

Abstract
Bovine paratuberculosis (PTB) is a chronic inflammatory disease caused by Mycobacterium avium susbp. paratuberculosis (MAP). Genome-wide association studies (GWAS) have identified single nucleotide polymorphisms (SNPs) significantly associated with susceptibility to bovine PTB. The main objective of this study was to identify quantitative trait loci (QTLs) associated with MAP infection in Spanish Holstein cows (N = 983) using combinations of diagnostic tests and imputed whole-genome sequence (WGS) data. The infection status of these animals was defined by three diagnostic methods including ELISA for MAP-antibodies detection, and tissue culture and PCR for MAP detection. The 983 cows included in this study were genotyped with the Bovine MD SNP50 Bead Chip, and the corresponding genotypes were imputed to WGS using the 1,000 Bull genomes reference population. In total, 33.77 million SNP variants per animal were identified across the genome. Linear mixed models were used to calculate the heritability (h2) estimates for each diagnostic test and test combinations. Next, we performed a case-control GWAS using the imputed WGS datasets and the phenotypes and combinations of phenotypes with h2 estimates > 0.080. After performing the GWAS, the test combinations that showed SNPs with a significant association (PFDR ≤ 0.05), were the ELISA-tissue PCR-tissue culture, ELISA-tissue culture, and ELISA-tissue PCR. A total of twelve quantitative trait loci (QTLs) highly associated with MAP infection status were identified on the Bos taurus autosomes (BTA) 4, BTA5, BTA11, BTA12, BTA14, BTA23, BTA24, and BTA28, and some of these QTLs were linked to immune-modulating genes. The identified QTLs on BTA23 spanning from 18.81 to 22.95 Mb of the Bos taurus genome overlapped with several QTLs previously found to be associated with PTB susceptibility, bovine tuberculosis susceptibility, and clinical mastitis. The results from this study provide more clues regarding the molecular mechanisms underlying susceptibility to PTB infection in cattle and might be used to develop national genetic evaluations for PTB in Spain.
Introduction
Paratuberculosis (PTB) or Johne´s disease is a chronic enteritis of domestic and wild ruminants caused by Mycobacterium avium susbp. paratuberculosis (MAP). PTB is a major problem for animal health and must be notified to the World Organization for Animal Health. In Europe and North America, PTB is considered endemic in dairy cattle, with herd prevalence estimates higher than 50% [1]. This can result in great economic losses to the dairy industry due to decreased milk production, weight loss, replacement cost, reduced slaughter value, a greater risk to other health problems, premature culling or death from the clinical disease, and the costs of veterinary expenses and control measures [2,3]. The most common clinical signs are progressive weight loss, diarrhea, and decreased milk yield [4]. However, most infected animals do not develop clinical disease, and microbiological and immunological diagnostic tests are not sensitive enough to identify them [5]. MAP is also a suspected cause of Crohn´s disease (CD) in humans, making it a potential zoonotic agent [6]. Colorectal cancer is a complication of the two forms of idiopathic inflammatory bowel disease (IBD); colonic CD and ulcerative colitis. Interestingly, MAP bacilli have been detected in the intestines of patients with CD, ulcerative colitis, and IBD-associated colorectal cancer [7,8].
Once a cow shows clinical signs of PTB, there is no effective treatment and the animal is culled. Commercial inactivated vaccines against bovine PTB are very successful in reducing MAP presence in feces and tissues, and in increasing both milk production and cattle productive life in infected farms [9,10]. However, PTB vaccination with heat-killed inactivated vaccines is not allowed in most European countries due to its interference with Mycobacterium bovis (Mb) detection tests [11]. Therefore, PTB control is currently based on testing and culling infected animals plus preventing MAP transmission to susceptible animals using voluntary hygiene and management practices [12,13]. PTB infection is commonly detected using Enzyme-Linked Immunosorbent Assay (ELISA) that detects serum and milk antibodies against MAP. Although serum ELISA is a simple, fast, and cost-effective method for diagnosis clinical PTB, it is known to have low sensitivity for MAP-infected animals that do not show clinical signs and could yield erroneous false-negative results [14]. Therefore, repeating testing or complementary methods (culture or PCR) are required to achieve reasonable sensitivity for PTB diagnosis.
Genomic selection could enhance natural resistance to MAP infection and complement existing control strategies [15]. Genetic improvement of disease resistance is a long-term process; however, the results are permanent and cumulative over generations and could result in disease eradication. Previous studies showed heritability (h2) estimates of susceptibility to MAP in Friesian cattle ranging from 0.03 to 0.27 [16–18]. Genome-wide association studies (GWAS) using genotyping of SNPs at low density (LD) [19], medium density (MD) [20–26], and high density (HD) [27–29], as well as imputed whole-genome sequences (WGS) [30–32], have identified quantitative trait loci (QTLs) in the Bos taurus genome associated with MAP infection. However, few QTLs have been identified with consistent association across studies due to multiple factors including differences in genetic backgrounds and variable disease prevalence in the studied populations, different samples size, different statistical models, differences in sensitivity of the various diagnostic methods used for infection status classification, different definitions for the cases and controls phenotypes, and the low heritable nature of these phenotypes. Given that different populations across the world can have differences in genetic backgrounds, it is entirely reasonable for some genes to be monomorphic in some populations (these would therefore not be associated with PTB in these populations) even when they have a major physiological role in the defense against PTB. Previous GWAS in Holstein cattle defined phenotypes based on ante-mortem tests such as serum ELISA, milk ELISA, MAP fecal culture, or fecal PCR. The ELISA test is most commonly used for the detection of MAP-specific antibodies in infected animals. This test is cost-effective and simple to perform as compared with detection by bacteriological culture. However, the drawback of this method is its low sensitivity for detection during the subclinical phase of infection and could yield false-negative results. Tissue culturing is considered to be the gold standard due to its increased sensitivity and ability to detect viable MAP in subclinical stages of PTB that can be missed with other diagnostic methods [33]. However, tissue PCR and culture are post-mortem tests that can be taken at a single point and are laborious to conduct on a large number of animals.
The first objective of the current study was to calculate the genetic parameters, variance, and h2 estimates, for MAP-specific humoral response and/or tissue MAP infection in Spanish Holstein cattle (N = 983) using different diagnostic methods including ELISA for MAP antibodies detection, and culture and PCR for MAP detection in gut tissues. The second objective was to identify genomic regions and candidate genes associated with MAP infection using imputed WGS data and the individual test results or test combinations with h2 estimates > 0.08.
Materials and methods
Ethics statement
Animals used in this study were not submitted to any in vivo experimentation before stunning for slaughter and, therefore, no specific ethics committee authorization was required. The cows were slaughtered in the Bilbao and Donostia municipal slaughterhouses (Basque Country, Spain) under the pertinent Basque (Basque Government Decree 454/1994), Spanish (Spanish Government Law 32/2007 and Royal decree 731/2007), and European (Council Regulation (EC) No 1099/2009) legislation on animal welfare.
Animals and PTB diagnosis
The Spanish Holstein population included in this study consisted of 983 culled Holstein cattle from several herds located in eight regions: Basque Country (N = 415, 42.21%), Catalonia (N = 220, 22.38%), Navarre (N = 204, 20.75%), Cantabria (N = 70, 7.12%), Aragon (N = 38, 3.87%), Castile and Leon (N = 25, 2.54%), La Rioja (N = 7, 0.71%) and Asturias (N = 4, 0.41%). Only cows (2 years or older, 5.6 years mean age) were included in the analyses as PTB has a long incubation period and older animals are, therefore, more likely to show clinical signs and positive diagnostic results. The cows were slaughtered in two abattoirs located in the Basque country from March 2007 to May 2010. The infectious status of the animals was determined by ELISA for MAP antibodies detection, and tissue culture and PCR for MAP detection [34]. Sampling was systematically performed once a week at the slaughterhouse. In each visit, the first 2 to 6 animals in the line satisfying the breed and age requirements were sampled. Duplicate blood samples were collected from the jugular vein into 10 ml Vacutainer EDTA tubes (BD, Franklin Lakes, NJ, USA) for genotyping and ELISA testing. Since the mucosa from the ileocecal valve-distal ileum and the jejunal caudal lymph nodes are the primary sites of MAP colonization, samples from these tissues were mixed and processed for tissue culture and PCR. Briefly, MAP isolation was performed in duplicate home-made Herrold´s egg yolk (Becton Dickinson, Franklin Lakes, NJ, USA) and Lowenstein-Jensen media (Difco, Detroit, MI, USA), both supplemented with 2 mg/L of mycobactin J (Allied Monitor, Fayette, MO, USA) as previously described [35]. A positive tissue culture result was considered if one or more MAP colonies were observed in any of the four medium slants. Individuals with 0 CFU in all culture samples were considered controls. A second aliquot from the same tissue homogenates was used for DNA isolation and MAP IS900 amplification using a combined extraction and amplification kit (Adiagene, Saint Brieuc, France). PCR amplifications were performed on an ABI Prism 7000 Sequence Detection System (Applied Biosystems, Foster City, CA, US). Samples showing amplification curves with a threshold cycle (Ct) below 40.0 were considered positive. Serum samples were tested for specific antibody production against MAP by using a two-step ELISA paratuberculosis antibody screening and verification kit (IDEXX Laboratories, Inc., Westbrook, ME, USA) according to the manufacturer´s instructions. The results were expressed as optical density values (OD) and categorized as positive according to the sample-to-positive control ratio defined by the manufacturer. Table 1 provides a summary of the number of animals with positive or negative results for each diagnostic test and test combinations. When combinations of diagnostics tests were used, animals were considered cases when they were positive to all the tests, and controls when they were negative to any (+/), or all of them (+/-).
Table 1. Number of cows with a positive and negative test result.
| Phenotype | Cases (%) | Controls (%) |
|---|---|---|
| ELISA (+/-) | 70 (7.12) | 913 (92.88) |
| Tissue PCR (+/-) | 266 (27.62) | 697 (72.38) |
| Tissue Culture (+/-) | 150 (15.26) | 833 (84.74) |
| ELISA-Tissue PCR (+/) | 59 (6.00) | 924 (94.00) |
| ELISA-Tissue Culture (+/) | 56 (5.70) | 927 (94.30) |
| Tissue PCR-Tissue Culture (+/) | 119 (12.11) | 864 (87.89) |
| ELISA-Tissue PCR-Tissue Culture (+/) | 55 (5.60) | 928 (94.40) |
| ELISA-Tissue PCR-Tissue Culture (+/-) | 55 (7.71) | 658 (92.29) |
When combinations of diagnostics tests were used, animals were considered cases when they were positive to all the tests and controls when they were negative to any (+/) or all of them (+/-).
Genotyping and imputation
Peripheral blood samples were taken at slaughter time and DNA was extracted using the QIAmp DNA Blood Mini Kit according to the manufacturer´s instructions (Qiagen, Hilden, Germany). Purified genomic DNA was quantified spectrophotometrically and subsequently genotyped with the Illumina Bovine MD SNP50 Bead Chip at the molecular genetic laboratory service of the Spanish Federation of Holstein Cattle (CONAFE) using the InfiniumTM iScan software for allele assignation (Illumina, San Diego, CA). Individual genotypes were phased using Eagle 2.4 [36] and imputed with minimac4 [37] to the Bovine HD Bead Chip using a reference panel of 1,278 Bos taurus from Run7.0 of the 1,000 Bull Genomes project and 581,712 SNPs (ASR-UCD1.2). Imputation to the WGS level was then undertaken using a reference population of 2,333 Bos taurus from Run6.0 of the 1,000 Bull Genomes project [38]. In total, 33.77 million SNPs per animal were identified across the genome. All the SNPs passed a call rate > 0.80. PTB-associated SNPs with minimum allele frequency (MAF) < 0.01 were removed. The number of SNPs kept in the analysis was 13,881,067.
Variance components and h2 estimates
The variance components, standard errors (SE), and h2 estimates for MAP infection status explained by all the SNPs were calculated using the genome-wide complex trait analysis (GCTA) software 1.93.2, according to the following formula: where σG is the additive genetic effect of the individuals and σe is the residual variance [39]. The variance components σG and σe in the equation were estimated by the genomic-relatedness-based restricted maximum-likelihood (GREML) approach implemented in GCTA. This approach estimates the proportion of phenotypic variation that can be explained by all genome-wide SNPs using an SNP-derived genetic relationship matrix. The concept behind this method is to fit all the SNPs simultaneously as random effects in a mixed linear model to estimate the variance explained by all the SNPs. GCTA implements the method in two steps; first generating the GRM between individuals and then estimating the variance explained by all SNPs by a Restricted Maximum Likelihood (REML) analysis of the phenotypes with the GRM.
Genome-wide association study (GWAS)
Imputed genotypes and the results from the diagnostic tests with h2 > 0.08 were analyzed in a case-control study using the mlma (mixed linear model) association analysis of the GCTA 1.93.2 which fits the effects of all the SNPs as random effects. The model is y = a + bx + g + e, where y is the phenotype, a is the mean term, b is the additive effect (fixed effect) of the candidate SNP to be tested for association, x is the SNP genotype indicator variable coded as 0, 1 or 2, g is the polygenic effect (random effect) i.e. the accumulated effect of all SNPs (as captured by the GRM calculated using all SNPs), and e is the residual [39]. Age was included as a covariate in the analysis. After the GWAS, all the SNPs had R2 values higher than 50% and were retained, with a mean R2 close to 90%. SNPs with R2 between 50 and 70% were scarce. To account for multiple testing, a 5% chromosome-wise false discovery rate (PFDR ≤ 0.05) was used to determine the probability that associations were not false positives. Uncorrected P-values between P = 5 × 10−5 and P = 5 × 10−7 provided a moderate significance level (α), and uncorrected P-values < 5 × 10−7 were used to identify highly significant associations (Wellcome Trust Case Control Consortium, 2007). The inflation factor (λ) and quantile-quantile plots were calculated to compare observed distributions of–log (P-values) to the expected distribution under the no association model for each phenotype. λ value close to 1 suggests appropriate adjustment for potential substructure and λ > 1.2 suggests population stratification.
The odds ratio (OR) and their 95% confidence intervals (CI) for the SNPs associated with a positive ELISA-tissue PCR-tissue culture diagnosis (P < 5 × 10−7) were calculated using logistic regression analysis with the WGassociation function of SNPassoc 1.9.2 under five different genetic models (co-dominant, dominant, recessive, over-dominant, and log-additive) [40]. The WGassociation function fits individual logistic regression models to each of the class variables (genotypes). For each SNP and genetic model, the function WGstats of SNPassoc 1.9.2. provides genotype frequencies, OR, and 95% CI with the major homozygous genotype deemed as the baseline.
SNP variants, QTLs, and candidate genes identification
The location of the significant SNPs was determined with biomaRt 2.44.1 for R [41] using the ARS-UCD1.2 reference genome. The genomic distribution of the identified SNPs was determined using the Ensembl Variant Effect Predictor (VEP). QTLs associated with PTB infection were defined based on SNPs on linkage disequilibrium patterns with SNPs that surpassed the suggestive significance threshold (P < 5 × 10−7) in a given chromosome. The beginning and end of each quantitative trait locus (QTL) were defined in a window of 500,000 base pairs upstream and downstream by the SNPs that were furthest upstream and downstream of the suggestive SNP. Overlapping QTLs were merged and considered as a single QTL. The defined QTLs were further investigated for the presence of candidate genes within 50,000 base pairs to each side of the defined QTL using Ensembl (https://www.ensembl.org). The identified QTLs and candidate genes were compared with the reported cattle QTLs and genes for PTB susceptibility, bovine tuberculosis (bTB) susceptibility, and clinical mastitis (http://www.animalgenome.org), and with human candidate genes for CD, IBD, and colorectal cancer (https://www.ebi.ac.uk/gwas/). In addition, we uploaded the list of candidate genes and search the Innate DB database for innate immunity genes (https://innatedb.com).
Results
Data and descriptive analysis
The infection status of each animal was investigated by serological and microbiological methods. Table 1 presents the descriptive statistics of the analyzed population. Although not included in Table 1, the mean (± SD) OD values were 0.178 ± 0.16 for negative, and 1.882 ± 0.80 for positive animals. ELISA OD values ranged from 0.028 to 3.108. The frequency of ELISA, tissue culture, and tissue PCR positive animals was 7.12, 15.26, and 27.62%, respectively. MAP was detected by tissue culture and PCR in the majority of ELISA-positive animals; 80% and 84%, respectively.
Variance components and h2 estimates
Variance components along with standard errors (SE) and h2 estimates were calculated for all the diagnostic tests and their combinations (Table 2). The h2 estimates ranged from 0.054 to 0.139 depending on the phenotype. The highest h2 estimate was obtained for the ELISA-tissue PCR-tissue culture (h2 = 0.139) followed by the tissue culture (h2 = 0.101), ELISA-tissue culture (h2 = 0.099), tissue PCR-tissue culture (h2 = 0.086) and ELISA-tissue PCR (h2 = 0.081). When the ELISA was treated as a positive/negative binary trait for MAP status, higher h2 estimates were obtained (h2 = 0.075) when compared with the ELISA (OD) h2 estimates (h2 = 0.057).
Table 2. Variance components, standard errors (SE), and h2 estimates for the diagnostic tests and their combinations.
| Phenotype | Additive genetic variance (σG) | SE | Residual variance (σe) | SE | Heritability (h2) |
|---|---|---|---|---|---|
| ELISA (+/-) | 0.004975 | 0.003905 | 0.060948 | 0.004611 | 0.075464 |
| ELISA (OD) | 0.014126 | 0.013584 | 0.232861 | 0.016736 | 0.057192 |
| Tissue PCR (+/-) | 0.010867 | 0.012009 | 0.189343 | 0.014418 | 0.054277 |
| Tissue Culture (+/-) | 0.013107 | 0.008410 | 0.115996 | 0.009433 | 0.101522 |
| ELISA-Tissue PCR (+/) | 0.004584 | 0.003268 | 0.051401 | 0.003860 | 0.081873 |
| ELISA-Tissue Culture (+/) | 0.005307 | 0.003154 | 0.047911 | 0.003653 | 0.099722 |
| Tissue PCR-Tissue Culture (+/) | 0.009149 | 0.006479 | 0.096878 | 0.007505 | 0.086287 |
| ELISA-Tissue PCR-Tissue Culture (+/) | 0.005741 | 0.003193 | 0.046567 | 0.003634 | 0.109759 |
| ELISA-Tissue PCR-Tissue Culture (+/-) | 0.009838 | 0.005710 | 0.060478 | 0.006215 | 0.139909 |
OD- Optical density. When combinations of diagnostics tests were used, animals were considered cases when they were positive to all the tests and controls when they were negative to any (+/) or all of them (+/-).
GWAS
To explore the genetic basis of MAP infection status, a GWAS using the imputed WGS datasets and the phenotypes and combinations of phenotypes with h2 > 0.08 was performed. After the GWAS, the phenotypes that showed SNPs with a significant association (PFDR ≤ 0.05) were the ELISA-tissue PCR-tissue culture, ELISA-tissue culture, and ELISA-tissue PCR. The number of SNPs that surpassed the moderate (between P = 5 × 10−5 and P = 5 × 10−7) and the high (P< 5 × 10−7) thresholds are presented in Fig 1.
Fig 1. Number of SNPs associated with PTB infection status.
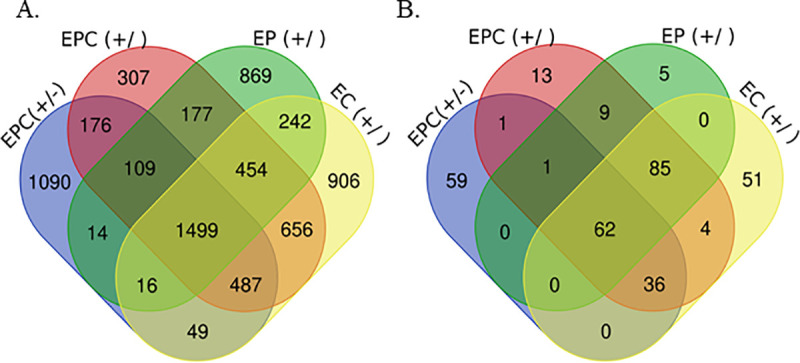
The total number of SNPs with moderate (A), or high (B) association with PTB infection status determined by ELISA (E), tissue PCR (P), and tissue culture (C) combinations. Animals were considered cases when they were positive to all the tests, and controls when they were negative to any (+/), or all of them (+/-). A moderate threshold for the association between a SNP and a phenotype was set at a significance level (α) between P = 5 × 10−5 and P = 5 × 10−7. The high threshold for evidence of an association was set at P < 5 × 10−7.
A total of 3,440 and 159 SNPs were found to be moderate and highly associated with the combination of the ELISA-tissue PCR-tissue culture results (+/-). In this test combination, animals were considered cases when they were positive to all the tests and controls when they were negative to all of them (+/-). Considering the results of the ELISA-tissue culture, the GWAS revealed 4,309 and 238 SNPs moderate, and highly associated with this combination, respectively. As shown in Fig 2 (Manhattan plots), various chromosomal regions were significantly associated with MAP infection according to the combinations of ELISA-tissue PCR-tissue culture (Fig 2A and 2B), ELISA-tissue culture (Fig 2C), and ELISA-tissue PCR (Fig 2D). In all the scenarios, the most significant peak was located on the BTA23, and most of the highly-associated SNPs were located in intronic regions (S1 Fig).
Fig 2. Manhattan plots showing–log10(P-values) of association between every single SNP and phenotype.
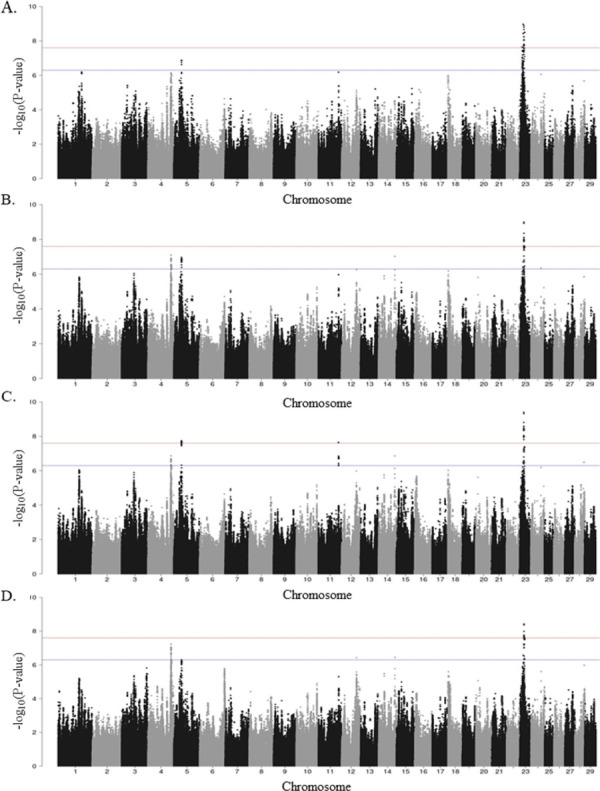
Each dot represents the result from the test association for a single SNP. Animals were considered cases when they were positive to all the tests and controls when they were negative to any (+/), or all of them (+/-). Chromosomes localization of the SNPs associated with ELISA-tissue culture-tissue PCR (+/-) (A), ELISA-tissue culture-tissue PCR (+/) (B), ELISA-tissue culture (+/) (C), and ELISA-tissue PCR (+/) (D) is indicated on the x-axis. The horizontal red line is drawn at–log10 (2.5 × 10−8), and the horizontal blue line at–log10 (5 × 10−7) to show the high level of significance.
The GWAS defined a total of 12 QTLs highly associated with MAP infection status on 8 chromosomes including BTA4, BTA5, BTA11, BTA12, BTA14, BTA23, BTA24, and BTA28. A description of the SNPs surpassing the threshold (P < 5 × 10−7), P-values, along with candidate genes located within the defined QTLs are shown in Table 3. Three QTLs on BTA5 and BTA23 were common to all the test combinations, with the most significant QTLs located on BTA23. Additional highly-associated QTLs were identified on BTA4, BTA11, BTA12, BTA14, BTA23, BTA24, and BTA28. As shown in Table 3, the 159 SNPs associated with the phenotype with the highest h2, the ELISA-tissue PCR-tissue culture results (+/-), resided within 5 QTLs which were distributed across BTA5 and BTA23. The BTA23 harbored 4 of the 5 QTL regions. The two QTLs that harbored the most genome-wide significantly-associated SNPs were located on BTA23 (15.09–16.86 and 18.81–22.95 Mb). Using the ELISA-Tissue PCR results, we identified the largest QTL (4.12 Mb) on BTA5. Two QTLs located on BTA11 and BTA28 were specifically associated with the combination ELISA-tissue culture, and one QTL on BTA12 was specifically associated with the ELISA-tissue PCR.
Table 3. Quantitative trait loci (QTLs) surpassing the significance threshold (P< 5 × 10−7) for evidence of an association with MAP infection status.
| Phenotype | BTA1 | QTL start (bp) | QTL end (bp) | P-value | SNP position2 | Positional candidate genes in QTL3 | SNPs in QTL |
|---|---|---|---|---|---|---|---|
| ELISA Tissue PCR Tissue Culture (+/-) | 23 | 15,060,376 | 16,864,597 | 1,06E-09 | 15,594,386 | NFYA, TREM1, TREM2, TREML1, TREML2, FOXP4, MDFI, TFEB, PGC, FRS6, USP49, MED20, TOMM6, BYSL,CCND3, TAF8, GUCA1A, MGC137036, C23H6orf132, GUCA1B, MRPS10, TERF1, U6, UBR2, PRPH2, 5S_rRNA, TBCC, BICRAL, RPL7L1, PEX6, MRPL2SRF, PTCRA, MEA1, CNPY3, KLHDC3, GNMT, RRP36, PPP2R5D, CUL7, KLC4, PTK7, CUL9, SLC22A7, DNPH1, TTBK1, CRIP3, ENSBTAG00000021359, ENSBTAG00000050887, ENSBTAG00000038916, ENSBTAG00000054479, ENSBTAG00000053124, ENSBTAG00000052790, ENSBTAG00000051935, ENSBTAG00000051409 | 13 |
| 23 | 18,812,221 | 20,933,699 | 1,26E-09 | 19,329,213 | RUNX2, U6, ENPP4, CLIC5, ENPP5, RCAN2, CYP39A1, SLC25A27, TDRD6, ADGRF5, PLA2G7, ANKRD66, ADGRF1, TNFRSF21, CD2AP, ADGRF2, ADGRF4, OPN5, ENSBTAG00000021415 | 63 | |
| 23 | 21,898,408 | 22,958,623 | 3,19E-09 | 22,401,117 | MMUT, CENPQ, GLYATL3, C23H6orf141, U6, RHAG, CRISP2, CRISP3, CRISP1, 7SK, DEFB114, DEFB110, ENSBTAG00000046237, ENSBTAG00000050850, ENSBTAG00000053808, ENSBTAG00000054396, ENSBTAG00000046711 | 6 | |
| 23 | 11,332,059 | 14,196,234 | 2,32E-08 | 11,900,592 | CMTR1, CCDC167, MDGA1, ZFAND3, BTBD9, GLO1, DNAH8, GLP1R, SAYSD1, KCNK5, KIF6, KCNK16, KCNK17, DAAM2, MOCS1, U6, bta-mir-2377, ENSBTAG00000050874 | 44 | |
| 5 | 35,035,017 | 36,111,179 | 1,35E-07 | 35,577,195 | ANO6, DBX2, NELL2, TMEM117, ENSBTAG00000026249, ENSBTAG00000032150 | 33 | |
| ELISA Tissue PCR Tissue Culture (+/) | 23 | 18,812,221 | 20,933,699 | 1,01E-09 | 19,331,156 | RUNX2, U6, ENPP4, CLIC5, ENPP5, RCAN2, CYP39A1, SLC25A27, TDRD6, ADGRF5, PLA2G7, ANKRD66, ADGRF1, TNFRSF21, CD2AP, ADGRF2, ADGRF4, OPN5, ENSBTAG00000021415 | 63 |
| 23 | 21,898,408 | 22,958,623 | 2,61E-08 | 22,401,993 | MMUT, CENPQ, GLYATL3, C23H6orf141, U6, RHAG, CRISP1, CRISP2, CRISP3, 7SK, DEFB114, DEFB110, ENSBTAG00000046237, ENSBTAG00000050850, ENSBTAG00000053808, ENSBTAG00000054396, ENSBTAG00000046711 | 6 | |
| 23 | 15,094,386 | 16,094,386 | 1,95E-07 | 15,594,386 | TREM1, TREM2, TREML1, TREML2, FOXP4, MDFI, TFEB, PGC, FRS6, USP49, MED20, TOMM6, BYSL, CCND3, TAF8, GUCA1A, MGC137036, C23H6orf132, GUCA1B, MRPS10, TRERF1, ENSBTAG00000050887, ENSBTAG00000038916, ENSBTAG00000054479, ENSBTAG00000053124 | 1 | |
| 23 | 13,196,234 | 14,196,234 | 3,98E-07 | 13,696,234 | KIF6, KCNK16, KCNK17, DAAM2, MOCS1, U6 | 1 | |
| 4 | 105,958,204 | 106,999,289 | 7,92E-08 | 106,492,268 | TRBV24-1, TRPV6, TRBV30, TRPV5, KEL, PRSS2, EPHB6, OR9A2, OR9A16, OR9A15, OR6V1, PIP, TAS2R39, TAS2R40, TMEM139, CLCN1, GSTK1, CASP2, FAM131B, ZYX, EPHA1, TAS2R60, OR2R3, OR10AC1, TCAF2, TCAF1, ENSBTAG00000049004, ENSBTAG00000027204, ENSBTAG00000050190, ENSBTAG00000053376, ENSBTAG00000053701, ENSBTAG00000053785, ENSBTAG00000054917, ENSBTAG00000051147, ENSBTAG00000047919, ENSBTAG00000034609, ENSBTAG00000052365, ENSBTAG00000050494, ENSBTAG00000031162 | 93 | |
| 4 | 108,183,737 | 109,219,627 | 3,24E-07 | 108,688,422 | 10 | ||
| 5 | 35,035,017 | 36,331,608 | 1,08E-07 | 35,580,801 | ANO6, DBX2, NELL2, TMEM117, 5S_rRNA, ENSBTAG00000026249, ENSBTAG00000032150 | 35 | |
| 14 | 74,743,059 | 75,743,059 | 9,47E-08 | 75,243,059 | MMP16, CNBD1, ENSBTAG00000051556 | 1 | |
| 24 | 45,728,397 | 46,728,397 | 4,49E-07 | 46,228,397 | EPG5, bta-mir6523a, PSTPIP2, ATP5F1A, HAUS1, U6, C24H1orf25, RNF165, LOXHD1, ST8SIA5, PIAS2, KATNAL2, ENSBTAG00000048422, ENSBTAG00000047360, ENSBTAG00000047547 | 1 | |
| ELISA Tissue Culture (+/) | 23 | 18,812,221 | 20,933,699 | 4,01E-10 | 19,331,156 | RUNX2, U6, ENPP4, CLIC5, ENPP5, RCAN2, CYP39A1, SLC25A27, TDRD6, ADGRF5, PLA2G7, ANKRD66, ADGRF1, TNFRSF21, CD2AP, ADGRF2, ADGRF4, OPN5, ENSBTAG00000021415 | 68 |
| 23 | 21,898,408 | 22,901,993 | 4,06E-08 | 22,401,993 | MMUT, CENPQ, GLYATL3, C23H6orf141, U6, RHAG, CRISP1, CRISP2, CRISP3, 7SK, DEFB114, DEFB110, ENSBTAG00000046237, ENSBTAG00000050850, ENSBTAG00000053808, ENSBTAG00000054396, ENSBTAG00000046711 | 5 | |
| 23 | 15,094,386 | 16,094,386 | 3,39E-07 | 15,594,386 | TREM1, TREM2, TREML1, TREML2, FOXP4, MDFI, TFEB, PGC, FRS6,USP49, MED20, TOMM6, BYSL, CCND3, TAF8, GUCA1A, MGC137036, C23H6orf132, GUCA1B, MRPS10, TRERF1, ENSBTAG00000050887, ENSBTAG00000038916, ENSBTAG00000054479, ENSBTAG00000053124 | 1 | |
| 11 | 91,785,241 | 93,618,888 | 2,24E-08 | 92,308,149 | GGTA1, DAB2IP, NDUFA8, MORN5, LHX6, RBM18, MRRF, PTGS1, bta-mir-10175, OR1L18, OR1L12, OR1L21,OR1L8H, OR1L8, OR1L8C, OR1L8E, OR1L8D, OR1L20, OR1N2, OR1J26, OR1J4E, OR1N1, OR1J2, ENSBTAG00000039201, ENSBTAG00000039186, ENSBTAG00000012827, ENSBTAG00000049150, ENSBTAG00000049268, ENSBTAG00000048686, ENSBTAG00000037739, ENSBTAG00000054916, ENSBTAG00000055261, ENSBTAG00000048884 | 43 | |
| 5 | 35,035,017 | 36,331,608 | 1,80E-08 | 35,580,801 | ANO6, DBX2, NELL2, TMEM117, 5S_rRNA, ENSBTAG00000026249, ENSBTAG00000032150 | 34 | |
| 28 | 42,484,673 | 43,484,698 | 3,23E-07 | 42,984,698 | FRMPD2, MAPK8, ARHGAP22, WDFY4, LRRC18, VSTM4, FAM170B, ENSBTAG00000006042 | 2 | |
| 14 | 74,743,059 | 75,743,059 | 1,38E-07 | 75,243,059 | MMP16, CNBD1, ENSBTAG00000051556 | 1 | |
| 4 | 105,958,204 | 106,999,289 | 1,35E-07 | 106,492,268 | TRBV24-1, TRPV6, TRBV30, TRPV5, KEL, PRSS2, EPHB6, OR9A2, OR9A16, OR9A15, OR6V1, PIP, TAS2R39, TAS2R40, TMEM139, CLCN1, GSTK1, CASP2, FAM131B, ZYX, EPHA1, TAS2R60, OR2R3, OR10AC1, TCAF2, TCAF1, ENSBTAG00000049004, ENSBTAG00000027204, ENSBTAG00000050190, ENSBTAG00000053376, ENSBTAG00000053701, ENSBTAG00000053785, ENSBTAG00000054917, ENSBTAG00000051147, ENSBTAG00000047919, ENSBTAG00000034609, ENSBTAG00000052365, ENSBTAG00000050494, ENSBTAG00000031162 | 84 | |
| ELISA Tissue PCR (+/) | 23 | 18,812,221 | 22,958,623 | 3,77E-09 | 19,331,156 | RUNX2, U6, ENPP4, CLIC5, ENPP5, RCAN2, CYP39A1, SLC25A27 TDRD6, ADGRF5 PLA2G7, ANKRD66,ADGRF1, TNFRSF21, CD2AP, ADGRF2, ADGRF4, OPN5, PTCHD, OR9G1, OR5M10C, 5S_rRNA, MEP1A, MMUT, CENPQ, GLYATL3, C23H6orf141,U6 RHAG, CRISP1, CRISP2, CRISP3, 7SK, DEFB114, DEFB110, ENSBTAG00000046237, ENSBTAG00000050850, ENSBTAG00000053808, ENSBTAG00000054396, ENSBTAG00000046711, ENSBTAG00000021415, ENSBTAG00000038677, ENSBTAG00000054235 | 61 |
| 23 | 15,094,386 | 16,094,386 | 2,66E-07 | 15,594,386 | TREM1, TREM2, TREML1, TREML2, FOXP4, MDFI, TFEB, PGC, FRS6, USP49, MED20, TOMM6, BYSL, CCND3, TAF8, GUCA1A, MGC137036, C23H6orf132, GUCA1B, MRPS10, TRERF1, ENSBTAG00000050887, ENSBTAG00000038916, ENSBTAG00000054479, ENSBTAG00000053124 | 1 | |
| 4 | 105,958,204 | 107,001,103 | 5,82E-08 | 106,492,268 | TRBV24-1, TRPV6, TRBV30, TRPV5, KEL, PRSS2, EPHB6, OR9A2, OR9A16, OR9A15, OR6V1, PIP, TAS2R39, TAS2R40, TMEM139, CLCN1, GSTK1, CASP2, FAM131B, ZYX, EPHA1, TAS2R60, OR2R3, OR10AC1, TCAF2, TCAF1, ENSBTAG00000049004, ENSBTAG00000027204, ENSBTAG00000050190, ENSBTAG00000053376, ENSBTAG00000053701, ENSBTAG00000053785, ENSBTAG00000054917, ENSBTAG00000051147, ENSBTAG00000047919, ENSBTAG00000034609, ENSBTAG00000052365, ENSBTAG00000050494, ENSBTAG00000031162 | 96 | |
| 5 | 35,080,801 | 36,080,801 | 4,92E-07 | 35,580,801 | DBX2, NELL2, TMEM117 | 1 | |
| 12 | 67,715,495 | 68,715,495 | 3,73E-07 | 68,215,495 | GPC6 | 1 | |
| 14 | 74,743,059 | 75,743,059 | 3,61E-07 | 75,243,059 | MMP16, CNBD1, ENSBTAG00000051556 | 1 |
1QTL location.
2 SNP location in the genome assembly.
3 Positional candidate genes are defined as genes that are located within 50 kb on either side of the identified QTL. When combinations of diagnostics tests were used, animals were considered cases when they were positive to all the tests and controls when they were negative to any (+/) or all of them (+/-).
Quantile-quantile plots comparing the observed distribution of–log (P-values) to the expected values under null hypothesis are shown in S2 Fig. The plots showed a distribution close to the expected distribution line for the following phenotypes: ELISA-tissue PCR-tissue culture (+/-) (λmedian = 1.007), ELISA-tissue PCR-tissue culture (+/) (λmedian = 1.004), ELISA-tissue culture (+/) (λmedian = 1.006), and ELISA-tissue PCR (+/) (λmedian = 1.006), indicating that significant values were not overestimated due to population stratification or cryptic relatedness. A slight deviation in the upper right tails from the y = x line suggested that some association was present in the data.
Table 3 also presents a list of candidate genes either containing highly-associated SNPs or within 50 kb of the surrounding genomic region of the highly-associated SNPs. We were able to identify candidate genes in all the QTLs except for one located in BTA4. One hundred and fourteen candidate genes were mapped within the 5 QTLs associated with the phenotype with the highest h2, the ELISA-tissue culture-tissue PCR (+/-), the majority located on BTA23. The OR calculated for all the SNPs associated with a positive ELISA-tissue PCR-tissue culture diagnosis (+/-) (P < 5 × 10−7) were > 1 indicating that the animals with the minor alleles were more likely to have a positive ELISA-tissue PCR-tissue culture result. The OR for the SNPs defining the peak of each QTL are shown in Table 4 under the codominant genetic model.
Table 4. Odds ratio (OR) for some the SNPs associated with a positive ELISA-tissue PCR-tissue culture diagnosis (+/-)(P < 5 x 10−7).
| Phenotype | BTA | SNP position | SNP ID | Genotype | Cases (%) | Controls (%) | OR (95% CI) |
|---|---|---|---|---|---|---|---|
| ELISA Tissue PCR Tissue Culture (+/-) | 23 | 15,594,386 | CCATGGGGT/CCATGGGGT | 29 (52.7) | 566 (86.0) | 1.0 | |
| C/CCATGGGGT | 24 (43.6) | 89 (13.5) | 4.7 (2.6–8.5) | ||||
| C/C | 2 (3.6) | 3 (0.5) | 17.5 (2.6–118.3) | ||||
| 23 | 19,329,213 | rs209106537 | C/C | 20 (36.4) | 461 (70.1) | 1.0 | |
| A/C | 25 (45.5) | 181 (27.5) | 3.2 (1.7–5.9) | ||||
| A/A | 10 (18.2) | 16 (2.4) | 13.2 (5.2–33.2) | ||||
| 23 | 22,401,117 | rs385614395 | G/G | 33 (60.0) | 578 (87.8) | 1.0 | |
| C/G | 18 (32.7) | 78 (11.9) | 3.8 (2.0–7.2) | ||||
| C/C | 4 (7.3) | 2 (0.3) | 43.3 (7.5–249.9) | ||||
| 23 | 11,900,592 | rs136621504 | G/G | 37 (67.3) | 607 (92.2) | 1.0 | |
| A/G | 18 (32.7) | 51 (7.8) | 5.5 (2.9–10.4) | ||||
| 5 | 35,577,195 | rs110029938 | T/T | 36 (65.5) | 583 (89.1) | 1.0 | |
| C/T | 17 (30.9) | 71 (10.8) | 4.2 (2.2–8.0) | ||||
| C/C | 2 (3.6) | 1 (0.2) | 44.1 (3.6–538.5) |
(+/-) Animals were considered cases when they were positive to all the tests and controls when they were negative to all of them.
Discussion
Previous studies that estimated genetic parameters for PTB in cattle indicated that susceptibility to MAP infection is heritable, with h2 estimates of susceptibility to the disease ranging from < 0.01 (16) to 0.2843 (18). This is comparable with the moderate heritability (h2 = 0.12) estimated for bTb infection even though the phenotype definitions and models used differed [42]. In our study population, the h2 estimates were also moderate (ranged from 0.054 to 0.139). Interestingly, we observed that the combination of multiple diagnostic tests increased the h2 estimates, with the highest h2 obtained for the combination of ELISA-tissue PCR-tissue culture (+/-) (h2 = 0.139). For this phenotype, all the common SNPs explained ~14% of the phenotypic variance. Except for the tissue culture (h2 = 0.101), the low h2 estimates obtained for the individual tests (h2 < 0.08) could be attributed to their lack of sensitivity for the detection of animals that are in the subclinical stages of MAP infection (false negatives). While cows testing ELISA-tissue PCR and tissue culture positive are very likely infected, animals could be test-negative because of a false-negative test result, lack of exposure to MAP, or be truly resistant. While a positive test result is almost certainly associated with disease susceptibility, a negative test does not always reflect resistance. In agreement with this, we observed that all the SNPs associated with the ELISA-tissue PCR-tissue culture diagnosis (+/-) had an OR > 1 and were, therefore, associated with disease susceptibility. This implies that susceptibility to MAP infection can be modified using genetic selection. In our study population, only 9 of the 70 ELISA-positive animals couldn´t be detected by tissue PCR and culture. As above, these cows could be tissue PCR and culture negative because of a false-negative test result or be truly resistant. PTB resistance defined as the ability of the host to clear the pathogen by mounting a protective immune response deserves further studies.
Our study is unique in the definition of cases and controls through the combination of multiple diagnostic tests including ELISA for detection of humoral responses against MAP, and culture and PCR detection of MAP in tissue samples. Moreover, the population size used (N = 983) was bigger than in a previous GWAS using the bovine SNP50 Bead Chip where cases were defined as tissue PCR-tissue culture positives (N = 459) [30]. Although a previous study combined the data from two GWAS to identify loci associated with MAP tissue infection and humoral immune response [20], our study provides the first comparison of the genetic effects associated with different phenotypic measurements or diagnostic definitions in a common set of samples using WGS data. Combination test interpretation (all tests negative equals non-infected and all tests positive equals infected) was used to increase the sensitivity of the ELISA-Tissue PCR-tissue culture combination. In addition, the use of imputed WGS increased the accuracy of the GWAS enabling the identification of 12 QTLs surpassing the high threshold (P < 5 × 10−7) for evidence of an association with MAP infection. These strategies increased the hereditability of the trait and reduced the risk of misclassification. Further studies will demonstrate whether the post-mortem examination of gut tissues and regional lymph nodes can improve, even more, the accuracy of the classification of naturally infected animals and uninfected controls, and provides high h2 estimates. Once a PTB-associated phenotype with a moderate/high h2 estimate is finally selected, genomic predictions will be developed in small, well-recorded reference populations using the genomic BLUP model, and then the prediction equation will be applied to predict genomic estimated breeding values of Holstein cattle that were genotyped but did not have PTB-associated phenotypes themselves.
Identifying significant SNPs and QTLs associated with PTB susceptibility is extremely important to understand the molecular mechanisms involved in the pathogenesis of the disease. Our results confirmed that susceptibility to PTB is polygenic with a large number of genetic variants each having a small effect on the regulation of the observed phenotype. Using combinations of diagnostic tests and genotypes imputed to the WGS level, we identified a total of 12 regions associated with PTB susceptibility (P < 5 x 10−7) on 8 chromosomes (BTA4, BTA5, BTA11, BTA12, BTA14, BTA23, BTA24, and BTA28). By examining the available cattle QTL database, we observed that the identified QTLs on BTA23 (18.81–22.95 Mb), BTA24 (45.72–46.72 Mb), and BTA12 overlapped with four regions previously associated with PTB susceptibility; QTL14876, QTL166685, QTL139831, and QTL211947 [25,29,32,43]. This finding provided additional evidence that genes within these regions such as the adhesion G protein-coupled receptor F1 (ADGRF1), adhesion G protein-coupled receptor F5 (ADGRF5), TNF receptor superfamily member 21 (TNFRSF21), defensin β110 (DEFB110) and defensin β114 (DEFB114) are likely associated with MAP infection. Previous studies supported a role for the β-defensins as important host defense effector molecules that are rapidly mobilized by the epithelium upon MAP infection [44]. ADGRF1and ADGRF5, also named GPR110 and GRP116, are G-protein-coupled receptors acting as important regulators in the progression and development of several human inflammatory diseases including IBD, hepatocellular carcinoma, lung cancer, gastric cancer, prostatic cancer, glioma, and colorectal cancer [45]. The TNFRSF21 is a member of the TNF/TNFR family and plays a critical role in pathogen recognition, immune response, inflammation, and tumor progression [46,47]. The encoded protein by this gene activates nuclear factor kappa-B (NF-κβ) and mitogen-activated protein kinase 8 (MAPK-8, also called c-Jun N-terminal kinase 1) and induces cell apoptosis. Fang et al. demonstrated that the level of TNFRSF21 was dramatically increased in bovine peripheral blood leucocytes of mastitis cows, which underlines its important regulatory role in bovine mastitis inflammation [48]. More recently, a role for the TNFRSF21 in the regulation of bovine mastitis susceptibility via GWAS and post-transcriptional analysis was discovered [49]. Interestingly, several PTB-associated QTLs identified in the current study were located on regions previously found to be associated with clinical mastitis including QTL30823, QTL30824, QTL30825, QTL30826, QTL161601, QTL161607, and QTL65863 [48–52].
Other candidate genes identified in BTA23 (18.81–22.95 Mb) include genes involved in the splicing process (U6), invasion and migration of tumors (CLIC5, ENPP4, ENPP5), and in the regulation of the calcineurin-nuclear factor of activated T cells (RCAN2). This region overlapped with QTLs on BTA23 associated with bTb infection in a multi-breed GWAS of Charolais, Limousin, and Holstein Friesian (genetic positions 19.44–22.72 Mb) [42]. Interestingly, this region included the RCAN2 gene as well. The RCAN2 is responsible for the regulation of calcineurin 2, and calcineurin activation promotes the survival of Mycobacterium tuberculosis (MTb) within its host by preventing phagocyte maturation which is required to destroy intracellular bacteria [53]. Our results, together with previous studies, suggested that regulation of calcineurin 2 may have a pivotal role in the susceptibility of cattle to both bTb and PTB. In addition, a second PTB-associated QTL on BTA23 (11.32–14.19 Mb) overlapped with a bTb-associated QTL (QTL96552) [54].
Several positional candidate genes that we identified in other QTLs were important transcriptional regulators, including the Forkhead BoxP4 (FOXP4), transcription factor EB (TFEB), the LIM/homeoboxprotein (LHX6), and the mediator of RNA polymerase II transcription subunit 20 (MED2). The TFEB acts as a master regulator of lysosomal biogenesis, autophagy, lysosomal exocytosis, lipid catabolism, energy metabolism, and immune response against intracellular pathogens including MTb [55]. Autophagy, an intracellular lysosomal degradation process, is an important cell-autonomous defense system involved in innate and adaptive immune responses and contributes to host defense against various intracellular microbes. Among the transcription factors implicated in autophagy, TFEB is a critical regulator of autophagic activation. In the current study, several candidate genes potentially associated with MAP infection were involved in the splicing process, including the U6 spliceosomal RNA and 7SKRNA. Further studies are needed to determine if the identified SNPs are affecting the positional candidate genes or other genes through cis- or trans-regulatory effects [56].
The candidate genes identified in this study are novel in the sense that they have not been associated with PTB risk in cattle before. Interestingly, some of the candidate genes identified in the current study were previously found associated with mastitis and bTb infection. For instance, the RCAN2 gene was found associated with bTb infection [42] and with 18 other bovine traits including somatic cell counts [57]. Although the TNFRSF21 has not been associated with PTB risk in cattle, other members of the TNF receptor superfamily such as TNFRSF18 and TNFRSF4 are known to stimulate the Th1 cell-mediated immune response and have been previously identified as PTB candidate genes [28]. As mentioned before, the TNFRSF21 was identified to be involved in the regulation of bovine mastitis susceptibility via GWAS [49].
The candidate genes identified in our study were also compared to candidate genes that were previously identified in CD, IBD, and colorectal cancer to determine if there was any overlap. Five of the candidate genes identified in the current study; the TNFRSF21, ADGRF1, FOXP4, Cyclin D3 (CCND3), and Transmembrane Protein 117 (TMEM117) were previously found to be associated with IBD [58]. FOXP4 and CCND3 were also found associated with CD. Recent association analysis identified a new risk locus harboring the TFEB for colorectal cancer susceptibility [59]. Using the candidate genes identified in our study, we could not find any enriched pathway underlying PTB susceptibility. However, several of the identified candidate genes matched genes in the Innate DB database with relevant innate immunity functions such as the triggering Receptor Expressed On Myeloid Cells 1 and 2 (TREM1, TREM2), triggering Receptor Expressed On Myeloid Cells like 1 and 2 (TREML1, TREML2), Canopy FGF Signaling Regulator 3 (CNPY3), Protein Phosphatase 2 Regulatory Subunit B’Delta (PPP2R5D), Cysteine Rich Secretory Protein 3 (CRISP3), Anoctamin 6 (ANO6), Protein Inhibitor of Activated STAT2 (PIAS2), DAB2 Interacting protein (DAB2IP), and MAPK8.
Conclusions
In summary, combining phenotypes and WGS into a joint GWAS improved the power for detecting genetic associations in Spanish Holstein cattle. Our results validated some previously reported associations and identified novel SNPs, QTLs, and candidate genes associated with the antibody response to MAP infection and with MAP detection in infected tissues. Furthermore, the addition to genotyping assays of the SNPs identified in the current study would allow producers to select cattle that are less susceptible to PTB and likely to other bovine diseases as well; ultimately reducing the spread of diseases, preventing further economic losses, and reducing antimicrobial use. Consequently, a reduction of the presence of MAP in the environment may also be beneficial to humans, especially if the link between MAP and human inflammatory diseases is confirmed.
Supporting information
The chart depicts the genomic distribution of the SNPs associated with (A) ELISA-tissue culture-tissue PCR (+/-), (B) ELISA-tissue culture-tissue PCR (+/), (C) ELISA-tissue culture (+/), and (D) ELISA-tissue PCR (+/) according to the Ensembl Variant Effect Predictor (VEP).
(TIF)
The plots showed a distribution close to the expected distribution line for the following phenotypes: ELISA-tissue PCR-tissue culture (+/-) (λmedian = 1.007), ELISA- tissue PCR-tissue culture (+/) (λmedian = 1.004), ELISA-tissue culture (+/) (λmedian = 1.006), and ELISA-tissue PCR (+/) (λmedian = 1.006). The red line is the slope expected under no inflation and no true association, the y-axis represents the observed–log (P-values), and the x-axis represents the expected–log (P-values), under the null hypothesis of no association.
(TIF)
Acknowledgments
We would like to thank Rosario Sánchez from the i2basque Research and Academic Network for computational support. We gratefully acknowledge the 1000 Bull Genomes Consortium for providing accessibility to WGS data that was used in this study.
Data Availability
Sequence data used in this study are owned by the 1000 Bull Genomes Project Consortium. The sequence variants for 1800 animals from Run8 are public at the European Variation Archive under project number PRJEB42783. The SNPs associated with the ELISA-tissue PCR-tissue culture results have been deposited in the animal genome database (https://www.animalgenome.org/cgi-bin/QTLdb/BT/pubtails?PUBMED_ID=ISU0121). Individual genotype data used in this study is managed by a third party, the Spanish Friesian Cattle National Federation (CONAFE). Requests for genotype data can be made to CONAFE, Ctra. de Andalucía, km. 23,600 - 28340 Valdemoro, Madrid, Spain; email: conafe@conafe.com; phone: +34 (91) 8952412; website: www.conafe.com. The authors confirm they had no special access privileges.
Funding Statement
Financial support for this study was provided by a grant from the Spanish Ministry of Science, Innovation, and Universities (MICINN, https://sede.micinn.gob.es/, project code: RTI2018-094192-R-C21) and by European Regional Development Funds (FEDER) to MAH. MC and GBB have been awarded fellowships from the National Institute for Agricultural Research (INIA) and MICINN, respectively. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Nielsen SS, Toft N. A review of prevalences of paratuberculosis in farmed animals in Europe. Prev Vet Med. 2009; 88(1):1–14. doi: 10.1016/j.prevetmed.2008.07.003 [DOI] [PubMed] [Google Scholar]
- 2.Ott SL, Wells SJ, Wagner BA. Herd-level economic losses associated with Johne’s disease on US dairy operations. Prev Vet Med. 1999; 40(3–4):179–92. doi: 10.1016/s0167-5877(99)00037-9 [DOI] [PubMed] [Google Scholar]
- 3.Hasonova L, Pavlik I. Economic impact of paratuberculosis in dairy cattle herds: A review. Vet Med. 2006; 51(5):193–211. doi: 10.17221/5539-VETMED [DOI] [Google Scholar]
- 4.Sweeney RW. Pathogenesis of paratuberculosis. Vet Clin North Am—Food Anim Pract. 2011; 27(3):537–46. doi: 10.1016/j.cvfa.2011.07.001 [DOI] [PubMed] [Google Scholar]
- 5.Bastida F, Juste RA. Paratuberculosis control: A review with a focus on vaccination. J Immune Based Ther Vaccines 2011; 9(1):8. doi: 10.1186/1476-8518-9-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Juste RA, Elguezabal N, Garrido JM, Pavon A, Geijo MV., Sevilla I, et al. On the prevalence of M. avium subspecies paratuberculosis DNA in the blood of healthy individuals and patients with inflammatory bowel disease. PLoS One. 2008; 3(7):3–8. doi: 10.1371/journal.pone.0002537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jeyanathan M, Boutros-Tadros O, Radhi J, Semret M, Bitton A, Behr MA. Visualization of Mycobacterium avium in Crohn’s tissue by oil-immersion microscopy. Microbes Infect. 2007; 9(14–15):1567–73. doi: 10.1016/j.micinf.2007.09.001 [DOI] [PubMed] [Google Scholar]
- 8.Pierce ES. Could Mycobacterium avium subspecies paratuberculosis cause Crohn’s disease, ulcerative colitis…and colorectal cancer? Infect Agent Cancer. 2018; 13(1):1–6. doi: 10.1186/s13027-017-0172-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Juste RA, Alonso-Hearn M, Molina E, Geijo M, Vázquez P, Sevilla IA, et al. Significant reduction in bacterial shedding and improvement in milk production in dairy farms after the use of a new inactivated paratuberculosis vaccine in a field trial. BMC Res Notes. 2009; 2. doi: 10.1186/1756-0500-2-233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alonso-Hearn M, Molina E, Geijo M, Vázquez P, Sevilla IA, Garrido JM, et al. Immunization of adult dairy cattle with a new heat-killed vaccine is associated with longer productive life prior to cows being sent to slaughter with suspected paratuberculosis. J Dairy Sci. 2012; 95(2):618–29. doi: 10.3168/jds.2009-2860 [DOI] [PubMed] [Google Scholar]
- 11.Garrido JM, Vázquez P, Molina E, Plazaola JM, Sevilla IA, Geijo MV., et al. Paratuberculosis vaccination causes only limited cross-reactivity in the skin test for diagnosis of bovine tuberculosis. PLoS One. 2013; 8(11):2–8. doi: 10.1371/journal.pone.0080985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Groenendaal H, Nielen M, Jalvingh AW, Horst SH, Galligan DT, Hesselink JW. A simulation of Johne’s disease control. Prev Vet Med. 2002; 54(3):225–45. doi: 10.1016/s0167-5877(02)00027-2 [DOI] [PubMed] [Google Scholar]
- 13.Garry F. Control of paratuberculosis in dairy herds. Vet Clin North Am—Food Anim Pract. 2011; 27(3):599–607. doi: 10.1016/j.cvfa.2011.07.006 [DOI] [PubMed] [Google Scholar]
- 14.Vázquez CB, Alonso-Hearn M, Juste RA, Canive M, Iglesias T, Iglesias N, et al. Detection of latent forms of Mycobacterium avium subsp. paratuberculosis infection using host biomarker-based ELISAs greatly improves paratuberculosis diagnostic sensitivity. PLoS One. 2020; 15(9 September 2020):1–21. doi: 10.1371/journal.pone.0236336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsairidou S, Allen A, Banos G, Coffey M, Anacleto O, Byrne AW, et al. Can we breed cattle for Lower Bovine TB infectivity? Front Vet Sci. 2018; 5(DEC):1–8. doi: 10.3389/fvets.2018.00310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koets AP, Adugna G, Janss LLG, Van Weering HJ, Kalis CHJ, Wentink GH, et al. Genetic variation of susceptibility to Mycobacterium avium subsp. paratuberculosis infection in dairy cattle. J Dairy Sci. 2000; 83(11):2702–8. doi: 10.3168/jds.S0022-0302(00)75164-2 [DOI] [PubMed] [Google Scholar]
- 17.Gonda MG, Chang YM, Shook GE, Collins MT, Kirkpatrick BW. Effect of Mycobacterium paratuberculosis infection on production, reproduction, and health traits in US Holsteins. Prev Vet Med. 2007; 80(2–3):103–19. doi: 10.1016/j.prevetmed.2007.01.011 [DOI] [PubMed] [Google Scholar]
- 18.Küpper JD, Brandt HR, Erhardt G. Genetic association between NOD2 polymorphism and infection status by Mycobacterium avium ssp. paratuberculosis in German Holstein cattle. Anim Genet. 2014; 45(1):114–6. doi: 10.1111/age.12097 [DOI] [PubMed] [Google Scholar]
- 19.Zare Y, Shook GE, Collins MT, Kirkpatrick BW. Genome-wide association analysis and genomic prediction of Mycobacterium avium subspecies paratuberculosis infection in US Jersey cattle. PLoS One. 2014; 9(2). doi: 10.1371/journal.pone.0088380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Minozzi G, Williams JL, Stella A, Strozzi F, Luini M, Settles ML, et al. Meta-analysis of two genome-wide association studies of bovine paratuberculosis. PLoS One. 2012; 7(3):1–8. doi: 10.1371/journal.pone.0032578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Settles M, Zanella R, McKay SD, Schnabel RD, Taylor JF, Whitlock R, et al. A whole genome association analysis identifies loci associated with Mycobacterium avium subsp. paratuberculosis infection status in US Holstein cattle. Anim Genet. 2009;40(5):655–62. doi: 10.1111/j.1365-2052.2009.01896.x [DOI] [PubMed] [Google Scholar]
- 22.Zanella R, Settles ML, McKay SD, Schnabel R, Taylor J, Whitlock RH, et al. Identification of loci associated with tolerance to Johne’s disease in Holstein cattle. Anim Genet. 2011; 42(1):28–38. doi: 10.1111/j.1365-2052.2010.02076.x [DOI] [PubMed] [Google Scholar]
- 23.Pant SD, Schenkel FS, Verschoor CP, You Q, Kelton DF, Moore SS, et al. A principal component regression based genome wide analysis approach reveals the presence of a novel QTL on BTA7 for MAP resistance in Holstein cattle. Genomics. 2010; 95(3):176–82. doi: 10.1016/j.ygeno.2010.01.001 [DOI] [PubMed] [Google Scholar]
- 24.Alpay F, Zare Y, Kamalludin MH, Huang X, Shi X, Shook GE, et al. Genome-wide association study of susceptibility to infection by Mycobacterium avium subspecies paratuberculosis in Holstein cattle. PLoS One. 2014; 9(12):1–14. doi: 10.1371/journal.pone.0111704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kirkpatrick BW, Shi X, Shook GE, Collins MT. Whole-Genome association analysis of susceptibility to paratuberculosis in Holstein cattle. Anim Genet. 2011; 42(2):149–60. doi: 10.1111/j.1365-2052.2010.02097.x [DOI] [PubMed] [Google Scholar]
- 26.van Hulzen KJE, Schopen GCB, van Arendonk JAM, Nielen M, Koets AP, Schrooten C, et al. Genome-wide association study to identify chromosomal regions associated with antibody response to Mycobacterium avium subspecies paratuberculosis in milk of Dutch Holstein-Friesians. J Dairy Sci. 2012; 95(5):2740–8. doi: 10.3168/jds.2011-5005 [DOI] [PubMed] [Google Scholar]
- 27.Brito LF, Mallikarjunappa S, Sargolzaei M, Koeck A, Chesnais J, Schenkel FS, et al. The genetic architecture of milk ELISA scores as an indicator of Johne’s disease (paratuberculosis) in dairy cattle. J Dairy Sci. 2018; 101(11):10062–75. doi: 10.3168/jds.2017-14250 [DOI] [PubMed] [Google Scholar]
- 28.Mallikarjunappa S, Sargolzaei M, Brito LF, Meade KG, Karrow NA, Pant SD. Short communication: Uncovering quantitative trait loci associated with resistance to Mycobacterium avium ssp. paratuberculosis infection in Holstein cattle using a high-density single nucleotide polymorphism panel. J Dairy Sci. 2018; 101(8):7280–6. doi: 10.3168/jds.2018-14388 [DOI] [PubMed] [Google Scholar]
- 29.Gao Y, Jiang J, Yang S, Cao J, Han B, Wang Y, et al. Genome-wide association study of Mycobacterium avium subspecies paratuberculosis infection in Chinese Holstein. BMC Genomics. 2018;19(1):1–10. doi: 10.1186/s12864-017-4368-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kiser JN, White SN, Johnson KA, Hoff JL, Taylor JF, Neibergs HL. Identification of loci associated with susceptibility to Mycobacterium avium subspecies paratuberculosis (Map) tissue infection in cattle. J Anim Sci. 2017; 95(3):1080–91. doi: 10.2527/jas.2016.1152 [DOI] [PubMed] [Google Scholar]
- 31.McGovern SP, Purfield DC, Ring SC, Carthy TR, Graham DA, Berry DP. Candidate genes associated with the heritable humoral response to Mycobacterium avium ssp. paratuberculosis in dairy cows have factors in common with gastrointestinal diseases in humans. J Dairy Sci. 2019; 102(5):4249–63. doi: 10.3168/jds.2018-15906 [DOI] [PubMed] [Google Scholar]
- 32.Sanchez MP, Guatteo R, Davergne A, Saout J, Grohs C, Deloche MC, et al. Identification of the ABCC4, IER3, and CBFA2T2 candidate genes for resistance to paratuberculosis from sequence-based GWAS in Holstein and Normande dairy cattle. Genet Sel Evol. 2020; 52(1):1–17. doi: 10.1186/s12711-019-0522-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pavlik I, Matlova L, Bartl J, Svastova P, Dvorska L, Whitlock R. Parallel faecal and organ Mycobacterium avium subsp. paratuberculosis culture of different productivity types of cattle. Vet Microbiol. 2000; 77(3–4):309–24. doi: 10.1016/s0378-1135(00)00316-3 [DOI] [PubMed] [Google Scholar]
- 34.Vázquez P, Ruiz-Larrañaga O, Garrido JM, Iriondo M, Manzano C, Agirre M, et al. Genetic association analysis of paratuberculosis forms in Holstein-Friesian cattle. Vet Med Int. 2014; 2014. doi: 10.1155/2014/321327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Juste RA, Marco JC, Sáez de Ocáriz C, Adúriz JJ. Comparison of different media for the isolation of small ruminant strains of Mycobacterium paratuberculosis. Vet Microbiol. 1991; 28(4):385–90. doi: 10.1016/0378-1135(91)90073-o [DOI] [PubMed] [Google Scholar]
- 36.Loh PR, Palamara PF, Price AL. Fast and accurate long-range phasing in a UK Biobank cohort. Nat Genet. 2016; 48(7):811–6. doi: 10.1038/ng.3571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Das S, Forer L, Schönherr S, Sidore C, Locke AE, Kwong A, et al. Next-generation genotype imputation service and methods. Nat Genet. 2016; 48(10):1284–7. doi: 10.1038/ng.3656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hayes BJ, Daetwyler HD. 1000 Bull Genomes project to map simple and complex genetic traits in cattle: Applications and Outcomes. Annu Rev Anim Biosci. 2019; 7:89–102. doi: 10.1146/annurev-animal-020518-115024 [DOI] [PubMed] [Google Scholar]
- 39.Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: A tool for genome-wide complex trait analysis. Am J Hum Genet. 2011; 88(1):76–82. doi: 10.1016/j.ajhg.2010.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.González JR, Armengol L, Solé X, Guinó E, Mercader JM, Estivill X, et al. SNPassoc: An R package to perform whole genome association studies. Bioinformatics. 2007; 23(5):644–5. doi: 10.1093/bioinformatics/btm025 [DOI] [PubMed] [Google Scholar]
- 41.Durinck S, Spellman PT, Birney E, Huber W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat Protoc. 2009; 4(8):1184–91. doi: 10.1038/nprot.2009.97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ring SC, Purfield DC, Good M, Breslin P, Ryan E, Blom A, et al. Variance components for bovine tuberculosis infection and multi-breed genome-wide association analysis using imputed whole genome sequence data. PLoS One. 2019; 14(2):1–24. doi: 10.1371/journal.pone.0212067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Del Corvo M, Luini M, Stella A, Pagnacco G, Ajmone-Marsan P, Williams JL, et al. Identification of additional loci associated with antibody response to Mycobacterium avium ssp. paratuberculosis in cattle by GSEA–SNP analysis. Mamm. Genome. 2017; 28, 520–527. doi: 10.1007/s00335-017-9714-7 [DOI] [PubMed] [Google Scholar]
- 44.Stolzenberg ED, Anderson GM, Ackermann MR, Whitlock RH, Zasloff M. Epithelial antibiotic induced in states of disease. Proc. Natl. Acad. Sci. 1997; U. S. A. 94, 8686–8690. doi: 10.1073/pnas.94.16.8686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ma B, Zhu J, Su J, Pan F, Ji Y, et al. The role of GPR110 in lung cancer progression. Ann Transl Med. 2020; 8(12):745. doi: 10.21037/atm-20-3146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: Integrating mammalian biology. Cell. 2001; 104(4):487–501. doi: 10.1016/s0092-8674(01)00237-9 [DOI] [PubMed] [Google Scholar]
- 47.Strilic B, Yang L, Albarrán-Juárez J, Wachsmuth L, Han K, Müller UC, et al. Tumour-cell-induced endothelial cell necroptosis via death receptor 6 promotes metastasis. Nature. 2016; 536(7615):215–8. doi: 10.1038/nature19076 [DOI] [PubMed] [Google Scholar]
- 48.Fang L, Sahana G, Su G, Yu Y, Zhang S, Lund MS, et al. Integrating sequence-based GWAS and RNA-Seq provides novel insights into the genetic basis of mastitis and milk production in dairy cattle. Sci Rep. 2017; 7(March):1–16. doi: 10.1038/srep45560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang F, Chen F, Li L, Yan L, Badri T, Lv C, et al. Three novel players: PTK2B, SYK, and TNFRSF21 were identified to be involved in the regulation of bovine mastitis susceptibility via GWAS and post-transcriptional analysis. Front Immunol. 2019; 10:1–20. doi: 10.3389/fimmu.2019.00001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wojdak-Maksymiec K, Szyda J, Strabel T. Parity-dependent association between TNF-α and LTF gene polymorphisms and clinical mastitis in dairy cattle. BMC Vet Res. 2013; 9. doi: 10.1186/1746-6148-9-114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cai Z, Guldbrandtsen B, Lund MS, Sahana G. Prioritizing candidate genes post-GWAS using multiple sources of data for mastitis resistance in dairy cattle. BMC Genomics. 2018; 19, 1–11. doi: 10.1186/s12864-017-4368-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tiezzi F, Parker-Gaddis KL, Cole JB, Clay JS & Maltecca C. A genome-wide association study for clinical mastitis in first parity US Holstein cows using single-step approach and genomic matrix re-weighting procedure. PLoS One. 205; 10, 1–15. doi: 10.1371/journal.pone.0114919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Trimble WS, Grinstein S. TB or not TB: Calcium regulation in mycobacterial survival. Cell. 2007; 130(1):12–4. doi: 10.1016/j.cell.2007.06.039 [DOI] [PubMed] [Google Scholar]
- 54.Richardson I. W. et al. A genome-wide association study for genetic susceptibility to Mycobacterium bovis infection in dairy cattle identifies a susceptibility QTL on chromosome 23. Genet. Sel. Evol. 2016; 48, 1–13. doi: 10.1186/s12711-015-0181-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim SH, Kim G, Han DH, Lee M, Kim I, Kim B, et al. Ezetimibe ameliorates steatohepatitis via AMP activated protein kinase-TFEB-mediated activation of autophagy and NLRP3 inflammasome inhibition. Autophagy. 2017; 13(10):1767–81. doi: 10.1080/15548627.2017.1356977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Canive M. et al. Identification of loci associated with susceptibility to bovine paratuberculosis and with the dysregulation of the MECOM, eEF1A2, and U1 spliceosomal RNA expression. Sci. Rep.2021; 11, 1–14. doi: 10.1038/s41598-020-79139-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cole JB, Wiggans GR, Ma L, Sonstegard TS, Lawlor TJ, Crooker BA, et al. Genome-wide association analysis of thirty one production, health, reproduction and body conformation traits in contemporary U.S. Holstein cows. BMC Genomics. 2011; 12:1–17. doi: 10.1186/1471-2164-12-408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kakuta Y, Kawai Y, Okamoto D, Takagawa T, Ikeya K, Sakuraba H, et al. NUDT15 codon 139 is the best pharmacogenetic marker for predicting thiopurine-induced severe adverse events in Japanese patients with inflammatory bowel disease: a multicenter study. J Gastroenterol. 2018; 53(9):1065–78. doi: 10.1007/s00535-018-1486-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Law PJ, Timofeeva M, Fernandez-Rozadilla C, Broderick P, Studd J, Fernandez-Tajes J, et al. Association analyses identify 31 new risk loci for colorectal cancer susceptibility. Nat Commun. 2019;10(1):1–15. doi: 10.1038/s41467-018-07882-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The chart depicts the genomic distribution of the SNPs associated with (A) ELISA-tissue culture-tissue PCR (+/-), (B) ELISA-tissue culture-tissue PCR (+/), (C) ELISA-tissue culture (+/), and (D) ELISA-tissue PCR (+/) according to the Ensembl Variant Effect Predictor (VEP).
(TIF)
The plots showed a distribution close to the expected distribution line for the following phenotypes: ELISA-tissue PCR-tissue culture (+/-) (λmedian = 1.007), ELISA- tissue PCR-tissue culture (+/) (λmedian = 1.004), ELISA-tissue culture (+/) (λmedian = 1.006), and ELISA-tissue PCR (+/) (λmedian = 1.006). The red line is the slope expected under no inflation and no true association, the y-axis represents the observed–log (P-values), and the x-axis represents the expected–log (P-values), under the null hypothesis of no association.
(TIF)
Data Availability Statement
Sequence data used in this study are owned by the 1000 Bull Genomes Project Consortium. The sequence variants for 1800 animals from Run8 are public at the European Variation Archive under project number PRJEB42783. The SNPs associated with the ELISA-tissue PCR-tissue culture results have been deposited in the animal genome database (https://www.animalgenome.org/cgi-bin/QTLdb/BT/pubtails?PUBMED_ID=ISU0121). Individual genotype data used in this study is managed by a third party, the Spanish Friesian Cattle National Federation (CONAFE). Requests for genotype data can be made to CONAFE, Ctra. de Andalucía, km. 23,600 - 28340 Valdemoro, Madrid, Spain; email: conafe@conafe.com; phone: +34 (91) 8952412; website: www.conafe.com. The authors confirm they had no special access privileges.