

Abstract
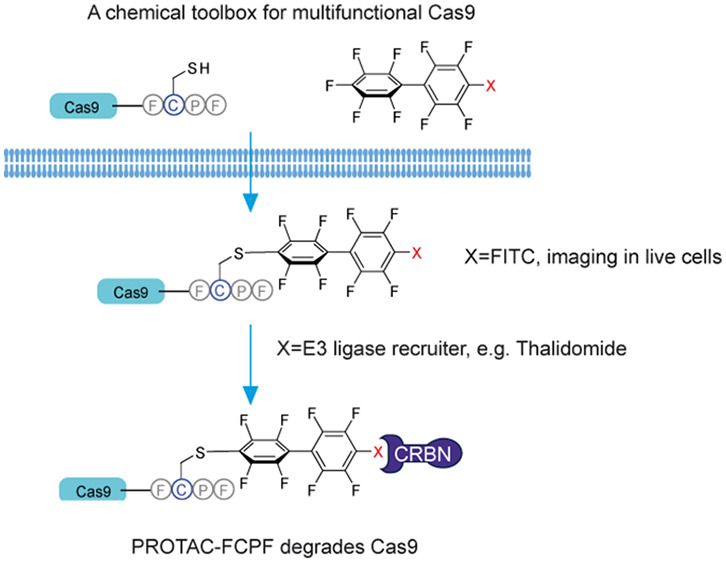
The discovery of clustered regularly interspaced short palindromic repeats and their associated proteins (Cas) has revolutionized the field of genome and epigenome editing. A number of new methods have been developed to precisely control the function and activity of Cas proteins, including fusion proteins and small-molecule modulators. Proteolysis-targeting chimeras (PROTACs) represent a new concept using the ubiquitin-proteasome system to degrade a protein of interest, highlighting the significance of chemically induced protein-E3 ligase interaction in drug discovery. Here, we engineered Cas proteins (Cas9, dCas9, Cas12, and Cas13) by inserting a Phe-Cys-Pro-Phe (FCPF) amino acid sequence (known as the π-clamp system) and demonstrate that the modified CasFCPF proteins can be (1) labeled in live cells by perfluoroaromatics carrying the fluorescein or (2) degraded by a perfluoroaromatics-functionalized PROTAC (PROTAC-FCPF). A proteome-wide analysis of PROTAC-FCPF-mediated Cas9FCPF protein degradation revealed a high target specificity, suggesting a wide range of applications of perfluoroaromatics-induced proximity in the regulation of stability, activity, and functionality of any FCPF-tagging protein.
Keywords: CRISPR/Cas9, genome editing, π-clamp, Cas9 inhibitor, PROTAC, chemical-induced proximity
Introduction
Clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated systems (Cas) constitute a family of nucleases responsible for adaptive immune responses in many bacteria and archaea against bacteriophages.1 In general, Cas enzymes use CRISPR RNA (crRNA), obtained from the incorporation of fragments of the invading genetic material, as a guide to target and cleave invading nucleic acids.1,2 These proteins have been classified into two classes (1 and 2) and six subtypes (I–VI) based on their molecular mechanisms. In comparison to class I proteins (types I, III, and IV), the class 2 Cas proteins (types II, V, and VI) utilize single nuclease effectors guided by crRNA to target nucleic acids. These nucleases have attracted attention among molecular biologists and biotechnologists.1,3 The most widely used type 2 protein is Cas9 from Streptococcus pyogenes Cas9 (SpyCas9, hereinafter referred to as Cas9); however, Cas9 proteins from other bacteria have also been shown to be effective.2 Cas9 proteins detect double-stranded DNA (dsDNA) and produce double-strand breaks (DSBs), resulting in genomic deletions. The dsDNA can then be repaired by either nonhomologous end-joining repair or homologous recombination, which can then mediate gene replacement or insertions.3 The most well-known example of type V proteins is Cas12 (isoform a and b), isolated from Prevotella buccae and Francisella novicida. The molecular mechanisms of Cas12 proteins display minimal differences to those of Cas9, although they both detect dsDNA and induce DSBs. Cas12 is also able to produce a collateral detection and trans-cleavage of single-stranded DNA (ssDNA), which makes it a powerful tool in several applications.4 In contrast, type VI proteins (Cas13a, Cas13b, and Cas13c) from Leptotrichia sp and Provetella sp, among others, are RNA-guided ribonucleases, by means of their two higher eukaryote and prokaryote nucleotide-binding domains that identify and degrade single-stranded RNA (ssRNA), thus allowing a post-transcriptional regulation of gene expression, nucleic acid detection, RNA knockdown, and transcript tracking in mammalian cells and plants.5−7
Although the first in-human phase I clinical study has reported that CRISPR-Cas9-edited T-cells are well-tolerated by patients,8 off-target effects are still the major concern in its clinical applications. Studies have shown that the precise regulation of Cas9 activity significantly reduces the off-target effect, hinting that a hyperactivation of Cas9 is most likely the reason for the observed off-target DNA cleavages.9−11 Recently, we and other groups showed the impact of small-molecule inhibitors on the activity of Cas9.12,13 However, chemical inhibitors usually have multiple targets, which may be associated with severe side effects. Liu and co-workers inserted a 4-hydroxytamoxifen-dependent intein into Cas9 and achieved a specific Cas9 activation.14 Indeed, fusing an activator/repressor into nuclease-dead Cas9 (dCas9) has become a general strategy for CRISPR-dCas9-mediated epigenome editing.15−17 One of the technical challenges is the delivery of such large chimeric proteins into living cells and organisms.18 Moreover, highly expressed exogenous epigenetic regulators may impact the nature of cells and induce significant side effects. Thus, it is urgently required to develop a new CRISPR-(d)Cas9 system for a more specific and flexible modulation.
Proteolysis-targeting chimera (PROTAC) represents a recent approach that uses the intracellular ubiquitin-proteasome system in order to degrade a protein of interest (POI). A PROTAC is a heterobifunctional small molecule, consisting of two ligands connected by a linker: one targeting a POI, and the other one recruiting an E3 ligase.19,20 The strategy of PROTAC to degrade a POI by inducing a POI-E3 ligase interaction offers a wide range of applications in the aspect of chemically induced proximity in drug development.21 Here, we inserted a sequence of four amino acids Phe(F)-Cys(C)-Pro(P)-Phe(F), also known as a π-clamp, into Cas proteins (herein referred to as CasFCPF). We demonstrate that this site-specific chemical modification made Cas9FCPF recognizable by perfluoroaromatics as reported previously by Zhang et al.,22 which could be utilized for labeling Cas9FCPF in living cells by a fusion of a fluorophore into perfluoroaromatics, or to degrade FCPF tagging Cas proteins, including Cas9FCPF, dCas9FCPF, Cas12FCPF, and Cas13FCPF, by a new synthetic perfluoroaromatics-containing PROTAC molecule (PROTAC-FCPF). This approach provides a new postexpression strategy to chemically regulate the stability, activity, and function of Cas and other proteins in genome editing and gene therapy.
Results
Labeling of Cas9FCPF with FITC-FCPF in Living Cells
The π-clamp system provides a useful approach to select FCPF-engineered proteins—for instance, the insertion of this short sequence has been used to form antibody-drug conjugates.22 To test if the π-clamp system can be applied for the recognition of exogenously expressed proteins, such as the Cas9 protein, we inserted FCPF at the C-terminus of Cas9, whose expression is controlled by a cytomegalovirus (CMV) promoter (Figure 1A). We first developed a simple labeling method to detect a Cas9FCPF expression by using a synthetic fluorescein (FITC)-conjugated perfluoroaromatic moiety, which we obtained by coupling perfluoro-1,1′-biphenyl with NHS-activated 6-fluorescein (referred to as FITC-FCPF, Figure 1A in green). The synthesis is comprehensively described in the Materials and Methods section. We observed the anticipated green fluorescence signal in HeLa cells expressing Cas9FCPF after an incubation with FITC-FCPF (10 μM) for 2 h, which was similar to that of Cas9GFP (Figure 1B and Figure S1A). A fluorescence-activated cell sorting (FACS) analysis confirmed that the intensity of the green fluorescence signal in cells, expressing Cas9FCPF and treated with FITC-FCPF labeling, was similar to that in cells expressing Cas9GFP (Figure 1C and Figure S1B). That intensity was almost 20-fold higher compared to FITC-FCPF treated cells expressing Cas9WT (Figure 1C and Figure S1B). We lysed FITC-FCPF-treated cells expressing either Cas9FCPF or Cas9WT using 6 M urea buffer and resolved the lysates in 8% sodium dodecyl sulfate poly(acrylamide) gel electrophoresis (SDS PAGE). An electrophoresis analysis confirmed that FITC-FCPF molecules specifically stained the Cas9FCPF protein as indicated by a strong signal at a molecular weight of ∼150 kDa when observed under UV light (Figure 1D and Figure S1C), which might indicate the formation of a stable irreversible covalent bond between FCPF and perfluoroaromatics.22
Figure 1.
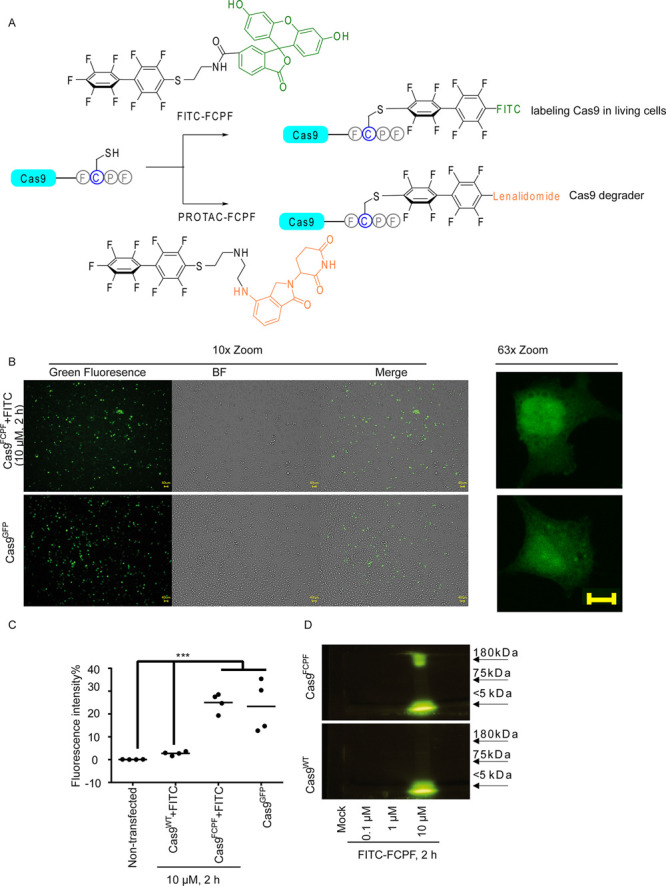
The application of Cas9FCPF for live cell imaging. (A) Overview of applications of Cas9FCPF for labeling (FITC-FCPF, green) and degradation (PROTAC-FCPF, orange). (B) Green fluorescence signal is detected in FCPF-FITC-treated HeLa cells expressing Cas9FCPF. HeLa cells expressing Cas9FCPF were treated with FITC-FCPF (10 μM) for 2 h. Cas9GFP was used as a positive control. The fluorescence images (10× and 63× enlarged) were taken from nontransfected cells, FITC-FCPF-treated cells expressing Cas9WT, and nontreated cells expressing Cas9FCPF. Images from Cas9GFP cells and Cas9FCPF cells treated with FITC-FCPF (10 μM, 2 h) were shown. Remaining images can be found in the Supporting Information (Figure S1A). Scale bar: 40 μm. (C) Comparison of fluorescence intensity of FITC-FCPF-treated cells expressing Cas9WT and Cas9FCPF HeLa cells expressing Cas9WT or Cas9FCPF were treated with FCPF-FITC (10 μM) for 2 h, and the fluorescence intensity was analyzed by FACS. Nontransfected cells and cells expressing Cas9GFP were used as negative and positive controls. The results were obtained from four independent biological replications. The center represents the median. Two-way ANOVA was performed. *** p < 0.001. (D) Electrophoresis analysis of FITC-FCPF-treated samples obtained from cells expressing either Cas9FCPF or Cas9WT. HeLa cells expressing Cas9WT or Cas9FCPF were treated with increasing concentrations of FITC-FCPF for 2 h. Proteins collected from a whole cell lysis were dissolved in 8% SDS PAGE. The original pictures with a protein loading marker can be found in the Supporting Information (Figure S1C).
PROTAC-FCPF Degrades Cas9FCPF
We designed a perfluoroaromatics-based PROTAC molecule to investigate if a heterobifunctional small molecule can induce an artificial Cas9FCPF-E3 ligase interaction for the regulation of Cas9FCPF stability. Several compounds have been previously reported as ligands of E3 ligases for the development of PROTACs, including thalidomide, which binds to cereblon (CRBN).20,23 The replacement of FITC with the thalidomide derivative lenalidomide (Figure 1A in orange) yielded a novel PROTAC molecule that we called PROTAC-FCPF (a PROTAC-targeting FCPF amino acid sequence). In HeLa cells treated with 10 μM PROTAC-FCPF we detected the reduction of Cas9FCPF as early as 6 h (Figure 2A and Figure S2A). The depletion lasted at least up to 72 h (Figure 2B and Figure S2B). Very recently, Sreekanth et al. developed a dTAG-system to degrade Cas9FKBP. They demonstrated that the small molecular degrader, dTAG-47, degraded Cas9 carrying two FK506-binding proteins (FKBPs, Cas9FKBP) at the N-terminal and loop region at concentrations of 0.1–1 μM in a 24 h treatment, which significantly increased the specificity of the CRISPR-Cas9 system.24 We evaluated the activity of PROTAC-FCPF for Cas9FCPF degradation in both 8 and 24 h treatments. We found that 10 μM PROTAC-FCPF robustly degraded the Cas9FCPF protein (Figure 2C and Figure S2C). A similar effect was observed at nanomolar concentrations in a 24 h treatment with an IC50 value of ∼150 nM (Figure 2D and Figure S2D). Of note, higher concentrations (>500 μM) resulted in an attenuated degradation (Figure 2C and Figure S2C). Douglass et al. developed a comprehensive mathematical model for analyzing a three-component system, which at least partially elucidated the autoinhibition in the formation of ternary complex.25 This so-called hook effect has been also repeatedly described in both reversible26 and irreversible covalent PROTACs.27 Preincubation with free lenalidomide (50 μM) inhibited PROTAC-FCPF-mediated Cas9FCPF degradation, which was probably caused by a competitive binding to CRBN E3 ligase (Figure 2E and Figure S2E). By contrast, the Cas9WT protein was not affected by PROTAC-FCPF (Figure 2A and Figure S2F), suggesting that the PROTAC-mediated degradation of Cas9 protein was FCPF-specific. Moreover, we found a dose-dependent degradation of Cas9FCPF induced by PROTAC-FCPF in HEK293T cells (IC50: 167.2 nM) similar to that in HeLa cells (IC50:143.4 nM), Figure 2D and Figure S2D). Because primary cells are more relevant to the application of CRISPR-Cas9-mediated genome editing, we tested PROTAC-FCPF-induced degradation in Jopaca-1 cells, a polymorphic primary cell line established from poorly differentiated ductal adenocarcinoma.28 The IC50 value was 72.0 nM in a 24 h treatment, which was slightly better than those in HeLa and HEK293T cells (Figure 2D and Figure S2D).
Figure 2.
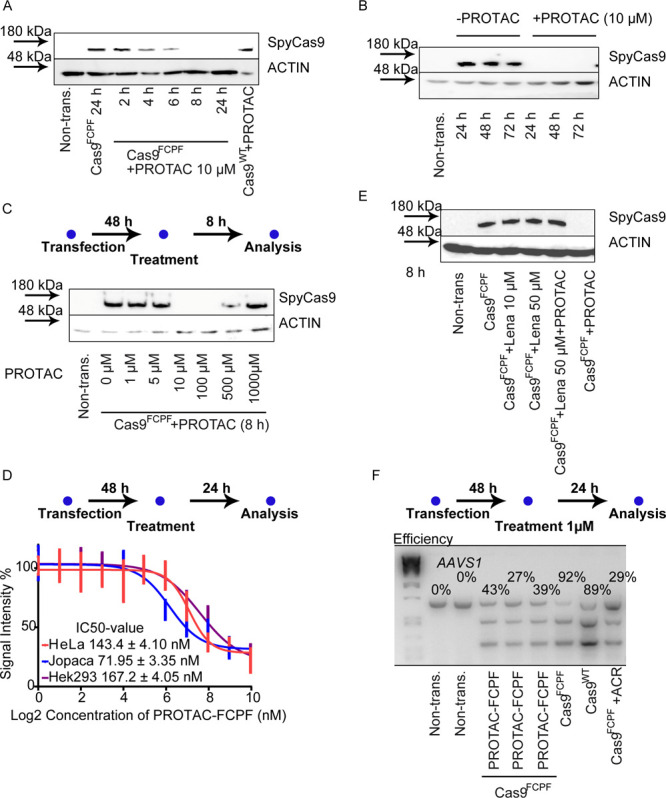
The PROTAC-FCPF inhibits Cas9-mediated genome editing through destabilizing Cas9FCPF protein in cells. (A) The PROTAC-FCPF induces Cas9FCPF protein degradation within 24 h and (B) over 72 h. HeLa cells expressing either Cas9WT or Cas9FCPF were treated with 10 μM PROTAC-FCPF for indicated time periods. ACTIN was used as a loading control. (C) The influence of various concentrations of PROTAC-FCPF on Cas9FCPF degradation. HeLa cells were transfected with Cas9FCPF plasmid for 48 h and treated with increasing concentrations of PROTAC-FCPF for 8 h. The residual Cas9 protein was detected by immunoblotting. (D) PROTAC-FCPF-mediated Cas9FCPF degradation in HeLa, Jopaca-1, and HEK293T cells for 24 h treatment. Cells were transfected with Cas9FCPF plasmid for 48 h and treated with increasing concentrations of PROTAC-FCPF for 24 h. The residual Cas9 protein was detected by immunoblotting. The results were obtained from four independent biological replications. The half-maximal inhibitory concentration (IC50) values were calculated from dose–response curves. Error bars ± SD. One original immunoblotting image as an example can be found in the Supporting Information. (E) The PROTAC-FCPF-induced Cas9FCPF protein degradation was rescued by lenalidomide. HeLa cells expressing Cas9FCPF were treated with lenalidomide (10 or 50 μM), PROTAC-FCPF (10 μM), or the combination of PRORAC-FCPF (10 μM) and lenalidomide (50 μM) for 8 h. (F) PROTAC-FCPF suppressed Cas9FCPF-mediated genome editing using T7E1 assay by an analysis of the AAVS1 gene. Cells were transfected with Cas9FCPF or Cas9WT for 48 h and treated with PROTAC-FCPF (1 μM) for 24 h. Anti-CRISPR-Cas9 protein AcrIIA4 (ACR) was cotransfected with Cas9FCPF and used as a positive control. The efficiency of Cas9FCPF varied from 27% to 42% in three independent experiments. The calculation of efficiency was described in the Materials and Methods section. The densitometric analysis of proteins in Figure 2A,B,C,E obtained from at least three independent experiments can be found in the Supporting Information with statistical analyses and an original picture as an example.
The T7E1 assay has been widely used to evaluate the efficiency of CRISPR-Cas9-mediated genome editing29,30 and was recruited to evaluate if PROTAC-FCPF interrupted the function of the Cas9 protein on the AAVS1 gene, as recently established and reported by Niopek’s lab.9 We detected similar efficiencies of delivery (Figure S2G) and genome editing for Cas9FCPF and Cas9WT (Figure 2F), implying that insertion of the FCPF sequence did not affect the biological activity of the Cas9 protein. As expected, we found that PROTAC-FCPF (1 μM) reduced the cleavage of target DNA in cells after a 24 h treatment, with similar activity as the anti-CRISPR/Cas9 protein (AcrIIA4), which was coexpressed with Cas9 and AAVS1 sgRNA.30 The result suggested that the PROTAC-FCPF-degraded Cas9FCPF efficiently repressed the biological function of the CRISPR-Cas9FCPF system in a time-controlled fashion. In agreement with the result reported by Sreekanth et al.,24 the incubation of PROTAC-FCPF (1 μM) for 24 h reduced the off-target effect from 12.1% to 3.56% (Figure S2H), implying that Cas9 degraders can reduce the off-target effects of a CRISPR/Cas9 system.
PROTAC-FCPF-Mediated Cas9FCPF Degradation Is Dependent on CRBN-Associated Ubiquitination
PROTAC-mediated target protein degradation requires the ubiquitination machinery.20 As expected, coincubation of PROTAC-FCPF (10 μM) with the proteasome inhibitor MG132 (5 μM), for 8 h, repressed the Cas9FCPF degradation (Figure 3A and Figure S3A). In comparison to Cas9WT cells, the level of polyubiquitination induced by PROTAC-FCPF in cells expressing Cas9FCPF was much higher compared to cells expressing Cas9WT (Figure 3A). Because lenalidomide recruits the CRBN E3 ligase for PROTAC-associated degradation,23,31 the PROTAC-FCPF-induced destabilization of the Cas9FCPF protein was considerably reduced in CRBN knockdown HeLa cells (CRBN KD) using a specific small interfering RNA against CRBN (Figure 3B and Figure S3B), while Cas9WT was not affected (Figure S3B). Next, we generated HeLa CRBNKO cells using a CRISPR-Cas9 knockout system as previously reported (Figure S3C).32 In total six CRBNKO cell lines CRBNKO showed the best knockout efficiency and were used for an in vitro ubiquitination assay (Figure S3C). We overexpressed Cas9WT or Cas9FCPF in this CRBNKO cell line and collected cell extracts for an in vitro ubiquitination assay. The reaction was initiated with the addition of purified FLAG-CRBN. The degradation of Cas9 proteins was compared in the presence or absence of PROTAC-FCPF (100 μM for 90 min). As shown in Figure 3C (Figure S3D), only the Cas9 with the FCPF sequence was degraded in the presence of CRBNWT and PROTAC-FCPF, while it was resistant to a loss-of-function mutation of CRBN (Y383A and W385A; referred to as CRBNm), as reported previously.32 Measuring adenosine triphosphate (ATP) levels revealed that the combination of CRBNWT and PROTAC-FCPF (100 μM for 90 min) elicited a significant ATP consumption, which was considerably reduced by pretreatment with lenalidomide (100 μM, Figure 3D). Lee reported that AMPK activity was associated with CRBN,33 which we were unable to demonstrate in our in vitro assay. DDB1 and CUL4A are essential components in CRBN-mediated degradation.32 We transiently transfected HeLa cells with CUL4A or DDB1 siRNA and detected a significant attenuation of PROTAC-FCPF-mediated (10 μM for 8 h) Cas9FCPF degradation, while no effect was found in cells lacking MDM2 or von Hippel-Lindau (VHL) (Figure 3E and Figure S3E). Moreover, the degradation was rescued, when cells were pretreated with PYR-41, an E1 inhibitor,34 or MLN4925, an NEDD8-activating enzyme, which is required for a fully active CUL4A/DDB1/CRBN complex (Figure 3F).35 Thus, the observed Cas9FCPF protein degradation was associated with functional CRBN-mediated ubiquitination.
Figure 3.
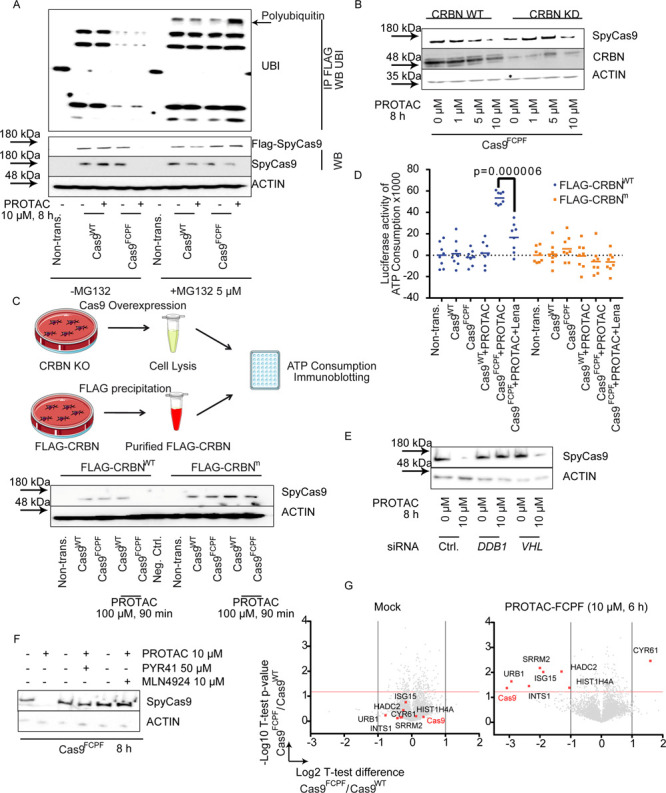
The PROTAC-FCPF-induced Cas9FCPFprotein degradation is ubiquitin-dependent. (A) MG132 inhibits PROTAC-FCPF-mediated Cas9FCPF protein degradation. HeLa cells expressing Cas9WT or Cas9FCPF were treated with PROTAC-FCPF (10 μM) in the absence or presence of MG132 (5 μM) for 8 h. DMSO was used as a control. Cas9 protein was immunoprecipitated, and immunoblotting was performed for detecting the ubiquitination. (B) Comparison of PROTAC-FCPF-induced Cas9FCPF in CRBN-WT or CRBN-KD HeLa cells. HeLa cells were cotransfected with Cas9FCPF and control siRNA or Cas9FCPF and CRBN siRNA for 48 h and treated with increasing concentrations of PROTAC-FCPF for 8 h. The control experiment in cells expressing Cas9WT can be found in Figure S3B. (C) PROTAC-FCPF-mediated Cas9FCPF protein degradation requires functional CRBN. (upper) Illustration of in vitro ubiquitination assay. In vitro degradation was performed using the whole CRBN-KO cell lysate, in which either FLAG-Cas9WT or FLAG-Cas9FCPF was expressed. FLAG-CRBNWT and FLAG-CRBNm were purified separately. The in vitro degradation was initiated by adding FLAG-CRBN into the cell lysate and incubated for 90 min at 37 °C in the presence or absence of PROTAC-FCPF (100 μM). The residual Cas9 protein was compared using immunoblotting, or (D) the ATP consumption was measured after an incubation with PROTAC-FCPF (100 μM) or the combination of PROTAC-FCPF (100 μM) and lenalidomide (100 μM) at 37 °C for 90 min. The result was obtained from at least three independent biological replications with at least two technical replications of each. Two-way ANOVA was performed. (E) PROTAC-FCPF degrades Cas9FCPF in cells lacking VHL but requires a DDB1 expression. HeLa cells were cotransfected with Cas9FCPF and DDB1 siRNA or VHL siRNA for 48 h and treated with PROTAC-FCPF (10 μM) for 8 h. (F) PROTAC-FCPF-mediated Cas9FCPF degradation was rescued in the presence of PYR41, an E1 inhibitor (50 μM), or MLN4924, an NEDD8-activating E1 enzyme (10 μM). HeLa cells expression of Cas9FCPF were treated with PROTAC-FCPF (10 μM) or in the combination of PYR41 or MLN4924 for 8 h. (G) Evaluation of specificity of PROTAC-FCPF-mediated Cas9FCPF degradation by a proteome-wide analysis. HeLa cells expressing either Cas9WT or Cas9FCPF were treated with PROTAC-FCPF (10 μM) for 6 h. DMSO was used as a mock. High specificity of PROTAC-FCPF-mediated Cas9FCPF degradation was reproducible from three independent experiments. Eight of a total of 4436 protein are highlighted due to their significant alternations in PROTAC-FCPF-treated cells expressing Cas9FCPF as compared with those in Cas9WT cells. Cas9 was highlighted in red. Detailed information regarding the experiment protocol and an analysis of proteomics can be found in Materials and Methods.
Recently, Bondeson et al. reported high-target specificity of the PROTAC-RIPK2 molecule, carrying hydroxyproline moieties resulting in the recruitment of the VHL E3 ligase and a chemical inhibitor of RIPK2, which specifically bound to RIPK2 and just one other unrelated protein kinase MAPKAPK3.26 To determine the cellular specificity of PROTAC-FCPF-mediated Cas9FCPF degradation, we treated HeLa cells expressing either Cas9WT or Cas9FCPF with dimethyl sulfoxide (DMSO) (mock) or PROTAC-FCPF (10 μM) for 6 h and performed a proteome-wide analysis. The experimental and analytic details were exclusively described in Materials and Methods. We could quantify in total 4436 proteins in all samples in our high-resolution mass spectrometry-based proteomics and compared them for a specificity analysis. As shown in Figure 3G, none of the proteins were significantly differentially expressed in mock-treated cells transfected with Cas9WT or Cas9FCPF (Figure 3G left), while adding PROTAC-FCPF, eight proteins were significantly up- or down-regulated, namely, Cas9, CYR61, HADC2, HIST1H4A, INTS1, ISG15, SRRM2, and URB1 (Figure 3G right). Among them Cas9 showed nearly an eightfold lower expression in cells expressing Cas9FCPF compared to cells expressing Cas9WT. Moreover, in Cas9WT cells no proteins were significantly affected by PROTAC-FCPF (Figure S3G left). By contrast, PROTAC-FCPF repressed the expression of Cas9FCPF and 10 other proteins in Cas9FCPF cells (Figure S3G right). Of note, ISG15, INTS1, and URB1 appeared synchronously with Cas9FCPF in both cases (Figure 3G and Figure S3G). It is well-known that ISG15 is a ubiquitin-like modifier and plays an important role in the antiviral immunity.36 Considering the original role of CRISPR-Cas9 in a host immune system, ISG15 might be involved in a PROTAC-FCPF-mediated Cas9FCPF degradation and related cellular responses. Lu et al. reported that lenalidomide recruited CRBN to degrade IKZF1,32 which was not detected in our experiment, because the expression of IKZF1 in HeLa cells is remarkably low according to our proteomic data.
Degradation of dCas9FCPF, Cas12FCPF, and Cas13FCPF with PROTAC-FCPF in Living Cells
Catalytically inactive Cas9 (dCas9) is widely used for epigenetic repression and activation. As expected, PROTAC-FCPF degraded dCas9FCPF in a similar manner to Cas9FCPF, confirming that the PROTAC-FCPF-mediated degradation is independent of catalytic activity (Figure 4A and Figure S4A), showing the important nature of FCPF-mediated degradation in comparison to classic chemical inhibitors, which inhibit protein activity by occupation in the catalytic binding pocket. Generally, a classic inhibitor development program would require a substantial synthetic effort for each targeted protein. For instance, Maji et al. reported BRD0539 as a Cas9 inhibitor, which could not inhibit other members of Cas enzyme demonstrated in their work.13 We inserted an FCPF amino acid sequence into HA-Cas12 and HA-Cas13 vectors. Gratifyingly, as shown in Figure 4B,C, we observed a time- and concentration-dependent degradation of Cas12FCPF and Cas13FCPF suggesting that this strategy presented here can be widely used to control the stability of various proteins tagging FCPF with the single PROTAC-FCPF molecule.
Figure 4.

The PROTAC-FCPF degrades dCas9FCPF, Cas12FCPF, and Cas13FCPF. The PROTAC-FCPF induces dCas9FCPF, Cas12FCPF, and Cas13FCPFdegradation. (A) PROTAC-FCPF degrades dCas9FCPF. HeLa cells expressing dCas9FCPF were treated with increasing concentrations of PROTAC-FCPF for 8 h. (B, C) PROTAC-FCPF degrades Cas12FCPF and Cas13FCPF in (B) concentration-dependent and (C) time-dependent manners. Cells expressing HA-Cas12aFCPF or HA-Cas13FCPF were treated with the PROTAC-FCPF as indicated. Immunoblotting was performed. We used HA antibodies to detect HA-tag proteins. We used ACTIN as a loading control. The densitometric analysis of proteins obtained from at least three independent experiments can be found in the Supporting Information (Figure S4), with one of the original pictures as an example.
Discussion
Despite the revolutionary role of CRISPR/Cas systems in biotechnology, in particular, in genetic manipulation and engineering, some limitations associated with this technology remain, including off-target mutations.37 To improve accuracy and reduce off-target activities, several research groups have been engaging in engineering Cas9 proteins and developing anti-CRISPR/Cas9 proteins.38−40 These anti-CRISPR proteins naturally occur in bacteria to block or diminish the activity of Cas9 proteins and, thus, regulate their function. Following this approach, anti-CRISPR/Cas12 inhibitors have also been identified.41 Moreover, BRD0539 and valproic acid have been identified as Cas9 small-molecular inhibitors from high-content screening, which opened a new window for the chemical regulation of Cas9 activity.12,13
Classic small-molecule inhibitors, such as various protein kinase inhibitors, inhibit the enzymatic activity of the POI, using a well-defined hydrophobic binding pocket.20 Numerous in vitro and in vivo results provide compelling evidence that an acquired drug resistance can develop during a treatment, such as mutations and activation of alternative compensatory downstream signaling pathways via the formation of homo/hetero-oligomers, which largely impacts the clinical outcome.42,43 PROTACs are a heterobifunctional molecule consisting of a chemical warhead, which brings small molecules to fast and short interaction with the POI and a ligand of E3 ligase, which leads to a ubiquitination and degradation of the POI.20 Thus, PROTACs can inhibit both catalytic-dependent and catalytic-independent activity of the target, which includes currently undruggable mechanisms.20
In the past decade, a series of PROTACs has been synthesized. However, few PROTACs were reported to degrade undruggable proteins, probably due to the lack of proper chemical warheads. Site-selective chemistry provides a means to target the POI at predesigned sites through postexpression modification.44 For instance, Virdee et al. incorporated a specific lysine into proteins to artificially generate an isopeptide bond with ubiquitin for degradation.45 Recently, Pentelute’s group developed the π-clamp system, in which recombinant proteins tagged by the FCPF amino acid sequence are selectively recognized by perfluoroaromatic compounds in a nucleophilic aromatic reaction (SNAr) at the thiol group of the cysteine residue (Cys) under physiological conditions.22 We showed that the π-clamp system can be applied to target exogenously expressed FCPF-engineered proteins, such as Cas9, dCas9, Cas12, and Cas13. After the perfluoroaromatic group is fused to lenalidomide, a ligand of the CRBN E3 ligase, the resulting PROTAC-FCPF selectively degraded FCPF-labeled proteins. In comparison, a substantial effort is required to develop a small-molecule inhibitor for each protein. Typically, chemical inhibitors require a well-defined hydrophobic binding pocket at an enzyme or receptor active site, which leads to unwanted off-target effects.20 Currently, two Cas9 small-molecule inhibitors have been reported.12,13 However, their specificity regarding Cas9 inhibition is not reported. PROTACs exhibit a relatively high target specificity.26,46 In line with this, our proteome-wide analysis showed that less than a dozen proteins of a total of 4436 were affected by PROTAC-FCPF.
Very recently, Choudhary and his co-workers developed a dTAG-based PROTAC system to degrade Cas9 tagging FKPB.24 They demonstrated significantly improved activity and specificity of Cas9FKPB-mediated genome editing under the control of a dTAG molecule. This new development highlights the potential application of Cas9 degraders in CRISPR-Cas9 genome editing. Using the π-clamp as an engineered sequence in Cas9 or other exogenously expressed proteins offers some advantages compared to other formats of PROTACs and approaches to target protein degradation. For instance, HALO- and dTAG-PROTACs require the insertion of a comparably large HALO tag or FKBP12 into the target protein. Such a large insertion could be problematic, as the large molecular weight increase (∼33 and 12 kDa, respectively) may interfere with protein interactions and other functions.26,47 In the case of Cas9FKBP, two insertions were required for a sufficient degradation.24 In contrast, an insertion of the FCPF sequence represents a minimal modification of the target protein. Therefore, multiple FCPF sequences could be inserted into a target protein, which makes this technology applicable for an endogenously expressed protein in combination with a CRISPR-Cas9 knockin system.
Conclusion
The CRISPR-Cas system is a powerful tool to manipulate the human genome for gene therapy. Although the first in-human phase I clinical trial confirmed more than 95% on-target efficiency, rare off-target edits on various transcription factors, such as ZNF609 and ZNF808, were still observed,8 which may lead to unexpected side effects. Compelling evidence has shown that the timely regulation of Cas9 protein activity significantly reduces the off-target effects. Using a combination of the π-clamp system and PROTAC technology, we demonstrated a novel PROTAC-FCPF molecule able to sufficiently suppress the activity of Cas proteins through the ubiquitin-proteasome pathway. Because the size of insertion is relatively small, this approach may be applicable to label and functionalize not only exogenously but also endogenously expressed proteins in a combination of the CRISPR-Cas9 knockin system with proper heterobifunctional small molecules. One of the disadvantages of PROTAC is the lipophilicity of relatively large bifunctional molecules. According to the classic Lipinski’s rule of five, PROTAC-FCPF is still drug-like with a LogP of 4.42. However, further modifications are required on both protein-FCPF and PROTAC-FCPF to achieve the effective regulation of FCPF-tagging protein activity within a short incubation time.
Experimental Section
Plasmids, siRNAs, and Chemicals for Bioassays
dCas9 (Addgene, No. 61355), Cas12 (hPdCpf1, Addgene No. 69990), Cas13 (LwCas13a, Addgene No. 91902), and CRBN KO (plenti-px330-CRBN-T1-pGK-Pur and plenti-px330-CRBN-T2-pGK-Pur, 107382 and 107383) were purchased from Addgene. Insertion of the FCPF amino acid sequence was performed and analyzed by Genscript. DDB1, CRBN, CUL4A, MDM2, and VHL siRNAs were purchased from Thermofisher Scientific. Cas9 (No. 19526) and FLAG (No. 8146) antibody were purchased from Cell Signaling Technologies. CRBN antibody was from Thermofisher Scientific (PA5-38037). ACTIN antibody was from Santa Cruz Biotechnology (No. SC-4778). CMV-SpyCas9 (Addgene No. 113033), sgRNA targeting AAVS1 (Addgene No. 128119) plasimd, and Anti-CRISPR protein (ACRIIA4, Addgene No. 113037) are gifts from Dr. Niopek (IPMB, Heidelberg).9,12 CRISPR-Cas9 CRBN double nickase knockout plasmid was purchased from Santa Cruz Biotechnology (SC-412142). Lenalidomide, MG132, PYR41, and MLN4924 were purchased from Biomol.
Cell Culture and Transfection
HeLa and HEK2943T cells were cultivated in Dulbecco’s Modified Eagle’s Medium (DMEM) (Gibco) containing 10% fetal bovine serum (FBS) (Gibco), as well as 100 Unit/mL penicillin and 100 μg/mL streptomycin (Gibco), at 37 °C in a humidified atmosphere under 5% CO2. Jopaca-1 cells were cultivated in RPMI 1640. Overexpression and knockdown were performed as previously described.48 Generally, lipofectamine 3000 was used for transfection. According to the manufacturer’s instructions, the mixture of 100 μL of lipofectamine 3000 and 100 μL of p3000 reagent with either 20 nM siRNA or 2500 ng of DNA was incubated at room temperature for 5 min. Cells with 60–70% of confluency in a 6-well plate were transiently transfected with the mixture for 24 h in medium without antibiotics and incubated with refreshed medium for another 24 h. For a T7E1 assay, Cas9WT or Cas9FCPF were cotransfected with sgRNA in the presence or absence of anti-CRISPR protein (ACRII4). The sequences of siRNAs can be found in the Supporting Information.
Degradation in Cells
HeLa, HeK293, or Jopaca-1 cells were transfected with Cas9FCPF for 48 h as above-described. Generally, 10 μM of PROTAC-FCPF was added to induce an acute Cas9FCPF degradation (less than 8 h). For a 24 h treatment, 1000 ng of DNA was transfected with Lipofectamine 3000 and incubated for 48 h. PROTAC-FCPF (1 μM) was used for 24 h treatment.
FITC-FCPF Staining
HeLa cells were transfected with Cas9FCPF for 48 h. FITC-FCPF (10 μM) was added and incubated for 2 h. Cells were washed at least three times with phosphate-buffered solution (PBS), maintained in PBS with 1% FBS, and monitored under a fluorescence microscope. Cas9WT and Cas9GFP were used as controls. For an FACS analysis, cells were trypsinized, centrifuged (200 g, 5 min), washed at least three times with PBS, and resuspended in PBS containing 1% BSA and 2 mM ethylenediaminetetraacetic acid (EDTA). Guava easyCyte was used as previously reported.49 For immunoblotting, cells were lysed in 6 M urea-lysis buffer with protease and phosphatase inhibitors (1 mM EDTA, 0.5% Triton X-100, 5 mM NaF, 6 M urea, 1 mM Na3VO4, 10 μg/mL pepstatin, 100 μM phenylmethylsulfonyl fluoride (PMSF), and 3 μg/mL aprotinin in PBS). Total protein was resolved on 8% SDS-PAGE gels for Cas9. Images were taken under visible or UV light.
In Vitro Ubiquitination Assay
CRBN KO cells were transfected with Cas9WT or Cas9FCPF plasmid as above-described and lysed with immunoprecipitation buffer (20 mM Tris-HCl, 150 mM NaCl, 1 mM Na2EDTA, 1 mM ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 μg/mL leupeptin, 1 mM PMSF, pH 7.5) freshly prepared before use. FLAG-CRBNWT or -CRBNm was expressed in HeLa cells and freshly isolated using a FLAG immunoprecipitation kit (Sigma-Aldrich, Merck, No. FLAGIPT1-1KT) according to the manufacturer’s instructions. Anti-FLAG M2 affinity gel (20 μL) was added into 500 μL of cell lysate and incubated at 4 °C overnight. The suspension was washed at least three times with a wash buffer (50 mM Tris HCl and 150 mM NaCl, pH 7.4) and eluted 100 μL of elution buffer (0.1 M glycine HCL, 1150 mM NaCl, pH 7.4). 10X Wash buffer was added to neutralize the pH value.
The assay was initiated with adding CRBN into cell lysis and incubated for 90 min at 37 °C in the presence or absence of PROTAC-FCPF (100 μM). The adenosine triphosphate (ATP) consumption was determined directly in ENLITEN ATP Assay (Promega, FF2000). Pretreatment of lenalidomide (100 μM) for 30 min sufficiently reduced the degradation of Cas9FCPF. The generation of CRBN KO cell lines can be found in the Supporting Information.
T7 Endonuclease I Assay
An EnGen mutation detection kit (NEB, No. E3321) was used to evaluate the efficiency of CRISPR-Cas9 in genome editing, which includes a positive control. HeLa cells were transfected with Cas9WT or Cas9FCPF or in a combination of Cas9FCPF and anti-CRISPR protein (ACR) in the presence of sgRNA AAVS1 for 48 h. CMV-SpyCas9 (Addgene No. 113033), sgRNA targeting AAVS1 (Addgene No. 128119) plasimd, and Anti-CRISPR protein (ACRIIA4, Addgene No. 113037) were gifts from Dr. Niopek, University Heidelberg). HeLa cells expressing Cas9FCPF were treated with PROTAC-FCPF (1 μM) for 24 h. Lucigen QuickExtract DNA Extraction Solution (Bioenzyme, No. MA150E) was used to collect DNA. HeLa cells were washed with PBS and trypsinized. The pellet of 500 000 cells was lysed with 500 μL of Lucigen QuickExtract DNA Extraction Solution and vortexed for 15 s. The lysis was first heated at 65 °C for 6 min and vortexed for 15 s and at 98 °C for 2 min. DNA can be stored at −70 °C for 6–8 weeks.
The target sequences of AAVS1 gene were amplified with Eppendorf Mastercycler. A mixture of Q5 Hot Start high-Fidelity 2x Master mix (12.5 μL), 10 μM forward and reverse primers (1.25 μL of each), and genomic DNA (100 ng) in nuclease-free water (25 μL in total) was prepared. The target sequence was amplified using the following setup: 1 cycle (98 °C for 30 min); 40 cycles (98 °C for 5 min, 70 °C for 10 min, and 72 °C for 20 min); and 1 cycle (72 °C for 2 min).
For heteroduplex formation between polymerase chain reaction (PCR) products with and without mutations 5 μL of PCR product was mixed with 2 μL of 10X NEBuffer2 in 12 μL of nuclease-free water. The mixture was denaturated and annealed using the following program: 95 °C for 5 min; 95–85 °C (−2 °C/sec) and 85–25 °C (−0.1 °C/sec). Afterward, 1 μL of EnGen T7 Endonuclease I was added into the annealed PCR product and incubated at 37 °C for 15 min. Proteinase K (1 μL) was added and incubated at 37 °C for another 5 min to inhibit the activity of endonuclease. The sample was analyzed on a 2% agarose gel. The signal was quantified by Aida image analysis software. We quantified band intensities by ImageJ software and calculated genome editing efficiency using the eq 100(1-(1-franction cleaved)1/2).50
The corresponding sequences can be found in the Supporting Information.
Statistics and Reproducibility
Data were analyzed with the GraphPad Prism software (v7.01). Two-way analysis of variance (ANOVA) was performed. All experiments except for proteomics were independently repeated at least three times, from which similar results were obtained. The standard deviation is noted as SD.
Acknowledgments
We thank the Quantitative Proteomics Unit at IBC2, Dr. Wölfl for his support, and Dr. Niopek for sharing the materials.
Glossary
Abbreviations
- CRISPR
clustered regularly interspaced short palindromic repeats
- crRNA
CRISPR RNA
- dCas9
nuclease-dead Cas9
- DSBs
double-strand breaks
- dsDNA
double-stranded DNA
- PROTAC
proteolysis-targeting chimera
- ssDNA
single-stranded DNA
- ssRNA
single-stranded RNA
- TLC
thin layer chromatography
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacsau.1c00007.
The application of Cas9FCPF for live cell imaging. The PROTAC-FCPF inhibits Cas9-mediated genome editing through destabilizing Cas9FCPF protein in cells. The PROTAC-FCPF-induced Cas9FCPF protein degradation is ubiquitin-dependent. PROTAC-FCPF degrades dCas9FCPF, Cas12FCPF, and Cas13FCPF (PDF)
Author Present Address
⊥ Institute for Tumor Biology and Experimental Therapy, Georg-Speyer-Haus, 60596 Frankfurt/Main, Germany.
Author Contributions
# (R.A.G.-B. and J.C.) These authors contributed equally. R.G.B. performed biological assessments and wrote the manuscript. J.C. synthesized compounds. V.N. helped with the chemical synthesis. Y.D. and S.K. helped with the experimental design and writing. G.T. and C.M. planned, executed, and analyzed the proteomics analyses. X.C. designed the experiments, performed experiments, and wrote the manuscript. All authors have given approval to the final version of the manuscript.
We thank DFG for the financial support (CH 1690/2-1 and 2-3). This work was supported in part by the LOEWE Center Frankfurt Cancer Institute funded by the Hessen State Ministry for Higher Education, Research and the Arts [III L 5-519/03/03.001-(0015)].
The authors declare no competing financial interest.
Supplementary Material
References
- Garcia-Doval C.; Jinek M. Molecular architectures and mechanisms of Class 2 CRISPR-associated nucleases. Curr. Opin. Struct. Biol. 2017, 47, 157–166. 10.1016/j.sbi.2017.10.015. [DOI] [PubMed] [Google Scholar]
- Esvelt K. M.; Mali P.; Braff J. L.; Moosburner M.; Yaung S. J.; Church G. M. Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nat. Methods 2013, 10 (11), 1116–21. 10.1038/nmeth.2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doudna J. A.; Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346 (6213), 1258096. 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- Li Y.; Li S.; Wang J.; Liu G. CRISPR/Cas Systems towards Next-Generation Biosensing. Trends Biotechnol. 2019, 37 (7), 730–743. 10.1016/j.tibtech.2018.12.005. [DOI] [PubMed] [Google Scholar]
- Wolter F.; Puchta H. The CRISPR/Cas revolution reaches the RNA world: Cas13, a new Swiss Army knife for plant biologists. Plant J. 2018, 94 (5), 767–775. 10.1111/tpj.13899. [DOI] [PubMed] [Google Scholar]
- O’Connell M. R. Molecular Mechanisms of RNA Targeting by Cas13-containing Type VI CRISPR-Cas Systems. J. Mol. Biol. 2019, 431 (1), 66–87. 10.1016/j.jmb.2018.06.029. [DOI] [PubMed] [Google Scholar]
- Cox D. B. T.; Gootenberg J. S.; Abudayyeh O. O.; Franklin B.; Kellner M. J.; Joung J.; Zhang F. RNA editing with CRISPR-Cas13. Science 2017, 358 (6366), 1019–1027. 10.1126/science.aaq0180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtmauer E. A.; Fraietta J. A.; Davis M. M.; Cohen A. D.; Weber K. L.; Lancaster E.; Mangan P. A.; Kulikovskaya I.; Gupta M.; Chen F.; Tian L.; Gonzalez V. E.; Xu J.; Jung I. Y.; Melenhorst J. J.; Plesa G.; Shea J.; Matlawski T.; Cervini A.; Gaymon A. L.; Desjardins S.; Lamontagne A.; Salas-Mckee J.; Fesnak A.; Siegel D. L.; Levine B. L.; Jadlowsky J. K.; Young R. M.; Chew A.; Hwang W. T.; Hexner E. O.; Carreno B. M.; Nobles C. L.; Bushman F. D.; Parker K. R.; Qi Y.; Satpathy A. T.; Chang H. Y.; Zhao Y.; Lacey S. F.; June C. H. CRISPR-engineered T cells in patients with refractory cancer. Science 2020, 367 (6481), eaba7365. 10.1126/science.aba7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aschenbrenner S.; Kallenberger S. M.; Hoffmann M. D.; Huck A.; Eils R.; Niopek D. Coupling Cas9 to artificial inhibitory domains enhances CRISPR-Cas9 target specificity. Sci. Adv. 2020, 6 (6), eaay0187 10.1126/sciadv.aay0187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempton H. R.; Qi L. S. When genome editing goes off-target. Science 2019, 364 (6437), 234–236. 10.1126/science.aax1827. [DOI] [PubMed] [Google Scholar]
- Zhang X. H.; Tee L. Y.; Wang X. G.; Huang Q. S.; Yang S. H. Off-target Effects in CRISPR/Cas9-mediated Genome Engineering. Mol. Ther.--Nucleic Acids 2015, 4, e264 10.1038/mtna.2015.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X. Valproic Acid Thermally Destabilizes and Inhibits SpyCas9 Activity. Mol. Ther. 2020, 28 (12), 2635–2641. 10.1016/j.ymthe.2020.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maji B.; Gangopadhyay S. A.; Lee M.; Shi M.; Wu P.; Heler R.; Mok B.; Lim D.; Siriwardena S. U.; Paul B.; Dancik V.; Vetere A.; Mesleh M. F.; Marraffini L. A.; Liu D. R.; Clemons P. A.; Wagner B. K.; Choudhary A. A High-Throughput Platform to Identify Small-Molecule Inhibitors of CRISPR-Cas9. Cell 2019, 177 (4), 1067–1079. 10.1016/j.cell.2019.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis K. M.; Pattanayak V.; Thompson D. B.; Zuris J. A.; Liu D. R. Small molecule-triggered Cas9 protein with improved genome-editing specificity. Nat. Chem. Biol. 2015, 11 (5), 316–8. 10.1038/nchembio.1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilton I. B.; D’Ippolito A. M.; Vockley C. M.; Thakore P. I.; Crawford G. E.; Reddy T. E.; Gersbach C. A. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol. 2015, 33 (5), 510–7. 10.1038/nbt.3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiani S.; Beal J.; Ebrahimkhani M. R.; Huh J.; Hall R. N.; Xie Z.; Li Y.; Weiss R. CRISPR transcriptional repression devices and layered circuits in mammalian cells. Nat. Methods 2014, 11 (7), 723–6. 10.1038/nmeth.2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakore P. I.; D’Ippolito A. M.; Song L.; Safi A.; Shivakumar N. K.; Kabadi A. M.; Reddy T. E.; Crawford G. E.; Gersbach C. A. Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat. Methods 2015, 12 (12), 1143–9. 10.1038/nmeth.3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adli M. The CRISPR tool kit for genome editing and beyond. Nat. Commun. 2018, 9 (1), 1911. 10.1038/s41467-018-04252-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersson M.; Crews C. M. Proteolysis TArgeting Chimeras (PROTACs) - Past, present and future. Drug Discovery Today: Technol. 2019, 31, 15–27. 10.1016/j.ddtec.2019.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondeson D. P.; Crews C. M. Targeted Protein Degradation by Small Molecules. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 107–123. 10.1146/annurev-pharmtox-010715-103507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanton B. Z.; Chory E. J.; Crabtree G. R. Chemically induced proximity in biology and medicine. Science 2018, 359 (6380), 1117. 10.1126/science.aao5902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C.; Welborn M.; Zhu T.; Yang N. J.; Santos M. S.; Van Voorhis T.; Pentelute B. L. Pi-Clamp-mediated cysteine conjugation. Nat. Chem. 2016, 8 (2), 120–8. 10.1038/nchem.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter G. E.; Buckley D. L.; Paulk J.; Roberts J. M.; Souza A.; Dhe-Paganon S.; Bradner J. E. DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348 (6241), 1376–81. 10.1126/science.aab1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreekanth V.; Zhou Q.; Kokkonda P.; Bermudez-Cabrera H. C.; Lim D.; Law B. K.; Holmes B. R.; Chaudhary S. K.; Pergu R.; Leger B. S.; Walker J. A.; Gifford D. K.; Sherwood R. I.; Choudhary A. Chemogenetic System Demonstrates That Cas9 Longevity Impacts Genome Editing Outcomes. ACS Cent. Sci. 2020, 6 (12), 2228–2237. 10.1021/acscentsci.0c00129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglass E. F.; Miller C. J.; Sparer G.; Shapiro H.; Spiegel D. A. A Comprehensive Mathematical Model for Three-Body Binding Equilibria. J. Am. Chem. Soc. 2013, 135 (16), 6092–6099. 10.1021/ja311795d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondeson D. P.; Mares A.; Smith I. E.; Ko E.; Campos S.; Miah A. H.; Mulholland K. E.; Routly N.; Buckley D. L.; Gustafson J. L.; Zinn N.; Grandi P.; Shimamura S.; Bergamini G.; Faelth-Savitski M.; Bantscheff M.; Cox C.; Gordon D. A.; Willard R. R.; Flanagan J. J.; Casillas L. N.; Votta B. J.; den Besten W.; Famm K.; Kruidenier L.; Carter P. S.; Harling J. D.; Churcher I.; Crews C. M. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 2015, 11 (8), 611–7. 10.1038/nchembio.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabizon R.; Shraga A.; Gehrtz P.; Livnah E.; Shorer Y.; Gurwicz N.; Avram L.; Unger T.; Aharoni H.; Albeck S.; Brandis A.; Shulman Z.; Katz B. Z.; Herishanu Y.; London N. Efficient Targeted Degradation via Reversible and Irreversible Covalent PROTACs. J. Am. Chem. Soc. 2020, 142 (27), 11734–11742. 10.1021/jacs.9b13907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X.; Kim J. Y.; Ghafoory S.; Duvaci T.; Rafiee R.; Theobald J.; Alborzinia H.; Holenya P.; Fredebohm J.; Merz K. H.; Mehrabi A.; Hafezi M.; Saffari A.; Eisenbrand G.; Hoheisel J. D.; Wolfl S. Methylisoindigo preferentially kills cancer stem cells by interfering cell metabolism via inhibition of LKB1 and activation of AMPK in PDACs. Mol. Oncol. 2016, 10 (6), 806–24. 10.1016/j.molonc.2016.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.; Kim D.; Cho S. W.; Kim J.; Kim J. S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014, 24 (6), 1012–9. 10.1101/gr.171322.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bubeck F.; Hoffmann M. D.; Harteveld Z.; Aschenbrenner S.; Bietz A.; Waldhauer M. C.; Borner K.; Fakhiri J.; Schmelas C.; Dietz L.; Grimm D.; Correia B. E.; Eils R.; Niopek D. Engineered anti-CRISPR proteins for optogenetic control of CRISPR-Cas9. Nat. Methods 2018, 15 (11), 924–927. 10.1038/s41592-018-0178-9. [DOI] [PubMed] [Google Scholar]
- Sievers Q. L.; Petzold G.; Bunker R. D.; Renneville A.; Slabicki M.; Liddicoat B. J.; Abdulrahman W.; Mikkelsen T.; Ebert B. L.; Thoma N. H. Defining the human C2H2 zinc finger degrome targeted by thalidomide analogs through CRBN. Science 2018, 362 (6414), 558. 10.1126/science.aat0572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu G.; Middleton R. E.; Sun H.; Naniong M.; Ott C. J.; Mitsiades C. S.; Wong K. K.; Bradner J. E.; Kaelin W. G. Jr. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 2014, 343 (6168), 305–9. 10.1126/science.1244917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K. M.; Jo S.; Kim H.; Lee J.; Park C. S. Functional modulation of AMP-activated protein kinase by cereblon. Biochim. Biophys. Acta, Mol. Cell Res. 2011, 1813 (3), 448–55. 10.1016/j.bbamcr.2011.01.005. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Kitagaki J.; Dai R. M.; Tsai Y. C.; Lorick K. L.; Ludwig R. L.; Pierre S. A.; Jensen J. P.; Davydov I. V.; Oberoi P.; Li C. C.; Kenten J. H.; Beutler J. A.; Vousden K. H.; Weissman A. M. Inhibitors of ubiquitin-activating enzyme (E1), a new class of potential cancer therapeutics. Cancer Res. 2007, 67 (19), 9472–81. 10.1158/0008-5472.CAN-07-0568. [DOI] [PubMed] [Google Scholar]
- Soucy T. A.; Smith P. G.; Milhollen M. A.; Berger A. J.; Gavin J. M.; Adhikari S.; Brownell J. E.; Burke K. E.; Cardin D. P.; Critchley S.; Cullis C. A.; Doucette A.; Garnsey J. J.; Gaulin J. L.; Gershman R. E.; Lublinsky A. R.; McDonald A.; Mizutani H.; Narayanan U.; Olhava E. J.; Peluso S.; Rezaei M.; Sintchak M. D.; Talreja T.; Thomas M. P.; Traore T.; Vyskocil S.; Weatherhead G. S.; Yu J.; Zhang J.; Dick L. R.; Claiborne C. F.; Rolfe M.; Bolen J. B.; Langston S. P. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 2009, 458 (7239), 732–6. 10.1038/nature07884. [DOI] [PubMed] [Google Scholar]
- Perng Y. C.; Lenschow D. J. ISG15 in antiviral immunity and beyond. Nat. Rev. Microbiol. 2018, 16 (7), 423–439. 10.1038/s41579-018-0020-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y.; Foden J. A.; Khayter C.; Maeder M. L.; Reyon D.; Joung J. K.; Sander J. D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31 (9), 822–6. 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinstiver B. P.; Prew M. S.; Tsai S. Q.; Topkar V. V.; Nguyen N. T.; Zheng Z.; Gonzales A. P.; Li Z.; Peterson R. T.; Yeh J. R.; Aryee M. J.; Joung J. K. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature 2015, 523 (7561), 481–5. 10.1038/nature14592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J. H.; Miller S. M.; Geurts M. H.; Tang W.; Chen L.; Sun N.; Zeina C. M.; Gao X.; Rees H. A.; Lin Z.; Liu D. R. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 2018, 556 (7699), 57–63. 10.1038/nature26155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slaymaker I. M.; Gao L.; Zetsche B.; Scott D. A.; Yan W. X.; Zhang F. Rationally engineered Cas9 nucleases with improved specificity. Science 2016, 351 (6268), 84–8. 10.1126/science.aad5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watters K. E.; Fellmann C.; Bai H. B.; Ren S. M.; Doudna J. A. Systematic discovery of natural CRISPR-Cas12a inhibitors. Science 2018, 362 (6411), 236–239. 10.1126/science.aau5138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holohan C.; Van Schaeybroeck S.; Longley D. B.; Johnston P. G. Cancer drug resistance: an evolving paradigm. Nat. Rev. Cancer 2013, 13 (10), 714–26. 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- Koppikar P.; Bhagwat N.; Kilpivaara O.; Manshouri T.; Adli M.; Hricik T.; Liu F.; Saunders L. M.; Mullally A.; Abdel-Wahab O.; Leung L.; Weinstein A.; Marubayashi S.; Goel A.; Gonen M.; Estrov Z.; Ebert B. L.; Chiosis G.; Nimer S. D.; Bernstein B. E.; Verstovsek S.; Levine R. L. Heterodimeric JAK-STAT activation as a mechanism of persistence to JAK2 inhibitor therapy. Nature 2012, 489 (7414), 155–U222. 10.1038/nature11303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krall N.; da Cruz F. P.; Boutureira O.; Bernardes G. J. Site-selective protein-modification chemistry for basic biology and drug development. Nat. Chem. 2016, 8 (2), 103–13. 10.1038/nchem.2393. [DOI] [PubMed] [Google Scholar]
- Virdee S.; Kapadnis P. B.; Elliott T.; Lang K.; Madrzak J.; Nguyen D. P.; Riechmann L.; Chin J. W. Traceless and site-specific ubiquitination of recombinant proteins. J. Am. Chem. Soc. 2011, 133 (28), 10708–11. 10.1021/ja202799r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adhikari B.; Bozilovic J.; Diebold M.; Schwarz J. D.; Hofstetter J.; Schroder M.; Wanior M.; Narain A.; Vogt M.; Dudvarski Stankovic N.; Baluapuri A.; Schonemann L.; Eing L.; Bhandare P.; Kuster B.; Schlosser A.; Heinzlmeir S.; Sotriffer C.; Knapp S.; Wolf E. PROTAC-mediated degradation reveals a non-catalytic function of AURORA-A kinase. Nat. Chem. Biol. 2020, 16 (11), 1179–1188. 10.1038/s41589-020-00652-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth S.; Fulcher L. J.; Sapkota G. P. Advances in targeted degradation of endogenous proteins. Cell. Mol. Life Sci. 2019, 76 (14), 2761–2777. 10.1007/s00018-019-03112-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X.; Haeberle S.; Shytaj I. L.; Gama-Brambila R. A.; Theobald J.; Ghafoory S.; Wolker J.; Basu U.; Schmidt C.; Timm A.; Taskova K.; Bauer A. S.; Hoheisel J.; Tsopoulidis N.; Fackler O. T.; Savarino A.; Andrade-Navarro M. A.; Ott I.; Lusic M.; Hadaschik E. N.; Wolfl S. NHC-gold compounds mediate immune suppression through induction of AHR-TGFbeta1 signalling in vitro and in scurfy mice. Commun. Biol. 2020, 3 (1), 10. 10.1038/s42003-019-0716-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabiri Y.; Gama-Brambila R. A.; Taskova K.; Herold K.; Reuter S.; Adjaye J.; Utikal J.; Mrowka R.; Wang J.; Andrade-Navarro M. A.; Cheng X. Imidazopyridines as Potent KDM5 Demethylase Inhibitors Promoting Reprogramming Efficiency of Human iPSCs. iScience 2019, 12, 168–181. 10.1016/j.isci.2019.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X.; Gao D.; Wang P.; Chen J.; Ruan J.; Xu J.; Xia X. Efficient homology-directed gene editing by CRISPR/Cas9 in human stem and primary cells using tube electroporation. Sci. Rep. 2018, 8 (1), 11649. 10.1038/s41598-018-30227-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.