

Abstract
Gene therapy is making a comeback. With its twin promise of targeting disease etiology and long-term correction, gene-based therapies (defined here as all forms of genome-manipulation) are particularly appealing for neurodegenerative diseases, where conventional pharmacologic approaches have been largely disappointing. The recent success of a viral-vector based gene therapy in spinal muscular atrophy (SMA) – promoting survival and motor function with a single intravenous injection – offers a paradigm for such therapeutic intervention, and a platform to build upon. Though challenges remain, the newfound optimism largely stems from advances in the development of viral vectors that can diffusely deliver genes throughout the CNS, as well as genome-engineering tools that can manipulate disease pathways in ways that were previously impossible. Surely SMA cannot be the only neurodegenerative disease amenable to gene therapy, and one can imagine a future where a clinician’s toolkit will include gene-based therapeutics. The goal of this review is to highlight advances in the development and application of gene-based therapies for neurodegenerative diseases and offer a prospective look into this emerging arena.
Over two decades have passed since the death of Jesse Gelsinger – a relatively healthy 18 year-old volunteer for a gene therapy trial – likely due to a fatal immune response triggered by adenovirus vectors1. The intervening years have seen public outcry, soul-searching, regulatory reforms, and even professional shunning of gene therapy researchers and advocates. Yet, work in the arena continued, including strategies to mitigate the viral vector-induced activation of immune responses that ultimately took Jesse’s life. More recently, advances in gene-delivery and gene-manipulation tools have re-energized the field (see timeline in Fig. 1a). A paradigm for such therapeutic intervention is AVXS-101 (onasemnogene abeparvovec), a gene therapy for SMA, where children destined to wheelchairs and early death show remarkable improvements in survival and motor function after a single intravenous injection of adeno-associated virus 9 (AAV9) carrying the SMN1 gene2. However, significant challenges need to be resolved before gene therapy for neurodegenerative diseases becomes widely accepted. In this review, we first discuss promises and challenges surrounding gene-based therapies for neurodegenerative diseases, and then summarize the repertoire of gene-manipulation tools available today. The final section gives examples of ongoing and pending gene-based clinical trials in neurodegeneration and discusses promising experimental strategies.
Figure 1: Timeline of marquee events in the gene therapy field.

a) Milestones in the development of gene therapy tools.
b) Timeline of key clinical trials in gene-based therapies for neurodegenerative diseases.
I). Promises and challenges of gene-based therapies for neurodegenerative diseases
The promise of gene therapy has always rested on two pillars: the ability to target etiology, and the capacity for “achieving a permanent correction”3. A single, long-lasting intervention (“one and done”) is particularly appealing for diseases affecting the CNS, because unlike other organs where repeated doses can readily achieve effective therapeutic concentrations, most peripherally administered agents are unable to cross the blood brain barrier, or only do so poorly. For instance, the human CSF to serum ratio of BAN2401 – a monoclonal antibody with some promise in Alzheimer’s disease (AD) – is only ~ 0.04%4. Nevertheless, small molecules and antibodies continue to be the mainstay of clinical trials in neurodegenerative diseases, and only a handful of gene therapy candidates have been tried (see timeline in Fig. 1b). Challenges surrounding gene-based therapies in the CNS are briefly outlined next (Table 1), and the reader is referred to recent reviews for more in-depth discussions5,6.
Table 1.
Comparing gene-therapy strategies for neurodegenerative diseases
| ASOs | AAVs/other viruses | Small molecules | DNA/RNA editing | |
|---|---|---|---|---|
| Risk/toxicity | • Intrathecal/invasive local CNS delivery is necessary. |
• Hepatotoxicity and dorsal root ganglion pathology after systemic delivery (particularly with high doses). • May need intrathecal/invasive local CNS delivery, though intravenous route is feasible. • Insertional mutagenesis. |
• Possible altered splicing/expression of off-target genes (e.g. high dose of SMN2 splicing modulator risdiplam affects FOXM1 splicing, leading to a protein product that blocks cell division). | • Irreversibility of DNA editing • Undesired on-target editing possible (large deletions and chromosomal rearrangements). • Possible immunogenicity from bacterial nucleases (Cas-proteins). |
| Off-target effects | Binding to off-target mRNA due to complete or partial complementarity. | Ectopic expression in peripheral tissues after systemic delivery. | Non-specific binding to off-target mRNA possible. | Binding to off-target site due to homologous sequence or mismatch tolerance. |
| Persistence of intervention | • Repeated administration necessary for persistent RNA modulation. | • Long-term transgene expression of AAV in postmitotic cells. • Long-term transgene expression of lentivirus in mitotically active and inactive cells. • Pre-existing neutralizing antibodies may reduce efficacy. • Immunogenicity of viral capsids and transgene products may affect long-term gene expression and repeated treatments. |
• Repeated oral administration necessary, typically low brain availability. | • Persistent genome manipulation by DNA-editing tools. |
Since correction of a single gene in a monogenic illness is the traditional view of gene therapy, its feasibility for illnesses with complex etiologies, such as neurodegenerative diseases, is sometimes questioned. However, almost all therapeutic strategies – gene-based or not – are focused on singular targets; for instance, almost all AD trials have targeted either amyloid-beta (Aβ) or tau7. Moreover, by modulating gene/protein levels or other physiologic properties of endogenous proteins, gene-based therapies are being applied to sporadic neurodegenerative diseases, as discussed later. The permanency of some gene-based therapies is both an advantage and risk. Once genomic modifications are installed in the DNA, they would be irreversible – particularly in post-mitotic neurons. Each gene therapy will also come with its own caveats, for example off-target effects with DNA/RNA editing, and the persistence of exogenous proteins such as bacterial Cas9. Careful consideration of risk-benefit will likely determine clinical application, as with other relatively permanent interventions such as surgical procedures.
The safe, efficient, and selective delivery of gene products into the CNS remains a challenge. Though non-viral delivery strategies such as nanoparticles and ribonucleoprotein complexes are being explored6, so far, their applicability to the CNS remains uncertain. Adeno-associated viral vectors (AAVs) are a clear frontrunner in this arena, with strong neuronal tropism, widespread distribution, and desirable safety profiles in many species, including non-human primates (NHPs)5 – though there are caveats, as discussed next. Development of engineered AAV-capsids to enhance neurotropism is also a rapidly emerging arena8. Many CNS gene therapies will require a sustained delivery of the exogenous transgene throughout life, and AAVs also seem suited for this. Unlike mitotically active cells where transduced AAVs are gradually lost because they fail to integrate into the host genome, AAV expression can persist for decades in postmitotic cells like neurons9, though the underlying mechanisms and extent of this persistence is not clear. However, our ability to detect the transgene expression in living patients is limited due to the lack of biomarkers and difficulties related to accessibility of brain-tissue.
Although AAVs have been used in over 200 human studies involving thousands of patients10 and are thought to be generally safe, several caveats have also emerged, mostly related to activation host immune responses by viral proteins. First, capsid proteins of recombinant AAV particles resemble capsids of natural viruses that humans are routinely exposed to, resulting in neutralizing host antibodies that can attenuate transduction efficiency11. In the first AVXS-101 trial, 16 patients were screened and one was excluded due to elevated anti-AAV9 antibody titer (>1:50)2. Exogenous AAV capsids can also activate cytotoxic T lymphocytes (CTLs) that can eliminate the transduced cells12 or generate neutralizing antibodies by triggering humoral immunity; precluding repeated administration13. Certain AAV serotypes like AAV1 and AAV5 have a high tropism for antigen-presenting cells and can trigger a greater adaptive immune response14. Another concern of AAVs relates to the activation of Toll-like receptors (TLRs) that sense the capsid/vector genome and induce innate immunity responses, producing pro-inflammatory cytokines15. If the transgene introduced by the gene therapy encodes a foreign protein – such as a non-human protein or a replacement protein in a null genomic background – both neutralizing antibodies and CTLs may be generated16. In a recent AAV-based gene therapy trial of myotubular myopathy, two out of three patients administered with a high-dose died due to severe hepatotoxicity, though encouraging results were seen in patients treated with a lower dose17. Another worrisome aspect highlighted by recent studies is that administration of AAVs by intrathecal or intravenous injections can cause dorsal root ganglion pathology, seemingly independent of immune responses18. The complex relationship between AAV vectors and host immune responses is being intensely investigated, along with mitigation strategies19.
II). Toolbox of gene manipulations
The ever expanding palette of genome manipulation tools has been the subject of recent reviews20, and here we will briefly discuss technologies relevant to therapeutics in neurodegenerative diseases (Box 1).
Box 1: Gene editing tools.
Canonical DNA editing
Comprised of two components – nucleases that cleave DNA, and synthetic peptide or RNA sequences that guide the nucleases to specific sites in the genome. Endogenous repair processes are error-prone, leading to disruption of the translational reading frame, nonsense decay of the mRNA, and effective gene knockout, called non-homologous end joining (NHEJ, Fig. 2a). Alternatively, in the presence of an exogenous donor-template, homology dependent repair (HDR) can insert desired sequences or point mutations into the host genome (Fig. 2b); though this is inefficient, particularly in neurons. ZFNs and TALENs use custom designed amino acids to direct nucleases, whereas CRISPR-based systems use synthetic guide RNA sequences (Fig. 2c-e).
Noncanonical DNA editing
Though gene editing typically leads to knockout of the gene/protein, CRISPRs can also be used to alter single-nucleotides and DNA-domains or modulate gene expression. For instance, CRISPR-guided domain-excision can remove pathologic mutations or small DNA stretches, if they lie in regions that are normally exempt from nonsense mRNA decay (Fig. 2f; see text for details). Transcriptional repressors or enhancers attached to catalytically dead Cas9 (dCas9) can be directed to specific genomic sites using guide RNAs, leading to gene inhibition (CRISPRi) or activation (CRISPRa); Figure 3a-b. Single DNA base-pairs in the genome can be altered by base-editing, using deaminase proteins tagged to dCas9, directed to specific sites by guide RNAs (Fig. 3c-d). Prime editing expands base editing by allowing broader transfer of genetic information to the host genome (Fig. 3e).
RNA editing
Unlike DNA-based editing, RNA editing is transient, reducing chances of undesirable long-term consequences, however the need for repeated administration is a drawback. Antisense oligonucleotides (ASOs) are synthetic sequences that bind RNA and influence their maturation in a variety of ways (Fig. 4). Cas13d is an outlier Cas enzyme that binds to and cleaves mRNA in mammalian cells, resulting in gene knockdown without the need for PAM sequences.
Gene expression:
Exogenous introduction of genes into the CNS is the most straightforward approach for gene therapy. Delivered genes can restore the loss of gene function due to pathologic mutations, as in the case of SMN1 gene for SMA2. Alternatively, genes can deliver neurotrophic factors to promote neuronal survival, as in the case of AD and PD; or metabolic enzymes to restore an imbalance of neurotransmitters, as in the case of PD – detailed later in this review.
DNA editing:
DNA editing tools can manipulate gene expression or correct pathogenic mutations, and are starting to enter the clinic21. In general, there are two key elements – DNA-binding domains that recognize specific genomic sequences, and nucleases that generate double-stranded breaks (DSBs). DSBs are repaired by non-homologous end-joining (NHEJ) which is an endogenous error-prone mechanism leading to sequence insertions and deletions (INDELs) in the reading frame that typically cause frameshift mutations and premature termination codons (PTCs), ultimately knocking out the targeted gene (Figure 2a). Alternatively, in the presence of an exogenous template, native homology dependent repair (HDR) mechanisms can be exploited to insert desired sequences or point mutations into the host genome (Figure 2b). The three main DNA-editing nucleases – zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and CRISPR-associated nucleases – are described below.
Figure 2. Mechanisms of genome editing by engineered nucleases.

a) Double-strand breaks (DSBs, arrowhead) in genomic DNA are repaired by non-homologous end joining (NHEJ), that introduces indel mutations (red dash lines), typically leading to a change of the reading frame, pre-mature termination codon (PTC), and disrupted gene function.
b) Alternately, in the presence of donor template, homology-directed repair (HDR) mechanisms lead to precise insertion or modification of DNA (green lines).
c) Zinc finger nucleases (ZFNs) are composed of three to six zinc finger domains (color circles), each recognizing three nucleotides. The zinc finger domains on opposite strands of DNA bring two Fok1 endonuclease domains together, inducing Fok1 dimerization and DSB.
d) Transcription activator-like effector nucleases (TALENs) have 16-20 TALE monomers, each recognizing a single nucleotide. A pair of TALENs induces Fok1 dimerization and DSB.
e) The CRISPR-Cas9 system contains a synthetic guide RNA and the nuclease Cas9. The guide RNA has two domains – a programmable crRNA sequence recognizing the genomic target, and a tracrRNA sequence for Cas9-binding.
f) Location of PTCs determine transcription fate. Only PTCs residing >50-55 nt upstream of the last exon-exon junction will lead to non-sense mediated decay (NMD) and degradation of the mRNA. The last exon is exempt from NMD, and PTCs in this “NMD-insensitive region” should lead to truncations (see article for details).
ZFNs and TALENs:
A ZFN is a fusion protein with two functional domains, a DNA-binding domain composed of three to six zinc fingers – each recognizing three DNA base-pairs in the host genome – and a DNA-cleaving domain of the endonuclease Fok1 (Figure 2c)22. As Fok1 is functional as a dimer, two ZFNs are designed to bind opposite strands of the targeted genomic DNA, which allow the Fok1 domains to dimerize and cleave DNA. TALEN contains a series of transcription activator-like effectors (TALEs) and the Fok1 DNA-cleavage domain (Figure 2d). Each TALE polypeptide comprises 33-34 amino acids, of which residues 12 and 13 recognize a specific DNA-base, allowing targeted DNA-binding by selecting a combination of modular TALEs23.
CRISPR-mediated canonical applications - NHEJ and HDR:
Unlike ZFNs and TALENs that use amino acids to direct the nuclease, the CRISPR system uses a custom-designed guide-RNA to target a Cas9 nuclease to target-specific sites in the host DNA. An additional requirement is the presence of PAM (protospacer adjacent motif) sites that are dispersed throughout the genome24. The guide-RNA for the most commonly used Cas9 – Streptococcus pyogenes Cas9 or SpCas9 – contains a CRISPR RNA (crRNA) that recognizes the genomic target sequence and a trans-activating CRISPR RNA (tracrRNA) that recruits Cas9 (Figure 2e). After Cas9-induced DSBs and repair by NHEJ, INDELs typically alter the reading frame and install a PTC. Typically, mRNAs containing the mutant PTCs are degraded by nonsense-mediated decay (NMD) – an endogenous surveillance mechanism – leading to a loss of the protein. In principle, CRISPR-guided HDR can be used to introduce any desired point mutation or insertions in the native genome via ‘donor-templates’. However, in most cells, HDR-mediated repair is less frequent than NHEJ, limiting recombination. Moreover, HDR naturally occurs during mitotic recombination, limiting application to post-mitotic neurons, though strategies are being developed to improve HDR efficiency in the brain25. One option is to use HDR-editing in an ex-vivo setting, and then transfer the edited cells in vivo, a bona-fide gene therapy option in sickle cell disease26. Although studies have explored stem cell transplant in neurodegenerative diseases27, the practicality of this approach seems limited.
CRISPR-mediated domain-excision:
Pathologic mutations within introns can be excised by designing two guide-RNAs on either side of the mutation – thereby removing the aberrant nucleotide. This strategy is being used in a Phase 1/2 clinical trial for Leber congenital amaurosis 10 (LCA10, NCT03872479) – an inherited retinal degenerative disease28. In LCA10, an intronic A to G mutation in the CEP290 mRNA creates a splice-donor site – adding an extra exon; and two guide-RNAs on either side of the mutation excises this defect, restoring normal mRNA splicing. A single AAV5 vector delivers the two gRNAs and a smaller Staphylococcus aureus Cas9 (SaCas9) into the eye by subretinal injections, making this the first CRISPR-based trial in humans21.
Instead of eliminating protein translation by triggering NMD of the PTC-containing mRNA, CRISPR-editing can also be used to selectively terminate translation at the PTC-site, effectively resulting in a truncated protein (Figure 2f). This ‘domain-excision’ strategy can be used to eliminate small pathogenic stretches – while preserving the majority of the protein and ensuing physiologic functions – as recently demonstrated by CRISPR-editing of the last ~ 36 aa of the amyloid precursor protein (APP); relevant to AD29. This strategy leverages transcriptional rules guiding NMD by introducing PTCs in the last exon that is known to be protected from nonsense decay (reviewed in30); also discussed later.
CRISPR-mediated transcriptional modulation:
In CRISPRi (inhibition), a catalytically dead Cas9 (dCas9) or a nickase Cas9 (nCas9) – only capable of generating single-strand breaks – is fused with transcription repressors to inhibit gene transcription (Figure 3a). Repression can also be achieved by enzymes that modify histones and alter chromatin structure by epigenome editing. Since decreasing gene expression is a logical strategy for many neurodegenerative diseases – for instance excessive α-synuclein, APP, tau and Huntingtin has been implicated – this is a promising approach. However, important issues such as safety and efficacy still need to be addressed. CRISPRa (activation) is an analogous strategy to activate endogenous genes by fusing dCas9 or nCas9 to transcription activators or epigenome modulators (Figure 3b). Though these large Cas9-effector proteins in CRISPRi/a poses a challenge for AAV-packaging and clinical development, this can be overcome by using the smaller SaCas931.
Figure 3. Non-canonical CRISPR tools.
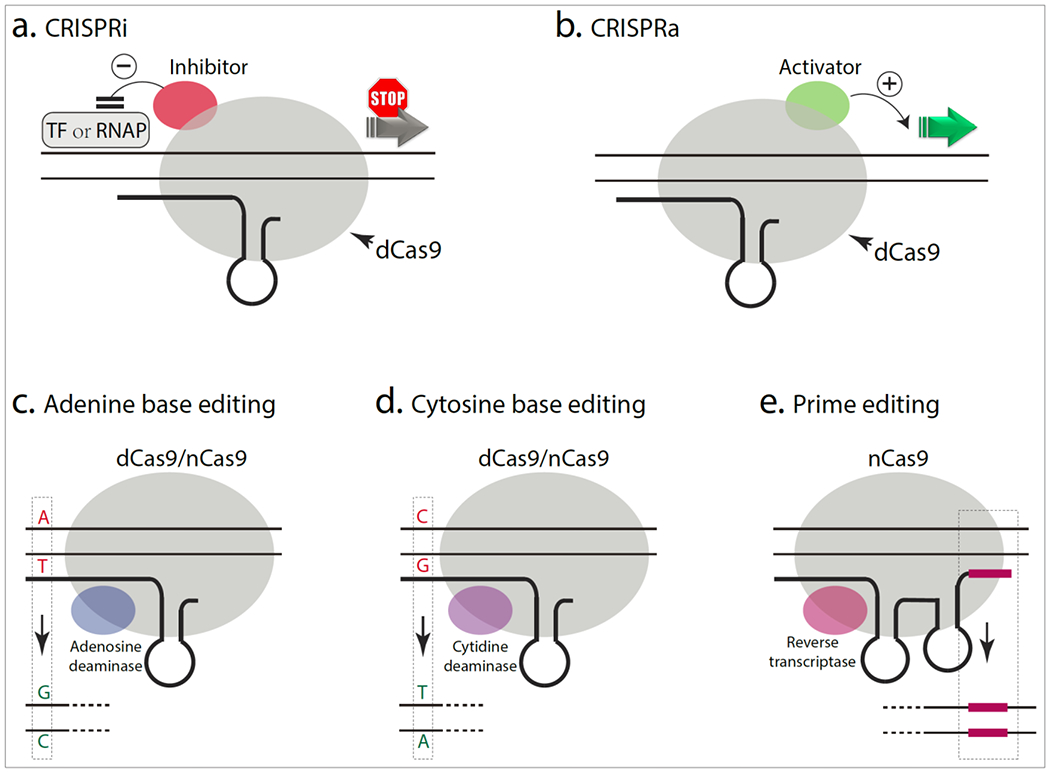
a,b) dCas9, a nuclease-deactivated Cas9, is fused to a transcriptional inhibitor (a) or activator (b) to inhibit or boost transcription, respectively.
c, d) The dCas9 or nCas9 (a Cas9 nickase) is fused to an adenine (c) or cytosine (d) deaminase that changes A:T base-pairs to G:C or C:G base-pairs to T:A, respectively.
e) The nCas9 is fused to a reverse transcriptase that copies the part of prime-editing guide RNA (pegRNA) sequence into the target site (red lines).
CRISPR-mediated base-editing and prime-editing:
Most human pathologic genetic variants – including inherited neurodegenerative diseases – are due to single base-pair point-mutations32. DNA base-editors are chimeric proteins composed of a dCas9 or nCas9 fused to a deaminase protein capable of deaminating and altering cytidine or adenine base-pairs: cytosine base editors (CBEs) mediating conversion of C:G to T:A, and adenine base editors (ABEs) converting A:T to G:C (Figure 3c-d)33,34. The application of base editing is constrained by the availability of suitable PAM sequences and undesirable nucleotide substitutions close to the target site, due to the wide activity-window of deaminases. Newer Cas9 variants with broader PAM compatibility and cytidine deaminase variants with narrower activity-windows have been developed35. Though the large size of base editors makes AAV-packaging for in vivo delivery challenging, a recent study used dual AAVs to deliver split base editors that were subsequently reconstituted in situ by trans-splicing inteins36. Concerning off-target effects – especially unwanted RNA editing – have been reported with base editing37, but newer base editors with reduced off-target activity have also been reported38. Prime editing expands the concept of base editing by allowing broader genomic targeting, more options for genetic remodeling, and increased precision. The prime editing guide-RNA (pegRNA) contains both the conventional DNA targeting sequence, as well as the desired editing sequence to replace targeted genomic DNA nucleotides39. The accompanying nCas9 is fused to a reverse transcriptase that primes the reverse transcription of pegRNA, transferring encoded genetic information from the pegRNA - including insertions, deletions and base conversions – to the targeted genome (Figure 3e).
RNA editing:
RNA-based editing alters gene expression at the transcript level. Since RNA is transient, there is lesser risk of permanent deleterious effects, which is a significant therapeutic appeal of this technology. However, the need for repeated administration – typically by intrathecal injections – is challenging in the clinical setting. Though antisense oligonucleotides (ASOs) were described over 40 years ago, clinical application of RNA-based therapeutics suffered numerous setbacks due to limited efficacy, lack of specificity, and deleterious off target effects40. However, numerous advances in chemical modifications, delivery vehicles, and RNA-editing tools have transformed the field, leading to several therapeutic agents that are in the clinic41. RNA-based editing technologies have been reviewed recently42, and here we discuss two strategies relevant to neurodegenerative diseases.
ASOs:
ASOs are synthetic nucleic acid sequences that bind RNA through Watson-Crick base pairing and influence the maturation of pre-mRNAs to mature mRNAs and proteins. Pre-mRNA splicing is a physiologic process by which introns are removed and exons are ligated, leading to a mature mRNA that is translated into a functional protein. This process is regulated by exonic or intronic splicing enhancers and silencers, where enhancers promote exon inclusion and silencers promote exon exclusion. ASOs can act in several ways to influence these processes; for instance a normally excised exon can be included – called splice inclusion – or exons that would normally be translated can be excluded, called splice exclusion or “exon skipping” (Figure 4).
Figure 4. Mechanisms of antisense oligonucleotides (ASOs).
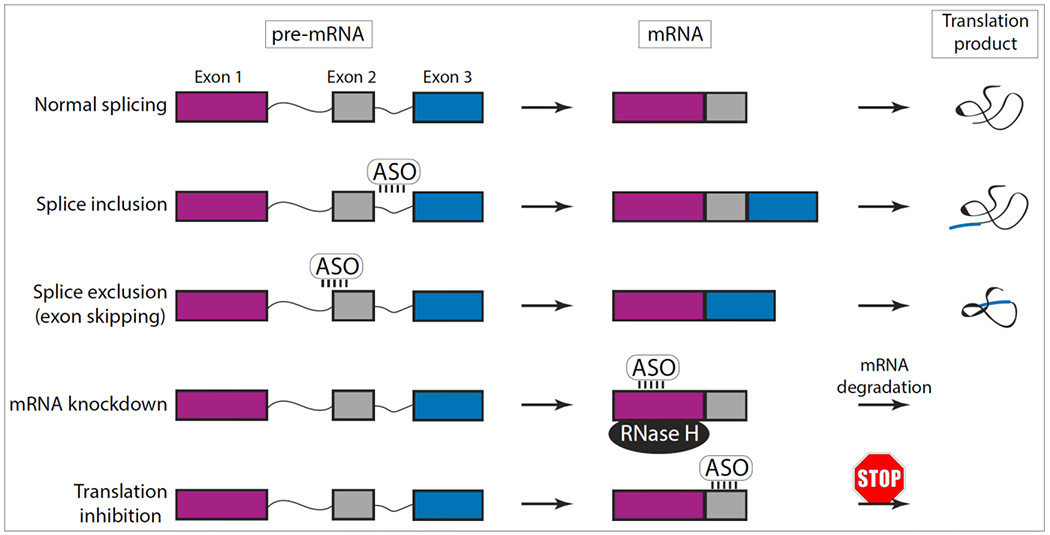
Normal RNA splicing: The newly synthesized precursor messenger RNA (pre-mRNA) becomes a mature mRNA by RNA-splicing – a process where introns (thin lines) are removed and exons (colored boxes) are ligated. Splice inclusion: ASOs bind to and block the activity of splicing enhancers on the pre-mRNA, leading to the inclusion of an exon that would normally be excluded; generating a modified protein with additional peptide sequences. Splice exclusion: ASOs bind to the exon-intron splicing junction on the pre-mRNA to skip an exon, leading to a truncated protein. mRNA knockdown: ASOs bind to mRNAs and activate RNase H, resulting in mRNA-cleavage and degradation. Translation inhibitor: ASOs bind to the 5’ UTR or coding regions of the mRNA and block translation.
Cas13d:
Unlike other Cas proteins that target DNA, Cas13 is an outlier enzyme that binds and cleaves RNA in mammalian cells, leading to gene knockdown. Specificity is conferred by spacer sequences complementary to target RNA, and a short hairpin that recruits Cas1343. Since there is no requirement for PAM sites, Cas13 is more flexible than other conventional Cas proteins. Of the four subtypes – Cas13a-d44 – Cas13d is the smallest, and can be packaged into a single AAV for in vivo applications45. Besides RNA knockdown, Cas13 enzymes have been adapted for RNA splicing-regulation and base editing. Catalytically inactive forms of Cas13d (dCas13d) have also been used to modulate splicing of endogenous tau transcripts and correct abnormal ratios of 4R/3R tau isoforms in frontotemporal dementia patient-derived iPSCs45. For RNA base editing, dCas13b is fused to deaminase enzymes for target nucleotide substitutions46.
III). Specific applications of gene-based therapies to neurodegenerative diseases
Ongoing gene therapy clinical trials and potential application of gene-based therapeutic strategies for selected neurodegenerative diseases are described below, also see Tables 2 and 3 for a list of trials.
Table 2.
Gene therapy trials for AD and PD
| Gene therapy | Delivery route | Identifier | Phase | Patients | Primary endpoint | Duration | Ref | ||
|---|---|---|---|---|---|---|---|---|---|
| Alzheimer’s disease | Growth factor | AAV2-NGF | IP (basal forebrain) | NCT00087789 | 1 | Mild to moderate AD | Safety and tolerability | 2004-2010 | 65,150 |
| AAV2-NGF | IP(basal forebrain) | NCT00876863 | 2 | Mild to moderate AD | Cognition | 2008-2015 | 64 | ||
| Enzymes | AAVrh.10-hAPOE2 | IP (basal forebrain) | NCT03634007 | 1 | MCI or mild to severe AD, APOE4 homozygotes,≥ 50 years of age | Safety | 2019-2021 | ||
| AAV-hTERT | IV + IT | NCT04133454 | 1 | AD or early dementia, ≥ 40 years of age | Safety and tolerability | 2019-2021 | |||
| Pathology | ASO-MAPT | IV | NCT03186989 | 1/2 | Mild AD, age 50-74 | Safety and tolerability | 2017-2022 | ||
| Parkinson’s disease | GABA | AAV2-GAD | IP (subthalamic nucleus) | NCT00195143 | 1 | Idiopathic PD, > 5 years of disease duration | Safety | 2003-2005 | 90 |
| AAV2-GAD | IP (subthalamic nucleus) | NCT00643890 | 2 | Idiopathic PD, > 5 years of disease duration | Disease severity and progression | 2008-2010 | 91,151 | ||
| Dopamine | Lentivirus-TH/AADC/CH1 | IP (striatum) | NCT00627588 | 1/2 | Bilateral idiopathic PD, age 48-65, > 5 years of disease duration | Safety | 2008-2012 | 86 | |
| Lentivirus-TH/AADC/CH1 | IP (striatum) | NCT01856439 | 1/2 | From NCT00627588 | Long-term safety and tolerability | 2011-2022 | 86 | ||
| AAV2-AADC | IP (striatum) | NCT00229736 | 1 | Mid- to late-stage PD, ≤ 75 years of age | Safety and tolerability | 2004-2013 | 83 | ||
| AAV2-AADC | IP (striatum) | NCT01973543 | 1 | Idiopathic PD, > 5 years of disease duration | Safety and tolerability | 2013-2020 | 152 | ||
| AAV2-AADC | IP (putamen) | NCT02418598 | 1/2 | Idiopathic PD, ≤ 75 years of age | Safety | 2015-2018 | |||
| AAV2-AADC | IP (striatum) | NCT03065192 | 1 | Idiopathic PD, > 5 years of disease duration | Safety and suicide risk | 2017-2021 | |||
| AAV2-AADC | IP (striatum) | NCT03562494 | 2 | PD, age 40-75, > 4 years of disease duration | Change of time in troublesome dyskinesia | 2018-2020 | 153 | ||
| Growth factors | AAV2-NTN | IP (putamen) | NCT00252850 | 1 | Bilateral idiopathic PD, age 35-75, > 5 years of disease duration | Safety and tolerability | 2005-2007 | 97 | |
| AAV2-NTN | IP (putamen) | NCT00400634 | 2 | Bilateral idiopathic PD, age 35-75, > 5 years of disease duration | Disease severity and progression | 2006-2008 | 98 | ||
| AAV2-NTN | IP (subthalamic nucleus + putamen) | NCT00985517 | 1/2 | Idiopathic PD, age 35-75 | Disease severity and progression | 2009-2017 | 100 | ||
| AAV2-GDNF | IP (putamen) | NCT01621581 | 1 | Idiopathic PD, > 5 years of disease duration | Safety and tolerability | 2012-2022 | 153 | ||
| AAV2-GDNF | IP (putamen) | NCT04167540 | 1 | PD, age 35-75, | Safety and tolerability | 2020-2026 | |||
| Lysosome | AAV9-GCase | IC | NCT04127578 | 1/2 | Moderate to severe PD, at least 1 pathogenic GBA1 mutation | Safety and immunogenicity | 2019-2026 | ||
Table 3.
Gene therapy trials for HD, SMA and ALS
| Gene therapy | Delivery route | Identifier | Phase | Patients | Primary endpoint | Duration | Ref | ||
|---|---|---|---|---|---|---|---|---|---|
| Huntington’s disease | Total HTT |
ASO-HTT | IT | NCT02519036 | 1/2 | Early manifest HD, age 25-65 | Safety and tolerability | 2015-2017 | 124,136,154 |
| ASO-HTT | IT | NCT03342053 | 2 | From NCT02519036 | Safety and tolerability | 2017-2019 | |||
| ASO-HTT | IT | NCT04000594 | 1 | Manifest HD, age 25-65 | Pharmacokinetics and pharmacodynamics | 2019-2021 | |||
| ASO-HTT | IT | NCT03761849 | 3 | Manifest HD, age 25-65 | Disease progression and total functional capacity | 2019-2022 | 124 | ||
| ASO-HTT | IT | NCT03842969 | 3 | Patients who participated in prior RG6042 (ASO-HTT) studies | Long-term safety and tolerability, suicide risk, cognition | 2019-2024 | |||
| AAV5-miHTT | IP (striatum) | NCT04120493 | 1/2 | Early manifest HD, ≤ 40 CAG repeats in HTT | Safety | 2019-2026 | |||
| Mutated HTT | ASO-mHTT | IT | NCT03225833 | 1/2 | Early manifest HD, carrying a targeted SNP rs362307, age 25-65 | Safety and tolerability | 2017-2020 | 136 | |
| ASO-mHTT | IT | NCT03225846 | 1/2 | Early manifest HD, carrying a targeted SNP rs362331, age 25-65 | Safety and tolerability | 2017-2020 | 136 | ||
| SMA | SMN1 | AAV9-SMN | IV | NCT02122952 | 1 | Type 1 SMA, ≤ 6 or 9 months of age | Safety | 2014-2019 | 2,55 |
| AAV9-SMN | IV | NCT03306277 | 3 | Type 1 SMA, ≤ 6 months of age | Independent sitting, survival | 2017-2019 | |||
| AAV9-SMN | IV | NCT03381729 | 1 | Type 2 SMA (onset at < 12 months of age), age 6 to 60 months | Safety and tolerability | 2017-2021 | |||
| AAV9-SMN | IV | NCT03461289 | 3 | Type 1 SMA, ≤ 6 months of age | Sitting without support | 2018-2020 | |||
| AAV9-SMN | IV | NCT03505099 | 3 | Pre-symptomatic SMA with 2 or 3 copies of SMN2, ≤ 6 weeks of age | Independent sitting, standing without support | 2018-2021 | |||
| AAV9-SMN | IV | NCT03837184 | 3 | Type 1 SMA, ≤ 6 months of age | Sitting without support | 2019-2033 | |||
| SMN2 | ASO-SMN2 | IT | NCT01494701 | 1 | Age 2-14 | Safety, tolerability, and pharmacokinetics | 2011-2013 | 155 | |
| ASO-SMN2 | IT | NCT01703988 | 1/2 | Age 2-15 | Safety and tolerability | 2012-2015 | 155 | ||
| ASO-SMN2 | IT | NCT01780246 | 1 | From NCT01494701 | Safety and tolerability | 2013-2014 | 155 | ||
| ASO-SMN2 | IT | NCT01839656 | 2 | Age ≤ 210 days, disease onset between 21 and 180 days of age | Motor milestones | 2013-2017 | 52,155 | ||
| ASO-SMN2 | IT | NCT02052791 | 1 | From NCT01703988or NCT01780246 | Safety and tolerability | 2014-2017 | 155 | ||
| ASO-SMN2 | IT | NCT02292537 | 3 | Disease onset at > 6 months of age, age 2-12 | Motor function | 2014-2017 | 155,156 | ||
| ASO-SMN2 | IT | NCT02386553 | 2 | SMA with 2 or 3 copies of SMN2, ≤ 6 weeks of age | Respiratory intervention, survival | 2015-2025 | 52 | ||
| ASO-SMN2 | IT | NCT02594124 | 3 | From NCT02193074, NCT02292537, NCT02052791, NCT01839656 | Long-term safety and tolerability | 2015-2023 | |||
| ASO-SMN2 | IT | NCT04050852 | 1 | Age 5-21 | Pulmonary function | 2019-2020 | |||
| ASO-SMN2 | IT | NCT04089566 | 2/3 | Child, adult, older adult | Safety and tolerability with high doses, neuromuscular function | 2020-2022 | |||
| ALS | SOD1 | ASO-SOD1 | IT | NCT01041222 | 1 | Familial ALS with SOD1 mutation, ≥18 years of age | Safety, tolerability, and pharmacokinetics | 2010-2012 | 141 |
| ASO-SOD1 | IT | NCT02623699 | 1/2 | Familial ALS with SOD1 mutation, ≥18 years of age | Safety, tolerability, Pharmacokinetics and progression of disability | 2016-2021 | 142 | ||
| ASO-SOD1 | IT | NCT03070119 | 3 | From NCT02623699 | Long-term safety and tolerability | 2017-2023 | |||
| ASO-SOD1 | IT | NCT03764488 | 1 | Age 18-65 | Drug distribution in CNS | 2018-2020 | |||
| C9orf72 | ASO-C9orf72 | IT | NCT03626012 | 1 | ALS with C9orf72 mutation, ≥18 years of age | Safety and tolerability | 2018-2021 | ||
| ASO-C9orf72 | IT | NCT04288856 | 1 | From NCT03626012 | Long-term safety and tolerability | 2020-2023 | |||
Intraparenchymal (IP)
Intracisternal (IC)
Intravenous (IV)
Intrathecal (IT)
Spinal muscular atrophy
SMA is a progressive, monogenic lower motor neuron disease, and one of the leading genetic cause of infant mortality. The genetics and clinical phenotypes are complex, but in most cases, SMA is caused by homozygous deletion of SMN1 gene that codes for the survival motor neuron (SMN) protein47, though the underlying reason for motor neuron vulnerability is still unclear. In humans, a paralog gene called SMN2 also produces the SMN protein, but at lower levels due to a single nucleotide substitution in the SMN-2 gene that alters splicing and excludes exon-7 from ~ 90% of the transcripts – resulting in a shortened mRNA and truncated SMN protein that is rapidly degraded48. Since SMA patients lack a functional SMN1 gene, the severity of their symptoms and the age of disease-onset is determined by copy-numbers of the SMN2 gene. SMA type-1 is the most common form of this disease with hypotonia and motor delay before 6 months of age, leading to death or need for mechanical ventilation by age 2. These patients tend to have two copies of SMN2 gene. Patients with three to four copies of SMN2 have milder symptoms that appear later (6 to 18 months of age for type-2, and ~3 years for type-3). Less than 5% of patients have four to eight copies of SMN2 gene (type-4) and have the mildest form of the disease with an adult onset. Recently, FDA approved three disease-modifying gene therapies that increase SMN protein-levels, either by enhancing the effectiveness of SMN2, or by introducing functional copies of SMN1.
Modulating SMN2:
In an RNA-based approach, ASOs were used to target exon-7 of the SMN2 gene, preventing its exclusion during splicing and subsequently increasing SMN protein levels49. After successful Phase 1/2 trials with intrathecal delivery of ASOs50, a Phase 3 trial initiated in 2014 showed significant motor improvement in 51% of SMA infants (37 out of 73)51, leading to the approval of an SMN2 ASO by FDA (called nusinersen). Patients with early treatment performed better, highlighting the importance of early diagnosis51. A Phase 2 trial also showed encouraging motor improvement in pre-symptomatic SMA infants52. The efficacy of nusinersen in older patients is currently being investigated. The requirement for repeated intrathecal administration is a limitation of ASOs in the clinical setting, and recently, an orally-administered small molecule designed to favorably modulate SMN2 splicing (risdiplam) was approved by the FDA53. Alternatively, DNA-based strategies like CRISPRs can install permanent changes to SMN2 to increase SMN expression. Though experimental, a recent study used a CRISPR/Cpf1 strategy in iPSCs to convert the SMN2 gene to an SMN1-like gene via HDR54; thus in principle, DNA-based strategies could also be used to modulate SMN2.
Expressing SMN1:
In a landmark gene therapy clinical trial, 15 infants with SMA type-1 received a single intravenous injection of an AAV9 vector carrying the SMN1 gene (AVXS-101/onasemnogene abeparvovec). All patients in this trial were alive and event-free at 20 months, compared to a historic survival rate of 8%2. Several patients getting a higher-dose showed unprecedented improvement in symptoms including the ability to walk unassisted, although they were treated at a younger age2. A subsequent two-year follow-up study in the high-dose cohort showed a reduced need for pulmonary and nutritional support and improved motor function55, and AVXS-101 was approved by FDA in 2019. Despite this optimism, caution is warranted with AAVs, and further clinical development of these vectors is critical. During the trial, four patients developed elevated liver transaminase levels, though no additional liver dysfunctions were reported2. Moreover, recent studies showed that intravenous injection of an AAVhu68 expressing SMN (an AAV-vector similar to the one used in the AVXS-101 trial) led to severe liver toxicity and dorsal root ganglion degeneration in NHPs and piglets, necessitating euthanasia in many cases18. The long-term therapeutic effects and complications of AVXS-101 are also unknown.
Alzheimer’s disease
Affecting nearly one in nine elderly people in the United States, with no disease-modifying therapy in sight, AD is perhaps the most poignant example of a neurodegenerative disease with an unmet need. Current FDA-approved treatments provide mild symptomatic relief at best, and many clinical trials with small molecules and antibodies have been disappointing7. Besides neurotrophins, three genes linked to AD can be potentially targeted for gene-based therapies – APP, MAPT and APOE. The APP gene encodes the amyloid precursor protein (APP) which is cleaved by β- and γ- secretases to eventually generate Aβ, and extensive evidence links APP to AD pathogenesis (reviewed in56). APP mutations and duplications are seen in familial AD, and an APP mutation (Icelandic) is protective in sporadic disease as well57. Recent studies suggest that other AD risk factors like APOE4 may upregulate APP transcription58, and evidence from human genetics in Down syndrome strongly implicate APP over-expression in AD linked to trisomy-2159; collectively make a strong case for the involvement of the APP gene in AD. MAPT encodes for the tau protein which is an integral component of neurofibrillary tangles, and is an established therapeutic target in AD60.
Expressing growth factors:
Gene delivery of NGF to the cholinergic nucleus basalis of Meynert (NBM) by direct, bilateral AAV2 injections into the brain was one of the first gene therapy clinical trials in AD61. Nerve growth factor (NGF) is an endogenous neurotrophic factor that regulates the growth and survival of cholinergic neurons by functional activation of the TrkA receptor62. Preclinical studies in animal models including NHPs showed that exogenous NGF was beneficial, provided a rationale for human trials63. However, while Phase 1/2 clinical trials in mild to moderate AD demonstrated safety and long-term expression of AAV2-NGF, there was no change in cognition64. Examination of autopsy brains from this study showed that the injected AAV2-NGF did not reach the targeted cholinergic neurons in the NBM65; thus the efficacy of this therapeutic option remains uncertain. Unlike human trials, the successful preclinical animal studies with NGF had used infusion pumps, and future studies administering brain derived neurotrophic factor (BDNF) into the entorhinal cortex (AAV2-BDNF) plans to use real-time MR guidance and convection-enhanced delivery66.
Targeting MAPT:
ASOs against the MAPT gene encoding tau have been used to reduce tau mRNA and protein levels in animal models. These ASOs attenuate tau phosphorylation and aggregation, prevent neuronal death, and extend survival in transgenic mice expressing a human mutant (P301S) tau; and also reduce CNS tau levels in NHPs67. A clinical trial using this strategy is currently ongoing (NCT03186989). Alternatively, ASO-mediated exon-skipping can also reduce tau mRNA/protein levels in cells and in vivo68. Acute knockdown of tau in adult mice showed deficits in learning and memory69, and there is an emerging role for tau in regulating presynaptic function70. Though these are potential concerning, it is unlikely that any strategy would cause a global depletion of tau, and toxicity studies would likely have to only contend with partial reductions.
Targeting APOE:
In humans, three apolipoprotein E (APOE) polymorphic alleles – E2, E3 and E4 – determine risk for AD. While the APOE4 variant is the single greatest risk factor for sporadic AD, APOE2 is protective71. Studies in animal models indicate that APOE4 is linked to both amyloid and tau pathology72,73. One strategy for gene therapy is to elevate protective APOE2 levels in the brain. Indeed, expressing APOE2 by viral vectors attenuates Aβ pathology in an amyloid-based mouse model74. AAV-mediated delivery of APOE2 was also effective and safe in NHPs75, and a Phase 1 trial of intrathecally-delivered AAV-APOE2 is expected to start soon (NCT03634007). Human APOE4 and APOE3 differ by only one amino acid residue at position 11271. Although existing technology cannot widely install single-nucleotide changes in the brain, studies converting APOE4 to APOE3 by gene-editing in iPSCs or cerebral organoids can rescue AD-linked phenotypes76 and may be an option for gene therapy in the future.
Targeting APP:
The APP gene has a central role in both sporadic and familial AD, and silencing or modulating APP is an appealing option for gene therapy. Using CRISPR-Cas9, a recent study selectively inactivated mutant familial APP-Swedish alleles in cells and in vivo, without affecting the corresponding WT alleles77. Another study used ASOs to skip the penultimate exon (exon-17) of APP78. Exon-17 encodes the γ-secretase cleavage site and about half of the transmembrane domain of APP, thus deleting this exon abolishes membrane-anchoring of APP and attenuates Aβ secretion in cells as well as in vivo (78 and Fig. 5a). However, removing the membrane-anchoring of APP is also expected to eliminate its physiologic functions. Also, APP-ASOs generate abnormal fusion proteins of APP that are rapidly secreted from the cell78 (Fig. 5a), and such non-native sequences may have detrimental functions. APP is evolutionarily conserved throughout the animal kingdom and has clear roles in synaptic plasticity79. A recent study used a NHEJ-based CRISPR-Cas9 ‘DNA-excision’ strategy to induce PTCs in the last exon (exon-18) of APP, truncating the last ~ 36 amino acids and eliminating a pentapeptide YENPTY endocytic motif that triggers the amyloidogenic pathway29. As noted previously, transcriptional rules render the last exon insensitive to NMD, thus PTCs in this region do not degrade the mRNA but lead to protein-truncations. The N-terminus and transmembrane domains of APP – thought to play roles in axonal and synaptic physiology – are intact in this setting, and physiologic functions appear unaffected (Fig. 5b). Moreover, the approach also up-regulated the α-cleavage pathway, known to play protective roles in AD79 (Fig. 5c-d). Coincidentally, the C-terminus of APP has extensive microduplications that lead to microhomology mediated end-joining (MMEJ)80 and precise corrections after gene editing, making this a suitable target. Expression of neuroprotective APP fragments such as sAPPα may also be a viable therapeutic option in AD81.
Figure 5. ASO and CRISPR-based editing strategies to modulate APP-cleavage products.
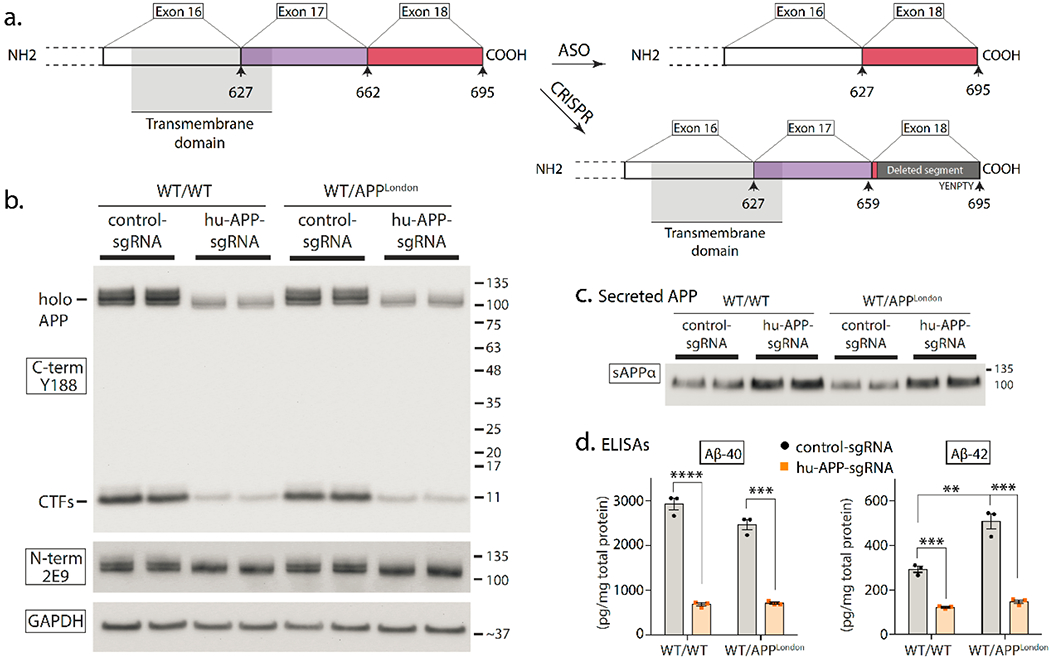
a)Left: Schematic showing the transmembrane domain and C-terminus of APP, with corresponding exons (small arrows with numbers denote amino acids). Right: The ASO strategy leads to “skipping” of APP exon 17, leading to a protein that lacks the γ-secretase cleavage site and a portion of the transmembrane domain, and consequently, less Aβ secretion81. Sun et al. used a NHEJ-based CRISPR-Cas9 strategy to introduce PTCs in the last exon (exon 18) of APP35. Since transcriptional rules dictate that the last exon is exempt from NMD, this method does not lead to mRNA decay, but effectively truncates the last ~ 36 amino acids of APP that includes an endocytic motif triggering APP β-cleavage (see35 for more details).
b-d) Data from editing of APP C-terminus with CRISPR showing attenuation of APP β-cleavage and upregulation of protective α-cleavage (figures reproduced from35, shared under a Creative Commons Attribution 4.0 International Licence).
b) Human iPSC-derived neurons (WT or isogenic APPV717I knock-in) were transduced with lentiviral vectors carrying APP-gRNA/Cas9 to edit the C-terminus of APP and immunoblotted with C- and N-terminus antibodies. Note selective attenuation of APP signal with C-terminus antibody Y188 after CRISPR-editing.
c) Immunoblotting of secreted sAPPα from the media of iPSC-neurons, treated as mentioned in (b). Note increased sAPPα in edited samples, indicating upregulation of protective APP α-cleavage.
d) ELISA of secreted Aβ from media, treated as mentioned in (b). Note decreased Aβ in CRISPR-treated samples.
Parkinson’s disease
Though Parkinson’s disease (PD) is a multi-system disorder, the classic symptoms of rigidity, tremor and hypokinesia are largely due to the loss of dopaminergic neurons in the substantia nigra (SN) pars-compacta, leading to an imbalance of inhibitory and excitatory pathways in nigrostriatal projections82. Current treatments only offer symptomatic relief, have significant side effects, and do not affect disease progression. The focal nature of SN pathology in PD has been long considered amenable to gene therapy, leading to approaches that can restore neurotransmitter imbalance, enhance neuronal survival, or target genes directly linked to the disease.
Modulating neuronal signaling:
AAV based gene therapies have been used to upregulate dopaminergic signaling in PD. The dopamine synthesis pathway is regulated by three rate-limiting enzymes GTP cyclohydrolase 1 (GCH1), tyrosine hydroxylase (TH) and aromatic amino acid dopa decarboxylase (AADC), with AADC as the final enzymatic step in dopamine production. Clinical trials with AAV2-AADC have demonstrated safety, stable expression for up to 4 years, and modest improvement in symptoms83,84. A more targeted delivery of AAV2-AADC in NHPs by real-time MRI-guidance was also safe and well tolerated85, and a human clinical trial (NCT03065192) was recently launched. Finally, a lentivirus-based gene therapy (called ProSavin) delivering all three rate-limiting enzymes (TH, AADC, and GCH1) was well-tolerated in Phase 1/2 trials, with follow-up studies reporting moderate improvements in motor function86. Decreased striatal dopaminergic tonus in PD leads to overactivity of the glutamatergic subthalamic nucleus (STN); and STN-infusion of agonist to GABA – the major inhibitory neurotransmitter in the brain – mitigates hyperactivity and PD-like symptoms in NHP disease models87. In preclinical studies, GABA activation by subthalamic overexpression of glutamate acid decarboxylase (GAD), an enzyme catalyzing the synthesis of GABA, diminishes PD-like symptoms in rat and NHP models88,89. Phase 1/2 clinical trials of AAV2-GAD injections in STN showed that the gene therapy was well tolerated and ameliorated PD symptoms90,91, with effects persisting for a year92.
Expressing neurotrophic and regenerative factors:
Two promising candidates supporting the survival of dopaminergic midbrain neurons are glial cell line-derived neurotrophic factor (GDNF) and neurturin (NRTN)93. Preclinical studies showed that AAV-GDNF injections into the putamen of rodent and NHP models were well tolerated 94–96, though bilateral SN-injections caused weight loss in aged monkeys96. Further Phase 1 clinical trials to determine the safety of bilateral AAV2-GDNF injections into the putamen are ongoing. Regarding NRTN, a Phase 1 clinical trial delivering AAV2-NRTN to the putamen was well-tolerated 97, but did not improve motor function98. Postmortem analyses showed that although NRTN expression was increased in the injected site at the putamen, there was no upregulation in the SN99 – likely due to the failure of retrograde NRTN transport, or inadequate transduction. Though a subsequent clinical trial delivered higher doses of AAV-NRTN directly into the SN, there was still no clinical improvement in motor function100. A possible reason for the limited success of GDNF and NRTN in PD may be that some PD patients show reduced expression of Ret – the receptor of both NRTN and GDNF – thus these trophic factors cannot confer protection through Ret101. Gene therapy can also be employed to generate new neurons, as demonstrated by a recent experimental study in a chemically induced model of PD, where ASO-based depletion of an RNA-binding protein PTB (also called PTB1) led to a conversion of resident astrocytes into neurons and amelioration of neurochemical and motor deficits102.
Targeting disease genes – SNCA, GBA and LRRK2:
Extensive evidence links SNCA – the gene encoding α-synuclein – in PD and related disorders; collectively called synucleinopathies (reviewed in103). SNCA mutations and genomic multiplications are seen in familial synucleinopathies, and genome-wide studies in sporadic populations identified SNCA variants that increase α-synuclein expression 104,105; providing a rationale for lowering α-synuclein levels in PD. Indeed knockdown of α-synuclein via shRNA or ASO prevents neurodegeneration in rodent models of PD106,107. CRISPR-mediated downregulation of α-synuclein also enhanced cell viability in human iPSC-derived dopaminergic neurons108. However, some in vivo studies have reported concerning phenotypes after α-synuclein knockdown, and this issue remains unresolved109,110.
Augmenting brain glucocerebrosidase (GCase) activity by gene therapy is a promising therapeutic approach in PD. GCase deficiency is the hallmark of Gaucher disease – an inherited lysosomal storage disorder due to mutations in the GBA1 gene – and interestingly, GBA1 mutations are also the most common risk factor for developing PD and other synucleinopathies111. Decreased GCase levels are also seen in cases of sporadic PD (without GBA1 mutations). Substantial evidence from PD animal models also indicate links between GCase and PD; collectively supporting a scenario where loss of GCase activity leads to stressed degradative pathways and increased risk of developing PD112. Direct injections of AAV-GBA1 in the brain decreased α-synuclein levels and pathology in rodent models113. Diffuse brain delivery of GBA1 by intravenous injections of AAV-PHP.B-GBA1 in an A53T α-synuclein mouse model attenuated α-synuclein pathology and led to significant behavioral recovery and extension of lifespan114. A Phase 1/2 clinical trial was recently launched to treat PD patients by intracisternal injection of AAV9-GBA1 (NCT04127578).
Variants in the leucine-rich repeat kinase 2 gene LRRK2 cause familial PD, and also increase the risk of developing idiopathic disease115. The most common PD risk variant G2019S lies in the catalytic site of LRRK2 and enhances its kinase activity – a property also shared by other PD-linked LRRK2-variants. Thus, attenuating LRRK2 expression is an appealing therapeutic option, and small molecule inhibitors of LRRK2 have been explored116. However, LRRK2 is also expressed in lungs, kidneys and spleen, and global inhibition of LRRK2 can lead to pathologic changes in these tissues117,118. Pulmonary complications are particularly concerning, though studies suggest that they may be reversible upon stopping treatment119. Gene therapy can be useful in this setting, by specifically inhibiting LRRK2 in the brain, and intracerebral injection of LRRK2 ASOs in the brain are effective in attenuating mRNA and protein levels, without obvious renal or pulmonary phenotypes120. A LRRK2 ASO (BIIB094, Ionis pharmaceuticals) is also undergoing Phase 1 clinical trial in PD patients, where it will be delivered by intrathecal injections (NCT03976349).
Huntington’s disease
Being a monogenic disorder, Huntington’s disease (HD) is an appealing target for gene therapy. HD patients have progressive motor dysfunction, psychiatric impairment, and cognitive decline121, with no disease-modifying therapy. HD is caused by a CAG trinucleotide repeat expansion in the huntingtin (HTT) gene, generating a mutant toxic HTT protein with abnormally long polyglutamine (polyQ) repeats121.
Global silencing of HTT:
ASOs targeting HTT mRNA reduces overall HTT protein level and delays disease progression in HD models122. In a Phase 1/2 trial, intrathecal administration of ASO-HTT decreased mutant HTT levels in the CSF without any adverse effects123, and a Phase 3 trial was recently started to determine efficacy124. Intracranial administration of AAV5 with a microRNA (miRNA) against HTT significantly reduced mutant HTT levels in transgenic HD minipig brain125, and a Phase 1/2 clinical trial has been initiated. RNA interference (RNAi) technology has also been used to decrease HTT. Several experimental studies in HD mouse models have shown that injection of AAV-HTT shRNA into the striatum significantly reduces HTT expression, improving motor and behavioral deficits without overt neurotoxicity126,127. Similar approaches are also reported to be safe in NHP128,129. An advantage of targeting total HTT is that in principle, a single therapeutic agent can work for all HD patients, regardless of the specific mutation. However, a downside is that wild-type protein – and any related physiological function – will also be suppressed. Indeed, depletion of endogenous HTT in adult mice can lead to progressive behavioral deficits, neuropathological changes, and acute pancreatitis130,131, which is concerning. Perhaps a balance between lowering mutant HTT and maintaining wild-type HTT need to be considered when designing non-selective HTT lowering therapies.
Selective silencing of HTT mutant alleles:
Experimental studies have selectively inactivated mutant HTT by targeting single nucleotide polymorphisms (SNPs) associated with the mutant allele, either by CRISPR/RNAi in cells derived from HD patients132,133, or by using ASOs in a humanized HD mouse model134. Since only three common HD-associated SNPs have been reported in over 75% of the patients in the United States and Europe135, only a few targeted gene-therapies are expected to benefit most patients, and a clinical trial using ASO-mutant-HTT is ongoing136. Although the strategy of targeting SNPs in the mutant allele circumvents the putative risk of depleting wild-type HTT, it also limits the availability of RNA binding sequences that can be targeted by ASOs. Recently, zinc finger protein transcription factors (ZFP-TFs) were also used to selectively disrupt the mutant HTT allele by targeting the CAG repeat region, and AAV-delivered ZFP-TFs were safe and efficacious in HD mouse models137.
Amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis (ALS) is caused by progressive degeneration of motor neurons, typically leading to respiratory failure and death within 3-5 years after disease onset. Up to 10% of ALS patients have a familial history of genetic mutations (fALS), while some mutations are also seen in apparently sporadic cases (sALS). Amongst the common ALS genes SOD1, C9orf72, TDP43, and FUS, gene therapies targeting SOD1 and C9orf72 mutations are being tested in clinical trials (for a recent review, see see138).
Targeting SOD1:
Around 10–20% of fALS and 1-2% of sALS patients have SOD1 mutations. Most SOD1 mutations alter protein conformation, leading to a gain of neurotoxicity139. Injection of ASO-SOD1 into CSF reduced SOD1 levels and prolonged the survival in a transgenic rat model carrying mutated human SOD1140. In a Phase 1 clinical trial, intrathecal administration of ASO-SOD1 was well tolerated in fALS patients with SOD1 mutations141. A subsequent Phase1/2 trial showed that ASO-SOD1 treatment reduces CSF SOD1 protein levels142, and a Phase 3 trial has been initiated. RNAi and CRISPRs have also been used to inactivate mutant SOD1. Intravenous delivery of AAV9-SOD1 shRNA reduced SOD1 levels, slowed disease progression, and prolonged lifespan in a transgenic SOD1 mouse model143,144. Disruption of SOD1 expression by CRISPR also improved motor function and prolonged survival of mutant SOD1 mice145. Recently, a proof-of concept study intrathecally delivered AAVs expressing miRNAs against SOD1 in two SOD1-ALS patients, and postmortem analysis showed suppression of SOD1 in spinal cord from one patient, demonstrating feasibility146.
Targeting C9orf72:
The G4C2 hexanucleotide repeat expansion (HRE) in intron 1 of C9orf72 is a frequent cause of ALS147. At least three primary mechanisms of HRE-induced neurotoxicity are recognized – reduced expression of normal C9orf72, RNA foci accumulation from sense and antisense transcripts of HRE, and dipeptide repeat proteins from HRE translation through a non-AUG (RAN) mechanism148. Intracerebroventricular injection of ASO-C9orf72 reduced C9orf72 mRNA foci and improved cognitive function in a transgenic mouse model expressing 450 G4C2 repeats in C9orf72 gene149. A Phase 1 trial of intrathecal ASO-C9orf72 administration in C9orf72-ALS patients is ongoing.
Conclusions
Once considered a pariah, gene therapy is now a reality. Traditionally, the widespread pathology of neurodegenerative diseases was considered unamenable to gene therapy, but advances in vector-technologies now allow diffuse delivery of genes into the CNS. Combined with contemporary genome-manipulation tools, gene-based therapies promise to alter the future clinical management of both inherited and sporadic neurodegenerative diseases – devastating illnesses that have few or no disease-modifying agents today. Even so, many challenges remain, and systematic development of gene-delivery vectors and rigorous evaluation of the safety of contemporary gene-manipulation tools, along with transparency and cooperation between various stakeholders will be critical in ushering this new era.
Acknowledgements:
The authors thank Leonardo Parra, Brent Aulston and James Brewer (all at UCSD) for comments on the manuscript. The authors apologize that due to space restrictions, primary papers could not be cited in most cases. Work in the Sun lab is supported by NSFC grants 82071193 and 32000673. Work in the Roy lab is supported by grants from the NIH (R01AG048218, R01NS11978, R01NS075233, R21AG052404, UG3NS111688), and the US-Israel Binational Science Foundation (BSF, #2019248).
Footnotes
Author disclosures: J. Sun and S. Roy have applied for patents related to gene editing in Alzheimer’s disease (US Patent App. 16251970). S. Roy is also scientific founder of, advisor to, and owns equity in CRISPRAlz.
References
- 1.Somanathan S, Calcedo R & Wilson JM Adenovirus-Antibody Complexes Contributed to Lethal Systemic Inflammation in a Gene Therapy Trial. Mol Ther 28, 784–793, doi: 10.1016/j.ymthe.2020.01.006 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mendell JR et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N Engl J Med 377, 1713–1722, doi: 10.1056/NEJMoa1706198 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Keeler CE Gene therapy. J Hered 38, 294–298 (1947). [PubMed] [Google Scholar]
- 4.Logovinsky V et al. Safety and tolerability of BAN2401--a clinical study in Alzheimer’s disease with a protofibril selective Abeta antibody. Alzheimers Res Ther 8, 14, doi: 10.1186/s13195-016-0181-2 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hudry E & Vandenberghe LH Therapeutic AAV Gene Transfer to the Nervous System: A Clinical Reality. Neuron 102, 263, doi: 10.1016/j.neuron.2019.03.020 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Doudna JA The promise and challenge of therapeutic genome editing. Nature 578, 229–236, doi: 10.1038/s41586-020-1978-5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang LK, Chao SP & Hu CJ Clinical trials of new drugs for Alzheimer disease. J Biomed Sci 27, 18, doi: 10.1186/s12929-019-0609-7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bedbrook CN, Deverman BE & Gradinaru V Viral Strategies for Targeting the Central and Peripheral Nervous Systems. Annu Rev Neurosci, doi: 10.1146/annurev-neuro-080317-062048 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Leone P et al. Long-term follow-up after gene therapy for canavan disease. Sci Transl Med 4, 165ra163, doi: 10.1126/scitranslmed.3003454 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ginn SL, Amaya AK, Alexander IE, Edelstein M & Abedi MR Gene therapy clinical trials worldwide to 2017: An update. J Gene Med 20, e3015, doi: 10.1002/jgm.3015 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Calcedo R, Vandenberghe LH, Gao G, Lin J & Wilson JM Worldwide epidemiology of neutralizing antibodies to adeno-associated viruses. J Infect Dis 199, 381–390, doi: 10.1086/595830 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mingozzi F et al. CD8(+) T-cell responses to adeno-associated virus capsid in humans. Nat Med 13, 419–422, doi: 10.1038/nm1549 (2007). [DOI] [PubMed] [Google Scholar]
- 13.Petry H et al. Effect of viral dose on neutralizing antibody response and transgene expression after AAV1 vector re-administration in mice. Gene Ther 15, 54–60, doi: 10.1038/sj.gt.3303037 (2008). [DOI] [PubMed] [Google Scholar]
- 14.Lu Y & Song S Distinct immune responses to transgene products from rAAV1 and rAAV8 vectors. Proc Natl Acad Sci U S A 106, 17158–17162, doi: 10.1073/pnas.0909520106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ashley SN, Somanathan S, Giles AR & Wilson JM TLR9 signaling mediates adaptive immunity following systemic AAV gene therapy. Cell Immunol 346, 103997, doi: 10.1016/j.cellimm.2019.103997 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mendell JR et al. Dystrophin immunity in Duchenne’s muscular dystrophy. N Engl J Med 363, 1429–1437, doi: 10.1056/NEJMoa1000228 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wilson JM & Flotte TR Moving Forward After Two Deaths in a Gene Therapy Trial of Myotubular Myopathy. Hum Gene Ther 31, 695–696, doi: 10.1089/hum.2020.182 (2020). [DOI] [PubMed] [Google Scholar]
- 18.Hinderer C et al. Severe Toxicity in Nonhuman Primates and Piglets Following High-Dose Intravenous Administration of an Adeno-Associated Virus Vector Expressing Human SMN. Hum Gene Ther 29, 285–298, doi: 10.1089/hum.2018.015 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nidetz NF et al. Adeno-associated viral vector-mediated immune responses: Understanding barriers to gene delivery. Pharmacol Ther 207, 107453, doi: 10.1016/j.pharmthera.2019.107453 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pickar-Oliver A & Gersbach CA The next generation of CRISPR-Cas technologies and applications. Nat Rev Mol Cell Biol 20, 490–507, doi: 10.1038/s41580-019-0131-5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mullard A First in vivo CRISPR candidate enters the clinic. Nat Rev Drug Discov 18, 656, doi: 10.1038/d41573-019-00140-6 (2019). [DOI] [PubMed] [Google Scholar]
- 22.Urnov FD, Rebar EJ, Holmes MC, Zhang HS & Gregory PD Genome editing with engineered zinc finger nucleases. Nat Rev Genet 11, 636–646, doi: 10.1038/nrg2842 (2010). [DOI] [PubMed] [Google Scholar]
- 23.Joung JK & Sander JD TALENs: a widely applicable technology for targeted genome editing. Nat Rev Mol Cell Biol 14, 49–55, doi: 10.1038/nrm3486 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sander JD & Joung JK CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol 32, 347–355, doi: 10.1038/nbt.2842 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Willems J et al. ORANGE: A CRISPR/Cas9-based genome editing toolbox for epitope tagging of endogenous proteins in neurons. PLoS Biol 18, e3000665, doi: 10.1371/journal.pbio.3000665 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park SH et al. Highly efficient editing of the beta-globin gene in patient-derived hematopoietic stem and progenitor cells to treat sickle cell disease. Nucleic Acids Res 47, 7955–7972, doi: 10.1093/nar/gkz475 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sugaya K & Vaidya M Stem Cell Therapies for Neurodegenerative Diseases. Adv Exp Med Biol 1056, 61–84, doi: 10.1007/978-3-319-74470-4_5 (2018). [DOI] [PubMed] [Google Scholar]
- 28.Maeder ML et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat Med 25, 229–233, doi: 10.1038/s41591-018-0327-9 (2019). [DOI] [PubMed] [Google Scholar]
- 29.Sun J et al. CRISPR/Cas9 editing of APP C-terminus attenuates beta-cleavage and promotes alpha-cleavage. Nat Commun 10, 53, doi: 10.1038/s41467-018-07971-8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Popp MW & Maquat LE Leveraging Rules of Nonsense-Mediated mRNA Decay for Genome Engineering and Personalized Medicine. Cell 165, 1319–1322, doi: 10.1016/j.cell.2016.05.053 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lau CH, Ho JW, Lo PK & Tin C Targeted Transgene Activation in the Brain Tissue by Systemic Delivery of Engineered AAV1 Expressing CRISPRa. Mol Ther Nucleic Acids 16, 637–649, doi: 10.1016/j.omtn.2019.04.015 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Landrum MJ et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res 42, D980–985, doi: 10.1093/nar/gkt1113 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Komor AC, Kim YB, Packer MS, Zuris JA & Liu DR Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424, doi: 10.1038/nature17946 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gaudelli NM et al. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 551, 464–471, doi: 10.1038/nature24644 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim YB et al. Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat Biotechnol 35, 371–376, doi: 10.1038/nbt.3803 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Levy JM et al. Cytosine and adenine base editing of the brain, liver, retina, heart and skeletal muscle of mice via adeno-associated viruses. Nat Biomed Eng 4, 97–110, doi: 10.1038/s41551-019-0501-5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grunewald J et al. Transcriptome-wide off-target RNA editing induced by CRISPR-guided DNA base editors. Nature 569, 433–437, doi: 10.1038/s41586-019-1161-z (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zuo E et al. A rationally engineered cytosine base editor retains high on-target activity while reducing both DNA and RNA off-target effects. Nat Methods 17, 600–604, doi: 10.1038/s41592-020-0832-x (2020). [DOI] [PubMed] [Google Scholar]
- 39.Anzalone AV et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157, doi: 10.1038/s41586-019-1711-4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rinaldi C & Wood MJA Antisense oligonucleotides: the next frontier for treatment of neurological disorders. Nat Rev Neurol 14, 9–21, doi: 10.1038/nrneurol.2017.148 (2018). [DOI] [PubMed] [Google Scholar]
- 41.Leavitt BR & Tabrizi SJ Antisense oligonucleotides for neurodegeneration. Science 367, 1428–1429, doi: 10.1126/science.aba4624 (2020). [DOI] [PubMed] [Google Scholar]
- 42.Chen G, Katrekar D & Mali P RNA-Guided Adenosine Deaminases: Advances and Challenges for Therapeutic RNA Editing. Biochemistry 58, 1947–1957, doi: 10.1021/acs.biochem.9b00046 (2019). [DOI] [PubMed] [Google Scholar]
- 43.Abudayyeh OO et al. RNA targeting with CRISPR-Cas13. Nature 550, 280–284, doi: 10.1038/nature24049 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Makarova KS et al. Evolutionary classification of CRISPR-Cas systems: a burst of class 2 and derived variants. Nat Rev Microbiol 18, 67–83, doi: 10.1038/s41579-019-0299-x (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Konermann S et al. Transcriptome Engineering with RNA-Targeting Type VI-D CRISPR Effectors. Cell 173, 665–676 e614, doi: 10.1016/j.cell.2018.02.033 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Abudayyeh OO et al. A cytosine deaminase for programmable single-base RNA editing. Science 365, 382–386, doi: 10.1126/science.aax7063 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lefebvre S et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80, 155–165, doi: 10.1016/0092-8674(95)90460-3 (1995). [DOI] [PubMed] [Google Scholar]
- 48.Han KJ et al. Ubiquitin-specific protease 9x deubiquitinates and stabilizes the spinal muscular atrophy protein-survival motor neuron. J Biol Chem 287, 43741–43752, doi: 10.1074/jbc.M112.372318 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Passini MA et al. Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci Transl Med 3, 72ra18, doi: 10.1126/scitranslmed.3001777 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Darras BT et al. Nusinersen in later-onset spinal muscular atrophy: Long-term results from the phase 1/2 studies. Neurology 92, e2492–e2506, doi: 10.1212/WNL.0000000000007527 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Finkel RS et al. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N Engl J Med 377, 1723–1732, doi: 10.1056/NEJMoa1702752 (2017). [DOI] [PubMed] [Google Scholar]
- 52.Finkel RS et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet 388, 3017–3026, doi: 10.1016/S0140-6736(16)31408-8 (2016). [DOI] [PubMed] [Google Scholar]
- 53.Naryshkin NA et al. Motor neuron disease. SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science 345, 688–693, doi: 10.1126/science.1250127 (2014). [DOI] [PubMed] [Google Scholar]
- 54.Zhou M et al. Seamless Genetic Conversion of SMN2 to SMN1 via CRISPR/Cpf1 and Single-Stranded Oligodeoxynucleotides in Spinal Muscular Atrophy Patient-Specific Induced Pluripotent Stem Cells. Hum Gene Ther 29, 1252–1263, doi: 10.1089/hum.2017.255 (2018). [DOI] [PubMed] [Google Scholar]
- 55.Al-Zaidy S et al. Health outcomes in spinal muscular atrophy type 1 following AVXS-101 gene replacement therapy. Pediatr Pulmonol 54, 179–185, doi: 10.1002/ppul.24203 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sun J & Roy S The physical approximation of APP and BACE-1: A key event in alzheimer’s disease pathogenesis. Dev Neurobiol 78, 340–347, doi: 10.1002/dneu.22556 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Das U et al. Visualizing APP and BACE-1 approximation in neurons yields insight into the amyloidogenic pathway. Nat Neurosci 19, 55–64, doi: 10.1038/nn.4188 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Huang YA, Zhou B, Wernig M & Sudhof TC ApoE2, ApoE3, and ApoE4 Differentially Stimulate APP Transcription and Abeta Secretion. Cell 168, 427–441 e421, doi: 10.1016/j.cell.2016.12.044 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Doran E et al. Down Syndrome, Partial Trisomy 21, and Absence of Alzheimer’s Disease: The Role of APP. J Alzheimers Dis 56, 459–470, doi: 10.3233/JAD-160836 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li C & Gotz J Tau-based therapies in neurodegeneration: opportunities and challenges. Nat Rev Drug Discov 16, 863–883, doi: 10.1038/nrd.2017.155 (2017). [DOI] [PubMed] [Google Scholar]
- 61.Tuszynski MH et al. A phase 1 clinical trial of nerve growth factor gene therapy for Alzheimer disease. Nat Med 11, 551–555, doi: 10.1038/nm1239 (2005). [DOI] [PubMed] [Google Scholar]
- 62.Mobley WC et al. Nerve growth factor increases choline acetyltransferase activity in developing basal forebrain neurons. Brain Res 387, 53–62, doi: 10.1016/0169-328x(86)90020-3 (1986). [DOI] [PubMed] [Google Scholar]
- 63.Nagahara AH et al. Long-term reversal of cholinergic neuronal decline in aged non-human primates by lentiviral NGF gene delivery. Exp Neurol 215, 153–159, doi: 10.1016/j.expneurol.2008.10.004 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rafii MS et al. Adeno-Associated Viral Vector (Serotype 2)-Nerve Growth Factor for Patients With Alzheimer Disease: A Randomized Clinical Trial. JAMA Neurol 75, 834–841, doi: 10.1001/jamaneurol.2018.0233 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Castle MJ et al. Postmortem Analysis in a Clinical Trial of AAV2-NGF Gene Therapy for Alzheimer’s Disease Identifies a Need for Improved Vector Delivery. Hum Gene Ther 31, 415–422, doi: 10.1089/hum.2019.367 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nagahara AH et al. MR-guided delivery of AAV2-BDNF into the entorhinal cortex of non-human primates. Gene Ther 25, 104–114, doi: 10.1038/s41434-018-0010-2 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.DeVos SL et al. Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci Transl Med 9, doi: 10.1126/scitranslmed.aag0481 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sud R, Geller ET & Schellenberg GD Antisense-mediated Exon Skipping Decreases Tau Protein Expression: A Potential Therapy For Tauopathies. Mol Ther Nucleic Acids 3, e180, doi: 10.1038/mtna.2014.30 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Velazquez R et al. Acute tau knockdown in the hippocampus of adult mice causes learning and memory deficits. Aging Cell, e12775, doi: 10.1111/acel.12775 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhou L et al. Tau association with synaptic vesicles causes presynaptic dysfunction. Nat Commun 8, 15295, doi: 10.1038/ncomms15295 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yu JT, Tan L & Hardy J Apolipoprotein E in Alzheimer’s disease: an update. Annu Rev Neurosci 37, 79–100, doi: 10.1146/annurev-neuro-071013-014300 (2014). [DOI] [PubMed] [Google Scholar]
- 72.Huang YA, Zhou B, Wernig M & Südhof TC ApoE2, ApoE3, and ApoE4 Differentially Stimulate APP Transcription and Aβ Secretion. Cell 168, 427–441.e421, doi: 10.1016/j.cell.2016.12.044 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shi Y et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549, 523–527, doi: 10.1038/nature24016 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hudry E et al. Gene transfer of human Apoe isoforms results in differential modulation of amyloid deposition and neurotoxicity in mouse brain. Sci Transl Med 5, 212ra161, doi: 10.1126/scitranslmed.3007000 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rosenberg JB et al. AAVrh.10-Mediated APOE2 Central Nervous System Gene Therapy for APOE4-Associated Alzheimer’s Disease. Hum Gene Ther Clin Dev 29, 24–47, doi: 10.1089/humc.2017.231 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lin YT et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron 98, 1294, doi: 10.1016/j.neuron.2018.06.011 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gyorgy B et al. CRISPR/Cas9 Mediated Disruption of the Swedish APP Allele as a Therapeutic Approach for Early-Onset Alzheimer’s Disease. Mol Ther Nucleic Acids 11, 429–440, doi: 10.1016/j.omtn.2018.03.007 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chang JL et al. Targeting Amyloid-beta Precursor Protein, APP, Splicing with Antisense Oligonucleotides Reduces Toxic Amyloid-beta Production. Mol Ther 26, 1539–1551, doi: 10.1016/j.ymthe.2018.02.029 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Muller UC, Deller T & Korte M Not just amyloid: physiological functions of the amyloid precursor protein family. Nat Rev Neurosci 18, 281–298, doi: 10.1038/nrn.2017.29 (2017). [DOI] [PubMed] [Google Scholar]
- 80.Iyer S et al. Precise therapeutic gene correction by a simple nuclease-induced double-stranded break. Nature 568, 561–565, doi: 10.1038/s41586-019-1076-8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fol R et al. Viral gene transfer of APPsalpha rescues synaptic failure in an Alzheimer’s disease mouse model. Acta Neuropathol 131, 247–266, doi: 10.1007/s00401-015-1498-9 (2016). [DOI] [PubMed] [Google Scholar]
- 82.Obeso JA et al. Functional organization of the basal ganglia: therapeutic implications for Parkinson’s disease. Mov Disord 23Suppl 3, S548–559, doi: 10.1002/mds.22062 (2008). [DOI] [PubMed] [Google Scholar]
- 83.Christine CW et al. Safety and tolerability of putaminal AADC gene therapy for Parkinson disease. Neurology 73, 1662–1669, doi: 10.1212/WNL.0b013e3181c29356 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mittermeyer G et al. Long-term evaluation of a phase 1 study of AADC gene therapy for Parkinson’s disease. Hum Gene Ther 23, 377–381, doi: 10.1089/hum.2011.220 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.San Sebastian W et al. Safety and tolerability of magnetic resonance imaging-guided convection-enhanced delivery of AAV2-hAADC with a novel delivery platform in nonhuman primate striatum. Hum Gene Ther 23, 210–217, doi: 10.1089/hum.2011.162 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Palfi S et al. Long-term safety and tolerability of ProSavin, a lentiviral vector-based gene therapy for Parkinson’s disease: a dose escalation, open-label, phase 1/2 trial. Lancet 383, 1138–1146, doi: 10.1016/S0140-6736(13)61939-X (2014). [DOI] [PubMed] [Google Scholar]
- 87.Baron MS, Wichmann T, Ma D & DeLong MR Effects of transient focal inactivation of the basal ganglia in parkinsonian primates. J Neurosci 22, 592–599 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Emborg ME et al. Subthalamic glutamic acid decarboxylase gene therapy: changes in motor function and cortical metabolism. J Cereb Blood Flow Metab 27, 501–509, doi: 10.1038/sj.jcbfm.9600364 (2007). [DOI] [PubMed] [Google Scholar]
- 89.Luo J et al. Subthalamic GAD gene therapy in a Parkinson’s disease rat model. Science 298, 425–429, doi: 10.1126/science.1074549 (2002). [DOI] [PubMed] [Google Scholar]
- 90.Kaplitt MG et al. Safety and tolerability of gene therapy with an adeno-associated virus (AAV) borne GAD gene for Parkinson’s disease: an open label, phase I trial. Lancet 369, 2097–2105, doi: 10.1016/S0140-6736(07)60982-9 (2007). [DOI] [PubMed] [Google Scholar]
- 91.LeWitt PA et al. AAV2-GAD gene therapy for advanced Parkinson’s disease: a double-blind, sham-surgery controlled, randomised trial. Lancet Neurol 10, 309–319, doi: 10.1016/S1474-4422(11)70039-4 (2011). [DOI] [PubMed] [Google Scholar]
- 92.Niethammer M et al. Long-term follow-up of a randomized AAV2-. JCI Insight 2, e90133, doi: 10.1172/jci.insight.90133 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Collier TJ & Sortwell CE Therapeutic potential of nerve growth factors in Parkinson’s disease. Drugs Aging 14, 261–287, doi: 10.2165/00002512-199914040-00003 (1999). [DOI] [PubMed] [Google Scholar]
- 94.Chen YH et al. MPTP-induced deficits in striatal synaptic plasticity are prevented by glial cell line-derived neurotrophic factor expressed via an adeno-associated viral vector. FASEB J 22, 261–275, doi: 10.1096/fj.07-8797com (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Eberling JL et al. Functional effects of AAV2-GDNF on the dopaminergic nigrostriatal pathway in parkinsonian rhesus monkeys. Hum Gene Ther 20, 511–518, doi: 10.1089/hum.2008.201 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Su X et al. Safety evaluation of AAV2-GDNF gene transfer into the dopaminergic nigrostriatal pathway in aged and parkinsonian rhesus monkeys. Hum Gene Ther 20, 1627–1640, doi: 10.1089/hum.2009.103 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Marks WJ et al. Safety and tolerability of intraputaminal delivery of CERE-120 (adeno-associated virus serotype 2-neurturin) to patients with idiopathic Parkinson’s disease: an open-label, phase I trial. Lancet Neurol 7, 400–408, doi: 10.1016/S1474-4422(08)70065-6 (2008). [DOI] [PubMed] [Google Scholar]
- 98.Marks WJ et al. Gene delivery of AAV2-neurturin for Parkinson’s disease: a double-blind, randomised, controlled trial. Lancet Neurol 9, 1164–1172, doi: 10.1016/S1474-4422(10)70254-4 (2010). [DOI] [PubMed] [Google Scholar]
- 99.Bartus RT et al. Bioactivity of AAV2-neurturin gene therapy (CERE-120): differences between Parkinson’s disease and nonhuman primate brains. Mov Disord 26, 27–36, doi: 10.1002/mds.23442 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bartus RT et al. Safety/feasibility of targeting the substantia nigra with AAV2-neurturin in Parkinson patients. Neurology 80, 1698–1701, doi: 10.1212/WNL.0b013e3182904faa (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Decressac M et al. α-Synuclein-induced down-regulation of Nurr1 disrupts GDNF signaling in nigral dopamine neurons. Sci Transl Med 4, 163ra156, doi: 10.1126/scitranslmed.3004676 (2012). [DOI] [PubMed] [Google Scholar]
- 102.Qian H et al. Reversing a model of Parkinson’s disease with in situ converted nigral neurons. Nature 582, 550–556, doi: 10.1038/s41586-020-2388-4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wong YC & Krainc D alpha-synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nat Med 23, 1–13, doi: 10.1038/nm.4269 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Soldner F et al. Parkinson-associated risk variant in distal enhancer of alpha-synuclein modulates target gene expression. Nature 533, 95–99, doi: 10.1038/nature17939 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Fuchs J et al. Genetic variability in the SNCA gene influences alpha-synuclein levels in the blood and brain. FASEB J 22, 1327–1334, doi: 10.1096/fj.07-9348com (2008). [DOI] [PubMed] [Google Scholar]
- 106.Zharikov AD et al. shRNA targeting α-synuclein prevents neurodegeneration in a Parkinson’s disease model. J Clin Invest 125, 2721–2735, doi: 10.1172/JCI64502 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Uehara T et al. Amido-bridged nucleic acid (AmNA)-modified antisense oligonucleotides targeting alpha-synuclein as a novel therapy for Parkinson’s disease. Sci Rep 9, 7567, doi: 10.1038/s41598-019-43772-9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kantor B et al. Downregulation of SNCA Expression by Targeted Editing of DNA Methylation: A Potential Strategy for Precision Therapy in PD. Mol Ther 26, 2638–2649, doi: 10.1016/j.ymthe.2018.08.019 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Benskey MJ et al. Silencing Alpha Synuclein in Mature Nigral Neurons Results in Rapid Neuroinflammation and Subsequent Toxicity. Front Mol Neurosci 11, 36, doi: 10.3389/fnmol.2018.00036 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gorbatyuk OS et al. In vivo RNAi-mediated alpha-synuclein silencing induces nigrostriatal degeneration. Mol Ther 18, 1450–1457, doi: 10.1038/mt.2010.115 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sidransky E et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med 361, 1651–1661, doi: 10.1056/NEJMoa0901281 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Do J, McKinney C, Sharma P & Sidransky E Glucocerebrosidase and its relevance to Parkinson disease. Mol Neurodegener 14, 36, doi: 10.1186/s13024-019-0336-2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sardi SP et al. Augmenting CNS glucocerebrosidase activity as a therapeutic strategy for parkinsonism and other Gaucher-related synucleinopathies. Proc Natl Acad Sci U S A 110, 3537–3542, doi: 10.1073/pnas.1220464110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Morabito G et al. AAV-PHP.B-Mediated Global-Scale Expression in the Mouse Nervous System Enables GBA1 Gene Therapy for Wide Protection from Synucleinopathy. Mol Ther 25, 2727–2742, doi: 10.1016/j.ymthe.2017.08.004 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Gandhi PN, Chen SG & Wilson-Delfosse AL Leucine-rich repeat kinase 2 (LRRK2): a key player in the pathogenesis of Parkinson’s disease. J Neurosci Res 87, 1283–1295, doi: 10.1002/jnr.21949 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hatcher JM, Choi HG, Alessi DR & Gray NS Small-Molecule Inhibitors of LRRK2. Adv Neurobiol 14, 241–264, doi: 10.1007/978-3-319-49969-7_13 (2017). [DOI] [PubMed] [Google Scholar]
- 117.Ness D et al. Leucine-rich repeat kinase 2 (LRRK2)-deficient rats exhibit renal tubule injury and perturbations in metabolic and immunological homeostasis. PLoS One 8, e66164, doi: 10.1371/journal.pone.0066164 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Fuji RN et al. Effect of selective LRRK2 kinase inhibition on nonhuman primate lung. Sci Transl Med 7, 273ra215, doi: 10.1126/scitranslmed.aaa3634 (2015). [DOI] [PubMed] [Google Scholar]
- 119.Baptista MA et al. Loss of leucine-rich repeat kinase 2 (LRRK2) in rats leads to progressive abnormal phenotypes in peripheral organs. PLoS One 8, e80705, doi: 10.1371/journal.pone.0080705 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhao HT et al. LRRK2 Antisense Oligonucleotides Ameliorate α-Synuclein Inclusion Formation in a Parkinson’s Disease Mouse Model. Mol Ther Nucleic Acids 8, 508–519, doi: 10.1016/j.omtn.2017.08.002 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ross CA & Tabrizi SJ Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol 10, 83–98, doi: 10.1016/S1474-4422(10)70245-3 (2011). [DOI] [PubMed] [Google Scholar]
- 122.Kordasiewicz HB et al. Sustained therapeutic reversal of Huntington’s disease by transient repression of huntingtin synthesis. Neuron 74, 1031–1044, doi: 10.1016/j.neuron.2012.05.009 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Tabrizi SJ et al. Targeting Huntingtin Expression in Patients with Huntington’s Disease. N Engl J Med 380, 2307–2316, doi: 10.1056/NEJMoa1900907 (2019). [DOI] [PubMed] [Google Scholar]
- 124.Rodrigues FB, Ferreira JJ & Wild EJ Huntington’s Disease Clinical Trials Corner: June 2019. J Huntingtons Dis 8, 363–371, doi: 10.3233/JHD-199003 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Evers MM et al. AAV5-miHTT Gene Therapy Demonstrates Broad Distribution and Strong Human Mutant Huntingtin Lowering in a Huntington’s Disease Minipig Model. Mol Ther 26, 2163–2177, doi: 10.1016/j.ymthe.2018.06.021 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Boudreau RL et al. Nonallele-specific silencing of mutant and wild-type huntingtin demonstrates therapeutic efficacy in Huntington’s disease mice. Mol Ther 17, 1053–1063, doi: 10.1038/mt.2009.17 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Stanek LM et al. Silencing mutant huntingtin by adeno-associated virus-mediated RNA interference ameliorates disease manifestations in the YAC128 mouse model of Huntington’s disease. Hum Gene Ther 25, 461–474, doi: 10.1089/hum.2013.200 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.McBride JL et al. Preclinical safety of RNAi-mediated HTT suppression in the rhesus macaque as a potential therapy for Huntington’s disease. Mol Ther 19, 2152–2162, doi: 10.1038/mt.2011.219 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Grondin R et al. Six-month partial suppression of Huntingtin is well tolerated in the adult rhesus striatum. Brain 135, 1197–1209, doi: 10.1093/brain/awr333 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wang G, Liu X, Gaertig MA, Li S & Li XJ Ablation of huntingtin in adult neurons is nondeleterious but its depletion in young mice causes acute pancreatitis. Proc Natl Acad Sci U S A 113, 3359–3364, doi: 10.1073/pnas.1524575113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Dietrich P, Johnson IM, Alli S & Dragatsis I Elimination of huntingtin in the adult mouse leads to progressive behavioral deficits, bilateral thalamic calcification, and altered brain iron homeostasis. PLoS Genet 13, e1006846, doi: 10.1371/journal.pgen.1006846 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Shin JW et al. Permanent inactivation of Huntington’s disease mutation by personalized allele-specific CRISPR/Cas9. Hum Mol Genet 25, 4566–4576, doi: 10.1093/hmg/ddw286 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.van Bilsen PH et al. Identification and allele-specific silencing of the mutant huntingtin allele in Huntington’s disease patient-derived fibroblasts. Hum Gene Ther 19, 710–719, doi: 10.1089/hum.2007.116 (2008). [DOI] [PubMed] [Google Scholar]
- 134.Southwell AL et al. In vivo evaluation of candidate allele-specific mutant huntingtin gene silencing antisense oligonucleotides. Mol Ther 22, 2093–2106, doi: 10.1038/mt.2014.153 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Pfister EL et al. Five siRNAs targeting three SNPs may provide therapy for three-quarters of Huntington’s disease patients. Curr Biol 19, 774–778, doi: 10.1016/j.cub.2009.03.030 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Rodrigues FB & Wild EJ Huntington’s Disease Clinical Trials Corner: February 2018. J Huntingtons Dis 7, 89–98, doi: 10.3233/JHD-189001 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zeitler B et al. Allele-selective transcriptional repression of mutant HTT for the treatment of Huntington’s disease. Nat Med 25, 1131–1142, doi: 10.1038/s41591-019-0478-3 (2019). [DOI] [PubMed] [Google Scholar]
- 138.Kim G, Gautier O, Tassoni-Tsuchida E, Ma XR & Gitler AD ALS Genetics: Gains, Losses, and Implications for Future Therapies. Neuron, doi: 10.1016/j.neuron.2020.08.022 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Bruijn LI et al. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science 281, 1851–1854, doi: 10.1126/science.281.5384.1851 (1998). [DOI] [PubMed] [Google Scholar]
- 140.Smith RA et al. Antisense oligonucleotide therapy for neurodegenerative disease. J Clin Invest 116, 2290–2296, doi: 10.1172/JCI25424 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Miller TM et al. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study. Lancet Neurol 12, 435–442, doi: 10.1016/S1474-4422(13)70061-9 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Miller T et al. Phase 1-2 Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N Engl J Med 383, 109–119, doi: 10.1056/NEJMoa2003715 (2020). [DOI] [PubMed] [Google Scholar]
- 143.Foust KD et al. Therapeutic AAV9-mediated suppression of mutant SOD1 slows disease progression and extends survival in models of inherited ALS. Mol Ther 21, 2148–2159, doi: 10.1038/mt.2013.211 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Iannitti T et al. Translating SOD1 Gene Silencing toward the Clinic: A Highly Efficacious, Off-Target-free, and Biomarker-Supported Strategy for fALS. Mol Ther Nucleic Acids 12, 75–88, doi: 10.1016/j.omtn.2018.04.015 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Gaj T et al. In vivo genome editing improves motor function and extends survival in a mouse model of ALS. Sci Adv 3, eaar3952, doi: 10.1126/sciadv.aar3952 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Mueller C et al. SOD1 Suppression with Adeno-Associated Virus and MicroRNA in Familial ALS. N Engl J Med 383, 151–158, doi: 10.1056/NEJMoa2005056 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.DeJesus-Hernandez M et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256, doi: 10.1016/j.neuron.2011.09.011 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Haeusler AR, Donnelly CJ & Rothstein JD The expanding biology of the C9orf72 nucleotide repeat expansion in neurodegenerative disease. Nat Rev Neurosci 17, 383–395, doi: 10.1038/nrn.2016.38 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Jiang J et al. Gain of Toxicity from ALS/FTD-Linked Repeat Expansions in C9ORF72 Is Alleviated by Antisense Oligonucleotides Targeting GGGGCC-Containing RNAs. Neuron 90, 535–550, doi: 10.1016/j.neuron.2016.04.006 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Tuszynski MH et al. Nerve Growth Factor Gene Therapy: Activation of Neuronal Responses in Alzheimer Disease. JAMA Neurol 72, 1139–1147, doi: 10.1001/jamaneurol.2015.1807 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Niethammer M et al. Long-term follow-up of a randomized AAV2-GAD gene therapy trial for Parkinson’s disease. JCI Insight 2, e90133, doi: 10.1172/jci.insight.90133 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Christine CW et al. Magnetic resonance imaging-guided phase 1 trial of putaminal AADC gene therapy for Parkinson’s disease. Ann Neurol 85, 704–714, doi: 10.1002/ana.25450 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.McFarthing K, Prakash N & Simuni T Clinical Trial Highlights: 1. Gene Therapy for Parkinson’s, 2. Phase 3 Study in Focus - Intec Pharma’s Accordion Pill, 3. Clinical Trials Resources. J Parkinsons Dis 9, 251–264, doi: 10.3233/JPD-199001 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Tabrizi SJ et al. Targeting Huntingtin Expression in Patients with Huntington’s Disease. N Engl J Med 380, 2307–2316, doi: 10.1056/NEJMoa1900907 (2019). [DOI] [PubMed] [Google Scholar]
- 155.Darras BT et al. An Integrated Safety Analysis of Infants and Children with Symptomatic Spinal Muscular Atrophy (SMA) Treated with Nusinersen in Seven Clinical Trials. CNS Drugs 33, 919–932, doi: 10.1007/s40263-019-00656-w (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Mercuri E et al. Nusinersen versus Sham Control in Later-Onset Spinal Muscular Atrophy. N Engl J Med 378, 625–635, doi: 10.1056/NEJMoa1710504 (2018). [DOI] [PubMed] [Google Scholar]