

Abstract
Rationale:
Methicillin resistant Staphylococcus aureus (MRSA) is prevalent and consequential in cystic fibrosis (CF). Whole genome sequencing (WGS) could reveal genomic differences in MRSA associated with poorer outcomes or detect MRSA transmission.
Objectives:
To identify MRSA genes associated with low lung function and potential MRSA transmission in CF.
Methods:
We collected 97 MRSA isolates from 74 individuals with CF from 2017 and performed short-read WGS. We determined sequence type (ST) and the phylogenetic relationship between isolates. We aligned accessory genes from 25 reference genomes to genome assemblies, classified isolates by accessory gene content, and correlated the accessory genome to clinical outcomes.
Results:
The most prevalent ST were ST5 (N=55), ST8 (N=15), and ST105 (N=14). Closely related MRSA strains were shared by family members with CF, but rarely between unrelated individuals. Three clusters of MRSA were identified by accessory genome content. Cluster A, including ST5 and ST105, was highly prevalent at all ages. Cluster B, including ST8, was more limited to younger patients. Cluster C included 6 distantly related strains. Patients 20 years old and younger infected with Cluster A had lower FEV1 and higher sputum biomass compared to similar-aged patients with Cluster B.
Conclusions:
In this CF cohort, we identified MRSA subtypes that predominate at different ages and differ by accessory gene content. The most prevalent cluster of MRSA, including ST5 and ST105, was associated with lower FEV1. ST8 MRSA was more common in younger patients and thus has the potential to rise in prevalence as these patients age.
Keywords: Staphylococcus aureus, Cystic Fibrosis, MRSA, Whole Genome Sequencing, Birth Cohort Effect
Introduction
In the United States, the majority of patients with cystic fibrosis (CF) develop chronic respiratory infections with Staphylococcus aureus,1 whereas countries with continuous anti-staphylococcal prophylaxis have lower S. aureus prevalence.2,3 S. aureus infections can begin in infancy and are difficult to eliminate with anti-staphylococcal antibiotics.4 Children infected with S. aureus have increased airway inflammation and can experience worsening lung function.5–7 Moreover, methicillin resistant S. aureus (MRSA) prevalence has increased significantly in the United States over the past two decades.1,8 Patients infected with MRSA require increased treatment and suffer worse outcomes.9–11 These observations raise questions about how MRSA is acquired in these patients and which bacterial virulence factors might accelerate disease progression.
Whole genome sequencing (WGS) may help us understand the origins of MRSA within this population. WGS facilitates the phylogenetic comparison of MRSA strains to reveal any unexpectedly strong relationship between bacteria isolated from unrelated individuals. Because CF is a genetic disorder, sibling dyads who live in the same household may share related strains of MRSA. The phylogenetic distance between MRSA strains cultured from siblings would provide a benchmark for identifying potential transmission of MRSA between unrelated patients with CF during clinical care.12–14 We hypothesized that WGS would identify person-to-person transmission of MRSA between unrelated individuals within our CF center.
Another advantage of WGS is that it allows classification of S. aureus isolates based on sequence type and provides detailed information about genomic content, including the presence or absence of virulence factors. We are particularly interested in the prognostic significance of genes encoding staphylococcal superantigens, as we have observed high prevalence of enterotoxin gene cluster superantigens in an earlier de-identified set of S. aureus isolates from this center.15 These secreted toxins have the potential to increase inflammation and interfere with the adaptive immune response to S. aureus, hindering clearance by the host.16,17 We hypothesized that strains of S. aureus that encode enterotoxin gene cluster superantigens may be associated with poorer clinical outcomes.
In this study, we used WGS to analyze MRSA strains isolated from children and adults attending CF clinic in the year 2017. We compared sequences to identify potential transmission of MRSA between patients. Additionally, we classified the MRSA isolates based on accessory gene content to determine whether differences in genomic content correlated with worsening clinical outcomes.
Methods
Ethics statement.
The Institutional Review Board (IRB) of the University of Iowa approved this study # 201905718. Informed consent was waived because the study was minimal risk.
Subjects.
To examine changes in incidence and prevalence of CF pathogens, we studied 337 patients diagnosed with CF and cared for in the adult or pediatric CF centers at the University of Iowa, as recently described.18 We additionally examined patients with CFTR related metabolic syndrome (CRMS) or patients with CF following lung transplant to assess for potential MRSA transmission, as these patients attended CF clinics and could be at risk of acquiring MRSA. However, we excluded patients with CRMS or post-lung transplant from calculations of incidence, prevalence, and outcomes such as FEV1.
Respiratory culture results.
We examined respiratory cultures for methicillin susceptible S. aureus (MSSA), MRSA, and Pseudomonas aeruginosa using the electronic medical record.18 Annualized prevalence of each organism was the number of individuals positive for an organism divided by the number of individuals tested during the same calendar year. The annualized incidence of MRSA was the number of individuals with a new MRSA infection divided by the number of individuals susceptible to MRSA during the same calendar year. We defined individuals as susceptible to MRSA if they were newborn or if they did not have a positive respiratory culture for MRSA during their first year of observation. Individuals who acquired an incident infection or had no further recorded data were removed from the denominator of susceptible individuals.
Spirometry and medications.
We obtained spirometry results from the electronic medical record. We determined the best FEV1 % predicted (with or without bronchodilator) for each patient in every year between 2011 – 2017. We examined electronic prescriptions for antibiotics and CFTR modulator drugs in the year 2017.
Selection of bacterial isolates.
S. aureus clinical isolates were previously stored in TSB with glycerol freezing medium by the clinical microbiology laboratory. Records from all 2017 banked S. aureus isolates were reviewed to determine if the patient was diagnosed with CF. S. aureus isolates were taken from 69 sputa, 24 oropharyngeal swabs, two bronchoalveolar lavages, one sinus culture, and one blood culture. The isolates from blood and sinus cultures were from patients following lung transplant.
Antibiotic susceptibility testing was performed in the clinical lab using VITEK 2 system (Biomérieux). We assumed that isolates cultured from the same patient with identical appearance and antimicrobial susceptibility represented the same strain. When several isolates from a patient had identical phenotypes, we selected the first isolate. When antimicrobial susceptibility was variable within a patient, we selected the first isolate for each unique antibiotic resistance phenotype to improve the likelihood of detecting strains shared between individuals. Thus, we selected 97 unique MRSA from 74 patients (56 patients - 1 isolate, 14 patients - 2 isolates, 3 patients - 3 isolates, and 1 patient - 4 isolates). One additional isolate was originally thought to be methicillin resistant S. aureus based on the clinical diagnosis. However, we learned that its genome sequence matched S. epidermidis, and it was removed from further analysis. Isolates were streaked on blood agar plates and isolated colonies were subcultured. DNA preparation was performed with the EZ1 DNA Tissue Kit on a Qiagen EZ1 instrument following manufacturer instructions.
Whole genome sequencing.
Library preparation was performed using Illumina Nextera DNA Flex. Paired-end sequencing was performed using Illumina MiSeq Reagent Kit. Phylogenetic analysis was completed using the Utah Public Health Laboratory (UPHL) reference free pipeline.19 This pipeline assembled genomes using SPAdes20 and produced alignments using Roary.21 Repetitive sequences and mobile genetic elements were removed, and the phylogenetic tree was produced using shared genes using a maximum likelihood approach. We used Lyve-SET22 to perform reference-based phylogenetic analysis of the largest clade, consisting of ST5 and ST105 strains, using N315 (NC_002745.2) as our genomic reference.
Analysis of strain sharing.
We used the Lyve-SET SNP matrix to identify strains with strong sequence similarity. Mobile genetic elements and repetitive elements were excluded from this calculation. We considered strains separated by ≤ 70 SNPs to be highly similar.23 We measured SNP distance between strains isolated from the same individual or from family dyads to estimate the distance between strains shared between individuals in close contact.
Sequence type, virulence factor, and antimicrobial resistance gene identification.
We uploaded sequences to the Staphopia API to obtain summaries for multilocus sequence type (MLST), presence of virulence factors, and predicted antibiotic resistance.24 We used Mykrobe 0.7.0 to identify predicted antimicrobial resistance genes.25 SCCmec elements were identified using SCCmecFinder 1.2.26 We re-tested strains lacking SCCmec using the Clearview PBP2a SA Culture Colony test (Abbott Laboratories) following manufacturer’s instructions.
Accessory gene content.
Sequence reads from FASTQ files were assembled de novo using SPAdes.20 We used 25 S. aureus genomes as our source of reference genes.27 To identify the presence of genes within our de novo assemblies, we used the sequence aligning software HISAT2,28 which can efficiently detect genes or transcripts in a variety of species.29,30 We used the detection of accessory genes to perform unsupervised hierarchical and K-means clustering of the MRSA isolates using R.
Statistical Analysis.
We used Poisson regression to test temporal trends in incidence and prevalence of CF pathogens and linear regression for trends in lung function. To compare two or more groups, we used non-parametric tests (Wilcoxon rank sum test or Kruskal-Wallis test, respectively) for continuous data and Fisher’s exact test for categorical data. We used R Studio Version 1.2.5001, Graphpad Prism Version 8.4.2, and SAS version 9.4 for statistical comparisons.
Results
Prevalence and incidence of respiratory infections.
Following the development of inhaled antibiotics for P. aeruginosa and improvements in infection control, others have noted a nationwide trend towards lower incidence and prevalence of P. aeruginosa infections in patients with CF.8,31 We observed similar trends within the University of Iowa CF center, with rising prevalence of MRSA and a slowly declining prevalence of P. aeruginosa, Supplemental Data 1–3. MRSA infections are persistent in CF;18 the rise in MRSA prevalence has occurred despite a slow decline in MRSA incidence.
We suspected that rising MRSA prevalence over the past decade may have been hastened by either person-to-person or healthcare worker-to-patient transmission. To test this possibility, we obtained clinical isolates of MRSA cultured from patients with CF in the year 2017 and compared them by WGS.
WGS typing and cluster analysis.
Using nucleotide variations, we constructed reference-free phylogenetic trees of the CF MRSA isolates. We observed three clades, Figure 1. 13 sequence types (ST) were represented. ST5 (57%) and ST105 (14%) were the most prevalent in clade 1 and ST8 (15%) in clade 2. ST for each isolate and linked subject numbers are given in Supplemental Data 4. One isolate was identified as livestock-associated ST398, possibly the first documented case in a North American patient with CF. We are not aware that this subject had any direct agricultural exposures that would have increased the risk of acquiring ST398 S. aureus.
Figure 1.
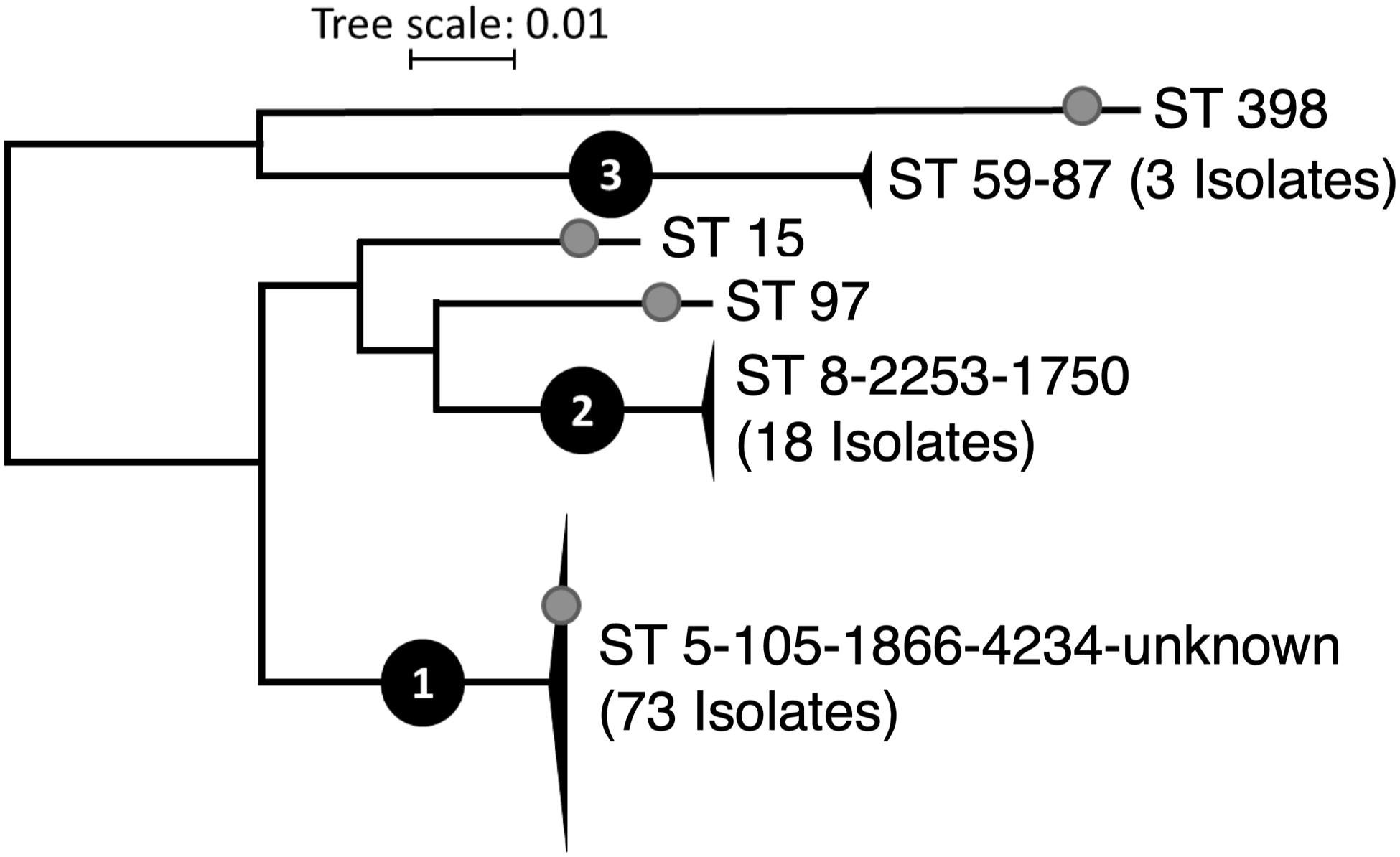
Phylogenetic relationship between 97 CF MRSA isolates using the UPHL reference-free pipeline. Sequence types (ST) found within each group are denoted. Black triangles represent groups of two or more isolates. Gray circles indicate four isolates predicted to be MSSA based on lack of detected SCCmec element. Tree scale indicates substitutions per site.
MRSA strain diversity within individuals.
We analyzed multiple MRSA isolates from 18 subjects, highlighted in Figure 2 and Supplemental Data 5. In 14 cases, strains isolated from the same individual had fewer than 60 SNPs difference. Examples of greater within-subject diversity included a subject whose isolates 094 and 090 were separated by 678 SNPs (indicated in blue brackets within Figure 2). Because these isolates had different MLST and were widely separated on the phylogenetic tree, we considered these as two distinct strains. Hypermutation was evident in multiple lineages, Supplemental Data 6. Isolates 043 and 091 had a large deletion removing both mutS and mutL, resulting in widespread mutations (up to 193 SNPs vs the subject’s non-mutator strains). This limited our ability to assign these strains to a MLST. The phylogenetic tree indicated these strains likely evolved within this subject from an ST5 ancestor. Four subjects had MRSA with mutL p.P340fs frameshift mutations, possibly accounting for increased genetic variation in these individuals.
Figure 2.
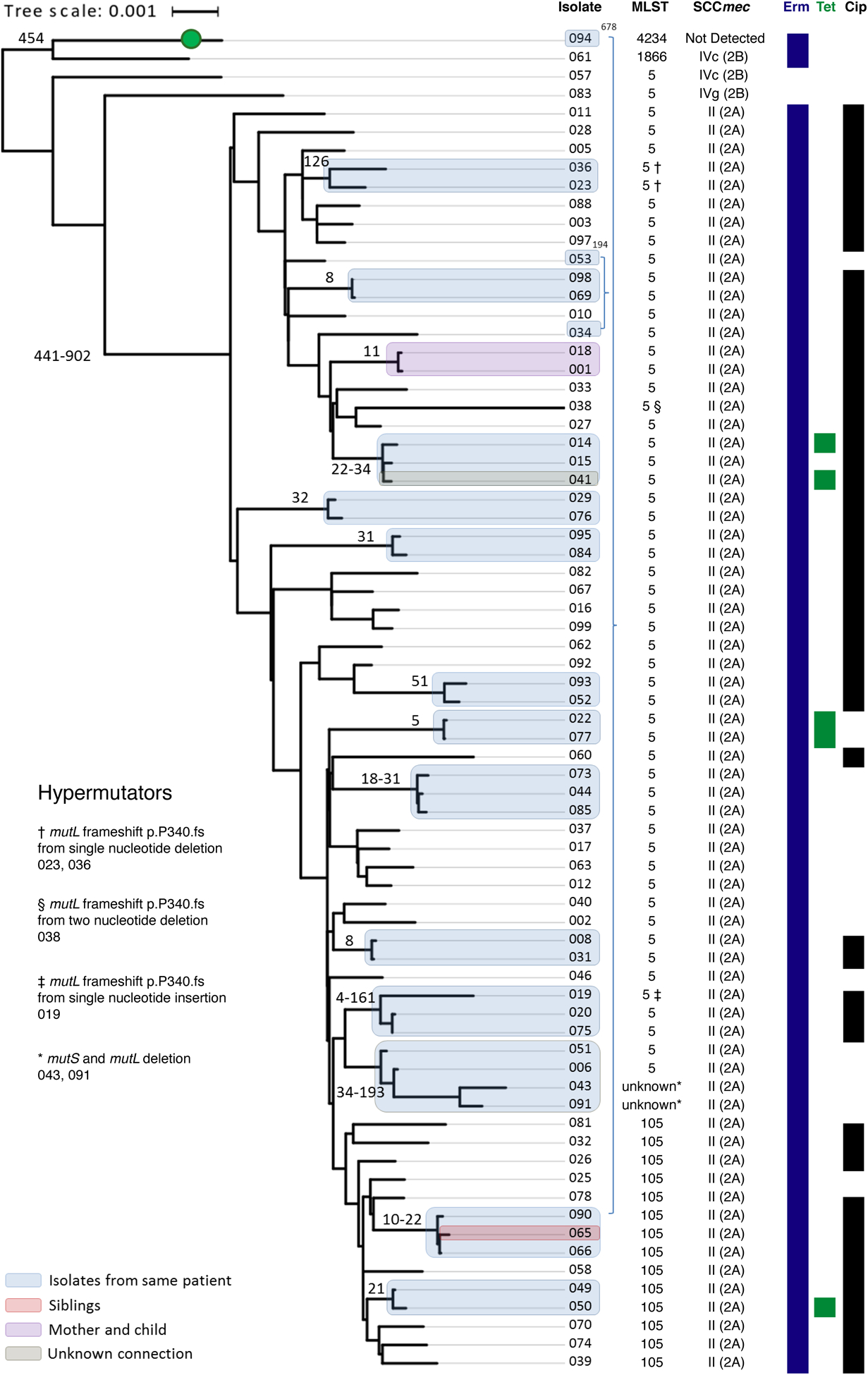
Lyve-SET Phylogenetic reconstruction for clade 1 reveals closely related strains within subjects or shared by different individuals. Sequence type (ST), SCCmec element, and predicted antibiotic resistance from genome sequence are given at right. A green circle indicates an isolate predicted to be MSSA based on lack of detection of SCCmec. Antibiotic susceptibilities denote isolates predicted to be resistant to erythromycin (Erm), tetracycline (Tet), or ciprofloxacin (Cip). Highly related isolates are indicated with shaded boxes, colored based on the relationship between patients. The SNP difference between these isolates is given as a number adjacent to the shaded box. Several isolates with increased genetic distance were found to have mutations in mutL and/or mutS. Tree scale represents substitutions per site
Sharing of MRSA strains between individuals.
WGS suggested possible MRSA transmissions between related individuals, including a parent and child (Figure 2, purple box) and siblings (red box). However, related individuals did not always share closely related MRSA. Isolates from a child (046) and the child’s parent’s sibling (041) had distantly related MRSA, with a difference of 238 SNPs. There was one instance in which unrelated patients (grey box) shared MRSA separated by only 34 SNPs. We did not identify shared hospital encounters between these unrelated individuals, but noted that they lived in geographic proximity.
Antibiotic resistance.
In S. aureus, methicillin resistance is usually conferred by an alternate penicillin binding protein, encoded by mecA within an SCCmec element.32 Other potential methicillin resistance mechanisms include the novel penicillin binding protein encoded by mecC or hypersecretion of beta-lactamases.32 In our cohort, 93 of 97 isolates were positive for mecA. Four isolates diagnosed as MRSA lacked an SCCmec element. These isolates were negative for PBP2a by antigen testing. Each of these isolates encoded the blaZ gene, which could confer resistance to methicillin when overexpressed. None of the isolates encoded mecC. Most of the CF MRSA isolates (97%) were resistant to erythromycin, Supplemental Data 7, consistent with the chronic use of azithromycin in CF. Eight isolates displayed resistance to tetracycline, also commonly prescribed for people with CF. Ciprofloxacin and aminoglycosides are commonly prescribed for treatment of P. aeruginosa. Ciprofloxacin resistance was identified in 81 isolates. Resistance was predicted by Mykrobe in 65 of these isolates, including 64 with S84L mutations in gyrA and 43 with mutations in grlA, Supplemental Data 8. By contrast, aminoglycoside resistance was uncommon; gentamicin resistance was detected in only 4 isolates.
Accessory gene content correlates with sequence type.
We considered the possibility that different S. aureus sequence types encode distinct repertoires of virulence factors in their accessory genomes. To determine the gene content of these isolates, we downloaded reference sequences for 25 annotated S. aureus strains.27 This data set consisted of 65,322 genes. To reduce redundancy, we combined genes having identical Genbank inference tag numbers, which indicate gene function. This reduced the size of the data set to 4,918 genes based on inference number. If at least one allele of a given gene was detected, we considered the gene to be present.
Of the 4,918 genes in our reference, 866 genes were not detected. 2,427 genes were detected in all 97 isolates. The remaining 1,625 genes that were detected at least once but not in every isolate composed our accessory genome set. We aligned this set of accessory genes to de novo genome assemblies for the 97 MRSA isolates. After determining which accessory genes were detected, we classified the isolates based on their gene content using hierarchical and K-means clustering, Figure 3A. As we observed when clustering based on SNPs, three distinct clusters of S. aureus emerged. The dominant cluster, Cluster A, included ST5 and ST105 isolates, Table 1. Cluster B was represented mainly by ST8 isolates.
Figure 3.
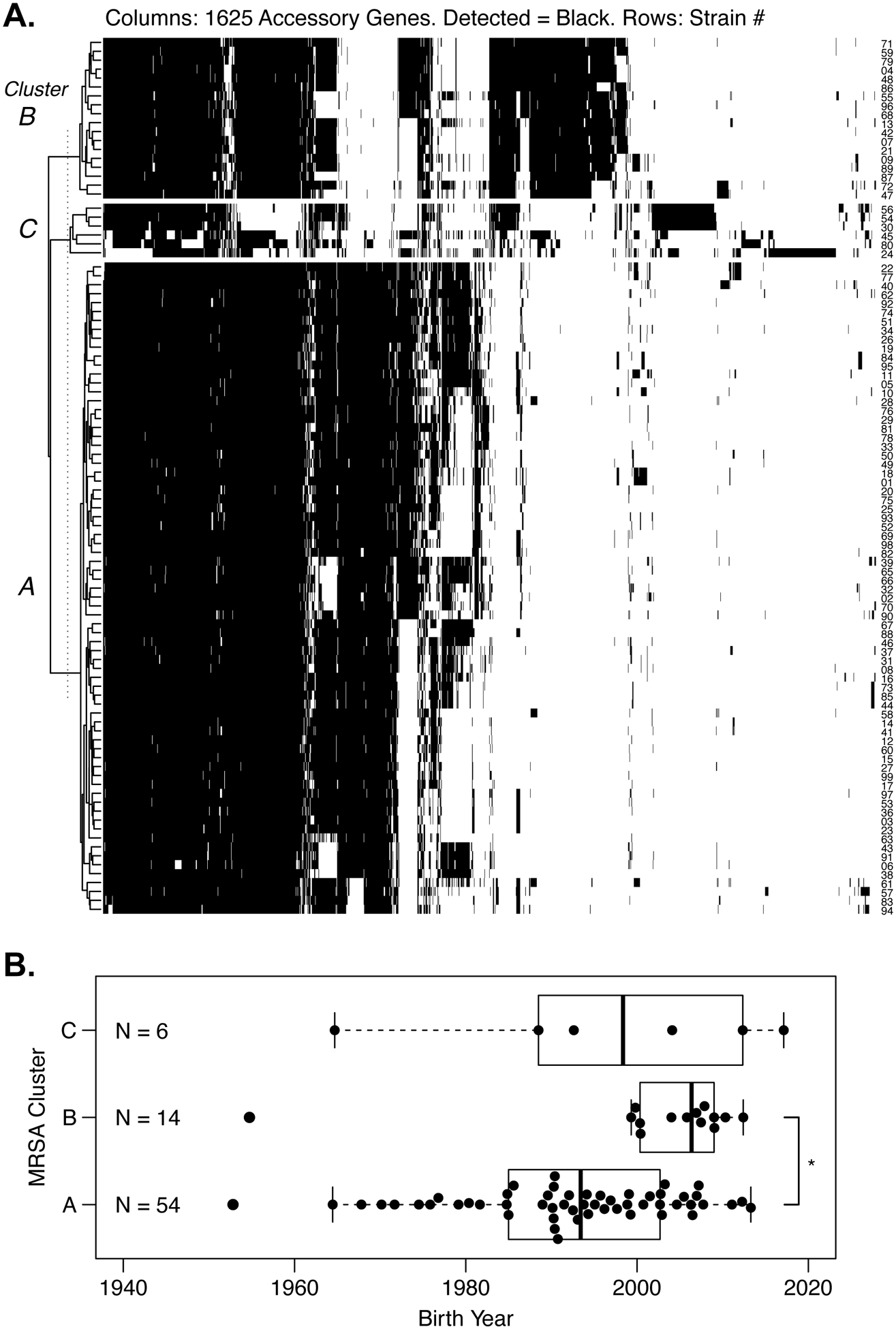
A. Clustering of 97 MRSA isolates based upon accessory gene content. Accessory genes were obtained from 25 reference genomes. Each row represents a CF MRSA isolate, and numbers are given at right. Each column is one of 1625 accessory genes; black color indicates detection of the accessory gene. The isolates clustered into three groups named at left. Clusters assigned on the basis of accessory genes matched the results from SNP-based clustering. Cluster A included ST5 and ST105 isolates. Cluster B included ST8 and related isolates. Cluster C included distantly related S. aureus, including a livestockassociated ST398 strain (#24). B. Unequal distribution of MRSA clusters by birth year. Cluster A was widely distributed across ages, whereas Cluster B was limited to more recent birth years. * P = 0.0009 by Wilcoxon rank sum test.
Table 1.
Cluster analysis of MRSA isolates analyzed by Whole Genome Sequencing.
| Number of Isolates (%) | |||
|---|---|---|---|
| Characteristic | Cluster A Isolates = 73 | Cluster B Isolates = 18 | Cluster C Isolates = 6 |
| Sequence Type | |||
| 5 | 55 (75.3%) | 0 | 0 |
| 105 | 14 (19.2%) | 0 | 0 |
| 1866 | 1 (1.4%) | 0 | 0 |
| 4234 | 1 (1.4%) | 0 | 0 |
| other/unknown | 2 (2.7%)* | 0 | 0 |
| 8 | 0 | 15 (77.8%) | 0 |
| 1750 | 0 | 1 (5.6%) | 0 |
| 2253 | 0 | 2 (5.6%) | 0 |
| 59 | 0 | 0 | 2 (33.3%) |
| 15 | 0 | 0 | 1 (16.7%) |
| 87 | 0 | 0 | 1 (16.7%) |
| 97 | 0 | 0 | 1 (16.7%) |
| 398 | 0 | 0 | 1 (16.7%) |
| SCCmec | |||
| II | 69 (94.5%) | 0 | 0 |
| IV | 3 (4.1%) | 18 (100%) | 3 (50%) |
| Not detected | 1 (1.4%) | 0 | 3 (50%) |
| Toxin | |||
| SEG | 71 (97.3%) | 0 | 0 |
| SEI | 71 (97.3%) | 0 | 0 |
| SEM | 73 (100%) | 0 | 0 |
| SEN | 72 (98.6%) | 0 | 0 |
| SEO | 73 (100%) | 0 | 0 |
| LukF | 0 | 9 (50%) | 0 |
| LukS | 0 | 9 (50%) | 0 |
| Resistance | |||
| Ciprofloxacin | 68 (93.2 %) | 12 (66.7 %) | 1 (16.7%) |
| Erythromycin | 72 (98.6 %) | 15 (83.3 %) | 5 (86.7%) |
| Rifampin | 10 (13.7%) | 0 | 0 |
| Tetracyclines | 6 (8.2 %) | 1 (5.6 %) | 1 (16.7%) |
| Trimethoprim/Sulfamethoxazole | 4 (5.5 %) | 0 | 0 |
These isolates share a common ancestor with ST5 isolates from the same subject.
The most prevalent MRSA cluster encodes SCCmec type II and EGC toxins.
We examined for genes that differed significantly between MRSA clusters, Table 1. Cluster A usually possessed SCCmec type II and was positive for toxin genes of the enterotoxin gene cluster, which we observed is also highly prevalent in CF-associated MSSA.15 By contrast, Cluster B was exclusively positive for SCCmec type IV, sometimes described as community acquired MRSA. The third cluster, Cluster C, had the fewest and most distantly related strains. Additional genes that were differentially detected by cluster are shown in Supplemental Data 9 and 10. Genes with high sequence polymorphism and multiple inference numbers (such as vwb) occasionally appeared in both lists.
Different MRSA clusters predominate at different ages.
We determined which S. aureus clusters were identified in each patient. Although some patients had multiple isolates, each subject was positive for only one of the three MRSA clusters during 2017. Patients infected with Cluster A MRSA were older on average and had a wide distribution of birth years, whereas all but one of the subjects with Cluster B were under 20 years old, Figure 3B. Genotype, sex, and Pseudomonas status were comparable between patients infected with different MRSA clusters, Supplemental Data 11.
Lung function and sputum biomass in patients infected by different MRSA clusters.
The lack of ST8 MRSA in older patients could indicate these patients were already colonized by a different strain or that they experience more rapid disease progression and earlier mortality. To examine the association between MRSA cluster and disease progression while controlling for the confounding effect of age, we examined the subset of patients born after January 1, 1997. These patients under age 20 years old historically experience the fastest declines in lung function.33 Because of the low number of subjects with cluster C, we compared lung function in subjects infected with clusters A and B, Figure 4A–B. Patients under age 20 with clusters A or B were similar in age, sex, genotype, sputum production, Pseudomonas status, and medications prescribed, Table 2. Young patients with cluster A MRSA had lower FEV1 compared to similarly aged patients with Cluster B MRSA. Patients positive for Cluster A MRSA had larger annual declines in FEV1 % predicted compared to patients with Cluster B.
Figure 4.
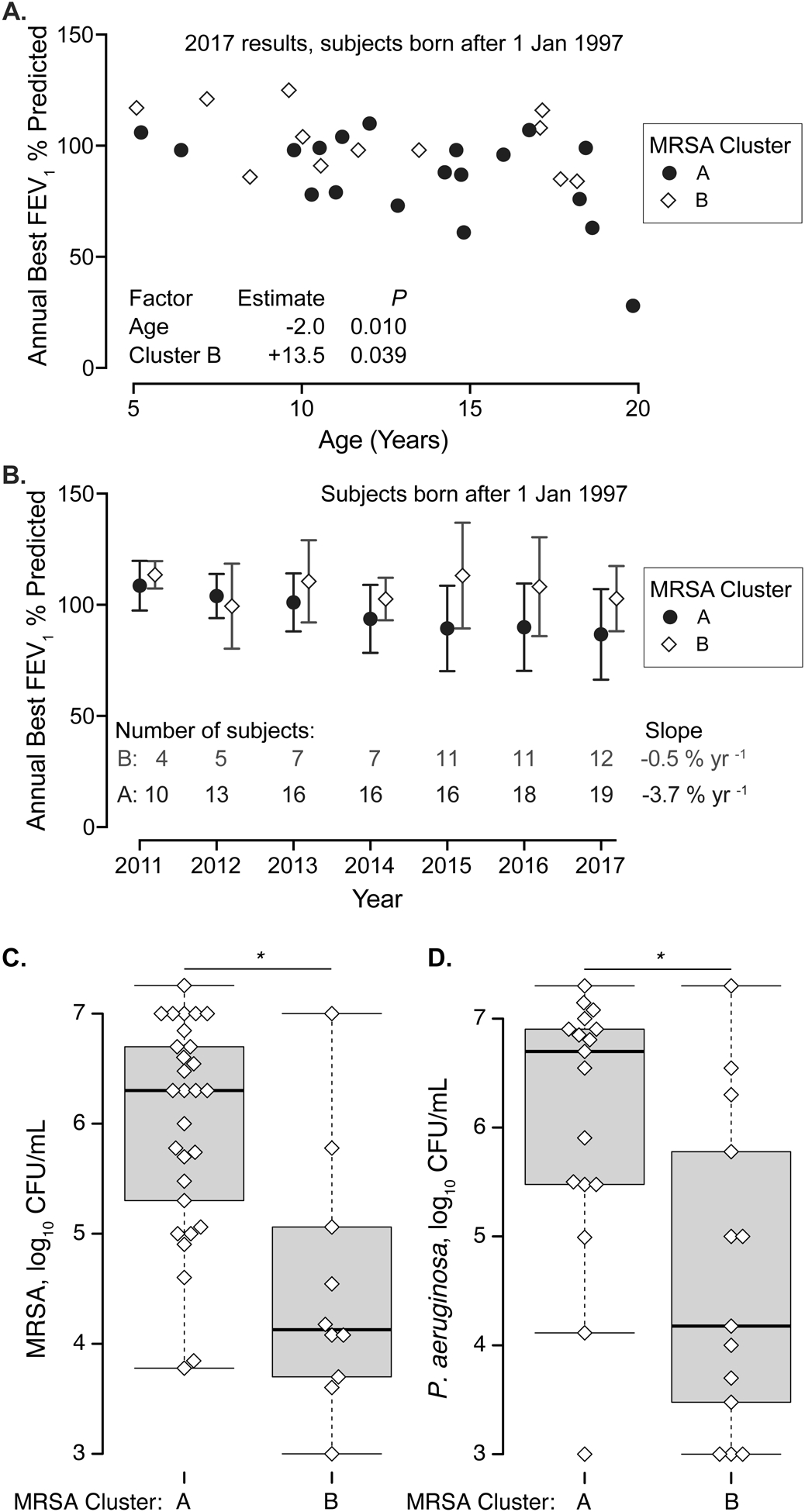
Lung function and sputum biomass in patients born after January 1, 1997 and infected by different MRSA clusters. A. Lung function in 2017 (annual best FEV1 % predicted) is shown as a function of age. Patients with Cluster A MRSA are depicted by circles, Cluster B by diamonds. Two subjects with Cluster C MRSA are omitted. Generalized linear regression results are at bottom; advancing age and Cluster A MRSA were associated with lower lung function. B. Trends in FEV1 % predicted by MRSA cluster between 2011 and 2017. Symbols indicate mean FEV1 % predicted by MRSA types, Cluster A (circles), Cluster B (diamonds). Bars indicate standard deviation. The number of patients examined per year is given at bottom. The average FEV1 % predicted for patients with Cluster A MRSA decreased with time (slope −3.7% yr −1, P < 0.001). The average FEV1 % predicted for patients infected with Cluster B MRSA was stable (slope −0.5 % yr −1, P = 0.72). C-D. Quantitation of CF microorganisms in sputum from young patients infected by MRSA belonging to cluster A or cluster B. Each dot represents a different sputum culture. C. MRSA, D. P. aeruginosa. * P < 0.01 by Wilcoxon rank sum test
Table 2.
Clinical characteristics of subjects born after 1997 infected with Cluster A or Cluster B MRSA.
| Characteristic | Cluster A N = 20 | Cluster B N = 12 | P |
|---|---|---|---|
| Age, years† Median [IQR] | 13.6 [10.4 – 16.4] | 11.1 [9.0 – 17.1] | 0.50* |
| MRSA acquisition date, Median [IQR] | 09/2009 [7/2007 – 02/2015] | 08/2013 [12/2011 – 03/2015] | 0.14* |
| Female Sex | 10 (50%) | 4 (33%) | 0.48‡ |
| Genotype | 0.62‡ | ||
| ΔF508/ΔF508 | 11 (55%) | 6 (50%) | |
| ΔF508/other | 7 (35%) | 6 (50%) | |
| Other/other | 2 (10%) | 0 | |
| Sputum production, N (%) | 15 (75%) | 10 (83%) | 0.68‡ |
| P. aeruginosa N (%) | 11 (55%) | 6 (50%) | 1.0‡ |
| Spirometry performed, N (%) | 19 (95%) | 12 (100%) | 1.0‡ |
| Medications in 2017 | |||
| Trimethoprim/Sulfamethoxazole | 12 (60%) | 10 (83%) | 0.25‡ |
| Linezolid | 14 (70%) | 5 (42%) | 0.15‡ |
| Doxycycline | 10 (50%) | 1 (8.3%) | 0.02‡ |
| Vancomycin IV | 9 (45%) | 6 (50%) | 1.0‡ |
| Tobramycin inhaled | 14 (70%) | 7 (58%) | 0.70‡ |
| Ciprofloxacin | 9 (45%) | 4 (33%) | 0.72‡ |
| Azithromycin | 19 (95%) | 11 (92%) | 1.0‡ |
| Ivacaftor | 2 (10%) | 2 (17%) | 0.62‡ |
| Lumacaftor/Ivacaftor | 11 (55%) | 5 (42%) | 0.72‡ |
Age determined on July 1, 2017. No subjects had lung transplant or CFTR related metabolic syndrome. P values determined by
Wilcoxon rank sum test or
Fisher’s exact test.
Quantitative respiratory culture studies were routinely ordered by the University of Iowa Pediatric CF center until 2017.18 We compared MRSA and P. aeruginosa culture density for patients with Cluster A MRSA vs. Cluster B MRSA, Figure 4C–D. Sputum samples collected from patients infected by Cluster A MRSA had a higher median density (in CFU/mL) of both MRSA and P. aeruginosa compared to sputum from patients infected with Cluster B MRSA of similar age.
Discussion
In this study of a CF center with high MRSA prevalence, we observed that most MRSA were unique to the individual as determined by WGS. Household contacts sometimes shared similar MRSA isolates differing by under 60 SNPs. We did not identify hospital-based transmission of MRSA, suggesting that these infections were most likely community-acquired.
MRSA strains collected from CF respiratory cultures displayed genetic diversity (13 sequence types and up to 24,400 SNPs difference). When isolates were classified by their accessory genome content, we found three distinct subpopulations of MRSA within this CF center. The most prevalent group included SCCmec type II isolates on ST5 and ST105 backgrounds. These isolates encoded the enterotoxin gene cluster, and were common across the age spectrum. Patients under 20 years old who were infected by cluster A MRSA had lower lung function, they experienced more rapid declines in lung function, and they had higher titers of both MRSA and P. aeruginosa in their sputum. The second cluster was mainly represented by ST8. Patients with this type of MRSA were younger and had higher average lung function. Although these differences in outcomes between MRSA clusters were intriguing, they require confirmation in larger studies. Our observation of differences in age between patients infected with different MRSA subtypes suggests a birth cohort effect—patients born in different eras likely had different environmental exposures, including the prevailing S. aureus strains in the community at the time they were initially infected by MRSA.
Most MRSA isolates cultured from the same patient were closely related (≤ 51 SNPs), similar to a previous report.23 However, we observed wider within-subject diversity when strains had mutations in DNA repair genes. Genome sequencing identified possible livestock-associated S. aureus in a patient with CF. ST398 is commonly isolated from pigs and can infect farm workers.34 Eight strains displayed resistance to tetracycline, an antibiotic used in both humans and livestock. Four isolates did not encode SCCmec elements. Other mechanisms could account for oxacillin resistance, such as over-production of beta-lactamases by the blaZ gene.32,35
Comparison to previous studies.
Our findings are consistent with a recent WGS study by Long and colleagues from the University of Washington,36 who found evidence of S. aureus strain sharing by siblings and similar sequence types of S. aureus account for MRSA in children with CF. As previously reported by others, the most prevalent SCCmec in our population was type II.11,14,37,38 SCCmec II typically appears on the ST5 background, whereas ST8 is associated with SCCmec IV.39 We found that adults and children with CF carry different MRSA strains, similar to a prior report from the University of North Carolina.11 In that study, patients with SCCmec II PVL-negative MRSA were older than those with SCCmec IV PVL-positive MRSA. We report that the subset of MRSA with SCCmec II is associated with lower FEV1 in young patients. Consistent with this finding, a multi-center study of pulmonary outcomes related to MRSA in children with CF, SCCmec II MRSA was associated with more pulmonary exacerbations.38
However, these observations raise several questions. Are the adverse outcomes experienced by patients infected with SCCmec II MRSA driven by the SCCmec type itself, by virulence factors encoded elsewhere in the genome, or are these MRSA subtypes simply markers of more intensive treatment and greater hospital exposure? Our dataset does not fully address these questions, but provides some context. MRSA that encode SCCmec II usually have different repertoires of accessory genes compared to MRSA with SCCmec IV, making it difficult to attribute differences in outcomes to the SCCmec type per se.
Advantages.
This study examined a cross section of both children and adults with CF, allowing us to identify how different age groups may harbor distinct types of MRSA. We used complementary analytical techniques of comparing both SNPs and gene content. Using our existing longitudinal database of respiratory cultures, we could estimate the duration of MRSA infections. Most of the patients had pulmonary function data, allowing us to examine potential associations of MRSA subtype with adverse clinical outcomes.
Limitations.
This study has limitations. Because it is a single-centered study, these patients could have different risk factors for MRSA acquisition and maintenance compared to patients in other centers. Because of the small number of observations, our statistical modeling of lung function by MRSA subtype could not address all sources of confounding, including the potential effects of S. aureus and P. aeruginosa biomass. Because the study design is observational and cross-sectional, we do not know whether differences in pulmonary function are caused by the type of MRSA.
Conclusions
The prevalence of MRSA in cystic fibrosis has increased over the previous decade. We observed two major clusters of MRSA within this cohort based on accessory genome content. The prevalence of each cluster depends upon age. Family dyads often shared MRSA isolates, but transmission between unrelated individuals was not common. Understanding the origins of MRSA in patients with CF could help limit acquisition of these resistant bacteria and improve patient outcomes.
Supplementary Material
Key Point:
Methicillin resistant Staphylococcus aureus (MRSA) causes persistent airway infections in cystic fibrosis. Children and adults with cystic fibrosis often have different MRSA strains, which encode distinctive repertoires of virulence factors in their accessory genomes.
Acknowledgments
We are grateful to Logan Fink (Colorado Department of Public Health and Environment), Jake Garfin (Minnesota Department of Health), Wes Hottel (State Hygienic Laboratory), Kelly Oakeson (Utah Public Health Laboratory) and Erin Young (Utah Public Health Laboratory) for their help installing data analysis pipelines on the high-performance computing cluster at the University of Iowa.
This study was funded in part by the CDC’s Advanced Molecular Detection, Epidemiology and Laboratory Capacity Cooperative Agreement (VR), a Cystic Fibrosis Foundation KBoost award (AJF), a career development award from NIH NHLBI K08 HL136927 (AJF), and the Cystic Fibrosis Foundation Research and Development Program (Bioinformatics Core) at the University of Iowa (AAP and ALT). Data storage on RedCap is supported by NIH and the University of Iowa CTSA grant number UL1TR002537.
Funding Sources:
NHLBI K08 HL136927 (AJF) University of Iowa CTSA UL1TR002537. Cystic Fibrosis Foundation K Boost (AJF) CFF Research and Development Program Bioinformatic Core, Iowa (AAP) CDC Advanced Molecular Detection, Epidemiology and Laboratory Capacity Cooperative Agreement (VR)
Footnotes
Conflicts of Interest:
Dr. Diekema has received consulting fees from OpGen, Inc. and grant funding from bioMerieux, Inc in the past 24 months. The remaining authors have no financial conflicts of interest to report.
Data Availability.
This Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank with BioProject accession number PRJNA650389 and accessions JACOAG000000000-JACODY000000000. Unassembled FASTQ files are available from the corresponding authors.
References:
- 1.Cystic Fibrosis Foundation 2016 Patient Registry Annual Data Report. 2017. URL: https://www.cff.org/research/researcher-resources/patient-registry/2016-patient-registry-annual-data-report.pdf, last accessedMay 10, 2021.
- 2.Cystic Fibrosis Trust Annual Report. 2019. URL: https://www.cysticfibrosis.org.uk/sites/default/files/2020-12/2019%20Registry%20Annual%20Data%20report_Sep%202020.pdf, last accessedMay 10, 2021
- 3.Australian Cystic Fibrosis Data Registry. 2017. URL: https://www.cysticfibrosis.org.au/getmedia/24e94d66-29fa-4e3f-8e65-21ee24ed2e5a/ACFDR-2017-Annual-Report_highres_singles.pdf.aspx, last accessedMay 10, 2021
- 4.Dezube R, Jennings MT, Rykiel M, et al. Eradication of persistent methicillin-resistant Staphylococcus aureus infection in cystic fibrosis. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society. 2018. [DOI] [PubMed] [Google Scholar]
- 5.Gangell C, Gard S, Douglas T, et al. Inflammatory responses to individual microorganisms in the lungs of children with cystic fibrosis. Clinical Infectious Diseases : an official publication of the Infectious Diseases Society of America. 2011;53(5):425–432. [DOI] [PubMed] [Google Scholar]
- 6.Sagel SD, Gibson RL, Emerson J, et al. Impact of Pseudomonas and Staphylococcus infection on inflammation and clinical status in young children with cystic fibrosis. The Journal of Pediatrics. 2009;154(2):183–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Junge S, Gorlich D, den Reijer M, et al. Factors Associated with Worse Lung Function in Cystic Fibrosis Patients with Persistent Staphylococcus aureus. PloS one. 2016;11(11):e0166220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Salsgiver EL, Fink AK, Knapp EA, et al. Changing Epidemiology of the Respiratory Bacteriology of Patients With Cystic Fibrosis. Chest. 2016;149(2):390–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dasenbrook EC, Checkley W, Merlo CA, Konstan MW, Lechtzin N, Boyle MP. Association between respiratory tract methicillin-resistant Staphylococcus aureus and survival in cystic fibrosis. JAMA : the Journal of the American Medical Association. 2010;303(23):2386–2392. [DOI] [PubMed] [Google Scholar]
- 10.Dasenbrook EC, Merlo CA, Diener-West M, Lechtzin N, Boyle MP. Persistent methicillin-resistant Staphylococcus aureus and rate of FEV1 decline in cystic fibrosis. American Journal of Respiratory and Critical Care Medicine. 2008;178(8):814–821. [DOI] [PubMed] [Google Scholar]
- 11.Muhlebach MS, Miller M, LaVange LM, Mayhew G, Goodrich JS, Miller MB. Treatment intensity and characteristics of MRSA infection in CF. Journal of Cystic Fibrosis : official journal of the European Cystic Fibrosis Society. 2011;10(3):201–206. [DOI] [PubMed] [Google Scholar]
- 12.Doring G, Jansen S, Noll H, et al. Distribution and transmission of Pseudomonas aeruginosa and Burkholderia cepacia in a hospital ward. Pediatric Pulmonology. 1996;21(2):90–100. [DOI] [PubMed] [Google Scholar]
- 13.Campana S, Taccetti G, Ravenni N, et al. Molecular epidemiology of Pseudomonas aeruginosa, Burkholderia cepacia complex and methicillin-resistant Staphylococcus aureus in a cystic fibrosis center. Journal of Cystic Fibrosis : official journal of the European Cystic Fibrosis Society. 2004;3(3):159–163. [DOI] [PubMed] [Google Scholar]
- 14.Harik NS, Com G, Tang X, Melguizo Castro M, Stemper ME, Carroll JL. Clinical characteristics and epidemiology of methicillin-resistant Staphylococcus aureus (MRSA) in children with cystic fibrosis from a center with a high MRSA prevalence. American Journal of Infection Control. 2016;44(4):409–415. [DOI] [PubMed] [Google Scholar]
- 15.Fischer AJ, Kilgore SH, Singh SB, et al. High Prevalence of Staphylococcus aureus Enterotoxin Gene Cluster Superantigens in Cystic Fibrosis Clinical Isolates. Genes (Basel). 2019;10(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Spaulding AR, Salgado-Pabon W, Kohler PL, Horswill AR, Leung DY, Schlievert PM. Staphylococcal and streptococcal superantigen exotoxins. Clinical Microbiology Reviews. 2013;26(3):422–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stach CS, Vu BG, Merriman JA, et al. Novel Tissue Level Effects of the Staphylococcus aureus Enterotoxin Gene Cluster Are Essential for Infective Endocarditis. PloS one. 2016;11(4):e0154762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fischer AJ, Singh SB, LaMarche MM, et al. Sustained Coinfections with Staphylococcus aureus and Pseudomonas aeruginosa in Cystic Fibrosis. American Journal of Respiratory and Critical Care Medicine. 2021;203(3):328–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oakeson KF, Wagner JM, Rohrwasser A, Atkinson-Dunn R. Whole-Genome Sequencing and Bioinformatic Analysis of Isolates from Foodborne Illness Outbreaks of Campylobacter jejuni and Salmonella enterica. Journal of Clinical Microbiology. 2018;56(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bankevich A, Nurk S, Antipov D, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19(5):455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Page AJ, Cummins CA, Hunt M, et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics (Oxford, England). 2015;31(22):3691–3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Katz LS, Griswold T, Williams-Newkirk AJ, et al. A Comparative Analysis of the Lyve-SET Phylogenomics Pipeline for Genomic Epidemiology of Foodborne Pathogens. Frontiers in Microbiology. 2017;8:375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ankrum A, Hall BG. Population Dynamics of Staphylococcus aureus in Cystic Fibrosis Patients To Determine Transmission Events by Use of Whole-Genome Sequencing. Journal of Clinical Microbiology. 2017;55(7):2143–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Petit RA 3rd, Read TD. Staphylococcus aureus viewed from the perspective of 40,000+ genomes. PeerJ. 2018;6:e5261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bradley P, Gordon NC, Walker TM, et al. Rapid antibiotic-resistance predictions from genome sequence data for Staphylococcus aureus and Mycobacterium tuberculosis. Nature Communications. 2015;6:10063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaya H, Hasman H, Larsen J, et al. SCCmecFinder, a Web-Based Tool for Typing of Staphylococcal Cassette Chromosome mec in Staphylococcus aureus Using Whole-Genome Sequence Data. mSphere. 2018;3(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chaves-Moreno D, Wos-Oxley ML, Jauregui R, Medina E, Oxley AP, Pieper DH. Application of a Novel “Pan-Genome”-Based Strategy for Assigning RNAseq Transcript Reads to Staphylococcus aureus Strains. PloS one. 2015;10(12):e0145861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nature Protocols. 2016;11:1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nature Biotechnology. 2019;37(8):907–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vila T, Kong EF, Montelongo-Jauregui D, et al. Therapeutic implications of C. albicans-S. aureus mixed biofilm in a murine subcutaneous catheter model of polymicrobial infection. Virulence. 2021;12(1):835–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Langton Hewer SC, Smyth AR. Antibiotic strategies for eradicating Pseudomonas aeruginosa in people with cystic fibrosis. The Cochrane database of systematic reviews. 2017;4:CD004197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Akil N, Muhlebach MS. Biology and management of methicillin resistant Staphylococcus aureus in cystic fibrosis. Pediatric Pulmonology. 2018;53(S3):S64–S74. [DOI] [PubMed] [Google Scholar]
- 33.Liou TG, Elkin EP, Pasta DJ, et al. Year-to-year changes in lung function in individuals with cystic fibrosis. Journal of Cystic Fibrosis : official journal of the European Cystic Fibrosis Society. 2010;9(4):250–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smith TC, Gebreyes WA, Abley MJ, et al. Methicillin-resistant Staphylococcus aureus in pigs and farm workers on conventional and antibiotic-free swine farms in the USA. PloS one. 2013;8(5):e63704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hryniewicz MM, Garbacz K. Borderline oxacillin-resistant Staphylococcus aureus (BORSA) - a more common problem than expected? Journal of Medical Microbiology. 2017;66(10):1367–1373. [DOI] [PubMed] [Google Scholar]
- 36.Long DR, Wolter DJ, Lee M, et al. Polyclonality, Shared Strains, and Convergent Evolution in Chronic CF S. aureus Airway Infection. American Journal of Respiratory and Critical Care Medicine. 2021;203(9):1127–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Muhlebach MS, Heltshe SL, Popowitch EB, et al. Multicenter Observational Study on Factors and Outcomes Associated with Various Methicillin-Resistant Staphylococcus aureus Types in Children with Cystic Fibrosis. Annals of the American Thoracic Society. 2015;12(6):864–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heltshe SL, Saiman L, Popowitch EB, et al. Outcomes and Treatment of Chronic Methicillin-Resistant Staphylococcus aureus Differs by Staphylococcal Cassette Chromosome mec (SCCmec) Type in Children With Cystic Fibrosis. Journal of the Pediatric Infectious Diseases Society. 2015;4(3):225–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Glikman D, Siegel JD, David MZ, et al. Complex molecular epidemiology of methicillin-resistant Staphylococcus aureus isolates from children with cystic fibrosis in the era of epidemic community-associated methicillin-resistant S. aureus. Chest. 2008;133(6):1381–1387. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank with BioProject accession number PRJNA650389 and accessions JACOAG000000000-JACODY000000000. Unassembled FASTQ files are available from the corresponding authors.