

Abstract
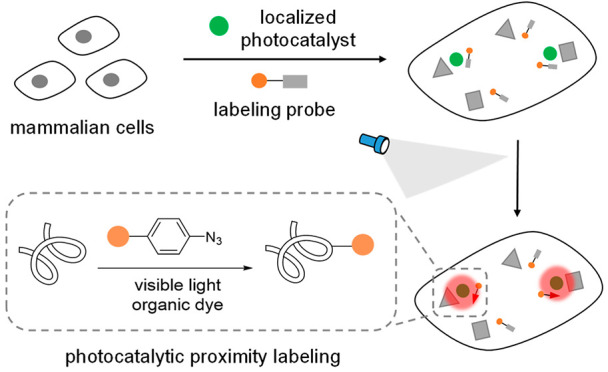
Biocompatible reactions are powerful tools to probe protein functions in their native environment. Due to the difficulty of penetrating the live-cell membrane and the complex intracellular environment, the biocompatible reactions inside live cells are challenging, especially at the subcellular level with spatial resolution. Here we report the first biocompatible photocatalytic azide conjugation reaction inside live cells to achieve the mitochondria-selective proteins labeling. The organic dyes acridine orange, fluorescein, and rhodamine 123 were developed as the biocompatible photocatalysts for the proteins labeling with aryl azides, which yielded benzazirines and ketenimines from triplet nitrenes for the protein nucleophilic residue trapping. The photocatalytic azide conjugation reaction with rhodamine 123 selectively labeled the mitochondrial proteins via the organic dye’s mitochondrial localization. In response to the mitochondrial stress induced by rotenone, this photocatalytic azide-promoted labeling method mapped the dynamic mitochondrial proteome changes with high temporal-spatial precision and identified several potential mitochondrial stress-response proteins for the first time. The high temporal-spatial precision of this photocatalytic azide-promoted labeling method holds excellent potential for intracellular protein network investigations.
Keywords: biocompatible reaction, visible light photocatalysis, protein labeling, live cells, mitochondria
Introduction
Proteins in cellular compartments undergo dynamic changes with the environmental stimuli or pathological process. To understand the regulated protein network, it is critical to map the temporal changes of these subcellular proteins.1,2 Equipped with the imaging- and mass-spectrometry-based analysis, the biocompatible reactions are valuable to probe protein functions with protein labeling reactions in their native environment.3−6 However, due to the difficulty of penetrating the live-cell membrane and the complex intracellular environment, it is challenging to investigate the intracellular protein network, especially at the subcellular level with spatial resolution. The intracellular protein networks’ elucidation has been greatly helped by the pioneer efforts in the subcellular protein labeling method development, which includes the enzymatic proximity labeling methods such as APEX and the chemical affinity labeling methods with organelle-targeting reactive probes.7−11 However, new intracellular protein labeling methods with precise temporal-spatial resolution, simple experimental operation and low biological disturbance are in increasingly high demands.7−11
Light provides high temporal-spatial precision while it disturbs the biological system relatively subtly, and the photoinducible chemical tools are valuable for cellular biomolecule studies.12−15 Compared to the traditional photochemical methods, the recently emerged photocatalytic reactions provide additional advantages: The reactive intermediates are generated in the proximity of the excited-state photocatalysts with light dependence.16−19 Using the antibody-conjugated iridium complex as the localized photocatalyst and the diazirine probe, the photocatalytic carbene insertion achieved the cell-membrane proteins labeling under blue light irradiation.20 Despite the impressive success in mapping the protein–protein interactions in the cell-membrane microenvironments, this antibody conjugation strategy was not suitable for the intracellular applications as the membrane impermeability of the iridium photocatalysts and its potential heavy-metal cytotoxicity.4,21
Compared to the transition-metal photocatalysts, organic photocatalysts derived from small-molecule organic dyes offer compelling advantages (Figure 1a).21,22 Organic dyes have historically been used for intracellular imaging and tracing due to their biocompatibility and have demonstrated excellent native subcellular localization.23−26 However, their photocatalytic reactivity in live cells remains elusive. As the commonly used photocatalysts cannot activate the diazirine probes with high triplet energy, the new labeling probes development will significantly enrich the photocatalytic proteins labeling applications. Herein we report the first biocompatible photocatalytic azide conjugation reaction inside live cells to achieve the mitochondria-selective protein labeling with organic dyes (Figure 1b).
Figure 1.

Photocatalytic proximity labeling with localized organic dyes and aryl azide probes inside live mammalian cells. (a) Representative transition-metal photocatalysts based on heavy metals ruthenium and iridium and organic photocatalysts acridine orange (AO), fluorescein (Fluo), and rhodamine 123 (Rh 123). (b) Stable aryl azide probes were activated to the reactive probes only in the proximity of the localized organic dyes for spatial protein labeling reactions under visible light irradiation.
Results and Discussion
Exploring Photocatalytic Proteins Labeling with Organic Dyes
Inspired by our recent discovery that organic dyes enabled the photocatalytic uncaging reactions inside live cells,27 we investigated fluorescein as the photocatalyst for the photocatalytic protein labeling reactions.28−31 The biotin-conjugated aryl azide, phenol, and acetophenone probes were synthesized, and their photolabeling reactivity was evaluated by the HRP-conjugated streptavidin via the Western blotting analysis (Figure 2a). The AzPh-biotin probe 1 showed clear bovine serum albumin (BSA) biotinylation in pH 7.4 PBS buffer after blue LED irradiation (468 nm, 5.8 mW/cm2) at 25 °C (Figure 2b). In contrast, the phenol-biotin probe 2 or the AcPh-biotin probe 3 resulted in a weak or little labeling signal. The photocatalytic labeling of BSA by the AzPh-biotin probe 1 showed clear light dependence. The t1/2 of the probe 1 conversion was around 24.8 min in the reaction conditions, and was not affected by the addition of BSA proteins (Figure S5). The control experiments of AzPh-biotin probe 1 indicated that the photocatalytic BSA labeling required both fluorescein and blue LED irradiation (Figure 2b).
Figure 2.

Photocatalytic labeling of bovine serum albumin (BSA) in vitro. (a) Structures of probes 1–3. The chemical labeling groups and linkers are shown in black, and the biotin groups for enrichment are shown in red. (b) Photocatalytic BSA labeling with biotin probes 1–3 using fluorescein (Fluo) under the blue LED irradiation (468 nm, 5.8 mW/cm2) at 25 °C for 60 min. Control experiments with AzPh-biotin probe 1 showed the fluorescein and the light irradiation were critical for the reaction. The biotinylated-BSA was analyzed by HRP-conjugated streptavidin (streptavidin-HRP), and the BSA proteins were detected by Coomassie brilliant blue staining (CBB staining). BSA (2 μM in pH 7.4 PBS buffer), fluorescein (100 μM), and probes 1–3 (100 μM). (c) Photocatalytic BSA labeling with AzPh-biotin probe 1 (100 μM) by different organic dyes (100 μM) in the air atmosphere or the nitrogen atmosphere under a Xe lamp source at 25 °C for 1 min (15 cm from Xe lamp, 19.8 mW/cm2 with 500 nm band-pass filter for lanes 1–4, 550 nm band pass filter for lanes 5 and 6).
Different organic dyes were next tested as potential photocatalysts for the protein labeling. Acridine orange (AO) and rhodamine 123 (Rh 123) effectively induced the BSA biotinylation with AzPh-biotin probe 1 under blue LED irradiation (Figure 2c), while no significant biotinylation was observed with probes 2 and 3 (Figures S12 and S13). As AO and Rh 123 absorbed both blue and green light, we also tested the photoconversion kinetics of AzPh-biotin probe 1 under blue and green LED irradiation and found they were similarly effective (Figures S7 and S8). In contrast, with mitochondrial dyes rhodamine B (Rh B) and tetramethylrhodamine ethyl ester (TMRE), no photolabeling was observed. The fact that the reaction performed effectively under both the air atmosphere and the nitrogen atmosphere suggested that oxygen was not required for the reaction. The protein labeling reactions with AO, Fluo, or Rh 123 could be observed within 1 min of irradiation at their maximal absorption, suggesting the excellent temporal resolution of this photocatalytic protein labeling reaction (Figure 2c).32
Mechanism Investigation of the Photocatalytic Azide-Promoted Protein Labeling
The UV-light-induced photoaffinity labeling with aryl azides was previously investigated extensively.33 However, the photocatalytic azide-promoted labeling mechanism remains unknown. In theory, there are two plausible photocatalytic reaction pathways to convert aryl azides: (i) The single electron transfer pathway yields the azide anion radical, followed by the nitrogen gas elimination to afford the aminyl radical;34 or (ii) the energy transfer pathway yields the triplet azide, followed by the nitrogen gas elimination to afford the triplet nitrene,35−37 which may further convert to ketenimines by the light irradiation (Figure 3a).38,39 We first measured the redox potentials of AO, Fluo, and Rh 123 (E1/2red > −1.22 V, vs SCE, Figure S1)22 and found that they had higher reduction potentials than those of aryl azides (Ep1/2red = −1.84 V, vs SCE, Figure S1). These results suggested that the single electron transfer pathway from organic dyes to aryl azides to yield the aminyl radical was unlikely. In addition, the aryl azides effectively quenched the fluorescence of AO, Fluo, and Rh 123 in the fluorescence quenching experiments (Figure S2), suggesting the photochemical pathways between AO, Fluo, and Rh 123 with aryl azides. The conversions of aryl azide 4 by organic dyes with different triplet energies were next evaluated (Figure 3b and Table S1). The organic dyes (AO, Fluo, and Rh 123) with the triplet energy greater than 1.9 eV effectively converted 4, while the organic dyes (TMRE and Rh B) with the triplet energy less than 1.9 eV did not convert 4. These results indicated that the triplet energy greater than 1.9 eV was the threshold for the efficient conversion of 4, which was consistent with the protein labeling results (Figure 2c).22
Figure 3.

Mechanistic studies of the photocatalytic azide-promoted protein labeling reaction. (a) Plausible photocatalysis reaction pathways (single electron transfer or energy transfer) to generate the reactive intermediates from aryl azides. (b) Conversion of 4-azidobenzoic acid 4 (R = H) (200 μM) after visible light irradiation at 25 °C for 1 h in PBS buffer (10 mM, pH 7.4) (xenon lamp equipped with band-pass filters, 19.8 mW cm–2) by organic dyes (50 μM) with different triplet energies. c) The formation of aniline 7, azepine 8, and thiolether-substituted aniline 10 supported the triplet nitrene, ketenimine, and benzazirine intermediates. The solution of 5 (0.10 mmol), Acridine Orange (0.020 mmol) and amine or thiols (5 mmol) in methanol were under blue LED (10.4 mW/cm2) irradiation at 25 °C for 40–48 h. (d) LC-MS/MS analysis of the AzPh-biotin probe 1 labeled nucleophilic amino acid residues on BSA protein. The identified sites are partially labeled on the crystal structure of BSA, and the whole identified sites are shown in Figure S28.
As the photocatalytic conversion of aryl azide 5 yielded complex reaction mixtures in the absence of proteins, we injected n-butylamine 6 to trap the potential intermediates of azide 5.40,41 With >95% conversion of 5, we observed the aniline 7 formation in 23% yield and butylamine-substituted azepine 8 in 61% yield, for which the structure was unambiguously assigned by the COSY and HSQC analysis (Figure 3c; see the SI for details). The ethanethiol 9 representing the amino acid thiol residues was next injected to the reaction of 5, and thiolether-substituted 10 was obtained in 16% yield together with aniline 7 in 59% yield (Figure 3c). We hypothesized that the aniline 7 was formed by the hydrogen abstraction, while thiolether-substituted 10 and butylamine-substituted azepine 8 originated from the benzazirine and the ketenimine intermediates, respectively.42,43 We further conducted the LC-MS/MS analysis of the AzPh-biotin probe 1 labeled BSA proteins induced by AO. Various nucleophilic residues including Lys, Tyr, Thr, Glu, Asp, Ser, Cys, and His were detected, whereas the less reactive amino acid residues such as Gln and Gly were not observed (Figures 3d and S28; Table S3). These results were consistent with the small molecule trapping results and together suggested the energy transfer pathway of aryl azides in the protein labeling reactions.37
Selective Mitochondrial Proteins Labeling Inside Live Cells
We next explored if this photocatalytic azide-promoted labeling reaction was compatible with the intracellular environment for live-cell applications. Various representative cellular reductants, reactive oxygen species, and metal ions were then added to the photocatalytic BSA labeling conditions. As a result, the addition of reactive oxygen species, metal ions, as well as ascorbates did not affect the protein labeling at the concentrations above the physiological relevance (Figures S15–S17). In contrast, cellular reductants including thiols, L-DOPA, β-carotene, and Trolox (water-soluble vitamin E) inhibited the labeling reactions (Figure S17). Among those, glutathione (GSH) and N-acetylcysteine (NAC) representing thiols showed relatively strong inhibition, which was expected from the reactive intermediate trapping experiments above. As GSH was abundant in live cells, we then tested how its addition affected the BSA labeling.44,45 With GSH added at the physiological concentrations, the photocatalytic BSA labeling efficiency was decreased but not completely inhibited (Figure S18). In contrast, the photoconversion of 4 was not significantly affected by the GSH additions (Table S2, Figure S6), suggesting GSH reacted with the labeling intermediates instead of the excited photocatalyst.46 We hypothesized that the quenching of the labeling intermediates by these natural reductants and nucleophiles may enable the proximity labeling in the intracellular environment.7,45
Besides cellular reductants, oxygen may also inhibit the intracellular photocatalytic reactions by quenching the excited-state photocatalysts.27,45 While the oxygen presence did not significantly affect the fluorescence of Fluo and Rh 123 in the fluorescent quenching experiments, it decreased the AO fluorescence by quenching the excited-state AO (Figure S3). As a result, AO demonstrated different reactivity of 4 photoconversion in the air and nitrogen atmosphere, consisting with the fluorescent quenching data (Figure 3b). Singlet oxygen generated in the photosensitization process was considered highly reactive and may cause protein degradation and cell damages.47,48 We also measured the quantum yields of AO, Fluo, and Rh 123 for oxygen sensitization and found they were not effective singlet oxygen sensitizers, suggesting their promising biocompatibility in live cells (Figure S4).
We next examined the photocatalytic proximity protein labeling inside live cells by organic dyes with expected biocompatibility and intracellular localization (Figure 4a). AO binds cellular nucleic acids and also targets acidic organelles, while Rh 123 is a mitochondria-specific dye.23 The AzPh-biotin probe 1 and AO were then incubated with the MCF-7 breast cancer cells under green light irradiation (515 nm, 2.9 mW/cm2) for 30 min, and the Cy5-conjugated streptavidin detected the labeled biotin signal from the confocal fluorescence microscope (Figure S20). However, cell damage was also observed. We further tested cell viability by performing the MTT assay and observed only 30% cell survival after the photocatalytic labeling by AO (Figure S25a). These results were inconsistent with the low quantum yield for the singlet oxygen generation by AO. To explain the difference, the quantum yield of singlet oxygen generation by AO was also measured in acidic conditions as AO tended to aggregate in acidic organelles (Figure S4).49,50 In fact, the AO-induced singlet oxygen generation under acidic conditions was higher than that under neutral conditions. In addition, the strong binding between AO and cellular nucleic acids also contributed to DNA damage induced by AO.51 From the LC-MS/MS analysis, the photocatalytic protein labeling with AO resulted in 453 enriched proteins and 199 proteins which (43.9%) were nucleus proteins (Figures S30 and S31). These results echoed the AO-induced labeling from the confocal fluorescence microscopy analysis (Figure S20)
Figure 4.

Photocatalytic labeling of the mitochondrial proteome by AzPh-biotin probe 1 in MCF-7 cells. (a) Photocatalytic mitochondrial proteome labeling scheme and confocal fluorescence microscopy analysis of AzPh-biotin probe 1 with Rh 123 in MCF-7 cells. The green dots represent the organic dye Rh 123, and the red circles represent the mitochondrial proteome labeled in the proximity of Rh 123. Cells were irradiated by the green LED with Rh 123 at 37 °C for 1 h (515 nm, 2.9 mW/cm2). TMRE (green) stains the mitochondria, and Cy5-streptavidin (red) detects the biotinylated proteins. The merged image shows good colocalization (R = 0.87, colocalization was quantified using Pearson’s R-value calculated with Coloc 2 in ImageJ). Scale bar: 20 μm. Rh 123 (20 μM), AzPh-biotin probe 1 (200 μM). (b) Proteome labeling in MCF-7 cells with AzPh-biotin probe 1 by green LED irradiation with Rh 123 for 1 h (515 nm, 2.9 mW/cm2) or UV light irradiation for 20 min (365 nm, 11.8 mW/cm2) at 37 °C. (c) MTT cell viability assay for the cytotoxicity of Rh 123 labeling using AzPh-biotin probe 1 under green LED irradiation at 37 °C for 1 h (515 nm, 2.9 mW/cm2). The statistical significance of the differences between groups was evaluated with the unpaired Student’s t test. A p-value of 0.05 and below was considered significant: p < 0.01 (**), p < 0.001 (***), N.S. is not statistically significant. All p-values were calculated with control cells that treated without light and small molecules. Error bars represent mean ± SD (n = 3). (d) Counts of the photocatalytic labeled MCF-7 mitochondrial proteins by the LC-MS/MS analysis with Rh 123 and the AzPh-biotin probe 1. The annotations were performed by the DAVID (167/333, 50.2%, DAVID) or by the combined databases including DAVID, Uniprot, and MitoCarta 2.0 (201/333, 60.4%, multiple).
In contrast, Rh 123 showed mitochondria-specific proteome labeling, and the Cy5-conjugated streptavidin showed biotinylation with excellent colocalization of the mitochondria dye TMRE (Figures 4b and S22, R = 0.87, Pearson’s R-value).52 We also performed UV-light-induced azide-promoted labeling for 20 min with a 365 nm UV lamp (11.8 mW/cm2) and observed no significant subcellular-specific protein labeling. The photocatalytic protein labeling by Rh 123 also demonstrated excellent biocompatibility with a greater than 85% cell survival rate even after 60 min of green LED irradiation (2.9 mW/cm2) (Figure 4c). We next analyzed the protein biotinylation by Western blot analysis with HRP-conjugated streptavidin and confirmed Rh 123 induced the proteome biotinylation (Figures S23 and S24). The control experiments indicated that both the organic dyes and the light irradiation were critical for the protein labeling reactions in live cells.
The Rh 123-induced labeling showed excellent mitochondria specificity that 201 proteins (60.4%) out of the 333 enriched proteins were mitochondrial proteins (Figure 4d, Table S5 for details). We further analyzed the submitochondrial specificity of the photocatalytic labeled proteome: 40% of the identified mitochondria proteins were the mitochondrial matrix (matrix), and 34% of that were the inner mitochondrial membrane (IMM) proteins (Figures S32 and S33). These results were consistent with the positive-charged Rh 123 localized to the negative-charged mitochondrial inner membrane at the submitochondrial level.52 In contrast, the intermembrane mitochondria space (IMS) proteins and the outer mitochondrial membrane (OMM) proteins were detected at a much lower frequency. Among the labeled mitochondria proteins, the mitochondrial respiratory chain complexes and the mitochondria protein-import machinery were all detected, which were critical mitochondria membrane protein complexes in the matrix and IMM (Table S5).
Mitochondrial Proteins Mapping for Stress-Response Proteins Identification
Mitochondria play a vital role in maintaining the redox homeostasis as the cellular respiration location and the energy powerhouse for cells. Mitochondrial dysfunction is known to be involved in many human diseases such as cancer and neurodegenerative diseases.53,54 As no genetic manipulation is required for the photocatalytic method, only a simple step of photocatalytic labeling is needed for the subsequent LC-MS/MS analysis and it allows for probing of the native mitochondria proteins. Rotenone is a widely used mitochondrial complex I inhibitor to investigate apoptosis and neurodegenerative diseases, which induces the decreased membrane potential (ΔΨm), ATP depletion, and ROS generation in mitochondria (Figure 5a).55−59 However, its specific impacts on mitochondrial proteome have not been fully explored.
Figure 5.

Photocatalytic proteome mapping of the mitochondria for the rotenone-induced stress response. (a) HeLa cells underwent mitochondrial stress response with the mild stress induced by the low-concentration of rotenone, while the depletion of mitochondrial mass was detected with the severe mitochondrial toxicity induced by the high-concentration of rotenone. (b) Volcano plot of the identified proteins by the high concentration (2 μM) of rotenone treatment and the changed protein counts. A significant portion of the decreased protein measurement was observed in mitochondria (26/31, 83.9%). (c) Volcano plot of the identified proteins by the low concentration (0.02 μM) of rotenone treatment and the changed protein counts. A significant portion of the increased protein measurement was observed in mitochondria (8/13, 61.5%). The decreased protein measurement is colored in blue, and the increased protein measurement is colored in red. FC: fold-change.
We then tested if the dynamic mitochondrial proteome changes could be assessed by the photocatalytic method combined with LC-MS/MS analysis (Tables S6–S9).2,53 The volcano plots were used to visualize the increased or decreased mitochondrial proteome measurements in response to the rotenone treatment (Figure 5b,c). With 2 μM rotenone, 31 proteins were significantly decreased in HeLa cells, 26 of which were mitochondrial proteins, including the mitochondrial matrix and IMM proteins (FC < 0.5, P < 0.05, see Figure 5b and Tables S4 and S5). Further TMRE staining of mitochondria suggested that the high concentration of rotenone induced the loss of mitochondrial membrane potential in cells (see Figures S26 and S27), which was consistent with the LC-MS/MS analysis results.56
With mild mitochondrial stress induced by 0.02 μM rotenone, only one out of seven decreased proteins were mitochondrial proteins (FC < 0.5, P < 0.05, Figure 5c and see Table S6). In contrast, 8 out of 13 increased proteins were mitochondrial proteins (FC > 2, P < 0.05, see Table S7). Among them, four (HSPE1,60 BCS1L,61 MTCH2,62 and TXNRD163) were reported to participate in mitochondrial functions but were not previously linked to the rotenone-induced mitochondrial responses. The other four mitochondrial proteins (PITRM1, IVD, ATAD3A, CCDC51) were not previously associated with the mitochondrial stress responses and may be worth further investigation (see Table S7). These results not only confirmed that the low concentration of rotenone-induced mitochondrial stress responses but also provided the detailed dynamic proteome information. Together, as showcased by rotenone here, this selective mitochondrial protein labeling method with simple experimental operation facilitates and casts novel insights into the mitochondrial biology study without the need of complicated genetic manipulations.
Conclusion
In conclusion, we have developed the first biocompatible photocatalytic azide conjugation reaction inside live cells for selective mitochondrial protein labeling. Readily available organic dyes were investigated for photochemical reactivity and biocompatibility to enable photocatalytic azide-promoted protein labeling for the first time. Mechanistic studies suggested that the nucleophilic protein residue labeling was attributed to the photocatalytic generation of triplet nitrenes for the benzazirine and ketenimine intermediates. The mitochondria-localized rhodamine 123 enabled the selective mitochondrial protein labeling with submitochondrial specificity via simple experimental procedures. The dynamic changes of the native mitochondrial proteomes upon external stimuli can be mapped without genetic manipulation in high temporal-spatial precision.
The biocompatibility and reactivity of the photocatalytic protein labeling reaction are the key parameters to consider for intracellular applications. The photolabeling reactions with low labeling efficiency may require extended light irradiation and cause phototoxicity. To this end, increasing the irradiation intensity may improve the photolabeling efficacy significantly. For example, the green LED irradiation with enhanced intensity (95 mW/cm2) led to sufficient AO-induced protein labeling in live cells in merely 1 min with decreased cytotoxicity (Figures S21 and S25b). Due to the intrinsic electrophilic reactivity of the labeling intermediates from aryl azides in photocatalysis, the nucleophilic residues in proteins were mostly detected by this method with limited labeling resolution (Figures 3d, S28, and S29; Table S3 and S4).64 Moreover, the diffusion distance of the labeling intermediates remained unclear as it was affected by the nucleophiles in different concentrations and cellular environments, which is worth further investigation.65 We envision that the development of new labeling probes with higher reactivity may increase the labeled protein residue coverage and labeling resolution. While Rh 123 was herein demonstrated to enable photocatalytic mitochondria protein labeling, we envision that organic dyes provide flexible chemical modification possibilities for other subcellular applications beyond mitochondria with their different localization properties. The photocatalytic labeling strategy is complementary to the traditional UV-light-induced methods for intracellular studies, given its use of visible light and the attractive localization capacity. This photocatalytic protein labeling method holds excellent potential for intracellular protein network investigations demanding high temporal-spatial resolution.
Acknowledgments
Financial support was provided by the National Natural Science Foundation of China 91753126, 21622207, 21602242, 31671428, and 31530041, CAS Interdisciplinary Innovation Team JCTD-2020-16, the National Key R&D Program of China 2016YFA0501904 and 2016YFA0501900, the Strategic Priority Research Program of the Chinese Academy of Sciences XDB20020200, and the Shanghai Municipal Science and Technology Major Project 2019SHZDZX02.
Glossary
Abbreviations
- AO
acridine orange
- Fluo
fluorescein
- Rh 123
rhodamine 123
- Rh B
rhodamine B
- TMRE
tetramethylrhodamine ethyl ester
- CBB staining
Coomassie brilliant blue staining
- BSA
bovine serum albumin
- GSH
glutathione
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacsau.1c00172.
Author Contributions
∇ H.W., Y.Z., K.Z., and J.Q. contributed equally. H.W., Y.Z., K.Z., Y.F., Y.Z., and Y.C. designed the research and wrote the manuscript. H.W., Y.Z., and K.Z. performed the experiments and analyzed the data. J.Q., Y.C., Y L., and Y.Z. performed the mass spectrometry analysis. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Yates J. R. 3rd; Gilchrist A.; Howell K. E.; Bergeron J. J. Proteomics of organelles and large cellular structures. Nat. Rev. Mol. Cell Biol. 2005, 6 (9), 702–714. 10.1038/nrm1711. [DOI] [PubMed] [Google Scholar]
- Lundberg E.; Borner G. H. H. Spatial proteomics: A powerful discovery tool for cell biology. Nat. Rev. Mol. Cell Biol. 2019, 20 (5), 285–302. 10.1038/s41580-018-0094-y. [DOI] [PubMed] [Google Scholar]
- Prescher J. A.; Bertozzi C. R. Chemistry in living systems. Nat. Chem. Biol. 2005, 1 (1), 13–21. 10.1038/nchembio0605-13. [DOI] [PubMed] [Google Scholar]
- Sletten E. M.; Bertozzi C. R. Bioorthogonal chemistry: Fishing for selectivity in a sea of functionality. Angew. Chem., Int. Ed. 2009, 48 (38), 6974–6998. 10.1002/anie.200900942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cravatt B. F.; Wright A. T.; Kozarich J. W. Activity-based protein profiling: From enzyme chemistry to proteomic chemistry. Annu. Rev. Biochem. 2008, 77, 383–414. 10.1146/annurev.biochem.75.101304.124125. [DOI] [PubMed] [Google Scholar]
- Tamura T.; Hamachi I. Chemistry for covalent modification of endogenous/native proteins: From test tubes to complex biological systems. J. Am. Chem. Soc. 2019, 141 (7), 2782–2799. 10.1021/jacs.8b11747. [DOI] [PubMed] [Google Scholar]
- Rhee H. W.; Zou P.; Udeshi N. D.; Martell J. D.; Mootha V. K.; Carr S. A.; Ting A. Y. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science 2013, 339 (6125), 1328–1331. 10.1126/science.1230593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasueda Y.; Tamura T.; Fujisawa A.; Kuwata K.; Tsukiji S.; Kiyonaka S.; Hamachi I. A set of organelle-localizable reactive molecules for mitochondrial chemical proteomics in living cells and brain tissues. J. Am. Chem. Soc. 2016, 138 (24), 7592–7602. 10.1021/jacs.6b02254. [DOI] [PubMed] [Google Scholar]
- Wiedner S. D.; Anderson L. N.; Sadler N. C.; Chrisler W. B.; Kodali V. K.; Smith R. D.; Wright A. T. Organelle-specific activity-based protein profiling in living cells. Angew. Chem., Int. Ed. 2014, 53 (11), 2919–2922. 10.1002/anie.201309135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujisawa A.; Tamura T.; Yasueda Y.; Kuwata K.; Hamachi I. Chemical profiling of the endoplasmic reticulum proteome using designer labeling reagents. J. Am. Chem. Soc. 2018, 140 (49), 17060–17070. 10.1021/jacs.8b08606. [DOI] [PubMed] [Google Scholar]
- Zhu H.; Tamura T.; Hamachi I. Chemical proteomics for subcellular proteome analysis. Curr. Opin. Chem. Biol. 2019, 48, 1–7. 10.1016/j.cbpa.2018.08.001. [DOI] [PubMed] [Google Scholar]
- Tanaka Y.; Bond M. R.; Kohler J. J. Photocrosslinkers illuminate interactions in living cells. Mol. BioSyst. 2008, 4 (6), 473–480. 10.1039/b803218a. [DOI] [PubMed] [Google Scholar]
- Fehrentz T.; Schonberger M.; Trauner D. Optochemical genetics. Angew. Chem., Int. Ed. 2011, 50 (51), 12156–12182. 10.1002/anie.201103236. [DOI] [PubMed] [Google Scholar]
- Brieke C.; Rohrbach F.; Gottschalk A.; Mayer G.; Heckel A. Light-controlled tools. Angew. Chem., Int. Ed. 2012, 51 (34), 8446–8476. 10.1002/anie.201202134. [DOI] [PubMed] [Google Scholar]
- Ankenbruck N.; Courtney T.; Naro Y.; Deiters A. Optochemical control of biological processes in cells and animals. Angew. Chem., Int. Ed. 2018, 57 (11), 2768–2798. 10.1002/anie.201700171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prier C. K.; Rankic D. A.; MacMillan D. W. Visible light photoredox catalysis with transition metal complexes: Applications in organic synthesis. Chem. Rev. 2013, 113 (7), 5322–5363. 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz D. M.; Yoon T. P. Solar synthesis: Prospects in visible light photocatalysis. Science 2014, 343 (6174), 1239176. 10.1126/science.1239176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu C.; Chen Y. Biomolecule-compatible chemical bond-formation and bond-cleavage reactions induced by visible light. Tetrahedron Lett. 2015, 56 (7), 884–888. 10.1016/j.tetlet.2014.12.034. [DOI] [Google Scholar]
- Ryu K. A.; Kaszuba C. M.; Bissonnette N. B.; Oslund R. C.; Fadeyi O. O. Interrogating biological systems using visible-light-powered catalysis. Nat. Rev. Chem. 2021, 5 (5), 322–337. 10.1038/s41570-021-00265-6. [DOI] [PubMed] [Google Scholar]
- Geri J. B.; Oakley J. V.; Reyes-Robles T.; Wang T.; McCarver S. J.; White C. H.; Rodriguez-Rivera F. P.; Parker D. L. Jr.; Hett E. C.; Fadeyi O. O.; Oslund R. C.; MacMillan D. W. C. Microenvironment mapping via dexter energy transfer on immune cells. Science 2020, 367 (6482), 1091–1097. 10.1126/science.aay4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravelli D.; Fagnoni M.; Albini A. Photoorganocatalysis. What for?. Chem. Soc. Rev. 2013, 42 (1), 97–113. 10.1039/C2CS35250H. [DOI] [PubMed] [Google Scholar]
- Romero N. A.; Nicewicz D. A. Organic photoredox catalysis. Chem. Rev. 2016, 116 (17), 10075–10166. 10.1021/acs.chemrev.6b00057. [DOI] [PubMed] [Google Scholar]
- Wang Y. L.; Taylor D. L.. Fluorescence microscopy of living cells in culture, part a: Fluorescent analogs, labeling cells and basic microscopy; Academic Press: 1989. [PubMed] [Google Scholar]
- Terai T.; Nagano T. Small-molecule fluorophores and fluorescent probes for bioimaging. Pfluegers Arch. 2013, 465 (3), 347–359. 10.1007/s00424-013-1234-z. [DOI] [PubMed] [Google Scholar]
- Dean K. M.; Palmer A. E. Advances in fluorescence labeling strategies for dynamic cellular imaging. Nat. Chem. Biol. 2014, 10 (7), 512–523. 10.1038/nchembio.1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W.; Zeng Z.; Jiang J.-H.; Chang Y.-T.; Yuan L. Discerning the chemistry in individual organelles with small-molecule fluorescent probes. Angew. Chem., Int. Ed. 2016, 55 (44), 13658–13699. 10.1002/anie.201510721. [DOI] [PubMed] [Google Scholar]
- Wang H.; Li W. G.; Zeng K.; Wu Y. J.; Zhang Y.; Xu T. L.; Chen Y. Photocatalysis enables visible-light uncaging of bioactive molecules in live cells. Angew. Chem., Int. Ed. 2019, 58 (2), 561–565. 10.1002/anie.201811261. [DOI] [PubMed] [Google Scholar]
- Raviv Y.; Salomon Y.; Gitler C.; Bercovici T. Selective labeling of proteins in biological systems by photosensitization of 5-iodonaphthalene-1-azide. Proc. Natl. Acad. Sci. U. S. A. 1987, 84 (17), 6103–6107. 10.1073/pnas.84.17.6103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fancy D. A.; Kodadek T. Chemistry for the analysis of protein-protein interactions: Rapid and efficient cross-linking triggered by long wavelength light. Proc. Natl. Acad. Sci. U. S. A. 1999, 96 (11), 6020–6024. 10.1073/pnas.96.11.6020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato S.; Nakamura H. Ligand-directed selective protein modification based on local single-electron-transfer catalysis. Angew. Chem., Int. Ed. 2013, 52 (33), 8681–8684. 10.1002/anie.201303831. [DOI] [PubMed] [Google Scholar]
- Qi L.; Chen Y. Polarity-reversed allylations of aldehydes, ketones, and imines enabled by hantzsch ester in photoredox catalysis. Angew. Chem., Int. Ed. 2016, 55 (42), 13312–13315. 10.1002/anie.201607813. [DOI] [PubMed] [Google Scholar]
- We also compared the efficiency of the organic-dye-induced photocatalytic protein labeling by AzPh-biotin probe 1 with other labeling methods (Figure S19). The photocatalytic labeling demonstrated similar or better labeling efficiency compared to the UV-light induced labeling with AzPh-biotin probe 1 or the iodoacetamide probe targeting the cysteine residues, both for the BSA protein labeling and the complex cell lysates labeling.
- Geurink P. P.; Prely L. M.; van der Marel G. A.; Bischoff R.; Overkleeft H. S.. Photoaffinity labeling in activity-based protein profiling. In Activity-based protein profiling; Sieber S. A., Ed.; Springer Berlin Heidelberg: Berlin, Heidelberg, 2012; pp 85–113. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Kamlet A. S.; Steinman J. B.; Liu D. R. A biomolecule-compatible visible-light-induced azide reduction from a DNA-encoded reaction-discovery system. Nat. Chem. 2011, 3 (2), 146–153. 10.1038/nchem.932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis F. D.; Saunders W. H. Sensitized photolysis of organic azides. Possible case of nonclassical energy transfer. J. Am. Chem. Soc. 1968, 90 (25), 7033–7038. 10.1021/ja01027a026. [DOI] [Google Scholar]
- Leyshon L. J.; Reiser A. Sensitized photodecomposition of phenyl azide and α-naphthyl azide. J. Chem. Soc., Faraday Trans. 2 1972, 68 (0), 1918–1927. 10.1039/F29726801918. [DOI] [Google Scholar]
- Zhou Q. Q.; Zou Y. Q.; Lu L. Q.; Xiao W. J. Visible-light-induced organic photochemical reactions through energy-transfer pathways. Angew. Chem., Int. Ed. 2019, 58 (6), 1586–1604. 10.1002/anie.201803102. [DOI] [PubMed] [Google Scholar]
- Leyva E.; Platz M. S.; Persy G.; Wirz J. Photochemistry of phenyl azide: The role of singlet and triplet phenylnitrene as transient intermediates. J. Am. Chem. Soc. 1986, 108 (13), 3783–3790. 10.1021/ja00273a037. [DOI] [Google Scholar]
- Platz M. S. Comparison of phenylcarbene and phenylnitrene. Acc. Chem. Res. 1995, 28 (12), 487–492. 10.1021/ar00060a004. [DOI] [Google Scholar]
- Li Y. Z.; Kirby J. P.; George M. W.; Poliakoff M.; Schuster G. B. 1,2-didehydroazepines from the photolysis of substituted aryl azides - analysis of their chemical and physical-properties by time-resolved spectroscopic methods. J. Am. Chem. Soc. 1988, 110 (24), 8092–8098. 10.1021/ja00232a022. [DOI] [Google Scholar]
- Nielsen P. E.; Buchardt O. Aryl azides as photoaffinity labels. A photochemical study of some 4-substituted aryl azides. Photochem. Photobiol. 1982, 35 (3), 317–323. 10.1111/j.1751-1097.1982.tb02568.x. [DOI] [Google Scholar]
- Gritsan N. P.; Platz M. S. Kinetics, spectroscopy, and computational chemistry of arylnitrenes. Chem. Rev. 2006, 106 (9), 3844–3867. 10.1021/cr040055+. [DOI] [PubMed] [Google Scholar]
- Inui H.; Sawada K.; Oishi S.; Ushida K.; McMahon R. J. Aryl nitrene rearrangements: Spectroscopic observation of a benzazirine and its ring expansion to a ketenimine by heavy-atom tunneling. J. Am. Chem. Soc. 2013, 135 (28), 10246–10249. 10.1021/ja404172s. [DOI] [PubMed] [Google Scholar]
- Wu G.; Fang Y. Z.; Yang S.; Lupton J. R.; Turner N. D. Glutathione metabolism and its implications for health. J. Nutr. 2004, 134 (3), 489–492. 10.1093/jn/134.3.489. [DOI] [PubMed] [Google Scholar]
- Winterbourn C. C. Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 2008, 4 (5), 278–286. 10.1038/nchembio.85. [DOI] [PubMed] [Google Scholar]
- Rizk M. S.; Shi X.; Platz M. S. Lifetimes and reactivities of some 1,2-didehydroazepines commonly used in photoaffinity labeling experiments in aqueous solutions. Biochemistry 2006, 45 (2), 543–551. 10.1021/bi0516632. [DOI] [PubMed] [Google Scholar]
- Sies H.; Berndt C.; Jones D. P. Oxidative stress. Annu. Rev. Biochem. 2017, 86, 715–748. 10.1146/annurev-biochem-061516-045037. [DOI] [PubMed] [Google Scholar]
- Dougherty T. J.; Gomer C. J.; Henderson B. W.; Jori G.; Kessel D.; Korbelik M.; Moan J.; Peng Q. Photodynamic therapy. J. Natl. Cancer Inst. 1998, 90 (12), 889–905. 10.1093/jnci/90.12.889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierzyńska-Mach A.; Janowski P. A.; Dobrucki J. W. Evaluation of acridine orange, lysotracker red, and quinacrine as fluorescent probes for long-term tracking of acidic vesicles. Cytometry, Part A 2014, 85 (8), 729–737. 10.1002/cyto.a.22495. [DOI] [PubMed] [Google Scholar]
- Thome M. P.; Filippi-Chiela E. C.; Villodre E. S.; Migliavaca C. B.; Onzi G. R.; Felipe K. B.; Lenz G. Ratiometric analysis of acridine orange staining in the study of acidic organelles and autophagy. J. Cell Sci. 2016, 129 (24), 4622–4632. 10.1242/jcs.195057. [DOI] [PubMed] [Google Scholar]
- Lin Y. C.; Lin J. F.; Tsai T. F.; Chen H. E.; Chou K. Y.; Yang S. C.; Tang Y. M.; Hwang T. I. Acridine orange exhibits photodamage in human bladder cancer cells under blue light exposure. Sci. Rep. 2017, 7 (1), 14103. 10.1038/s41598-017-13904-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobs S. High resolution imaging of live mitochondria. Biochim. Biophys. Acta, Mol. Cell Res. 2006, 1763 (5–6), 561–575. 10.1016/j.bbamcr.2006.04.004. [DOI] [PubMed] [Google Scholar]
- Wisnovsky S.; Lei E. K.; Jean S. R.; Kelley S. O. Mitochondrial chemical biology: New probes elucidate the secrets of the powerhouse of the cell. Cell. Chem. Biol. 2016, 23 (8), 917–927. 10.1016/j.chembiol.2016.06.012. [DOI] [PubMed] [Google Scholar]
- Lin M. T.; Beal M. F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443 (7113), 787–795. 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- Lenaz G.; Fato R.; Genova M. L.; Bergamini C.; Bianchi C.; Biondi A. Mitochondrial complex i: Structural and functional aspects. Biochim. Biophys. Acta, Bioenerg. 2006, 1757 (9), 1406–1420. 10.1016/j.bbabio.2006.05.007. [DOI] [PubMed] [Google Scholar]
- Li N.; Ragheb K.; Lawler G.; Sturgis J.; Rajwa B.; Melendez J. A.; Robinson J. P. Mitochondrial complex i inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J. Biol. Chem. 2003, 278 (10), 8516–8525. 10.1074/jbc.M210432200. [DOI] [PubMed] [Google Scholar]
- Guan Q.; Wang X.; Jiang Y.; Zhao L.; Nie Z.; Jin L. Rna-seq expression analysis of enteric neuron cells with rotenone treatment and prediction of regulated pathways. Neurochem. Res. 2017, 42 (2), 572–582. 10.1007/s11064-016-2112-9. [DOI] [PubMed] [Google Scholar]
- Jin J.; Davis J.; Zhu D.; Kashima D. T.; Leroueil M.; Pan C.; Montine K. S.; Zhang J. Identification of novel proteins affected by rotenone in mitochondria of dopaminergic cells. BMC Neurosci. 2007, 8 (1), 67. 10.1186/1471-2202-8-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiaradia E.; Renzone G.; Scaloni A.; Caputo M.; Costanzi E.; Gambelunghe A.; Muzi G.; Avellini L.; Emiliani C.; Buratta S. Protein carbonylation in dopaminergic cells exposed to rotenone. Toxicol. Lett. 2019, 309, 20–32. 10.1016/j.toxlet.2019.04.002. [DOI] [PubMed] [Google Scholar]
- Samali A.; Cai J.; Zhivotovsky B.; Jones D. P.; Orrenius S. Presence of a pre-apoptotic complex of pro-caspase-3, hsp60 and hsp10 in the mitochondrial fraction of jurkat cells. EMBO J. 1999, 18 (8), 2040–2048. 10.1093/emboj/18.8.2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamai S.; Iida H.; Yokota S.; Sayano T.; Kiguchiya S.; Ishihara N.; Hayashi J.; Mihara K.; Oka T. Characterization of the mitochondrial protein letm1, which maintains the mitochondrial tubular shapes and interacts with the aaa-atpase bcs1l. J. Cell Sci. 2008, 121 (15), 2588–2600. 10.1242/jcs.026625. [DOI] [PubMed] [Google Scholar]
- Gross A. Bcl-2 family proteins as regulators of mitochondria metabolism. Biochim. Biophys. Acta, Bioenerg. 2016, 1857 (8), 1243–1246. 10.1016/j.bbabio.2016.01.017. [DOI] [PubMed] [Google Scholar]
- Arner E. S.; Holmgren A. Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 2000, 267 (20), 6102–6109. 10.1046/j.1432-1327.2000.01701.x. [DOI] [PubMed] [Google Scholar]
- The nucleophilic protein residues Cys, Thr, Tyr, Asp, Lys, and Ser were detected in the MCF-7 cells photolabeling (Figure S29, Table S4).
- Zhu H.; Tamura T.; Fujisawa A.; Nishikawa Y.; Cheng R.; Takato M.; Hamachi I. Imaging and profiling of proteins under oxidative conditions in cells and tissues by hydrogen-peroxide-responsive labeling. J. Am. Chem. Soc. 2020, 142 (37), 15711–15721. 10.1021/jacs.0c02547. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.