

Abstract
Glucosamine (GlcN) is a naturally occurring amino-monosaccharide with putative chondroprotective activity. Optimum responses to GlcN are achieved at concentrations which are impractical with oral dosing. Intra-articular delivery of a bolus dose of GlcN is one way to overcome these pharmacokinetic obstacles. In this study we report the effects of exposing primary human chondrocytes to a bolus dose of GlcN. We also locally administered GlcN in the context of a meniscal transection model of rat osteoarthritis (OA). The knees of male rats were subjected to medial meniscal transection and developed arthritic changes over 4 weeks. Treatment groups were then given thrice weekly 100 μL injections of 35 μg, 350 μg, 1.8 mg, or 3.5 mg of GlcN dissolved in normal saline. Gross images, modified Mankin scores, and histomorphometric measurements were used as outcome measures. The 350 μg dosage of GlcN had the most significant positive impact on all components of the modified Mankin score. Together, these findings suggest the local delivery of high concentrations of GlcN is well tolerated and can suppress experimental OA through influences on both bone and cartilage.
Keywords: glucosamine, osteoarthritis, rat arthritis, animal models
Osteoarthritis is one of the most common joint disorders afflicting humans, and it is estimated that over 60% of the United States population develop radio-graphic evidence of OA by 55 years of age.1 The pathologic hallmarks of OA include progressive degeneration of articular cartilage and changes in subchondral bone architecture. OA is a disease of the entire joint organ including cartilage, subchondral bone, synovium, ligament, and resident stem cells.2–5 The etiology of OA is not fully understood. At the cellular level, disease progression has been divided into a biosynthetic and degenerative phase.6,7 The biosynthetic phase begins with extracellular matrix (ECM) damage that the chondrocytes fail to adequately repair. During the degenerative phase, ECM production is inhibited and the chondrocytes release proteolytic enzymes which accelerate cartilage loss. The degraded ECM products diffuse throughout the joint ultimately leading to inflammatory changes which perpetuate disease progression.3,6,8 Currently accepted pharmacologic therapy for OA is centered on symptomatic relief and is limited to analgesic medications, anti-inflammatory agents, and viscosupplimentation. These interventions provide temporary relief from pain and improve function; however, they have little effect on the structural degradation of joint tissue.9
The utility of putative chondroprotective agents such as chondrotin sulfate and glucosamine (GlcN) are controversial and under intense investigation. The molecular mechanisms of GlcN remain poorly defined; however, numerous in vitro studies demonstrate GlcN affects multiple cell types within the joint organ including chondrocytes, mesenchymal stem cells, synoviocytes, osteoblasts, and osteoclasts.10–14 The beneficial effects of GlcN in vitro are frequently seen at millimolar concentrations, which are typically 103–104 fold above those obtained with standard oral dosing.15 One potential solution to achieve higher concentrations of GlcN in vivo is through an intra-articular injection. This clearly represents a different pharmacokinetic profile than continuously exposing cells to high concentrations. Nevertheless, with appropriate dosing, it is feasible that multiple high dose exposures could have a significant effect. To our knowledge, there are no published studies examining the effects of a single bolus exposure to GlcN.
The multi-tissue involvement of OA requires the development of in vivo models. Animal models of OA can be achieved through natural aging processes, genetic approaches, pharmacologic manipulation, or through surgically induced joint instability.16–20 No animal models fully mimic all facets of human OA and the relevance of these models is based primarily on the histopathological similarities to human disease.19 In this study we characterize a surgical destabilization technique similar to Glasson et al.21 in which the medial meniscus of the rat is fully transected and the medial collateral ligament is spared (MMx). Menisci provide congruency and increase the contact area of articulating bones which distributes contact forces over greater areas. Surgical destabilization of the knee focuses mechanical stresses to smaller areas and results in arthritic changes that closely represent OA. These models are widely accepted for evaluating both the pathogenesis and pharmacological management of OA.
This study evaluated the anti-arthritic efficacy of locally injected GlcN within the context of a rat knee surgically destabilized through MMx. Three related sets of experiments were carried out. First, primary human OA chondrocytes were exposed in vitro to a bolus dose of GlcN within a poly(ethylene glycol) diacrylate (PEGDA) hydrogel. Hydrogels are ideal candidates for in vitro culture of chondrocytes due to their high water content and tissue-like elasticity.22 Outcome measures for the in vitro study include biochemical and gene expression analysis for commonly accepted markers of cartilage formation. The second study characterized the progression of untreated OA at 4 and 8 weeks in a surgically destabilized rat knee. Lastly, following 4 weeks of OA progression in the rat, GlcN was delivered intra-articularly with weekly injections for 3 weeks. Outcome measures employed to characterize joint disease included gross morphology, semiquantitative histopathology, and histomorphometry. The experimental design is summarized in Figure 1A and B. Taken together, our results demonstrate the delivery of small molecules through intra-articular injection is feasible, and that GlcN is efficacious for OA in surgically destabilized rat hind limbs.
Figure 1.
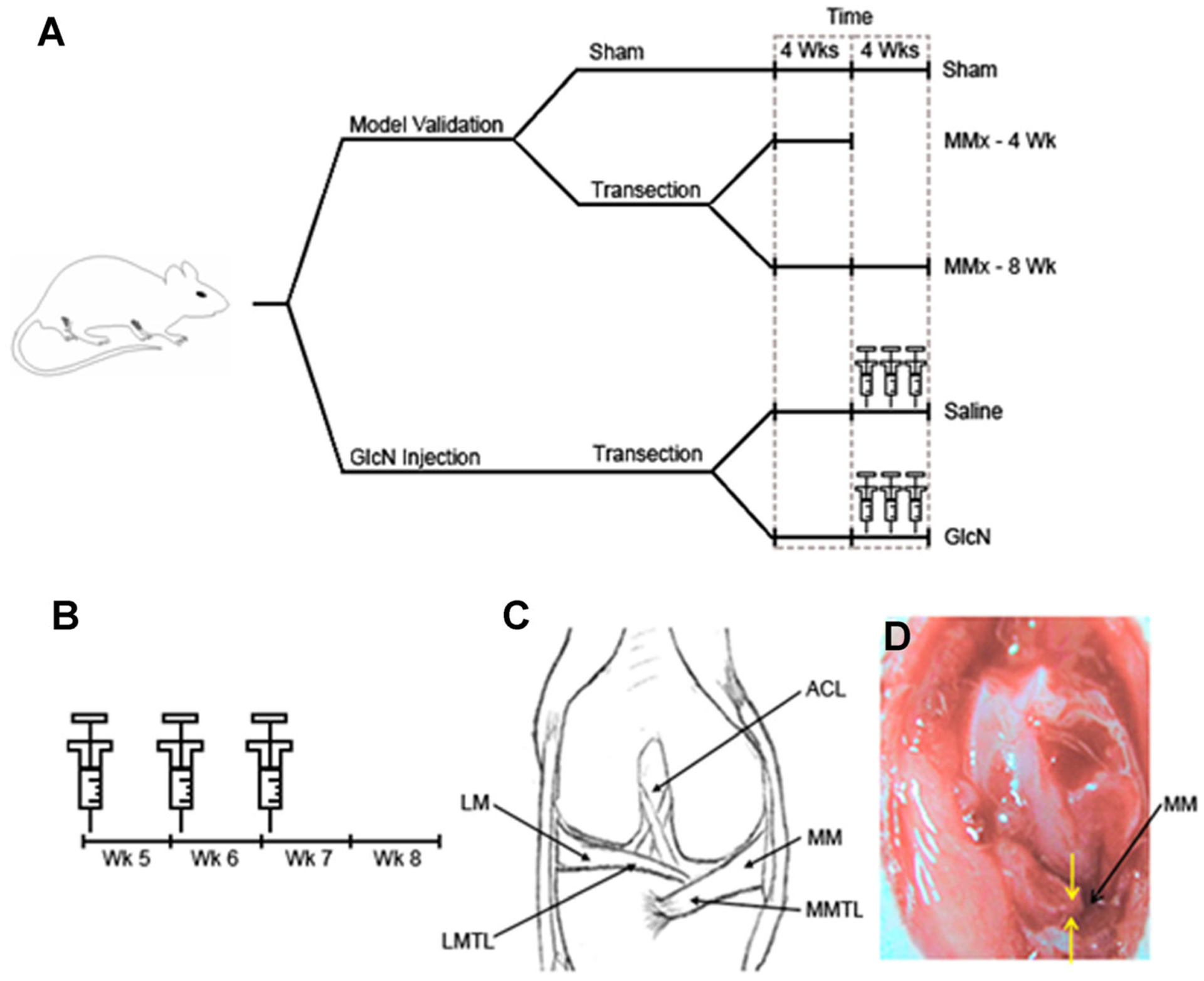
Experimental design for establishing the rat medial meniscal transeciton (MMx) model and evaluating the efficacy of intra-articular delivered GlcN. Separate experimental arms were established to validate the arthritic changes following MMx at both 4 and 8 weeks post-operatively (A). GlcN is administered through single weekly injections for 3 weeks at the beginning of weeks 5, 6, and 7 (B). Representative surgical fields and landmarks for the sham and MMx are depicted in (C) and (D), respectively. MMTL, medial meniscotibial ligament; LMTL, lateral meniscotibial ligament; MM, medial meniscus; ACL, anterior cruciate ligament.
METHODS
Chondrocyte Isolation
Human osteoarthritic chondrocytes were enzymatically isolated as previously described.23 Briefly, the cells were isolated from tissue obtained during total knee arthroplasty. The knee joints were supplied by National Disease Research Interchange (Philadelphia, PA). Cartilage explants were digested with agitation for 16 h at 37°C in a 0.17% w/v Type II Collagenase solution (Worthington Biochemical Corporation, Lakewood, NJ) containing high-glucose Dulbecco’s Modified Eagle Medium (DMEM; Gibco, Grand Island, NY) with 6% fetal bovine serum (FBS; Hyclone, Logan, UT). The resulting filtrate was passed through a 70-μm filter, and cells were rinsed thoroughly with phosphate buffered saline (PBS) containing 100 U/ml penicillin and 100 μg/ml streptomycin (Gibco). Before encapsulation, primary chondrocytes were passaged once in monolayer to obtain adequate cell numbers.
Media Conditions and Cell Encapsulation
Chondrocyte growth media (CCM) consisted of high-glucose DMEM (Gibco), 10% fetal bovine serum, 0.1 mM MEM nonessential amino acids solution (Invitrogen, Carlsbad, CA), 10 mM HEPES (Invitrogen), 50 μg/ml ascorbic acid, 0.4 mM proline (Sigma), 1 mM sodium pyruvate (Sigma), and 100 U/ml penicillin and 100 μg/ml streptomycin. Constructs were cultured in CCM for 3 weeks in a humidified 37°C environment with 5% CO2 and media was changed every 2–3 days until time of harvest.
Three-dimensional constructs were formed, using 10% poly(ethylene glycol) diacrylate(PEGDA) (Sun-Bio, Orinda, CA) dissolved in PBS. The photoinitiatorIrgacure D2959 (Ciba Specialty Chemicals, Tarytown, NY) was added to the PEGDA solution and mixed thoroughly to achieve a final 0.05% w/v concentration. Passage 1 chondrocytes were suspended within the precursor solution at a density of 2 × 107 cells/ml and then transferred into a sterile cylindrical mold followed by exposure to long wavelength 365 nm light at 4.5 mW/cm2 (Glowmark Systems, Upper Saddle River, NJ) for 5 min to achieve complete gelation. To mimic a bolus dose of GlcN, GlcN was dissolved in the precursor solution at 1 mg/ml before polymerization. The constructs were analyzed at 21 days as described below.
The initial concentration of GlcN in the construct was 1 mg/ml, and when diluted into 1.5 ml of media, this corresponds to 50 μg/ml or 0.23 mM. This assumes GlcN was distributed equally throughout the media, and ignores cellular uptake. With these same assumptions, the first media change lowers the GlcN concentration by a factor of 20 and occurred 36–48 h after the initial encapsulation.
Biochemical Analysis
Six constructs per treatment group were collected for biochemical studies. To quantify glycosaminoglycan (GAG) content, constructs were lyophilized, weighed, and crushed using a pellet pestle mixer (Kimble/Kontes, Vineland, NJ). The resulting material was digested in a 125 μg/ml papainase solution (Worthington Biomedical, Lakewood, NJ) for 18 h at 60°C. The DNA content was determined through fluorophotometry using Hoechst 33258 dye (Hoefer, Holliston, MA) as previously described.12 GAG content was measured using a dimethylene blue (DMMB) spectrophotometric assay at an absorbance of 525 nm. Standard curves were generated using chondroitin sulfate. Collagen content was determined using the hydroxyproline assay as previously described.12 Samples were hydrolyzed for 16 h at 115°C in 6 N hydrochloric acid. The resulting solutions containing 4 hydroxyproline were neutralized, oxidized with chloramine-T hydrate, and reacted with p-dimethylaminobenzaldehyde. Absorbance was measured at 550 nm and trans-4-hydroxy-L-proline (Sigma–Aldrich) was used as a standard.
Gene Expression
Total RNA was extracted from the 3D constructs using TRIzol (Invitrogen) according to the manufacturer’s instructions. Complementary DNA was formed using the reverse transcriptase Superscript First-Strand Synthesis kit (Invitrogen). Quantitative real-time PCR was conducted on the following genes: aggrecan (F: CAC GAT GCC TTT CAC CAC GAC; R: TGC GGG TCA ACA GTG CCT ATC), type II collagen (F: GAA ACC ATC AAT GGT GGC TTCC; R: CGA TAA CAG TCT TGC CCC ACTT), MMP-1 (F: CTG AAG GTG ATG AAG CAG CC; R: AGT CCA AGA GAA TGG CCG AG), MMP-2 (F: GCG ACA AGA AGT ATG GCT TC; R: TGC CAA GGT CAA TGT CAG GA), MMP-3 (F: CTC ACA GAC CTG ACT CGG TT; R: CAC GCC TGA AGG AAG AGA TG), and MMP-13 (F: CTA TGG TCC AGG AGA TGA AG; R: AGA GTC TTG CCT GTA TCC TC), MT1-MMP (F: CAA CAC TGC CTA CGA GAG GA; R: GTT CTA CCT TCA GCT TCT GG), and normalized to β-actin (F: TGG CAC CAC ACC TTC TAC AAT GAGC; R: GCA CAG CTT CTC CTT AAT GTC ACGC). All genes were analyzed in triplicates, using the method. The quantification was conducted on the ABI Prism 7700 Sequence Detection System with the SYBR Green PCR Master Mix (Invitrogen).
Surgical Induction of Joint Instability
All procedures were performed according to the Institutional Animal Care and Use Committee at Johns Hopkins University School of Medicine. Six-week old male Sprague-Dawley rats (Charles River, Germantown, MD) weighing approximately 250 g were used in these studies. Medial meniscal transection was performed based on a modification of the procedure utilized in the mouse by Glasson et al.21 Animals were housed individually for the duration of the study in 15 by 30 inch cages and allowed to move without restriction. Standard chow and water were provided ad libitum. Rats were anesthetized with 3–5% isoflurane per standard methods and the knees were prepped for aseptic surgery. The knee joint was exposed by a 1 cm longitudinal incision medial to the quadriceps tendon from the proximal patella to the proximal tibial plateau. The quadriceps tendon was subluxed laterally and joint capsule opened with a #10 scalpel. Recalcitrant bleeding was controlled with electrocautery. The medial meniscus was fully transected and released 5 mm medial to the insertion of the medial meniscobtibial ligament. The joint capsule, overlying muscle, and quadriceps tendon were closed with continuous 4–0 tapered Vicryl suture. Buprenorphrine (Reckitt and Coleman, Kingston-Upon-Hull, UK) was administered perioperatively at 0.05 mg/kg and the rats regained full mobility within 3 h.
Sham surgeries were performed as described above only the medial meniscus was spared. Rats were allocated randomly to the experimental groups depicted in Figure 1A. Control animals that underwent sham surgeries were sacrificed at 8 weeks and are referred to as sham. For OA model validation, rats were sacrificed at 4 and 8 weeks postoperatively and referred to as MMx 4-week or MMx 8-week, respectively.
Intra-articular Delivery of Glucosamine
Injection therapy was initiated at the beginning of week 5 following the MMx. Each rat then received a single weekly injection for 3 weeks as illustrated in Figure 1B. For example, a rat allocated to the GlcN 3.5 mg group would undergo MMx at the beginning of week 1 and injections at the beginning of weeks 5, 6, and 7. The animal would then be sacked at the beginning of week 8.
Practice injections on cadaveric rats with Indian ink helped to establish this technique, ensure there were no leaks into the surrounding tissues, and also confirmed the joint capsule had healed from the initial surgery. Rats were anesthetized as above, and the knee joint was exposed by a 5 mm longitudinal skin incision medial to the quadriceps tendon. This provided adequate visualization of the knee and surrounding musculature to ensure accurate placement. A 100 μl dose of glucosamine hydrochloride (Sigma) dissolved in normal saline at the indicated concentrations was administered intra articularly with a 1″ 27G needle using a shallow Z track injection path medial to the trochlea. Following injection, the knee was fully flexed and extended three times while monitoring for leakage of injected fluid into surrounding tissues.
This large injection volume provides a slight resistance as the fluid is pushed into the joint capsule. With practice, this resistance provides confirmation of the correct placement as the injection is difficult to administer directly into muscle or surrounding fascia. For a given knee, if the first injection attempt failed it was not feasible to try a second attempt because the fluid would leak out through the hole created in the first attempt. All solutions were filter sterilized before use. The skin was closed as above.
Gross Visualization and Histopathological Evaluation
The distal femur and proximal tibia were harvested en bloc with retention of 3–4 cm of the femur and tibia. The articular surfaces of were cleaned of menisci and loose tissue. Images of the articular surfaces were obtained using a Zeiss dissection scope equipped with an Axiovision MRC5 digital camera. Following imaging, samples were fixed in 10% formalin, and decalcified with 10% formic acid. The progress of the decalcification was followed with calcium oxalate precipitation. Samples were dehydrated with ethanol and embedded in paraffin. Coronal sections 7 μm thick were collected at 100 μm throughout the entire articular surface in order to capture arthritic changes throughout the entire joint. These sections were stained with safranin O/fast green per standard laboratory procedures. The degree of arthritic changes were quantified with the modified Mankin Score at least every 200 μm throughout the weight bearing articular surface and averaged.18,24 Higher scores are indicative of more sever arthritic changes while low scores are consistent with minor disease.
Cartilage thickness was quantified using the ImageJ analysis program (National Institutes of Health, Bethesda, MD). Images of the articular cartilage and subchondral bone were examined using a Nikon Eclipse TE200 microscope with a Nikon DXM1200 camera and 10× objective. Histomorphometric measurements of the uncalcified and calcified cartilage thickness were acquired from midline coronal sections of the medial tibial plateau. For this analysis the cartilage thickness of five different tissue sections from each individual sample (n = 6) were quantified at 10 uniformly spaced intervals.
Statistical Analysis
For in vitro studies all analyses were either done with n = 3 or n = 4, and analyzed with Student’s t-test for pair-wise comparison. For in vivo studies, all treatment groups were performed with n = 6. Mankin scores were analyzed using the nonparametric Wilcoxon rank sum. The data are expressed as the mean ± SEM and statistical significance was noted for all p-values <0.05.
RESULTS
Response of Primary Human Chondrocytes to a Bolus Dose of GlcN
There were no significant changes in either GAG or total collagen matrix production when OA chondrocytes were exposed to GlcN (Fig. 2A). There were also no differences in DNA content of the constructs (data not shown). The presence of GlcN resulted in a significant up-regulation in type IX collagen expression and down-regulation in MMP-13 and MT1-MMP (Fig. 2B).
Figure 2.
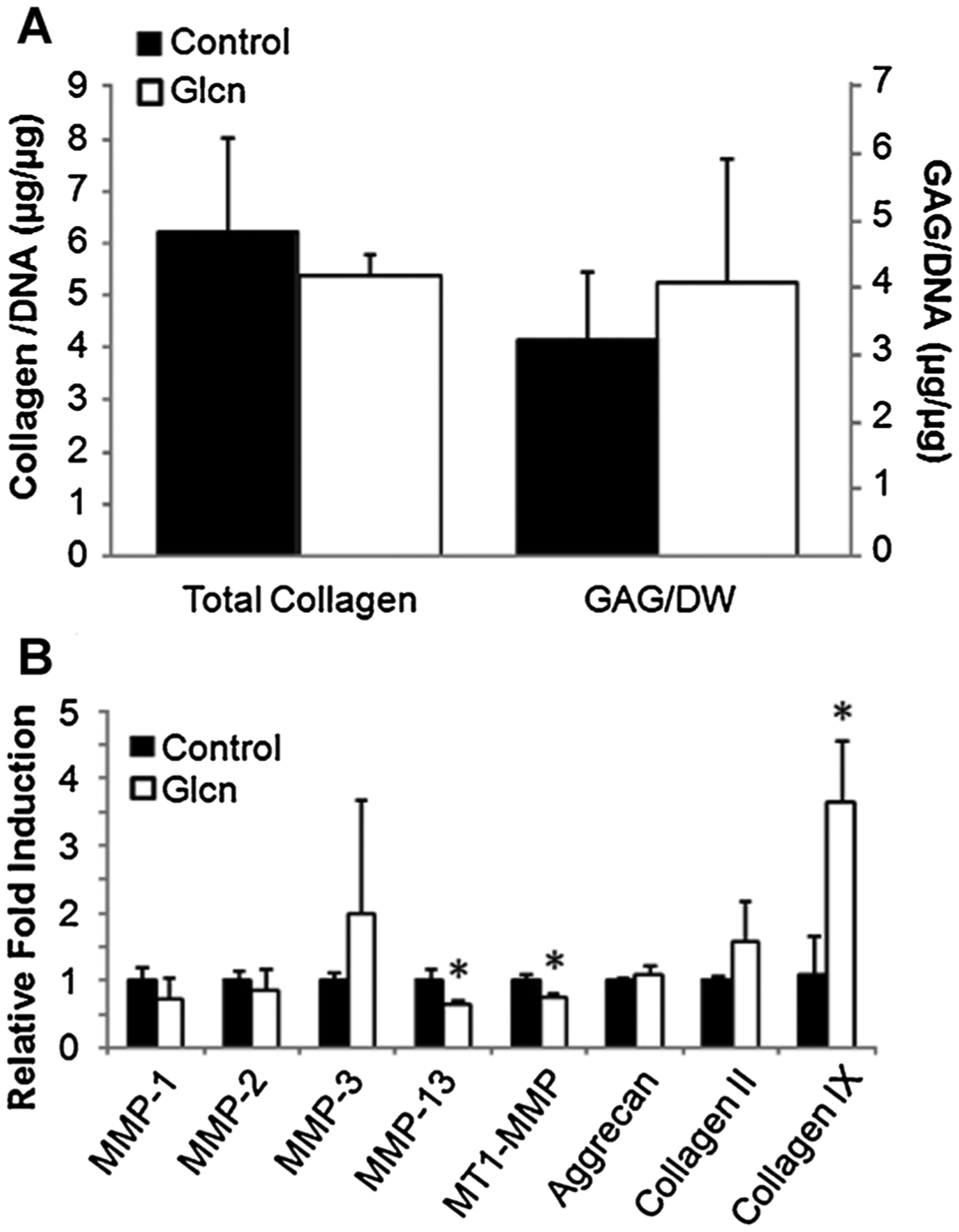
Response of primary human OA chondrocytes to a GlcN bolus within a PEGDA hydrogel. There were no significant changes in GAG or total collagen levels (A). GlcN treatment resulted in the up-regulation in type IX collagen and down-regulation in MMP-13 and MT1-MMP (B). *p < 0.05 compared to control.
Cartilage Pathology Following Meniscal Transection
Meniscal transection was performed as described above and the animals were sacrificed at 4 and 8 weeks post-operatively. There were no immediate post-surgical complications. No macroscopic changes representative of OA were observed in the appearance of the sham operated joints at either 4 or 8 weeks (Fig. 3A and B). In contrast, 4 weeks post-operatively, the medial compartment developed focal cartilage erosions, osteophytes, and subchondral bone sclerosis on both the tibia and femur (Fig. 3C and D). After 8 weeks of destabilization, severe bi-compartmental involvement with bone eburnation was observed (Fig. 3E and F).
Figure 3.
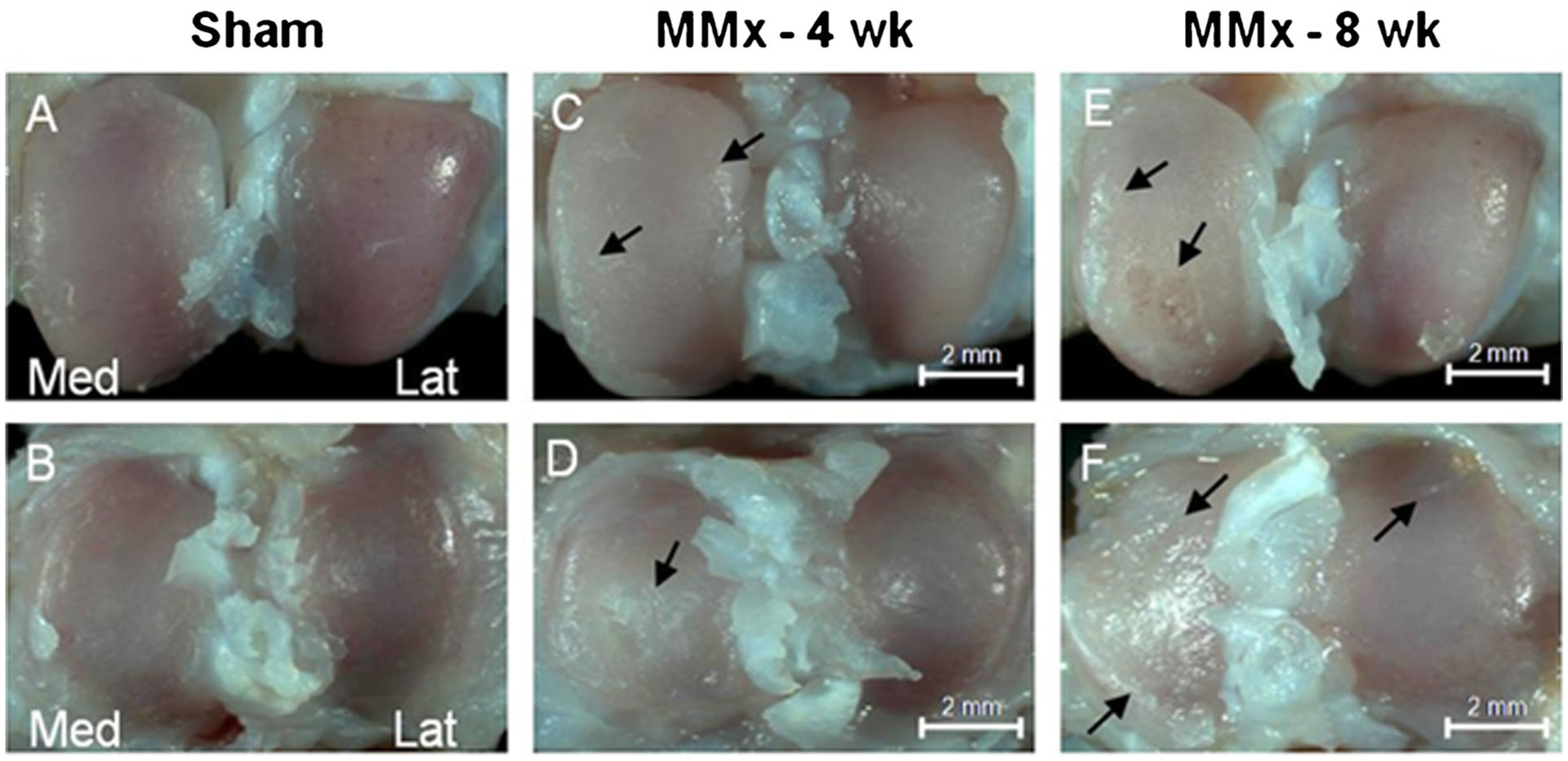
Representative gross morphological findings of the knee articular surface 4 and 8 weeks post-operatively. Sham operated joints showed no significant changes on either the femoral condyles or tibial plateau (A and B, respectively). Four weeks after destabilization pathology was limited to the medial compartment and consisted of and osteophyte formation (C, arrows) opaque changes, and fibrillations, (D, arrow). Both the medial and lateral compartments developed osteoarthritic lesions following 8 weeks of destabilization (E and F, arrows).
The sham-operated joints had a normal gross and histological appearance with a thin, uninterrupted superficial layer and no evidence of proteoglycan loss or cellularity changes. For both time points, the medial compartment consistently developed more severe and reproducible pathology. By 4 weeks, focal cartilage defects with fibrillation, blistering, and proteoglycan loss was evident (Fig. 4B). Low magnification micrographs allow the comparison of overall joint changes to the gross pathology while higher magnification demonstrates safraninophilia, cloning, and hypocellularity (Fig. 4E). Severe arthritic changes were readily apparent following 8 weeks of destabilization (Fig. 4C and F).
Figure 4.
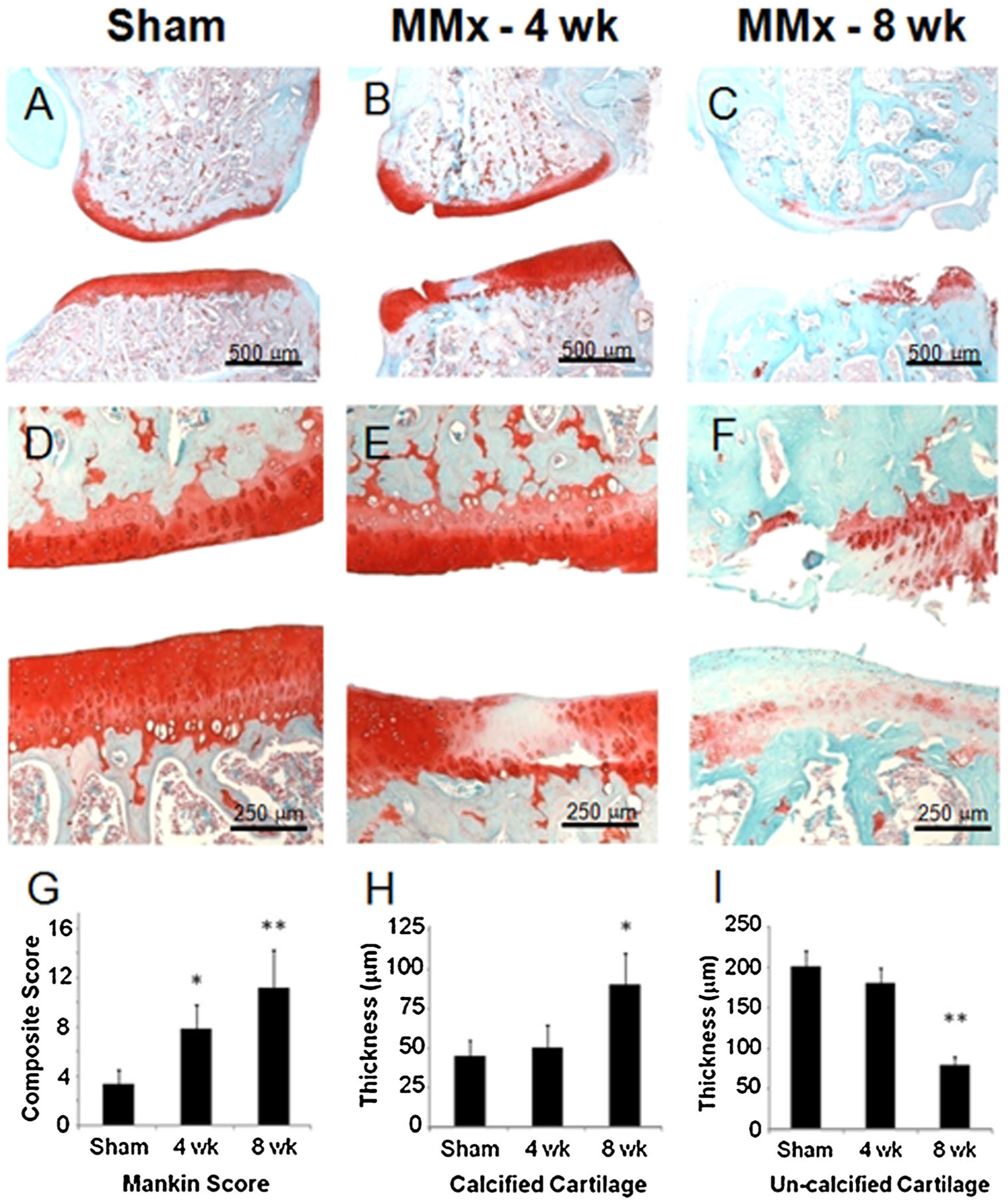
Histological analysis of arthritic changes and quantitative assessment of cartilage thickness in the rat meniscal transection model of osteoarthritis. Representative micrographs of the medial knee compartment stained with Safranin O. Sham operated knees showed no arthritic changes evident at low or high magnification (A and D, respectively). Osteophytes and chondrophytes are commonly observed in the periarticular region and increase in size and severity from 4 to 8 weeks post-operatively (B and C, respectively). Cellular changes consistent with OA are depicted at higher magnification (E and F). The semiquantative modified Mankin scores indicates that OA severity increases with time (G). Consistent with OA progression, increases in calcified cartilage thickness (H) and decreases in un-calcified cartilage thickness were also observed. Statistical significance is indicated as *p < 0.05 compared to sham and **p < 0.05 compared to 4-week time point.
Arthritic changes occurred sequentially in a time-dependent fashion as determined by the modified Mankin scores (Fig. 4G). In this model, the modified Mankin score was significantly higher than sham at both 4 and 8 weeks post-operatively. We used histomorphometric analysis to examine cartilage thickness. Calcified cartilage thickness increased in a time dependent fashion and this parameter was statistically greater than sham knees at 8 weeks (Fig. 4H). Conversely, the un-calcified cartilage decreased in thickness (Fig. 4I). Taken together, the data demonstrated this model is of tunable severity as joint degeneration increases with time.
Inhibition of OA With the Local Delivery of GlcN
There were no complications or post-surgical irregularities in animals from any groups and there were no obvious systemic effects of GlcN treatment. Treatment groups were given thrice weekly 100 μl injections of 35 μg, 350 μg, 1.8 mg, or 3.5 mg of GlcN dissolved in normal saline 4 weeks after the initial meniscal transection. Total time between initial surgery and sacrifice was 8 weeks and this included 4 weeks for arthritic changes to develop followed by 4 weeks of treatment. Controls consisted of sham operations and destabilized knees injected with saline alone. There was variability in both the response to meniscal transection and treatment efficacy, most likely due to differences in the activity level and biomechanics of the individual rats, which required a minimum of six rats per group to obtain statistical significance.
The local delivery of 350 μg of GlcN had the most significant positive impact on the overall modified Mankin score (Fig. 5A). The modified Mankin score includes parameters for surface structure, proteoglycan content, and cellularity changes. The positive effects of locally delivering GlcN can be attributed to changes in each of these parameters (Fig. 5B and D) indicating GlcN improves the overall tissue quality at both the cellular and organ level. GlcN also reduces the calcified cartilage thickness and increases the uncalcified cartilage thickness when compared to vehicle injections (Fig. 5E and F).
Figure 5.
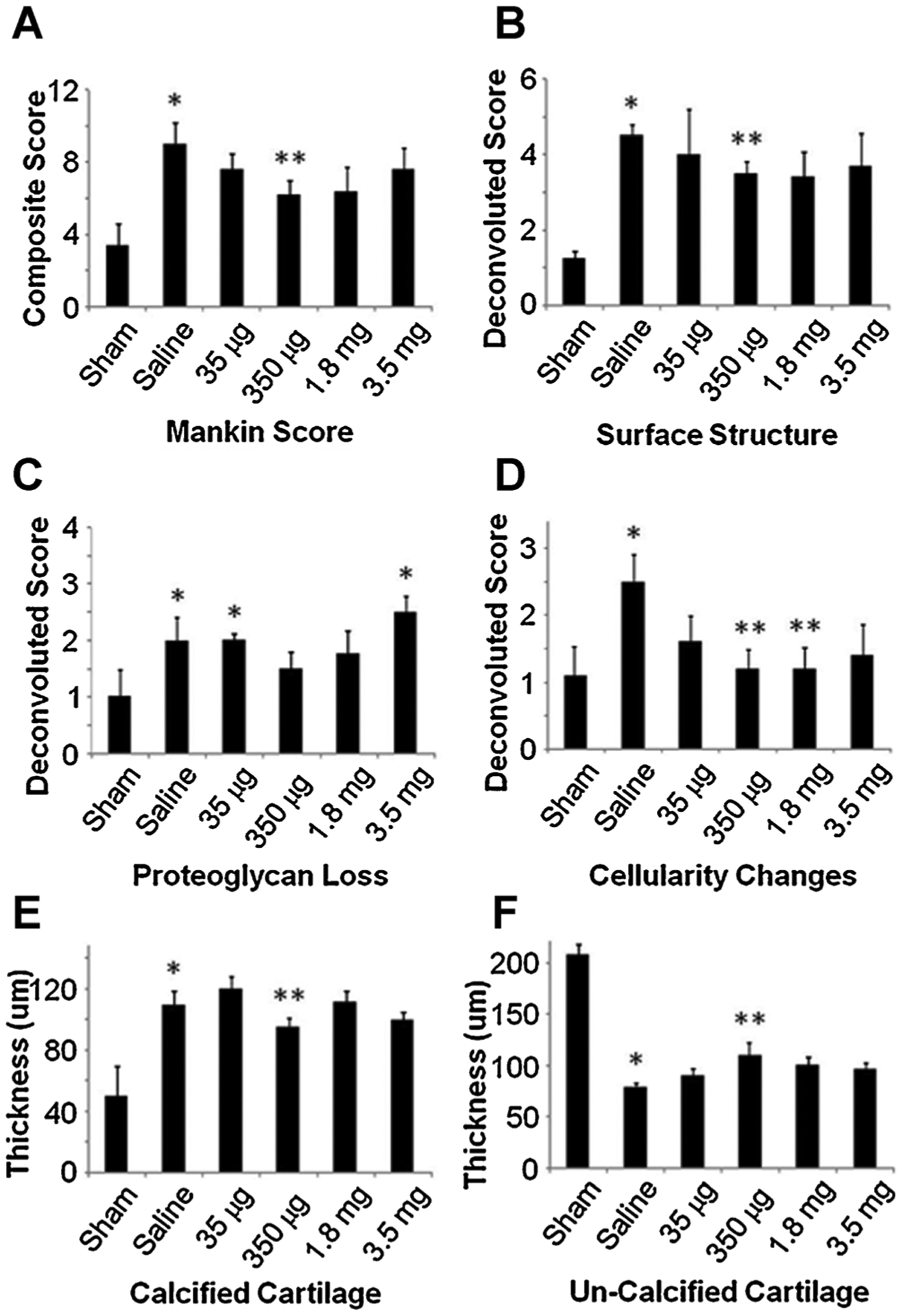
Semiquantative histopathological analysis of the medial tibia following the intra-articular administration of GlcN. The arthritic changes due to MMx are not altered through saline injections; however, locally delivered GlcN at 350 μg lowers the overall Mankin score (A). The deconvoluted Mankin scores indicate that GlcN has favorable impacts on surface structure and cellularity changes (B and D, respectively). The reduction of proteoglycan loss failed to reach statistical significance (C) As quantitatively depicted in (E and F) GlcN also reduces the calcified cartilage thickness and increases the uncalcified cartilage thickness. Statistical significance is indicated as *p < 0.05 compared to sham and **p < 0.05 compared to saline.
DISCUSSION
Glucosamine is a putative chondroprotective agent with reported efficacy in the treatment of osteoarthritis. The clinical efficacy of GlcN remains controversial, and it is difficult to reconcile discrepancies between clinical studies because of small sample sizes, varied patient populations, short follow-ups and differing GlcN preparations.25–32
The intra-articular delivery of GlcN is a feasible alternative to overcome the pharmacokinetic obstacles associated with the oral dosing of GlcN. Our data demonstrate that this approach is both feasible and well tolerated. Also, the data presented in this study suggest that primary human OA chondrocytes can respond to a bolus dose of GlcN, and that this response is lasting, and can be detected 21 days after the initial exposure. To our knowledge, this is the first report to characterize the cellular response to this pharmacokinetic profile. In contrast, there are several published studies where cells are continuously exposed to elevated concentrations of GlcN for extended periods of time. A single exposure to 1 mg/ml GlcN significantly reduced the gene expression of MMP-13 and MT1-MMP, two commonly studied catabolic mediators known to play a role in human OA.24 Similarly, GlcN exposure significantly upregulated the expression of type IX collagen which is highly expressed in embryonic cartilage, crosslinks type II collagen fibers, and its absence is associated with a fibroblastic morphology.24–32
In vivo models are necessary to evaluate potential OA disease modifying interventions. Intra-articular administration of GlcN in a Vitamin A induced rabbit OA model reduced some aspects OA progression.33 After treatment with GlcN, the articular cartilage exhibited more homogenous cellularity and increased staining of the cartilage matrix with Alcian Blue although histological scoring was not possible. However, Vitamin A induces only localized and variable OA, in contrast to a mechanical disruption model. In our study, we utilized a rat meniscal transection model of OA, a mimic of posttraumatic OA. Similar surgical destabilization models in the rat have been used to characterize the disease modifying activity for broad-spectrum MMP inhibitors, selective ADAMTS inhibitors, FGF-18.28–29 The potential disease modifying activity of locally administered GlcN has not been evaluated in this system. Severe cartilage degeneration occurs in rats after meniscal transection and the cartilage lesions are morphologically and biochemically similar to those that occur in humans. The rapid disease progression with this model sets a high threshold for testing therapeutic efficacy.28 Also, this rapid progression contributes to the inherent variability discussed above.
The roles of subchondral bone and more recently calcified cartilage in cartilage degradation are currently under investigation. Cartilage loss during OA progression can be initiated both at the articular surface through erosion and the osteochondral interface through chondrocyte calcification and hypertrophy. In this study, we analyzed the calcified and uncalcified cartilage thickness, which increased and decreased as the arthritic changes developed. These changes also occur in other models of OA and the data contained herein is the second study to document these changes in the rat.25 Interestingly, these trends are reversed in a concentration dependent manner by GlcN treatment. There are published in vitro studies demonstrating GlcN influences both chondrocytes, osteoblasts, and osteoclasts. The contributions of these cell types to OA progression remain to be determined.
A definitive mechanism to explain the complex actions of GlcN remains elusive and the multiple modes of biological activity emanating from GlcN compounds this challenge. To explain briefly, the metabolic fate of GlcN is salvage by the hexosamine biosynthetic pathway (HBP), thereby increasing cellular levels of UDP-GlcNAc. UDP-GlcNAc is a versatile metabolic intermediate that potentially can provide chondreoprotective benefits in several ways. First, it can be used as substrate for GAG production; recent evidence, however, where the incorporation of radio-labeled substrates into ECM components was monitored, has downplayed the importance of this mechanism.34–35 More promisingly, UDP-GlcNAc is the rate-determining substrate for O-GlcNAc protein modification, which is a post-translational modification of hundreds of nucleocytosolic proteins. O-GlcNAcylation plays a pro-survival role at low to moderate levels until a threshold is crossed above which it contributes to apoptotic cascades. Finally, elevated UDP-GlcNAc levels contribute to the formation of highly-branched N-glycans, a class of surface displayed epitopes that function as ligands for galectins, proteins that produce a cell surface lattice that sequesters signaling molecules and alters their activity.36 Recent studies have convincingly implicated this mechanism in chondrogenesis and OA through fibronectin matrix remodeling and activation of signaling through the epidermal growth factor receptor and focal adhesions.37–38 Deciphering the relative contributions of these facets of glucosamine activity remains a significant challenge for ultimately understanding and fully exploiting this sugar in tissue engineering and regenerative medicine.
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health Grants R01 EB 05517 (to J.H.E.), F31 AG 033999 (to J.M.C.), and F30 AG 034807 (to M.G.).
Grant sponsor:
National Institutes of Health; Grant numbers: R01 EB 05517, F31 AG 033999, F30 AG 034807.
Footnotes
Conflicts of interest: None.
REFERENCES
- 1.Elders MJ. 2000. The increasing impact of arthritis on public health. J Rheumatol Suppl 60:6–8. [PubMed] [Google Scholar]
- 2.Martin JA, Buckwalter JA. 2002. Human chondrocyte senescence and osteoarthritis. Biorheology 39:145–152. [PubMed] [Google Scholar]
- 3.Hunziker EB. 2002. Articular cartilage repair: basic science and clinical progress. A review of the current status and prospects. Osteoarthritis Cartilage 10:432–463. [DOI] [PubMed] [Google Scholar]
- 4.Manicourt DH, Altman RD, Williams JM, et al. 1999. Treatment with calcitonin suppresses the responses of bone, cartilage, and synovium in the early stages of canine experimental osteoarthritis and significantly reduces the severity of the cartilage lesions. Arthritis Rheum 42:1159–1167. [DOI] [PubMed] [Google Scholar]
- 5.Noth U, Steinert AF, Tuan RS. 2008. Technology insight: adult mesenchymal stem cells for osteoarthritis therapy. Nat Clin Pract Rheumatol 16:1501–1508. [DOI] [PubMed] [Google Scholar]
- 6.Mollenhauer JA. 2008. Perspectives on articular cartilage biology and osteoarthritis. Injury 39:S5–S12. [DOI] [PubMed] [Google Scholar]
- 7.Aigner T, Haag J, Martin J, et al. 2007. Osteoarthritis: aging of matrix and cells—going for a remedy. Curr Drug Targets 8:325–331. [DOI] [PubMed] [Google Scholar]
- 8.Martin JA, Brown TD, Heiner AD, et al. 2004. Chondrocyte senescence, joint loading and osteoarthritis. Clin Orthop Relat Res S96–S103. [DOI] [PubMed] [Google Scholar]
- 9.Dieppe P 1995. Therapeutic targets in osteoarthritis. J Rheumatol Suppl 43:136–139. [PubMed] [Google Scholar]
- 10.Ilic MZ, Martinac B, Samiric T, et al. 2008. Effects of glucosamine on proteoglycan loss by tendon, ligament and joint capsule explant cultures. Osteoarthritis Cartilage 4:371–380. [DOI] [PubMed] [Google Scholar]
- 11.Derfoul A, Miyoshi AD, Freeman DE, et al. 2007. Glucosamine promotes chondrogenic phenotype in both chondrocytes and mesenchymal stem cells and inhibits MMP-13 expression and matrix degradation. Osteoarthritis Cartilage 15:646–655. [DOI] [PubMed] [Google Scholar]
- 12.Varghese S, Theprungsirikul P, Sahani S, et al. 2007. Glucosamine modulates chondrocyte proliferation, matrix synthesis, and gene expression. Osteoarthritis Cartilage 15:59–68. [DOI] [PubMed] [Google Scholar]
- 13.Wang SX, Laverty S, Dumitriu M, et al. 2007. The effects of glucosamine hydrochloride on subchondral bone changes in an animal model of osteoarthritis. Arthritis Rheum 56:1537–1548. [DOI] [PubMed] [Google Scholar]
- 14.Terry DE, Rees-Milton K, Smith P, et al. 2005. N-acylation of glucosamine modulates chondrocyte growth, proteoglycan synthesis, and gene expression. J Rheumatol 32:1775–1786. [PubMed] [Google Scholar]
- 15.Matheson AJ, Perry CM. 2003. Glucosamine: a review of its use in the management of osteoarthritis. Drugs Aging 20:1041–1060. [DOI] [PubMed] [Google Scholar]
- 16.Castro RR, Feitosa JP, da Cunha PL, et al. 2007. Analgesic activity of a polysaccharide in experimental osteoarthritis in rats. Clin Rheumatol 26:1312–1319. [DOI] [PubMed] [Google Scholar]
- 17.Castro RR, Cunha FQ, Silva FS Jr, et al. 2006. A quantitative approach to measure joint pain in experimental osteoarthritis—evidence of a role for nitric oxide. Osteoarthritis Cartilage 14:769–776. [DOI] [PubMed] [Google Scholar]
- 18.Henson FMD, Vincent TA. 2008. Alterations in the vimentin cytoskeleton in response to single impact load in an in vitro model of cartilage damage in the rat. BMC Musculoskelet Disord 9:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bendele AM. 2001. Animal models of osteoarthritis. J Musculoskelet. Neuronal Interact 1:363–376. [PubMed] [Google Scholar]
- 20.Bendele AM, White SL, Hulman JF. 1989. Osteoarthrosis in guinea pigs: histopathologic and scanning electron microscopic features. Lab Anim Sci 39:115–121. [PubMed] [Google Scholar]
- 21.Glasson SS, Blanchet TJ, Morris EA. 2007. The surgical destabilization of the medial meniscus (DMM) model of osteoarthritis in the 129/SvEv mouse. Osteoarthritis Cartilage 15:1061–1069. [DOI] [PubMed] [Google Scholar]
- 22.Sharma B, Williams CG, Khan M, et al. 2007. In vivo chondrogenesis of mesenchymal stem cells in a photopolymerized hydrogel. Plast Reconstr Surg 119:112–120. [DOI] [PubMed] [Google Scholar]
- 23.Yonenaga K, Nishizawa S, Fujihara Y, et al. 2010. The optimal conditions of chondrocyte isolation and its seeding in the preparation for cartilage tissue engineering. Tissue Eng Part C Methods 16:1461–1469. [DOI] [PubMed] [Google Scholar]
- 24.Kleemann RU, Krocker D, Cedraro A, et al. 2005. Altered cartilage mechanics and histology in knee osteoarthritis: relation to clinical assessment (ICRS Grade). Osteoarthritis Cartilage 13:958–963. [DOI] [PubMed] [Google Scholar]
- 25.Rozendaal RM, Koes BW, van Osch GJ, et al. 2008. Effect of glucosamine sulfate on hip osteoarthritis: a randomized trial. Ann Intern Med 148:268–277. [DOI] [PubMed] [Google Scholar]
- 26.Tat SK, Pelletier JP, Verges J, et al. 2007. Chondroitin and glucosamine sulfate in combination decrease the proresorptive properties of human osteoarthritis subchondral bone osteoblasts: a basic science study. Arthritis Res Ther 9:R117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wollheim FA. 2007. Prescription of glucosamine for osteoarthritis: does it work and is it safe? Nat Clin Pract Rheumatol 3:364–365. [DOI] [PubMed] [Google Scholar]
- 28.Clegg DO, Reda DJ, Harris CL, et al. 2006. Glucosamine, chondroitin sulfate, and the two in combination for painful knee osteoarthritis. N Engl J Med 354:795–808. [DOI] [PubMed] [Google Scholar]
- 29.Mroz PJ, Silbert JE. 2004. Use of 3H-glucosamine and 35S-sulfate with cultured human chondrocytes to determine the effect of glucosamine concentration on formation of chondroitin sulfate. Arthritis Rheum 50:3574–3579. [DOI] [PubMed] [Google Scholar]
- 30.Dodge GR, Jimenez SA. 2003. Glucosamine sulfate modulates the levels of aggrecan and matrix metalloproteinase-3 synthesized by cultured human osteoarthritis articular chondrocytes. Osteoarthritis Cartilage 11:424–432. [DOI] [PubMed] [Google Scholar]
- 31.Naito K, Watari T, Furuhata A, et al. 2010. Evaluation of the effect of glucosamine on an experimental rat osteoarthritis model. Life Sci 86:538–543. [DOI] [PubMed] [Google Scholar]
- 32.Wen ZH, Tang CC, Chang YC, et al. 2010. Glucosamine sulfate reduces experimental osteoarthritis and nociception in rats: association with changes of mitogen-activated protein kinase in chondrocytes. Osteoarthritis Cartilage 18: 1192–1202. [DOI] [PubMed] [Google Scholar]
- 33.Scotto d’Abusco A, Corsi A, Grillo MG, et al. 2008. Effects of intra-articular administration of glucosamine and a peptidyl-glucosamine derivative in a rabbit model of experimental osteoarthritis: a pilot study. Rheumatol Int 28: 437–443. [DOI] [PubMed] [Google Scholar]
- 34.Shikhman AR, Brinson DC, Valbracht J, et al. 2009. Differential metabolic effects of glucosamine and N-acetylglucosamine in human articular chondrocytes. Osteoarthritis Cartilage 17:1022–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lippiello L 2003. Glucosamine and chondroitin sulfate: biological response modifiers of chondrocytes under simulated conditions of joint stress. Osteoarthritis Cartilage 11: 335–342. [DOI] [PubMed] [Google Scholar]
- 36.Kleine TO, Baumann HJ. 1990. In-vivo biosynthesis of acid glycosaminoglycans with radiosulfate and [3H]glucosamine in single joints from rats of different age. Z Gerontol 23: 123–125. [PubMed] [Google Scholar]
- 37.de Mattei M, Pellati A, Pasello M, et al. 2002. High doses of glucosamine-HCl have detrimental effects on bovine articular cartilage explants cultured in vitro. Osteoarthritis Cartilage 10:816–825. [DOI] [PubMed] [Google Scholar]
- 38.Persiani S, Roda E, Rovati LC, et al. 2005. Glucosamine oral bioavailability and plasma pharmacokinetics after increasing doses of crystalline glucosamine sulfate in man. Osteoarthritis Cartilage 13:1041–1049. [DOI] [PubMed] [Google Scholar]