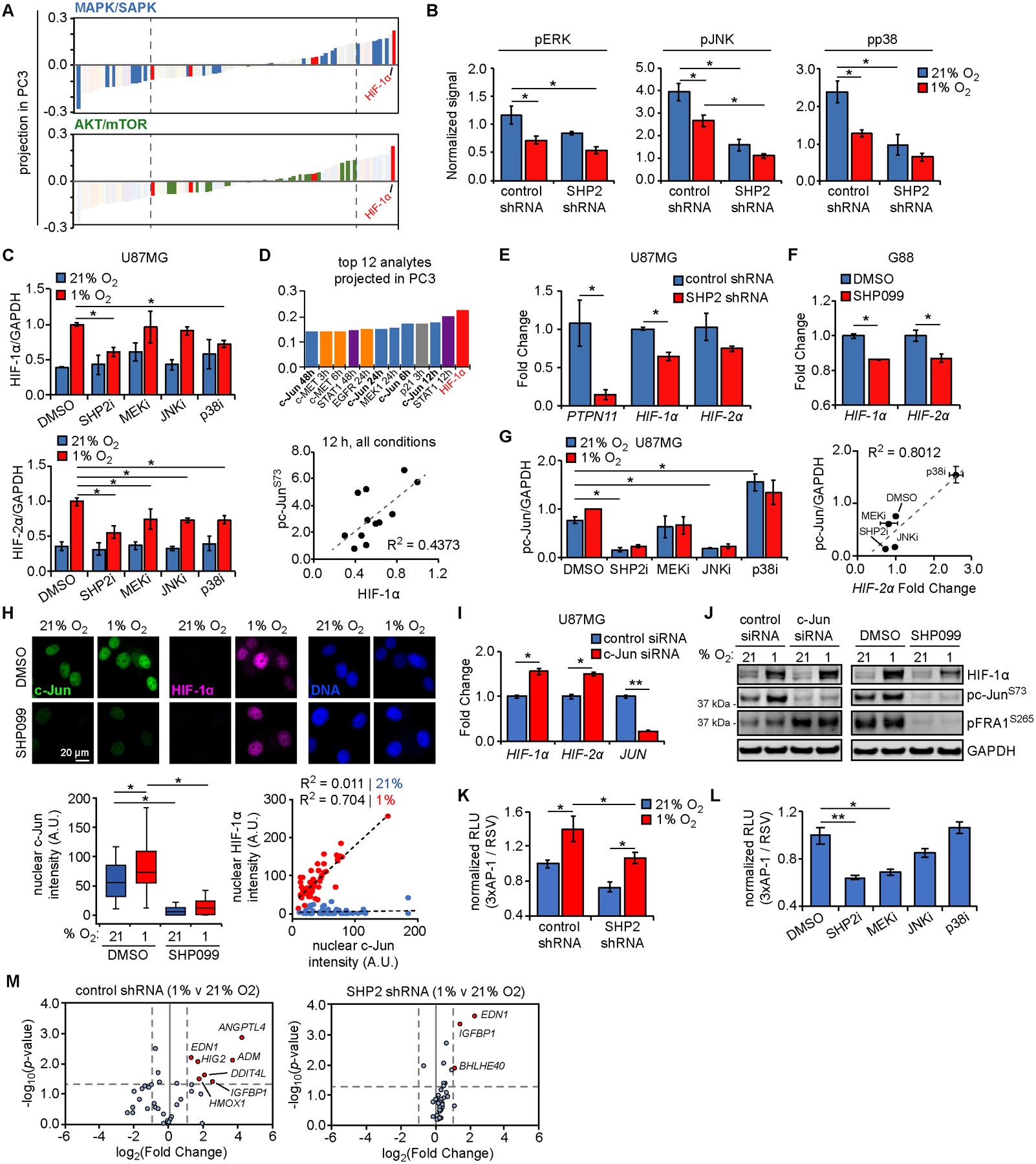
(A) Projection of signals and phenotypes (smallest to largest) in principal component 3 (PC3) with highlighted signals from MAPK/SAPK and AKT/mTOR Luminex kits. Dashed lines indicate highly negatively or positively projected analytes. (B) Luminex signals (normalized to control shRNA at 0 h) for DMSO treatment in 21% or 1% oxygen, for phosphorylated ERK (T185/Y187; 48 h), JNK (T183/Y185; 48 h), and p38 (T180/Y182; 48 h). (C) U87MG cells were pre-treated for 24 h with 10 μM SHP099 (SHP2i), 5 μM CI-1040 (MEKi), 20 μM SP600125 (JNKi), 20 μM SB203580 (p38i), or DMSO, followed by exposure to 21% or 1% oxygen for 6 h. Lysates were analyzed by western blotting, with densitometry for normalized HIF-1α and HIF-2α. Representative blot images in Fig. S4H. (D) The 12 signals or phenotypes with largest positive projection in PC3 are shown, with c-Jun loadings highlighted (bold). Normalized phosphorylated c-Jun (S73) Luminex values are plotted versus HIF-1α expression at 12 h, across all conditions. (E) RNA was extracted from U87MG cells expressing control or SHP2 shRNA. qRT-PCR was performed using primers against indicated transcripts; PTPN11 = SHP2 gene symbol. (F) G88 cells were treated for 48 h with 10 μM SHP099 or DMSO, and RNA was extracted. qRT-PCR was performed using primers for indicated transcripts. (G) Quantification of phosphorylated c-Jun for blots described in (C). U87MG cells similarly pre-treated with MAPK inhibitors in 21% oxygen were lysed and RNA extracted. qRT-PCR was performed using primers for HIF-2α. Fold changes shown in Fig. S4L. Normalized pc-Jun signals are plotted versus HIF-2α fold changes, across treatments with MAPK inhibitors in 21% oxygen. (H) U87MG cells were pre-treated for 24 h with 10 μM SHP099 or DMSO, followed by exposure to 21% or 1% oxygen for 6 h. Cells were stained for c-Jun, HIF-1α, and DNA. Scale bar = 20 μm. Nuclear c-Jun and HIF-1α intensities were quantified for n > 100 cells across three biological replicates. Scatter plots for cells in 21% (blue) or 1% oxygen (red) were also created. (I) U87MG cells were transfected with control or c-Jun siRNA for 48 h. qRT-PCR was performed using extracted RNA and primers for indicated transcripts. (J) Cells transfected in parallel were exposed to 21% or 1% oxygen for 6 h. Lysates were analyzed by western blotting using antibodies against indicated proteins. Lysates from cells treated with SHP099 or DMSO, as described in (C), were re-blotted using antibodies against indicated proteins. (K) U87MG cells expressing control or SHP2 shRNA were transfected with plasmids encoding AP-1- or Rous sarcoma virus (RSV)-regulated luciferase expression, followed by 21% or 1% oxygen exposure for 6 h, addition of D-luciferin, and bioluminescence measurement. (L) U87MG cells transfected as in (K) were treated for 24 h with 10 μM SHP099 (SHP2i), 5 μM CI-1040 (MEKi), 20 μM SP600125 (JNKi), 20 μM SB203580 (p38i), or DMSO, prior to D-luciferin addition and bioluminescence measurement. (M) U87MG cells expressing control or SHP2 shRNA were exposed to 21% or 1% oxygen for 72 h, and hypoxia-regulated transcripts were measured by qPCR array. Volcano plots comparing gene expression between 21% and 1% oxygen for control or SHP2 shRNA are shown. Red dots indicate genes with p-value and fold-change above indicated thresholds (dashed lines). Throughout the panels, representative images are shown, and error bars indicate mean ± s.e.m. for three replicates, unless otherwise indicated. For box plots, median, first/third quartiles, minimum/maximum, and outlier values are displayed for indicated cell numbers across replicates. Statistical comparisons were made by Tukey’s post-hoc testing following two-way ANOVA (panels B, C, G, K, M), Tukey’s post-hoc testing following one-way ANOVA (panels H, L), or Student’s t-test (panels E, F, I). * p < 0.05 and ** p < 0.01 for indicated comparisons.
Returning to the PLSR model, MAPK/SAPK analytes projected more strongly in principal component 3 than others (Fig. 4A, S4G), and ERK, JNK, and p38 phosphorylation decreased in response to SHP2 knockdown and hypoxia (Fig. 4B). Previous studies have suggested that hypoxia can positively or negatively regulate MAPKs, and that MAPKs can regulate HIF expression and transcriptional activity (34–36). To test the importance of individual MAPKs, we pretreated U87MG cells with MEK, JNK, p38, or SHP2 inhibitors, and evaluated HIF expression (Fig. 4C, S4H). HIF-1α and/or HIF-2α expression in hypoxia was significantly impaired by all MAPK inhibitors. At the concentrations used, SHP099 reduced HIF-1α and HIF-2α expression more than any MAPK inhibitor, potentially because SHP099 more effectively inhibited ERK or because SHP2 inhibition targets multiple MAPKs. HIF-1α expression at 1% O2 was not influenced by PI3K or mTOR inhibition (Fig. S4I).
In seeking to determine if multiple SHP2-regulated MAPKs influence HIF expression, we noted that c-Jun, which is regulated by ERK, p38, and JNK (37,38), projected strongly in principal component 3 (Fig. 4D) and that c-Jun has been implicated in HIF-2α transcription in GBM (39). In differentiated GBM cells and GSCs, SHP2 knockdown or inhibition reduced HIF-1/2α transcript abundance (Fig. 4E, 4F, S4J, S4K). Moreover, the extent to which individual inhibitors reduced c-Jun phosphorylation correlated with HIF-2α transcript abundance (Fig. 4G, S4L). Interestingly, SB203580 has been reported to promote phosphorylation of ERK, JNK, and c-Jun (40), consistent with our observations (Fig. S4H), and potentially explaining why SB203580 increased HIF-2α transcripts. When cells were treated with proteasome inhibitor MG132, SHP2 knockdown-mediated differences in HIF-1α expression were also apparent in normoxia (Fig. S4M).
We further noted that nuclear c-Jun abundance (an indirect indicator of c-Jun transcriptional potential) correlated with nuclear HIF-1α expression in hypoxia but was reduced by SHP2 inhibition (Fig. 4H). However, siRNA-mediated c-Jun knockdown did not decrease HIF-1/2α transcripts (Fig. 4I). Given that c-Jun carries out its role in AP-1 complexes with other Jun or Fos family members (41), we screened cells with c-Jun knockdown for phosphorylation of another AP-1 member, fos-related antigen 1 (FRA1), to test for possible compensatory phosphorylation (Fig. 4J). Indeed, c-Jun knockdown, but not SHP2 inhibition, promoted FRA1 phosphorylation. These data suggest that SHP2-dependent differences in HIF-1/2α transcripts may be generally due to AP-1 complexes. Importantly, SHP2-regulated MAPKs regulate multiple AP-1 complex members (42). Furthermore, promoters of HIF1A and EPAS1 (which encodes HIF-2α) contain AP-1 binding sites, a subset of which preferentially bind Jun/Fos heterodimers (Fig. S4N). Using an AP-1 DNA binding reporter, we observed that AP-1 transcriptional activity decreased with SHP2 knockdown, consistent with trends in HIF-1/2α transcripts (Fig. 4K). Similarly, AP-1 transcriptional activity in response to SHP2 or MAPK inhibitors mirrored HIF-2α transcript levels (Fig. 4L, S4L). A qPCR array revealed broader SHP2 regulation of hypoxia-regulated genes (Fig. 4M). SHP2 inhibition also influenced transcriptional response to hypoxia, as demonstrated by VEGF expression (Fig. S4O).
Highlighted model inference (III): SHP2 inhibition promotes resistance to temozolomide through PTEN-dependent DNA damage repair.
In Fig. 1, we learned that SHP2 antagonism promotes resistance to temozolomide. To understand this, we generated a version of the filtered PLSR model shown in Fig. 2F displaying only analytes that were significantly and substantially perturbed by SHP2 knockdown (p < 0.05 and log2 fold change > 0.5 or < −0.5 between control and SHP2 shRNA conditions) when temozolomide was present (Fig. 5A). Recalling that several AKT/mTOR analytes projected antiparallel to cell death when screening based on response to RTK inhibitors, it was striking that a single AKT/mTOR pathway member, PTEN, strongly projected antiparallel to cell death for temozolomide (Fig. S5A). Inspection of the Luminex data used to generate the PLSR model revealed that temozolomide promoted PTEN S380 phosphorylation, which was augmented by SHP2 knockdown (Fig. 5B). While PTEN is best known as a lipid phosphatase, it can also participate in DNA damage repair (43). Phosphorylation of PTEN S380, and other C-terminal serine/threonine residues, can promote PTEN nuclear translocation and participation in DNA damage repair by facilitating Rad 51 chromatin loading (44–47). Interestingly, PTEN S380 phosphorylation correlated best with AKT S473 and GSK3β S9 phosphorylation for control treatment (Pearson’s correlation coefficients of 0.615 and 0.605, respectively). In the presence of temozolomide, correlation coefficients of the DNA damage indicators CHK1 S345 phosphorylation and MDM2 expression (0.319 and 0.289, respectively) exceeded those for AKT S473 and GSK3β S9 (0.017 and 0.059, respectively) (Fig. 5C).
Figure 5. SHP2 antagonism promotes resistance to temozolomide via PTEN-mediated DNA damage repair.
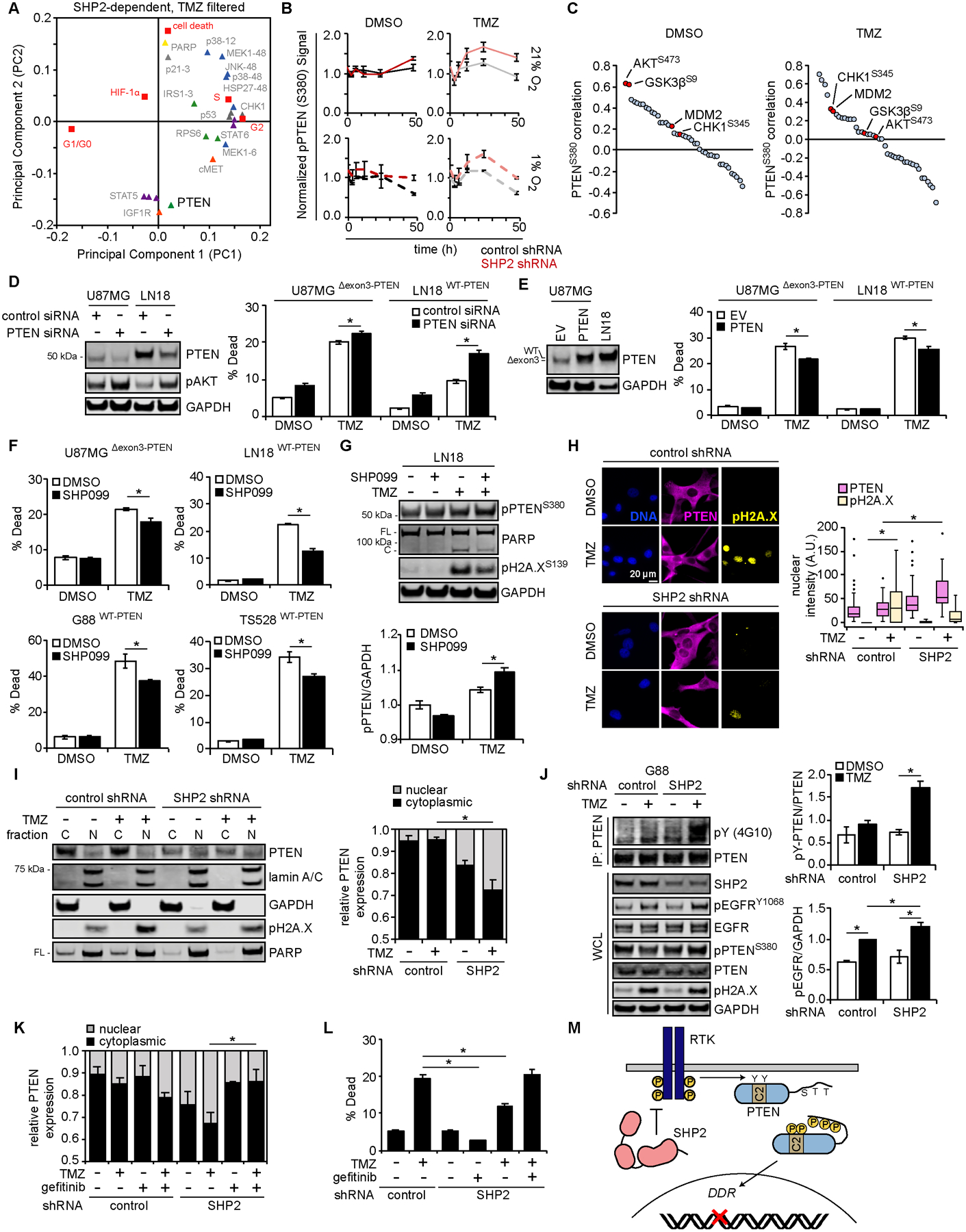
(A) Loadings plot of signals differentially regulated by SHP2 for samples treated with temozolomide (TMZ), highlighting AKT/mTOR analytes projecting opposite cell death in principal component 2 (PC2). (B) Phosphorylated PTEN (S380) signals (normalized to control shRNA at 0 h) are shown for DMSO or TMZ treatment, in 21% or 1% oxygen. (C) Pearson’s correlation coefficients for PTEN S380 phosphorylation with other analytes, across all DMSO or TMZ treatment conditions, are plotted largest to smallest. Red points highlight analytes of interest. (D) U87MG and LN18 cells were transfected with control or PTEN siRNA for 48 h. Lysates were analyzed by western blotting using antibodies against indicated proteins, with densitometry shown in Fig. S5B. Cells transfected in parallel were treated for 48 h with 500 μM TMZ or DMSO, and cell death was measured by flow cytometry. (E) U87MG and LN18 cells were transduced with a vector encoding PTEN or empty vector (EV). Lysates were analyzed by western blotting using antibodies against indicated proteins. The same cells were treated for 72 h (U87MG) or 96 h (LN18) with 500 μM TMZ or DMSO, and cell death was measured by flow cytometry. (F) U87MG, LN18, G88, and TS528 cells were treated for ≤ 96 h with SHP099, TMZ, SHP099+TMZ or DMSO, and cell death was measured by flow cytometry [U87MG: 10 μM SHP099, 500 μM TMZ; LN18: 10 μM SHP099, 500 μM TMZ; G88: 5 μM SHP099, 100 μM TMZ; TS528: 5 μM SHP099, 500 μM TMZ]. (G) LN18 cells were treated for 48 h with 10 μM SHP099, 500 μM TMZ, 10 μM SHP099 + 500 μM TMZ, or DMSO. Lysates were analyzed by western blotting using antibodies against indicated proteins, with pPTEN (S380) densitometry normalized to DMSO-treated values. (H) U87MG cells expressing control or SHP2 shRNA were treated for 24 h with 500 μM TMZ or DMSO. Cells were stained for PTEN, pH2A.X (S139), and DNA. Scale bar = 20 μm. Nuclear PTEN was quantified for n >100 cells across three biological replicates. (I) U87MG cells expressing control or SHP2 shRNA were treated for 24 h with 500 μM TMZ or DMSO. Cytoplasmic (C) and nuclear (N) fractions were prepared for western blotting using antibodies against indicated proteins, with PTEN densitometry performed. Western blots of matched whole-cell lysates are shown in Fig. S5F. (J) G88 cells expressing control or SHP2 shRNA were treated for 24 h with 100 μM TMZ or DMSO. Whole-cell lysates (WCL) or PTEN immunoprecipitates (IP) were analyzed by western blotting using antibodies against indicated proteins, with normalized tyrosine-phosphorylated PTEN and pEGFR signals quantified by densitometry. (K) U87MG cells expressing control or SHP2 shRNA were treated for 24 h with 500 μM TMZ, 10 μM gefitinib, 500 μM TMZ + 10 μM gefitinib, or DMSO. Lysates from cytoplasmic (C) and nuclear (N) fractions were analyzed by western blotting, with densitometry for PTEN. Representative blot images are shown in Fig. S5M. (L) Cells treated as in (K) were treated for 72 h, and cell death was measured by flow cytometry. (M) Schematic demonstrating SHP2-dependent regulation of PTEN phosphorylation and localization, through regulation of RTKs. Throughout the panels, representative blot images are shown, and error bars indicate mean ± s.e.m. for three replicates, unless otherwise indicated. For box plots, median, first/third quartiles, minimum/maximum, and outlier values are displayed for indicated number of cells across replicates. Statistical comparisons were made by Tukey’s post-hoc testing following two-way ANOVA (panels D, E, F, G, I, J, L), Tukey’s post-hoc testing following one-way ANOVA (panel H), or Tukey’s post-hoc testing following three-way ANOVA (panel K). * p < 0.05 for indicated comparisons.
To test the importance of PTEN, we transfected cells with control or PTEN-targeting siRNA and measured temozolomide-induced cell death (Fig. 5D, S5B). In U87MG [PTEN exon-deletion mutant retaining phosphatase activity (48)] and LN18 cells (wild-type PTEN), PTEN knockdown promoted AKT phosphorylation but also promoted sensitivity to temozolomide. In both cell lines, ectopic wild-type PTEN promoted resistance to temozolomide (Fig. 5E). Moreover, SHP2 inhibition promoted resistance to temozolomide in differentiated GBM lines and GSCs with intact PTEN expression (Fig. 5F, S5C), but did not alter response to temozolomide in T3691 GSCs, which express little PTEN (Fig. S5D). In LN18 cells, SHP2 inhibition promoted PTEN S380 phosphorylation and reduced PARP cleavage and H2A.X phosphorylation (Fig. 5G, S5E). SHP2 knockdown also promoted PTEN nuclear localization in response to temozolomide (Fig. 5H, 5I, S5F).
Regulation of PTEN serine phosphorylation by SHP2 (a tyrosine phosphatase) is likely indirect, but we hypothesized that relevant PTEN tyrosines could be phosphorylated in response to DNA damage. PTEN phosphorylation at Y240 by fibroblast growth factor receptor (FGFR) has been implicated in GBM radioresistance (49), and Y240 and Y315 reside in the PTEN C2 domain, which regulates membrane binding (50). We suspected that their phosphorylation could promote PTEN membrane dissociation, as observed with C-terminal serine phosphorylation. In fact, in a prior study of radiation-induced DNA damage, tyrosine-phosphorylated PTEN was detected exclusively in the nucleus, and Y240F PTEN mutants displayed impaired nuclear localization (49). Using ScanSite (51), we identified EGFR and PDGFR as possible drivers of PTEN Y240 and Y315 phosphorylation (Fig. S5G). We previously identified EGFRvIII as a possible SHP2 substrate (15), and others have shown that SHP2 can dephosphorylate RTKs (52). In addition, our data showed that SHP2 knockdown promoted EGFR, FGFR1, and PDGFRβ phosphorylation (Fig. S5H). Because we observed that temozolomide promoted EGFR phosphorylation in G88 cells (Fig. S5I), we hypothesized that EGFR could be an SHP2-regulated RTK that promotes PTEN tyrosine phosphorylation. Consistent with this, SHP2 knockdown promoted EGFR and PTEN tyrosine phosphorylation and survival in cells treated with temozolomide (Fig. 5J, S5J, S5K, S5L). In SHP2 knockdown cells, EGFR inhibition prevented temozolomide-induced PTEN nuclear localization (Fig. 5K, S5M) and restored cell death response to temozolomide to levels observed in SHP2-replete cells (Fig. 5L), consistent with the proposed mechanism (Fig. 5M).
Tumor xenografts and GBM patient data support key model predictions.
To assess response to SHP099 in vivo, G88 cells expressing luciferase were orthotopically implanted in BALB/c SCID mice. Longitudinal bioluminescence imaging of mice treated with SHP099 (daily, 10 or 50 mg/kg, 5 days after implantation) revealed a dose-dependent decrease in tumor size and disease burden (determined by overall weight loss) in response to SHP099 (Fig. 6A, Fig. S6A, S6B). Expected differences in SHP2 Y542 phosphorylation (53), a proxy for SHP2 activity (1), and percentage of cells expressing Ki67 (Fig. 6B, S6C) were observed.
Figure 6. Model-predicted relationships are recapitulated in vivo.
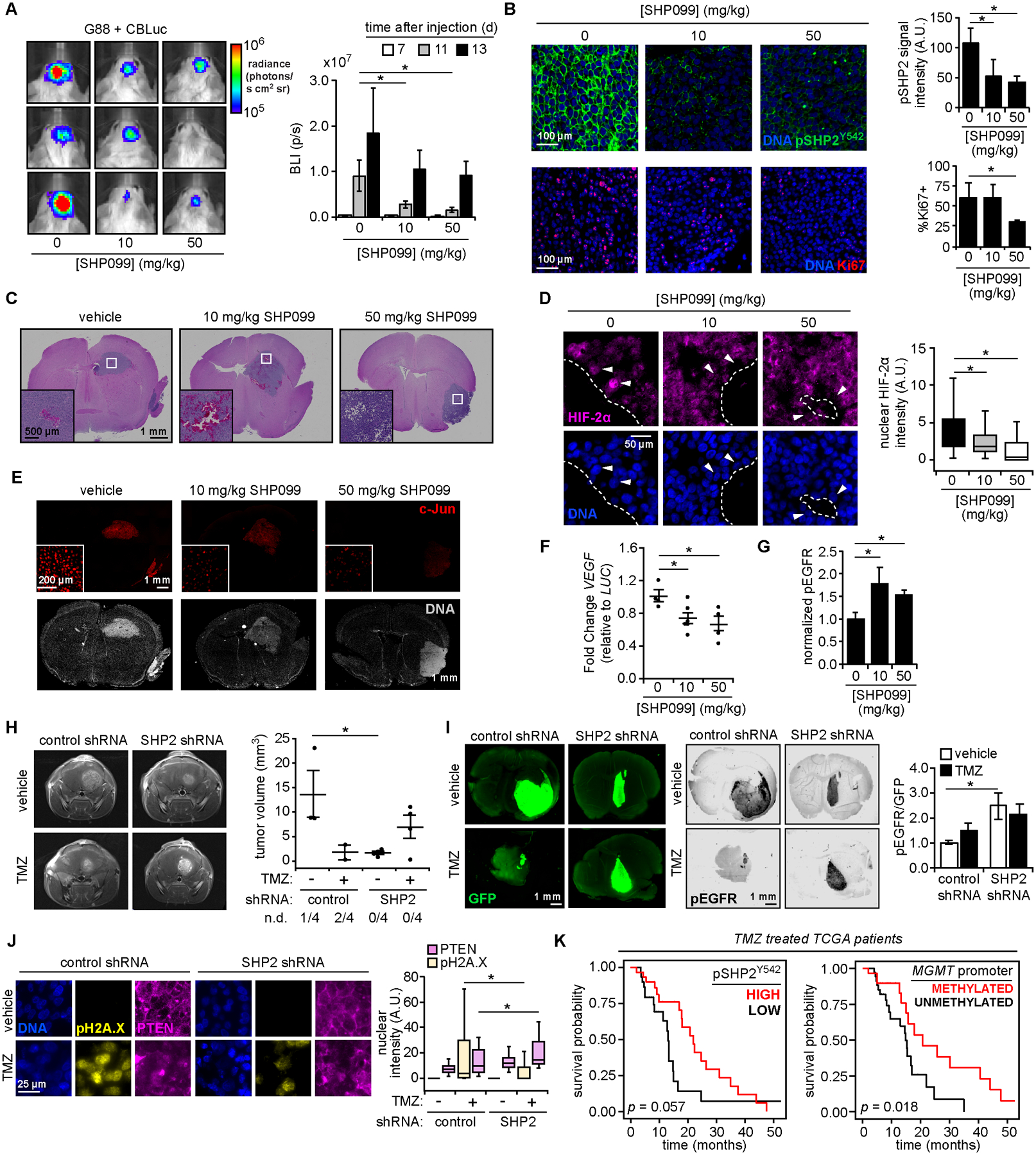
(A) Orthotopic mouse xenograft tumors were generated using G88 cells expressing click beetle luciferase (CBLuc). Mice were treated SHP099 and imaged at indicated times (n = 5 for vehicle and 50 mg/kg SHP099; n = 6 for 10 mg/kg SHP099; see Fig. S6A for timeline). Representative images 11 days (d) after injection are shown, with bioluminescence (photons per second, p/s) plotted for all times. (B) Tumor sections were stained for indicated proteins and DNA. Scale bar = 100 μm. pSHP2 Y542 signal quantification was performed for >3 fields-of-view per tumor. Ki67 positivity was determined for n >1000 cells per tumor. (C) Hematoxylin and eosin stained tumors for mice treated as in (A). Scale bar = 1 mm. White boxes highlight features resembling hypoxic GBM tumors, enlarged as inset. Scale bar = 500 μm. (D) Tumor sections from SHP099-treated mice were stained for HIF-2α and DNA. Nuclear HIF-2α intensities for n >150 cells, across three tumors per group, that neighbor low oxygen regions, as identified in (C), are plotted. Arrows highlight nuclei adjacent to void regions with differences in nuclear HIF-2α. Boundaries between these regions and tumor indicated by dashed line. Scale bar = 50 μm. (E) For tumors described in (C), sections were stained for c-Jun and DNA. Scale bar = 1 mm, inset scale bar = 200 μm. (F) Using tumor-extracted RNA, qRT-PCR was performed with primers for indicated transcripts (n = 4 for vehicle and 50 mg/kg SHP099; n = 5 for 10 mg/kg SHP099). (G) Tumor sections stained with antibodies against pEGFR Y1068 or human COXIV, plus Alexa Fluor 647 secondary antibodies, were imaged on an infrared scanner with densitometry performed. Normalized pEGFR values are shown (n = 3 per group), with representative sections in Fig. S6D. (H) 13 days after orthotopic injection of G88 cells expressing control or SHP2 shRNA, and treatment with 15 mg/kg temozolomide (TMZ) or vehicle as described in Fig. S6E (n = 5 for vehicle groups, n = 6 for TMZ groups), MRI was performed (n = 4 mice per group for MRI). Tumor volumes are plotted, with number of non-detected (n.d.) tumors indicated. See Fig. S6F for complete MRI data. (I) Tumor sections from panel (H) stained with antibodies against GFP and pEGFR, plus Alexa Fluor 647 secondary antibodies, were scanned with an infrared scanner, with densitometry. Normalized pEGFR values are shown (n = 3 per group), with representative sections. (J) Tumor sections were stained for indicated proteins and DNA. Scale bar = 25 μm. Quantified nuclear pH2A.X and PTEN intensities for n >100 cells across three tumors per group are plotted. (K) TCGA data were analyzed to calculate survival probability versus time for GBM patients treated with temozolomide, classified by pSHP2 Y542 RPPA levels (stratified by patients with n = 30 highest or lowest signal) or MGMT-promoter methylation status (n = 29 methylated, n = 34 unmethylated). P-values from log-rank test for survival comparisons are shown. Throughout the panels, representative images are shown, and error bars indicate mean ± s.e.m for indicated number of replicates. For box plots, median, first/third quartiles, and minimum/maximum values are displayed for indicated cell numbers across replicates. Statistical comparisons were made by Tukey’s post-hoc testing following one-way ANOVA (panels A, B, D, F, G, J) or Tukey’s post-hoc testing following two-way ANOVA (panel H). * p < 0.05 for indicated comparisons.
To interrogate relationships between SHP2 and HIF, we identified tissue void regions resembling necrotic zones found in human GBM (54) that contained most HIF-2α-positive nuclei (Fig. 6C, 6D). Nuclear HIF-2α expression was decreased in tumors treated with SHP099 (Fig. 6D). Consistent with in vitro observations, SHP099 reduced nuclear c-Jun abundance (Fig. 6E) and VEGF transcripts (Fig. 6F) and elevated EGFR phosphorylation (Fig. 6G, S6D).
To investigate SHP2 effects on temozolomide response, G88 cells expressing control or SHP2 shRNA were used in an orthotopic model (daily temozolomide, starting 10 days after implantation; see Fig. S6E). After 3 days of temozolomide, MRI revealed that tumors expressing SHP2 shRNA were smaller but less responsive than controls (Fig. 6H, S6F). Tumor sizes were consistent with overall disease burden (Fig. S6G). In fact, mice with SHP2 shRNA tumors treated with temozolomide lost as much weight as untreated controls. Ex vivo analyses supported MRI-based comparisons of tumor size (Fig. 6I, S6H, S6I), and revealed decreased ERK phosphorylation with SHP2 knockdown (Fig. S6J). Consistent with the proposed mechanism of temozolomide resistance, SHP2 knockdown tumors displayed elevated EGFR phosphorylation, elevated nuclear PTEN, and decreased H2A.X phosphorylation in response to temozolomide (Fig. 6I, 6J). Tumor sizes were consistent with Ki67 staining (Fig. S6K).
To further explore SHP2 regulation of DNA damage response, we analyzed TCGA data by pairing survival outcomes with phosphorylated SHP2 (Y542) measurements from reverse phase protein arrays (55). While SHP2 phosphorylation was not prognostic for all GBM patients (p = 0.22 by log-rank test comparing survival probabilities; Fig. S6L), a potential survival difference did emerge for patients treated with temozolomide (p = 0.057; Fig. 6K). Temozolomide-treated patients whose tumors exhibited high (above median) SHP2 phosphorylation had a median survival of 21.9 months, compared to 13.0 months for those with low (below median) SHP2 phosphorylation. Furthermore, stratifying tumors elevated SHP2 Y542 phosphorylation corresponded to elevated c-Jun phosphorylation and expression of CD31 (tumor vascularization marker) and VEGF (Fig. S6M), consistent with other model inferences described in Results.
Discussion
The data-driven model developed here predicted multiple SHP2-dependent signaling events that influence SHP2 inhibitor efficacy in GBM (Fig. 7). Predictions developed from a single cell line were validated in multiple GBM cell lines, with differences among cell lines reinforcing model-predicted relationships (e.g., effects of PTEN expression). While SHP2 inhibitor responses can be studied purely experimentally [e.g., (53)], data-driven models, which have been used to study effects of kinase inhibitors (20,56), may be particularly useful for understanding multivariate effects of phosphatase inhibitors, given the substrate promiscuity of phosphatases (57). Such models can also directly inform clinical trial design, given that our findings can be readily incorporated into single-agent and combination trials for GBM patients. Whether inferences from our study are applicable outside the GBM context remains to be seen.
Figure 7. SHP2 inhibitor efficacy depends on a network of SHP2-regulated signaling responses.
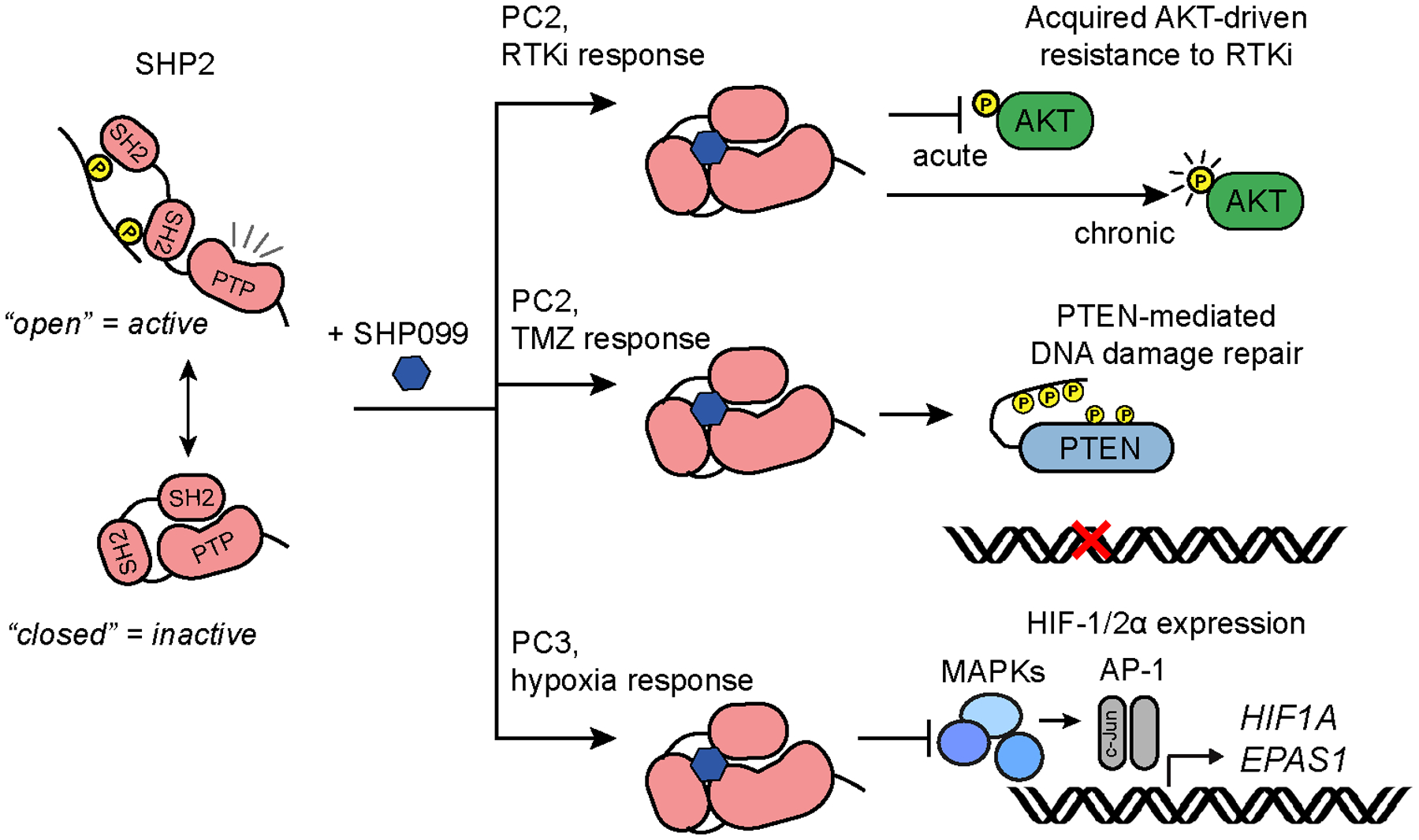
Pursuit of three inferences from the model developed here identified three unanticipated consequences of SHP2 inhibition, summarized in the schematic.
One limitation revealed by pursuing model inference (I) was the model’s inability to predict qualitatively different effects of short- versus long-term SHP2 inhibition on AKT signaling. Such limitations can potentially be overcome by tracking both signaling and phenotypes over time. The substantial increase in data dimensionality of such problems can be efficiently addressed by tensor PLSR (58). It may also eventually be possible to integrate data-driven models with mechanistic models of SHP2 regulation (59) to predict the impact of targeting proteins involved in SHP2 regulation on cancer phenotypes.
The finding that SHP2 inhibition impaired HIF expression in GSC-derived orthotopic tumors has important implications given the roles of HIFs in GSC self-renewal and tumorigenic potential (60) and ongoing efforts to therapeutically target HIFs (e.g., trial of HIF-2α inhibitor PT2977, NCT02974738). If SHP2 broadly controls HIF expression through effects of multiple MAPKs on AP-1 complex members, as our data suggest, SHP2 inhibitors may be better candidates for targeting hypoxia response than individual kinase inhibitors [NCT01497444, (61)]. While our studies focused on SHP2 control of AP-1, non-transcriptional mechanisms may also be relevant. Indeed, MAPKs can phosphorylate HIFs in ways that impact protein stability and transcriptional activity (62,63). Ultimately, for inhibitors of SHP2 and HIFs, evaluation in immunocompetent GBM models will be necessary as both proteins have immune cell roles (64,65). In fact, an ongoing Phase Ib clinical trial of SHP2 inhibitor TNO155 in solid tumor malignancies includes combination treatment with PD1 monoclonal antibody spartalizumab (NCT04000529).
The unanticipated effects of SHP2 inhibition on GBM response to temozolomide suggest a potential need for tumor molecular profiling in deploying SHP2 inhibitors. Indeed, our findings could translate directly to a clinical trial combining an SHP2 inhibitor with temozolomide in GBM patients, with PTEN status indicated by genetic profiling. Results of data-driven studies such as these will be increasingly applicable clinically, given growing availability of genetic profiling of GBMs and other cancers through Foundation Medicine and other providers. The effects we observed appeared independent of other indicators of temozolomide response, such as MGMT promoter methylation, based on characteristics of the cells studied (Fig. S5C, Table S1). Moreover, SHP2 inhibitors may broadly antagonize DNA damaging therapies, given that PTEN-dependent DNA damage repair in GBM was originally identified with ionizing radiation (49). Interestingly, SHP099 plus temozolomide has been reported to be an effective combination in mouse orthotopic GBM xenografts (14). One potential explanation is the unknown PTEN-status of the tumor model. Ultimately, further preclinical evaluation of combination therapies involving SHP2 inhibitors, potentially using staggered or periodic dosing to minimize undesirable consequences of simultaneous or continuous treatments, will be needed to maximize therapeutic benefit of SHP2 inhibitors for GBM patients.
Supplementary Material
Statement of Significance.
Findings demonstrate that allosteric SHP2 inhibitors have multivariate and context-dependent effects in glioblastoma that may make them useful components of some combination therapies but not others.
Acknowledgments
We thank UVA core facility staff for technical assistance, Dr. Frank Furnari for the pY240-PTEN antibody, Sara Adair for technical support, and Dr. Marieke Jones for assistance with statistics. This work was supported by NSF CBET 1511853 (M.J. Lazzara), American Cancer Society RSG-15-010-01-CDD (M.J. Lazzara), NSF GRFP DGE-1321851 (E.K. Day), and UVA Cancer Center Support Grant from the National Cancer Institute (P30CA044579).
Grant support:
This work was supported by NSF CBET Grant No. 1511853 (M.J. Lazzara), American Cancer Society Research Scholar Grant No. RSG-15-010-01-CDD (M.J. Lazzara), NSF GRFP Grant No. DGE-1321851 (E.K. Day), and UVA Cancer Center Support Grant from the National Cancer Institute (P30CA044579).
Footnotes
Potential conflicts of interest: Since the first submission of this manuscript, Evan Day began a postdoctoral position at Novartis (Cambridge, MA). SHP099 was used in this study and was developed by Novartis.
References
- 1.Araki T, Nawa H, Neel BG. Tyrosyl phosphorylation of Shp2 is required for normal ERK activation in response to some, but not all, growth factors. J Biol Chem 2003;278:41677–84 [DOI] [PubMed] [Google Scholar]
- 2.Buonato JM, Lan IS, Lazzara MJ. EGF augments TGFbeta-induced epithelial-mesenchymal transition by promoting SHP2 binding to GAB1. J Cell Sci 2015;128:3898–909 [DOI] [PubMed] [Google Scholar]
- 3.Chan G, Kalaitzidis D, Neel BG. The tyrosine phosphatase Shp2 (PTPN11) in cancer. Cancer Metastasis Rev 2008;27:179–92 [DOI] [PubMed] [Google Scholar]
- 4.Mullard A Phosphatases start shedding their stigma of undruggability. Nat Rev Drug Discov 2018;17:847–9 [DOI] [PubMed] [Google Scholar]
- 5.Chen YN, LaMarche MJ, Chan HM, Fekkes P, Garcia-Fortanet J, Acker MG, et al. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 2016;535:148–52 [DOI] [PubMed] [Google Scholar]
- 6.Fedele C, Ran H, Diskin B, Wei W, Jen J, Geer MJ, et al. SHP2 Inhibition Prevents Adaptive Resistance to MEK Inhibitors in Multiple Cancer Models. Cancer Discov 2018;8:1237–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roccograndi L, Binder ZA, Zhang L, Aceto N, Zhang Z, Bentires-Alj M, et al. SHP2 regulates proliferation and tumorigenicity of glioma stem cells. J Neurooncol 2017;135:487–96 [DOI] [PubMed] [Google Scholar]
- 8.Sturla LM, Zinn PO, Ng K, Nitta M, Kozono D, Chen CC, et al. Src homology domain-containing phosphatase 2 suppresses cellular senescence in glioblastoma. Br J Cancer 2011;105:1235–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhan Y, O’Rourke DM. SHP-2-dependent mitogen-activated protein kinase activation regulates EGFRvIII but not wild-type epidermal growth factor receptor phosphorylation and glioblastoma cell survival. Cancer Res 2004;64:8292–8 [DOI] [PubMed] [Google Scholar]
- 10.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005;352:987–96 [DOI] [PubMed] [Google Scholar]
- 11.Snuderl M, Fazlollahi L, Le LP, Nitta M, Zhelyazkova BH, Davidson CJ, et al. Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell 2011;20:810–7 [DOI] [PubMed] [Google Scholar]
- 12.Szerlip NJ, Pedraza A, Chakravarty D, Azim M, McGuire J, Fang Y, et al. Intratumoral heterogeneity of receptor tyrosine kinases EGFR and PDGFRA amplification in glioblastoma defines subpopulations with distinct growth factor response. Proc Natl Acad Sci U S A 2012;109:3041–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Furnari FB, Cloughesy TF, Cavenee WK, Mischel PS. Heterogeneity of epidermal growth factor receptor signalling networks in glioblastoma. Nat Rev Cancer 2015;15:302–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sang Y, Hou Y, Cheng R, Zheng L, Alvarez AA, Hu B, et al. Targeting PDGFRalpha-activated glioblastoma through specific inhibition of SHP-2-mediated signaling. Neuro Oncol 2019;21:1423–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Furcht CM, Buonato JM, Skuli N, Mathew LK, Munoz Rojas AR, Simon MC, et al. Multivariate signaling regulation by SHP2 differentially controls proliferation and therapeutic response in glioma cells. J Cell Sci 2014;127:3555–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bard-Chapeau EA, Li S, Ding J, Zhang SS, Zhu HH, Princen F, et al. Ptpn11/Shp2 acts as a tumor suppressor in hepatocellular carcinogenesis. Cancer Cell 2011;19:629–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Han T, Xiang DM, Sun W, Liu N, Sun HL, Wen W, et al. PTPN11/Shp2 overexpression enhances liver cancer progression and predicts poor prognosis of patients. J Hepatol 2015;63:651–60 [DOI] [PubMed] [Google Scholar]
- 18.Kumar N, Wolf-Yadlin A, White FM, Lauffenburger DA. Modeling HER2 effects on cell behavior from mass spectrometry phosphotyrosine data. PLoS Comput Biol 2007;3:e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heijink AM, Everts M, Honeywell ME, Richards R, Kok YP, de Vries EGE, et al. Modeling of Cisplatin-Induced Signaling Dynamics in Triple-Negative Breast Cancer Cells Reveals Mediators of Sensitivity. Cell Rep 2019;28:2345–57 e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee MJ, Ye AS, Gardino AK, Heijink AM, Sorger PK, MacBeath G, et al. Sequential application of anticancer drugs enhances cell death by rewiring apoptotic signaling networks. Cell 2012;149:780–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Day EK, Sosale NG, Xiao A, Zhong Q, Purow B, Lazzara MJ. Glioblastoma Cell Resistance to EGFR and MET Inhibition Can Be Overcome via Blockade of FGFR-SPRY2 Bypass Signaling. Cell Rep 2020;30:3383–96 e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Walsh AM, Kapoor GS, Buonato JM, Mathew LK, Bi Y, Davuluri RV, et al. Sprouty2 Drives Drug Resistance and Proliferation in Glioblastoma. Mol Cancer Res 2015;13:1227–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Law HC, Lagundzin D, Clement EJ, Qiao F, Wagner ZS, Krieger KL, et al. The Proteomic Landscape of Pancreatic Ductal Adenocarcinoma Liver Metastases Identifies Molecular Subtypes and Associations with Clinical Response. Clin Cancer Res 2020;26:1065–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim HD, Meyer AS, Wagner JP, Alford SK, Wells A, Gertler FB, et al. Signaling network state predicts twist-mediated effects on breast cell migration across diverse growth factor contexts. Mol Cell Proteomics 2011;10:M111 008433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cunnick JM, Meng S, Ren Y, Desponts C, Wang HG, Djeu JY, et al. Regulation of the mitogen-activated protein kinase signaling pathway by SHP2. J Biol Chem 2002;277:9498–504 [DOI] [PubMed] [Google Scholar]
- 26.Zhang SQ, Tsiaras WG, Araki T, Wen G, Minichiello L, Klein R, et al. Receptor-specific regulation of phosphatidylinositol 3’-kinase activation by the protein tyrosine phosphatase Shp2. Mol Cell Biol 2002;22:4062–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu D, Qu CK. Protein tyrosine phosphatases in the JAK/STAT pathway. Front Biosci 2008;13:4925–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Agazie YM, Hayman MJ. Molecular mechanism for a role of SHP2 in epidermal growth factor receptor signaling. Mol Cell Biol 2003;23:7875–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ziegler DS, Wright RD, Kesari S, Lemieux ME, Tran MA, Jain M, et al. Resistance of human glioblastoma multiforme cells to growth factor inhibitors is overcome by blockade of inhibitor of apoptosis proteins. J Clin Invest 2008;118:3109–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Caporali S, Falcinelli S, Starace G, Russo MT, Bonmassar E, Jiricny J, et al. DNA damage induced by temozolomide signals to both ATM and ATR: role of the mismatch repair system. Mol Pharmacol 2004;66:478–91 [DOI] [PubMed] [Google Scholar]
- 31.Worley B, Powers R. Multivariate Analysis in Metabolomics. Curr Metabolomics 2013;1:92–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seidel S, Garvalov BK, Wirta V, von Stechow L, Schanzer A, Meletis K, et al. A hypoxic niche regulates glioblastoma stem cells through hypoxia inducible factor 2 alpha. Brain 2010;133:983–95 [DOI] [PubMed] [Google Scholar]
- 33.Heddleston JM, Li Z, McLendon RE, Hjelmeland AB, Rich JN. The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle 2009;8:3274–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sang N, Stiehl DP, Bohensky J, Leshchinsky I, Srinivas V, Caro J. MAPK signaling up-regulates the activity of hypoxia-inducible factors by its effects on p300. J Biol Chem 2003;278:14013–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhan L, Yan H, Zhou H, Sun W, Hou Q, Xu E. Hypoxic preconditioning attenuates neuronal cell death by preventing MEK/ERK signaling pathway activation after transient global cerebral ischemia in adult rats. Mol Neurobiol 2013;48:109–19 [DOI] [PubMed] [Google Scholar]
- 36.Huelsemann MF, Patz M, Beckmann L, Brinkmann K, Otto T, Fandrey J, et al. Hypoxia-induced p38 MAPK activation reduces Mcl-1 expression and facilitates sensitivity towards BH3 mimetics in chronic lymphocytic leukemia. Leukemia 2015;29:981–4 [DOI] [PubMed] [Google Scholar]
- 37.Pulverer BJ, Kyriakis JM, Avruch J, Nikolakaki E, Woodgett JR. Phosphorylation of c-jun mediated by MAP kinases. Nature 1991;353:670–4 [DOI] [PubMed] [Google Scholar]
- 38.Humar M, Loop T, Schmidt R, Hoetzel A, Roesslein M, Andriopoulos N, et al. The mitogen-activated protein kinase p38 regulates activator protein 1 by direct phosphorylation of c-Jun. Int J Biochem Cell Biol 2007;39:2278–88 [DOI] [PubMed] [Google Scholar]
- 39.Mathew LK, Skuli N, Mucaj V, Lee SS, Zinn PO, Sathyan P, et al. miR-218 opposes a critical RTK-HIF pathway in mesenchymal glioblastoma. Proc Natl Acad Sci U S A 2014;111:291–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Henklova P, Vrzal R, Papouskova B, Bednar P, Jancova P, Anzenbacherova E, et al. SB203580, a pharmacological inhibitor of p38 MAP kinase transduction pathway activates ERK and JNK MAP kinases in primary cultures of human hepatocytes. Eur J Pharmacol 2008;593:16–23 [DOI] [PubMed] [Google Scholar]
- 41.Hess J, Angel P, Schorpp-Kistner M. AP-1 subunits: quarrel and harmony among siblings. J Cell Sci 2004;117:5965–73 [DOI] [PubMed] [Google Scholar]
- 42.Karin M The regulation of AP-1 activity by mitogen-activated protein kinases. Philos Trans R Soc Lond B Biol Sci 1996;351:127–34 [DOI] [PubMed] [Google Scholar]
- 43.Lee YR, Chen M, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor: new modes and prospects. Nat Rev Mol Cell Biol 2018;19:547–62 [DOI] [PubMed] [Google Scholar]
- 44.Chang CJ, Mulholland DJ, Valamehr B, Mosessian S, Sellers WR, Wu H. PTEN nuclear localization is regulated by oxidative stress and mediates p53-dependent tumor suppression. Mol Cell Biol 2008;28:3281–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Misra S, Mukherjee A, Karmakar P. Phosphorylation of PTEN at STT motif is associated with DNA damage response. Mutat Res 2014;770:112–9 [DOI] [PubMed] [Google Scholar]
- 46.Vazquez F, Matsuoka S, Sellers WR, Yanagida T, Ueda M, Devreotes PN. Tumor suppressor PTEN acts through dynamic interaction with the plasma membrane. Proc Natl Acad Sci U S A 2006;103:3633–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.He J, Kang X, Yin Y, Chao KS, Shen WH. PTEN regulates DNA replication progression and stalled fork recovery. Nat Commun 2015;6:7620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Furnari FB, Lin H, Huang HS, Cavenee WK. Growth suppression of glioma cells by PTEN requires a functional phosphatase catalytic domain. Proc Natl Acad Sci U S A 1997;94:12479–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ma J, Benitez JA, Li J, Miki S, Ponte de Albuquerque C, Galatro T, et al. Inhibition of Nuclear PTEN Tyrosine Phosphorylation Enhances Glioma Radiation Sensitivity through Attenuated DNA Repair. Cancer Cell 2019;36:690–1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Georgescu MM, Kirsch KH, Kaloudis P, Yang H, Pavletich NP, Hanafusa H. Stabilization and productive positioning roles of the C2 domain of PTEN tumor suppressor. Cancer Res 2000;60:7033–8 [PubMed] [Google Scholar]
- 51.Obenauer JC, Cantley LC, Yaffe MB. Scansite 2.0: Proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res 2003;31:3635–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ahmed Z, Lin CC, Suen KM, Melo FA, Levitt JA, Suhling K, et al. Grb2 controls phosphorylation of FGFR2 by inhibiting receptor kinase and Shp2 phosphatase activity. J Cell Biol 2013;200:493–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ahmed TA, Adamopoulos C, Karoulia Z, Wu X, Sachidanandam R, Aaronson SA, et al. SHP2 Drives Adaptive Resistance to ERK Signaling Inhibition in Molecularly Defined Subsets of ERK-Dependent Tumors. Cell Rep 2019;26:65–78 e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rong Y, Durden DL, Van Meir EG, Brat DJ. ‘Pseudopalisading’ necrosis in glioblastoma: a familiar morphologic feature that links vascular pathology, hypoxia, and angiogenesis. J Neuropathol Exp Neurol 2006;65:529–39 [DOI] [PubMed] [Google Scholar]
- 55.Li J, Lu Y, Akbani R, Ju Z, Roebuck PL, Liu W, et al. TCPA: a resource for cancer functional proteomics data. Nat Methods 2013;10:1046–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Niepel M, Hafner M, Pace EA, Chung M, Chai DH, Zhou L, et al. Profiles of Basal and stimulated receptor signaling networks predict drug response in breast cancer lines. Sci Signal 2013;6:ra84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sacco F, Perfetto L, Castagnoli L, Cesareni G. The human phosphatase interactome: An intricate family portrait. FEBS Lett 2012;586:2732–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chitforoushzadeh Z, Ye Z, Sheng Z, LaRue S, Fry RC, Lauffenburger DA, et al. TNF-insulin crosstalk at the transcription factor GATA6 is revealed by a model that links signaling and transcriptomic data tensors. Sci Signal 2016;9:ra59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Furcht CM, Buonato JM, Lazzara MJ. EGFR-activated Src family kinases maintain GAB1-SHP2 complexes distal from EGFR. Sci Signal 2015;8:ra46. [DOI] [PubMed] [Google Scholar]
- 60.Li Z, Bao S, Wu Q, Wang H, Eyler C, Sathornsumetee S, et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 2009;15:501–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu LP, Ho RL, Chen GG, Lai PB. Sorafenib inhibits hypoxia-inducible factor-1alpha synthesis: implications for antiangiogenic activity in hepatocellular carcinoma. Clin Cancer Res 2012;18:5662–71 [DOI] [PubMed] [Google Scholar]
- 62.Mylonis I, Chachami G, Samiotaki M, Panayotou G, Paraskeva E, Kalousi A, et al. Identification of MAPK phosphorylation sites and their role in the localization and activity of hypoxia-inducible factor-1alpha. J Biol Chem 2006;281:33095–106 [DOI] [PubMed] [Google Scholar]
- 63.Kwon SJ, Song JJ, Lee YJ. Signal pathway of hypoxia-inducible factor-1alpha phosphorylation and its interaction with von Hippel-Lindau tumor suppressor protein during ischemia in MiaPaCa-2 pancreatic cancer cells. Clin Cancer Res 2005;11:7607–13 [DOI] [PubMed] [Google Scholar]
- 64.Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med 2012;209:1201–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Palazon A, Goldrath AW, Nizet V, Johnson RS. HIF transcription factors, inflammation, and immunity. Immunity 2014;41:518–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.