

Abstract
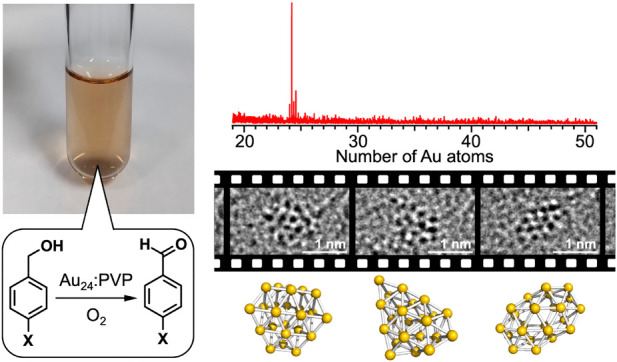
An unprecedented magic number cluster, Au24Clx (x = 0–3), was selectively synthesized by the kinetically controlled reduction of the Au precursor ions in a microfluidic mixer in the presence of a large excess of poly(N-vinyl-2-pyrrolidone) (PVP). The atomic structure of the PVP-stabilized Au24Clx was investigated by means of aberration-corrected transmission electron microscopy (ACTEM) and density functional theory (DFT) calculations. ACTEM video imaging revealed that the Au24Clx clusters were stable against dissociation but fluctuated during the observation period. Some of the high-resolution ACTEM snapshots were explained by DFT-optimized isomeric structures in which all the constituent atoms were located on the surface. This observation suggests that the featureless optical spectrum of Au24Clx is associated with the coexistence of distinctive isomers. X-ray photoelectron spectroscopy and Fourier-transform infrared spectroscopy of CO adsorbates revealed the electron-rich nature of Au24Clx clusters due to the interaction with PVP. The Au24Clx:PVP clusters catalyzed the aerobic oxidation of benzyl alcohol derivatives without degradation. Hammett analysis and the kinetic isotope effect indicated that the hydride elimination by Au24Clx was the rate-limiting step with an apparent activation energy of 56 ± 3 kJ/mol, whereas the oxygen pressure dependence of the reaction kinetics suggested the involvement of hydrogen abstraction by coadsorbed O2 as a faster process.
Keywords: magic number gold cluster, poly(N-vinyl-2-pyrrolidone), isomerization, aerobic alcohol oxidation, aberration-corrected transmission electron microscopy, density functional theory calculation
Introduction
Metal clusters with diameters smaller than the critical dimension of ∼2 nm are known to show unique chemical reactivities that cannot be predicted by a simple scaling law deduced from those of the extended surfaces and nanoparticles (NPs) of the corresponding metal.1−10 The emergence of the novel catalytic properties in metal clusters is ascribed to the quantized electronic structures, specific surface structures, and structural fluxionality. To understand the origin of novel catalysis and to optimize the catalytic performances of metal clusters, it is crucial to control their sizes with atomic precision. Atomically precise synthesis of Au clusters has been achieved using protecting ligands such as thiolates, alkynyls, halides, phosphines, and N-heterocyclic carbenes.2,3,11 Successful synthesis relies on size-focusing by chemical etching and subsequent isolation by chromatography and fractional precipitation. The main advantage of using atomically precise Au clusters for establishing the correlation between structures and catalytic performances is that their geometrical structures are determined by single-crystal X-ray diffraction (SCXRD).2,3 Nevertheless, these ligand-protected Au clusters are in general not suitable for catalytic applications because all the surface atoms of the metal core are masked by the ligands, although ligand-induced promotion of catalysis has been reported for some reactions.12
For the catalytic application of metal clusters, stabilization by linear polymers is suitable, since the cluster surface is inevitably exposed owing to the steric repulsion between the polymers.13−21 For example, it has been demonstrated that small (<3 nm) Au clusters stabilized by poly(N-vinyl-2-pyrrolidone) (PVP) show size-specific and high catalytic activities for oxidation and coupling reactions under aerobic conditions.22−26 The average diameter of the Au NPs can be tuned using conventional wet-chemical methods such as seed-mediated growth.27 It was reported that the catalytic activity of Au:PVP was monotonically enhanced with the decrease in the average diameter.22,24 However, postsynthetic size-focusing methods,2 size-selective fractionation,28 and SCXRD are not applicable because of the weak Au–polymer interactions and polydisperse structures of the polymer layers. Thus, major challenges for the catalytic studies of polymer-stabilized Au clusters are (1) atomically precise control of the cluster size and (2) determination of the atomic structure. The first challenge is tackled by controlling the formation kinetics of Au clusters in the presence of an excess amount of PVP using a microfluidic mixer,29 followed by matrix-assisted laser desorption/ionization (MALDI) mass spectrometry. Previous studies have shown that Au:PVP prepared by microfluidic mixing were a mixture of magic number Au clusters such as Au35±1, Au43±1, and Au58±1.30,31 Therefore, there is a possibility of producing atomically precise Au:PVP through further control of the formation kinetics. The second issue can be addressed by using aberration-corrected transmission electron microscopy (ACTEM). A three-dimensional (3D) atomic structure can be determined from the 2D projection image with the help of density functional theory (DFT) calculations.32,33 In addition, ACTEM video imaging with a high frame rate allows us to monitor the dynamic change in the structures at an atomic resolution.
In this study, we achieved atomically precise synthesis of an unprecedented magic number cluster, Au24Clx, stabilized by PVP through the kinetically controlled reduction of Au precursor ions in the presence of a large amount of PVP. ACTEM video imaging with the help of DFT calculations revealed the coexistence of structural isomers in which all the Au atoms constituted the cluster surface. Au 4f X-ray photoelectron spectroscopy (XPS) and Fourier-transform infrared (FT-IR) spectroscopy of adsorbed CO indicated that Au24Clx was negatively charged. Kinetic studies were conducted for the aerobic oxidation of benzyl alcohol derivatives catalyzed by Au24Clx. The clear kinetic isotope effect (KIE) and negative reaction constant in the Hammett plot indicated that the hydride elimination by Au24Clx was rate-limiting: the apparent activation energy was estimated to be 56 ± 3 kJ/mol by the Arrhenius equation. The reaction kinetics under pure oxygen (1 atm) suggested the involvement of direct hydrogen abstraction from adsorbed alkoxide by coadsorbed O2. We believe that this work will open up a new research paradigm for atomically precise nanocatalysts.
Results and Discussion
Atomically Precise Synthesis of Au24Clx
Three samples of Au:PVP (a–c) were obtained at the [Au]:[PVP] mixing ratios of 1:40, 1:100, and 1:200, respectively. The size distributions of a–c were evaluated by MALDI mass spectrometry, based on our previous observations that Au clusters could be desorbed in the naked anionic form from the PVP matrix as a result of the MALDI processes.30,31Figure 1 shows typical MALDI mass spectra of a–c in the negative ion mode recorded under minimal laser fluence. Sample a contained Au34Clx– (x = 0–4) and Au43Clx– (x = 0–4), whereas sample b was a mixture of Au24Clx– (x = 0–3), Au33Clx– (x = 0–2), and Au34Clx– (x = 0–3). Most notably, the mass spectrum of sample c is dominated by the progression of peaks assigned to Au24Clx– (x = 0–3). According to classical nucleation theory,34 formation of atomically monodisperse Au clusters in sample c is attributed to a kinetically controlled nucleation of Au atoms and a suppression of the subsequent growth. Such a condition was achieved by instantaneous and homogeneous reduction of all the AuCl4– precursors by mixing with the strong reducing agent (BH4–) in the micromixer. The trend in Figure 1 indicates that PVP also plays an important role in atomically precise synthesis by retarding the nucleation of Au atoms. The ligation by Cl originating from AuCl4– has been frequently observed for small Au clusters such as Au34Cln, Au43Cln, [Au11(PPh3)8Cl2]+, and Au55(PPh3)12Cl6.31,35,36 Elemental analysis of sample c determined the average number of the Cl ligands on Au24 to be 9.4, suggesting that the MALDI process is accompanied by dissociation of the Cl ligands. Such selective dissociation suggests that the Cl ligands are weakly bound to Au24 as electron withdrawing ligands as implied by weak magic behavior at x = 1 and 3 (Figure 1c). Noncontamination of sample c with Au NPs larger than 2 nm was confirmed by the following results: (1) absence of the localized surface plasmon resonance (LSPR) band at ∼520 nm in the optical absorption spectrum (Figure 2a); (2) observation of a single broad peak in the powder X-ray diffraction (PXRD) pattern (Figure 2b); (3) small coordination number (CN) of the Au–Au bond (4.2 ± 0.5) obtained by the curve-fitting analysis of the Au L3-edge extended X-ray absorption fine structure (EXAFS) (Figure 2c, Table S1); and (4) mean diameter of the Au clusters estimated to be 1.0 ± 0.2 nm by ACTEM (Figure 2d). We concluded from these results that single-sized Au24Clx was selectively formed in sample c. In the following, we focus on sample c, which is hereafter referred to as Au24:PVP.
Figure 1.
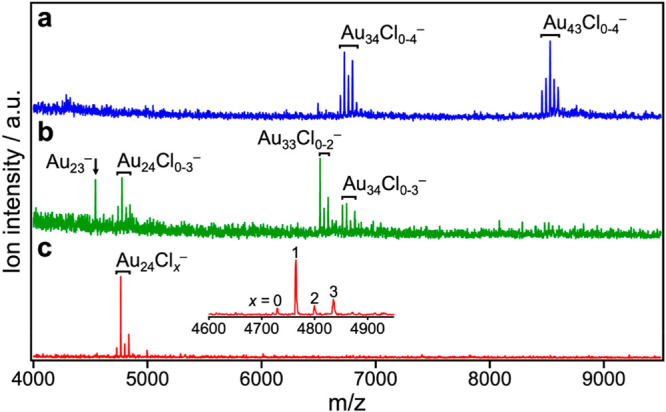
Negative-ion MALDI mass spectra of Au:PVP (a–c).
Figure 2.
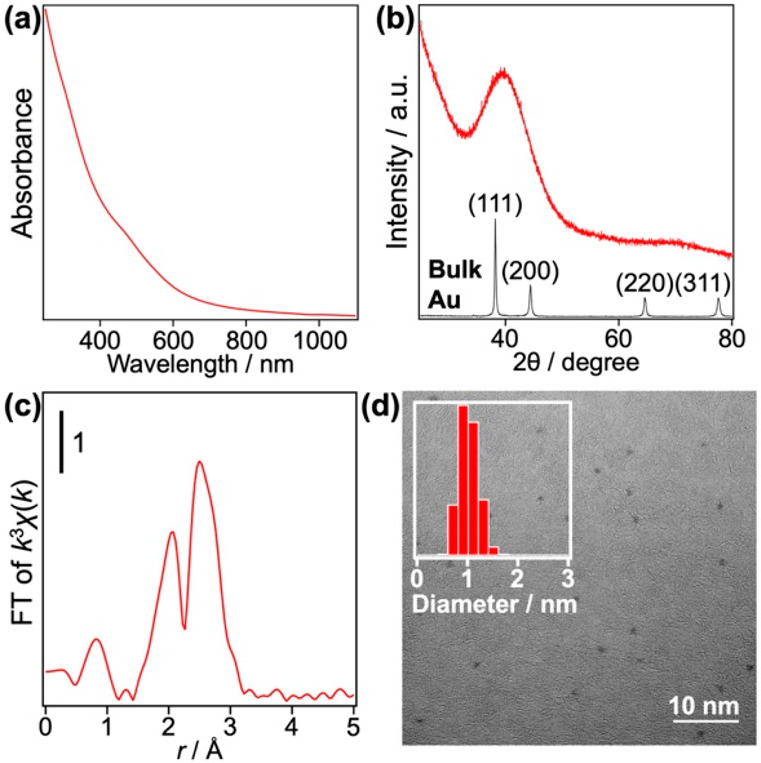
(a) Optical absorption spectrum in water, (b) PXRD pattern, (c) Au L3-edge FT-EXAFS spectrum, and (d) representative ACTEM image with low magnification of Au:PVP (sample c). Results of the curve fitting analysis of Au L3-edge EXAFS are summarized in Table S1 in the Supporting Information. The inset of panel (d) indicates the diameter distribution of 388 randomly chosen particles.
Geometrical Structure of Au24:PVP
It is well-known that the magic stability of naked Au clusters is governed by electronic shell closure based on a jellium model.37 The formation of Au34 and Au58 in PVP reported in our previous studies30,31 is explained by the same model. In contrast, Au24Clx in Au24:PVP corresponds to an unprecedented magic cluster. This result implies that the Au24Clx cluster does not have a spherical shape and does not provide a spherical potential to confine the valence electrons. To gain insight into the atomic structure of Au24Clx, we first conducted DFT calculations on Cl-free Au24 clusters. Figure 3 lists the DFT-optimized structures of Au24 (1–7) in the order of the relative stability. The frontier Kohn–Sham orbitals of 1–7 are depicted in Figure S1. The relative energies (ΔE) and energy gaps between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) are summarized in Table 1. Interestingly, all the constituent Au atoms of structures 1–6 are located on the surface: 1 and 3 have flat cage structures; 4 and 6 have empty tubular shapes; and 2 is constructed by attaching an Au4 unit to the well-known pyramidal Au20 magic cluster.38 In contrast, the least stable structure 7 contains two Au atoms inside the Au22 cage. The dimensions of 1–7 fall within the diameter distribution shown in Figure 2d. The projection of 4 or 6 along the longitudinal axis gave the smallest diameter (∼0.7 nm), whereas that of 1 or 3 from the flattened surface normal gave the largest diameter (∼1.3 nm) (Figure S3). This semiquantitative agreement supports the validity of the structural models in Figure 3. These results suggest that various disordered structures such as 1–6, in which all the constituent Au atoms constitute the cluster surface, are energetically accessible. Then, the effect of Cl ligation on the geometrical structures of Au24 was studied by using Au24Cl4 with 20 valence electrons as a model. Figure 4 lists the isomeric structures 1Cl–6Cl obtained by optimizing the initial structures in which four Cl ligands are bonded to low coordination sites of low energy isomers 1–6 in Figure 3, respectively. The frontier Kohn–Sham orbitals of 1Cl–6Cl are depicted in Figure S2. Cl ligation induced the slight distortion of the Au frameworks and as a result the relative stability of the isomers was altered. Nevertheless, it is safe to conclude that Au24Cl4 can take isomeric structures in which all the Au atoms are located on the surface, similarly to the naked Au24 clusters. The CNs and average lengths of the Au–Au bonds in 1–7 and 1Cl–6Cl are summarized in Table S2. The CN values of the Au–Au bonds in 1–7 (5.0–5.4) and 1Cl–6Cl (4.2–4.8) qualitatively agreed with that (4.2 ± 0.5) determined by Au L3-edge EXAFS analysis (Table S1), while the calculated Au–Au bond lengths of 1–7 (2.87–2.90 Å) and 1Cl–6Cl (2.85–2.87 Å) are longer than that experimentally determined (2.71 ± 0.01 Å). In the following discussion of the geometrical structures, we ignore the Cl ligands.
Figure 3.
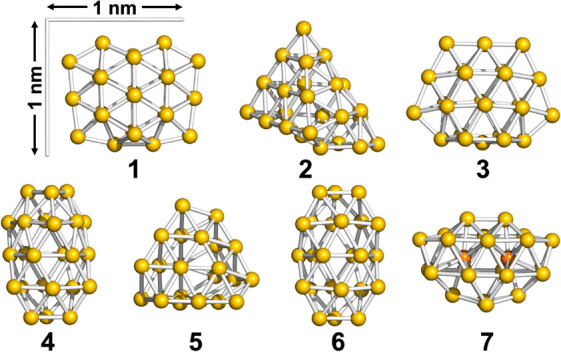
DFT-optimized model structures of Au24.
Table 1. Relative Energies and HOMO–LUMO Gaps of Model Structures of Au24 and Au24Cl4.
| Au24 |
Au24Cl4 |
||||
|---|---|---|---|---|---|
| model | ΔE/eVa | HLG/eVb | model | ΔE/eVa | HLG/eVb |
| 1 | 0 | 1.56 | 1Cl | 0.33 | 1.10 |
| 2 | 0.02 | 1.28 | 2Cl | 1.05 | 1.64 |
| 3 | 0.30 | 1.12 | 3Cl | 0.14 | 1.64 |
| 4 | 0.31 | 1.60 | 4Cl | 0 | 1.60 |
| 5 | 0.33 | 1.11 | 5Cl | 0.72 | 1.25 |
| 6 | 0.61 | 1.29 | 6Cl | 0.18 | 1.40 |
| 7 | 1.64 | 1.20 | – | ||
Relative energy.
HOMO–LUMO gap.
Figure 4.
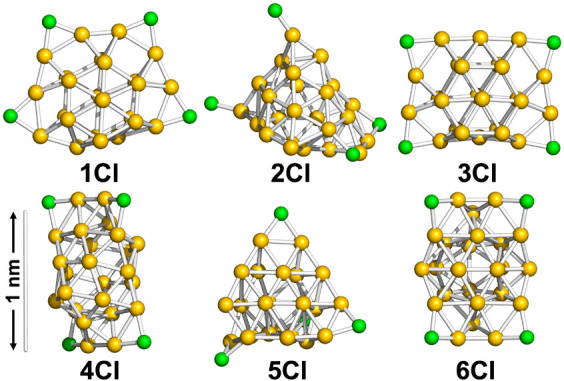
DFT-optimized model structures of Au24Cl4 (Au, yellow; Cl, green).
Theoretical prediction of energetically comparable isomers suggests that Au24 clusters in PVP are composed of distinct structural isomers and/or interconvert among isomers under ambient temperature. To address this interesting issue, atomic structures were probed by ACTEM video imaging at 0.04 s per frame. The ACTEM images of Au24:PVP did not show any signs of atom ejection during continuous observation for several minutes. Figure 5 shows representative ACTEM snapshots of different particles (#1–#4) of Au24:PVP. These images could not be reproduced by considering a single isomeric form in Figure 3 but could be by using different isomers 1, 2, 2, and 5, respectively. Other snapshots of different particles (#5–#7) in Figure S4 reasonably agree with the images simulated for isomers 4, 5, and 7, respectively. No images could be assigned to the icosahedral nor face-centered cubic structures observed in the selenolate (RSe)- or thiolate (RS)-protected gold clusters Au25(SeR)18 and Au144(SR)60.39 Furthermore, the images of a single particle (#8) of Au24:PVP fluctuated every 0.08–0.4 s during the observation period, as exemplified in Figure S5 and Movies S1–S3. Figure 6 shows snapshots of particle #8 taken from the images in Figure S5 at different times (t1–t4). Comparison with a set of images simulated by systematically changing the projection directions revealed that the fluctuation of the images was associated with structural isomerization rather than simple rotation of a single isomer: the images of Figure 6a–d are assigned to the distinct isomers 5, 2, 6, and 6 (Figure 6e–h), respectively. Thus, we concluded that the individual particles of Au24:PVP interconverted among structural isomers possibly due to migration and/or dissociation of the Cl ligands40 as well as thermal energy. The coexistence of isomers with hollow tubular structures has also been proposed for the naked cluster anions, Au24–, in the gas phase.41 At present, the selective formation of Au24:PVP could not be explained in terms of the electronic nor geometrical shell closure. We speculate that the cavity volume created by the multiple PVP chains determines the preferable and smallest size of Au clusters to be stabilized.
Figure 5.

Representative ACTEM snapshots of different particles of Au24:PVP (#1–#4) and simulated TEM images of model structures of Au24 in Figure 3. Scale bars correspond to 1 nm.
Figure 6.

(a–d) Representative ACTEM images of a single particle of Au24:PVP (#8) at different times from the beginning of the video recording (t1, 29.6 s; t2, 64.4 s; t3, 69.1 s; t4, 91.3 s) and (e–h) simulated images of model structures in Figure 3. Scale bars correspond to 1 nm.
Electronic Structure of Au24:PVP
Although Au24Clx was synthesized with atomic precision, the optical absorption spectrum of Au24:PVP in Figure 2a exhibits a featureless profile. This phenomenon was attributed to the coexistence of structural isomers given that the HOMO–LUMO gaps of 1–6 varied in the range of 1.1–1.6 eV depending on the atomic structures (Table 1). The electronic structure of Au24:PVP was further characterized by XPS and FT-IR spectroscopy of the adsorbed 12CO with the help of DFT calculations. The XP spectrum of Au24:PVP (Figure 7a) shows an Au 4f7/2 peak at 82.7 eV, which is red-shifted with respect to that of bulk Au (84.0 eV). This result indicates that Au24Clx is negatively charged due to the electron donation from PVP,24 although DFT calculations showed that the Cl ligands of Au24Cl4 withdrew the electronic charge from Au24: the natural charge of the Cl ligand was ∼ –0.5. It is known that the stretching frequency of the adsorbed CO reflects the electron density of the adsorption site on Au clusters.42−45 FT-IR spectra exhibited a broad peak of 12CO adsorbed on Au24:PVP at 2106 cm–1 in addition to that of free 12CO at 2136 cm–1 (Figure 7b). The redshift is ascribed to the π back-donation from the electron-rich Au sites of Au24Clx.45 To gain deeper insight into the geometrical and electronic structures of Au24:PVP, CO-adsorbed structures were theoretically studied for two isomers 1 and 4 with charge states of 0 and −1. DFT-optimized structures are illustrated in Figure S6: structural isomers 1COa0–1COh0 and 1COa––1COh– with different CO adsorption sites were obtained for neutral and anionic 1, respectively, whereas 4COa0–4COc0 and 4COa––4COc– were for neutral and anionic 4, respectively. The adsorption energy and stretching frequency of CO adsorbed on different sites of 1 and 4 are summarized in Tables S3 and S4, respectively. The binding energies are in the range of 0.1–0.4 eV regardless of the structures, charge states, and adsorption sites of Au24 clusters. The CO stretching frequencies on neutral 1 and 4 (green plots in Figure 7b) differ only slightly (<20 cm–1) depending on the adsorption sites and can explain the experimental result. On the other hand, the CO stretching frequencies on anionic 1 and 4 (blue plots in Figure 7b) are significantly red-shifted compared to those for neutral 1 and 4. Although these results could not identify the structure of the Au24Clx core and adsorption site of CO, they imply that the negative charge on the Au24Clx core is smaller than 1e as a result of a balance between the electron-withdrawing nature of Cl and the electron-donating nature of PVP.
Figure 7.
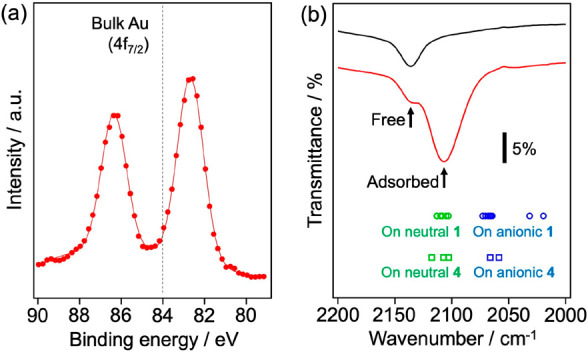
(a) XP spectrum of Au24:PVP. (b) FT-IR spectra of 12CO-saturated dichloromethane (black), 12CO-saturated colloidal dispersion of Au24:PVP (red), and DFT-calculated stretching frequencies of 12CO adsorbed on different adsorption sites of 1 (circle) or 4 (square) with a total charge of 0 (green) or −1 (blue). Details are shown in Tables S3, S4, and Figure S6.
Aerobic Oxidation Catalysis of Au24:PVP
Since Haruta’s discovery of CO oxidation catalyzed by Au NPs on metal oxide supports,46 the catalytic properties of nanosized Au have attracted considerable interest in nanoscience.47−50 In particular, alcohol oxidation is catalyzed efficiently by Au-based catalysts5,51,52 and has been studied as a model reaction to investigate the effects of structural parameters on the catalysis. For example, we have demonstrated that Au:PVP shows size-specific and high catalytic activities for aerobic alcohol oxidation.22,24 However, these studies were conducted using monodisperse Au clusters but with distributions in the number of constituent atoms. Au24:PVP synthesized here provides a novel opportunity to study atomistic details of the catalytic mechanism.
Kinetic measurements were conducted for the aerobic oxidation of p-substituted benzyl alcohol X-C6H4CH2OH (X = OCH3, CH3, H, CF3) catalyzed by Au24:PVP (Figure 8a). The selectivity of products between the aldehydes and carboxylic acids depended on the substituent group X (Figure S7). The pseudo-first-order rate constant was determined from the time course of the alcohol concentration (Figure S8). The effect of X on the rate constant was analyzed using the Hammett plot (Figure 8b). The negative reaction constant (ρ = −1.2 ± 0.2) indicates that the benzylic carbon has cationic characteristics at the transition state of the rate-limiting step. Furthermore, a large KIE (kH/kD = 4.1) was observed for α-deuterated benzyl alcohol (C6H5CD2OH). Thus, it was concluded that the C–H bond cleavage at the benzylic position was the rate-limiting step and that the H atom was abstracted as a hydride. The apparent activation energy for X = H was estimated to be 56 ± 3 kJ/mol from the Arrhenius plot (Figure 8c).
Figure 8.
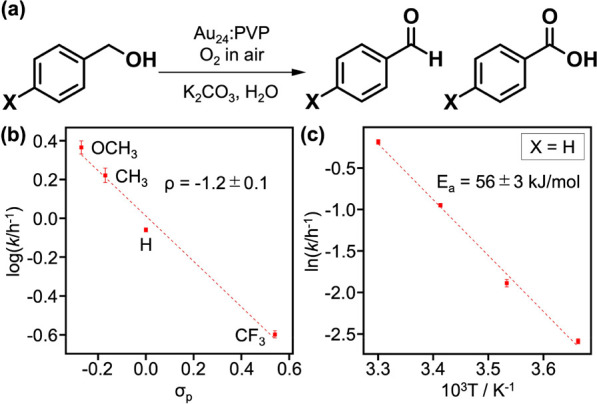
(a) Aerobic oxidation of p-substituted benzyl alcohol X–C6H4CH2OH (X = OCH3, CH3, H, CF3) catalyzed by Au24:PVP. (b) Hammett plot for the aerobic oxidation of p-substituted benzyl alcohol catalyzed by Au24:PVP dispersed in H2O.53 (c) Arrhenius plot for the benzyl alcohol oxidation (X = H). Reaction conditions: substrate 80 μmol; Au24:PVP 5 atom %; K2CO3 300 mol %; H2O 20 mL; air; 303 K (unless specified).
Importantly, MALDI-MS, optical spectroscopy, and Au L3-edge EXAFS confirmed that the cluster size of Au24:PVP was retained during the catalytic reactions under the present conditions (Figures S9–S19). In contrast, MALDI-MS and Au L3-edge EXAFS suggested the loss of Cl ligands during the reaction (Figures S9–S19). These results indicate that the catalysis is governed by Au24 and not affected by the Cl ligands. The negligible effect of the Cl ligands on the catalysis was supported by the fact that different batches of Au24:PVP having a different number of Cl ligands showed a similar activity (Figures S9 and S15). It was also reported that the Cl ligands of PVP-stabilized Au34Cln and Au43Cln were easily released during the reactions and did not prevent the catalytic alcohol oxidation.31
Two different mechanisms have been proposed for aerobic alcohol oxidation in which O2 plays different roles. We have proposed that O2 is reductively activated upon adsorption onto the negatively charged Au:PVP based on the correlation between the electron affinities of naked Aun and the reactivity to O2.24 In this mechanism, the resulting superoxo-like species abstracts hydrogen from the α-carbon of coadsorbed alkoxide (Scheme 1a).25 A similar mechanism has been proposed by Ebitani in the alcohol oxidation by hydrotalcite-supported metal (AuPd, AuPt or Pt) NPs costabilized by polymers such as PVP and starch.54−56 The other mechanism, proposed by Kobayashi, Chechik, and co-workers is based on the observation of an Au–H intermediate during the alcohol oxidation on Au NPs stabilized by styrene-based copolymers (Au:PS): hydride is directly transferred to the Au NP surface (Scheme 1b) and adsorbed hydrogen is removed by O2.57 This hydride elimination mechanism has been accepted for alcohol oxidation by Au NPs supported on metal oxides such as TiO2, CeO2, and Al2O3.58−63 A similar reaction mechanism has been proposed by Kaneda for alcohol oxidation catalyzed by hydroxyapatite-supported Pd clusters.64
Scheme 1. Previously Proposed Reaction Mechanisms of Alcohol Oxidation Catalyzed by (a) Au:PVP and (b) Au:PS.
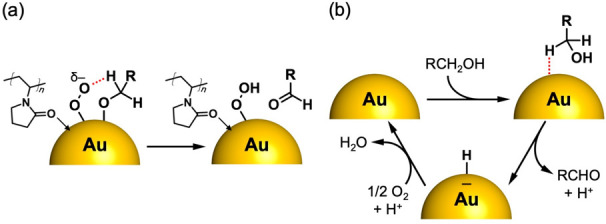
The reaction constant for Au24:PVP determined by the Hammett plot under air (Figure 8b, −1.2 ± 0.2) is comparable to those reported for Au NPs supported on metal oxides (−0.2 to −1.4).58,59,61,63 This fact also lends support for the hydride elimination mechanism by Au24 (Scheme 1b). To gain further insight into the mechanism by Au24:PVP, the effect of O2 pressure was investigated. The time course of the alcohol oxidation under 1 atm of O2 (Figure 9a) showed two characteristic behaviors: (1) for t > 0.05 h, the rate constant under pure O2 (1.18 h–1) was larger than that under air (O2: 0.21 atm, 0.87 h–1), although the rate constants were not proportional to the oxygen pressure; (2) the rate constant at the initial stage (t < 0.05 h) was apparently larger than that for t > 0.05 h. The former can be explained by the hydride elimination mechanism (Scheme 1b) with the assumption that the catalyst recovery by O2 is not much faster than the hydride elimination. However, it was difficult to rationalize the latter only by hydride elimination mechanism. We propose that hydrogen abstraction by O2 (Figure 9b) is involved in addition to the rate-limiting hydride elimination by Au24 (Figure 9c). Involvement of the two mechanisms was supported by a recent theoretical study on the oxidation of p-hydroxybenzyl alcohol by a model cluster Au38L3 (L = 2-pyrrolidone) reported by Okumura et al.65 It was concluded that the most energetically plausible catalytic cycle is composed of two consecutive steps: hydrogen abstraction by O2 (step 1) and hydride elimination by Au38 (step 2):
Figure 9.
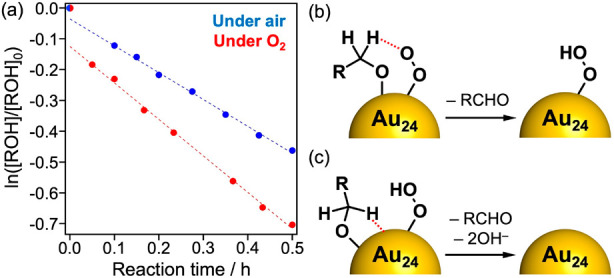
(a) Natural logarithm of benzyl alcohol concentration (normalized by initial concentration) during the catalytic oxidation reaction under air (blue, O2: 0.21 atm) or pure O2 (red, O2: 1 atm). Conditions other than O2 pressure were the same as those for Figure 8. Two alcohol oxidation pathways on Au24:PVP: (b) hydrogen abstraction by O2 and (c) hydride elimination by Au24.
Step 1: ArCH2O– + O2 + [Au38L3] → ArCHO + [(HOO)Au38L3]−
Step 2: ArCH2O– + [(HOO)Au38L3]− → ArCHO + 2OH– + [Au38L3]
The cooperation of two oxidation steps was ascribed to the order of the reactivities of oxidants: (O2 on Au38) > (Au38 surface) > (HOO on Au38).
Conclusion
In summary, a novel magic number cluster, Au24:PVP, was serendipitously and reproducibly obtained by homogenizing the formation process using a micromixer and limiting the growth step by the presence of a large amount of PVP. ACTEM video imaging revealed the polydispersity and fluxionality of the atomic structure of Au24:PVP. Noncrystalline structures with all constituent Au atoms on the surface are suggested by DFT calculations. We speculate that the observation of an unprecedented magic number is associated with the cavity size created by assemblies of PVP in which Au clusters can be confined. Au24:PVP catalyzed the aerobic oxidation of benzyl alcohol derivatives while retaining the size. Kinetics studies revealed that the hydride elimination from benzylic carbon by Au24 corresponds to the rate-limiting step with an apparent activation energy of 56 ± 3 kJ/mol. It was also suggested from the O2 pressure dependence on the reaction kinetics that the hydrogen abstraction by the adsorbed O2 is involved as well. We propose that the oxidation is initiated by the hydrogen abstraction by O2 adsorbed on Au24, followed by the rate-limiting hydride elimination by Au24.
Methods
Chemicals
All chemicals were commercially available and were used without further purification. Hydrogen tetrachloroaurate(III) tetrahydrate, sodium borohydride, PVP (K30, average molecular weight: ∼40 000), benzyl alcohol, p-methylbenzyl alcohol, p-methoxybenzyl alcohol, and potassium carbonate were purchased from Wako Pure Chemical Industries. trans-2-[3-(4-tert-Butylphenyl)-2-methyl-2-propenylidene]malononitrile (DCTB) and p-(trifluoromethyl)benzyl alcohol were purchased from Tokyo Chemical Industry. Deuterated benzyl alcohol (C6H5CD2OH) was purchased from Sigma-Aldrich. Deionized water used was Milli-Q grade (>18 MΩ cm).
Synthesis
Au clusters were synthesized by microfluidic mixing of two aqueous solutions of Au precursors and a reducing agent as reported previously.29−31 The ice-cooled aqueous solution of HAuCl4 and PVP was mixed with that of NaBH4 and PVP at a flow rate of 200 mL/h in a micromixer (SIMM-V2, IMM GmbH) placed in an ice bath. Three samples of Au:PVP (a–c) were obtained by tuning the [Au]:[PVP] mixing ratios as listed in Table 2. The resulting brown solution was collected in an Erlenmeyer flask, stirred for 1 h at 273 K, and then deionized four times at 273 K by using a centrifugal ultrafiltration concentrator (Vivaspin20, Vivascience) having a membrane filter with a cutoff molecular weight of 10 kDa (for a) or 100 kDa (for b and c). The deionized solution was lyophilized to obtain Au:PVP in a powder form. The yields of a–c were 79, 73, and 79%, respectively, based on the amount of Au. Optical absorption spectroscopy and thermogravimetry showed that the amount of PVP was reduced to ∼40 equiv (monomer unit to Au atom) by ultrafiltration. PVP was added to adjust the equivalence to 50 in the catalytic tests.
Table 2. Conditions of Synthesizing Au:PVP.
| Au precursor |
reducing agent |
||||
|---|---|---|---|---|---|
| sample | [HAuCl4]/mM | [PVP]/mMa | [NaBH4]/mM | [PVP]/mMa | [Au]:[NaBH4]:[PVP]a |
| a | 10 | 200 | 50 | 200 | 1:5:40 |
| b | 10 | 500 | 50 | 500 | 1:5:100 |
| c | 10 | 1000 | 50 | 1000 | 1:5:200 |
Concentrations of monomer unit.
Characterization
MALDI mass spectra were recorded using a time-of-flight mass spectrometer (AXIMA-CFR, Shimadzu): DCTB was used as a matrix. The specimens for MALDI-MS were prepared by drop-casting a methanol dispersion of Au:PVP and DCTB ([Au]:[DCTB] = 1:18) onto a sample plate. Optical absorption spectra were recorded by using a spectrophotometer (V-670, JASCO). The PXRD pattern was obtained using a diffractometer with CuKα radiation (SmartLab 3, Rigaku). ACTEM was carried out on a JEM-ARM200F instrument (JEOL) at an acceleration voltage of 80 kV, under 1 × 10–5 Pa at 298 K in the specimen column. A series of ACTEM images were continuously recorded at 25 frames per second (fps) on a CMOS camera (Gatan OneView with in situ option, output image size: 2,048 × 2,048 pixels operated in the binning 2 mode, pixel resolution: 0.01 nm at ×2 000 000 magnification). We adjusted the spherical aberration value, electron dose rate, and defocus value to 1–3 μm, 2.7–5.6 × 106 e– nm–2 s–1, approximately −4 nm (underfocus), respectively. Acquired images were filtered by a bandpass filter using ImageJ software;66 structures larger than 40 pixels or smaller than 3 pixels were filtered with the tolerance of direction of 5%. The specimens for ACTEM were prepared by drop-casting a methanol dispersion of Au:PVP onto a thin carbon-coated copper grid (SHR-C075, Okenshoji). The average diameter was determined by measuring the diameters of 388 particles in low magnification (×800 000) images. XPS was carried out using a PHI-5000 VersaProbe (ULVAC-PHI) instrument at an energy resolution of 0.2 eV. Electron binding energy was calibrated referring to the Au 4f7/2 peak of bulk Au (84.0 eV). FT-IR spectra of carbon monoxide adsorbed on Au:PVP were measured by using a spectrophotometer (FTIR-4200, Jasco) in the transmission mode at a resolution of 2 cm–1. For FT-IR measurements, Au:PVP (Au 15 μmol) was dispersed in dichloromethane (3 mL) under CO (1.0 atm) and the colloidal dispersion was introduced into the IR cell (CaF2 window, 0.5 mm path length) after the stirring for 50 min. Au L3-edge X-ray absorption spectroscopy (XAS) was conducted in the transmission mode using the BL01B1 beamline at SPring-8 of the Japan Synchrotron Radiation Research Institute (JASRI). The incident X-ray beam was monochromatized by a Si (111) double crystal monochromator. The spectral data were analyzed by using the REX2000 program (Rigaku). The k3-weighted χ spectra in the k range of 3–16.5 Å–1 were Fourier-transformed into the r space. The Au–Au and Au–Cl bonds were considered in the curve-fitting analysis over the r range of 1.7–3.0 Å.
Calculations and Simulations
DFT calculations of the model structures of Au24, Au24Cl4, and Au24(CO)1 were performed using the Gaussian 09 program.67 B3LYP was used as a functional.68,69 A double-ζ basis set with scalar relativistic effective core potential (ECP), LANL2DZ, was applied for Au and the 6-31+G(d) basis set was used for C, O, and Cl. Structures of Au24 were obtained by reoptimization of those reported previously.70,71 PVP was not included in the calculations since the impact on the atomic structure of Au clusters is negligibly small.72 Neutral clusters (Au24, Au24(CO)1, and Au24Cl4) were optimized with the singlet spin state, whereas the doublet spin state was applied for the anionic cluster, Au24(CO)1–. The vibrational frequencies were computed for the optimized structures to ensure that they corresponded to the local minima of the potential energy surface. The relative energy of the clusters presented in this study includes the zero-point energy. For the stretching frequencies of CO adsorbed on Au24, a scaling factor of 0.970 was applied so that the calculated frequency of isolated CO (2203 cm–1) reproduced that of free CO in dichloromethane (2136 cm–1).
TEM simulation images of DFT-optimized structures were generated by using a multislice procedure implemented in a BioNet elbis software:73 DFT-optimized structures were converted to a series of simulated TEM images with the observation direction varying every 10° around the x and y axes, producing 18 × 36 = 648 images.74 A defocus value of −4 nm and a spherical aberration coefficient (Cs) of −3 μm were applied for the simulation. To quantify the similarity of the experimental and simulated images, a cross-correlation analysis was conducted.75,76 The cross-correlation function of two images (a and b), γ(a, b), is defined as follows:
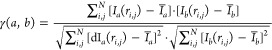 |
where I, ri,j, and I̅ represent the intensity of the image, pixel position, and mean intensity, respectively. The value of this function is an index of the similarity of images; γ of 1 or 0 corresponds to a perfect match or no match, respectively. Therefore, the cross-correlation function of the experimental and simulated images was calculated for the efficient screening of the large number of simulated images. Candidate images obtained by the screening were then carefully compared to the experimental images.
Catalytic Test
Catalytic properties of Au:PVP for aerobic oxidation of benzyl alcohol derivatives in water were studied by using a temperature-controlled personal organic synthesizer, PPS-2510 (Eyela). The catalytic reaction was initiated by adding the aqueous dispersion (8 mL) of Au:PVP (4 μmol) to the aqueous solution (12 mL) of alcohol (80 μmol) and K2CO3 (240 μmol) magnetically stirred at 800 rpm. Aliquots (5 mL) were sampled at the given time and hydrochloric acid (HCl 600 μmol in 150 μL) was added to quench the reaction. Reaction products were extracted by ethyl acetate (1 mL) three times. The extract thus collected was dried over Na2SO4 and analyzed by a gas chromatograph (GC-2014, Shimadzu) with a flame ionization detector. Rate constants for the oxidation reaction were obtained by linear fitting of the natural logarithm of the alcohol concentration using ≥7 data points at which the conversion was typically between 10% and 40%. Since the reaction proceeds faster in the initial stage (conv. < 10%), the zero point was excluded for the linear fitting.
Acknowledgments
The authors thank Mr. Ko Kamei (The University of Tokyo) for supporting TEM simulation. This research was financially supported by JST CREST Grant Number JPMJCR20B2, the Elements Strategy Initiative for Catalysts & Batteries (ESICB) (Grant No. JPMXP0112101003) and the Nanotechnology Platform (Project No. 12024046) of MEXT, a Grant-in-Aid for Scientific Research (A) (No. 20H00370), and a JSPS Research Fellow Grant (No. JP19J22154) from JSPS. The synchrotron radiation experiments were performed under the approval of JASRI (Proposal No. 2020A0672). Calculations were partly performed using the Research Center for Computational Science, Okazaki, Japan.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacsau.1c00102.
Supplementary tables and figures for Au L3-edge XAS, DFT calculations, ACTEM, and catalytic tests (PDF)
Movie S1: ACTEM video of a single particle (#8) of Au24:PVP at 24.04–36.00 s (AVI)
Movie S2: ACTEM video of a single particle (#8) of Au24:PVP at 60.04–72.00 s (AVI)
Movie S3: ACTEM video of a single particle (#8) of Au24:PVP at 84.04–96.00 s (AVI)
The authors declare no competing financial interest.
Supplementary Material
References
- Yamazoe S.; Koyasu K.; Tsukuda T. Nonscalable Oxidation Catalysis of Gold Clusters. Acc. Chem. Res. 2014, 47, 816–824. 10.1021/ar400209a. [DOI] [PubMed] [Google Scholar]
- Jin R.; Zeng C.; Zhou M.; Chen Y. Atomically Precise Colloidal Metal Nanoclusters and Nanoparticles: Fundamentals and Opportunities. Chem. Rev. 2016, 116, 10346–10413. 10.1021/acs.chemrev.5b00703. [DOI] [PubMed] [Google Scholar]
- Chakraborty I.; Pradeep T. Atomically Precise Clusters of Noble Metals: Emerging Link between Atoms and Nanoparticles. Chem. Rev. 2017, 117, 8208–8271. 10.1021/acs.chemrev.6b00769. [DOI] [PubMed] [Google Scholar]
- Ye R.; Zhukhovitskiy A. V.; Deraedt C. V.; Toste F. D.; Somorjai G. A. Supported Dendrimer-Encapsulated Metal Clusters: Toward Heterogenizing Homogeneous Catalysts. Acc. Chem. Res. 2017, 50, 1894–1901. 10.1021/acs.accounts.7b00232. [DOI] [PubMed] [Google Scholar]
- Liu L.; Corma A. Metal Catalysts for Heterogeneous Catalysis: From Single Atoms to Nanoclusters and Nanoparticles. Chem. Rev. 2018, 118, 4981–5079. 10.1021/acs.chemrev.7b00776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y.; Sheng H.; Astruc D.; Zhu M. Atomically Precise Noble Metal Nanoclusters as an Efficient Catalysts: A Bridge between Structure and Properties. Chem. Rev. 2020, 120, 526–622. 10.1021/acs.chemrev.8b00726. [DOI] [PubMed] [Google Scholar]
- Li Z.; Ji S.; Liu Y.; Cao X.; Tian S.; Chen Y.; Niu Z.; Li Y. Well-Defined Materials for Heterogeneous Catalysis: From Nanoparticles to Isolated Single-Atom Sites. Chem. Rev. 2020, 120, 623–682. 10.1021/acs.chemrev.9b00311. [DOI] [PubMed] [Google Scholar]
- Yamamoto K.; Imaoka T.; Tanabe M.; Kambe T. New Horizon of Nanoparticle and Cluster Catalysis with Dendrimers. Chem. Rev. 2020, 120, 1397–1437. 10.1021/acs.chemrev.9b00188. [DOI] [PubMed] [Google Scholar]
- Kawawaki T.; Negishi Y.; Kawasaki H. Photo/Electrocatalysis and Photosensitization Using Metal Nanoclusters for Green Energy and Medical Applications. Nanoscale Adv. 2020, 2, 17–36. 10.1039/C9NA00583H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin R.; Li G.; Sharma S.; Li Y.; Du X. Toward Active-Site Tailoring in Heterogeneous Catalysis by Atomically Precise Metal Nanoclusters with Crystallographic Structures. Chem. Rev. 2021, 121, 567–648. 10.1021/acs.chemrev.0c00495. [DOI] [PubMed] [Google Scholar]
- Omoda T.; Takano S.; Tsukuda T. Toward Controlling the Electronic Structures of Chemically Modified Superatoms of Gold and Silver. Small 2020, 2001439. 10.1002/smll.202001439. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Wan X.-K.; Ren L.; Su H.; Li G.; Malola S.; Lin S.; Tang Z.; Häkkinen H.; Teo B. K.; Wang Q. M.; Zheng N. Atomically Precise Alkynyl-Protected Metal Nanoclusters as a Model Catalyst: Observation of Promoting Effect of Surface Ligands on Catalysis by Metal Nanoparticles. J. Am. Chem. Soc. 2016, 138, 3278–3281. 10.1021/jacs.5b12730. [DOI] [PubMed] [Google Scholar]
- Narayanan R.; El-Sayed M. A. Effect of Catalysis on the Stability of Metallic Nanoparticles: Suzuki Reaction Catalyzed by PVP-Palladium Nanoparticles. J. Am. Chem. Soc. 2003, 125, 8340–8347. 10.1021/ja035044x. [DOI] [PubMed] [Google Scholar]
- Grass M. E.; Zhang W.; Butcher D. R.; Park J. Y.; Li Y.; Bluhm H.; Bratlie K. M.; Zhang T.; Somorjai G. A. A Reactive Oxide Overlayer on Rhodium Nanoparticles during CO Oxidation and Its Size Dependence Studied by in situ Ambient-Pressure X-ray Photoelectron Spectroscopy. Angew. Chem., Int. Ed. 2008, 47, 8893–8896. 10.1002/anie.200803574. [DOI] [PubMed] [Google Scholar]
- Santiago González B.; Rodríguez M. J.; Blanco C.; Rivas J.; López-Quintela M. A.; Martinho J. M. G. One Step Synthesis of the Smallest Photoluminescent and Paramagnetic PVP-Protected Gold Atomic Clusters. Nano Lett. 2010, 10, 4217–4421. 10.1021/nl1026716. [DOI] [PubMed] [Google Scholar]
- Balcha T.; Strobl J. R.; Fowler C.; Dash P.; Scott R. W. J. Selective Aerobic Oxidation of Crotyl Alcohol Using AuPd Core-Shell Nanoparticles. ACS Catal. 2011, 1, 425–436. 10.1021/cs200040a. [DOI] [Google Scholar]
- Zhang H.; Watanabe T.; Okumura M.; Haruta M.; Toshima N. Catalytically Highly Active Top Gold Atom on Palladium Nanocluster. Nat. Mater. 2012, 11, 49–52. 10.1038/nmat3143. [DOI] [PubMed] [Google Scholar]
- Yuan Y.; Yan N.; Dyson P. J. Advances in the Rational Design of Rhodium Nanoparticle Catalysts: Control via Manipulation of the Nanoparticle Core and Stabilizer. ACS Catal. 2012, 2, 1057–1069. 10.1021/cs300142u. [DOI] [Google Scholar]
- Kusada K.; Kitagawa H. A Route for Phase Control in Metal Nanoparticles: A Potential Strategy to Create Advanced Materials. Adv. Mater. 2016, 28, 1129–1142. 10.1002/adma.201502881. [DOI] [PubMed] [Google Scholar]
- Axet M. R.; Philippot K. Catalysis with Colloidal Ruthenium Nanoparticles. Chem. Rev. 2020, 120, 1085–1145. 10.1021/acs.chemrev.9b00434. [DOI] [PubMed] [Google Scholar]
- Hasegawa S.; Tsukuda T. Exploring Novel Catalysis Using Polymer-Stabilized Metal Clusters. Bull. Chem. Soc. Jpn. 2021, 94, 1036–1044. 10.1246/bcsj.20200377. [DOI] [Google Scholar]
- Tsunoyama H.; Sakurai H.; Negishi Y.; Tsukuda T. Size-Specific Catalytic Activity of Polymer-Stabilized Gold Nanoclusters for Aerobic Alcohol Oxidation in Water. J. Am. Chem. Soc. 2005, 127, 9374–9375. 10.1021/ja052161e. [DOI] [PubMed] [Google Scholar]
- Sakurai H.; Tsunoyama H.; Tsukuda T. Oxidative Homo-Coupling of Potassium Aryltrifluoroborates Catalyzed by Gold Nanocluster under Aerobic Conditions. J. Organomet. Chem. 2007, 692, 368–374. 10.1016/j.jorganchem.2006.04.054. [DOI] [Google Scholar]
- Tsunoyama H.; Ichikuni N.; Sakurai H.; Tsukuda T. Effect of Electronic Structures of Au Clusters Stabilized by Poly(N-vinyl-2-pyrrolidone) on Aerobic Oxidation Catalysis. J. Am. Chem. Soc. 2009, 131, 7086–7093. 10.1021/ja810045y. [DOI] [PubMed] [Google Scholar]
- Tsukuda T.; Tsunoyama H.; Sakurai H. Aerobic Oxidations Catalyzed by Colloidal Nanogold. Chem. - Asian J. 2011, 6, 736–748. 10.1002/asia.201000611. [DOI] [PubMed] [Google Scholar]
- Haesuwannakij S.; Kimura T.; Furutani Y.; Okumura K.; Kokubo K.; Sakata T.; Yasuda H.; Yakiyama Y.; Sakurai H. The Impact of the Polymer Chain Length on the Catalytic Activity of Poly(N-vinyl-2-pyrrolidone)-Supported Gold Nanoclusters. Sci. Rep. 2017, 7, 9579. 10.1038/s41598-017-10165-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunoyama H.; Sakurai H.; Tsukuda T. Size Effect on the Catalysis of Gold Clusters Dispersed in Water for Aerobic Oxidation of Alcohol. Chem. Phys. Lett. 2006, 429, 528–532. 10.1016/j.cplett.2006.08.066. [DOI] [Google Scholar]
- Niihori Y.; Yoshida K.; Hossain S.; Kurashige W.; Negishi Y. Deepening the Understanding of Thiolate-Protected Metal Clusters Using High-Performance Liquid Chromatography. Bull. Chem. Soc. Jpn. 2019, 92, 664–695. 10.1246/bcsj.20180357. [DOI] [Google Scholar]
- Tsunoyama H.; Ichikuni N.; Tsukuda T. Microfluidic Synthesis and Catalytic Application of PVP-Stabilized ∼ 1 nm Gold Clusters. Langmuir 2008, 24, 11327–11330. 10.1021/la801372j. [DOI] [PubMed] [Google Scholar]
- Tsunoyama H.; Tsukuda T. Magic Numbers of Gold Clusters Stabilized by PVP. J. Am. Chem. Soc. 2009, 131, 18216–18217. 10.1021/ja908188f. [DOI] [PubMed] [Google Scholar]
- Ishida R.; Arii S.; Kurashige W.; Yamazoe S.; Koyasu K.; Negishi Y.; Tsukuda T. Halogen Adsorbates on Polymer-Stabilized Gold Clusters: Mass Spectrometric Detection and Effects on Catalysis. Chin. J. Catal. 2016, 37, 1656–1661. 10.1016/S1872-2067(16)62501-9. [DOI] [Google Scholar]
- Bals S.; Van Aert S.; Romero C.P.; Lauwaet K.; Van Bael M.J.; Schoeters B.; Partoens B.; Yucelen E.; Lievens P.; Van Tendeloo G. Atomic Scale Dynamics of Ultrasmall Germanium Clusters. Nat. Commun. 2012, 3, 897. 10.1038/ncomms1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambie S. G.; Weal G. R.; Blackmore C. E.; Palmer R. E.; Garden A. L. Contrasting Motif Preferences of Platinum and Gold Nanoclusters between 55 and 309 atoms. Nanoscale Adv. 2019, 1, 2416–2425. 10.1039/C9NA00122K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Mer V. K. Nucleation in Phase Transitions. Ind. Eng. Chem. 1952, 44, 1270–1277. 10.1021/ie50510a027. [DOI] [Google Scholar]
- Schmid G. The Relevance of Shape and Size of Au55 Clusters. Chem. Soc. Rev. 2008, 37, 1909–1930. 10.1039/b713631p. [DOI] [PubMed] [Google Scholar]
- McKenzie L. C.; Zaikova T. O.; Hutchison J. E. Structurally Similar Triphenylphosphine-Stabilized Undecagolds, Au11(PPh3)7Cl3 and [Au11(PPh3)8Cl2]Cl, Exhibit Distinct Ligand Exchange Pathways with Glutathione. J. Am. Chem. Soc. 2014, 136, 13426–13435. 10.1021/ja5075689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Heer W. A. The Physics of Simple Metal Clusters: Experimental Aspects and Simple Models. Rev. Mod. Phys. 1993, 65, 611–676. 10.1103/RevModPhys.65.611. [DOI] [Google Scholar]
- Li J.; Li X.; Zhai H.-J.; Wang L. S. Au20: A Tetrahedral Cluster. Science 2003, 299, 864–867. 10.1126/science.1079879. [DOI] [PubMed] [Google Scholar]
- Takahata R.; Yamazoe S.; Maehara Y.; Yamazaki K.; Takano S.; Kurashige W.; Negishi Y.; Gohara K.; Tsukuda T. Electron Microscopic Observation of an Icosahedral Au13 Core in Au25(SePh)18 and Reversible Isomerization between Icosahedral and Face-Centered Cubic Cores in Au144(SC2H4Ph)60. J. Phys. Chem. C 2020, 124, 6907–6912. 10.1021/acs.jpcc.9b11412. [DOI] [Google Scholar]
- Kang X.; Zhu M. Structural Isomerism in Atomically Precise Nanoclusters. Chem. Mater. 2021, 33, 39–62. 10.1021/acs.chemmater.0c03979. [DOI] [Google Scholar]
- Khetrapal N. S.; Bulusu S. S.; Zeng X. C. Structural Evolution of Gold Clusters Aun– (n = 21–25) Revisited. J. Phys. Chem. A 2017, 121, 2466–2474. 10.1021/acs.jpca.7b00367. [DOI] [PubMed] [Google Scholar]
- Yoon B.; Häkkinen H.; Landman U.; Wörz A. S.; Antonietti J.-M.; Abbet S.; Judai K.; Heiz U. Charging Effects on Bonding and Catalyzed Oxidation of CO on Au8 Clusters on MgO. Science 2005, 307, 403–407. 10.1126/science.1104168. [DOI] [PubMed] [Google Scholar]
- Sterrer M.; Yulikov M.; Fischbach E.; Heyde M.; Rust H.-P.; Pacchioni G.; Risse T.; Freund H.-J. Interaction of Gold Clusters with Color Centers on MgO(001) Films. Angew. Chem., Int. Ed. 2006, 45, 2630–2632. 10.1002/anie.200504443. [DOI] [PubMed] [Google Scholar]
- Fielicke A.; von Helden G.; Meijer G.; Pedersen D. B.; Simard B.; Rayner D. M. Gold Cluster Carbonyls: Saturated Adsorption of CO on Gold Cluster Cations, Vibrational Spectroscopy, and Implications for Their Structures. J. Am. Chem. Soc. 2005, 127, 8416–8423. 10.1021/ja0509230. [DOI] [PubMed] [Google Scholar]
- Chen M.; Goodman D. W. Catalytically Active Gold: From Nanoparticles to Ultrathin Films. Acc. Chem. Res. 2006, 39, 739–746. 10.1021/ar040309d. [DOI] [PubMed] [Google Scholar]
- Haruta M.; Kobayashi T.; Sano H.; Yamada N. Novel Gold Catalysts for the Oxidation of Carbon Monoxide at a Temperature Far Below 0 °C. Chem. Lett. 1987, 16, 405–408. 10.1246/cl.1987.405. [DOI] [Google Scholar]
- Mitsudome T.; Kaneda K. Gold Nanoparticle Catalysts for Selective Hydrogenation. Green Chem. 2013, 15, 2636–2654. 10.1039/c3gc41360h. [DOI] [Google Scholar]
- Takale B. S.; Bao M.; Yamamoto Y. Gold Nanoparticle (AuNPs) and Gold Nanopore (AuNPore) Catalysts in Organic Synthesis. Org. Biomol. Chem. 2014, 12, 2005–2027. 10.1039/c3ob42207k. [DOI] [PubMed] [Google Scholar]
- Ishida T.; Murayama T.; Taketoshi A.; Haruta M. Importance of Size and Contact Structure of Gold Nanoparticles for the Genesis of Unique Catalytic Processes. Chem. Rev. 2020, 120, 464–525. 10.1021/acs.chemrev.9b00551. [DOI] [PubMed] [Google Scholar]
- Sankar M.; He Q.; Engel R. V.; Sainna M. A.; Logsdail A. J.; Roldan A.; Willock D. J.; Agarwal N.; Kiely C. J.; Hutchings G. J. Role of the Support in Gold-Containing Nanoparticles as Heterogeneous Catalysts. Chem. Rev. 2020, 120, 3890–3938. 10.1021/acs.chemrev.9b00662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abad A.; Concepción P.; Corma A.; García H. A Collaborative Effect between Gold and a Support Induces the Selective Oxidation of Alcohols. Angew. Chem., Int. Ed. 2005, 44, 4066–4069. 10.1002/anie.200500382. [DOI] [PubMed] [Google Scholar]
- Enache D. I.; Edwards J. K.; Landon P.; Solsona-Espriu B.; Carley A. F.; Herzing A. A.; Watanabe M.; Kiely C. J.; Knight D. W.; Hutchings G. J. Solvent-Free Oxidation of Primary Alcohols to Aldehydes Using Au-Pd/TiO2 Catalysts. Science 2006, 311, 362–365. 10.1126/science.1120560. [DOI] [PubMed] [Google Scholar]
- Hansch C.; Leo A.; Taft R. W. A Survey of Hammett Substituent Constants and Resonance and Field Parameters. Chem. Rev. 1991, 91, 165–195. 10.1021/cr00002a004. [DOI] [Google Scholar]
- Nishimura S.; Yakita Y.; Katayama M.; Higashimine K.; Ebitani K. The Role of Negatively Charged Au States in Aerobic Oxidation of Alcohols over Hydrotalcite Supported AuPd Nanoclusters. Catal. Sci. Technol. 2013, 3, 351–359. 10.1039/C2CY20244A. [DOI] [Google Scholar]
- Tongsakul D.; Nishimura S.; Ebitani K. Platinum/Gold Alloy Nanoparticles-Supported Hydrotalcite Catalyst for Selective Aerobic Oxidation of Polyols in Base-Free Aqueous Solution at Room Temperature. ACS Catal. 2013, 3, 2199–2207. 10.1021/cs400458k. [DOI] [Google Scholar]
- Tongsakul D.; Nishimura S.; Ebitani K. Effect of Stabilizing Polymers on Catalysis of Hydrotalcite-Supported Platinum Nanoparticles for Aerobic Oxidation of 1,2-Propanediol in Aqueous Solution at Room Temperature. J. Phys. Chem. C 2014, 118, 11723–11730. 10.1021/jp501836a. [DOI] [Google Scholar]
- Conte M.; Miyamura H.; Kobayashi S.; Chechik V. Spin Trapping of Au–H Intermediate in the Alcohol Oxidation by Supported and Unsupported Gold Catalysts. J. Am. Chem. Soc. 2009, 131, 7189–7196. 10.1021/ja809883c. [DOI] [PubMed] [Google Scholar]
- Abad A.; Corma A.; García H. Catalyst Parameters Determining Activity and Selectivity of Supported Gold Nanoparticles for the Aerobic Oxidation of Alcohols: The Molecular Reaction Mechanism. Chem. - Eur. J. 2008, 14, 212–222. 10.1002/chem.200701263. [DOI] [PubMed] [Google Scholar]
- Fristrup P.; Johansen L. B.; Christensen C. H. Mechanistic Investigation of the Gold-Catalyzed Aerobic Oxidation of Alcohols. Catal. Lett. 2008, 120, 184–190. 10.1007/s10562-007-9301-8. [DOI] [Google Scholar]
- Zope B. N.; Hibbitts D. D.; Neurock M.; Davis R. J. Reactivity of the Gold/Water Interface During Selective Oxidation Catalysis. Science 2010, 330, 74–78. 10.1126/science.1195055. [DOI] [PubMed] [Google Scholar]
- Kumar G.; Tibbitts L.; Newell J.; Panthi B.; Mukhopadhyay A.; Rioux R. M.; Pursell C. J.; Janik M.; Chandler B. D. Evaluating Differences in the Active-Site Electronics of Supported Au Nanoparticle Catalysts Using Hammett and DFT Studies. Nat. Chem. 2018, 10, 268–274. 10.1038/nchem.2911. [DOI] [PubMed] [Google Scholar]
- Muñoz-Santiburcio D.; Camellone M. F.; Marx D. Solvation-Induced Changes in the Mechanism of Alcohol Oxidation at Gold/Titania Nanocatalysts in the Aqueous Phase versus Gas Phase. Angew. Chem., Int. Ed. 2018, 57, 3327–3331. 10.1002/anie.201710791. [DOI] [PubMed] [Google Scholar]
- Mahdavi-Shakib A.; Sempel J.; Babb L.; Oza A.; Hoffman M.; Whittaker T. N.; Chandler B. D.; Austin R. N. Combining Benzyl Alcohol Oxidation Saturation Kinetics and Hammett Studies as Mechanistic Tools for Examining Supported Metal Catalysts. ACS Catal. 2020, 10, 10207–10215. 10.1021/acscatal.0c02212. [DOI] [Google Scholar]
- Mori K.; Hara T.; Mizugaki T.; Ebitani K.; Kaneda K. Hydroxyapatite-Supported Palladium Nanoclusters: A Highly Active Heterogeneous Catalyst for Selective Oxidation of Alcohols by Use of Molecular Oxygen. J. Am. Chem. Soc. 2004, 126, 10657–10666. 10.1021/ja0488683. [DOI] [PubMed] [Google Scholar]
- Ato Y.; Hayashi A.; Koga H.; Kawakami T.; Yamanaka S.; Okumura M. Theoretical Study of Aerobic Oxidation of Alcohols over Au38 Nanocluster by a Two-Step-Modeling Approach. Chem. Phys. Lett. 2019, 724, 115–121. 10.1016/j.cplett.2019.03.060. [DOI] [Google Scholar]
- Schneider C.; Rasband W.; Eliceiri K. NIH Image to ImageJ: 25 Years of Image Analysis. Nat. Methods 2012, 9, 671–675. 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Keith T.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam J. M.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas O.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 09; Gaussian, Inc.: Wallingford, CT, 2009. [Google Scholar]
- Lee C.; Yang W.; Parr R. G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B: Condens. Matter Mater. Phys. 1988, 37, 785. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- Becke A. D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]
- Luo C.; Fa W.; Dong J. Adsorption of O2 on Tubelike Au24 and Au24– Clusters. J. Chem. Phys. 2006, 125, 084707. 10.1063/1.2338810. [DOI] [PubMed] [Google Scholar]
- Nhat P. V.; Si N. T.; Nguyen M. T. Structural Evolution and Stability Trend of Small-Sized Gold Clusters Aun (n = 20–30). J. Phys. Chem. A 2020, 124, 1289–1299. 10.1021/acs.jpca.9b09287. [DOI] [PubMed] [Google Scholar]
- Matsuo A.; Hasegawa S.; Takano S.; Tsukuda T. Electron-Rich Gold Clusters Stabilized by Polyvinylpyridines as Robust and Active Oxidation Catalysts. Langmuir 2020, 36, 7844–7849. 10.1021/acs.langmuir.0c00812. [DOI] [PubMed] [Google Scholar]
- Hosokawa F.; Shinkawa T.; Arai Y.; Sannomiya T. Benchmark Test of Accelerated Multi-Slice Simulation by GPGPU. Ultramicroscopy 2015, 158, 56–64. 10.1016/j.ultramic.2015.06.018. [DOI] [PubMed] [Google Scholar]
- Kamei K.; Shimizu T.; Harano K.; Nakamura E. Aryl Radical Addition to Curvatures of Carbon Nanohorns for Single-Molecule-Level Molecular Imaging. Bull. Chem. Soc. Jpn. 2020, 93, 1603–1608. 10.1246/bcsj.20200232. [DOI] [Google Scholar]
- Gorgoll R. M.; Yücelen E.; Kumamoto A.; Shibata N.; Harano K.; Nakamura E. Electron Microscopic Observation of Selective Excitation of Conformational Change of a Single Organic Molecule. J. Am. Chem. Soc. 2015, 137, 3474–3477. 10.1021/jacs.5b00511. [DOI] [PubMed] [Google Scholar]
- Du K.; von Hochmeister K.; Phillipp F. Quantitative Comparison of Image Contrast and Pattern between Experimental and Simulated High-Resolution Transmission Electron Micrographs. Ultramicroscopy 2007, 107, 281–292. 10.1016/j.ultramic.2006.08.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.