

Summary
Congenital abnormalities of the kidney and urinary tract are amongst the most common birth defects affecting 3% of newborns. The human kidney forms around a million nephrons from a pool of nephron progenitors over a 30-week period of development. To establish a framework for human nephrogenesis, we spatially resolved a stereotypical process by which equipotent nephron progenitors generate a nephron anlagen, then applied data-driven approaches to construct three-dimensional protein maps on anatomical models of the nephrogenic program. Single cell RNA sequencing identified progenitor states which were spatially mapped to the nephron anatomy enabling the generation of functional gene-networks predicting interactions within and between nephron cell-types. Network mining identified known developmental disease genes and predicts targets of interest. The spatially resolved nephrogenic program made available through the Human Nephrogenesis Atlas (https://sckidney.flatironinstitute.org/) will facilitate an understanding of kidney development and disease, and enhance efforts to generate new kidney structures.
Keywords: Human kidney, development, disease, single cell, spatial mapping, nephrogenesis, gene regulatory networks, machine learning, image registration
Graphical Abstract
eTOC Blurb
Lindström et al., show mouse and human nephrogenesis follow evolutionary conserved and stereotypical programs. Spatial mapping of transcriptional profiles predicts cell intermediates and regulatory interactions at play in development and disease. A web-interface facilitates viewing and analysis of these findings.
Introduction
Birth defects across all organ systems often manifest as gross anatomical and cellular changes. These are difficult to link to specific developmental events or cell-types. While single-cell-omic approaches now facilitate the dissection and cataloguing of adult organs into their cellular components at an RNA and chromatin level (Macosko et al., 2015; Han et al., 2018; Schaum et al., 2018), performing equivalent analyses on developmental and disease processes is challenging. Tools that extrapolate developmental trajectories from fluid cell profiles differentiating from one state to the next do not provide key spatial information critical to a mechanistic understanding of the developmental program
The importance of understanding human developmental programs - i.e. the sequential series of differentiation steps by which progenitors and precursors give rise to their mature counterparts - is well illustrated by malformations of the genitourinary system (Figure 1A). Around 20–30% of all neonatal anomalies map to the genitourinary system accounting for 50% of pediatric end-stage kidney disease (Schedl, 2007; Hildebrandt, 2010). Once formed, the kidney controls homeostasis through blood pressure regulation and filtration, metabolites are excreted and solutes recovered predominantly through the actions of highly specialized cell types within distinct segments of the nephron (Lee et al., 2015). All nephron cell types are thought to arise from an equipotential nephron progenitor pool established at the onset of kidney development (Boyle et al., 2008; Kobayashi et al., 2008; McMahon, 2016). Recent single cell sequencing and spatial mapping studies indicate there are at least 23 cell-types within the nephrons of the adult male and female mouse kidney (Ransick et al., 2019). Considerable cell diversity has been reported for the human kidney (Lake et al., 2019) and it is likely this will expand as the depth and resolution of adult human studies increases.
Figure 1 – Assembly of the Human Nephrogenesis Atlas.
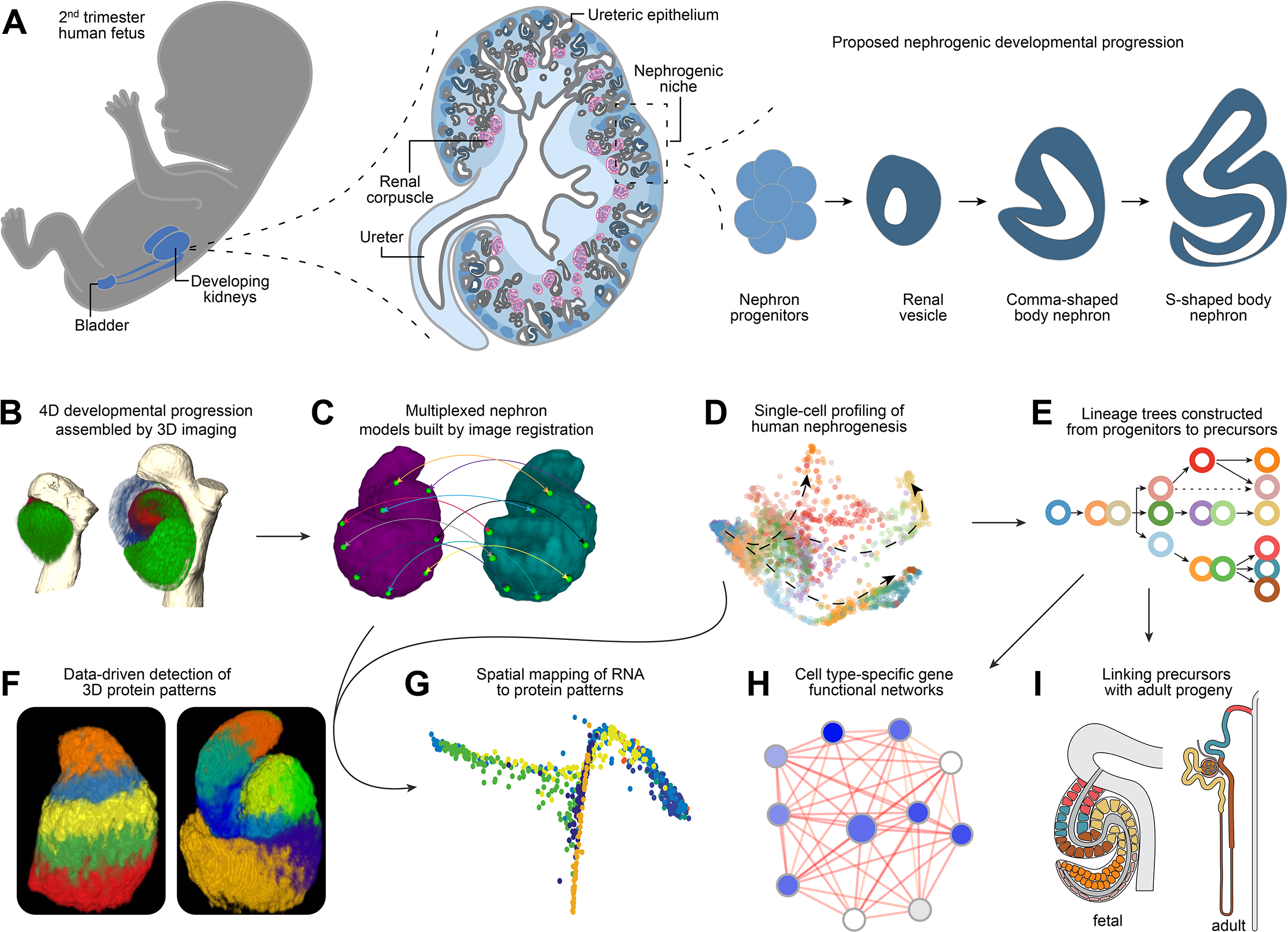
Schematics and data illustrating: (A) Basic principles of human nephrogenesis. Schematic depicts genitourinary system in human fetus (left), a cross section of a developing human kidney (middle), and the proposed nephrogenic program (right). Dashed lines indicate magnified portions shown in a left to right direction. Shades of blue from lightest to darkest in kidney cross section indicate inner medulla, outer medulla, cortex, nephron progenitors, developing nephrons. Ureteric epithelium is shown in grey and maturing nephrons beyond the S-shaped body stage in dark grey. Renal corpuscles as indicated (pink/blue). (B) Visualization of immunofluorescently labelled nephrons in 3D. (C) Image registration of nephron protein data into multiplexed models. (D) Single cell RNA profiling of human nephrogenesis. (E) Establishing a lineage tree for nephrogenesis. (F) Computational approaches to predict protein patterns in 3D space. (G) Spatial mapping of RNA to protein models. (H) Building cell-type specific functional gene networks. (I) Linking precursors with their mature progeny.
The nephrogenic program has been well studied in mammalian model systems, principally the mouse (Saxen, 1987; McMahon, 2016; Figure 1A). Mesenchymal progenitor cells for the nephron and interstitial cell lineages overlie epithelial progenitors of the kidney’s collecting system, generating a highly interactive mobile nephrogenic niche that drives kidney assembly. Wnt9b signaling to mesenchymal metanephric nephron progenitor cells triggers commitment to the nephrogenic program (Davies and Garrod, 1995; Carroll et al., 2005; Kuure et al., 2007; Park et al., 2007). Nephron progenitors cluster into pretubular aggregates, then epithelialize to form cystic renal vesicles, which transition through morphologically distinct stages (comma and S-shaped bodies), interconnecting with the epithelium of the developing collecting system, to establish the kidney’s fluid transporting epithelial network (Huber, 1905; Osathanondh and Potter, 1963; Oliver, 1968; Little et al., 2007; Georgas et al., 2009; Kao et al., 2012; Short et al., 2014). Though precursor-product relationships are not well understood, growth and morphogenesis of the nephron anlagen is coupled with the emergence of dynamic profiles of gene expression along a proximal-distal axis of epithelial specialization that prefigures and determines the molecular and cellular organization of the adult nephron (Stark et al., 1994; Nakai et al., 2003; Carroll et al., 2005; Grieshammer et al., 2005; Cheng et al., 2007; Kobayashi et al., 2008; Georgas et al., 2009; Karner et al., 2011; Lindström et al., 2018b, 2018a).
In previous efforts to resolve the diversity of nephron precursors, we and others have applied single cell RNA sequencing (scRNA-seq) to human nephrogenesis (Lindström et al., 2018a; Menon et al., 2018; Hochane et al., 2019; Tran et al., 2019). When cell clusters are compared with in situ and immunolocalization studies (Lindström et al., 2018b, 2018a), distinct cell clusters are likely an amalgamation of multiple related precursor types. Delineating the stepwise program of differentiation by which nephron progenitors generate nephrons will facilitate our understanding of disease origins. Further, the application of insight to stem cell systems will enable both the modelling of kidney disease in vitro, and de novo generation of kidney cell types and kidney structures.
To establish the spatial organization, diversity, and gene expression profiles of human nephron precursors we adopted and developed several approaches that are broadly applicable to other developmental systems. The data are brought together as a cohesive map of protein distribution, gene expression, and gene networks within a webinterface (https://sckidney.flatironinstitute.org/) that facilitates study of the human kidney.
Results
An overview of the workflow and projected goals underlying the generation of an atlas of human nephrogenesis is provided in Figure 1. To provide a template for each method and their integration, these are detailed in the STAR Methods and a walk-through for the use of the Human Nephrogensis Atlas is provided online. Confocal imaging and image segmentation of the human nephrogenic zone with distinct antibody sets enabled the assembly of a 4-dimensional map of human nephrogenesis (Figure 1A, B). Image registration with these data was then used to generate multiplexed nephron models (Figure 1C). Extensive scRNA-seq analysis was performed on the nephrogenic zone (Figure 1D), transitional cell types verified in vivo and compared against previous validation (Figure 1E), and multiplex protein patterns (Figure 1F) were employed for relational mapping of transcriptomic data to a computational model of a developing nephron (Figure 1G). Cell type-specific gene functional networks were built using an integrative machine learning framework to examine genes associated with congenital abnormalities and kidney disease, and to predict potential disease associated partners (Figure 1H). Finally, predictive precursor-product relationships were framed from the analysis of developmental and adult scRNA-seq datasets (Figure 1I). The following sections provide a detailed insight into the approaches, and the collection, analysis and interpretation of data, at each step.
Visualizing progressive development of the human nephron
To visualize how mammalian nephrons form, we performed whole-mount immunolabeling of mouse and human nephrogenic niches with JAG1 antibodies as a reference landmark across nephrons, experiments and samples, in combination with antibodies recognizing eighteen other informative regional protein markers, incorporating DAPI labelling to resolve nuclei (STAR Methods). Selection of antibody targets was designed to maximize detection of cell diversity in conjunction with nephron patterning and morphogenesis based on prior studies of nephron patterning (Georgas et al., 2009; Lindström et al., 2018c, 2018b, 2018a; Menon et al., 2018; Hochane et al., 2019), and to link development to disease given the known developmental roles of many of these proteins, e.g. POU3F3, HNF1B, JAG1, PAX2, and LHX1 in mouse and human kidney studies (Spinner et al., 2001; Nakai et al., 2003; Kobayashi, 2005; Grote et al., 2007; Heliot et al., 2013). Two hundred and fifty-one human (sampled from 14 kidneys ranging between 13 and 17 weeks) and one hundred and seventy-seven mouse nephrons (sampled from 16 E15.5 C57BL/6J kidneys) were captured at a subcellular resolution by confocal imaging (voxel resolution: 0.1μm × 0.1μm × 0.35μm z-resolution through whole nephrons), digitally isolated from image stacks using Amira, and categorized into developmental stages based on gradual morphological changes, pattern, and size. Together, these data resolved in 3-D detail the developmental progression for human and mouse nephrogenesis to the S-shaped body stage (Figure 2A; Supplementary Figure 1A–C).
Figure 2 -. The developmental progression of human nephrogenesis.
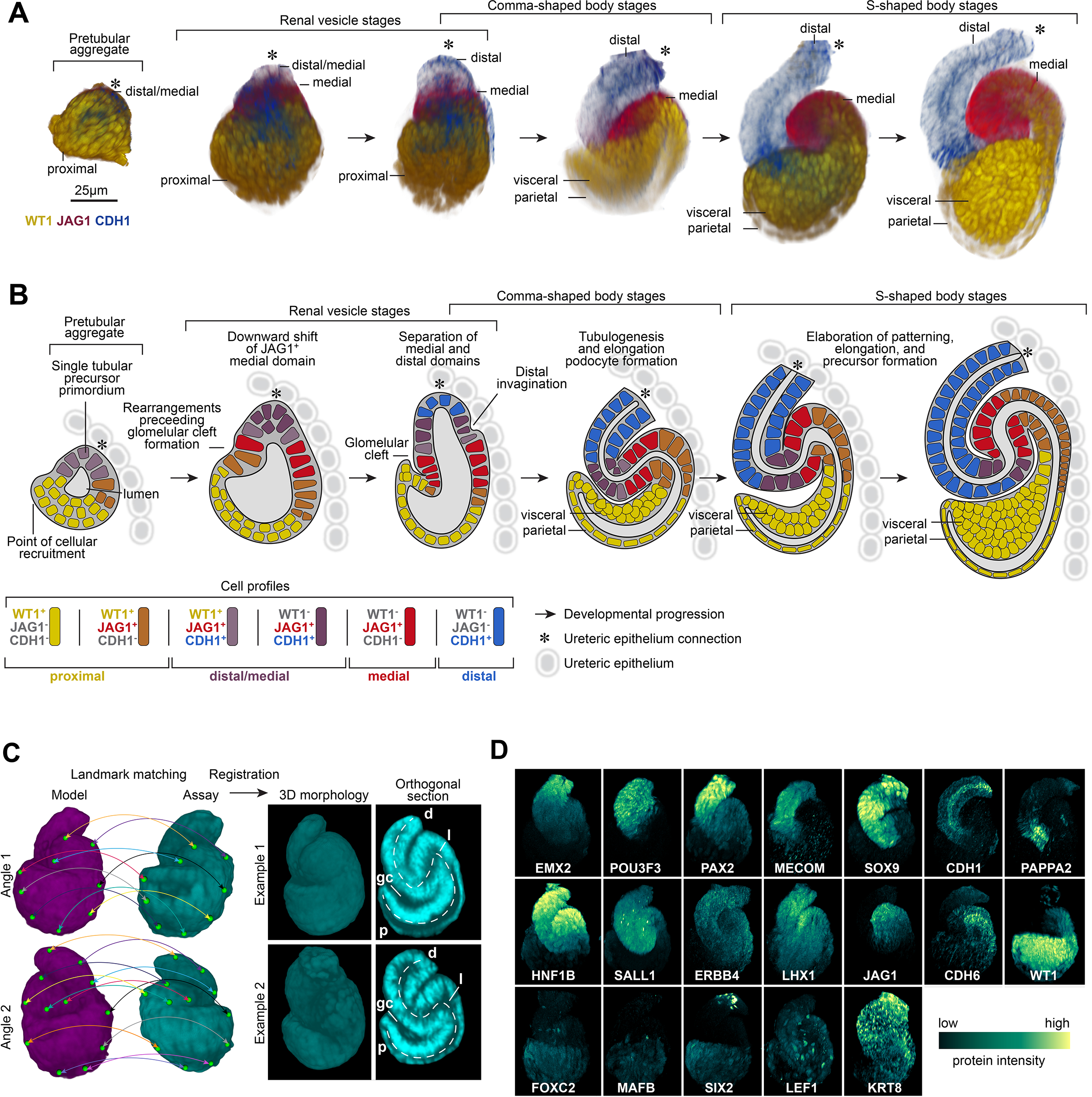
(A) Immunofluorescently labelled human nephrons rendered in 3D shown from most immature stages (left) to most mature stages of S-shaped body nephrons (right). (B) Schematic for nephrogenesis for an equivalent range of nephrons as shown in A, displaying separation of precursor identities over time. Immunofluorescent labels, features, and developmental stages as indicated on fields. Key defines cell profiles and markers. (C) Strategy for image registration and examples of registered 3D S-shaped body nephrons. A multi-stage registration process that utilized landmarks (green marks and colored lines linking matching landmarks), a general affine transform, and an elastic (ANTs) transform. Successfully registered examples of two S-shaped body nephrons shown at as orthogonal sections through the middle at two angles. Dotted lines indicate proximal distal axes. p: proximal. d: distal. gc: glomerular cleft. l: lumen. (D) Average localization patterns for proteins based on registered nephrons. Immunofluorescent labels as indicated on fields.
Nephron development initiates within a pretubular aggregate of tightly packed nephron progenitors, likely tuned by complex expression of partially redundant cadherins (Cho et al., 1998; Mah et al., 2000). We opted to visualize the acquisition of polarity and epithelial characteristics using CDH1, deposited in a distal-to-proximal direction, generating an apical epithelial surface shaped like an inverted funnel opening into a space that later will incorporate the kidney’s glomerular filter. The gradual nature of nephrogenesis (Lindström et al., 2018a) is mirrored in the progressive distal-to-proximal acquisition of epithelial characteristics. In human nephrogenesis, epithelialization begins in a distal JAG1+/ CDH1+ domain in pretubular aggregates where proximal recruitment of mesenchymal cell types is still taking place over an extended period of time relative to the mouse kidney (Figure 2A). In the mouse kidney, the renal vesicle is generally though to represent the first epithelialized nephrogenic structure (Supplementary Figure 1C; Little et al., 2007; Georgas et al., 2009; McMahon, 2016). We reserve the term renal vesicle to describe a fully epitheliarized nephron progenitor for both species. In the human and mouse epithelial renal vesicle, the distal half resolves into two domains comprising distal CDH1+/JAG1− cells and medial CDH1+/JAG1+ cells (Figure 2A–B; Supplementary Figure 1C). An additional medial CDH1−/JAG1+ domain emerges during the transition to comma- and S-shaped bodies (Figure 2A–B). During the transition from renal vesicle to comma-shape body, inward folding between the medial and distal segments results in a distal orientated bend in the nephron anlagen, while proximal epithelial movements establish the glomerular cleft. Vascular endothelial cells invade the cleft to establish the glomerular vascular supply for renal filtration.
A schematic model for human nephrogenesis based on the cumulative view of these data (Figure 2B) is consistent with a gradual recruitment of cells in a distal-to-proximal order followed by progressive formation of nephron domains (Lindström et al., 2018a). Development of the mouse nephron over a similar developmental time course was similar, aside from the forementioned epithelialization dynamics (Supplementary figure 1B–C), and human nephrons were approximately twice the size of their mouse counterparts with two-fold greater cellularity (Supplementary figure 1D). At an individual marker/domain level, CDH1+/Cdh1+ and JAG1+/Jag1+ cell numbers matched this two-fold difference and displayed similar levels of cell variation (Supplementary figure 1E, F); indicative of linear scaling of domains. The 3-D morphologies and protein patterns were consistently sharper in the human nephron across more than twenty-five proteins analyzed. The size difference and the slower developmental pace of human nephrogenesis, roughly eight times slower than the mouse kidney to the S-shaped body stage (Lindström et al., 2018d), likely contribute to the enhanced resolution of human nephrogenesis in these data.
Image registration predicts stereotyped anatomies
Nephron formation within and across mammalian species follows a highly consistent early developmental progression at different stages of kidney development though there are clear differences in the final pattern and cell composition depending on the time of nephron formation (Ransick et al., 2019). To determine whether nephrogenesis is stereotypical, we digitally isolated renal vesicles and S-shaped bodies from large confocal stacks from mammalian kidneys of distinct developmental ages using Amira. To combine protein localization data, we developed a semi-automated image processing pipeline applying ANTs and Woolz to process nephron image stacks, register stacks to nephron models, and thus by applying an affine transform and elastic registration, multiplexed protein localization models were generated with a subcellular resolution (STAR methods). Nephron anatomies and fine anatomical details were preserved, following registration: similar cellular domains occupied the same voxel space with minimal distortions to morphology or patterning (Figure 2C). The lumen space (a very narrow anatomical feature, 20–30 μm), the forming glomerular cleft within the S-shaped body, and even distinct podocyte morphologies were maintained in the registered nephrons. Together, these data highlight the low morphological variability in the early nephrogenic program. Importantly, the ability to co-register different nephron precursors within a kidney, and between kidney samples at different ages (13 to 17 weeks), shows human nephrogenesis is a stereotypical process following a tightly controlled molecular and cellular program. Interestingly, S-shaped body nephrons displayed a left- or right-handed chirality depending on their orientation to the adjacent ureteric epithelium (Supplementary figure 1G). As expected, proteins were distributed in distinct distal-to-proximal patterns (Figure 2D). The multiplex nephron models for both renal vesicles and S-shaped body nephrons in themselves provide high-resolution views of the exact positioning of proteins with known roles in genetically linked developmental renal syndromes such as CAKUT (congenital anomalies of the kidney and urinary tract; (Schedl, 2007); including POU3F3, PAX2, HNF1B, SALL1, LHX1, and WT1. In addition, each distinct contribution enabled the creation of a high-resolution map of protein-defined, cellular diversity (Supplementary figure 1H–I), which is predicted to correlate with underlying differences in gene expression for each cognate gene within distinct cell populations.
Single-cell transcriptomes identified for human nephrogenesis
As the next step to both integrate scRNA-seq datasets to the spatial maps, and to resolve a fuller set of S-shaped body transcriptional signatures, we performed independent scRNA-seq analyses on two week-14 kidneys. We specifically focused on the nephrogenic niche and not more mature cell-populations, profiling 24,157 cells within a median of 2,644 genes/cell cells within the cortical nephrogenic zone (Supplementary figure 2A). To focus on the most informative cells, a gene/cell count threshold of greater than 3,000 identified 8,316 cells of which 2893 cell comprised non-cycling subsets.
This high-quality cell-set was evaluated further through reiterative unbiased clustering and gene enrichment analyses (see STAR Methods for details and Supplementary figure 2A–J; Supplementary table 1). Eighteen distinct transcriptional states were identified using SEURAT (Satija et al., 2015; Stuart et al., 2019) and by examining differential expression of key marker genes on tSNE analysis (Figure 3A; Supplementary table 2). To establish the range of sampled transcriptomes in clusters, we analyzed expression profiles for genes that decrease slowly after the progenitor state (O’Brien et al., 2016: SIX1), genes that are transiently upregulated in pretubular aggregates through to S-shaped body nephrons and then down-regulated (Lindström et al., 2018d, 2018b; Tran et al., 2019: CAPN6, CRYM, OLFM3, JAG1; Supplementary figure 3A), and genes that are first activated in S-shaped body nephrons (Lindström et al., 2018d, 2018b; Tran et al., 2019: NPHS1, CDH6, HNF4A, MECOM, GATA3, PAPPA2; Supplementary figure 3B). While false negatives can challenge interpretations of individual cells in scRNA-seq data, each of these genes were detected robustly at a cluster level. The sampled cells ranged from nephron progenitors to S-shaped body nephron cells. As expected, cell clusters formed gradual transitions from one state to another with the most distinct states being the nephron progenitors and the six clusters identified as S-shaped body nephron cells (Supplementary figure 3C). The S-shaped body cells displayed deep transcriptional differences and analyses of the top fifty differentially expressed genes per group highlighted the specificity and unique gene expression signatures emerging within each cell-type (Supplementary figure 3D). The 18 clusters expressed on average 12,900 genes - likely a good representation of full transcriptomes. Importantly, three-fold more clusters were detected for the S-shaped body compared to previously reported data (Lindström et al., 2018a; Menon et al., 2018; Hochane et al., 2019; Tran et al., 2019) and the depth and breadth of the resulting transcriptomic data enabled subtle gene expression changes to be visualized within scarce transitioning cell-states.
Figure 3 -. Single cell analyses and spatial mapping resolve precursor transcriptomes.

(A) tSNE plot of 18 cell clusters of cells from human nephrogenic lineage with cell-types and marker genes indicated. (B) Graph-based dimensionality reduction plots of all cell transcriptomes (clusters 1–18) color coded according to cell clustering in Figure 3A. Marker genes for each endpoint shown as inserts. (C, D) Lineage tree model for nephrogenesis placing cell transcriptomes in clusters 1–18 into a tree formation and into an anatomical schematic for the nephrogenic niche. Colors and numbers correspond to cluster colors in Figure 3A. Arrows indicate direction of differentiation. Dotted arrow indicates uncertain relationship. Stages and precursor regions or identities as annotated. White areas with small grey dots in D mark the ureteric epithelium and the interstitium which did not form part of these analyses.
Resolving changing expression profiles across time and anatomy
Assigning cell clusters to precise anatomical bins remains a significant challenge in single-cell omic analyses. To unbiasedly resolve cell states between nephron progenitors and the S-shaped body cell populations we used graph-based dimensionality reduction (DRGraph) (Menon et al., 2018). Three main trajectories were evident, starting from nephron progenitors (clusters 1 and 2) and ending in cells displaying markers of distal (cluster 16, 17, 18), proximal (cluster 12), podocyte (cluster 8), and parietal epithelium (cluster 9) (Figure 3B; Supplementary figure 4A–D). These cluster relationships agreed with correlation analyses of all variable genes (12995) across the clusters (Supplementary figure 4E) which similarly grouped clusters into distal, proximal, and podocyte/parietal categories. Parietal cells were projected as an intermediate state between podocyte and proximal lineages. Thus, DRGraph trajectories, correlation analyses of all variable genes across all cells, and SWNE projections (Supplementary figure 2J) suggested a consistent developmental ordering of the clusters.
While computed developmental trajectories are useful in predicting how cells differentiate and their relationships in developmental programs, we used RNAscope and secondary validation to simultaneously visualize the expression of multiple genes to annotate cells with accurate developmental and anatomical terms. The genes used for secondary validation were predicted to display dynamic expression profiles across anatomically distinct developmental stages, facilitating the assignment of precise anatomical bins (Supplementary figure 5A–D; Supplementary figure 5E–J). Emerging proximal and distal clusters from pretubular aggregate to renal vesicles were annotated by TMEM72/SLC39A8/LAMP5 and COL4A4/TMEM72/SLC39A8, respectively. In the S-shaped body, SLC39A8/LAMP5/HNF4A facilitated resolving proximal cell identities, comprised predominantly of HNF4A/LAMP5, and occasional SLC39A8/HNF4A, positive cells. SLC39A8 straddles the border between distal and medial S-shaped body domains. COL4A4/HNF4A/EFNB2 identified cell fates formed during the differentiation of distal, proximal and podocyte fates, respectively (Supplementary figure 5A–J). Anatomical and developmental annotation resulted in an anatomical model for the 18 transcriptomes that predicts hierarchical relationships amongst the cell types and potential relationships to the adult nephron (see later data and discussion; Figure 3C–D). Collectively, these data have been incorporated into a spatially resolved and searchable Human Nephrogenesis Atlas (NephMap - https://sckidney.flatironinstitute.org/).
Unsupervised protein clustering predicts precursor populations
Twelve distinct transcriptomes characterized the progression of nephrogenesis from renal vesicles to S-shaped bodies (Figure 3C–D). To determine if protein combinations in the nephron models could be formalized into distinct combinations of protein abundance corresponding to precursor populations, we performed k-means clustering treating each voxel within the multiplexed 3D models as an n-dimensional vector in which each dimension corresponds to the abundance of a single protein. For renal vesicles we based clustering on the localization of 18 proteins, while for the S-shaped body nephrons we performed clustering on 19 proteins (Figure 4A). In renal vesicles, patterns formed increasing numbers of parallel layers along the distal-proximal axis as increasing k-values were used as the input, thus indicating proteins were distributed in gradients along the proximal distal axis (Supplementary figure 6A). By the S-shaped body stage, eight patterns were predicted in separate distinct regions along the proximal-distal axis. These patterns were stable across increasing k-values, suggesting each consisted of a unique protein combination with defined boundaries consistent with previously postulated precursor domains (Figure 4A; Supplementary figure 6B; Lindström et al., 2018b). The protein patterns and abundance of proteins within each pattern support the model that the tubular portion of the nephron, excluding podocytes and parietal epithelial cells of the renal filter, forms from a single precursor domain, which separates as development progresses into distal and medial domains (Figure 4B–C). For example, initially CDH1 and JAG1 overlap, then separate into distal and medial domains (Figure 4B–C; Supplementary figure 6E; Figure 2B). However, other proteins such as EMX2 and HNF1B remain co-localized (Figure 4B–C; Supplementary figure 6E).
Figure 4 -. Unsupervised clustering approaches predict precursor populations.

(A) Workflow of computational approach used to apply k-means clustering to 3D multi-dimensional voxel-based data and resulting predictions of combinatorial protein patterns. (B, C) Protein abundance across predicted protein patterns in renal vesicles and S-shaped body nephrons. (D) Schematic of mapping of cells to 8 protein patterns in the S-shaped body nephron and proportion of cells of the selected clusters mapping to each protein pattern. (E) Graph-based dimensionality reduction plots of all cell transcriptomes (clusters 1–18) showing cells spatially mapped to S-shaped body patterns. Colors in E correspond to protein patterns as shown in D. CNT: connecting tubule. D/M: distal/medial. P: Proximal. DT: distal tubule. aLOH: analage of loop of Henle. dLOH: putative analage of descending loop of Henle. PTd: analage of proximal tubule. PTp: analage of proximal tubule and parietal epithelium. RC: renal corpuscle.
Three-dimensional spatial mapping of single-cell transcriptomes
We examined whether mRNA levels could broadly approximate levels of encoded protein products by mapping cell gene expression profiles to the 3D protein models based on gene-to-protein relationships (Figure 4D). Three hundred and thirty-two S-shaped body cells mapped to eight regional domains within the S-shaped body with >11,000 genes detected per pattern. Differentiation trajectories were predicted with DRGraph for cells transitioning from the pretubular aggregate to S-shape body stage and mapped cell-groupings were projected onto these trajectories, with cells mapping to each pattern defining distinct regions of the trajectory (Figure 4E; Supplementary figure 6F). As protein and mRNA abundance can vary we scrutinized gene enrichment in cells mapped to patterns 1–8, and compared these against the conventional scRNA-seq clustering algorithms (Supplementary figure 7A vs. Supplementary figure 3D). Mapped cells displayed similar gene enrichment to the conventional clustering with signatures associated with proximal-distal nephron cells, but with distinct differences. Validation by in situ hybridization for genes of interest showed co-expression of in cell population highlighting additional cell states e.g. COL4A4 and GATA3, TMEM72 and CAPN6, and EFNB2 and PCDH9 (Supplementary figure 7B–G).
In reconciling the gene expression profiles generated by spatial mapping to S-shape body nephrons with those from conventional clustering algorithms (Supplementary figure 7A vs. Supplementary figure 3D), several genes highlighted different views reached by these methods. SLC39A8, encoding a transmembrane zinc transporter reported to exhibit preweaning lethality in mouse mutants (International Mouse Phenotyping Consortium). Through conventional gene enrichment analysis, SLC39A8 was differentially expressed in clusters 13, 14, 15, (Supplementary table 2) but not cluster 16 (Figure 3D and Figure 4A, D). However, differential gene expression based on spatially mapped cells showed SLC39A8 to be expressed in a distal subdomain of the S-shaped body nephron equivalent to pattern 2; RNA-Scope in situ hybridization confirmed this distribution in vivo (Supplementary figure 7A, H). The increased temporal resolution provided by spatial mapping of transcriptomes therefore accurately identified SLC39A8 as a differentially expressed gene marking a previously uncharacterized region of the S-shaped body nephron. Likewise, transcriptomic analyses of LAMP5, a lysosome-associated membrane glycoprotein required for normal neural activity (Tiveron et al., 2016), showed enrichment in clusters 7, 9, and 11 but not in S-shaped body clusters. However, spatial mapping predicted expression and LAMP5 transcripts mapped to a narrow region of the proximal S-shaped body (pattern 6; Supplementary figure 7I). A striking example showing complementary strengths of applying spatial transcriptomics comes from the analysis of HNF4A which encodes a transcriptional regulator required for proximal tubule development and linked to CAKUT (Marable et al., 2018, 2020). HNF4A expression was predicted to overlap with LAMP5 in the proximal S-shaped body in pattern 6, despite divergent expression at earlier stages of nephrogenesis. This prediction was borne out mapping expression in vivo (Supplementary figure 7J), with HNF4A mapping to a small domain within the S-shaped body (discussed further in Figure 6). In summary, mapping of RNA profiles to protein domains provides a temporal resolution of gene expression patterns identifying distinct subdomains and gives detailed molecular insight into cell diversification underlying nephron differentiation.
Figure 6 -. Linking precursors and progeny.

(A) Correlation of gene expression between adult mouse clusters and human developmental clusters in S-shaped body. (B) Proposed model for relationships between S-shaped body precursors and adult nephron cell-types based on correlation analyses. (C) Expression of transcription factors in the adult mouse nephron. (D) Immunofluorescent stains for transcription factors in human nephrons. White arrowheads indicate putative macula densa precursors. Schematics indicating the area each protein is detected in S-shaped body nephrons. Schematics also summarize data in Supplementary figure 11. (E-F) Immunofluorescent labelling of human nephrons during serial stages of development and schematic indicating relationship between macula densa precursors and mature derivatives. MD: macula densa. CLSN: capillary loop stage nephron. Labelling as indicated on fields.
Beyond the single gene: gene networks in development and disease
To build a richer view of gene expression environments and facilitate rapid data mining to pinpoint cell-disease/dysgenesis relationships, we applied a regularized Bayesian machine learning approach building cell type-specific and nephron lineage-based functional gene networks, combining the scRNA-seq data with ~1500 publicly available human genomic datasets (Greene et al., 2015; Wong et al., 2018) (Figure 5A; https://sckidney.flatironinstitute.org/genenetwork).
Figure 5 -. Cell-type specific functional gene networks in development and disease.
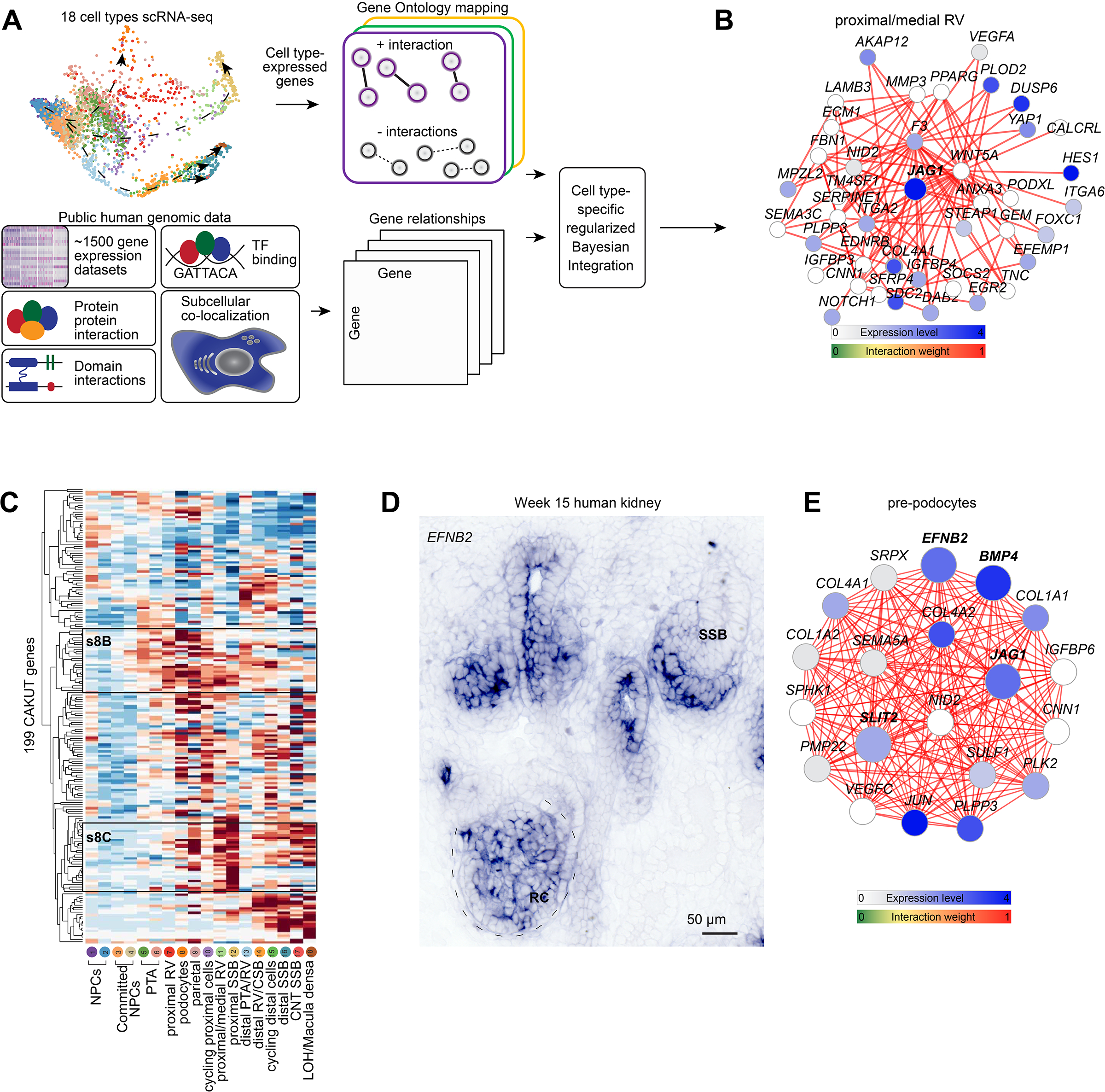
(A) Schematic for building cell-type specific functional gene networks. (B) Functional gene network for JAG1 (in bold) in the proximal/medial renal vesicle (cluster 11). (C) Hierarchical clustering of 199 CAKUT genes across cell cluster identities 1–18. High/Low as indicated. S8B and s8C annotations indicate regions highlighted in Supplementary figure 8B, C. (D) In situ hybridization for EFNB2 on human fetal kidney. (E) Example of quadruple gene network for EFNB2, JAG1, SLIT2, and BMP4 – in bold. Scales and stains as indicated. Expression levels and edge weights as indicated by scales on panels. PTA: pretubular aggregate. RV: renal vesicle. CSB: comma-shaped body nephron. SSB: S-shaped body nephron. CNT: connecting tubule. LOH/MD: loop of Henle/Macula densa precursor. RC: renal corpuscle. Gene network thresholds set for visualization purposes to ensure network simplicity.
As a first step, we constructed cell type-specific/non-specific gene interactions. For each cell type, all genes were analyzed for cell type-specific expression and functional relatedness. Gene-pairs were labelled as cell type-specific (categorized as ‘positive’) when both genes were differentially expressed in the same cell type and co-annotated in a shared GO biological process term. Gene-pairs not co-expressed in the same cell type but with similar or different functional roles were used as negatives. These labelled positive and negative gene pairs allowed us to develop a supervised approach to predict cell-type specific functional relatedness for all pairs of genes by training a regularized Naive Bayesian classifier (Greene et al., 2015; Wong et al., 2018) to distinguish between positive and negative gene pairs across 1,541 genome-scale data sets (Supplementary table 5, Star Methods). Using the trained classifier for each cell type, we predicted cell-type specific functional relatedness for every pair of genes as an edge score (0~1) in a weighted gene functional network, and generated a total of 18 cell type networks.
To validate these networks, we first explored genes with known expression, function and interacting partners. JAG1, which is active in proximal nephron development (Liu et al., 2013), was identified as being tightly connected to Notch pathway components NOTCH1 and HES1 in the proximal/medial renal vesicle network where JAG1 was strongly expressed (Figure 5B) but as expected not in the distal most connecting tubule region where JAG1 expression was absent (Supplementary figure 8A). To focus on renal dysgenesis and disease, genes published and linked to kidney disease (KD) - with an emphasis on CAKUT (Hildebrandt, 2010; Vivante et al., 2014) - were hierarchically clustered based on their cell type-enriched gene expression in the Human Nephrogenesis Atlas. Of the 272 genes, 199 (~75%) were enriched in distinct cell-types (Figure 5C). Proximal epithelial cells abutting the podocytes showed strong expression of the lipid and solute transporter proteins LRP2, SLC7A9, SLC3A1, and SLC5A2, which are components of adult proximal tubule cells (Supplementary figure 8B). PAX8 and WNT4 defined early differentiation and epithelialization stages, Other gene sets exhibiti cell-type specific expression of multiple pathway components; for example, expression of the Notch pathway genes JAG1, NOTCH2 and LFNG was proximal biased (Supplementary figure 8C), consistent with published mouse and human studies (Chen and Al-Awqati, 2005; Cheng et al., 2007; Chung et al., 2016; Lindström et al., 2018b).
Genes linked to a range of renal disease categories (nephrotic syndrome, renal tubular acidosis, kidney stone, CAKUT) have previously been mapped to adult mouse cell transcriptomes (Park et al., 2018). Analysis of the 20 CAKUT genes evaluated by Park et al. highlights the complementary insight from comparisons to developmental data. As examples, EYA1 and SALL1 are required for nephron progenitor self-renewal (Basta et al., 2014; Xu et al., 2014) and show expression in nephron progenitors consistent with this role. However, in the adult mammalian kidney, EYA1 and SALL1 map to podocytes and unknown cell-types, that may not relate to CAKUT (Park et al., 2018; Ransick et al., 2019) (Supplementary figure 8D). In contrast, JAG1 is known to have multiple functions in the kidney. JAG1 is required for patterning of the nephron (Cheng et al., 2007; Liu et al., 2013) and in the differentiation and maintenance of collecting duct cell-types in the adult kidney (Chen et al., 2017). Developmental and adult datasets identify each cell population (Supplementary figure 8D). Collectively, direct comparison of the CAKUT gene subset showed a stronger association with relevant cell types within the developmental data, as expected for a developmental category of disease genes (Supplementary figure 8E).
The nephrogenic cell type-specfic gene functional networks have the power to predict disease-associated gene candidates (those with high functional relatedness to known disease genes) (Krishnan et al., 2016). We explored disease-associated genes by integrating the nephrogenic cell-type specific functional gene networks and gene expression, previously verified CAKUT/KD causative genes, and genome-wide association studies (GWAS) covering 966,864 individuals with reduced glomerular filtration rate (GFR) (Hwang et al., 2007; Köttgen et al., 2009; Pattaro et al., 2016; Wuttke et al., 2019). The GWAS studies provide information complementary to the developmental kidney networks. It provides a pool of disease gene candidates but no information on tissue or cell type-specificity or about gene expression. We prioritize these candidates based on their act in the developmental functional networks, selecting genes with connectivity patterns that are more similar to CAKUT/KD genes than non-CAKUT/KD genes (OMIM database) (p < 0.05, Wilcoxon test).
Forty genes, across ten cell-types, were finally selected (Supplementary table 4). Ephrin B2 (EFNB2) was identified in the pre-podocyte cluster (cluster 6) as a close neighbor to JAG1, SLIT2, and BMP4 (in situ hybridization also shown in Supplementary figure 7F). JAG1 has strong links to Alagille syndrome and kidney abnormalities (Li et al., 1997; Spinner et al., 2001), and the regulation of angiogenesis (Benedito et al., 2009). SLIT2/ROBO2 ligand receptor signalling is important at several stages of kidney organogenesis (Grieshammer et al., 2004), and this pathway is required in the renal corpuscle for normal assembly of the slit-diaphragm regulating podocyte foot-process assembly (Fan et al., 2016). BMP4 is upregulated during early human renal corpuscle formation (Kim et al., 2019), mutations to BMP4 have been linked to CAKUT (Weber et al., 2008), and Bmp4 is required for normal renal corpuscle formation (Ueda et al., 2008). While there is no current data on the role of EFNB2 in human congenital kidney abnomalies, Efnb2 has been associated with glomerular vascular assembly (Takahashi et al., 2001) and is linked to angiogenesis outside of the kidney (Wang et al., 2010). SNPs were associated with EFNB2 in all of the four GWAS studies and we confirmed EFNB2 expression in the developing renal corpuscle and vasculature (Figure 5D; Supplementary figure 8F). The combined EFNB2, JAG1, SLIT2, BMP4 network in the pre-podocyte cells was enriched for GO terms indicating vascular and tubular development (Figure 5E; data not shown) suggesting the mixture of known and putative KD/CAKUT genes regulate the formation of the renal corpuscle and its vascular architecture, pointing to a developmental category of vascular abnormalities in renal corpuscle/glomerulus assembly linked to EFNB2 dysregulation.
Expression of a second set of genes with strong connections to KD/CAKUT genes - PAPPA2, TNFRSF11B, CDH3 - coincide in cluster 18. Pappa2 is connected to salt-induced hypertension in rats (Cowley et al., 2016), and TNFRSF11B, also known as Osteoprotegerin, is linked to hypertension and chronic kidney disease (Bernardi et al., 2017), while CDH3 has not previously been published as a KD/CAKUT gene or renal dysfunction. TNFRSF11B was confirmed to be strongly expressed in the macula densa and also more broadly in the distal tubule (Supplementary figure 8G, H). Strikingly, PAPPA2 was robustly connected to Renin (REN) a key component of the Renin Angiotensis System (RAS), while CDH3 was connected to both Angiotensin I Converting Ensyme (ACE) and Angiotensinogen (AGT), two critical components of the production of Angiotensin. The link between cluster 18 and predicted gene/disease associations to the macula densa, raises the possibility that the PAPPA2, TNFRSF11B, CDH3 gene set may couple to functional macula densa/RAS programs. Collectively, these illustrative examples highlight how intersecting single-cell transcriptomics and functional gene networks can provide potential insight into gene associations linked to development and disease.
Exploring precursor-product relationships in the developing nephron
Precursor-product relationships are poorly understood in the nephron and the complexity of gene expression patterns, convoluted morphogenesis, and activation of genes at multiple times in different nephron segments have prevented genetic lineage tracing.
To explore precursor-product relationships, we performed whole transcriptome correlation analyses between all genes expressed in each of the six developmental S-shaped body cell-types and all the orthologs expressed in the twenty-two resolved final fates found in the adult mouse nephron (Little et al., 2007; Ransick et al., 2019). Adult podocytes, parietal epithelium, proximal tubule, distal portions of the loop of Henle, the distal tubule, and connecting tubule showed positive correlation and with the same spatial ordering of cells observed in the S-shaped body (Figure 6A). Adult cell signatures from the inner medullary region and from specialized cells in the connecting tubule correlated poorly to the developmental transcriptomes.
Given that transcription factors impart and maintain cell fates, we examined if their transcriptional boundaries found in the adult mouse nephron or in the S-shaped body are forming or maintained in positional relative domains (Supplementary figure 9A, B). Transcription factor domains resembling those of the adult nephron were evident in the S-shaped body, and vice versa. Proximally, adult mouse podocyte markers MAFB, WT1, and CREB3L2 were enriched in developing human podocytes (cluster 8 - human annotations used), proximal tubule genes HNF4A, HNF4G, and HNF1A were detected in putative proximal precursors (cluster 12). Shifting distally, IRX1, IRX2, and MECOM, normally enriched in the loop of Henle and macula densa, were expressed in cluster 18, and distal and connecting tubule genes EMX1, TFAP2A, and TFCP2L1 were expressed in clusters 16 and 17. Further, broad transcription factor domains predicted from the adult mouse nephron (Pou3f3, Irx1, Hnf4a) (Figure 6C) were identifiable in the developing nephron from their onset of their expression in the S-shaped body through later developmental stages (Figure 6D). These data are consistent with an anatomical pre-positioning of precursor populations along the proximal distal S-shaped body axis that relate to mature progeny. Hnf4a, one of few genes demonstrated to be functionally required for mature proximal tubule formation (Marable et al., 2018, 2020), was first detected at a protein level in a narrow cell-layer between proximal WT1+ and medial JAG1+ S-shaped body cells (Figure 4D, Supplementary figure 7J; Supplementary figure 10A) raising the possibility that the entire proximal convoluted tubule arises from a small cell-population in the proximal S-shaped body.
We further examined the predicted domain structures in the S-shaped body in light of the conservation in nephron morphogenesis for evidence that the S-bends may bring into juxtaposition progenitor cell types fated to interact in regulation of kidney function in the adult kidney. The macula densa is an asymmetric plaque of 20–30 cells in the distal nephron abutting the renal corpuscle, regulating renal blood flow and filtration within the renal corpuscle through local and systemic signaling (Mundel et al., 1992; Wilcox et al., 1992; Peti-Peterdi and Harris, 2010). Interestingly, PAPPA2 highlights a small population of cells in the distal limb of the S-shaped body in which PAPPA2 is asymetrically positioned towards the forming podocytes (Figure 2D). Careful temporal analysis of the PAPPA2+ domain across developmental stages of nephrons showed PAPPA2 persists in this configuration maintaining close contact with the developing renal corpuscle through to mature stages where PAPPA2 highlights an anatomically identifiable macula densa (Figure 6E). These data are consistent with the early interaction of physiologically linked cell regions of the nephron enabled by the morphogenesis of the nephron anlagen.
In cluster 18, PAPPA2 is co-expressed with locally expressed transcriptional regulators including IRX1, IRX2, and ERBB4. These factors are more broadly expressed than the macula densa, in the distal loop of Henle and distal tubule, suggesting cluster 18 may contain progenitors for multiple distal cell identities (Supplementary figure 3D, Supplementary figure 9A; (Reggiani et al., 2007; Alarcón et al., 2008; Veikkolainen et al., 2012). Re-analyzing week 17 kidney scRNA-seq data, extending to more mature stages of nephrons (Tran et al., 2019), suggests PAPPA2+/NOS1+ macula densa cells emerge after the S-shaped body stage (Supplementary figure 10B–D). Genes tightly correlating with PAPPA2, and expressed in the mature macula densa in the week 17 data (Supplementary figure 10E, F), could be detected already in the in the S-shaped body. These include many members of the Iroquois-class homeodomain family of transcription factors IRX1, IRX2, IRX3, IRX5, and IRX6 (but not IRX4). Further, examining the distribution in the S-shaped body of six transcription factors strongly correlated with PAPPA2 in the scRNA-seq data (IRX1, TFAP2A, TFAP2B, EMX2, TFCP2L1, MECOM), confirmed an overlap with PAPPA2. Their overlap highlighted a transcriptionally distinct, narrow subset of POU3F3+/CDH1+ cells in the distal S-shaped body as a potential precursor to the macula densa (Figure 6E,F; Supplementary figure 11A–F).
Discussion
Next-generation views of biological systems need to integrate detailed single cell approaches with the spatial organization of cells in the developing and functional anatomy. Three leading paths to delineate spatial expression patterns include the capture of mRNA from spatially defined regions, multiplexed sequential in situ hybridizations, and spatial mapping of profiled transcriptomes to anatomical origins. Commercial systems are now available for the capture of mRNA from groups of cells on tissue sections (10XGenomics Spatial Transcriptomics), making a significant advance but not yet achieving a cellular resolution. Several groups have also analysed spatial transcriptional signatures of thousands of genes in sections and cultured cells using sequential and multiplexed in situ hybridization, albeit primarily with a two-dimensional resolution (Lignell et al., 2017; Xia et al., 2019). However, when biological structures adopt complex morphologies through time and space, across hundreds of micrometers or milimeters of nontransparent tissue, resolving spatial relationships for cell transcriptomes and epigenomes remains a challenge. In Drosophila development, this challenge was met by first generating a spatial gene expression atlas at a relatively high voxel resolution, deeply profiling cell transcriptomes using scRNA-seq, then through probabilistic mapping, aligning transcriptomes to their inferred expression domains along the embryonic axes (Karaiskos et al., 2017). Adopting a similar approach here – integrating spatial, transcriptomic and protein information sources – generates a model for cell relationships as nephron assembly progresses. The data describes a range of previously uncharacterized transient transcriptomic and protein states at each point of the differentiation trajectories, insights consolidated within the framework of the Human Nephrogenesis Atlas (https://sckidney.flatironinstitute.org/). This serves as a valuable resource that can be leveraged with excellent existing data collections such as those harboured in the Genitourinary Developmental Molecular Anatomy Project (GUDMAP: Little et al., 2007; McMahon et al., 2008).
Our studies have led to a testable model for nephrogenesis (Figure 2B; Supplementary figure 1C) where the morphological progression is depicted against the localization of markers, and a developmental lineage map is proposed, assigning cluster cell identities (Figure 3C). The annotation of clusters to precise anatomical bins is based on extensive validation of early expression patterns in human nephrogenesis (Lindström et al., 2018d, 2018c, 2018b, 2018a; Kim et al., 2019; Tran et al., 2019) in combination with work performed in the mouse (Kobayashi, 2005; Grote et al., 2007; Georgas et al., 2009). A notable difference in the developmental progression of the earliest stages of mouse and human nephrogenesis challenges current ontological descriptions of mammalian nephrogenesis. The classical definition of a pretubular aggregate based on model systems, predominantly the mouse and rat, is a non-epithelial cell-aggregate which rapidly transitions to a fully epithelial renal vesicle encasing a lumen (Saxen, 1987; Georgas et al., 2009; Mugford et al., 2009; Gao et al., 2017). In human nephrogenesis, the pretubular aggregate is a less transient structure and shows evidence of a partial epithelialization, with the initiation of the synthesis and deposition of CDH1 in an apical-distal polarized manner in distal cells adjacent to the ureteric epithelium (Lindström et al., 2018a). Additionally, nephron progenitors can exhibit plasticity after initial inductive responses, reverting back to a nephron progenitor state (Lawlor et al., 2019). Both the plasticity of progenitors and the step-wise acquisition of epithelial characteristics present challenges to ontological descriptions of cells undergoing these early transitions. While we have annotated cell profiles in this study as best fits the accepted nomenclature, considering the renal vesicle as fully epithelial structure with an enclosed lumen, we recognize this may not capture the fluidity of transitional states that are present and note this in the lineage-map. Further, there is no current understanding of developmental plasticity, determination and resulting cell fates, amongst epithelial progenitor states, identified during subsequent morphogenesis of the renal vesicle, though the activation of transcriptional determinants demarcating particular cell-types in the adult nephron (e.g., HNF4A for the proximal tubule) are consistent with the specification of some adult cell fates by the S-shaped body stage. Consequently, the cell cluster to adult fate map relationships we propose in this study should not be regarded with a fixed, deterministic view, but as a reasonable set of predictions that provides a coherent framework for the current data which will undoubtedly be rigorously explored and challenged in future experimental studies.
Nephron progenitors display complex cell behaviors associated with cell division including the detaching and reattaching to the collecting duct progenitors located at the tips of the growing epithelium (O’Brien et al., 2018), while cell cycling regulated by Fgf8 controls emerging distal nephron identitites (Grieshammer et al., 2005). Close scrutiny of cell cycling within the nephrogenic lineage will be required to determine its impact on differentiaton. Overall, the data point to the emergence of cell-fates within a stereotypical program of epithelial morphogenesis which prefigures a progressive increase in cellular complexity. Consistent with lineage tracing studies in mouse nephron progenitor cells (Boyle et al., 2008; Kobayashi et al., 2008), single cell analysis points to a common precursor of distal and proximal epithelial tubular segments in the human nephron. Distinct proximal-distal programs correlate with proximal-distal positioning within the renal vesicle and later structures, which is itself dependent on the time of recruitment of committed cells into the renal vesicle and likely differential signaling networks (Lindström et al., 2018a). Many of the precursor populations begin to show some characteristics of transcription factor expression and function-associated gene expression profiles pre-figuring adult cell types by the S-shaped body stage (Supplementary figure 9), for instance HNF4A+ cells in the medial limb of the S-shaped body. While previous work has indicated Hnf4a deficient kidneys perturb proximal tubule development (Marable et al., 2018, 2020), there are as yet no formal genetic fate-mapping from Hnf4a+ cells in the S-shaped body to their adult counterparts. The potential links from precursors to their physiologically active progeny will facilitate optimization and analysis of synthetic strategies to generate adult cell types from stem cell systems.
A cell-type of particular biomedical significance is the macula densa. Twenty-six percent of the world’s population is affected by hypertension (Kearney et al., 2005). The macula densa regulates blood pressure systemically through the renin-angiotensin system (Peti-Peterdi and Harris, 2010). It comprises around 20–30 cells on the glomerular facing side of the distal nephron tubule, a critical positioning for local regulation of renal filtration (Peti-Peterdi and Harris, 2010). Macula densa cells secrete local signals in response to elevated Na+ levels to constrict nearby afferent arterioles, reducing blood flow into the glomerulus, decreasing renal filtration. Interestingly, cells in population 18 in the S-shaped body (Figure 6A–E), characterized by a narrow asymmetric domain of PAPPA2, share several features of cells of the adult macula densa. Putative macula densa progenitors postioned at the second hair-pin bend along the proximal to distal trajectory of the convoluted nephron anlagen, have a close spatial association with the developing renal corpuscle, presaging an adult functional, anatomical association. Thus, the highly conserved morphogenesis evident from comparing the mouse and human developmental models, may reflect the early generation of cellular relationships essential to critical physiological regulation in the functioning kidney. Considering the size of this cell population and that of adjacent podocytes at the S-shaped body stage, there may be little further replication of either cell type to the adult kidney, stabilizing their interactions, and local assembly of renal filtration systems. This model predicts a large component of nephron diversity, all proximal tubule segments, and the ascending and descending loops of Henle, emerges from a progenitor pool within proximally adjacent populations of the S-shaped body. (Figure 6B, F). Recent data on the role of Tfap2b in distalizing the mouse and zebrafish nephron (Chambers et al., 2019, 2020; Marneros, 2020) suggest a gradual acquitision and fine-tuning of cell identitites consistent with our data. Though we focus on the nephrogenic lineage, the distal nephron shares several important characteristics with the ureteric epitheliuam including Hoxb7 driven transgene expression (Lindström et al., 2018a; Combes et al., 2019a) and likely the ureteric epithelium generates intercalated and principal cell types (Combes et al., 2019a; Ransick et al., 2019). Strikingly, this can be recapitulated within in vitro systems (Howden et al., 2021) and serves as an important reminder for careful scrutiny of normal development.
Cell fate analyses in the developing mouse kidney provide additional insights in support of this view. Lgr5-expressing cells in the distal S-shaped body have been fate-mapped to multiple distal segments, namely the thick ascending limb of the loop of Henle, the distal convoluted tubule segment, and the macula densa (Barker et al., 2012). The scRNA-seq data shows LGR5 is expressed broadly in the distal portion of renal vesicles and in S-shaped body nephrons in populations 14, 15, 16, 17, 18 (Supplementary figure 3D; strongest in population 16 in the S-shaped body nephron, Figure 6B). The human single cell data also predict an overlap with five members of the IRX family of transcriptional regulators (Kim et al., 2012), of which Irx1 and Irx2 show a similar configuration in the putative macula densa anlagen to the macula densa of the adult mouse kidney (Supplementary figure 9). The question of plasticity and clonality in nephrogenesis has been examined in the mouse nephron using random clonal cell labelling concluding that there is little regional intermixing within the epithelialized nephron (Rinkevich et al., 2014). The current study highlights regionally expressed target genes that could be vehicles for further insightful lineage analysis to establish when lineage decisions are locked in within the developing mouse nephron.
Cell identities and their behavior are underpinned by complex gene interaction networks. We utilized the power of predicting gene networks and applied this to the resolved cell transcriptomes, analyzing gene networks for over 200 genes involved in KD/CAKUT. These were further compared against 4 GWAS studies totaling ~1M individuals with data on altered GFR. The predicted KD/CAKUT genes were expressed as predicted and combinations of new and known genes were identified within networks. The integration of existing public data with single cell omic analyses represents a step forward for predicting gene interactions and genes likely responsible for disease. Importantly, disease manifesting in the adult may have altered developmental gene activity as a root cause. The data here will facilitate evaluating genetic linkage of human disease associated genes to developmental programs in the human kidney.
In vitro kidney-like cell that are generated in organoid systems imperfectly recapitulate normal kidney cell types (Combes et al., 2019b; Tran et al., 2019). However, implanting organoids into the kidney can normalize some deficiencies (van den Berg et al., 2018; Low et al., 2019; Tran et al., 2019). The Human Nephrogenesis Atlas benchmarks the early developmental steps where proximal-distal nephron lineages likely develop, precisely outlines the transcriptional profiles of each precursor state, and as such provides a blueprint for building next-generation organoid models.
In summary, our analysis, integrating space and time across across scRNA-seq and three-dimensional protein imaging datasets of the human nephrogenic program, generates the most comprehensive view to date of mammalian nephrogenesis. Spatial modelling and transcriptional analysis support an early commitment of some cell types, while others maintain a more extended progenitor capability for distinct regions of the kidney. The data made broadly accessible through the Human Nephrogenesis Atlas (https://sckidney.flatironinstitute.org/) will serve as a valuable resource for mining regulatory pathways generating nephron cell types, facilitating approaches to generate functional nephron structures in vitro and identifying potential developmental relationships in gene associations linked to kidney disease.
Limitations of the Study
To label and visualize proteins in the nephron forming program, we leveraged clearing by benzyl benzoate and benzyl alcohol (BABB) against antibodies and secondary fluorescent labelling. Intact human and mouse nephrons were thus analyzed within their niche and post-digital extraction, the voxel-based data was suitable for image registration and machine-learning to scrutnize patterns. Dehydration steps and treatment with clearing agents can induce tissue deformation (Richardson and Lichtman, 2015). We cannot eliminate the possibility of subtle variations created by the protocol though these would likely be fairly minimal given the successful registration of data collected from different nephron progenitor samples.
The protein data highlighted eight major domains in the S-shaped body (Figure 4A) whereas previous two-dimensional analysis of sectioned material has resolved nine to ten (Lindström et al., 2018b). Post image-registration with WT1, MAFB, FOXC2, and SIX2 labelling did not separate pattern 8 into its sub-types; parietal and visceral cells. Integrating complementary approaches such as metal-conjugated multiplex imaging mass cytometry (Singh et al., 2019), multiplexed in situ hybridization (Lignell et al., 2017; Xia et al., 2019), multiomic single cell analyses (Miao et al., 2021), and genetic fate-mapping (Kobayashi et al., 2008) will likely refine the lineage maps and cell-states, and help determine how the patterns estimate cell-states within a diversifying lineage-tree.
Signicant variability has been documented between mRNA expression and the cellular profiles of encoded protein protein which could compromise the mapping of scRNA-seq data to protein maps of nephron precursors (Liu et al., 2016). Though the data successfully predicted gene/cell/structure relationships, spatial transcriptomics such as CITE-seq (Stoeckius et al., 2017) and seq-FISH (Lignell et al., 2017) will likely provide additional resolution. Perhaps the most significant limitation is the inability to experimentally address questions of cell commitment and cell fate in the human kidney. Human kidney organoid models may provide one option though it is not clear whether organoid systems replicate the stereotypical early nephron morphogenesis reported here. Parallel genetic analysis in the mouse is a reasonable approach given similarities between mouse and human programs.
STAR Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by Nils O. Lindström (nils.lindstrom@med.usc.edu), Olga Troyanskaya (ogt@genomics.princeton.edu) and Andrew P. McMahon (amcmahon@med.usc.edu).
Experimental Model and Subject Details
Data code and availability
Single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. For microscopy or other additional data reported in this paper contact Dr. Lindström. All original code will be made available at Zenodo. DOIs and URLs are listed in the key resources table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | ||
|---|---|---|
| Antibodies and Probes | SOURCE | IDENTIFIER |
| anti-Ap-2-α | Cell Signaling | 3215S |
| anti-Cdh1 | BD Transduction laboratories | 610182 |
| anti-Cdh6 | R&D | AF2715 |
| anti-Cldn5 | Novus Biologicals | NB120–15107 |
| anti-Emx2 | R&D | AF6470 |
| anti-Erbb4 | R&D | MAB1131 |
| anti-Foxc2 | R&D | AF6989 |
| anti-Hnf1b | Santa Cruz | sc-22840 |
| anti-Hnf4a | R&D | MAB4605 |
| anti-Irx1 | SIGMA | HPA043160 |
| anti-Jagged1 | R&D | AF599 |
| anti-Krt8 | DSHB | TROMA-1 |
| anti-Lef1 | Cell Signaling | 2230 |
| anti-Lhx1 | Santa Cruz | sc-19341 |
| anti-Mafb | R&D | MAB3810 |
| anti-Mafb | Santa Cruz | sc-10022 |
| anti-Mecom | R&D | MAB75061 |
| anti-Pappa2 | R&D | AF1668 |
| anti-Pax2 | Biolegend | 901001 |
| anti-Pax8 | Abcam | ab189249 |
| anti-Podxl | R&D | AF1658 |
| anti-Pou3f3 | Thermo Scientific | PA5–64311 |
| anti-Pou3f3 | Novus Biologicals | NBP1–49872 |
| anti-Sall1 | R&D | PP-K9814–00 |
| anti-Six2 | Mybiosource | MBS610128 |
| anti-Six2 | ProteinTech | 11562–1-AP |
| anti-Sox9 | Abcam | ab185230 |
| anti-Tfcp2l1 | Novus Biologicals | AF5726 |
| anti-Tfap2a | Cell Signaling | 3215 |
| anti-Tfap2b | Cell Signaling | 2509S |
| anti-Wt1 | Abcam | ab89901 |
| RNA-Scope SLC39A8 | ACD | 556831 |
| RNA-Scope TMEM72 | ACD | 556851-C2 |
| RNA-Scope COL4A4 | ACD | 557261-C3 |
| RNA-Scope LAMP5 | ACD | 487691-C3 |
| RNA-Scope HNF4A | ACD | 442921-C2 |
| RNA-Scope EFNB2 | ACD | 430651 |
| Deposited data | GEO accession number | Reference |
| Human week 14 scRNA-seq data | GSE139280 | (this study) |
| Zonal Human week 17 scRNA-seq data | GSE127344 | (Tran et al., 2019) |
| Adult mouse nephron scRNA-seq | GSE129798 | (Ransick et al., 2019) |
| Experimental models: Organisms/strains | ||
| Swiss Webster mice | Lab stock | |
| Software and algorithms | URL | DOI/Reference |
| ANTs | https://github.com/ANTsX/ANTs | 10.5281/zenodo.5138159 |
| Woolz | https://github.com/ma-tech/Woolz | 10.5281/zenodo.5138133 |
| Registration pipeline | https://github.com/ma-tech/RenalObjectRegistrationPipeline | 10.5281/zenodo.5138175 |
| Amira versions 6.1.1, 6.2, and 6.3 | Themofisher | |
| CellRanger version 2.1 | 10xgenomix | |
| Code for 3D protein patterns & single-cell + spatial data integration | https://github.com/flatironinstitute/fetal_kidney_clusters | 10.5281/zenodo.5138196 |
| Code for Bayesian integration | https://functionlab.github.io/sleipnir-docs/index.html | (Huttenhower et al., 2008) |
| Seurat v2, 3, 4 | https://satijalab.org/seurat/ | (Butler et al., 2018; Stuart et al., 2019) |
| Additional resources | URL | Reference |
| Human Nephrogenesis Atlas | https://sckidney.flatironinstitute.org | this study |
Animal studies
All animal work performed in this study was reviewed and approved by the Institutional Animal Care and Use Committees (IACUC) at the University of Southern California. All work adhered to institutional guidelines. Embryos were recovered at embryonic day 15.5 following timed matings of Swiss Webster mice. The sex of embryos was not known.
Human samples
Consented, anonymized, human fetal kidney tissue was obtained from elective terminations following review of the study by Keck School of Medicine of the University of Southern California’s Institutional Review Board. Kidney samples ranging in age from 14 to 17 weeks of gestation were provided by collaborators at Family Planning Associates. Gestational age was determined per guidelines specified by the American College of Obstetricians and Gynecologists using ultrasound, heel to toe, and crown to rump measurements following published Carnegie Stages (O’Rahilly and Müller, 2010; O’Rahilly et al., 1987; Strachan et al., 1997). The sex of the specimen was not reported. Consented samples were received immediately after elective terminations and transported from the Children’s Hospital of Los Angeles on ice at 4°C in 10% fetal bovine serum, 25mM Hepes, high glucose DMEM (SigmaAldrich). Only kidneys that were intact within the kidney capsule were analyzed.
Immunofluorescent labelling of mouse and human kidneys
Whole embryonic day 15.5 Swiss Webster kidneys were dissected, decapsulated in 1xPBS by standard dissection tools and fixed for 20 min at 4°C in 4% PFA without shaking. Human kidneys were carefully decapsulated in 1xPBS, placed on surgical gauze in 10cm Petri dish submerged in 1xPBS. Blunt-ended forceps were used to hold the kidney and 3 mm thick cortical slices were sliced off manually using a scalpel. Human kidney slices were fixed 4% PFA in 1xPBS for 45 minutes at 4°C without shaking. Human and mouse kidneys were subsequently washed with several rounds of 1xPBS.
For preparation for immunofluorescent staining slices and kidneys were blocked for 1hr in 1xPBS with 2% SEA Block and 0.1% TritonX100 at 4°C with gentle movement. The primary antibodies were resuspended in blocking solution and kidney tissue incubated in primary antibody at 4°C with gentle movement for 48 hr. Samples were washed up to 8 hr through several rounds of 1xPBS with 0.1% TritonX100. Secondary antibodies were resuspended in blocking solution and tissue incubated at 4°C with gentle movement for 48 hr. Washing steps for primary antibody were repeated. Tissue was counterstained in 1 μg/ml Hoechst 33342 in 1xPBS for 2 hr prior to final wash in 1xPBS. For dehydration steps prior to clearing steps, the tissue was passed through a series of increasing concentrations of methanol (50%, 75%, 100%) 1 hr for each step. To clear tissue the dehydrated tissue was immersed in 50% Benzyl alcohol, Benzyl benzoate (BABB) / 50% methanol solution for a 1 hr. Full clearing was achieved in 100% BABB. The tissue was stored at 4°C in the dark.
Antibodies used were validated on frozen sections of mouse and human kidneys and are detailed in the Key Resource Table.
In situ hybridization and RNA-scope
RNAscope probes were designed and purchased (Advanced Cell Diagnostics) and reactions performed on human kidney cryosections according to manufacturer’s recommendations. The Multiplex Fluorescent Reagent kit v2 was used. Probes were used for SLC39A8, TMEM72, COL4A4, LAMP5, HNF4A, EFNB2 and are detailed in the Key Resource Table.
All in situ hybridization stains were performed on human kidney cryosections as described in detail at the GUDMAP website: https://www.gudmap.org/Research/Protocols/McMahon.html and as used previously on human sections (Lindström et al., 2018b).
Image acquisition
Imaging of cortical slices (3D) was performed on a Leica SP8 using a 40X objective (40x/1.30 Oil HC PL APO CS2). 1024×1024 images were captured at 0.35 μm optical intervals with a 1.5x zoom for human samples and 2x zoom for mouse kidneys. Channels were captured sequentially. Images were captured at 12-bit depth over a 0–4095 range. Care was taken to ensure voxels were not saturated.
Image Processing Pipeline
Confocal image stacks were loaded into Amira (versions 6.1.1, 6.2, and 6.3), nephrons volumes were digitally marked by hand using the segmentation panel and paint brush tool, extracted at full resolution into individual volumes and channels, and scrutinized. 251 human nephrons were extracted from 14 separate kidneys ranging between 13 and 17 weeks. 177 mouse nephrons were extracted from 16 separate E15.5 C57BL/6J kidneys). Nephrons were binned into anatomical terms: pretubular aggregate, renal vesicle, comma-shaped body, s-shaped body based on size, morphology, and protein patterns. For visualization of the nephrogenic developmental progression e.g. Supplementary figure 1A-B, the adjacent ureteric tip and stalk were extracted to provide orientation. The developmental progressions for human and mouse nephrogenesis as depicted in Figure 2A and Supplementary figure 1C, respectively, are representative for each species. Nuclei, and cells positive for JAG1 and CDH1 were manually counted within stacks.
Image Data Preparation for 3D Mapping
The image processing pipeline, ANTs and Woolz are freely available from the GitHub repositories:
Registration pipeline: https://github.com/ma-tech/RenalObjectRegistrationPipeline
ANTs: https://github.com/ANTsX/ANTs
Woolz: https://github.com/ma-tech/Woolz
To process, register and combine protein localization assay images from both human and mouse renal objects allowing quantitative comparison of protein abundance a mainly automated image processing pipeline was developed. The pipeline, written in Python, makes extensive use of both the Advanced Normalization Tools (ANTs) (Tustison et al., 2014) and the Woolz image processing systems – the latter redeveloped for the Mouse Atlas Databases www.emouseatlas.org. ANTs was used for elastic and some affine image registration; Woolz for all other image processing tasks including the affine registration of S-shaped body-stage nephrons.
Confocal images were organized into assays, with each assay composed of the set of segmented images obtained from a single nephron. In all cases each assay contained the DAPI and JAG1 channel images for reference, but the assays also contained a selection of two other channels from the set CDH1, LEF1, SALL1, SIX2, SOX9, TROMA1, WT1, LHX2, PAX2, HNF1B, FOXC2, MECOM, EMX2, TFAP2A, POU3F3, ERBB4, PAPPA2, CDH6, MAFB, and CLDN5. Four classes were used to group the assays of nephron objects, namely human and mouse renal vesicles and human and mouse s-shaped body stage nephrons.
The confocal images were rescaled using rescaling factors of 0.4103 within the image plane and a factor of 0.9889 across planes to achieve cubic voxels. Composite DAPI and JAG1 channel rescaled images were created by combining the voxel values of these two channel with a JAG1:DAPI weighting ratio of 4:1. The composite images were created to combine the broad coverage of the DAPI channel with the directional coverage of the JAG1 channel. A Gaussian blurred version of each of the rescaled images was also generated for visualization and registration using an isotropic Gaussian filter with a sigma value of two voxels.
Image Registration
For each of the nephron object classes a representative assay was selected as the basis of the class model and each assay was then registered to the corresponding model using a multi-stage registration process. First a general affine transform was established to align each assay object with its model using the Gaussian blurred combined DAPI - JAG1 images. While ANTs was used for renal vesicle affine registration, ANTs proved unreliable when establishing an initial affine registration of S-shaped bodies due to their morphology, so landmarks manually placed using WoolzWarp were used to identify the proximal, medial, and distal compartments. From these landmarks, least squares affine transforms were computed using Woolz. With the transformations that oriented and scaled assay images to their corresponding models established, ANTs was used to elastically register combined (but not blurred) DAPI – JAG1 images to their models using a cross-correlation similarity metric. The resulting affine and elastic transforms were then used to register all channels of all assays to their models. With all assays of all models aligned, model images were subsequently composed from the combined registered single channel assay images.
Extracting 3D protein patterns from registered models
For each protein with imaging data, we first found the average protein expression at each voxel across all images for the protein in both the renal vesicle and S-shaped body image sets. We then normalized across all voxels so that the intensities of each protein had a mean 0 and standard deviation of 1 within both the renal vesicle and the S-shaped body datasets. Selecting the set of 18 or 19 proteins with imaging data for renal vesicle and S-shaped body, respectively, we performed k-means clustering on each set of voxels, treating each voxel as an 18 or 19 -dimensional point in genomic space. We use the k-means implementation in the stats R package for clustering, and tested values of k ranging from 2 to 20. We then visualized the resulting spatial patterns using Fiji, manually removing patterns that corresponded to background noise. The code for generating 3D protein patterns and integrating across single-cell and spatial data is available at https://github.com/flatironinstitute/fetal_kidney_clusters.
Capture, sequencing, and processing of single-cell RNA transcriptomes
Single cells were isolated from two replicate week 14 kidneys as previously described (Lindström et al., 2018a). The nephrogenic niche was dissociated by enzymatic digestion by placing the decapsulated but intact whole kidneys in collagenase A/ pancreatin, an enzymatic cocktail described elsewhere (Brown et al., 2015) that biased cell dissociation to the nephrogenic niche. The kidneys were kept incubated at 37°C in a nutator and shaken at 450rpm to release cells from the nephrogenic niche over a 40–50min period. Released cells were stained for DRAQ5+ (ThermoFisher Scientific) and DAPI (ThermoFisher Scientific) to identify live and intact and dead cells, respectively. DRAQ5+/DAPI- cells were selected by FACS. Per repeat, 7000 cells were input into the 10X Chromium system and processed for single-cell library construction as per 10x Genomics instructions and as we describe previously (Lindström et al., 2018a). Cell transcriptomes were sequenced by Hi-Seq (Illumina) in 8 separate runs with ~ 120,000 reads per cell. 24,254 cells were sequenced to a mean of 2644 genes per cell (Supplementary figure 2A).
Quality control, mapping (to GRCh37.p13) and count table assembly of the library was performed using the CellRanger pipeline version 2.1 (as consistent with 10x Genomics guidelines) and as described in our previous work (Lindström et al., 2018a).
Computational isolation of nephrogenic lineage
To begin analyzing the cell transcriptomes we assessed and filtered transcriptome qualities using Seurat (Satija et al., 2015). Cells with more than 3000 genes per cell, fewer than 5% of reads mapping to mitochondrial genes, and a Good-Turing estimate greater than 0.7 (Good, 1953) were kept for downstream analyses. We deliberately set the gene content to 3,000 in order to keep only cells that displayed very rich information. Analyses with a 1000–6000 gene range provided greater cell-numbers (24008 cells), but made resolving putative S-shaped body cell-types challenging (analyses not shown). We therefore focused on the 8,316 cells with a gene content greater than 3000 and clustered these using the Seurat R package. We ran Principal Component Analysis on the dataset and used 39 principal components based on the JackStraw test (p < 0.05) and clustered the cells using the Seurat FindClusters function with 39 PCs and default remaining parameters. We found 39 resulting clusters (Supplementary figure 2D). Based on the differential expression test (FindAllMarkers function, bimod test) and expression of marker genes we determined that 24 of the clusters, a total of 6,667 cells, were nephrogenic (Supplementary figure 2E).
Genes linked to cell-cycling (e.g. TOP2A and MKI67) were expressed strongly in several clusters (Supplementary Figure 2E) and on closer scrutiny against genes with known periodicity during cell- cycle phases (Dominguez et al., 2016), clusters containing cells in the cell-cycle were categorized based on cycle-stage (Supplementary figure 2F). Non-cycling nephrogenic cells (2893 cells) were selected for downstream analyses (nephrogenic lineage clusters in Supplementary figure 2F). To maximally resolve precursor populations, the nephron progenitor cells were initially removed (CITED1+, SIX2+), and differentiating cells (clusters 20,27,19,25,18,17,26,5) reiteratively reclustered and identified based on marker gene expression and gene enrichment analyses (Supplementary figure 2G, H). The subclustering was repeated as described above (using 30 principal components, and clustering explored using a range of nearest neighbour values ranging between 4 and 30), analyzing the differentially expressed genes between resulting clusters for segregation of known marker genes of the predicted subdomains. We identified 3 clusters displaying a very early nephron signature, which we termed pretubular aggregate clusters. Similarly, we identified 5 clusters that consisted of transcriptomes from renal vesicle cells, and 6 clusters from S-shape body nephrons (see Results section for details of annotations). Note that cluster numbers in Supplementary figure 2G, H do not correspond to those in Supplementary figure 2C–F). Resolved transcriptomes of differentiating cells were then remerged with the nephron progenitors (Figure 3A) and cell relationships explored by Similarity Weighted Nonnegative Embedding (SWNE) (Supplementary figure 2J) (Wu et al., 2018) which showed three main trajectories corresponding to distal, proximal, and podocyte fates, with the clusters distributed along the trajectories in an order reflecting developmental progression. The SWNE projection was consistent with the tSNE plot (Supplementary figure 2J vs. Figure 3A). Based on marker gene analysis (Supplementary table 2) and work outlined in the Results section, the cluster annotations 1 to 18 were assigned to the resolved clusters based on developmental relationships.
Single cell RNA sequencing data availability
The human week 14 scRNA-seq data presented in this work is GEO accession number GSE139280.
Zonal human week 17 scRNA-seq data was obtained as described (Tran et al., 2019) GSE127344.
Adult mouse nephron scRNA-seq data was obtained as described (Ransick et al., 2019) GSE129798.
Computing cell relationships from single cell RNA sequencing data
Cell relationships were computed by graph-based dimensionality reduction (Menon et al., 2018) and Zhou and Troyanskaya https://www.biorxiv.org/content/10.1101/2020.04.12.022806v1).
Mapping single cells to voxel data
To map single cells to protein patterns, we imputed gene expression for only those genes with matching proteins confocal imaging data using the SAVER algorithm (Huang et al., 2018). All other genes were not imputed or altered as described below. Similarly, the protein information was not imputed but rather used the k-means predicted protein patterns. For each gene with matching protein data, we normalized its expression across the single-cell data. Genes were scaled to give each gene mean expression of 0 and standard deviation 1 across all cells. We next selected the fourteen genes with matching confocal imaging data that were highly expressed in the S-shaped body scRNA-seq clusters (EMX2, MECOM, POU3F3, SOX9, PAX2, PAPPA2, HNF1B, LHX1, ERBB4, KRT8, CDH6, JAG1, MAFB, and WT1). We further selected the 8 protein patterns identified (see Results) and added a null pattern with zero expression of all proteins. We computed the Euclidean distance between the vector of highly expressed genes and the corresponding vector of pattern centers for each cell and each pattern center. Each cell was assigned to the closest pattern center. We identify genes differentially expressed in the cells assigned to each pattern using the FindAllMarkers function in the Seurat package.
Annotating cell type-specific and functionally related genes
To establish genes’ functional relatedness in a cell type-specific manner, we first constructed gold standards by labelling gene pairs as cell type-specific (positive examples) if both genes were differentially expressed within a cell type (fold change > 0, Wilcoxon test adjusted p-value < 0.05), or non-specific if at least one gene did not fit the criteria. These positive gene pairs were further filtered by determining their relatedness using a set of 618 expert-selected GO biological process terms (GO evidence codes: EXP, IDA, IPI, IMP, IGI and IEP). Pairs of genes that were co-annotated were treated as positive examples (i.e., functionally related). Gene pairs that were not both expressed in the same cell type (could still be functionally related or not but here treated as not fitting our criteria) were used as negatives. These gold standards allowed us to train a classifier to predict gene pairs that were both cell-type specific and functionally related.
positive: if both genes were differential expressed in the current cell type and functionally related;
negative: if both genes are from other cell types and functionally related; or if both genes are from other cell types and not functionally related.
Building developmental kidney gene functional networks
To predict functional relatedness for pairwise genes, for each developmental kidney cell type, we build a classifier by integrating labeled cell type-specific genes pairs with 1,541 genome-scale data sets (including 1533 human gene expression data sets from GEO, interaction data from BioGRID, IntAct, MINT and MIPs, transcription factor co-regulation from JASPAR, genetic perturbation (c2:CGP) and microRNA target (c3:MIR) from MSigDB) (Supplementary 5).
We downloaded 1533 human gene expression data sets from GEO (https://www.ncbi.nlm.nih.gov/geo/). The Pearson correlation was calculated for each gene pair, and further normalized with Fisher’s z transform. The resulting z scores were discretized into bins ((−infinity, −1.5), [–1.5, −0.5), [–0.5, 0.5), [0.5, 1.5), [1.5, 2.5), [2.5, 3.5), [3.5, infinity)).
BioGRID interactions (https://thebiogrid.org/) were discretized into five bins, labeled 0 to 4, where the bin number reflected the number of protein, genetic and/or chemical experiments supporting the gene interaction. For the remaining databases, edges were discretized into the presence or absence of an interaction.
To estimate shared transcription factor regulation, binding motifs were downloaded from JASPAR (http://jaspar.genereg.net/). Genes were scored for the presence of transcription factor binding motif at their promoter regions using MEME (Bailey et al., 2009). Specifically, for each transcription factor, FIMO (Grant et al., 2011) was used to scan for significant motif matches within the 1-kb sequence upstream of each gene. Motif matches were treated as binary scores (present if P < 0.001). The final score for each gene pair was obtained by calculating the Pearson correlation between the motif association vectors for the two genes.
Chemical and genetic perturbation (c2:CGP) and microRNA target (c3:MIR) profiles were downloaded from MSigDB (http://www.gsea-msigdb.org/gsea/msigdb/index.jsp). Each gene pair’s score was the sum of shared profiles weighted by the specificity of each profile (1/len(genes)). The resulting scores were converted to z scores and discretized into bins ((−infinity, −1.5), [−1.5, −0.5), [–0.5, 0.5), [0.5, 1.5), [1.5, 2.5), [2.5, 3.5), [3.5, 4.5), [4.5, infinity)).
A regularized naive Bayesian classifier was trained between positive and negative gene pairs, using gene co-expression or interaction in each data set as feature inputs. A C++ library (Huttenhower et al., 2008) that implements Bayesian integration can be accessed at https://functionlab.github.io/sleipnir-docs/index.html. Using the learned classifier, for every gene pair, we predicted its cell-type specific functional relatedness as an edge score (0 ~ 1) in a gene functional network.
We assessed the network performance on CAKUT disease genes. We curated 272 genes involved in congenital and/or kidney disease from public literature review. Using OMIM genes that are not associated with kidney diseases as background, the median network prediction accuracy (in terms of AUC) on CAKUT disease genes is 0.72 (with the highest performance of 0.77), significantly higher than the mean prediction AUC of 0.67 on kidney disease genes (p-value = 1.2e-5, Wilcoxon rank sum test).
Identifying genes functionally associated with kidney disease using gene functional networks
To identify candidate genes functionally related to KD/CAKUT, we integrated literature-curated KD/CAKUT-associated genes with cell type-specific gene functional networks. In each functional network, among genes differentially expressed in that cell type, we selected genes whose interactions to KD/CAKUT genes were significantly stronger than the interactions to 4,029 non-KD disease genes in the OMIM database (Wilcoxon test, p-value <0.05). To further strengthen the disease potential of each predicted gene, we intersected the candidate gene list against the GFR GWAS enrichment of each gene. The GFR GWAS association suggested kidney abnormal function. Using each of four GWAS traits, we calculated the enrichment of GWAS signals in the gene bodys using Vegas2 (Mishra and Macgregor, 2015) and filtered the candidate gene list by selecting significant ones (p-value < 0.05) in at least two studies.
Additional resources
The Human Nephrogenesis Atlas (https://sckidney.flatironinstitute.org/) displays scRNA-seq data in three displays (1) as a lineage tree with expression levels averaged across the cluster, (2) as an anatomical diagram of the nephrogenic niche using the same values as in the lineage tree, and (3) as a tSNE plot. The atlas enables individual or multi-gene searches for gene networks within each cell-type as defined in the 18 clusters. Further, 3D rendering views are presented for the predicted protein patterns as well as the protein abundance for each of the proteins used for the construction of the S-shaped body nephron.
Supplementary Material
Supplementary table 1 - Related to Figure 3 and Supplementary figure 2 - Differential gene expression in whole week 14 kidneys
Differential gene expression analyses between all cells in replicate 1 and replicate 2 of the week 14 human fetal cortex cell isolations as shown in Supplementary figure 2A.
Supplementary table 2 - Related to Figure 3 - Differential gene expression during nephrogenesis
Differential gene expression analyses between cells in clusters 1-18 as shown in Figure 3A.
Supplementary table 3 - Related to Figure 4 - Differential gene expression between mapped transcriptomes
Differential gene expression analyses between cells mapping to patterns 1-8 as shown in Figure 4D.
Supplementary table 4 - Related to Figure 5 - KD/CAKUT genes expression and functionally related gene prediction
Sheet1. 272 KD/CAKUT genes from literature. Sheet 2. 199 KD/CAKUT genes are active in at least one cell type (expressed in >10% cells). Normalized gene expression value per gene per cell type is shown. Sheet 3. Summary for connectivity of KD/CAKUT genes in each of the 18 cell type-specific functional networks. Sheet 4. Network-based prediction of cell type-specific genes that are functionally related to KD/CAKUT, validated against GWAS.
Supplementary table 5 - Related to Figure 5 - Genome-scale data sets used for generating gene functional networks.
Sheet 1. Dataset Name, Title, Description, and Version Download Link for each dataset used in generating the gene functional networks.
Research highlights.
Mouse and human kidneys undergo a conserved stereotypical developmental program.
Transcriptomes were mapped to protein-based developmental models.
Data predict lineage relationships in early human nephron development.
View lineage and interaction predictions: https://sckidney.flatironinstitute.org/
Acknowledgments
We thank current and past members of the McMahon lab for helpful discussions and feedback. We would also like to thank Melissa L. Wilson (Department of Preventive Medicine, University of Southern California) and Family Planning Associates for coordinating fetal tissue collection. BH and CA wish to thank Richard Baldock of the MRC Human Genetics Unit, IGMM, University of Edinburgh for supporting this project and providing financial support enabling travel for CA to visit USC and NOL at the outset of the project. We thank Friedhelm Hildebrandt for discussion and informational sources on known KD/CAKUT genes and Pietro Cippa for helpful discussions on GWAS studies.
Funding
Work in APM’s laboratory was supported by grants from the National Institutes of Health (DK107350, DK094526, DK110792) and the California Institute for Regenerative Medicine (LA1–06536).Work in OT’s laboratory was supported by NIH/NIDDK grants U24DK100845, UGDK114907, U2CDK114886 and NIH grant UH3TR002158 to O.G.T. O.G.T. is a senior fellow of the Genetic Networks program of the Canadian Institute for Advanced Research (CIFAR).
Footnotes
Declaration of Interests
APM is a scientific advisor on kidney research at Novartis, TRESTLE Therapeutics, eGENESIS and IVIVA Medical.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alarcón P, Roadríguez-Seguel E, Fernández-González A, Rubio R, and Gómez-Skarmeta JL (2008). A dual requirement for Iroquois genes during Xenopus kidney development. Development 135, 3197–3207. [DOI] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, and Noble WS (2009). MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 37, W202–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker N, Rookmaaker MB, Kujala P, Ng A, Leushacke M, Snippert H, van de Wetering M, Tan S, Van Es JH, Huch M, et al. (2012). Lgr5+ve Stem/Progenitor Cells Contribute to Nephron Formation during Kidney Development. Cell Rep. 2, 540–552. [DOI] [PubMed] [Google Scholar]
- Basta JM, Robbins L, Kiefer SM, Dorsett D, and Rauchman M (2014). Sall1 balances self-renewal and differentiation of renal progenitor cells. Development 141, 1047–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedito R, Roca C, Sörensen I, Adams S, Gossler A, Fruttiger M, and Adams RH (2009). The Notch Ligands Dll4 and Jagged1 Have Opposing Effects on Angiogenesis. Cell 137, 1124–1135. [DOI] [PubMed] [Google Scholar]
- van den Berg CW, Ritsma L, Avramut MC, Wiersma LE, van den Berg BM, Leuning DG, Lievers E, Koning M, Vanslambrouck JM, Koster AJ, et al. (2018). Renal Subcapsular Transplantation of PSC-Derived Kidney Organoids Induces Neo-vasculogenesis and Significant Glomerular and Tubular Maturation In Vivo. Stem Cell Reports 10, 751–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi S, Toffoli B, Bossi F, Candido R, Stenner E, Carretta R, Barbone F, and Fabris B (2017). Circulating osteoprotegerin is associated with chronic kidney disease in hypertensive patients. BMC Nephrol. 18, 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle S, Misfeldt A, Chandler KJ, Deal KK, Southard-Smith EM, Mortlock DP, Baldwin HS, and de Caestecker M (2008). Fate mapping using Cited1-CreERT2 mice demonstrates that the cap mesenchyme contains self-renewing progenitor cells and gives rise exclusively to nephronic epithelia. Dev. Biol. 313, 234–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AC, Muthukrishnan SD, and Oxburgh L (2015). A Synthetic Niche for Nephron Progenitor Cells. Dev. Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll TJ, Park JS, Hayashi S, Majumdar A, and McMahon AP (2005). Wnt9b plays a central role in the regulation of mesenchymal to epithelial transitions underlying organogenesis of the mammalian urogenital system. Dev. Cell 9, 283–292. [DOI] [PubMed] [Google Scholar]
- Chambers BE, Gerlach GF, Clark EG, Chen KH, Levesque AE, Leshchiner I, Goessling W, and Wingert RA (2019). Tfap2a is a novel gatekeeper of nephron differentiation during kidney development. Development 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers BE, Clark EG, Gatz AE, and Wingert RA (2020). Kctd15 regulates nephron segment development by repressing Tfap2a activity. Development. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, and Al-Awqati Q (2005). Segmental expression of Notch and Hairy genes in nephrogenesis. Am. J. Physiol. - Ren. Physiol 288, F939–F952. [DOI] [PubMed] [Google Scholar]
- Chen L, Lee JW, Chou CL, Nair AV, Battistone MA, Păunescu TG, Merkulova M, Breton S, Verlander JW, Wall SM, et al. (2017). Transcriptomes of major renal collecting duct cell types in mouse identified by single-cell RNA-seq. Proc. Natl. Acad. Sci. U. S. A. 114, E9989–E9998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H-T, Kim M, Valerius MT, Surendran K, Schuster-Gossler K, Gossler A, McMahon AP, and Kopan R (2007). Notch2, but not Notch1, is required for proximal fate acquisition in the mammalian nephron. Development 134, 801–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho EA, Patterson LT, Brookhiser WT, Mah S, Kintner C, and Dressler GR (1998). Differential expression and function of cadherin-6 during renal epithelium development. Development 125, 803–812. [DOI] [PubMed] [Google Scholar]
- Chung E, Deacon P, Marable S, Shin J, and Park JS (2016). Notch signaling promotes nephrogenesis by downregulating Six2. Dev. 143, 3907–3913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combes AN, Phipson B, Lawlor KT, Dorison A, Patrick R, Zappia L, Harvey RP, Oshlack A, and Little MH (2019a). Single cell analysis of the developing mouse kidney provides deeper insight into marker gene expression and ligand-receptor crosstalk. Development 146. [DOI] [PubMed] [Google Scholar]
- Combes AN, Zappia L, Er PX, Oshlack A, and Little MH (2019b). Single-cell analysis reveals congruence between kidney organoids and human fetal kidney. Genome Med. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowley AW, Yang C, Kumar V, Lazar J, Jacob H, Geurts AM, Liu P, Dayton A, Kurth T, and Liang M (2016). Pappa2 is linked to salt-sensitive hypertension in Dahl S rats. Physiol. Genomics 48, 62–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies JA, and Garrod DR (1995). Induction of Early Stages of Kidney Tubule Differentiation by Lithium Ions. Dev. Biol. 167, 50–60. [DOI] [PubMed] [Google Scholar]
- Dominguez D, Tsai Y-H, Gomez N, Jha DK, Davis I, and Wang Z (2016). A high-resolution transcriptome map of cell cycle reveals novel connections between periodic genes and cancer. Cell Res. 26, 946–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X, Yang H, Kumar S, Tumelty KE, Pisarek-Horowitz A, Rasouly HM, Sharma R, Chan S, Tyminski E, Shamashkin M, et al. (2016). SLIT2/ROBO2 signaling pathway inhibits nonmuscle myosin IIA activity and destabilizes kidney podocyte adhesion. JCI Insight 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, Yang Z, Hiremath C, Zimmerman SE, Long B, Brakeman PR, Mostov KE, Bryant DM, Luby-Phelps K, and Marciano DK (2017). Afadin orients cell division to position the tubule lumen in developing renal tubules. Development 144, 3511 LP–3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgas K, Rumballe B, Valerius MT, Chiu HS, Thiagarajan RD, Lesieur E, Aronow BJ, Brunskill EW, Combes AN, Tang D, et al. (2009). Analysis of early nephron patterning reveals a role for distal RV proliferation in fusion to the ureteric tip via a cap mesenchyme-derived connecting segment. Dev. Biol. 332, 273–286. [DOI] [PubMed] [Google Scholar]
- Good IJ (1953). The Population Frequencies of Species and the Estimation of Population Parameters. Biometrika 40, 237. [Google Scholar]
- Grant CE, Bailey TL, and Noble WS (2011). FIMO: scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene CS, Krishnan A, Wong AK, Ricciotti E, Zelaya RA, Himmelstein DS, Zhang R, Hartmann BM, Zaslavsky E, Sealfon SC, et al. (2015). Understanding multicellular function and disease with human tissue-specific networks. Nat. Genet. 47, 569–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieshammer U, Le Ma, Plump AS, Wang F, Tessier-Lavigne M, and Martin GR (2004). SLIT2-Mediated ROBO2 Signaling Restricts Kidney Induction to a Single Site. Dev. Cell 6, 709–717. [DOI] [PubMed] [Google Scholar]
- Grieshammer U, Cebrián C, Ilagan R, Meyers E, Herzlinger D, and Martin GR (2005). FGF8 is required for cell survival at distinct stages of nephrogenesis and for regulation of gene expression in nascent nephrons. Development 132, 3847–3857. [DOI] [PubMed] [Google Scholar]
- Grote D, Narlis M, Gaitan Y, Boualia SK, and Bouchard M (2007). Pax2 and Pax8 regulate branching morphogenesis and nephron differentiation in the developing kidney. Dev. Biol. 306, 413. [DOI] [PubMed] [Google Scholar]
- Han X, Wang R, Zhou Y, Fei L, Sun H, Lai S, Saadatpour A, Zhou Z, Chen H, Ye F, et al. (2018). Mapping the Mouse Cell Atlas by Microwell-Seq. Cell 172, 1091–1107.e17. [DOI] [PubMed] [Google Scholar]
- Heliot C, Desgrange A, Buisson I, Prunskaite-Hyyryläinen R, Shan J, Vainio S, Umbhauer M, and Cereghini S (2013). HNF1B controls proximal-intermediate nephron segment identity in vertebrates by regulating Notch signalling components and <em>Irx1/2</em> Development 140, 873 LP–885. [DOI] [PubMed] [Google Scholar]
- Hildebrandt F (2010). Genetic kidney diseases. Lancet 375, 1287–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochane M, van den Berg PR, Fan X, Bérenger-Currias N, Adegeest E, Bialecka M, Nieveen M, Menschaart M, Chuva de Sousa Lopes SM, and Semrau S (2019). Single-cell transcriptomics reveals gene expression dynamics of human fetal kidney development. PLoS Biol. 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howden SE, Wilson SB, Groenewegen E, Starks L, Forbes TA, Tan KS, Vanslambrouck JM, Holloway EM, Chen Y-H, Jain S, et al. (2021). Plasticity of distal nephron epithelia from human kidney organoids enables the induction of ureteric tip and stalk. Cell Stem Cell 28, 671–684.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M, Wang J, Torre E, Dueck H, Shaffer S, Bonasio R, Murray JI, Raj A, Li M, and Zhang NR (2018). SAVER: Gene expression recovery for single-cell RNA sequencing. Nat. Methods. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber CG (1905). On the development and shape of uriniferous tubules of certain of the higher mammals. Am. J. Anat. 4, 1–98. [Google Scholar]
- Huttenhower C, Schroeder M, Chikina MD, and Troyanskaya OG (2008). The Sleipnir library for computational functional genomics. Bioinformatics 24, 1559–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang SJ, Yang Q, Meigs JB, Pearce EN, and Fox CS (2007). A genome-wide association for kidney function and endocrine-related traits in the NHLBI’s Framingham Heart Study. BMC Med. Genet. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao RM, Vasilyev A, Miyawaki A, Drummond IA, and McMahon AP (2012). Invasion of distal nephron precursors associates with tubular interconnection during nephrogenesis. J. Am. Soc. Nephrol. 23, 1682–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaiskos N, Wahle P, Alles J, Boltengagen A, Ayoub S, Kipar C, Kocks C, Rajewsky N, and Zinzen RP (2017). The Drosophila embryo at single-cell transcriptome resolution. Science (80-. ). [DOI] [PubMed] [Google Scholar]
- Karner CM, Das A, Ma Z, Self M, Chen C, Lum L, Oliver G, and Carroll TJ (2011). Canonical Wnt9b signaling balances progenitor cell expansion and differentiation during kidney development. Development 138, 1247–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearney PM, Whelton M, Reynolds K, Muntner P, Whelton PK, and He J (2005). Global burden of hypertension: Analysis of worldwide data. Lancet 365, 217–223. [DOI] [PubMed] [Google Scholar]
- Kim AD, Lake BB, Chen S, Wu Y, Guo J, Parvez RK, Tran T, Thornton ME, Grubbs B, McMahon JA, et al. (2019). Cellular Recruitment by Podocyte-Derived Pro-migratory Factors in Assembly of the Human Renal Filter. IScience 20, 402–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KH, Rosen A, Bruneau BG, Hui CC, and Backx PH (2012). Iroquois homeodomain transcription factors in heart development and function. Circ. Res. 110, 1513–1524. [DOI] [PubMed] [Google Scholar]
- Kobayashi A (2005). Distinct and sequential tissue-specific activities of the LIM-class homeobox gene Lim1 for tubular morphogenesis during kidney development. Development 132, 2809–2823. [DOI] [PubMed] [Google Scholar]
- Kobayashi A, Valerius MT, Mugford JW, Carroll TJ, Self M, Oliver G, and McMahon AP (2008). Six2 Defines and Regulates a Multipotent Self-Renewing Nephron Progenitor Population throughout Mammalian Kidney Development. Cell Stem Cell 3, 169–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köttgen A, Glazer NL, Dehghan A, Hwang SJ, Katz R, Li M, Yang Q, Gudnason V, Launer LJ, Harris TB, et al. (2009). Multiple loci associated with indices of renal function and chronic kidney disease. Nat. Genet. 41, 712–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan A, Zhang R, Yao V, Theesfeld CL, Wong AK, Tadych A, Volfovsky N, Packer A, Lash A, and Troyanskaya OG (2016). Genome-wide prediction and functional characterization of the genetic basis of autism spectrum disorder. Nat. Neurosci. 19, 1454–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuure S, Popsueva A, Jakobson M, Sainio K, and Sariola H (2007). Glycogen Synthase Kinase-3 Inactivation and Stabilization of β-Catenin Induce Nephron Differentiation in Isolated Mouse and Rat Kidney Mesenchymes. J. Am. Soc. Nephrol. 18, 1130–1139. [DOI] [PubMed] [Google Scholar]
- Lake BB, Chen S, Hoshi M, Plongthongkum N, Salamon D, Knoten A, Vijayan A, Venkatesh R, Kim EH, Gao D, et al. (2019). A single-nucleus RNA-sequencing pipeline to decipher the molecular anatomy and pathophysiology of human kidneys. Nat. Commun 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawlor KT, Zappia L, Lefevre J, Park J-S, Hamilton NA, Oshlack A, Little MH, and Combes AN (2019). Nephron progenitor commitment is a stochastic process influenced by cell migration. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JW, Chou CL, and Knepper M. a. (2015). Deep sequencing in microdissected renal tubules identifies nephron segment-specific transcriptomes. J. Am. Soc. Nephrol. 26, 2669–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Krantz ID, Deng Y, Genin A, Banta AB, Collins CC, Qi M, Trask BJ, Kuo WL, Cochran J, et al. (1997). Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for notch1. Nat. Genet. 16, 243–251. [DOI] [PubMed] [Google Scholar]
- Lignell A, Kerosuo L, Streichan SJ, Cai L, and Bronner ME (2017). Identification of a neural crest stem cell niche by Spatial Genomic Analysis. Nat. Commun. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindström NO, De Sena Brandine G, Tran T, Ransick A, Suh G, Guo J, Kim AD, Parvez RK, Ruffins SW, Rutledge EA, et al. (2018a). Progressive Recruitment of Mesenchymal Progenitors Reveals a Time-Dependent Process of Cell Fate Acquisition in Mouse and Human Nephrogenesis. Dev. Cell 45, 651–660.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindström NO, Tran T, Guo J, Rutledge E, Parvez RK, Thornton ME, Grubbs B, McMahon JA, and McMahon AP (2018b). Conserved and divergent molecular and anatomic features of human and mouse nephron patterning. J. Am. Soc. Nephrol. 29, 825–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindström NO, Guo J, Kim AD, Tran T, Guo Q, De Sena Brandine G, Ransick A, Parvez RK, Thornton ME, Basking L, et al. (2018c). Conserved and divergent features of mesenchymal progenitor cell types within the cortical nephrogenic niche of the human and mouse kidney. J. Am. Soc. Nephrol. 29, 806–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindström NO, McMahon JA, Guo J, Tran T, Guo Q, Rutledge E, Parvez RK, Saribekyan G, Schuler RE, Liao C, et al. (2018d). Conserved and divergent features of human and mouse kidney organogenesis. J. Am. Soc. Nephrol. 29, 785–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little MH, Brennan J, Georgas K, Davies JA, Davidson DR, Baldock RA, Beverdam A, Bertram JF, Capel B, Chiu HS, et al. (2007). A high-resolution anatomical ontology of the developing murine genitourinary tract. Gene Expr. Patterns. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Beyer A, and Aebersold R (2016). On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 165, 535–550. [DOI] [PubMed] [Google Scholar]
- Liu Z, Chen S, Boyle S, Zhu Y, Zhang A, Piwnica-Worms DR, Ilagan MXG, and Kopan R (2013). The extracellular domain of notch2 increases its cell-surface abundance and ligand responsiveness during kidney development. Dev. Cell 25, 585–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low JH, Li P, Chew EGY, Zhou B, Suzuki K, Zhang T, Lian MM, Liu M, Aizawa E, Rodriguez Esteban C, et al. (2019). Generation of Human PSC-Derived Kidney Organoids with Patterned Nephron Segments and a De Novo Vascular Network. Cell Stem Cell 25, 373–387.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, et al. (2015). Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161, 1202–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mah SP, Saueressig H, Goulding M, Kintner C, and Dressler GR (2000). Kidney Development in Cadherin-6 Mutants: Delayed Mesenchyme-to-Epithelial Conversion and Loss of Nephrons. Dev. Biol. 223, 38–53. [DOI] [PubMed] [Google Scholar]
- Marable SS, Chung E, Adam M, Potter SS, and Park J-SS (2018). Hnf4a deletion in the mouse kidney phenocopies Fanconi renotubular syndrome. JCI Insight 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marable SS, Chung E, and Park J-S (2020). Hnf4a Is Required for the Development of Cdh6-Expressing Progenitors into Proximal Tubules in the Mouse Kidney. J. Am. Soc. Nephrol. 31, 2543–2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marneros AG (2020). AP-2β/KCTD1 Control Distal Nephron Differentiation and Protect against Renal Fibrosis. Dev. Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon AP (2016). Development of the Mammalian Kidney. In Current Topics in Developmental Biology, pp. 31–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon AP, Aronow BJ, Davidson DR, Davies JA, Gaido KW, Grimmond S, Lessard JL, Little MH, Potter SS, Wilder EL, et al. (2008). GUDMAP: the genitourinary developmental molecular anatomy project. J. Am. Soc. Nephrol. 19, 667–671. [DOI] [PubMed] [Google Scholar]
- Menon R, Otto EA, Kokoruda A, Zhou J, Zhang Z, Yoon E, Chen YC, Troyanskaya O, Spence JR, Kretzler M, et al. (2018). Single-cell analysis of progenitor cell dynamics and lineage specification in the human fetal kidney. Dev. 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao Z, Balzer MS, Ma Z, Liu H, Wu J, Shrestha R, Aranyi T, Kwan A, Kondo A, Pontoglio M, et al. (2021). Single cell regulatory landscape of the mouse kidney highlights cellular differentiation programs and disease targets. Nat. Commun. 12, 2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mugford JW, Yu J, Kobayashi A, and McMahon AP (2009). High-resolution gene expression analysis of the developing mouse kidney defines novel cellular compartments within the nephron progenitor population. Dev. Biol. 333, 312–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundel P, Bachmann S, Bader M, Fischer A, Kummer W, Mayer B, and Kriz W (1992). Expression of nitric oxide synthase in kidney macula densa cells. Kidney Int. 42, 1017–1019. [DOI] [PubMed] [Google Scholar]
- Nakai S, Sugitani Y, Sato H, Ito S, Miura Y, Ogawa M, Nishi M, Jishage KI, Minowa O, and Noda T (2003). Crucial roles fo Brn1 in distal tubule formation and function in mouse kidney. Development. [DOI] [PubMed] [Google Scholar]
- O’Brien LL, Guo Q, Lee YJ, Tran T, Benazet JD, Whitney PH, Valouev A, and McMahon AP (2016). Differential regulation of mouse and human nephron progenitors by the six family of transcriptional regulators. Dev. 143, 595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien LL, Combes AN, Short KM, Lindström NO, Whitney PH, Cullen-McEwen LA, Ju A, Abdelhalim A, Michos O, Bertram JF, et al. (2018). Wnt11 directs nephron progenitor polarity and motile behavior ultimately determining nephron endowment. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver J (1968). A Quantitative Study of Development and Evolutionary Mammalian Renal Architectonics (New York: Harper & Row; ). [Google Scholar]
- Osathanondh V, and Potter EL (1963). DEVELOPMENT OF HUMAN KIDNEY AS SHOWN BY MICRODISSECTION. III. FORMATION AND INTERRELATIONSHIP OF COLLECTING TUBULES AND NEPHRONS. Arch. Pathol. 76, 290–302. [PubMed] [Google Scholar]
- Park J-S, Valerius MT, and McMahon AP (2007). Wnt/ -catenin signaling regulates nephron induction during mouse kidney development. Development 134, 2533–2539. [DOI] [PubMed] [Google Scholar]
- Park J, Shrestha R, Qiu C, Kondo A, Huang S, Werth M, Li M, Barasch J, and Suszták K (2018). Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science (80-. ). 360, 758–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattaro C, Teumer A, Gorski M, Chu AY, Li M, Mijatovic V, Garnaas M, Tin A, Sorice R, Li Y, et al. (2016). Genetic associations at 53 loci highlight cell types and biological pathways relevant for kidney function. Nat. Commun. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peti-Peterdi J, and Harris RC (2010). Macula densa sensing and signaling mechanisms of renin release. J. Am. Soc. Nephrol. 21, 1093–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransick A, Lindström NO, Liu J, Zhu Q, Guo JJ, Alvarado GF, Kim AD, Black HG, Kim J, and McMahon AP (2019). Single-Cell Profiling Reveals Sex, Lineage, and Regional Diversity in the Mouse Kidney. Dev. Cell 51, 399–413.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reggiani L, Raciti D, Airik R, Kispert A, and Brändli AW (2007). The prepattern transcription factor Irx3 directs nephron segment identity. Genes Dev. 21, 2358–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson DS, and Lichtman JW (2015). Clarifying Tissue Clearing. Cell 162, 246–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinkevich Y, Montoro DT, Contreras-Trujillo H, Harari-Steinberg O, Newman AM, Tsai JM, Lim X, Van-Amerongen R, Bowman A, Januszyk M, et al. (2014). In vivo clonal analysis reveals lineage-restricted progenitor characteristics in mammalian kidney development, maintenance, and regeneration. Cell Rep. 7, 1270–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satija R, Farrell JA, Gennert D, Schier AF, and Regev A (2015). Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 33, 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxen L (1987). Organogenesis of the Kidney (Cambridge University Press; ). [Google Scholar]
- Schaum N, Karkanias J, Neff NF, May AP, Quake SR, Wyss-Coray T, Darmanis S, Batson J, Botvinnik O, Chen MB, et al. (2018). Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 562, 367–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schedl A (2007). Renal abnormalities and their developmental origin. Nat. Rev. Genet. 8, 791–802. [DOI] [PubMed] [Google Scholar]
- Short KM, Combes AN, Lefevre J, Ju AL, Georgas KM, Lamberton T, Cairncross O, Rumballe BA, McMahon AP, Hamilton NA, et al. (2014). Global quantification of tissue dynamics in the developing mouse kidney. Dev. Cell. [DOI] [PubMed] [Google Scholar]
- Singh N, Avigan ZM, Kliegel JA, Shuch BM, Montgomery RR, Moeckel GW, and Cantley LG (2019). Development of a 2-dimensional atlas of the human kidney with imaging mass cytometry. JCI Insight 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinner NB, Colliton RP, Crosnier C, Krantz ID, Hadchouel M, and Meunier-Rotival M (2001). Jagged1 mutations in Alagille syndrome. Hum. Mutat. 17, 18–33. [DOI] [PubMed] [Google Scholar]
- Stark K, Vainio S, Vassileva G, and McMahon AP (1994). Epithelial transformation of metanephric mesenchyme in the developing kidney regulated by Wnt-4. Nature 372, 679–683. [DOI] [PubMed] [Google Scholar]
- Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, Satija R, and Smibert P (2017). Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 14, 865–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Takahashi K, Gerety S, Wang H, Anderson DJ, and Daniel TO (2001). Temporally compartmentalized expression of ephrin-B2 during renal glomerular development. J. Am. Soc. Nephrol. 12, 2673–2682. [DOI] [PubMed] [Google Scholar]
- Tiveron MC, Beurrier C, Céni C, Andriambao N, Combes A, Koehl M, Maurice N, Gatti E, Abrous DN, Kerkerian-Le Goff L, et al. (2016). LAMP5 fine-tunes GABAergic synaptic transmission in defined circuits of the mouse brain. PLoS One 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran T, Lindström NO, Ransick A, De Sena Brandine G, Guo Q, Kim AD, Der B, Peti-Peterdi J, Smith AD, Thornton M, et al. (2019). In Vivo Developmental Trajectories of Human Podocyte Inform In Vitro Differentiation of Pluripotent Stem Cell-Derived Podocytes. Dev. Cell 50, 102–116.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tustison NJ, Cook PA, Klein A, Song G, Das SR, Duda JT, Kandel BM, van Strien N, Stone JR, Gee JC, et al. (2014). Large-scale evaluation of ANTs and FreeSurfer cortical thickness measurements. Neuroimage 99, 166–179. [DOI] [PubMed] [Google Scholar]
- Ueda H, Miyazaki Y, Matsusaka T, Utsunomiya Y, Kawamura T, Hosoya T, and Ichikawa L (2008). Bmp in podocytes is essential for normal glomerular capillary formation. J. Am. Soc. Nephrol. 19, 685–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veikkolainen V, Naillat F, Railo A, Chi L, Manninen A, Hohenstein P, Hastie N, Vainio S, and Elenius K (2012). ErbB4 modulates tubular cell polarity and lumen diameter during kidney development. J. Am. Soc. Nephrol. 23, 112–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivante A, Kohl S, Hwang DY, Dworschak GC, and Hildebrandt F (2014). Single-gene causes of congenital anomalies of the kidney and urinary tract (CAKUT) in humans. In Pediatric Nephrology, p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Nakayama M, Pitulescu ME, Schmidt TS, Bochenek ML, Sakakibara A, Adams S, Davy A, Deutsch U, Lüthi U, et al. (2010). Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature 465, 483–486. [DOI] [PubMed] [Google Scholar]
- Weber S, Taylor JC, Winyard P, Baker KF, Sullivan-Brown J, Schild R, Knüppel T, Zurowska AM, Caldas-Alfonso A, Litwin M, et al. (2008). SIX2 and BMP4 Mutations Associate With Anomalous Kidney Development. J. Am. Soc. Nephrol. 19, 891–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox CS, Welch WJ, Murad F, Gross SS, Taylor G, Levi R, and Schmidt HHHW (1992). Nitric oxide synthase in macula densa regulates glomerular capillary pressure. Proc. Natl. Acad. Sci. U. S. A. 89, 11993–11997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong AK, Krishnan A, and Troyanskaya OG (2018). GIANT 2.0: Genome-scale integrated analysis of gene networks in tissues. Nucleic Acids Res. 46, W65–W70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Tamayo P, and Zhang K (2018). Visualizing and Interpreting Single-Cell Gene Expression Datasets with Similarity Weighted Nonnegative Embedding. Cell Syst. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuttke M, Li Y, Li M, Sieber KB, Feitosa MF, Gorski M, Tin A, Wang L, Chu AY, Hoppmann A, et al. (2019). A catalog of genetic loci associated with kidney function from analyses of a million individuals. Nat. Genet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia C, Fan J, Emanuel G, Hao J, and Zhuang X (2019). Spatial transcriptome profiling by MERFISH reveals subcellular RNA compartmentalization and cell cycle-dependent gene expression. Proc. Natl. Acad. Sci. 116, 19490–19499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Wong EYM, Cheng C, Li J, Sharkar MTK, Xu CY, Chen B, Sun J, Jing D, and Xu PX (2014). Eya1 interacts with Six2 and Myc to regulate expansion of the nephron progenitor pool during nephrogenesis. Dev. Cell 31, 434–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary table 1 - Related to Figure 3 and Supplementary figure 2 - Differential gene expression in whole week 14 kidneys
Differential gene expression analyses between all cells in replicate 1 and replicate 2 of the week 14 human fetal cortex cell isolations as shown in Supplementary figure 2A.
Supplementary table 2 - Related to Figure 3 - Differential gene expression during nephrogenesis
Differential gene expression analyses between cells in clusters 1-18 as shown in Figure 3A.
Supplementary table 3 - Related to Figure 4 - Differential gene expression between mapped transcriptomes
Differential gene expression analyses between cells mapping to patterns 1-8 as shown in Figure 4D.
Supplementary table 4 - Related to Figure 5 - KD/CAKUT genes expression and functionally related gene prediction
Sheet1. 272 KD/CAKUT genes from literature. Sheet 2. 199 KD/CAKUT genes are active in at least one cell type (expressed in >10% cells). Normalized gene expression value per gene per cell type is shown. Sheet 3. Summary for connectivity of KD/CAKUT genes in each of the 18 cell type-specific functional networks. Sheet 4. Network-based prediction of cell type-specific genes that are functionally related to KD/CAKUT, validated against GWAS.
Supplementary table 5 - Related to Figure 5 - Genome-scale data sets used for generating gene functional networks.
Sheet 1. Dataset Name, Title, Description, and Version Download Link for each dataset used in generating the gene functional networks.