

Abstract
This study reveals a new method for the preparation of 1,4-oxazinone derivatives by Staudinger reductive cyclization of functionalized vinyl azide precursors. The resulting oxazinone derivatives prepared in this manner were intercepted with terminal alkyne substrates through an intermolecular cycloaddition/cycloreversion sequence to afford polysubstituted pyridine products. Alkyne substrates bearing propargyl oxygen substitution showed good regioselectivity in the cycloaddition operation selectively affording 2,4,6-substituted pyridines. Application of this chemistry to the synthesis of an ErbB4 receptor inhibitor is also described.
Graphical Abstract

INTRODUCTION
The synthesis of polysubstituted pyridine structures through cycloaddition and cycloreversion strategies is a topic of historic importance1 as well as the focus of current research.2 Through these efforts, a variety of reactive aromatic heterocycles have emerged as valid cycloaddition/cycloreversion precursors capable of constructing diverse pyridine derivatives.3 The isomeric 1,2,4- and 1,2,3-triazines 2 and 3 remain the most commonly explored substrates for cycloaddition/cycloreversion sequences directed toward pyridine products (Figure 1).4,5 Cycloaddition with triazine substrates proceeds favorably within the inverse electron demand paradigm, most often with enamine precursors. Alkyne 2π components have been shown to be competent in the intermolecular cycloaddition with triazines in only a handful of specialized cases.4a,6
Figure 1.
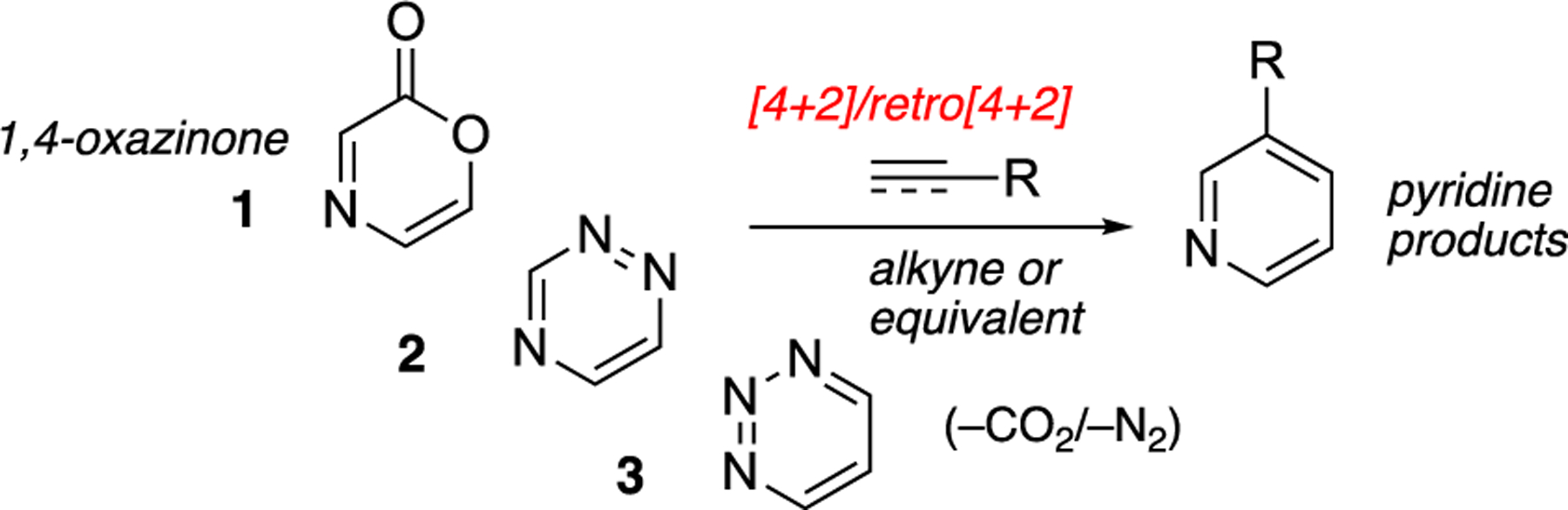
Diels–Alder/retro Diels–Alder sequence with 1,4-oxazinone and triazines.
1,4-Oxazinones (represented by general structure 1) are complementary substrates in [4 + 2]/retro[4 + 2] tandem sequences leading to pyridines. Recent computational efforts suggest that oxazinones are the most reactive precursors for the [4 + 2] reaction component (of the tandem cycloaddition/cycloreversion sequence) and proceed readily with either electron-rich or electron-deficient substrates to yield pyridine products after loss of carbon dioxide.7 Because of the increased reactivity, 1,4-oxazinones are attractive substrates that can enable the use of less reactive 2π cycloaddition reaction components and/or permit use of milder reaction conditions.8 In this way, ubiquitous alkyne precursors may be used routinely and at reaction temperatures that are operationally convenient for the practicing chemist. Pyridine synthesis using oxazinone precursors was established from the foundational work of Hoornaert9 and co-workers but remains obscure in comparison to use of triazines.
We hypothesized that the underutilized nature of oxazinones related to 1 was attributable not to reactivity but to the limited methods available to prepare them. Excluding two examples,10 1,4-oxazinones have been prepared from cyanohydrin derivatives in the presence of excess oxalyl chloride at elevated temperatures (>90 °C).9,11 This method, though direct, suffers from use of toxic cyanohydrin starting materials, harsh reaction conditions, and modest yields for some derivatives (in particular 4a) and necessarily produces the 3,5-dichloro substitution (eq 1, Figure 2). Although further derivation of 3,5-dichlorooxazinones is possible at the more reactive 3-position12 through palladium or SNAr chemistries, room for improvement and additional methods for the construction of 1,4-oxazinones is evident.
Figure 2.
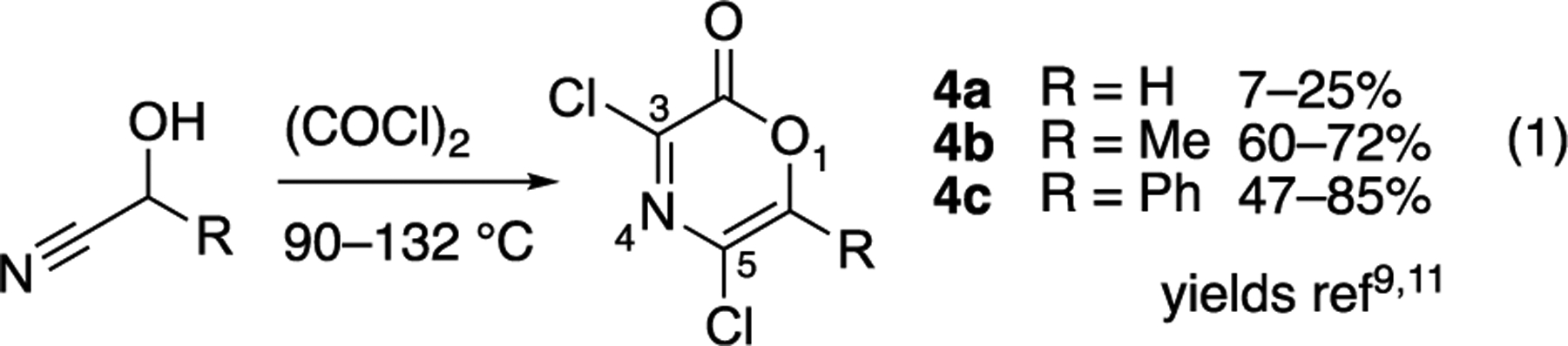
Method for preparation of substituted oxazinone derivatives.
We sought to explore an alternative route that utilized α-amino acid derivatives as starting materials for the construction of 3,5-carbon-substituted oxazinone substrates. Because precedent for cycloaddition of such carbon-substituted oxazinones is limited, we hoped a new synthesis of these derivatives would enable exploration of the reactivity and selectivity in the tandem cycloaddition/cycloreversion sequence.11 To this end, the goals of pyridine synthesis were to confirm the reactivity of substituted oxazinones with terminal alkyne precursors and discern baseline regioselectivity trends.
Because oxazinones bearing 6-H substitution (e.g., 4a) are some of the most challenging to construct using existing methods, we elected to first direct our attention to the construction of these substrates: 1,4-oxazinones with C3, C5, and H6 substitution.
RESULTS AND DISCUSSION
We initiated the synthesis with aldol condensation between anisaldehyde and azidoacetate 5 to give the azidoacrylate 6 (Scheme 1).13 Hydrolysis of 6 followed by direct alkylation of the derived carboxylate with bromoacetophenone cleanly delivered product 8. Staudinger reduction with PPh3 was performed at reflux in toluene (for 12 or more hours) in order to affect the subsequent intramolecular cyclization of the derived aza-Wittig intermediate on the pendant ketone. The concentration of the reaction mixture revealed both oxazinone isomers 9 and 10. The observed mixture of isomers 9 and 10 (~1:1) appears to be the thermodynamic equilibrium ratio; efforts to perturb this ratio with additional time, heat, or additives (e.g., catalytic NEt3) did not alter the isomeric ratio. Given this observation, we elected to explore ‘one-pot’ reaction conditions whereby Staudinger reductive cyclization, alkene isomerization, cycloaddition (with the alkyne), and cycloreversion (extrusion of CO2) would be executed in a single reaction vessel. In order to facilitate a more rapid isomerization of the exocyclic-alkene in 9 to the Diels–Alder-reactive endocyclic oxazinone 10, catalytic NEt3 was added in conjunction with the alkyne. These reaction conditions proved effective in constructing the isomeric 2,3- and/or 2,4-pyridine derivatives 11a and 11b and formed the basis of our initial investigations into this tandem reaction sequence.
Scheme 1.

Synthesis of 1,4-Oxazinone and Tandem [4 + 2]/Retro[4 + 2] Reaction to Give Substituted Pyridines
Using the reaction conditions highlighted above, we began exploring the reaction scope of the key intermediate oxazinone 10 in order to demonstrate both cycloaddition reactivity with several alkyne components and to establish meaningful regioselectivity trends with these substrates. Table 1 describes our preliminary results evaluating a series of terminal alkyne reaction components 12a–f. Notably, aryl substitution on the alkyne (phenylacetylene 12a, entry 1) showed favorable 9:1 selectivity for predominantly the 2,3-regioisomer 13a. A reversal in regioselectivity was observed when the reaction sequence was executed with trimethylsilylacetylene (12b, entry 2). Accordingly, 2,4-substituted pyridine 14b was isolated as the major product (regioselection 1:7). Pyridine yield in this case is diminished, an observation we attribute to the high volatility of alkyne 12b (bp 53 °C), which both lowered the internal reaction reflux temperature and led to some evaporation of the precursor during the course of reaction. Consistent with this hypothesis, a small amount of unreacted oxazinone mix 9 and 10 (21% yield, ca. 1:1 isomeric mixture) was also observed in this case using the standard reaction conditions. Separation of the isomeric mixture 14a and 14b was challenging, and a mixed fraction (ca. 1:1 isomeric mixture, 22% yield) was also obtained. When the sequence was performed with propargyl alcohol (12c, entry 3), the major product was again the 2,4-regioisomer and a 1:5 ratio of pyridines 15a and 15b was obtained in 69% combined yield. Success of this reaction demonstrates that the overall reaction sequence leading to pyridine products is tolerant of primary alcohol functional groups, a welcome feature given the proclivity of lactone functionalities to undergo ring opening and polymerization.
Table 1.

|
Key: Isolated yield of the major isomer.
An additional 22% yield containing mixed 14a,b and an unreacted oxazinone precursor (21% yield) was obtained.
Isomers inseparable by chromatography. Yield refers to the combined yield of both isomers.
Reaction conditions: a mixture of 8 (1 equiv) and PPh3 (1 equiv) were heated to reflux in toluene. Following completion of the cyclization, the alkyne and catalytic NEt3 were subsequently added and the reaction returned to reflux.
Because useful cycloaddition regioselectivity was achieved with propargyl alcohol, we selected modified precursors bearing propargyl heteroatom substitution in order to evaluate subtle electronic and steric components. Propargyl ether 12d (entry 4) performed similarly to the aforementioned propargyl alcohol, producing a 1:4 ratio of the 2,3- and 2,4-regioisomeric pyridines 16a and 16b was obtained in 70% combined yield. We anticipated that propargyl acetal 12e would offer enhanced regioselection. To this end, acetal 12e (entry 5) was employed and analysis of the resulting unpurified reaction mixture revealed a 1:14 ratio mixture of the pyridine products. The major 2,4-pyridine 17b was isolated in 78% yield.
More electron-deficient substrates, such as methyl propiolate 12f, were poor substrates for the tandem reaction sequence. When propiolate 12f was employed (entry 6), the reaction was poorly regioselective (2:1) and led to low yields of desired pyridine products, which could not be effectively separated, as well as other undesired unidentified products. We do not have an explanation for the low regioselectivity in this case; however, the low yield is likely attributed to other reaction pathways accessible to electron-deficient alkynes, such as a Michael reaction and other conjugate addition processes.
Given the favorable regioselection for the formation of 1,3-pyridine isomers with alkynes bearing propargylic oxygen functionalization, we sought to apply this method to the construction of target molecule 22, a selective ErbB4 inhibitor that was discovered as part of a library synthesis.14 Although the related membrane receptor kinases ErbB1 and ErbB2 receptors are well-characterized targets for cancer therapeutics, much less is understood about ErbB4 and its effects on disease. Gene expression and genetic analysis suggest that the receptor is involved in the progression of breast cancer and other disorders, and the ErbB4 gene is overexpressed or mutated in several solid tumors.15 Screening efforts identified pyridine 22 as a potent inhibitor of the ErbB4 receptor, with an EC50 of 0.30 μM.14 Development of a versatile synthetic sequence for the preparation of inhibitor 22 (and related structures) would potentially facilitate exploration on the role of the ErbB4 receptor and the development of novel treatments.
With minor changes to the sequence previously described (in Scheme 1), we devised a modified route intercepting oxazinone 20 for the cycloaddition/cycloreversion operation that would provide access to pyridine 22. To this end, the route was initiated with aldol condensation with trimethoxybenzaldehyde (Scheme 2) and further elaboration to the azido cinnamate precursor 19 was accomplished in 50% yield over the three steps. Adhering to the ‘one pot’ conditions previously described, PPh3 was added, which led to Staudinger reductive cyclization. The derived isomerized oxazinone intermediate 20 (not isolated) was engaged in productive cycloaddition/cycloreversion with the propargylic pentynol 21a. In this way, pyridine 22 was afforded as the major isomer (regioselection 1:8 as judged by 1H NMR) and was isolated in 78% yield following careful chromatographic separation on silica gel. We hypothesized that a more electron-withdrawing propargyl ester would potentially enhance the observed ratio of the 2,4-isomer relative to the isomeric 2,3-isomer. Accordingly, the analogous sequence was performed with propargylic benzoate 21b. A similar efficiency (79% yield) was observed in the reaction sequence; however, our hypothesis regarding regioselection was not realized and a slightly lower 1:6 ratio was observed also favoring the 2,4 isomer.
Scheme 2.

Synthesis of the ErbB4 Inhibitor from the 1,4-Oxazinone Intermediate
In conclusion, we have devised a new method for the synthesis of Diels–Alder/retro-Diels–Alder reactive 1,4-oxazinone structures and have exploited this intermediate in the construction of substituted pyridine derivatives. The key reaction was executed in a single reaction vessel, whereby a Staudinger reductive cyclization precedes an alkene isomerization to the reactive oxazinone, and is intercepted by intermolecular [4 + 2] cycloaddition and tandem cycloreversion (−CO2). Several terminal alkyne reaction partners were evaluated in the reaction sequence to afford trisubstituted pyridine derivatives. We determined that phenylacetylene favored the 2,3-isomer and derivatives with polar heteroatom functionalization at the propargylic site show good regioselection for the pyridine bearing the alternate 2,4-substitution pattern. Application of this chemistry to the synthesis of a known ErbB4 inhibitor was realized. Further efforts to advance this methodology in the selective preparation of other complex pyridine derivatives, including tetra- and penta-substituted variants, are ongoing.
EXPERIMENTAL SECTION
General Experimental Considerations.
All reactions were carried out under an atmosphere of nitrogen in flame-dried or oven-dried glassware with magnetic stirring unless otherwise indicated. Acetonitrile, THF, toluene, and Et2O were degassed with argon and purified by passage through a column of molecular sieves and a bed of activated alumina. Dichloromethane was distilled from CaH2 prior to use. All reagents were used as received unless otherwise noted. Flash column chromatography was performed using SiliCycle siliaflash P60 silica gel (230–400 mesh). Analytical thin layer chromatography was performed on SiliCycle 60 Å glass plates. Visualization was accomplished with UV light, anisaldehyde, ceric ammonium molybdate, potassium permanganate, or ninhydrin followed by heating. Film infrared spectra were recorded using a Digilab FTS 7000 FTIR spectrophotometer. Optical rotations were determined using a Perkin-Elmer 341 polarimeter at 25 °C. 1H NMR spectra were recorded on a Varian Mercury 400 (400 MHz) spectrometer and are reported in ppm using solvent as an internal standard (CDCl3 at 7.26 ppm) or tetramethylsilane (0.00 ppm). Proton-decoupled 13C-NMR spectra were recorded on a 100 MHz spectrometer and are reported in ppm using solvent as an internal standard (CDCl3 at 77.00 ppm). All compounds were judged to be homogeneous (>95% purity) by 1H and 13C NMR spectroscopy. Mass spectral data analysis was performed through positive electrospray ionization (w/NaCl) using a Bruker 12 Tesla APEX–Qe FTICR-MS with an Apollo II ion source.
Methyl (Z)-2-azido-3-(4-methoxyphenyl)acrylate (6).
A dry flask was charged with sodium metal (1.00 g, 43.5 mmol) and allowed to dissolve in MeOH (22 mL). After 1 h at room temperature, the NaOMe solution was cooled to −10 °C. 4-Methoxybenzaldehyde and azidomethyl acetate were inserted into an addition funnel with MeOH (22 mL), and the reagents were added dropwise to the cold NaOMe solution in MeOH over 15 min. The reaction was let to stir at −10 °C for 5.5 h and then warmed to 5 °C over 1 h. The reaction was quenched with 1 M HCl and partitioned between a 1:1 mixture of Et2O/H2O (150 mL each). The organic layer was removed, and the aqueous layer was extracted with Et2O (3 × 30 mL). The combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated. The resulting residue was purified by flash column chromatography on silica gel (gradient elution: 0% EtOAc to 20% EtOAc in hexanes) to afford acrylate 6 (3.82 g, 16.4 mmol, 66% yield) as bright yellow crystals: TLC (20% EtOAc in hexane), Rf: 0.70 (CAM). Spectral data for the azido acrylate 6 are in agreement with published data.16
2-Oxo-2-phenylethyl (Z)-2-azido-3-(4-methoxyphenyl)-acrylate (8).
A dry flask was charged with acrylate 6 (1.00 g, 4.3 mmol) and dissolved in warm MeOH (25 mL). In a 25 mL Erlenmeyer flask, LiOH (360 mg, 8.5 mmol) was dissolved in water (17 mL). The aqueous solution was added to the reaction flask, and the vessel was fitted with a Vigreux condenser and heated on an aluminum heating block to 40 °C. After stirring for 15 h, the reaction was cooled to room temperature, acidified with 1 M HCl, and extracted with EtOAc (50 mL × 4). The combined organic layers were washed with brine, dried over Na2SO4, and filtered. Concentration in vacuo yielded the intermediate acrylic acid, which was dissolved in butanone (14 mL). The flask was fitted with a Vigreux condenser, and bromoacetophenone (824 mg, 4.1 mmol) and K2CO3 (571 mg, 4.1 mmol) were added in one portion at room temperature. The reaction was stirred for 3 h at 40 °C and then cooled slightly prior to dilution of the mixture with 1:1 Et2O/hexanes (20 mL) and filtration through Celite. The filtrate was concentrated in vacuo, and the resulting solid was washed with cold 1:3 Et2O/hexanes (10 mL × 3) using a Buchner filter to remove the small amount of excess bromoacetophenone. After drying the solid under vacuum, the resulting product 8 (1.09 g, 3.2 mmol, 78% yield) was obtained as a light yellow solid that was used without further purification: mp: 113.4–115.8 °C; TLC (20% EtOAc in hexane) Rf: 0.50 (CAM); IR (film) 2114, 1693, 1595, 1259, 1176, 1026, 835, 732, 646, cm−1; 1H NMR (400 MHz, CDCl3) 7.95 (dd, J1 = 7.0 Hz, J2 = 1.6 Hz, 2H), 7.84 (d, J = 8.6 Hz, 2H), 7.64 (t, J = 7.4 Hz, 1H), 7.52 (ap. T, J = 7.63 Hz, 2H), 7.09 (s, 1H), 6.92 (d, J = 9.0, 2H), 5.55 (s, 2H), 3.86 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 191.4, 163.2, 160.6, 134.1, 134.0, 132.6, 128.9, 127.8, 126.8, 125.9, 122.4, 113.9, 66.9, 55.3; exact mass calcd for C18H15N3O4Na+ 360.0954, found 360.0945.
2-(4-Methoxybenzyl)-3,6-diphenylpyridine (13a) and 2-(4-Methoxybenzyl)-4,6-diphenylpyridine (13b).
A dry flask was charged with acrylate 8 (58.2 mg, 0.17 mmol) and dissolved in PhMe (2 mL). The flask was fitted with a water condenser, and PPh3 (45.2 mg, 0.17 mmol) was added in one portion at room temperature. After gas evolution steadied, the reaction mixture was heated to reflux for 20 h using an aluminum heating block. The reaction was allowed to cool to 90 °C before adding phenylacetylene (35.7 mg, 0.35 mmol) and one drop (10 μL) of triethylamine. The reaction temperature was heated to reflux again and allowed to stir for 5 h. The reaction mixture was cooled to room temperature and concentrated in vacuo. 1H NMR analysis of the unpurified mixture revealed a 9:1 mixture of regioisomeric pyridine products 13a and 13b. This mixture was purified by flash column chromatography on silica gel (gradient elution: 5% EtOAc to 40% EtOAc in hexanes) to afford pyridine 13a (41.8 mg, 0.12 mmol, 70% yield) and pyridine 13b (5 mg, 0.01 mmol, 8% yield), both as yellow oils.
Data for compound 13a: TLC (20% EtOAc in hexane), Rf: 0.65 (CAM); IR (film) 3057, 3030, 2951, 2833, 1608, 1508, 1244, 759, 731, cm−1; 1H NMR (400 MHz, CDCl3) 8.09–8.07 (m, 2H), 7.64 (d, J = 7.8 Hz, 1H), 7.57 (d, J = 8.2 Hz, 1H), 7.50–7.46 (m, 2H), 7.43–7.39 (m, 4H), 7.26–7.24 (m, 2H), 7.05 (d, J = 8.6 Hz, 2H), 6.74 (d, J = 8.6 Hz, 2H), 4.14 (s, 2H), 3.75 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 157.8, 157.7, 155.6, 139.8, 139.3, 138.5, 135.6, 132.3, 129.8, 129.3, 128.8, 128.7, 128.6, 128.3, 127.4, 126.9, 117.7, 113.5, 55.2, 40.9. Exact mass calcd for C25H21NONa + 374.1516, found 374.1516.
Data for compound 13b: TLC (20% EtOAc in hexane), Rf: 0.60 (CAM); IR (film) 3057, 3030, 2951, 2833, 1608, 1508, 1244, 759, 731, cm−1; 1H NMR (400 MHz, CDCl3) 8.08–8.06 (m, 2H), 7.74 (d, J = 1.6 Hz, 1H), 7.62–7.60 (m, 2H), 7.51–7.41 (m, 6H), 7.30 (d, J = 8.6, 2H), 7.22 (d, J = 1.2 Hz, 1H), 6.87 (d, J = 9.0 Hz, 2H), 4.24 (s, 2H), 3.80 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 161.8, 158.2, 157.5, 149.8, 139.8, 138.9, 131.8, 130.2, 129.0, 128.84, 128.82, 128.7, 127.14, 127.11, 119.5, 116.4, 114.0, 55.2, 44.1. Exact mass calcd for C25H21NONa + 374.1516, found 374.1516.
2-(4-Methoxybenzyl)-6-phenyl-4-(trimethylsilyl)pyridine (14b).
A dry flask was charged with acrylate 8 (100 mg, 0.3 mmol) and dissolved in PhMe (1 mL). The reaction flask was fitted with a water condenser, and PPh3 (78 mg, 0.3 mmol) was added in one portion at room temperature. After gas evolution steadied, the reaction mixture was heated to reflux for 2 h using an aluminum heating block. The reaction was allowed to cool to 60 °C before adding TMS acetylene (206 mg, 2.1 mmol) and one drop (10 μL) of triethylamine. The reaction temperature was heated to reflux and allowed to stir for an additional 16 h. The reaction mixture was cooled to room temperature and concentrated in vacuo. 1H NMR analysis of the unpurified mixture revealed a 1:6 mixture of regioisomeric pyridine products. This mixture was purified by flash column chromatography on silica gel (gradient elution: 0% EtOAc to 30% EtOAc in hexanes), and the major isomeric pyridine 14b (42 mg, 0.12 mmol, 40% yield) was obtained as a yellow oil. An additional amount (22 mg, 22% yield) of mixed fractions containing both 14a and 14b was also obtained (1:1 ratio). A small amount of unreacted oxazinone precursors 9 and 10 (26 mg, 21% yield, ca. 1:1 ratio) was also isolated. Data for compound 14b: TLC (20% EtOAc in hexane), Rf: 0.80 (CAM); IR (film) 2974, 2929, 2895, 2883, 1607, 1510, 1246, 1053, 1033, 692 cm−1; 1H NMR (400 MHz, CDCl3) 8.00–7.98 (m, 2H), 7.62 (s, 1H), 7.48–7.45 (m, 2H), 7.41–7.39 (m, 1H), 7.29–7.25 (m, 2H), 7.15 (s, 1H), 6.85 (d, J = 8.0 Hz, 2H), 4.16 (s, 2H), 3.79 (s, 3H), 0.28–0.26 (m, 9H); 13C{1H} NMR (100 MHz, CDCl3) δ 159.8, 158.0, 155.8, 151.3, 10.1, 132.1, 130.0, 128.6, 128.5, 127.1, 125.7, 122.4, 113.8, 55.2, 43.9. −1.6; exact mass calcd for C22H25NOSiNa+ 370.1598, found 370.1598.
(2-(4-Methoxybenzyl)-6-phenylpyridin-3-yl)methanol (15a) and (2-(4-Methoxybenzyl)-6-phenylpyridin-4-yl)methanol (15b).
A dry flask was charged with acrylate 8 (50 mg, 0.15 mmol) and dissolved in PhMe (1.0 mL). The flask was fitted with a water condenser, and PPh3 (39.3 mg, 0.15 mmol) was added in one portion at room temperature. After gas evolution steadied, the reaction mixture was heated to reflux for 8 h using an aluminum heating block. The reaction was cooled to 90 °C before addition of propargyl alcohol (16.8 mg, 0.3 mmol) and one drop (10 μL) of triethylamine. The reaction temperature was heated to reflux and stirred for 6 h. After cooling to room temperature, the reaction mixture was concentrated in vacuo. 1H NMR analysis of the unpurified mixture revealed a 1:5 mixture of regioisomeric pyridine products 15a and 15b. This mixture was purified by flash column chromatography on silica gel (gradient elution: 5% EtOAc to 45% EtOAc in hexanes). The resulting pyridines 15a and 15b (31.6 mg, 0.10 mmol, 69% yield) proved inseparable and were obtained as a yellow oil: TLC (20% EtOAc in hexane), Rf: 0.10 (CAM); IR (film) 3289, 3059, 2908, 2833, 1608, 1560, 1508, 1417, 1244, 1176, 1029, 771 cm−1; 1H NMR (400 MHz, CDCl3, data for 15a) 8.01–7.99 (m, 2H), 7.51 (s, 1H), 7.48–7.37 (m, 3H), 7.27–7.24 (m, 2H), 6.96 (s, 1H), 6.84 (d, J = 8.6 Hz, 2H), 4.66 (s, 2H), 4.16 (s, 2H) 3.78 (s, 3H); (1H NMR data for 15b) 8.05–8.03 (m, 2H), 7.74 (d, J = 8.2 Hz, 1H), 7.60 (d, J = 7.8 Hz, 1H), 7.48–7.37 (m, 3H), 7.19 (d, J = 8.6 Hz, 2H), 6.80 (d, J = 8.6 Hz, 2H), 4.66 (s, 2H), 4.23 (s, 2H), 3.75 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3, data for mixture of 15a and 15b) δ 161.5, 158.1, 157.1150.9, 139.5, 136.7, 131.7, 131.3, 130.2, 129.6, 128.81, 128.77, 128.7, 128.6, 127.0, 126.9, 118.6, 118.4, 115.5, 113.92, 113.91, 63.8, 61.9, 55.22, 55.21, 43.9, 41.0; exact mass calcd for C20H19NO2Na+ 328.1308, found 328.1307.
3-((Benzyloxy)methyl)-2-(4-methoxybenzyl)-6-phenylpyridine (16a) and 4-((Benzyloxy)methyl)-2-(4-methoxybenzyl)-6-phenylpyridine (16b).
A dry flask was charged with acrylate 8 (85 mg, 0.25 mmol) and dissolved in PhMe (1.5 mL). The flask was fitted with a water condenser, and PPh3 (66 mg, 0.25 mmol) was added in one portion at room temperature. After gas evolution steadied, the reaction mixture was heated to reflux for 16 h using an aluminum heating block. The reaction was allowed to cool to 90 °C before addition of benzyl propargyl ether (74 mg, 0.5 mmol) and one drop (10 μL) of triethylamine. The reaction temperature was returned to reflux and stirred for 5 h. The reaction mixture was cooled to room temperature and concentrated in vacuo. 1H NMR analysis of the unpurified mixture revealed a 1:4 mixture of regioisomeric pyridine products 16a and 16b. This mixture was purified by flash column chromatography on silica gel (gradient elution: 5% EtOAc to 20% EtOAc in hexanes). The resulting pyridines 16a and 16b (83 mg, 0.21 mmol, 70% yield) proved inseparable and were obtained as a yellow oil: TLC (20% EtOAc in hexane), Rf: 0.50 (CAM); IR (film) 3057, 3032, 2833, 1603, 1558, 1510, 1244, 1178, 819, 777, 731, 694 cm−1; 1H NMR (400 MHz, CDCl3, data for 16a) 8.03–8.01 (m, 2H), 7.54 (s, 1H), 7.48–7.30 (m, 8H), 7.27–7.25 (m, 2H), 6.98 (s, 1H), 6.86 (d, J = 8.6 Hz, 2H), 4.56 (s, 2H), 4.53 (s, 2H), 4.17 (s, 2H) 3.79 (s, 3H); (1H NMR data for 16b) 8.06–8.05 (m, 2H), 7.74 (d, J = 7.8 Hz, 2H), 7.59 (d, J = 7.8 Hz, 2H), 7.48–7.30 (m, 8H), 7.13 (d, J = 8.6 Hz, 2H), 6.77 (d, J = 8.6 Hz, 2H), 4.53 (d, J = 6.7 Hz, 2H), 4.21 (s, 2H), 3.76 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3, data for the mixture of 16a and 16b) δ 161.5, 158.2, 157.1, 148.6, 139.6, 137.7, 137.5, 131.8, 131.4, 130.2, 130.0, 129.8, 128.8, 128.76, 128.7, 128.5, 128.48, 127.9, 127.88, 127.84, 128.82, 127.1, 126.9, 119.5, 118.1, 116.3, 113.9, 113.7, 72.7, 72.65, 70.7, 68.9, 55.3, 55.2, 44.0, 40.8; exact mass calcd for C27H25NO2Na+ 418.1778, found 418.1778.
4-(Diethoxymethyl)-2-(4-methoxybenzyl)-6-phenylpyridine (17b).
A dry flask was charged with acrylate 8 (150 mg, 0.44 mmol) and dissolved in PhMe (3 mL). The flask was fitted with a water condenser, and PPh3 (116.5 mg, 0.44 mmol) was added in one portion at room temperature. After gas evolution steadied, the reaction mixture was heated to reflux for 6 h using an aluminum heating block. The reaction was cooled to 90 °C before addition of propargylaldehyde diethyl acetal (85.5 mg, 0.67 mmol) and one drop (10 μL) of triethylamine. The reaction temperature was returned to reflux and stirred for 16 h, cooled to room temperature, and concentrated in vacuo. 1H NMR analysis of the unpurified mixture revealed a 1:14 mixture of regioisomeric pyridine products. This mixture was purified by flash column chromatography on silica gel (gradient elution: 2% EtOAc to 20% EtOAc in hexanes) to afford pyridine 17b (129.5 mg, 0.34 mmol, 78% yield) as a yellow oil. Additionally, a small amount of the mixed product containing both 17a and 17b was also obtained (7 mg, 4% yield). Data for compound 17b: TLC (20% EtOAc in hexane), Rf: 0.50 (CAM); IR (film) 2974, 2929, 2895, 2883, 1607, 1510, 1246, 1053, 1033, 692 cm−1; 1H NMR (400 MHz, CDCl3) 8.05–8.02 (m, 2H), 7.65 (d, J = 0.8 Hz, 1H), 7.48–7.44 (m, 2H), 7.41–7.39 (m, 1H), 7.26 (d, J = 8.6 Hz, 2H), 7.11 (s, 1H), 6.85 (d, J = 8.6 Hz, 2H), 5.46 (s, 1H), 4.18 (s, 2H), 3.79 (s, 3H), 3.62–3.51 (m, 4H), 1.23 (t, J = 7.0 Hz, 6H); 13C{1H} NMR (100 MHz, CDCl3) δ 161.5, 158.1, 157.1, 148.8, 139.6, 131.8, 130.1, 128.8, 128.6, 119.3, 115.9, 113.9, 100.2, 61.3, 55.3, 44.0; exact mass calcd for C24H27NO3H+ 378.2064, found 378.2063.
Methyl (Z)-2-azido-3-(3,4,5-trimethoxyphenyl)acrylate.
A dry flask was charged with sodium metal (0.5 g, 21.8 mmol) and dissolved in MeOH (10 mL) with stirring at room temperature for 1 h and cooled to −10 °C. 2,3,4-Trimethoxybenzaldehyde and azidomethyl acetate were inserted into an addition funnel and diluted with MeOH (10 mL), and the reagents were added dropwise to the NaOMe/MeOH mixture over 20 min. The reaction was stirred at −10 °C for 6 h and warmed to 5 °C over the course of 1 h. The reaction was quenched with 1 M HCl and partitioned between a 1:1 mixture of Et2O/H2O (150 mL). The aqueous portion was removed and extracted with additional Et2O (20 mL × 3). The combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated. The resulting residue was purified by flash column chromatography on silica gel (gradient elution: 2% EtOAc to 35% EtOAc) to afford the acrylate product (1.28 g, 4.37 mmol, 43% yield) as a light yellow crystal: TLC (20% EtOAc in hexane), Rf: 0.45 (CAM). Spectral data for the azido acrylate product are in agreement with published data.17
2-Oxo-2-phenylethyl (Z)-2-azido-3-(3,4,5-trimethoxyphenyl)acrylate (19).
A dry flask was charged with methyl (Z)-2-azido-3-(3,4,5-trimethoxyphenyl)acrylate, the azidoacrylate (767 mg, 2.62 mmol), and dissolved in warm MeOH (5 mL) and THF (10 mL). In a 25 mL Erlenmeyer flask, LiOH (274 mg, 6.5 mmol) was dissolved in water (13 mL). The aqueous solution was added to the flask, and the reaction vessel was fitted with a Vigreux condenser and stirred at 55 °C for 1.5 h using an aluminum heating block. After cooling to room temperature, the reaction mixture was acidified with 1 M HCl and extracted with EtOAc (30 mL × 4). The combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentration in vacuo. The resulting acrylic acid (700 mg, 2.5 mmol) was dissolved in butanone (11 mL), and the reaction flask was fitted with a Vigreux condenser. To this reaction flask, bromoacetophenone (636 mg, 3.2 mmol) and K2CO3 (450 mg, 3.3 mmol) were added, and the reaction was warmed to 40 °C using an aluminum heating block. After stirring for 7 h, the reaction was cooled slightly and diluted with 1:1 Et2O/hexanes (20 mL) and filtered using a glass frit and Celite. The filtrate was concentrated in vacuo, and the resulting solid was washed with cold 1:3 Et2O/hexanes (3 × 10 mL) using a Buchner filter to remove a small amount of excess bromoacetophenone. The resulting acrylate 19 (801 mg g, 2.0 mmol, 50% yield) was obtained as a light yellow solid and used without further purification: mp: 117.1–119.2 °C; TLC (20% EtOAc in hexane) Rf: 0.25 (CAM); IR (film) 2938, 2122, 1712, 1695, 1417, 1242, 1221, 1124, 748, 688, 669 cm−1; 1H NMR (400 MHz, CDCl3) 7.97–7.94 (m, 2H), 7.67–7.63 (m, 1H), 7.55–7.51 (m, 2H), 7.14 (s, 2H), 7.05 (s, 1H), 5.57 (s, 2H), 3.91 (s, 9H); 13C{1H} NMR (100 MHz, CDCl3) δ 191.2, 163.0, 152.8, 139.5, 134.1, 133.9, 128.9, 128.4, 127.7, 126.7, 123.8, 108.2, 67.0, 60.9, 56.1; exact mass calcd for C20H19N3O6Na+ 420.1166, found 420.1165.
1-(2-Phenyl-6-(3,4,5-trimethoxybenzyl)pyridin-4-yl)propan-1-ol (22).
A dry flask was charged with acrylate 19 (100 mg, 0.25 mmol) and dissolved in PhMe (1.75 mL). The flask was fitted with a water condenser, and PPh3 (66 mg, 0.25 mmol) was added in one portion at room temperature. After gas evolution steadied, the reaction mixture was heated to reflux for 16 h using an aluminum heating block. The reaction was cooled to 90 °C before adding 1-pentyn-3-ol (42 mg, 0.5 mmol) and one drop (10 μL) of triethylamine. The reaction temperature was returned to reflux and stirred for 6 h. The reaction mixture was cooled to room temperature and concentrated in vacuo. 1H NMR analysis of the unpurified mixture revealed a 1:8 mixture of regioisomeric pyridine products. This mixture was purified by flash column chromatography on silica gel (gradient elution: 5% EtOAc to 40% EtOAc in hexanes) to afford pyridine 22 (76.7 mg, 0.20 mmol, 78% yield) as a yellow oil: TLC (40% EtOAc in hexane), Rf: 0.30 (CAM); IR (film) 3403, 2933, 1589, 1504, 1417, 1236, 1122, 972, 777.3, 723.3, 694.4 cm−1; 1H NMR (400 MHz, CDCl3) 8.02 (d, J = 8.4 Hz, 2H), 7.55 (s, 1H), 7.49–7.40 (m, 3H), 7.02 (s, 1H), 6.59 (s, 2H), 4.62 (t, J = 6.5 Hz, 1H), 4.15 (s, 2H), 3.83 (s, 9H), 2.02 (bs, 1H) 1.78–1.73 (m, 2H), 0.94 (t, J = 7.4 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 160.8, 157.1, 154.6, 153.1, 139.5, 136.4, 135.3, 128.8, 128.7, 127.0, 118.6, 115.4, 106.2, 74.6, 60.8, 56.1, 45.1, 31.7, 9.8; exact mass calcd for C24H27NO4Na+ 416.1832, found 416.1830.
Pent-1-yn-3-yl benzoate (21b).
A dry flask was charged with 1-pentyn-3-ol (890 mg, 10.6 mmol) and dissolved in CH2Cl2 (45 mL). Triethylamine and DMAP were added, and the reaction was allowed to stir for 5 min. The reaction temperature was cooled to 0 °C, and benzoyl chloride was added dropwise. The mixture stirred for 30 min, warmed to room temperature, and stirred for 1 h. The reaction mixture was acidified with 1 M HCl (11 mL) and water (15 mL) and extracted with CH2Cl2 (30 mL × 4). The combined organic layers were washed with sat. aqueous NaHCO3 (10 mL × 2), brine, dried over Na2SO4, and filtered and concentrated. The resulting residue was purified by flash column chromatography on silica gel (gradient elution: 0% EtOAc to 20% EtOAc). The resulting alkyne product 21b (1.61 g, 8.55 mmol, 81% yield) was obtained as a colorless oil: TLC (10% EtOAc in hexanes), Rf: 0.60 (CAM). Spectral data for the alkyne 21b are in agreement with published data.18
1-(2-Phenyl-6-(3,4,5-trimethoxybenzyl)pyridin-4-yl)propyl benzoate (23).
A dry flask was charged with acrylate 19 (100 mg, 0.25 mmol) and dissolved in PhMe (0.85 mL). The flask was fitted with a water condenser, and PPh3 (66 mg, 0.25 mmol) was added in one portion at room temperature. After gas evolution steadied, the reaction mixture was heated to reflux for 6 h using an aluminum heating block. The reaction was cooled to 90 °C before adding benzoate 21b (140 mg, 0.75 mmol) and one drop (10 μL) of triethylamine. The reaction temperature was returned to reflux and stirred for 16 h. The reaction mixture was cooled to room temperature and concentrated in vacuo. 1H NMR analysis of the unpurified mixture revealed a 1:6 mixture of regioisomeric pyridine products. This mixture was purified by flash column chromatography on silica gel (gradient elution: 5% EtOAc to 40% EtOAc in hexanes) to afford pyridine 23 (98.8 mg, 0.20 mmol, 79% yield) as a yellow oil. Data for compound 23: TLC (20% EtOAc in hexane), Rf: 0.30 (CAM); IR (film) 2966, 2936, 1716, 1589, 1450, 1419, 1269, 1240, 1122, 1107, 1070, 711, 694 cm−1; 1H NMR (400 MHz, CDCl3) 8.05 (d, J = 7.0 Hz, 2H), 8.02 (d, J = 7.1 Hz, 2H), 7.61–7.57 (m, 1H), 7.55 (s, 1H), 7.48–7.40 (m, 5H), 7.05 (s, 1H), 6.55 (s, 2H), 5.90 (t, J = 6.4 Hz, 1H), 4.17 (s, 2H), 3.82 (s, 3H), 3.79 (s, 6H), 2.04–1.98 (m, 2H), 0.99 (t, J = 7.4 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 165.8, 161.1, 157.3, 153.2, 150.8, 139.5, 136.5, 135.0, 133.3, 129.9, 129.6, 128.9, 128.7, 128.5, 127.1, 118.7, 115.6, 106.2, 76.5, 60.9, 56.0, 45.0, 29.3, 9.7; exact mass calcd for C31H31NO5Na+ 520.2094, found 520.2094.
Supplementary Material
ACKNOWLEDGMENTS
The authors acknowledge support from the National Institutes of Health (R15GM107702 to J.R.S.). N.A.C.V. acknowledges the Arnold and Mabel Beckman Foundation for research support.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.1c00288.
Experimental procedures, characterization data, and 1H and 13C NMR spectra for all new compounds (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.joc.1c00288
The authors declare no competing financial interest.
Contributor Information
Nicole A. Carrillo Vallejo, Department of Chemistry, The College of William & Mary, Williamsburg, Virginia 23187, United States
Jonathan R. Scheerer, Department of Chemistry, The College of William & Mary, Williamsburg, Virginia 23187, United States;.
REFERENCES
- (1).(a) Rickborn B The retro–Diels–Alder reaction. Part I. C-C Dienophiles. Org. React 1998, 52, 1–393. [Google Scholar]; (b) Rickborn B The Retro-Diels–Alder Reaction. Part II. Dienophiles with One or MoreHeteroatom. Org. React 1998, 53, 223–630. [Google Scholar]; ((c)) van der Plas HC In Advances in Heterocyclic Chemistry; Katrizky AR, Ed.; Academic Press, 2003; 84, 31–70. [Google Scholar]; (d) Boger DL Diels-Alder Reactions of Heterocyclic Azadienes-Scope and Applications. Chem. Rev 1986, 86, 781–793. [Google Scholar]
- (2).(a) Bartko SG; Hamzik PJ; Espindola L; Gomez C; Danheiser RL Synthesis of Highly Substituted Pyridines via 4+2 Cycloadditions of Vinylallenes and Sulfonyl Cyanides. J. Org. Chem 2020, 85, 548–563. [DOI] [PubMed] [Google Scholar]; (b) Osano M; Jhaveri DP; Wipf P Formation of 6-Azaindoles by Intramolecular Diels-Alder Reaction of Oxazoles and Total Synthesis of Marinoquinoline A. Org. Lett 2020, 22, 2215–2219. [DOI] [PubMed] [Google Scholar]; (c) Le Fouler V; Chen Y; Gandon V; Bizet V; Salome C; Fessard T; Liu F; Houk KN; Blanchard N Activating Pyrimidines by Pre-distortion for the General Synthesis of 7-Aza-indazoles from 2-Hydrazonylpyrimidines via Intramolecular Diets-Alder Reactions. J. Am. Chem. Soc 2019, 141, 15901–15909. [DOI] [PubMed] [Google Scholar]; (d) Duret G; Le Fouler V; Bisseret P; Bizet V; Blanchard N Diels-Alder and Formal Diels-Alder Cycloaddition Reactions of Ynamines and Ynamides. Eur. J. Org. Chem 2017, 6816–6830. [Google Scholar]; (e) Duret G; Quinlan R; Martin RE; Bisseret P; Neuburger M; Gandon V; Blanchard N Inverse Electron-Demand 4+2 -Cycloadditions of Ynamides: Access to Novel Pyridine Scaffolds. Org. Lett 2016, 18, 1610–1613. [DOI] [PubMed] [Google Scholar]
- (3).(a) Duret G; Quinlan R; Yin B; Martin RE; Bisseret P; Neuburger M; Gandon V; Blanchard N Intramolecular Inverse Electron-Demand 4+2 Cycloadditions of Ynamides with Pyrimidines: Scope and Density Functional Theory Insights. J. Org. Chem 2017, 82, 1726–1742. [DOI] [PubMed] [Google Scholar]; (b) Foster RAA; Willis MC Tandem inverse-electron-demand hetero–/retro-Diels-Alder reactions for aromatic nitrogen heterocycle synthesis. Chem. Soc. Rev 2013, 42, 63–76. [DOI] [PubMed] [Google Scholar]; (c) Kotha S; Banerjee S Recent developments in the retro-Diels–Alder reaction. RSC Adv. 2013, 3, 7642–7666. [Google Scholar]; (d) Linder I; Gerhard M; Schefzig L; Andra M; Bentz C; Reissig HU; Zimmer R A Modular Synthesis of Functionalized Pyridines through Lewis-Acid-Mediated and Microwave-Assisted Cycloadditions between Azapyrylium Intermediates and Alkynes. Eur. J. Org. Chem 2011, 2011, 6070–6077. [Google Scholar]
- (4).(a) Select examples of 1,2,4-triazine chemistryBachollet SPJT; Vivat JF; Cocker DC; Adams H; Harrity JPA Development of a Mild and Versatile Directed Cycloaddition Approach to Pyridines. Chem. – Eur. J 2014, 20, 12889–12893. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Prokhorov AM; Kozhevnikov DN Reactions of triazines and tetrazines with dienophiles. Chem. Heterocycl. Compd 2012, 48, 1153–1176. [Google Scholar]; ((c)) Raw SA; Taylor RJK, Recent Advances in the Chemistry of 1,2,4-Triazines. In Advances in Heterocyclic Chemistry; Katrizky AR, Ed. Elsevier Academic Press, 2010; 100, 75–100. [Google Scholar]; (d) Padwa A; Bur SK The domino way to heterocycles. Tetrahedron 2007, 63, 5341–5378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).(a) Select examples of 1,2,3-triazine chemistryAnderson ED; Duerfeldt AS; Zhu KC; Glinkerman CM; Boger DL Cycloadditions of Noncomplementary Substituted 1,2,3-Triazines. Org. Lett 2014, 16, 5084–5087. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Anderson ED; Boger DL Inverse Electron Demand Diels-Alder Reactions of 1,2,3-Triazines: Pronounced Substituent Effects on Reactivity and Cycloaddition Scope. J. Am. Chem. Soc 2011, 133, 12285–12292. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Anderson ED; Boger DL Scope of the Inverse Electron Demand Diels-Alder Reactions of 1,2,3-Triazine. Org. Lett 2011, 13, 2492–2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).(a) Horner KA; Valette NM; Webb ME Strain-Promoted Reaction of 1,2,4-Triazines with Bicyclononynes. Chem. – Eur. J 2015, 21, 14376–14381. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Diring S; Retailleau P; Ziessel R A rational protocol for the synthesis of arylated bipyridine ligands via a cycloaddition pathway. J. Org. Chem 2007, 72, 10181–10193. [DOI] [PubMed] [Google Scholar]
- (7).Rooshenas P; Hof K; Schreiner PR; Williams CM 1,2,4-Triazine vs. 1,3-and 1,4-Oxazinones in Normal- and Inverse-Electron-Demand Hetero-Diels-Alder Reactions: Establishing a Status Quo by Computational Analysis. Eur. J. Org. Chem 2011, 2011, 983–992. [Google Scholar]
- (8).Delaney PM; Huang J; Macdonald SJF; Harrity JPAA 4+2 cycloaddition strategy to pyridine boronic ester derivatives. Org. Lett 2008, 10, 781–783. [DOI] [PubMed] [Google Scholar]
- (9).(a) Meerpoel L; Deroover G; Van Aken K; Lux G; Hoornaert G Diels-Alder Reactions of 6-Alkyl-3,5-Dichloro-2h-1,4-Oxazin-2-ones with Alkynes - Synthesis of 3,5-Disubstituted 2,6-Dichloropyridines. Synthesis-Stuttgart 1991, 765–768. [Google Scholar]; (b) Meerpoel L; Hoornaert G Synthesis of 3,5-Dihalogeno-2H-1,4-oxazin-2-ones from Cyanohydrins. Tetrahedron Lett. 1989, 30, 3183–3186. [Google Scholar]
- (10).(a) Williamson J; Smith E; Scheerer J A Merged Aldol Condensation, Alkene Isomerization, Cycloaddition/Cycloreversion Sequence Employing Oxazinone Intermediates for the Synthesis of Substituted Pyridines. Synlett 2017, 28, 1170–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chinchilla R; Falvello LR; Galindo N; Nájera C New chiral didehydroamino acid derivatives from a cyclic glycine template with 3,6-dihydro-2H-1,4-oxazin-2-one structure: Applications to the asymmetric synthesis of nonproteinogenic alpha-amino acids. J. Org. Chem 2000, 65, 3034–3041. [DOI] [PubMed] [Google Scholar]
- (11).Afarinkia K; Bahar A; Bearpark MJ; Garcia-Ramos Y; Ruggiero A; Neuss J; Vyas M Investigation of the Diels-Alder cycloadditions of 2(H)-1,4-oxazin-2-ones. J. Org. Chem 2005, 70, 9529–9537. [DOI] [PubMed] [Google Scholar]
- (12).(a) van Aken KJ; Lux GM; Deroover GG; Meerpoel L; Hoornaert GJ The Synthesis of 3-Functionalized 5-Chloro-6-Methyl-2h-1,4-Oxazin-2-ones and of Pyridines from Cycloaddition-Elimination Reactions with Substituted Acetylenic Compounds. Tetrahedron 1994, 50, 5211–5224. [Google Scholar]; (b) Fannes CC; Hoornaert GJ Cycloadditions of 3-Amino-2h-1,4-Oxazin-2-Ones with Olefins - Generation of 5,6-Dihydro-2-Oxo-2h-Pyran-6-Carbonitriles. Tetrahedron Lett. 1992, 33, 2049–2052. [Google Scholar]
- (13).Stokes BJ; Dong H; Leslie BE; Pumphrey AL; Driver TG Intramolecular C-H amination reactions: Exploitation of the Rh-2(II)-Catalyzed decomposition of azidoacrylates. J. Am. Chem. Soc 2007, 129, 7500–7501. [DOI] [PubMed] [Google Scholar]
- (14).Gray BL; Wang X; Brown WC; Kuai L; Schreiber SL Diversity synthesis of complex pyridines yields a probe of a neurotrophic signaling pathway. Org. Lett 2008, 10, 2621–2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).(a) Veikkolainen V; Vaparanta K; Halkilahti K; Iljin K; Sundvall M; Elenius K Function of ERBB4 is determined by alternative splicing. Cell Cycle 2011, 10, 2647–2657. [DOI] [PubMed] [Google Scholar]; (b) Sundvall M; Iljin K; Kilpinen S; Sara H; Kallioniemi OP; Elenius K Role of ErbB4 in breast cancer. J. Mammary Gland Biol. Neoplasia 2008, 13, 259–268. [DOI] [PubMed] [Google Scholar]; (c) Junttila TT; Sundvall M; Lundin M; Lundin J; Tanner M; Harkonen P; Joensuu H; Isola J; Elenius K Cleavable ErbB4 isoform in estrogen receptor-regulated growth of breast cancer cells. Cancer Res. 2005, 65, 1384–1393. [DOI] [PubMed] [Google Scholar]
- (16).Chen L; Yang T; Cui H; Zhang L; Su C-Y A porous metal–organic cage constructed from dirhodium paddle-wheels: synthesis, structure and catalysis. J. Mater. Chem 2015, 3, 20201–20209. [Google Scholar]
- (17).Dong G; Wu S; Sheng C Scaffold Diversity Inspired by the Natural Product Evodiamine: Discovery of Highly Potent and Multitargeting Antitumor Agents. J. Med. Chem 2020, 58, 6678–6696. [DOI] [PubMed] [Google Scholar]
- (18).Nakamura K; Takenaka K Tetrahedron: Asymmetry 2002, 13, 415–422. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.