

Abstract
Perovskite oxides based on earth-abundant transition metals have been extensively explored as promising oxygen evolution reaction (OER) catalysts in alkaline media. The (electro)chemically induced transformation of their initially crystalline surface into an amorphous state has been reported for a few highly active perovskite catalysts. However, little knowledge is available to distinguish the contribution of the amorphized surface from that of the remaining bulk toward the OER. In this work, we utilize the promoting effects of two types of Fe modification, i.e., bulk Fe dopant and Fe ions absorbed from the electrolyte, on the OER activity of SrCoO3−δ model perovskite to identify the active phase. Transmission electron microscopy and X-ray photoelectron spectroscopy confirmed the surface amorphization of SrCoO3−δ as well as SrCo0.8Fe0.2O3−δ after potential cycling in Fe-free KOH solution. By further cycling in Fe-spiked electrolyte, Fe was incorporated into the amorphized surface of SrCoO3−δ (SrCoO3−δ + Fe3+), yielding approximately sixfold increase in activity. Despite the difference in remaining perovskites, SrCoO3−δ + Fe3+ and SrCo0.8Fe0.2O3−δ exhibited remarkably similar activity. These results reflect that the in situ developed surface species are directly responsible for the measured OER activity, whereas the remaining bulk phases have little impact.
Keywords: surface reconstruction, oxygen evolution, perovskite, oxides, cobalt, iron
1. Introduction
Large-scale hydrogen production from water electrolysis has been proposed as a means to satisfy ever-increasing demands for sustainable energy and to reduce the emission of greenhouse gases. To advance this technology to commercial viability, a breakthrough is needed to accelerate the rate of the oxygen evolution reaction (OER), the sluggish kinetics of which restricts the overall efficiency of water splitting.1 For this reason, the development of efficient and robust electrocatalysts for OER has been pursued by many researchers over the past decades.2,3
Oxides based on earth-abundant transition metals (TMs) such as Fe, Co, and Ni have been demonstrated to be strong candidates for OER catalysis, owing to their low price and promising activity.4−11 Among them, perovskite-type oxides with the general formula of ABO3 (A = alkaline-earth and/or rare-earth metals; B = TMs) open many avenues for catalyst design due to the versatility of the structure. By tailoring chemical composition and/or crystal structure, a few highly efficient perovskites have been developed, and their superior intrinsic activities have been attributed to optimizing the binding strength of intermediates associated with the elementary steps of the four-electron oxidation reaction.4,12,13 At the same time, the rationale for tailoring the bulk structure of the perovskite oxides has been challenged by reports that their surface is not stable under OER conditions.14−20 For example, May et al. found that the largely crystalline surface of Ba0.5Sr0.5Co0.8Fe0.2O3−δ (BSCF), a state-of-the-art perovskite-type OER catalyst, was rapidly converted into an amorphous form during OER in alkaline solution, accompanied by the leaching of the A-site cations Ba and Sr.14 We note that significant surface amorphization has also been reported for other highly active perovskites, e.g., SrCo0.8Fe0.2O3−δ (SCF02), while the surface of less active perovskites such as LaCoO3 appears to be more stable.14,15 More recently, with the aid of operando techniques, Fabbri et al. identified a positive correlation between the measured OER activity and the extent of surface self-reconstruction during OER, specifically finding the growth of TM (oxy)hydroxide layer on the oxide surface.17 Accordingly, they hypothesized that the reconstruction-derived surface, instead of the initial perovskite, is the real catalyst.
Experimentally, it has been difficult to isolate the role of in situ developed TM (oxy)hydroxide from the underlying bulk perovskite, and hence, it remains unclear whether the measured OER activity is solely (or even partially) determined by the reconstructed surface layer or is influenced by the structure of the underlying bulk phase. Answering this question is critical to the improvement of catalyst design.
Resolving this issue requires a method to evaluate the relative contributions of the amorphized surface and the bulk in situ. In light of previous observations that Fe ions in the alkaline electrolyte can be incorporated into Co- or Ni-based (oxy)hydroxides and positively affect the OER catalytic performance,21−23 it is reasonable to speculate that the TM (oxy)hydroxide in situ derived from a Co/Ni-based perovskite catalyst is also sensitive to the presence of Fe in the electrolyte, whereas the underlying bulk phase is insensitive, which thus offers the possibility to decouple the electrochemical responses of the reconstructed surface and the remaining perovskite.
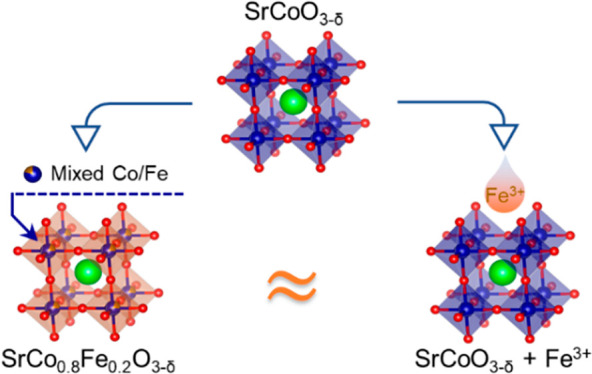
In this work, we employed this method using cubic SrCoO3−δ (SC) perovskite and Fe-modified SC perovskites as the model catalyst. This choice is based on the following three considerations. First, SC with a cubic perovskite structure has been theoretically and experimentally demonstrated to be highly active.24,25 Second, although, to our knowledge, the surface reconstruction of cubic SC has not been considered, the relatively high solubility of A-site cation (e.g., Sr) has been suggested to be a key to inducing the surface reconstruction of perovskites,17,26 and the surface restructuring after OER measurements has been reported for SC with brownmillerite- or hexagonal-type perovskite structure.27,28 Third, Fe can be stabilized in the solid solution of SC perovskite, which modulates the bulk electronic properties and thus provides a contrast to the introduction of Fe into the surface of SC from the electrolyte. As illustrated in Figure 1a, Fe is incorporated into SC in two different ways. As a bulk dopant, Co and Fe cations are uniformly distributed within the perovskite lattice (SCF02). In a surface decorating pathway, Fe3+ ions spiked into the electrolyte are expected to be incorporated onto the surface of SC during oxygen evolution (denoted as SC + Fe3+). Our results show that the surfaces of both SC and SCF02 were amorphized and Sr-leached after potential cycling in 0.1 M KOH, while their bulk perovskite structures were well-retained. Upon further cycling in Fe-spiked KOH, SC exhibited significantly enhanced catalytic performance, which thus is comparable to that of SCF02. The structure–activity relationship found between SC, SC + Fe3+, and SCF02 demonstrates that the in situ reconstructed surface governs the apparent OER activity, while the remaining bulk phase is only a minor contributor.
Figure 1.

(a) Schematic illustration of the two ways to incorporate Fe into cubic-structured SC perovskite. Sr, Co, and O atoms are shown as green, blue, and red balls, respectively. Rietveld refined XRD patterns of as-synthesized (b) SC and (c) SCF02. HRTEM images of pristine (d) SC and (e) SCF02.
2. Results and Discussion
2.1. Bulk and Surface Characterization of As-Synthesized SC and SCF02
SC perovskite was synthesized by a conventional sol–gel method with a modified thermal treatment process, i.e., high-temperature calcination followed by quenching and annealing (see Experimental Section for details). As confirmed by the Rietveld refined X-ray diffraction (XRD) profile in Figure 1b, SC prepared in this work crystallizes in a cubic structure with the space group of Pm3̅m and the lattice parameters of a = b = c = 3.8451 Å (Table S1). Partial substitution of B-site Co with Fe cations maintains the cubic phase well (Figure 1c and Figure S1), which is consistent with previous findings that an appropriate amount of Fe doping stabilizes the cubic structure of SC perovskite.29,30Figure 1d, e presents the high-resolution transmission electron microscopy (HRTEM) images of as-synthesized SC and SCF02, respectively, revealing that their surfaces and bulk are highly crystalline. The interplane distances of 0.27 nm for SC and 0.22 nm for SCF02 correspond to the [110] and [111] crystal plane of the cubic perovskite structure, respectively. Moreover, as displayed by scanning electron microscope (SEM) images (Figure S2), there is little difference in the morphology and particle size of undoped and Fe-doped SC, which is in agreement with their similarity in Brunauer–Emmett–Teller (BET) specific surface area (Figure S3 and Table S2).
2.2. OER Catalytic Behaviors and Surface Evolution of SC and SCF02 in Fe-Free KOH
The OER catalytic activities of undoped and Fe-doped SC were evaluated by cyclic voltammetry (CV) at a scan rate of 10 mVs–1 in 0.1 M Fe-free KOH solution (see Experimental Section for details).31 As shown in Figure S4, the incorporation of Fe into the lattice of SC increases OER activity, and the enhancement effect approaches the maximum when the value of x in SrCo1–xFexO3−δ equals 0.2. This result is consistent with previous reports about the iron-substitution effect on cobalt-based oxides and (oxy)hydroxides.6,22,29 Among the investigated Fe-doped SC catalysts, SCF02 exhibited the highest activity and thus was selected for further study. Figure 2a, b presents the CV cycles of SC and SCF02 in 0.1 M KOH electrolyte, respectively. In the positive-going scan of the first cycle, both SC and SCF02 exhibited a precatalytic oxidation process that is different from the redox behaviors in subsequent scans, indicative of irreversible changes occurring on the perovskite surfaces. Starting from the second cycle, two pairs of redox waves prior to the onset of OER, one between 1.0–1.3 V vs RHE and another between 1.35–1.52 V vs RHE, are discernible, which can be attributed to the Co2+/3+ and the Co3+/4+ redox couples, respectively.32 Along with the precatalytic redox peaks being more prominent, the OER activities of both perovskites gradually increased during successive cycling (Figure S5). These changes are similar to those observed in the CV scans of BSCF perovskite,14,15 in which the near-surface region was found to be amorphized, and thus imply that SC and SCF02 probably underwent similar restructuring. After 50 cycles, the OER currents as well as the capacitive currents of both SC and SCF02 approached stable values, suggesting that their restructured surfaces reached relatively steady states.
Figure 2.

Cyclic voltammograms of (a) SC and (b) SCF02 at a scan rate of 10 mVs–1 in 0.1 M Fe-free KOH, showing the 1st, 2nd, 50th, and 100th cycles. Insets show the capacitive region of CV scans, and the shaded areas indicate the redox waves. O 1s XPS spectra of (c) SC and (d) SCF02 before and after OER tests. Sr 3d XPS spectra of (e) SC and (f) SCF02 before and after OER tests. Dashed lines indicate the Sr 3d5/2 peaks. (g) HRTEM images of cycled SC and SCF02. The boundaries between the crystalline and amorphous regions are divided by white dashed lines. (h) The concentration of Sr dissolving in the electrolyte per cycle at different stages, i.e., during 1–10, 10–50, and 50–100 cycles.
To probe the potential surface changes during OER, the surface chemistry of pristine and cycled SC and SCF02 was analyzed by X-ray photoelectron spectroscopy (XPS) (Figure 2c–f and Figure S6). Related peak-fitting parameters are summarized in Tables S3 and S4. O 1s XPS spectra of SC and SCF02 before and after electrochemical cycling are shown in Figure 2c, d, respectively. For either pristine SC or SCF02, the O 1s spectrum can be mainly deconvoluted into two oxygen components, i.e., the low-energy component (529.5–529.8 eV) that is attributed to the lattice oxygen from the perovskite oxide and the high-energy component (∼531.7 eV) that is assigned to the oxygen from surface strontium carbonates and/or hydroxides.33−35 The minor peak at ∼533.1 eV in the O 1s spectrum of pristine SC can be assigned to the molecular water adsorbed on the surface.36,37 After OER measurements, the peak intensity of the oxygen from the perovskite lattice greatly reduced, suggesting that the surfaces of SC and SCF02 lost the initial perovskite structure. The additional peak appearing at ∼535.6 eV in the O 1s spectra of cycled perovskites corresponds to the oxygen from Nafion (the electrode binder).38 Sr 3d XPS spectra displayed in Figure 2e, f also provide evidence for the surface rearrangements of SC and SCF02. For as-prepared perovskites, each Sr 3d spectrum can be well-fitted with two sets of Sr 3d doublets (Sr 3d5/2 and 3d3/2 with splitting energy of ∼1.8 eV). The low-energy set (Sr 3d5/2: 131.4–132.0 eV) is related to the Sr from perovskite lattice and the high-energy set (Sr 3d5/2: 133.1–133.7 eV) is associated with the Sr from surface carbonates/hydroxides.33−35 Like the changes found in O 1s spectra, the relative intensities of Sr from the perovskite lattice became significantly weaker (for SCF02) or even absent (for SC) after electrochemical tests.
HRTEM imaging further revealed the reconstruction-derived structures for cycled SC and SCF02. As shown in Figure 2g, an amorphous layer with a depth of ∼10 nm was observed in the near-surface region of SC as well as SCF02 after 100 CV cycles. Under the surface region, the crystalline bulk phases of both SC and SCF02 were found to be well-maintained, as evidenced by the interplane distances in line with the XRD Rietveld refinement results (Table S1). Additionally, inductively coupled plasma mass spectrometry (ICP-MS) analysis (Figure 2h and Tables S5 and S6) demonstrated that the surface amorphization of SC and SCF02 was accompanied by the loss of A-site metals (i.e., Sr), as has been detected for other perovskites subjected to reconstruction under OER conditions (e.g., BSCF).14,17 The leaching rates of Sr during 1–10, 10–50, and 50–100 cycles were assessed by normalizing the concentration of Sr dissolving in the electrolyte at each stage to the times of CV scan (Figure 2h and Table S5). It can be noted that the Sr leaching rate in the first 10 CV cycles was approximately an order of magnitude higher than those in the subsequent cycles, suggesting that surface changes on SC and SCF02 were most drastic during initial cycles. This is in good consistency with the changing trend observed in CV profiles (Figure 2a, b and Figure S5); that is, the prominent increment of OER activity and capacitance induced by surface restructuring mainly took place in the first 10 cycles.
2.3. Oxygen Evolution of SC in Fe-Spiked KOH Electrolyte
After cycling in 0.1 M KOH 100 times, SC attained a relatively stable state (Figure 2a) and then was immediately tested in 0.1 M KOH electrolyte spiked with Fe3+ (Fe3+spiked). The CV profiles of SC in Fe-spiked KOH demonstrated a sharp increase in OER currents, which remained nearly constant during 10 continuous CV scans (Figure 3a). The concentration of Fe3+spiked is optimized to be 0.1 mM, which results in the maximum activity enhancement (Figure S7) with only a negligible influence on pH (see Experimental Section). As presented in Figure 3b, the capacitive regions of CV scans in 0.1 M KOH and 0.1 M KOH + 0.1 mM Fe3+spiked are almost overlapped, implying little variation in the surface area accessible to the electrolyte and the redox behaviors of Co atoms. As further evidence, double layer capacitance (Cdl), an indicator of electrochemical active surface area (ECSA),39 was measured for SC in the two kinds of electrolytes, and the nearly identical value of Cdl suggests that the performance improvement cannot be attributed to the change in ECSA (Figure 3c and Figure S8).
Figure 3.

(a) The 2nd and 10th CV scans of SC in 0.1 M KOH + 0.1 mM Fe3+spiked in comparison to the 100th CV scan of SC in 0.1 M KOH. Before cycling in Fe-spiked KOH electrolyte, SC underwent 100 CV scans in Fe-free KOH solution to steady its electrochemical behavior. (b) The zoomed-in capacitive region of the above CV scans. (c) Linear fitting curves of the capacitive currents versus CV scan rates for SC in 0.1 M KOH and 0.1 M KOH + 0.1 mM Fe3+spiked. (d) The 1st, 10th, and 50th CV scans of α-FeOOH (containing AB) at a scan rate of 10 mVs–1 in 0.1 M KOH. (e) Geometric current densities at 1.6 V vs RHE of blank GCE, pure AB carbon, and SC (containing AB) in Fe-free and Fe-spiked 0.1 M KOH electrolytes.
Thus, the positive effect of Fe3+spiked on the activity of SC is taken into consideration. On one hand, due to the low equilibrium solubility of Fe3+ as Fe(OH)4– at pH 13 (ca. 10–6 M),40 most of the Fe3+spiked in 0.1 M KOH should precipitate as FeOxHy, which is confirmed to be α-FeOOH by Rietveld refined XRD pattern (Figure S9). Hence, the observed activity increment could be attributed to α-FeOOH precipitate adsorbed onto the catalyst electrode. However, as shown in Figure 3d, α-FeOOH exhibited poor OER activity and quick deactivation during 50 CV scans, indicating that the precipitate of α-FeOOH should not be the cause of the boosted OER activity. On the other hand, Fe3+spiked ions also could be deposited onto the catalyst electrode and thus gave rise to OER activity. This is evidenced by the enhanced OER currents of pure acetylene black (AB) carbon and blank glassy carbon electrode (GCE) cycling in Fe-spiked KOH electrolyte (Figure S10). Meanwhile, it is noteworthy that the absence of SC leads to negligible OER currents and inferior enhancement effect (Figure S10 and Figure 3e), suggesting that the interaction between Fe3+spiked ions and SC surface is critical to the observed high activity.
As previously has been proposed, the surface amorphization of a perovskite oxide during oxygen evolution in alkaline solution is resulted from the dissolution of metal cations (both A-site and B-site cations) and the quick redeposition of almost insoluble B-site cations on the oxide surface.15−17 Accordingly, it is speculated that the Fe3+spiked ions accumulated in the vicinity of SC amorphous surface participate in the dissolution/redeposition cycle along with Co ions,23 yielding Co/Fe (oxy)hydroxide akin to the reconstructed surface layer of SCF02. This speculation is further supported by the almost unchanged OER activity of SCF02 cycling in Fe-spiked KOH (Figure S11).
2.4. Activity Comparison between SC, SC + Fe3+, and SCF02
To estimate the specific activity (is), the OER current is normalized to the oxide surface area as determined by BET measurement. As shown in Figure 4a, SC demonstrated a specific activity superior to that of rutile IrO 2 nanoparticles,41 in agreement with preceding reports that cubic SC perovskite is a promising OER catalyst.24,25 Incorporation of Fe either by bulk doping (SCF02) or surface decorating (SC + Fe3+) markedly enhanced the OER activity of SC, resulting in catalytic performance comparable to that of BSCF.12 Besides, Fe incorporation reduces the Tafel slope of SC from 75 to 60 mVdec–1 for SCF02 and 49 mVdec–1 for SC + Fe3+. The decrease in Tafel slope can be explained by that the presence of Fe sites, which have recently been recognized to be catalytically active for OER,21,22,42,43 varies the rate-determining step of OER on Co-based catalyst.44,45 As has been found for Co1–xFexOOH that the Tafel slope is sensitive to the Fe content,22 the small difference in the Tafel slope between SC + Fe3+ and SCF02 may be due to that the Fe content present in the surface of SC + Fe3+ is slightly different from that of SCF02 (Figure S12). Moreover, as reflected in the electrochemical impedance spectra (EIS) (inset of Figure 4a), the charge transfer resistance of SC was steeply decreased after Fe incorporation, indicative of an increase in charge transfer rate.6,46
Figure 4.

(a) Tafel plots for SC (100th cycle), SCF02 (100th cycle), and SC + Fe3+ (2nd cycle in Fe-spiked KOH) normalized to the oxide surface area. The benchmark catalysts, IrO241 and BSCF,12 are included for comparison. Error bars represent the standard deviation from three independent measurements. The inset shows the EIS of SC, SCF02, and SC + Fe3+ recorded at 1.6 V vs RHE. (b) Schematic illustration of the structure–activity relationship between SC, SC + Fe3+, and SCF02. The specific activity at 1.6 V vs RHE is used as the metric for activity comparison. Sr, Co, and Fe atoms are shown as green, blue, and gold balls, respectively. For simplicity, the residual Sr in the surface layers and oxygen atoms in the surface as well as the bulk are omitted.
As discussed in previous sections, the ordered surface structures of SC and SCF02 after 100 CV scans in 0.1 M KOH evolved into disordered (oxy)hydroxides rich in B-site cations, while their bulk phases remained crystalline. When amorphized SC undergoes further cycling in Fe-spiked KOH, Fe3+spiked ions in the electrolyte can be incorporated into the CoOxHy surface layer via involvement in the dissolution/redeposition process of hardly soluble TM cations,15−17 leading to (Co/Fe)OxHy resembling the amorphous surface of cycled SCF02. Therefore, the structure–activity relationship between SC, SC + Fe3+, and SCF02 is depicted by the schematic shown in Figure 4b. Merely with Fe ions decorated on the surface, SC + Fe3+ exhibited specific activity (∼3.4 mA cmoxide–2 at 1.6 V vs RHE) superior to that of SC (∼0.5 mA cmoxide at 1.6 V vs RHE), indicating that the OER catalysis is highly sensitive to the surface environment as opposed to the remaining bulk phase. This conclusion is further supported by the comparison between SC + Fe3+ and SCF02. Given their similarity in measured OER activity and surface (oxy)hydroxide layer, their remaining perovskites (SC and SCF02), which are different from each other in B-site composition and bulk electronic structure (Figure S13), have little difference in influencing the OER activity and thus are expected to play a negligible role. Based on the above discussion, new insights possibly need to be brought into the correlation ever found between the OER activity and the bulk properties of initial catalysts. For perovskite oxides undergoing reconstruction, the real active phase can be different from the as-synthesized one, and it is more reasonable to explore the links between the bulk chemistry of the precatalyst, the behaviors of surface evolution, and the observed catalytic performance.
3. Conclusions
In summary, both SC and SCF02 perovskites underwent surface amorphization during electrochemical cycling in 0.1 M KOH, along with the mass loss of Sr. With the spiking of 0.1 mM Fe3+ into the electrolyte, the apparent OER activity of SC was boosted due to the incorporation of Fe3+spiked ions into the amorphized surface of SC. This observation emphasizes the decisive role of the oxide–electrolyte interface in governing the catalytic performance. In addition, SC + Fe3+ and SCF02 catalytic systems, with similar surface Co/Fe (oxy)hydroxide layer and different bulk phase, exhibited comparable activities, further evidencing the little impact of the remaining perovskite on the OER activity. Our study highlights the importance of understanding factors controlling reconstruction behaviors for future catalyst design, as the in situ derived species account for the activity. The approach used in this study is also applicable for investigating the roles of reconstructed surface and remaining bulk for other oxide families such as spinel.
4. Experimental Section
Material Synthesis
SrCo1–xFexO3−δ (x = 0, 0.05, 0.2 and 0.5) perovskites were synthesized via a sol–gel method. In brief, stoichiometric amounts of strontium nitrate (Sr(NO3)2), cobalt nitrate (Co(NO3)2·6H2O) and iron nitrate (Fe(NO3)3·9H2O) were dissolved into a solution containing ethylenediaminetetraacetic acid (EDTA), ammonium hydroxide (NH4OH), and citric acid (C6H8O7). After being heated and continuously stirred at 260 °C for several hours, the solution became a homogeneous gel, which subsequently was heated at 200 °C in an oven to form a precursor. Next, the precursors of SrCo1–xFexO3−δ (x = 0.05, 0.2, and 0.5) were calcined at 1000 °C for 5 h in air. For the successful synthesis of SrCoO3−δ with cubic phase, its precursor underwent calcination at 1000 °C for 12 h and subsequently quenching in air. After that, the SrCoO3−δ sample was further annealed under an oxygen atmosphere at 300 °C for 96 h.47
Physical Characterization
XRD patterns of samples were collected using a Bruker D8 Advance XRD with Cu Kα radiation (λ = 1.5418 Å) at a scan rate of 2° min–1. The Rietveld refinement of XRD patterns was performed with the open-source software GSAS and EXPGUI interface.48 HRTEM was performed on a JEOL 2010F transmission electron microscope at 200 kV. SEM images of samples were recorded on a JEOL 6340F at 5 kV accelerating voltage. The BET specific surface areas of perovskite oxides were measured with ASAP Tristar II 3020. XPS spectra were collected on PHI-5400 equipment using Al Kα beam source (250 W) and position-sensitive detector. ICP-MS analysis was conducted on an Elan DRC-e ICP-MS.
Density Functional Theory Calculation
Density of states (DOSs) were calculated using the Vienna ab initio simulation package (VASP)49,50 with the projector-augmented wave (PAW) approach51 and the Perdew–Burk–Ernzerhof (PBE) exchange-correlation functional.52 To account for the strongly localized d-electrons, effective Hubbard U parameters of 3.3 and 4.0 eV were used for Co and Fe, respectively.53 Structural models for calculation were 2 × 2 × 2 cubic perovskite supercells. The Brillouin zone was sampled in a 6 × 6 × 6 mesh, and the energy cutoff was set to be 520 eV.
Electrode Preparation and Electrochemical Characterization
For the preparation of oxide electrodes, 5 mg of oxide catalysts, 2 mg of AB carbon, 750 μL of deionized water, 225 μL of isopropanol, and 25 μL of Nafion were mixed and ultrasonicated for 30 min to prepare the homogeneous ink of catalysts. Next, 10 μL of ink was drop-casted onto a GCE with the geometric surface area of 0.196 cm2, yielding a mass loading of 255 μgoxide cm–2disk. The preparation of pure carbon (AB) electrode followed the above procedures except that the oxide catalyst was excluded from the ink.
Electrochemical measurements were conducted in a three-electrode plastic cell on a Biologic SP150 potentiostat. A GCE, a platinum wire, and a saturated calomel reference electrode (SCE) were used as the working electrode, the counter electrode, and the reference electrode, respectively. The 0.1 M KOH electrolyte used for electrochemical measurements was prepared from KOH pellets (Sigma-Aldrich, 99.99%) and Milli-Q water (18.2 MΩ·cm) to avoid the interference from Fe impurity.31 According to ICP-MS analysis, the trace Fe in the 0.1 M KOH solution was less than 1 ppb. Magnetic stirring was used to remove bubbles from the working electrode surface and make the Fe3+spiked homogeneously dispersed in the electrolyte. The OER currents measured by CV at a scan rate of 10 mV s–1 were capacitance-corrected by averaging the forward and backward CV scans. The applied potentials were iR-corrected with the measured current i and the uncompensated ohmic electrolyte resistance R (∼45 ohm) determined by EIS measurements. All EIS measurements were recorded at 0.6 V (vs SCE) with frequencies ranging from 105 to 10–1 Hz and an AC voltage amplitude of 10 mV. All potentials were referenced to the reversible hydrogen electrode (RHE) scale (RHE = SCE + 0.245 + 0.059 × pH) unless otherwise stated.
Influence of Fe3+ Spiking on the pH of 0.1 M KOH Electrolyte
0.1 M aqueous Fe(NO3)3 solution was prepared from Fe(NO3)3·9H2O and Milli-Q water. For the preparation of 0.1 mM Fe3+-spiked KOH electrolyte, 0.05 mL of 0.1 M Fe(NO3)3 solution was spiked into 50 mL of 0.1 M KOH solution. The addition of aqueous solution changed the overall OH– concentration to be
| 1 |
Then, supposing that all of the Fe3+spiked ions were precipitated, the OH– concentration was reduced to be
| 2 |
Hence, the pH of 0.1 mM Fe3+-spiked KOH electrolyte was changed from pH 13 to be
| 3 |
Accordingly, the spiking of 0.1 mM (or less) Fe3+ into 0.1 M KOH solution results in a negligible variation of the pH value.
Even if the concentration of Fe3+ spiked into 0.1 M KOH solution is as high as 1 mM, the pH will be changed to 12.98245 (the calculation process follows the above steps). This minor variation in pH value (0.01755) will make the applied potential (Eapplied vs RHE) reduced by ∼1 mV, which is small enough to be not corrected for.
Acknowledgments
Authors thank the Facility for Analysis, Characterization, Testing and Simulation (FACTS) at the Nanyang Technological University for materials characterizations. This research is supported by the Singapore National Research Foundation under its Campus for Research Excellence and Technological Enterprise (CREATE) programme, through the Singapore Berkeley Research Initiative for Sustainable Energy (SinBeRISE), The Cambridge Center for Carbon Reduction in Chemical Technology (C4T), and eCO2EP programs. The work is partially supported by the Singapore Ministry of Education Tier 2 Grant (MOE2018-T2-2-027) and Tier 1 grant (2019-T1-002-125).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacsau.0c00022.
Additional data for physical characterization, including XPS, SEM, BET, XPS, and ICP-MS analysis, electrochemical tests, and DOS calculation (PDF)
Author Contributions
# H.L. and Y.C. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Jiao Y.; Zheng Y.; Jaroniec M.; Qiao S. Z. Design of Electrocatalysts for Oxygen- and Hydrogen-Involving Energy Conversion Reactions. Chem. Soc. Rev. 2015, 44, 2060–2086. 10.1039/C4CS00470A. [DOI] [PubMed] [Google Scholar]
- Suen N. T.; Hung S. F.; Quan Q.; Zhang N.; Xu Y. J.; Chen H. M. Electrocatalysis for the Oxygen Evolution Reaction: Recent Development and Future Perspectives. Chem. Soc. Rev. 2017, 46, 337–365. 10.1039/C6CS00328A. [DOI] [PubMed] [Google Scholar]
- Song J.; Wei C.; Huang Z. F.; Liu C.; Zeng L.; Wang X.; Xu Z. J. A Review on Fundamentals for Designing Oxygen Evolution Electrocatalysts. Chem. Soc. Rev. 2020, 49, 2196–2214. 10.1039/C9CS00607A. [DOI] [PubMed] [Google Scholar]
- Suntivich J.; May K. J.; Gasteiger H. A.; Goodenough J. B.; Shao-Horn Y. A Perovskite Oxide Optimized for Oxygen Evolution Catalysis from Molecular Orbital Principles. Science 2011, 334, 1383–1385. 10.1126/science.1212858. [DOI] [PubMed] [Google Scholar]
- Yagi S.; Yamada I.; Tsukasaki H.; Seno A.; Murakami M.; Fujii H.; Chen H.; Umezawa N.; Abe H.; Nishiyama N.; et al. Covalency-Reinforced Oxygen Evolution Reaction Catalyst. Nat. Commun. 2015, 6, 8249. 10.1038/ncomms9249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y.; Zhou W.; Chen Y.; Yu J.; Liu M.; Shao Z. A High-Performance Electrocatalyst for Oxygen Evolution Reaction: LiCo0.8Fe0.2O2. Adv. Mater. 2015, 27, 7150–7155. 10.1002/adma.201503532. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Dastafkan K.; Zhao C. Design Strategies for Non-Precious Metal Oxide Electrocatalysts for Oxygen Evolution Reactions. Curr. Opin. Electrochem. 2018, 10, 16–23. 10.1016/j.coelec.2018.03.015. [DOI] [Google Scholar]
- Wu T.; Sun S.; Song J.; Xi S.; Du Y.; Chen B.; Sasangka W. A.; Liao H.; Gan C. L.; Scherer G. G.; et al. Iron-Facilitated Dynamic Active-Site Generation on Spinel CoAl2O4 with Self-Termination of Surface Reconstruction for Water Oxidation. Nat. Catal. 2019, 2, 763–772. 10.1038/s41929-019-0325-4. [DOI] [Google Scholar]
- Chen R. R.; Sun Y.; Ong S. J. H.; Xi S.; Du Y.; Liu C.; Lev O.; Xu Z. J. Antiferromagnetic Inverse Spinel Oxide LiCoVO4 with Spin-Polarized Channels for Water Oxidation. Adv. Mater. 2020, 32, e1907976 10.1002/adma.201907976. [DOI] [PubMed] [Google Scholar]
- Ren X.; Wei C.; Sun Y.; Liu X.; Meng F.; Meng X.; Sun S.; Xi S.; Du Y.; Bi Z.; Shang G.; Fisher A. C.; Gu L.; Xu Z. J. Constructing an Adaptive Heterojunction as a Highly Active Catalyst for the Oxygen Evolution Reaction. Adv. Mater. 2020, 32, e2001292 10.1002/adma.202001292. [DOI] [PubMed] [Google Scholar]
- Sun Y.; Liao H.; Wang J.; Chen B.; Sun S.; Ong S. J. H.; Xi S.; Diao C.; Du Y.; Wang J.-O.; Breese M. B. H.; Li S.; Zhang H.; Xu Z. J. Covalency Competition Dominates the Water oxidation Structure–Activity Relationship on Spinel Oxides. Nat. Catal 2020, 3, 554–563. 10.1038/s41929-020-0465-6. [DOI] [Google Scholar]
- Grimaud A.; May K. J.; Carlton C. E.; Lee Y. L.; Risch M.; Hong W. T.; Zhou J.; Shao-Horn Y. Double Perovskites as a Family of Highly Active Catalysts for Oxygen Evolution in Alkaline Solution. Nat. Commun. 2013, 4, 2439. 10.1038/ncomms3439. [DOI] [PubMed] [Google Scholar]
- Zhu Y.; Zhou W.; Chen Z. G.; Chen Y.; Su C.; Tadé M. O.; Shao Z. SrNb0.1Co0. 7Fe0.2O3−δ Perovskite as a Next-Generation Electrocatalyst for Oxygen Evolution in Alkaline Solution. Angew. Chem., Int. Ed. 2015, 54, 3897–3901. 10.1002/anie.201408998. [DOI] [PubMed] [Google Scholar]
- May K. J.; Carlton C. E.; Stoerzinger K. A.; Risch M.; Suntivich J.; Lee Y.-L.; Grimaud A.; Shao-Horn Y. Influence of Oxygen Evolution During Water Oxidation on the Surface of Perovskite Oxide Catalysts. J. Phys. Chem. Lett. 2012, 3, 3264–3270. 10.1021/jz301414z. [DOI] [Google Scholar]
- Risch M.; Grimaud A.; May K. J.; Stoerzinger K. A.; Chen T. J.; Mansour A. N.; Shao-Horn Y. Structural Changes of Cobalt-Based Perovskites Upon Water Oxidation Investigated by EXAFS. J. Phys. Chem. C 2013, 117, 8628–8635. 10.1021/jp3126768. [DOI] [Google Scholar]
- Binninger T.; Mohamed R.; Waltar K.; Fabbri E.; Levecque P.; Kotz R.; Schmidt T. J. Thermodynamic Explanation of the Universal Correlation between Oxygen Evolution Activity and Corrosion of Oxide Catalysts. Sci. Rep. 2015, 5, 12167. 10.1038/srep12167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbri E.; Nachtegaal M.; Binninger T.; Cheng X.; Kim B. J.; Durst J.; Bozza F.; Graule T.; Schaublin R.; Wiles L.; et al. Dynamic Surface Self-Reconstruction Is the Key of Highly Active Perovskite Nano-Electrocatalysts for Water Splitting. Nat. Mater. 2017, 16, 925–931. 10.1038/nmat4938. [DOI] [PubMed] [Google Scholar]
- Tong Y.; Wu J.; Chen P.; Liu H.; Chu W.; Wu C.; Xie Y. Vibronic Superexchange in Double Perovskite Electrocatalyst for Efficient Electrocatalytic Oxygen Evolution. J. Am. Chem. Soc. 2018, 140, 11165–11169. 10.1021/jacs.8b06108. [DOI] [PubMed] [Google Scholar]
- Kim B. J.; Fabbri E.; Abbott D. F.; Cheng X.; Clark A. H.; Nachtegaal M.; Borlaf M.; Castelli I. E.; Graule T.; Schmidt T. J. Functional Role of Fe-Doping in Co-Based Perovskite Oxide Catalysts for Oxygen Evolution Reaction. J. Am. Chem. Soc. 2019, 141, 5231–5240. 10.1021/jacs.8b12101. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Li H.; Wang J.; Du Y.; Xi S.; Sun Y.; Sherburne M.; Ager J. W. 3rd; Fisher A. C.; Xu Z. J. Exceptionally Active Iridium Evolved from a Pseudo-Cubic Perovskite for Oxygen Evolution in Acid. Nat. Commun. 2019, 10, 572. 10.1038/s41467-019-08532-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trotochaud L.; Young S. L.; Ranney J. K.; Boettcher S. W. Nickel-Iron Oxyhydroxide Oxygen-Evolution Electrocatalysts: The Role of Intentional and Incidental Iron Incorporation. J. Am. Chem. Soc. 2014, 136, 6744–6753. 10.1021/ja502379c. [DOI] [PubMed] [Google Scholar]
- Burke M. S.; Kast M. G.; Trotochaud L.; Smith A. M.; Boettcher S. W. Cobalt-Iron (Oxy)hydroxide Oxygen Evolution Electrocatalysts: The Role of Structure and Composition on Activity, Stability, and Mechanism. J. Am. Chem. Soc. 2015, 137, 3638–3648. 10.1021/jacs.5b00281. [DOI] [PubMed] [Google Scholar]
- Chung D. Y.; Lopes P. P.; Farinazzo Bergamo Dias Martins P.; He H.; Kawaguchi T.; Zapol P.; You H.; Tripkovic D.; Strmcnik D.; Zhu Y.; et al. Dynamic Stability of Active Sites in Hydr(oxy)oxides for the Oxygen Evolution Reaction. Nat. Energy 2020, 5, 222–230. 10.1038/s41560-020-0576-y. [DOI] [Google Scholar]
- Man I. C.; Su H. Y.; Calle-Vallejo F.; Hansen H. A.; Martínez J. I.; Inoglu N. G.; Kitchin J.; Jaramillo T. F.; Nørskov J. K.; Rossmeisl J. Universality in Oxygen Evolution Electrocatalysis on Oxide Surfaces. ChemCatChem 2011, 3, 1159–1165. 10.1002/cctc.201000397. [DOI] [Google Scholar]
- Li X.; Wang H.; Cui Z.; Li Y.; Xin S.; Zhou J.; Long Y.; Jin C.; Goodenough J. B. Exceptional Oxygen Evolution Reactivities on CaCoO3 and SrCoO3. Sci. Adv. 2019, 5, eaav6262 10.1126/sciadv.aav6262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z. J. Transition Metal Oxides for Water Oxidation: All About Oxyhydroxides?. Sci. China Mater. 2020, 63, 3–7. 10.1007/s40843-019-9588-5. [DOI] [Google Scholar]
- Zhu Y.; Zhou W.; Sunarso J.; Zhong Y.; Shao Z. Phosphorus-Doped Perovskite Oxide as Highly Efficient Water Oxidation Electrocatalyst in Alkaline Solution. Adv. Funct. Mater. 2016, 26, 5862–5872. 10.1002/adfm.201601902. [DOI] [Google Scholar]
- Han B.; Grimaud A.; Giordano L.; Hong W. T.; Diaz-Morales O.; Yueh-Lin L.; Hwang J.; Charles N.; Stoerzinger K. A.; Yang W.; et al. Iron-Based Perovskites for Catalyzing Oxygen Evolution Reaction. J. Phys. Chem. C 2018, 122, 8445–8454. 10.1021/acs.jpcc.8b01397. [DOI] [Google Scholar]
- Wygant B. R.; Jarvis K. A.; Chemelewski W. D.; Mabayoje O.; Celio H.; Mullins C. B. Structural and Catalytic Effects of Iron- and Scandium-Doping on a Strontium Cobalt Oxide Electrocatalyst for Water Oxidation. ACS Catal. 2016, 6, 1122–1133. 10.1021/acscatal.5b02429. [DOI] [Google Scholar]
- Nagai T.; Ito W.; Sakon T. Relationship between Cation Substitution and Stability of Perovskite Structure in SrCoO3−δ -Based Mixed Conductors. Solid State Ionics 2007, 177, 3433–3444. 10.1016/j.ssi.2006.10.022. [DOI] [Google Scholar]
- Bak J.; Bin Bae H.; Chung S. Y. Atomic-Scale Perturbation of Oxygen Octahedra Via Surface Ion Exchange in Perovskite Nickelates Boosts Water Oxidation. Nat. Commun. 2019, 10, 2713. 10.1038/s41467-019-10838-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H. Y.; Hung S. F.; Chen H. Y.; Chan T. S.; Chen H. M.; Liu B. Operando Identification of Geometrical-Site-Dependent Water Oxidation Activity of Spinel Co3O4. J. Am. Chem. Soc. 2016, 138, 36–39. 10.1021/jacs.5b10525. [DOI] [PubMed] [Google Scholar]
- Van Der Heide P. A. W. Systematic X-Ray Photoelectron Spectroscopic Study of La1-xSrx-Based Perovskite-Type Oxides. Surf. Interface Anal. 2002, 33, 414–425. 10.1002/sia.1227. [DOI] [Google Scholar]
- Mutoro E.; Crumlin E. J.; Biegalski M. D.; Christen H. M.; Shao-Horn Y. Enhanced Oxygen Reduction Activity on Surface-Decorated Perovskite Thin Films for Solid Oxide Fuel Cells. Energy Environ. Sci. 2011, 4, 3689–3696. 10.1039/c1ee01245b. [DOI] [Google Scholar]
- Zhu Z.; Shi Y.; Aruta C.; Yang N. Improving Electronic Conductivity and Oxygen Reduction Activity in Sr-Doped Lanthanum Cobaltite Thin Films: Cobalt Valence State and Electronic Band Structure Effects. ACS Appl. Energy Mater. 2018, 1, 5308–5317. 10.1021/acsaem.8b00931. [DOI] [Google Scholar]
- Wang Y.; Ren J.; Wang Y.; Zhang F.; Liu X.; Guo Y.; Lu G. Nanocasted Synthesis of Mesoporous LaCoO3 Perovskite with Extremely High Surface Area and Excellent Activity in Methane Combustion. J. Phys. Chem. C 2008, 112, 15293–15298. 10.1021/jp8048394. [DOI] [Google Scholar]
- Zhu Y.; Zhou W.; Yu J.; Chen Y.; Liu M.; Shao Z. Enhancing Electrocatalytic Activity of Perovskite Oxides by Tuning Cation Deficiency for Oxygen Reduction and Evolution Reactions. Chem. Mater. 2016, 28, 1691–1697. 10.1021/acs.chemmater.5b04457. [DOI] [Google Scholar]
- Friedman A. K.; Shi W.; Losovyj Y.; Siedle A. R.; Baker L. A. Mapping Microscale Chemical Heterogeneity in Nafion Membranes with X-Ray Photoelectron Spectroscopy. J. Electrochem. Soc. 2018, 165, H733–H741. 10.1149/2.0771811jes. [DOI] [Google Scholar]
- Wei C.; Sun S.; Mandler D.; Wang X.; Qiao S. Z.; Xu Z. J. Approaches for Measuring the Surface Areas of Metal Oxide Electrocatalysts for Determining Their Intrinsic Electrocatalytic Activity. Chem. Soc. Rev. 2019, 48, 2518–2534. 10.1039/C8CS00848E. [DOI] [PubMed] [Google Scholar]
- Cornell R. M.; Schwertmann U.. The Iron Oxides: Structure, Properties, Reactions, Occurrences and Uses; John Wiley & Sons, 2003. [Google Scholar]
- Lee Y.; Suntivich J.; May K. J.; Perry E. E.; Shao-Horn Y. Synthesis and Activities of Rutile IrO2 and RuO2 Nanoparticles for Oxygen Evolution in Acid and Alkaline Solutions. J. Phys. Chem. Lett. 2012, 3, 399–404. 10.1021/jz2016507. [DOI] [PubMed] [Google Scholar]
- He Q.; Xie H.; Rehman Z. u.; Wang C.; Wan P.; Jiang H.; Chu W.; Song L. Highly Defective Fe-Based Oxyhydroxides from Electrochemical Reconstruction for Efficient Oxygen Evolution Catalysis. ACS Energy Lett. 2018, 3, 861–868. 10.1021/acsenergylett.8b00342. [DOI] [Google Scholar]
- Garlyyev B.; Liang Y.; Xue S.; Watzele S.; Fichtner J.; Li W.-J.; Ding X.; Bandarenka A. S. Theoretical and Experimental Identification of Active Electrocatalytic Surface Sites. Curr. Opin. Electrochem. 2019, 14, 206–213. 10.1016/j.coelec.2018.09.002. [DOI] [Google Scholar]
- Shinagawa T.; Garcia-Esparza A. T.; Takanabe K. Insight on Tafel Slopes from a Microkinetic Analysis of Aqueous Electrocatalysis for Energy Conversion. Sci. Rep. 2015, 5, 13801. 10.1038/srep13801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S.; Sun Y.; Zhou Y.; Shen J.; Mandler D.; Neumann R.; Xu Z. J. Switch of the Rate-Determining Step of Water Oxidation by Spin-Selected Electron Transfer in Spinel Oxides. Chem. Mater. 2019, 31, 8106–8111. 10.1021/acs.chemmater.9b02737. [DOI] [Google Scholar]
- Zhao B.; Zhang L.; Zhen D.; Yoo S.; Ding Y.; Chen D.; Chen Y.; Zhang Q.; Doyle B.; Xiong X.; et al. A Tailored Double Perovskite Nanofiber Catalyst Enables Ultrafast Oxygen Evolution. Nat. Commun. 2017, 8, 14586. 10.1038/ncomms14586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda Y.; Kanno R.; Takada T.; Yamamoto O.; Takano M.; Bando Y. Phase Relation and Oxygen-Non-Stoichiometry of Perovskite-Like Compound SrCoOx (2.29 < x > 2.80). Z. Anorg. Allg. Chem. 1986, 540, 259–270. 10.1002/zaac.19865400929. [DOI] [Google Scholar]
- Toby B. H. EXPGUI, A Graphical User Interface for GSAS. J. Appl. Crystallogr. 2001, 34, 210–213. 10.1107/S0021889801002242. [DOI] [Google Scholar]
- Kresse G.; Hafner J. Ab Initio Molecular Dynamics for Liquid Metals. Phys. Rev. B: Condens. Matter Mater. Phys. 1993, 47, 558–561. 10.1103/PhysRevB.47.558. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Furthmüller J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B: Condens. Matter Mater. Phys. 1996, 54, 11169–11186. 10.1103/PhysRevB.54.11169. [DOI] [PubMed] [Google Scholar]
- Blochl P. E. Projector Augmented-Wave Method. Phys. Rev. B: Condens. Matter Mater. Phys. 1994, 50, 17953–17979. 10.1103/PhysRevB.50.17953. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- Lee Y.-L.; Kleis J.; Rossmeisl J.; Shao-Horn Y.; Morgan D. Prediction of Solid Oxide Fuel Cell Cathode Activity with First-Principles Descriptors. Energy Environ. Sci. 2011, 4, 3966–3970. 10.1039/c1ee02032c. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.