

Abstract
The recently developed narrow-band blue-emitting organoboron chromophores based on the multiple-resonance (MR) effect have now become one of the most important components for constructing efficient organic light emitting diodes (OLEDs). While they basically emit through fluorescence, they are also known for showing substantial thermally activated delayed fluorescence (TADF) even with a relatively large singlet–triplet gap (ΔEST). Indeed, understanding the reverse intersystem crossing (RISC) dynamics behind this peculiar TADF will allow judicious molecular designs toward achieving better performing OLEDs. Explaining the underlying nonadiabatic spin-flip mechanism, however, has often been equivocal, and how the sufficiently fast RISC takes place even with the sizable ΔEST and vanishingly small spin–orbit coupling is not well understood. Here, we show that a vibronic resonance, namely the frequency matching condition between the vibration and the electronic energy gap, orchestrates three electronic states together and this effect plays a major role in enhancing RISC in a typical organoboron emitter. Interestingly, the mediating upper electronic state is quite high in energy to an extent that its thermal population is vanishingly small. Through semiclassical quantum dynamics simulations, we further show that the geometry dependent non-Condon coupling to the upper triplet state that oscillates with the frequency ΔEST/ℏ is the main driving force behind the peculiar resonance enhancement. The existence of an array of vibrational modes with strong vibronic rate enhancements provides the ability to sustain efficient RISC over a range of ΔEST in defiance of the energy gap law, which can render the MR-emitters peculiar in comparison with more conventional donor–acceptor type emitters. Our investigation may provide a new guide for future blue emitting molecule developments.
Keywords: thermally activated delayed fluorescence, multiple-resonance effect, spin−vibronic coupling, vibronic resonance, exciton transfer, coherence
Introduction
Thermally activated delayed fluorescence (TADF) has recently emerged
as a promising and pragmatic alternative to more traditional phosphorescence
in the fabrication of efficient organic light emitting diodes (OLEDs).1−4 Via triplet-to-singlet upconversion, so-called reverse intersystem
crossing (RISC), TADF permits singlet emission of nearly 100% internal
quantum efficiency (IQE) in OLEDs. Due to considerably weak spin–orbit
couplings (SOCs) in purely organic emitters, an important parameter
for TADF performance has been the energy gap between the singlet and
the triplet excited states (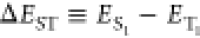 ), which is related to the exchange interaction K between electrons in the frontier orbitals with ΔEST ≃ 2K.5 In this regard, conventional TADF emitters often adopt
the donor–acceptor (DA) structure for spatial separation of
the frontier orbitals, thereby reducing K and ΔEST. However, such DA-type emitters have inherently
a dominant charge transfer character in the S1 state. Therefore,
they entail broad emission features, typically with a full-width-at-half-maximum
(fwhm) > 50 nm,6 resulting in low color
purity. This aspect has indeed been one of the major hurdles to overcome
for commercialization. For instance, the fwhm of the blue emission
spectrum from the latest OLED display is ∼30 nm,7 and meeting this specification will require to
implement a narrow-band color filter. This will inescapably compromise
the efficiency and damage the enhanced performance via TADF. Of course,
this drawback will be especially serious for battery powered displays.
), which is related to the exchange interaction K between electrons in the frontier orbitals with ΔEST ≃ 2K.5 In this regard, conventional TADF emitters often adopt
the donor–acceptor (DA) structure for spatial separation of
the frontier orbitals, thereby reducing K and ΔEST. However, such DA-type emitters have inherently
a dominant charge transfer character in the S1 state. Therefore,
they entail broad emission features, typically with a full-width-at-half-maximum
(fwhm) > 50 nm,6 resulting in low color
purity. This aspect has indeed been one of the major hurdles to overcome
for commercialization. For instance, the fwhm of the blue emission
spectrum from the latest OLED display is ∼30 nm,7 and meeting this specification will require to
implement a narrow-band color filter. This will inescapably compromise
the efficiency and damage the enhanced performance via TADF. Of course,
this drawback will be especially serious for battery powered displays.
With regard to achieving high color purity in the blue region, the molecular design concept that arose recently by utilizing the multiple-resonance (MR) effect is indeed revolutionary and has already attracted significant attention.8−11 It is realized by placing electron-donating nitrogen atoms at para-positions to a central electron-accepting boron atom in a π-conjugation scaffold as in 5,9-diphenyl-5,9-diaza-13b-boranaphtho[3,2,1-de]anthracene (DABNA, Figure 1).8 Hence, the distributions of the highest-occupied molecular orbital (HOMO) and the lowest-unoccupied molecular orbital (LUMO) alternate within the same rigid molecular unit. As a result, a relatively fast emission (>107 s–1) with an ultrapure blue spectrum (fwhm < 30 nm) results, owing to the local-excitation character and the structural rigidity of the S1 state.
Figure 1.
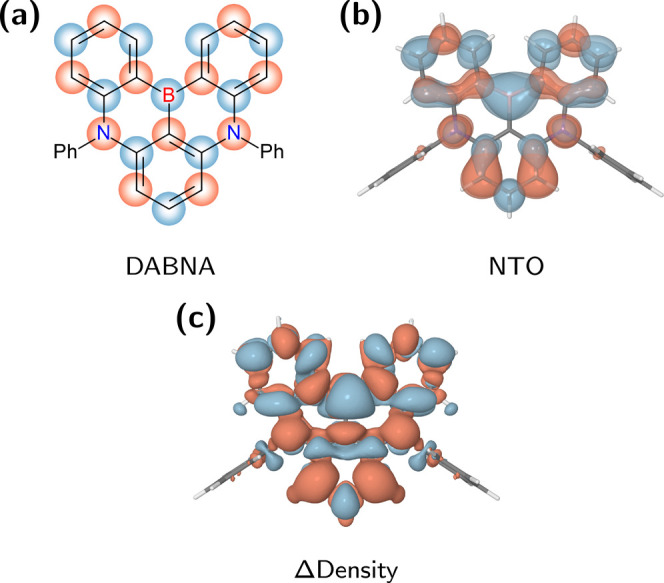
Narrow-band blue TADF emitter, DABNA. (a) Chemical structure with an illustration of the multiple-resonance effect (HOMO/LUMO in red/blue). (b) Hole/particle wave functions shown in red/blue, as represented by the natural transition orbitals from ω*B97X calculations. (c) Difference density plot upon S0–S1 excitation calculated at the SCS-ADC(2) level where the decreased/increased electron density is shown in red/blue. Notice that the transition densities include correlation effects from double excitation terms and appear somewhat differently from b.
Interestingly, in MR-type emitters, although the spin–orbit coupling (SOC) between the S1 and the T1 states is insignificantly small and ΔEST is non-negligibly large, it is known that the RISC process also participates in generating the S1 state during electroluminescence. While the ensuing delayed fluorescence is not remarkably swift (e.g., ∼104 s–1 for DABNA8), the existence of this noticeable amount of TADF is important in many respects. Indeed, the RISC process not only improves the light emitting efficiency but suppresses exciton quenching (e.g., triplet–triplet annihilation and triplet–polaron quenching), while also helping the emitters remain chemically intact during the shortened T1 state lifetime.12 From this perspective, understanding the molecular factors behind RISC of MR-TADF emitters is of central importance to ultimately improve their excited state behaviors. However, RISC processes are generally nonradiative and nonadiabatic in nature, and accordingly, experimental and computational assessments tend to be notoriously difficult. Even greater complications are added to already-difficult computations when vibronic contributions to RISC dynamics in the form of spin–vibronic coupling are considered,13−15 requiring a model that can handle this complex state mixing.16,17 Recently, researchers have been actually tackling this difficult but important system theoretically.15,18,19 For example, Northey and Penfold15 employed the multiconfiguration time-dependent Hartree (MCTDH) quantum dynamics simulation method with a model Hamiltonian derived from the time-dependent density functional theory (TDDFT) to highlight the importance of the spin–vibronic mechanism in the ISC process from S1 to T1 and T2, facilitated by nonadiabatic coupling within the singlet manifold. However, they could not observe S1–T1 ISC even with a sizable SOC between the two states, likely due to the overestimation of ΔEST from TDDFT (∼0.6 eV) and an accidental quasi-degeneracy between S1 and T2. Although some spatial separation of the frontier orbitals occurs due to the alternating HOMO/LUMO distribution, the relatively small ΔEST of an MR-type emitter (<0.2 eV) is induced mainly by higher-order electron correlations.19−21 Pershin et al.19 employed a high level ab initio method to correctly investigate the theoretical origin of the reduced ΔEST in DABNA by explicitly considering the electron correlation effects that renders the local density rearrangement in spatially alternating HOMO–LUMO regions. In fact, achieving a correct energy gap structure is important not just for estimating the S1–T1 gap. If the T1 energy is underestimated or the S1 energy is overestimated, the close-by T2 state15 or even T3 state18 may mistakenly get into energetic resonance with S1, rendering the T2–S1 or T3–S1 intersystem crossing (ISC) estimated to be unphysically too fast. Such artifacts will of course distort the prediction on the interconversion kinetics among all involved electronic states. Therefore, one can easily imagine that combining high level ab initio methods and kinetics and/or dynamics simulation tools will be important in unraveling the processes that are happening in DABNA.
In this work, with such purposes in our mind, we unravel the RISC dynamics of DABNA as a prototype of an ultrapure blue MR-emitter with TADF. Based on high-level quantum chemical calculations, we first evaluate the RISC rate constant (kRISC) through Fermi’s golden rule between T1 and S1 by perturbatively expanding the two involved states within the full singlet and triplet spin manifolds. We also perform semiclassical quantum dynamics simulations to investigate a superexchange behavior in its RISC. Then, by comparing with DA emitters, we discuss distinctive characteristics in the RISC of DABNA, where the spin crossover in a range of ΔEST is effectively mediated by successive and overlapping appearances of the spin–vibronic enhancements provided by an array of vibrational modes. This characterization leads to a new design rule for the development of efficient MR-TADF emitters.
Results and Discussion
Reverse Intersystem Crossing Rate
We reiterate that low-cost correlated excited state methods such as linear response TDDFT do not predict ΔEST well for DABNA, as electron correlations in the form of double excitations tend to reduce the gap.21 We therefore employed a relatively higher level of theory, SCS-CC2/TZVPP//SCS-ADC(2)/SVP. Its spin-component scaling can also improve the accuracy of the excited state potential energy surface22 and, therefore, aids the normal-mode analyses toward obtaining mode-mixing rotations and displacements between pairs of excited states.23 In addition, reliably calculating the energy gaps between excited states improves the quality of perturbation expansions.
We estimated the RISC upconversion kinetics with the golden rule formula, as it can provide a quantitative basis for understanding physical characteristics governed by thermally populated vibrational states of the initial T1 state. Presuming an instantaneous thermalization among the substates of T1 with M = 0 and ±1, the rate constant of RISC is given by24
| 1 |
where the δ-function
imposes energy conservation with the T1 and the S1 vibrational energies Eν and Eν′. The thermal population at temperature T is described by Boltzmann distribution Pν. The nonadiabatic interaction between the two
electronic states is represented by 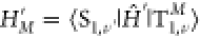 originating from the spin–orbit
(SO) and the non-Born–Oppenheimer (nBO) couplings,
originating from the spin–orbit
(SO) and the non-Born–Oppenheimer (nBO) couplings, 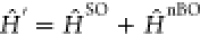 . After perturbatively expanding both T1 and S1 state vectors with respect to
. After perturbatively expanding both T1 and S1 state vectors with respect to 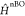 within their respective spin manifolds
T′ and S′, one can show that the coupling matrix element
can be expressed as a sum-over-state expansion:25,26
within their respective spin manifolds
T′ and S′, one can show that the coupling matrix element
can be expressed as a sum-over-state expansion:25,26
| 2 |
The first term represents first-order direct spin–orbit (DSO) coupling driven by the electronic characters of S1 and T1, while the second and the third terms serve as second-order spin–vibronic (SV) coupling, where intermediary states in the triplet and the singlet manifolds assist the spin–orbit coupling. See also Supporting Information (SI) Text 1 for the derivation details on the coupling matrix elements of the full second-order SV terms.
The rate constant kRISC can be analytically
solved from a correlation function formalism combined with a path-integral
expression27−30 as briefly summarized in SI Text 1. Because kRISC involves 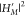 , it includes pure DSO and SV contributions
from the squares of the corresponding terms in eq 2. We will respectively refer to them as kDSO and kSV. Of
course, an SV–DSO cross term will also arise, but it vanishes
due to time-reversal symmetry.30 The calculated
rate constants are listed in Table 1 together with other related photophysical properties
obtained by quantum chemical calculations. One can see that our computed kRISC of 1.05 × 104 s–1 agrees quite well with the experimental value of 0.99 × 104 s–1 measured in a thin film condition.8 Of course, care must be taken in assessing the
degree of agreement as both theory and experiment will surely have
limitations when estimating rate constants.
, it includes pure DSO and SV contributions
from the squares of the corresponding terms in eq 2. We will respectively refer to them as kDSO and kSV. Of
course, an SV–DSO cross term will also arise, but it vanishes
due to time-reversal symmetry.30 The calculated
rate constants are listed in Table 1 together with other related photophysical properties
obtained by quantum chemical calculations. One can see that our computed kRISC of 1.05 × 104 s–1 agrees quite well with the experimental value of 0.99 × 104 s–1 measured in a thin film condition.8 Of course, care must be taken in assessing the
degree of agreement as both theory and experiment will surely have
limitations when estimating rate constants.
Table 1. Calculated Photophysical Properties of DABNA and its Related Moleculesa.
| SOC
(cm–1)e |
NAC (bohr–1)f | ||||||
|---|---|---|---|---|---|---|---|
| molecule | kRISCb (104 s–1) | kSV contribution (%) | ΔESTc (eV) | S1d (eV) | S1/T1 | S1/T3 | T1/T3 |
| DABNA | 1.05 | 99.1 | 0.144 (0.15g) | 3.04 (2.71g) | 0.035 | 0.804 | 8.56 |
| TBN-TPA | 0.94 | 95.4 | 0.137 (0.14h) | 3.03 (2.64h) | 0.040 | 0.698 | 9.06 |
| TABNA | 0.16 | 99.9 | 0.161 (0.21i) | 3.36 (3.11i) | 0.005 | 0.439 | 14.23 |
Average rate in a small range around ΔEST as explained in the text.
Adiabatic singlet–triplet energy difference.
Vertical emission energy calculated at the S1 geometry.
Triplet substate-averaged spin–orbit coupling.
Norm of the derivative coupling
vector, 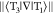 .
.
Measured in ethanol,
PMMA/1 wt % thin film, or in
toluene.
Direct Spin–Orbit Coupling versus Second–Order Spin–Vibronic Coupling
In fact, what is much more important than observing a numerical agreement is analyzing the driving force for the spin crossover. The spin–orbit coupling between the T1 and the S1 states of DABNA is merely ∼0.04 cm–1, which is clearly insufficient to incur any meaningful RISC unless it is matched with an unphysically very small ΔEST (<0.05 eV). From Table 1, one can see that kSV affected by intermediary spin states actually dominates kRISC. To gain further insights, we parametrically varied ΔEST in the golden rule estimation and subsequently recalculated kRISC. Figure 2a displays the RISC rate constants thus obtained. The single ΔEST value as obtained with the adopted SCS-CC2 method is marked with a vertical line, together with experimental ΔEST values measured under two different conditions.8 Surprisingly, unlike typical organic molecules, the trend of the rate constants deviates substantially from the conventional energy gap law, which would manifest as a linearly decaying line in our log-scaled plot. Based on a deconvolution into kDSO and kSV portions also shown in Figure 2a, this unusual kinetic behavior can be attributed mainly to the latter spin–vibronic part.
Figure 2.

Vibronic enhancement of RISC. (a) Total RISC rate as a function of ΔEST and its decomposition into DSO and SV contributions. Some notable ΔEST values are marked as vertical lines: the computational (SCS-CC2) result at 0.144 eV; the experimentally observed gaps in ethanol solvent and in 3,3′-di(9H-carbazol-9-yl)-1,1′-biphenyl (mCBP) host at 0.15 and 0.18 eV. (b) Deconvolution of the SV component into each normal mode contribution in the ΔEST range of 0.14–0.18 eV. Rate contributions greater than 1000 s–1 are shown. The vertical scale is visually supplemented by the coloring scheme shown at the bottom. The contribution sum is depicted by the gray projection. The vibrational frequencies are represented along the axis denoted with ℏω.
Not only is RISC dynamics completely governed by SV coupling, but kSV also remains nearly constant within an order
of magnitude when ΔEST is in 0.14–0.18
eV. This range actually covers all ΔEST values predicted by both theory and experiment and was used for
calculating average rates reported in Table 1. Beyond this range, kSV decays in a monotonous manner following the energy gap law.
We have further broken down the rate components associated with each
vibrational normal mode as 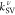 (Figure 2b). We immediately see that, in this range, multiple
modes can contribute quite significantly to enhancing the RISC rate
in a successive and overlapping manner as long as their frequencies
match ΔEST closely. For instance,
if ΔEST is 0.148 eV (close to both
theoretical and experimental values), the calculated total rate of
1.95 × 104 s–1 is mainly governed
by a single vibrational mode “ν106”
at 1279 cm–1. Without the SV contribution, kRISC would have been as small as kDSO = 56 s–1. Similar analyses were
also performed for other gaps and are shown in Table S1.
(Figure 2b). We immediately see that, in this range, multiple
modes can contribute quite significantly to enhancing the RISC rate
in a successive and overlapping manner as long as their frequencies
match ΔEST closely. For instance,
if ΔEST is 0.148 eV (close to both
theoretical and experimental values), the calculated total rate of
1.95 × 104 s–1 is mainly governed
by a single vibrational mode “ν106”
at 1279 cm–1. Without the SV contribution, kRISC would have been as small as kDSO = 56 s–1. Similar analyses were
also performed for other gaps and are shown in Table S1.
At first sight, as accelerations in exciton transfers by gap-matching vibrations have been continuously reported,31−34 this RISC enhancement may appear trivial: spin crossover via a resonance between S1,ν′=0 and T1,ν=1. In the present case, however, T1–S1 SOC is very small, and kDSO is still small even in the existence of a resonant vibration. Indeed, from Figure 2a, one can see that RISC by only DSO would have been 2 orders of magnitude slower even with the vibronic resonance. RISC becomes sufficiently fast only when the process is mediated by another electronic state, and the mediation is somehow strongly influenced by the vibrational frequency. Thus, we now turn our attention to which intermediary state plays the most important role in enhancing RISC rate in DABNA.
Contributions by Individual Electronic States
The successive vibronic enhancements by an array of vibrational modes serve to sustain the RISC rate for a plausibly expected range of ΔEST in DABNA. When we focus on three normal modes, ν106, ν116, and ν118, which preeminently contribute to the total kRISC as shown in Figure 3a, SV coupling through the triplet manifold originating from the second term in eq 2 is dominant as denoted by kSV,T in Figure 3b. On the other hand, the contribution from the singlet manifold, kSV,S, is the least significant. Interestingly, the singlet manifold contributes more substantially via the cross-product of the second and the third terms, shown with kSV,T-S in Figure 3b. To identify important upper excited states that vibronically mediate T1 and S1, Figures 3c and S1 show the convergence profiles of the sum-over-states expansions in eq 2 again with respect to ν106, ν116, and ν118. In the triplet manifold, which often acts as an important route toward spin–vibronic mixing, the nonadiabatic coupling effect between the T1–T3 pair is the most noticeable. For example, the nonadiabatic coupling between these states caused by a displacement corresponding to the normalized ν118 mode vector amounts to 12.5 cm–1 (Figure 4). This is enormously larger than the conventional SOC size in organic molecules. In addition, SOC between S1 and T3 is approximately 23 times larger than that between S1 and T1 (Table 1). This confirms that it is the T3 state, which strongly mixes with the T1 state via the “ΔEST-matching” vibrational mode, that enhances kSV.
Figure 3.

Analysis of kRISC in DABNA. (a) Contributions
by vibrational modes with strong resonance enhancements, namely ν106, ν116, and ν118. The
ΔEST values at which 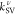 become maxima are marked as vertical lines.
(b) Contributions by each vibrational mode to kSV at the vertically marked energies in a. The contribution
is further partitioned into spin-manifold components according to eq 4. (c) Convergence profiles
of the sum-over-electronic-states expansion for each mode in the triplet
manifold. Similar profiles in the singlet manifold are provided in Figure S1. Here, the excited state with a dominant
contribution (T3) can be easily identified as marked with
gray spheres. Convergence was reached with 20 excited states as denoted
with dashed lines and stars. The T1 energy was used as
the zero energy reference. (d) Natural transition orbital pairs from
the ω*B97X calculations at the S1 geometry. Hole
(red) and particle (blue) wave functions are represented by frontier
orbitals with the largest weights, shown in parentheses. The S1 and the T1 energies are from the SCS-CC2 level
calculations. (e) Nonadiabatic T1–T3 and
S1–S6 coupling vectors responsible for
RISC.
become maxima are marked as vertical lines.
(b) Contributions by each vibrational mode to kSV at the vertically marked energies in a. The contribution
is further partitioned into spin-manifold components according to eq 4. (c) Convergence profiles
of the sum-over-electronic-states expansion for each mode in the triplet
manifold. Similar profiles in the singlet manifold are provided in Figure S1. Here, the excited state with a dominant
contribution (T3) can be easily identified as marked with
gray spheres. Convergence was reached with 20 excited states as denoted
with dashed lines and stars. The T1 energy was used as
the zero energy reference. (d) Natural transition orbital pairs from
the ω*B97X calculations at the S1 geometry. Hole
(red) and particle (blue) wave functions are represented by frontier
orbitals with the largest weights, shown in parentheses. The S1 and the T1 energies are from the SCS-CC2 level
calculations. (e) Nonadiabatic T1–T3 and
S1–S6 coupling vectors responsible for
RISC.
Figure 4.

Mode–projected nonadiabatic couplings between T2 and T1 (blue) and between T3 and T1 (red). Projections were carried out onto each mode vector, normalized
in the atomic unit (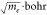 ). Each mode is supplemented with its vibrational
frequency (gray). Modes at 3000 cm–1 and above are
stretches involving terminal hydrogen atoms.
). Each mode is supplemented with its vibrational
frequency (gray). Modes at 3000 cm–1 and above are
stretches involving terminal hydrogen atoms.
It is noteworthy that the T2 state, on the other hand, even with a reduced energy gap, has relatively insignificant contribution to the total coupling due to little nonadiabatic component along the same ΔEST-matching vibrational mode. This might be surprising when one considers that the T2–T1 nBO coupling is slightly larger than the T3–T1 coupling at least along a few vibrational modes (Figure 4). At this point however, we emphasize that, from the perturbative expansion in eq 2, the total coupling responsible for RISC is determined by the product of nBO coupling and SOC terms between vibronic states. This combined coupling will undoubtedly be the decisive factor and revealing the important electronic state should be judged based on that combined factor. Given that T2–S1 SOC is only ∼1/3 of T3–S1 SOC (Figure S2), T2 will not be able to outcompete T3 with slight advantages that are only occasionally found in Figure 4. Namely, the combination of nBO coupling and SOC renders T2 not as important toward promoting resonantly enhanced vibronic RISC. Even the mode ν103, along which the T2–T1 coupling has the largest component in Figure 4, afforded at best 12.5% of the kRISC at ΔEST = 0.146 eV, where the spin crossover was mostly steered by the T3–T1 coupling along the ν106 direction (Figure 3a).
To reason why the T3–T1 nBO coupling dominates, we may resort to the relationship between the spatial motions of three gap-matching vibrational modes and the nature of the electronic transitions. The atomic motions of the modes are in general along in-plane ring distortions (Figure S3) and will match well with the changes in conjugation patterns. The electronic change characterized by the switch from the T1 particle wave function to the T3 one shown in Figure 3d indeed shares a similar character. However, care must be taken with this reasoning. In the case of the T2–T1 pair, because the two states share almost the same particle wave functions (Figure S4), the electronic change may be characterized by the switch from the T1 hole wave function to the T2 one. In fact, this change is also with a conjugation switch. This aspect also cautions us against overusing the natural transition orbitals in reasoning for three-state-involving transition properties. As the nature of electronic transitions is determined by the involved many-electron wave functions, reasoning by visualizing one electron orbitals may become oversimplifications at times. Though computationally demanding, verifying with numerical data as presented here can form the clearest evidence.
The fact that T2 contributes little is in line with an earlier report with MCTDH simulations,15 where no appreciable T2–T1 internal conversions were found. In fact, if we had considered only T1 and T2 as in that report, we would have observed that the singlet manifold would contribute more than the triplet one by inferring from, for instance, a largish singlet coupling for ν118 (Figure S1) that is bigger than the T2-coupling (Figure 3c). In addition, we believe that the cross-product term kSV,T-S is particularly enhanced because there is an electronic state that strongly resembles T3 in the singlet manifold. Namely, the electronic character of S6 is very similar (Figure 3d), and accordingly, the directionality of the nonadiabatic coupling vectors between the S1–S6 and the T1–T3 pairs will also be similar (Figure 3e). As a result, the second-order spin–vibronic transition paths between the T1 and the S1 states that involve the two intermediary states will likely cooperate.
Resonance Enhancement that Involves Three States
Now
that we identified T3 as the most important intermediary
state for DABNA, we can infer that there is a strong and peculiar
resonance enhancement in kSV from a vibration
(1) because it oscillates at a frequency that matches ΔEST and (2) because it induces strong T1–T3 coupling. This vibronic resonance is peculiar
and is different from earlier observations31−34 in that it involves more than
two electronic states. We also stress that this superexchange process
involving T3 should be distinguished from a stepwise conversion
including the “reverse internal conversion” (RIC) from
T1 to T3. The calculated gap between the two
electronic states is much larger than the thermal energy with 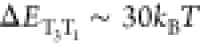 (Table S2),
and as a result the equilibrium constant between T1 and
T3 will be diminutive. Furthermore, S1 and T3 are not isoenergetic with
(Table S2),
and as a result the equilibrium constant between T1 and
T3 will be diminutive. Furthermore, S1 and T3 are not isoenergetic with 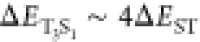 , which will also significantly prohibit
the spin conversion between these states. Should the RISC process
take place via the spin crossover as T3 → S1 after a pre-equilibrium involving RIC as T1 ⇌
T3, the overall rate will be too slow. One may also wonder
about the role of T3 hot excitons that can form transiently
during electroluminescence. For DABNA, a direct hot exciton transfer
from T3 to S1 is appraised as an unlikely event,
because with the strong vibronic coupling between the two triplets,
the T3 → T1 transition will prevail and
lead to an almost complete and instant population transfer to the
lowest triplet.
, which will also significantly prohibit
the spin conversion between these states. Should the RISC process
take place via the spin crossover as T3 → S1 after a pre-equilibrium involving RIC as T1 ⇌
T3, the overall rate will be too slow. One may also wonder
about the role of T3 hot excitons that can form transiently
during electroluminescence. For DABNA, a direct hot exciton transfer
from T3 to S1 is appraised as an unlikely event,
because with the strong vibronic coupling between the two triplets,
the T3 → T1 transition will prevail and
lead to an almost complete and instant population transfer to the
lowest triplet.
To further substantiate the peculiar resonance, we have also employed a semiclassical quantum dynamics simulation approach, Poisson bracket mapping equation (PBME-nH).35,36 Based on the observations described in the above, we have chosen {S1, T1, T3} as our electronic state space. These states and one underdamped vibration corresponding to ν106 form our minimal model for exhibiting the resonance. The underdamped mode actually vibronically couples T1 and T3, and the coupling strength changes in proportion to the normal mode displacement (see Computational Methods). To further probe the resonant behavior by ν106, we scanned its frequency around its physical value. Figure 5 displays how kRISC changes as a function of this frequency and we can indeed directly observe a resonant behavior again near the physical frequency, 1279 cm–1 (= 0.159 eV). As is shown in the same figure, when we fortuitously set the T1–T3 coupling as constant in time with the ν106 mode still preserved in the system, the S1 population did not grow any faster than what was allowed by the small SOC between T1 and S1, even when the constant T1–T3 coupling was rather strong and the ν106 frequency matched ΔEST. Thus, the fluctuating nature of the T1–T3 coupling is the key to the RISC enhancement, which is maximized when the fluctuating frequency is in a near-resonant condition with ΔEST.
Figure 5.
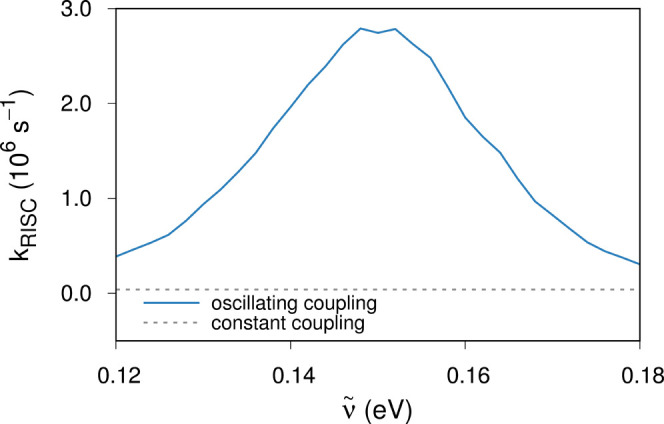
Resonance enhancement of RISC by a coupled vibration. The blue
line denotes the rate estimated with PBME-nH quantum dynamics simulations
with an underdamped emitter vibration with a frequency 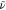 that induces T1–T3 coupling in a non-Condon manner. The gray line shows the
rate with a strong constant coupling but without this non-Condon effect,
that induces T1–T3 coupling in a non-Condon manner. The gray line shows the
rate with a strong constant coupling but without this non-Condon effect, 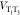 = 500 cm–1.
= 500 cm–1.
It is intriguing to observe that the vibrational resonance involves
three electronic states, with the frequency matching both the energy
gap for the T1–S1 pair and the coupling
oscillation for the T1–T3 pair. Thus,
RISC enhancement through SV coupling bears a strong superexchange
character.37 When this peculiar resonance
condition is met, the RISC process can take place even when ΔEST is rather substantial as was already noted
with Figure 2a. This
interesting behavior can be explained in the following manner. The
oscillating non-Condon T1–T3 coupling
(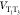 ) forces the electronic characters of a
triplet state to alternate in time. Namely, a state initially at T1 will become a superposition of T1 and T3, whose composition will vary with the vibrational motion. Because
the T3–S1 SOC is relatively large (Table 1), the new superposition
state can transfer the population to S1 with a much larger
probability than the pure T1. In addition, because
) forces the electronic characters of a
triplet state to alternate in time. Namely, a state initially at T1 will become a superposition of T1 and T3, whose composition will vary with the vibrational motion. Because
the T3–S1 SOC is relatively large (Table 1), the new superposition
state can transfer the population to S1 with a much larger
probability than the pure T1. In addition, because 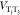 oscillates in time, the T3 composition
will keep fluctuating and sustain the transfer until all triplet population
is consumed. Surprisingly, if
oscillates in time, the T3 composition
will keep fluctuating and sustain the transfer until all triplet population
is consumed. Surprisingly, if 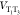 is constant in time, i.e., Condon coupling,
the electronic character of the triplet state quickly settles to a
steady state, from which the RISC process is not as effective. In
fact, the constant coupling adopted with Figure 5 was twice larger than the root-mean-squared
average of the oscillating coupling. The fact that the two RISC kinetics
shown in the figure are drastically different evidently exhibits the
importance of the oscillating nature of the vibronic coupling, with
the matching resonance condition with ΔEST. Perhaps, the resonantly fluctuating coupling is taking
a role of replenishing the fragile coherence31 between T1 and T3. We note that the time scale
of state transitions in OLED materials is often much longer than that
of light harvesting systems, and it is striking that there can still
be a process with commonly shared characters31 between the two different groups.
is constant in time, i.e., Condon coupling,
the electronic character of the triplet state quickly settles to a
steady state, from which the RISC process is not as effective. In
fact, the constant coupling adopted with Figure 5 was twice larger than the root-mean-squared
average of the oscillating coupling. The fact that the two RISC kinetics
shown in the figure are drastically different evidently exhibits the
importance of the oscillating nature of the vibronic coupling, with
the matching resonance condition with ΔEST. Perhaps, the resonantly fluctuating coupling is taking
a role of replenishing the fragile coherence31 between T1 and T3. We note that the time scale
of state transitions in OLED materials is often much longer than that
of light harvesting systems, and it is striking that there can still
be a process with commonly shared characters31 between the two different groups.
The physical foundation of the spin–vibronically enhanced
RISC at substantial ΔEST can be
additionally explained with Figure 6a, where molecular factors affecting kSV are schematically displayed. Because both the S1 and the T1 states share the identical local excitation
character, the potential energy surfaces are quite similar in shape
to each other. In addition, structural rigidity stemming from extensive
conjugation in combination with planarity resembling nanographene
restricts the T1–S1 geometric distortion
as 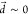 (Figure S5).
These aspects combine to produce only slightly shifted vibrational
frequencies and a diagonal-dominant Duschinsky matrix from T1 to S1 (Figure S6), resulting
in no normal mode mixing upon state switching. RISC dynamics can therefore
be reduced to independent mode-by-mode dynamics (Figure 6a). When ΔEST ≠ 0, the δ-function in eq 1 and hence kRISC will reach a maximum as ΔEST approaches the vibrational gap ℏω. In addition, as discussed above, the spin crossover process is
undertaken through the state mixing with T3 especially
when
(Figure S5).
These aspects combine to produce only slightly shifted vibrational
frequencies and a diagonal-dominant Duschinsky matrix from T1 to S1 (Figure S6), resulting
in no normal mode mixing upon state switching. RISC dynamics can therefore
be reduced to independent mode-by-mode dynamics (Figure 6a). When ΔEST ≠ 0, the δ-function in eq 1 and hence kRISC will reach a maximum as ΔEST approaches the vibrational gap ℏω. In addition, as discussed above, the spin crossover process is
undertaken through the state mixing with T3 especially
when 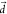 is along the T1–T3 coupling direction. Therefore, for an optimal kRISC,
is along the T1–T3 coupling direction. Therefore, for an optimal kRISC, 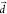 along ν106 must remain
small to maximize the Franck–Condon factor and the derivative
integral between the v = 0 and the v′ = 1 vibrational wave functions (SI Text 1). The effect of the displacement can be easily verified numerically
by increasing the quantum chemically obtained
along ν106 must remain
small to maximize the Franck–Condon factor and the derivative
integral between the v = 0 and the v′ = 1 vibrational wave functions (SI Text 1). The effect of the displacement can be easily verified numerically
by increasing the quantum chemically obtained 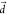 into fortuitously larger
into fortuitously larger 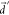 . Indeed, the vibronic enhancement in kSV almost disappears at large
. Indeed, the vibronic enhancement in kSV almost disappears at large 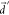 in the golden rule rate estimation (Figure 6b), and a similar
behavior is also observed from PBME-nH simulations (Figure S7). We also note that at larger ΔEST, increasing
in the golden rule rate estimation (Figure 6b), and a similar
behavior is also observed from PBME-nH simulations (Figure S7). We also note that at larger ΔEST, increasing 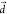 will lead to a slower decay of kDSO (Figure S8).
The temperature effect was also investigated (Figure S9), affirming that kSV becomes more prominent at higher temperature because the transition
originates from a thermal population at v = 1.
will lead to a slower decay of kDSO (Figure S8).
The temperature effect was also investigated (Figure S9), affirming that kSV becomes more prominent at higher temperature because the transition
originates from a thermal population at v = 1.
Figure 6.

Mechanism of vibrational enhancement of RISC. (a) Schematic representation
of the vibronic spin-flip transition. The vibrational wave functions
are illustrated with filled curves (T1, blue; S1, red). (b) Displacement dependence of the second-order (spin–vibronic)
RISC rates at varying ΔEST as measured
by fortuitously increasing the original displacement vector 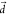 by some integers,
by some integers, 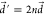 . See SI Text 1 for the mathematical formulation of
. See SI Text 1 for the mathematical formulation of 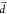 .
.
Behaviors of DABNA Variants
We further applied the same golden-rule rate analysis to a few more emitting molecules related to DABNA (Table 1 and Figure S5). First, TBN-TPA with a carbazole group in a para-position to boron and six additional tert-butyl groups on peripheral phenyl rings was investigated. As shown in the table, the added carbazole moiety only slightly reduces ΔEST by ∼0.01 eV. At this energy gap, predominance of the SV contribution was retained (Figure S10a). If we assume that we can tune the energy gap by changing the doping conditions, our calculations indicate that it will be possible to obtain a maximum kRISC at 1.07 × 104 s–1 appearing at ΔEST ≃ 0.15 eV. The average rate over a small range (±0.1 eV) of ΔEST is estimated as 0.94 × 104 s–1. This is comparable to DABNA, allowing us to speculate that the reported higher PLQY (98%) from an earlier experiment38 is due to the bulky tert-butyl groups that physically hinder external quenching factors. TABNA with an added phenylamino group onto the DABNA core was also considered, even though its emission is excessively blue-shifted with λmax = 399 nm.39 In this case, while kSV still governed the RISC dynamics because T1–S1 SOC was almost negligible, the absolute value of kSV was smaller by an order of magnitude, resulting in no vibrational resonance peaks (Figure S10b). This is mainly due to a much larger geometric difference between T1 and S1 states, corresponding to a reorganization energy of λ = 434 cm–1 by the emitter itself. This reaffirms that a small geometric change is needed to observe vibrational enhancement of kSV (Figure 6).
Finally, we note that the energy gap between the vibronically coupled triplet pair (the T1–T3 gap for DABNA) will also be important for efficient and practical RISC. This is easily discernible both with the golden rule and PBME formalisms. When the gap is too large, the contribution of T3 in the perturbation expansion for T1 diminishes. Likewise, T1–T3 mixing in PBME weakens under the same condition and the effect of superexchange becomes unimportant. Therefore, reducing this gap should also be a tactic of consideration if one attempts to optimize kRISC through kSV. Of course, achieving this without disturbing electronic structure properties, such as the MR effect, will be important, as breaking it can return the emitter properties to conventional DA-type behaviors.40 Indeed, we have already seen that TABNA does not exhibit a resonantly enhanced RISC rate even with the reduced T1–T3 gap (Table S3) and subsequently increased nonadiabatic coupling (Table 1). This is because the electronic structure consistency between S1 and T1 is broken with the added modification from DABNA.
Donor–Acceptor Type versus Multiple–Resonance Type Emitters
Due to conflicting preferences for the exchange
energy K,41 DA-emitters
tend to have different natures in their lowest excited states: charge-transfer
(CT) S1 vs locally excited (LE) T1 states.42,43 This entails relatively large geometric differences (Table S4) and, subsequently, large reorganization
energies during ISC between them. While a small displacement can be
expected for DA type emitters when both the S1 and the
T1 states correspond to the same CT character, the displacement
will likely be still larger than in DABNA. This is because there should
be another LE triplet state whose energy is close to the T1 energy to facilitate the RISC process. With the neighboring LE state,
due to inevitable electronic mixing, T1 will still have
a non-negligible amount of LE character, which will subsequently induce
a noticeable size of displacement from the S1 geometry.
As we observed in Figure 6b, the vibrational rate enhancement in kSV disappears when 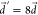 . The reorganization energy of
. The reorganization energy of 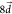 amounts to 2128 cm–1 (
amounts to 2128 cm–1 (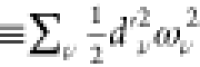 ), which is close to the reorganization
energies of DA-TADF emitters. Thus, it explains why a drastic vibrational
enhancement in kSV was not observed in
more flexible DA-TADF emitters in an earlier study.30 In addition, El-Sayed-allowed DSO between a charge transfer
S1 state and a locally excited T1 state provides
the primary pathway to RISC, with reducing ΔEST working as the widely accepted design principle.4,42,43 Hence, for DA-TADF emitters,
the DSO portion will play a major role with a minor enhancement from
SV coupling.30 We should, however, note
that by exploiting the disparate responses of CT and LE states to
the surrounding polarity, the 1CT–3CT
pair can be (quasi-)degenerate or be even more stable than the 3LE state in a polar medium. In such cases, the SV coupling
can be elevated for DA-TADF emitters at the expense of a redshift
in the emission wavelength.13
), which is close to the reorganization
energies of DA-TADF emitters. Thus, it explains why a drastic vibrational
enhancement in kSV was not observed in
more flexible DA-TADF emitters in an earlier study.30 In addition, El-Sayed-allowed DSO between a charge transfer
S1 state and a locally excited T1 state provides
the primary pathway to RISC, with reducing ΔEST working as the widely accepted design principle.4,42,43 Hence, for DA-TADF emitters,
the DSO portion will play a major role with a minor enhancement from
SV coupling.30 We should, however, note
that by exploiting the disparate responses of CT and LE states to
the surrounding polarity, the 1CT–3CT
pair can be (quasi-)degenerate or be even more stable than the 3LE state in a polar medium. In such cases, the SV coupling
can be elevated for DA-TADF emitters at the expense of a redshift
in the emission wavelength.13
Owing to the similarity between the electronic structures of T1 and S1, ΔEST of DABNA, albeit being reduced by electron correlations, remains relatively large. In addition, the DSO coupling between T1 and S1 is suppressed by the El-Sayed rule.44 However, such an electronic structure similarity gives rise to a small geometric displacement in the spin crossover. If there is a vibrational mode that energetically matches ΔEST and effectively induces the coupling between the lowest excited state and higher-lying intermediary state, then such a SV coupling can speed up the RISC process dramatically. It is evident that, from the existence of multiple modes with such properties, the vibronic enhancement can be contiguous over extended ΔEST values. These aspects will defy the energy gap law that essentially governs the RISC processes in DA-TADF molecules.
With the above comparisons, it will be interesting to further consider how the present case differs from the recently discussed spin–vibronic mechanism with a third mediating state in a DA-TADF system DPTZ-DPTO2.13,14 As a matter of fact, the spin–vibronic mechanism of state switching involving two different spin multiplicities has been known for quite some time (for instance, ref (25)), and there can be many different possibilities through which the mechanism can manifest itself. In the case of DPTZ-DPTO2, the energy resonance condition between the involved electronic states was discussed to be crucial, and the “Type II” case with 1CT, 3CT, and 3LE states gathered together at near-degeneracy was suggested as an ideal system for TADF. When that condition is met, because the spin–orbit coupling is much larger for the 3LE–1CT pair than for 3CT–1CT, and because 3LE is thermally quite accessible, RISC via 3LE–1CT will play a dominant role either through a direct transfer or through mediation involving 3CT. In this case, a vibration plays only a nonadiabatic coupling role for the 3LE–3CT pair but not for the energy resonance. Our results with the MR-TADF system, whose S1/T1/T3 states play the somewhat analogous roles of 1CT/3CT/3LE of the DA-TADF system, show different behaviors in a few crucial ways.
-
1.
The T1 energy is noticeably lower than the S1 energy in MR-TADF. Thus, a vibration should be combined to satisfy the energy resonance condition to facilitate the T1–S1 conversion. Indeed, we have verified the associated RISC rate enhancement as shown in Figures 2 and 5. Again, the vibrational frequency resonance is a key to MR-TADF but not for DA-TADF. Also, there are multiple vibrations that can resonantly enhance the state switching rate.
-
2.
The mediating T3 state in DABNA is quite high in energy and is thermally inaccessible for hopping or reverse internal conversion. On the other hand, in DA-TADF systems, the mediating 3LE state tends to lie below the 3CT level especially when ΔEST is large and a significant spin–vibronic effect is expected. This difference induces many different aspects: for example, the extraordinarily large non-Condon effect, which appears important for the RISC rate enhancement with DABNA.
-
3.
The geometric distortion associated with the T1 → S1 switch is quite small with DABNA, whereas the equivalent distortion with a DA-TADF molecule tends to be large when T1 is 3LE, with different characters from 1CT as S1 (Table S4). Indeed, we found that a large distortion will simply erase the resonant vibronic RISC enhancement (Figure 6b), and the vibrational rate enhancement as in DABNA will not be observed in DA-TADF systems.
This leads us to an important alternative of designing effective MR-TADF emitters: the optimal SV coupling through vibronic resonance. For certain molecules like DABNA, lowering ΔEST can often be challenging. Sometimes, peripheral substitutions for improving other functionalities such as intermolecular interactions and charge transport behaviors should often be conducted even at the cost of increasing ΔEST. In such cases, it will be ideal if the increase in ΔEST can be compensated by the vibrational resonance effect. One may be concerned that the resonance would be too fragile as the gap energy can be easily changed by the emitter environment and matching the frequency might be too restrictive. We believe this will not be too serious as there may be multiple vibrations that can play the enhancing role. With DABNA, indeed, three modes with enough overlaps with the T1–T3 derivative coupling (Figure S3) can take their turns when ΔEST was varied (Figure 2). In addition, the resonance peaks are actually quite broad (Figures 2 and 5), and strictly matching the frequency is not a necessity.34
Conclusion
We have applied a spin–vibronic model and a semiclassical quantum dynamics simulation tool to elucidate the RISC dynamics of MR-TADF organoboron emitters with superior color purity. We found that it is highly driven by a spin–vibronic effect, which emerges as a second-order process with the golden rule description and as a superexchange event from the semiclassical treatment. More importantly, we observed a peculiar three-state involving resonance with which the RISC rate is enhanced significantly when the frequency of a vibration that induces a non-Born–Oppenheimer coupling between the T1–T3 pair matches the gap energy between the T1–S1 pair. We also showed that multiple vibrational modes exist so that they can resonantly enhance exciton transfers over a wide enough range of ΔEST.
The fact that the spin–vibronic effect plays a major role in the considered MR-TADF emitters is in stark contrast to the aspect observed with more conventional DA-TADF emitters, where the dynamics could be explained largely by the first-order direct spin–orbit coupling.30 Perhaps, the widely accepted design principle for achieving superior TADF emitters by minimizing ΔEST may not be the only path to consider at least for the emitters with rigid core structures, and maximizing the spin–vibronic coupling through vibronic resonance can be additionally sought for. Nonetheless, predicting in an a priori fashion whether a vibration will participate to resonantly enhance RISC will not be a simple task as it requires comprehensively computing the emitter properties with the rather complicated network of excited states, as attempted in our present work. Even still, we conclude that treating spin–vibronic coupling is of utmost importance and should be included in a regular basis, as well-defined design principles based on second-order effects will surely benefit the development of efficient emitters. As is often the case with any excited state tuning, predicting the relationships between multiple modification aspects will be a complicated undertaking, but a viable solution can still be reached by adopting advanced tools as screening measures. We hope that our approaches described here can serve such a purpose in future molecular designs.
Computational Methods
Electronic Structure Calculations
Electronic structure calculations were performed with the TURBOMOLE 7.345 and Q-Chem 5.146 program packages. With the original spin-component scaling parameters,47 the excited state geometries were optimized by the strict variant of the second-order algebraic diagrammatic construction (SCS-ADC(2)) method.48 This was followed by the approximate coupled-cluster singles and doubles (SCS-CC2) method49 for the energy evaluations. The resolution-of-the-identity approximation for the electron repulsion integrals were used in treating correlations. For normal mode calculations, numerical Hessians were employed with a frequency scaling factor of 0.9631.50 The optimized geometries and the corresponding vibrational frequencies are provided in Tables S5 and S6. Because the nonadiabatic and spin–orbit couplings were not available at the ADC(2) or CC2 level, single-point time-dependent DFT calculations under the Tamm–Dancoff approximation was adopted instead, with the gap-tuned long-range corrected ω*B97X functional.51 Because the nuclear screening two-electron term is often small and is less important in the context of spin–vibronic model with the more contributing vibronic coupling, we have employed the one-electron Breit–Pauli spin–orbit Hamiltonian.52 Solvent effects were treated with the polarizable continuum model in the integral equation formalism (IEF-PCM).53 The amorphous molecular solid film condition was imitated by the parameters of ϵ = 2.84 and n = 1.7, derived from an archetypal blue host material 1,3-bis(N-carbazolyl)benzene (mCP). Natural transition orbital (NTO) analyses were postprocessed by TheoDORE 2.0 package.54 The def2-SVP basis set was used for all calculations. A number of progressively larger basis sets were tested in order to evaluate the basis set effect on ΔEST and only slight shifts were observed. See SI Text 2 for other details.
Golden Rule Rates
Fermi’s golden rule rate calculations with the time-correlation function formalism30 were carried out through the discrete Fourier transform with the FFTW 3.3.6 library.55 The time integration was performed in the domain of [−6553.6:6553.6] fs and the time correlation functions were discretized in steps of 12.5 ps, which corresponded to 219 points in the positive time direction. Of course, the rate expression is obtained with the square modulus of eq 2. Because the rate in eq 1 includes infinity from the δ-function, damping is often introduced to avoid divergence in kRISC.56,57 Physically, the damping time scale is related to the dephasing time of the involved excited state and is often difficult to deduce, which leads to a certain degree of ambiguity in the calculated rate. In our case with DABNA, there was no ambiguity because the rate change was quite insignificant for any damping time chosen in a wide range of 1 ps ≤ γ ≤ 100 ns. See SI Text 3 for more details.
Because the cross-product of the DSO and the SV terms vanishes due to the time-reversal symmetry,30 the total RISC rate can be neatly partitioned into individual components:
| 3 |
Because there are two SV terms in eq 2, the spin cross-product from the second and the third terms in that equation does not vanish and contributes to kSV. Still, we can separately compare and contrast the contributions by the spin manifolds to gain insights into the details of the process:
| 4 |
as displayed in Figure 3b. This decomposition is feasible because, in the nonrelativistic regime, the singlet and the triplet excited states are computed independently from each other.
PBME-nH Simulations
The electronic subsystem Hamiltonian
was defined by the two gap energies (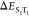 = ΔEST and
= ΔEST and 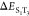 ) and two SOC elements (
) and two SOC elements (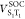 and
and 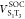 ). One harmonic underdamped mode (ν106) was set to induce the nonadiabatic coupling between T1 and T3. Parameters for forming this Hamiltonian
elements were taken from the quantum chemical data as listed in Tables 1, S3, S6, and S7. As the nonadiabatic coupling is dictated by
the derivative coupling ⟨T1|∇|T3⟩ and fluctuates with geometric distortions by the ν106 normal mode motion, it is intrinsically non-Condon. The
diagonal energy coupling by the normal vibrations of the emitter were
included by considering the mode-specific reorganization energies
as specified in Table S8. A thermal bath
representing the emitter environment was also added as a Debye spectral
density with a reorganization energy λph = 20 cm–1. Starting with 100% population at T1 initially,
multiple trajectory simulations were performed for 1 ps durations
and kRISC was obtained by monitoring the
S1 population growth in an extrapolating manner. More details
about the quantum dynamics simulations can be found in SI Text 4.
). One harmonic underdamped mode (ν106) was set to induce the nonadiabatic coupling between T1 and T3. Parameters for forming this Hamiltonian
elements were taken from the quantum chemical data as listed in Tables 1, S3, S6, and S7. As the nonadiabatic coupling is dictated by
the derivative coupling ⟨T1|∇|T3⟩ and fluctuates with geometric distortions by the ν106 normal mode motion, it is intrinsically non-Condon. The
diagonal energy coupling by the normal vibrations of the emitter were
included by considering the mode-specific reorganization energies
as specified in Table S8. A thermal bath
representing the emitter environment was also added as a Debye spectral
density with a reorganization energy λph = 20 cm–1. Starting with 100% population at T1 initially,
multiple trajectory simulations were performed for 1 ps durations
and kRISC was obtained by monitoring the
S1 population growth in an extrapolating manner. More details
about the quantum dynamics simulations can be found in SI Text 4.
Acknowledgments
The authors thank Prof. Yoon Sup Lee (KAIST) for helpful discussions. D.K. and Y.M.R. acknowledge Samsung Electronics Co. Ltd. for financial support (IO 190115-05888-01). D.K.’s contribution was additionally supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (MSIT) of the Korea government (No. 2018R1D1A1A09084233). Y.M.R. is grateful for the support from another NRF grant funded by MSIT (No. 2020R1A5A1019141). Computational resources were provided by the Supercomputing Center of Samsung Electronics and by KAIST.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacsau.1c00179.
Details about the adopted computational methods and the proof of the finiteness of kRISC; Tables with normal mode specific analyses, quantum chemical data, S1–T1 geometric differences of MR and DA type emitters, and kRISC dependence on the damping time parameter; Figures analyzing various nonadiabatic coupling, spin–crossover rates, and the related data based on normal modes and natural transition orbitals (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Uoyama H.; Goushi K.; Shizu K.; Nomura H.; Adachi C. Highly efficient organic light-emitting diodes from delayed fluorescence. Nature 2012, 492, 234–238. 10.1038/nature11687. [DOI] [PubMed] [Google Scholar]
- Hirata S.; Sakai Y.; Masui K.; Tanaka H.; Lee S. Y.; Nomura H.; Nakamura N.; Yasumatsu M.; Nakanotani H.; Zhang Q.; Shizu K.; Miyazaki H.; Adachi C. Highly efficient blue electroluminescence based on thermally activated delayed fluorescence. Nat. Mater. 2015, 14, 330–336. 10.1038/nmat4154. [DOI] [PubMed] [Google Scholar]
- Wong M. Y.; Zysman-Colman E. Purely Organic Thermally Activated Delayed Fluorescence Materials for Organic Light-Emitting Diodes. Adv. Mater. 2017, 29, 1605444. 10.1002/adma.201605444. [DOI] [PubMed] [Google Scholar]
- Chen X. K.; Kim D.; Brédas J. L. Thermally Activated Delayed Fluorescence (TADF) Path toward Efficient Electroluminescence in Purely Organic Materials: Molecular Level Insight. Acc. Chem. Res. 2018, 51, 2215–2224. 10.1021/acs.accounts.8b00174. [DOI] [PubMed] [Google Scholar]
- Kaji H.; Suzuki H.; Fukushima T.; Shizu K.; Suzuki K.; Kubo S.; Komino T.; Oiwa H.; Suzuki F.; Wakamiya A.; Murata Y.; Adachi C. Purely organic electroluminescent material realizing 100% conversion from electricity to light. Nat. Commun. 2015, 6, 8476. 10.1038/ncomms9476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z.; Mao Z.; Xie Z.; Zhang Y.; Liu S.; Zhao J.; Xu J.; Chi Z.; Aldred M. P. Recent advances in organic thermally activated delayed fluorescence materials. Chem. Soc. Rev. 2017, 46, 915–1016. 10.1039/C6CS00368K. [DOI] [PubMed] [Google Scholar]
- Light Spectra for the Samsung Galaxy S10, released in March 2019; http://www.displaymate.com/Spectra_47S.html (accessed 2021/04/26).
- Hatakeyama T.; Shiren K.; Nakajima K.; Nomura S.; Nakatsuka S.; Kinoshita K.; Ni J.; Ono Y.; Ikuta T. Ultrapure Blue Thermally Activated Delayed Fluorescence Molecules: Efficient HOMO-LUMO Separation by the Multiple Resonance Effect. Adv. Mater. 2016, 28, 2777–2781. 10.1002/adma.201505491. [DOI] [PubMed] [Google Scholar]
- Kondo Y.; Yoshiura K.; Kitera S.; Nishi H.; Oda S.; Gotoh H.; Sasada Y.; Yanai M.; Hatakeyama T. Narrowband deep-blue organic light-emitting diode featuring an organoboron-based emitter. Nat. Photonics 2019, 13, 678–682. 10.1038/s41566-019-0476-5. [DOI] [Google Scholar]
- Mellerup S. K.; Wang S. Boron-Doped Molecules for Optoelectronics. Trends Chem. 2019, 1, 77–89. 10.1016/j.trechm.2019.01.003. [DOI] [Google Scholar]
- Jiang H.; Cao Y.; Yang Q.; Xian L.; Tao Y.; Chen R.; Huang W. Organic Resonance Materials: Molecular Design, Photophysical Properties, and Optoelectronic Applications. J. Phys. Chem. Lett. 2020, 11, 7739–7754. 10.1021/acs.jpclett.0c01571. [DOI] [PubMed] [Google Scholar]
- Vázquez R. J.; Yun J. H.; Muthike A. K.; Howell M.; Kim H.; Madu I. K.; Kim T.; Zimmerman P.; Lee J. Y.; III T. G. New Direct Approach for Determining the Reverse Intersystem Crossing Rate in Organic Thermally Activated Delayed Fluorescent (TADF) Emitters. J. Am. Chem. Soc. 2020, 142, 8074–8079. 10.1021/jacs.0c01225. [DOI] [PubMed] [Google Scholar]
- Etherington M. K.; Gibson J.; Higginbotham H. F.; Penfold T. J.; Monkman A. P. Revealing the spin-vibronic coupling mechanism of thermally activated delayed fluorescence. Nat. Commun. 2016, 7, 13680. 10.1038/ncomms13680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson J.; Monkman A. P.; Penfold T. J. The Importance of Vibronic Coupling for Efficient Reverse Intersystem Crossing in Thermally Activated Delayed Fluorescence Molecules. ChemPhysChem 2016, 17, 2956–2961. 10.1002/cphc.201600662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northey T.; Penfold T. J. The intersystem crossing mechanism of an ultrapure blue organoboron emitter. Org. Electron. 2018, 59, 45–48. 10.1016/j.orgel.2018.04.038. [DOI] [Google Scholar]
- Penfold T. J.; Gindensperger E.; Daniel C.; Marian C. M. Spin-Vibronic Mechanism for Intersystem Crossing. Chem. Rev. 2018, 118, 6975–7025. 10.1021/acs.chemrev.7b00617. [DOI] [PubMed] [Google Scholar]
- Marian C. M. Understanding and Controlling Intersystem Crossing in Molecules. Annu. Rev. Phys. Chem. 2021, 72, 617–640. 10.1146/annurev-physchem-061020-053433. [DOI] [PubMed] [Google Scholar]
- Lin L.; Fan J.; Cai L.; Wang C. K. Excited state dynamics of new-type thermally activated delayed fluorescence emitters: theoretical view of light-emitting mechanism. Mol. Phys. 2018, 116, 19–28. 10.1080/00268976.2017.1362119. [DOI] [Google Scholar]
- Pershin A.; Hall D.; Lemaur V.; Sancho-Garcia J. C.; Muccioli L.; Zysman-Colman E.; Beljonne D.; Olivier Y. Highly emissive excitons with reduced exchange energy in thermally activated delayed fluorescent molecules. Nat. Commun. 2019, 10, 597. 10.1038/s41467-019-08495-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D. A theoretical understanding of the energy difference between singlet and triplet states of oligoacene molecules. Int. J. Quantum Chem. 2016, 116, 651–655. 10.1002/qua.25047. [DOI] [Google Scholar]
- De Silva P. Inverted Singlet-Triplet Gaps and Their Relevance to Thermally Activated Delayed Fluorescence. J. Phys. Chem. Lett. 2019, 10, 5674–5679. 10.1021/acs.jpclett.9b02333. [DOI] [PubMed] [Google Scholar]
- Tajti A.; Szalay P. G. Accuracy of spin-component-scaled CC2 excitation energies and potential energy surfaces. J. Chem. Theory Comput. 2019, 15, 5523–5531. 10.1021/acs.jctc.9b00676. [DOI] [PubMed] [Google Scholar]
- Reimers J. R. A practical method for the use of curvilinear coordinates in calculations of normal-mode-projected displacements and Duschinsky rotation matrices for large molecules. J. Chem. Phys. 2001, 115, 9103–9109. 10.1063/1.1412875. [DOI] [Google Scholar]
- Peng Q.; Fan D.; Duan R.; Yi Y.; Niu Y.; Wang D.; Shuai Z. Theoretical Study of Conversion and Decay Processes of Excited Triplet and Singlet States in a Thermally Activated Delayed Fluorescence Molecule. J. Phys. Chem. C 2017, 121, 13448–13456. 10.1021/acs.jpcc.7b00692. [DOI] [Google Scholar]
- Henry B. R.; Siebrand W. Between singlet and triplet states in benzene and naphthalene. J. Chem. Phys. 1971, 54, 1072–1085. 10.1063/1.1674940. [DOI] [Google Scholar]
- Parker S. M.; Shiozaki T. Quasi-diabatic states from active space decomposition. J. Chem. Theory Comput. 2014, 10, 3738–3744. 10.1021/ct5004753. [DOI] [PubMed] [Google Scholar]
- Peng Q.; Niu Y.; Shi Q.; Gao X.; Shuai Z. Correlation function formalism for triplet excited state decay: Combined spin-orbit and nonadiabatic couplings. J. Chem. Theory Comput. 2013, 9, 1132–1143. 10.1021/ct300798t. [DOI] [PubMed] [Google Scholar]
- Baiardi A.; Bloino J.; Barone V. General time dependent approach to vibronic spectroscopy including Franck-Condon, Herzberg-Teller, and Duschinsky effects. J. Chem. Theory Comput. 2013, 9, 4097–4115. 10.1021/ct400450k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Souza B.; Farias G.; Neese F.; Izsák R. Predicting Phosphorescence Rates of Light Organic Molecules Using Time-Dependent Density Functional Theory and the Path Integral Approach to Dynamics. J. Chem. Theory Comput. 2019, 15, 1896–1904. 10.1021/acs.jctc.8b00841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I.; Jeon S. O.; Jeong D.; Choi H.; Son W. J.; Kim D.; Rhee Y. M.; Lee H. S. Spin-Vibronic Model for Quantitative Prediction of Reverse Intersystem Crossing Rate in Thermally Activated Delayed Fluorescence Systems. J. Chem. Theory Comput. 2020, 16, 621–632. 10.1021/acs.jctc.9b01014. [DOI] [PubMed] [Google Scholar]
- Scholes G. D.; et al. Using coherence to enhance function in chemical and biophysical systems. Nature 2017, 543, 647–656. 10.1038/nature21425. [DOI] [PubMed] [Google Scholar]
- Lee M. K.; Bravaya K. B.; Coker D. F. First-Principles Models for Biological Light-Harvesting: Phycobiliprotein Complexes from Cryptophyte Algae. J. Am. Chem. Soc. 2017, 139, 7803–7814. 10.1021/jacs.7b01780. [DOI] [PubMed] [Google Scholar]
- Jumper C. C.; Van Stokkum I. H.; Mirkovic T.; Scholes G. D. Vibronic Wavepackets and Energy Transfer in Cryptophyte Light-Harvesting Complexes. J. Phys. Chem. B 2018, 122, 6328–6340. 10.1021/acs.jpcb.8b02629. [DOI] [PubMed] [Google Scholar]
- Kim C. W.; Lee W. G.; Kim I.; Rhee Y. M. Effect of Underdamped Vibration on Excitation Energy Transfer: Direct Comparison between Two Different Partitioning Schemes. J. Phys. Chem. A 2019, 123, 1186–1197. 10.1021/acs.jpca.8b10977. [DOI] [PubMed] [Google Scholar]
- Kim H.; Nassimi A.; Kapral R. Quantum-classical Liouville dynamics in the mapping basis. J. Chem. Phys. 2008, 129, 084102. 10.1063/1.2971041. [DOI] [PubMed] [Google Scholar]
- Kim H. W.; Rhee Y. M. Improving long time behavior of Poisson bracket mapping equation: A non-Hamiltonian approach. J. Chem. Phys. 2014, 140, 184106. 10.1063/1.4874268. [DOI] [PubMed] [Google Scholar]
- Scholes G. D. Long-Range Resonance Energy Transfer in Molecular Systems. Annu. Rev. Phys. Chem. 2003, 54, 57–87. 10.1146/annurev.physchem.54.011002.103746. [DOI] [PubMed] [Google Scholar]
- Liang X.; Yan Z. P.; Han H. B.; Wu Z. G.; Zheng Y. X.; Meng H.; Zuo J. L.; Huang W. Peripheral Amplification of Multi-Resonance Induced Thermally Activated Delayed Fluorescence for Highly Efficient OLEDs. Angew. Chem., Int. Ed. 2018, 57, 11316–11320. 10.1002/anie.201806323. [DOI] [PubMed] [Google Scholar]
- Nakatsuka S.; Gotoh H.; Kinoshita K.; Yasuda N.; Hatakeyama T. Divergent Synthesis of Heteroatom-Centered 4,8,12-Triazatriangulenes. Angew. Chem., Int. Ed. 2017, 56, 5087–5090. 10.1002/anie.201701246. [DOI] [PubMed] [Google Scholar]
- Ahn D. H.; Kim S. W.; Lee H.; Ko I. J.; Karthik D.; Lee J. Y.; Kwon J. H. Highly efficient blue thermally activated delayed fluorescence emitters based on symmetrical and rigid oxygen-bridged boron acceptors. Nat. Photonics 2019, 13, 540–546. 10.1038/s41566-019-0415-5. [DOI] [Google Scholar]
- Kim D. Effects of Intermolecular Interactions on the Singlet–Triplet Energy Difference: A Theoretical Study of the Formation of Excimers in Acene Molecules. J. Phys. Chem. C 2015, 119, 12690–12697. 10.1021/acs.jpcc.5b02021. [DOI] [Google Scholar]
- Lee K.; Kim D. Local-excitation versus charge-transfer characters in the triplet state: Theoretical insight into the singlet-triplet energy differences of carbazolyl-phthalonitrile-based thermally activated delayed fluorescence materials. J. Phys. Chem. C 2016, 120, 28330–28336. 10.1021/acs.jpcc.6b10161. [DOI] [Google Scholar]
- Samanta P. K.; Kim D.; Coropceanu V.; Brédas J. L. Up-Conversion Intersystem Crossing Rates in Organic Emitters for Thermally Activated Delayed Fluorescence: Impact of the Nature of Singlet vs Triplet Excited States. J. Am. Chem. Soc. 2017, 139, 4042–4051. 10.1021/jacs.6b12124. [DOI] [PubMed] [Google Scholar]
- El-Sayed M. A. Spin-orbit coupling and the radiationless processes in nitrogen heterocyclics. J. Chem. Phys. 1963, 38, 2834–2838. 10.1063/1.1733610. [DOI] [Google Scholar]
- Furche F.; Ahlrichs R.; Hättig C.; Klopper W.; Sierka M.; Weigend F. Turbomole. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2014, 4, 91–100. 10.1002/wcms.1162. [DOI] [Google Scholar]
- Shao Y.; et al. Advances in molecular quantum chemistry contained in the Q-Chem 4 program package. Mol. Phys. 2015, 113, 184–215. 10.1080/00268976.2014.952696. [DOI] [Google Scholar]
- Gerenkamp M.; Grimme S. Spin-component scaled second-order Møller-Plesset perturbation theory for the calculation of molecular geometries and harmonic vibrational frequencies. Chem. Phys. Lett. 2004, 392, 229–235. 10.1016/j.cplett.2004.05.063. [DOI] [Google Scholar]
- Schirmer J. Beyond the random-phase approximation: A new approximation scheme for the polarization propagator. Phys. Rev. A: At., Mol., Opt. Phys. 1982, 26, 2395–2416. 10.1103/PhysRevA.26.2395. [DOI] [Google Scholar]
- Christiansen O.; Koch H.; Jørgensen P. The second-order approximate coupled cluster singles and doubles model CC2. Chem. Phys. Lett. 1995, 243, 409–418. 10.1016/0009-2614(95)00841-Q. [DOI] [Google Scholar]
- Friese D. H.; Törk L.; Hättig C. Vibrational frequency scaling factors for correlation consistent basis sets and the methods CC2 and MP2 and their spin-scaled SCS and SOS variants. J. Chem. Phys. 2014, 141, 194106. 10.1063/1.4901725. [DOI] [PubMed] [Google Scholar]
- Chai J. D.; Head-Gordon M. Systematic optimization of long-range corrected hybrid density functionals. J. Chem. Phys. 2008, 128, 084106. 10.1063/1.2834918. [DOI] [PubMed] [Google Scholar]
- Marian C. M. Spin-orbit coupling and intersystem crossing in molecules. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 187–203. 10.1002/wcms.83. [DOI] [Google Scholar]
- Tomasi J.; Mennucci B.; Cancès E. The IEF version of the PCM solvation method: An overview of a new method addressed to study molecular solutes at the QM ab initio level. J. Mol. Struct.: THEOCHEM 1999, 464, 211–226. 10.1016/S0166-1280(98)00553-3. [DOI] [Google Scholar]
- Plasser F.; Bäppler S. A.; Wormit M.; Dreuw A. New tools for the systematic analysis and visualization of electronic excitations. II. Applications. J. Chem. Phys. 2014, 141, 024107. 10.1063/1.4885820. [DOI] [PubMed] [Google Scholar]
- Frigo M.; Johnson S. G. The design and implementation of FFTW3. Proc. IEEE 2005, 93, 216–231. 10.1109/JPROC.2004.840301. [DOI] [Google Scholar]
- Niu Y.; Peng Q.; Deng C.; Gao X.; Shuai Z. Theory of Excited State Decays and Optical Spectra: Application to Polyatomic Molecules. J. Phys. Chem. A 2010, 114, 7817–7831. 10.1021/jp101568f. [DOI] [PubMed] [Google Scholar]
- Etinski M.; Tatchen J.; Marian C. M. Time-dependent approaches for the calculation of intersystem crossing rates. J. Chem. Phys. 2011, 134, 154105. 10.1063/1.3575582. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.