

Abstract
The coronavirus disease-2019 (COVID-19) pandemic has caused an enormous loss of lives. Various clinical trials of vaccines and drugs are being conducted worldwide; nevertheless, as of today, no effective drug exists for COVID-19. The identification of key genes and pathways in this disease may lead to finding potential drug targets and biomarkers. Here, we applied weighted gene co-expression network analysis and LIME as an explainable artificial intelligence algorithm to comprehensively characterize transcriptional changes in bronchial epithelium cells (primary human lung epithelium (NHBE) and transformed lung alveolar (A549) cells) during severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection. Our study detected a network that significantly correlated to the pathogenicity of COVID-19 infection based on identified hub genes in each cell line separately. The novel hub gene signature that was detected in our study, including PGLYRP4 and HEPHL1, may shed light on the pathogenesis of COVID-19, holding promise for future prognostic and therapeutic approaches. The enrichment analysis of hub genes showed that the most relevant biological process and KEGG pathways were the type I interferon signaling pathway, IL-17 signaling pathway, cytokine-mediated signaling pathway, and defense response to virus categories, all of which play significant roles in restricting viral infection. Moreover, according to the drug–target network, we identified 17 novel FDA-approved candidate drugs, which could potentially be used to treat COVID-19 patients through the regulation of four hub genes of the co-expression network. In conclusion, the aforementioned hub genes might play potential roles in translational medicine and might become promising therapeutic targets. Further in vitro and in vivo experimental studies are needed to evaluate the role of these hub genes in COVID-19.
Keywords: COVID-19, SARS-CoV-2, transcriptional profiling, WGCNA, bronchial epithelium cells, treatment, explainable artificial intelligence
1. Introduction
Since December 2019, millions of people have been directly or indirectly affected by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) global pandemic. Until 12 February 2021, more than 107 million people had already been reported to be infected, with 2,373,733 mortalities (https://coronavirus.jhu.edu/map.html, accessed on 12 February 2021). SARS-CoV-2 is a single-stranded RNA virus that belongs to the Coronaviridae family and can infect mammals and birds [1]. Clinical researchers have defined COVID-19 as an acute respiratory tract infection with diverse symptoms, including fever, dry cough, fatigue, myalgia, conjunctivitis, and pneumonia. However, some patients develop severe illnesses, including pneumonia, acute respiratory distress syndrome, pulmonary edema, acute kidney injury, or quick multiple organ failure [2,3,4,5]. Despite the high infection and mortality rates, the host’s immune response to infection with SARS-CoV-2 remains poorly understood [6,7]. In multiple cases, the involvement of the lungs suggests viral dissemination after the initial infection. Viral RNA has been identified in symptomatic patients in the upper airways, with greater viral loads in nasal swabs than those collected from the throat [8]. Comparable viral loads have been observed in an asymptomatic patient, suggesting that the nasal epithelium is a significant portal for initial infection and may act as the main reservoir for viral dissemination across the respiratory mucosa, an essential viral transmission mediating locus. To improve diagnostic and therapeutic approaches, it is critical to recognize not only the cells and mechanisms that host viruses and enable viral replication, but also what contributes to the inflammation and pathogenesis of the disease.
Dysregulated host immune response and inflammatory cytokine production are believed to correlate with the severity of the disease and poor prognosis in two other coronavirus-related infections, SARS-CoV and MERS-CoV [9,10]. Nevertheless, the underlying molecular mechanisms of the anomalous inflammatory responses under SARS-CoV-2 infection are still unclear.
Since the COVID-19 outbreak, numerous transcriptomic studies of various specimens of COVID-19 patients alongside experimental models have been attempted to further elucidate the dynamics of the host immune response along with gene regulatory networks. In this regard, Xiong et al. conducted a transcriptome sequencing of the RNAs isolated from bronchoalveolar lavage fluid as well as peripheral blood mononuclear cell (PBMC) samples of COVID-19 patients. Their results revealed host inflammatory cytokine profiles to SARS-CoV-2 infection in patients and highlighted the association between COVID-19 pathogenesis and excessive cytokine releases such as CCL2/MCP-1, CXCL10/IP-10, CCL3/MIP-1A, and CCL4/MIP1B [11]. In another study, Ren et al. used single-cell RNA sequencing (scRNA-seq) analysis to evaluate the expression and distribution of angiotensin-converting enzyme 2 (ACE2) and type II transmembrane serine protease (TMPRSS) genes in kidney cell components. They found that podocytes and proximal straight tubule cells were potential host cells targeted by SARS-CoV-2, resulting in acute kidney injury (AKI) caused by the SARS-CoV-2-induced cytopathic effect [12]. Given the critical role of chromatin factors such as topoisomerase I in regulating the transcriptional response to SARS-CoV-2 infection, Ho et al. evaluated transcriptional and epigenetic changes in the human alveolar basal epithelial carcinoma (A549) cell line expressing the human SARS-CoV-2 entry receptor Ace2 (A549-ACE2) infected with SARS-CoV-2 followed by the depletion of topoisomerase I and discovered promising effects of epigenetic therapy in modifying aberrant genome restructuring and selective suppression of inflammatory and anti-viral genes involved in severe COVID-19 [13]. In an effort to investigate the repertoire of viral epitopes promoting T cell-mediated immunity, Weingarten-Gabbay et al. uncovered the stable representation of SARS-CoV-2 HLA-I peptides over the time course of infection and the probable immune evasion mechanisms of SARS-CoV-2 by interfering with the IFN signaling and HLA class I-mediated antigen processing of highly expressed SARS proteins, as shown by dynamic transcriptional profiling of SARS-CoV-2-infected human lung A549 cells [14]. Interestingly, gene expression profiling of SARS-CoV-2-infected A549-ACE2 cells treated with potent anti-viral compounds allowed the exploration of the cholesterol biosynthesis pathway as the key mechanism underlying the blocking of viral replication, which holds the promise of providing indications for capable biomarkers and therapeutic targets [15]. More recently, RNA-seq analysis of SARS-CoV-2-infected A549-ACE2 cell line treated with calcium-channel blocker, amlodipine, identified that pre-treatment with amlodipine would significantly up-regulate the cholesterol biosynthesis-related gene pathway, which results in a marked decrease in SARS-CoV-2 viral load [16].
Due to the large-scale transcriptome data produced by high-throughput technologies such as RNA-seq and microarray, new appropriate approaches are required to efficiently extract meaningful associations from highly multivariate datasets. Several bioinformatics tools and machine learning algorithms, such as support vector machines, random forests, and extremely randomized trees classifiers, have been used to better understand complex essential gene interactions involved in disease development [2,17]. One large-scale genome-wide association approach named weighted correlation network analysis (WGCNA) is a tool for exploring hidden relationships between gene modules and disease traits [18]. WGCNA has been widely applied to construct a scale-free network alongside especially identifying functional modules of highly correlated genes to provide unbiased targets for genetic testing and potential therapies and illustrate the unraveling complexities of the disease by efficiently analyzing gene expression datasets [19,20,21,22,23].
In this study, we applied weighted gene co-expression network analysis (WGCNA) as well as LIME as an explainable artificial intelligence algorithm to investigate the transcriptional changes in primary human lung epithelium (NHBE) and transformed lung alveolar (A549) cell lines after SARS-CoV-2 infection in the hope of shedding light on the pathogenesis of this virus.
2. Materials and Methods
2.1. Gene Expression Dataset Acquisition and Preprocessing
Data analysis procedures are illustrated in Figure 1. The RNA-seq gene expression dataset GSE147507 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE147507, accessed on 8 March 2021) was obtained and downloaded from the Gene Expression Omnibus (GEO) database, which is based on Illumina NextSeq 500 platform. In total, the above dataset contains a raw read count of 20 samples of human lung epithelial cells, including independent biological triplicates of NHBE and A549 cells. The NHBE cells were mock-treated or infected with SARS-CoV-2 (USA-WA1/2020), and independent biological duplicates of A549 cells were mock-treated or infected with SARS-CoV-2, human respiratory syncytial virus (RSV), or seasonal influenza A virus (IAV) [24,25]. Following the mapping of the probe to gene IDs, the expression coefficient of variance was calculated to obtain a list with decreasing coefficient of variance for all processed expression data, and the first 4000 genes with a large coefficient of variance expression were selected.
Figure 1.

Flowchart of data preparation, processing, and analysis in this study.
2.2. Identification of Differentially Expressed Genes
Differentially expressed genes (DEGs) were considered by using the DESeq2 package v1.18.1. Genes were considered DEGs when they met the following criteria: FDR < 0.05, and |log2FC| ≥ 2.
2.3. WGCNA Network Construction and Identification of Significant Modules
Gene co-expression network of treated and control groups was reconstructed using WGCNA [26]. Briefly, the gene expression profile matrix was converted into the matrix of pairwise gene similarity according to the Pearson test, followed by conversion into the matrix of adjacency. According to the already represented scale-free gene co-expression topological algorithm, a minimum possible β value was considered so that the adjacency matrix could meet the scale-free topology criteria. For the next step, the topological overlap matrix (TOM) and dissimilarity TOM (dissTOM) were created using TOM similarity and dissimilarity modules. Finally, a module identification was performed by the dynamic tree cut, while the minimum module size was defined as 30. Modules with high similarity scores were merged with a threshold of 0.25. Moreover, the values of gene significance (G.S) were used to calculate the association of individual genes with COVID-19. In addition, the Module Membership (M.M) was defined as the ME correlation as well as the gene expression profile for each module. If the G.S and M.M are strongly associated, it can be stated that the most important (central) elements in the modules are also closely related to the trait. They can also be used to create a network and identify the hub genes.
2.4. Explain the Gene Importance by SP-LIME
Explainable AI (XAI) methods allow us to explain and interpret the result of machine learning models and reveal the importance of features in the model predictions [27,28]. LIME is an XAI algorithm that can explain the predictions of any classifier or regressor in a faithful way by approximating it locally with an interpretable model. Although an explanation of a single prediction provides some understanding of the reliability of the classifier to the user, it is not sufficient to evaluate and assess trust in the model as a whole. An extension of LIME is Submodular Pick LIME (SP-LIME), which selects a set of representative instances with explanations to denote the global importance of features in the explanation space via submodular optimization [29]. Algorithm 1 explains the SP-LIME procedures in more detail.
| Algorithm 1: SP-LIME algorithm. |
| Inputs: : The set of samples : The set of features : The set of instances for explanations
Feature importance Instances that cover the important features |
2.5. Functional Enrichment of Significant Modules
The ClueGO (version 2.2.5) Plug-in tool on Cytoscape (version 3.6.0) was used to identify and visualize the enriched Gene Ontology (GO), KEGG pathway, and biological pathways in interesting gene modules (kappa score = 0.4).
2.6. Hub Gene Detection and Co-Expression Network Reconstruction
Differentially expressed genes with the highest G.S and M.M were chosen as hub genes. Venn diagram was generated using the freely available “Venny” v 2.1 software (http://bioinfogp.cnb.csic.es/tools/venny/, accessed on 7 March 2021). Functional networks were constructed by GeneMANIA (https://genemania.org, accessed on 7 March 2021) and visualized using Cytoscape v 3.0 software [30].
2.7. Evaluation of Selected Hub Genes’ Behavior in Other Virus-Based Infections
To evaluate the expression behavior of selected hub genes in other virus-based infections, including SARS-CoV-2, RSV, IAV, and IAV-ΔNS1, we performed DEG analysis on related datasets from GSE147507.
2.8. Evaluation of Selected Hub Genes’ Behavior in Lung Tissues and Secretions Isolated from COVID-19 Patients
To further characterize selected gene signatures’ expression level changes in respiratory specimens extracted from COVID-19 patients, we performed DEG analysis on related datasets including GSE147507, GSE163426, GSE150316, GSE163151, and GSE154770.
2.9. Evaluation of Selected Hub Genes’ Behavior in COVID-19 Patients with Severe Pneumonia Varied by Viral Load
In order to illustrate the hub gene expression differences between lung samples by viral load strata, we used GSE150316, which included postmortem lung sections of individuals who succumbed to severe SARS-CoV-2 infection with definitive viral load labels. Additionally, transcriptomic changes in selected hub genes during disease resolution and viral clearance were investigated using DEG analysis on GSE154770.
2.10. Identification of Candidate Regulatory Drugs
The firmly established Drug–Gene Interaction Database (DGIDB) (http://www.dgidb.org, accessed on 15 March 2021) was used to predict functional and drug-able hub genes with the list of commercially available or clinical trial drugs [31].
3. Results
3.1. Preprocessing and DEG Analysis
We performed quantile normalization to reduce the effects of technical noises. No outliers were observed in dataset samples by sample clustering. Thus, all samples were included in the analysis (Figure S1). Based on DEG analysis between SARS-CoV-2-treated and mock-treated cell lines, there were a total of 20 genes in A549 cells (all up-regulated) and 42 genes in NHBE cells (34 up-regulated, eight down-regulated) (Figure S2). The DEGs were then selected for subsequent analysis. The GO and KEGG pathway analysis of DEGs is represented in Figure 2.
Figure 2.

DEG characterization. Biological process identification and KEGG analysis of A549 and NHBE DEGs (A549 (A) and NHBE (B) cell lines). ** p-value < 0.01.
3.2. Identification of WGCNA Modules
Based on the variance of expression values, a total of 4000 genes were included in WGCNA. Afterward, the β value equal to 12 and 24 was considered for A549 and NHBE cells, respectively, and a weighted gene co-expression network of treated and mocked samples was reconstructed (Figure S3). The result of the dynamic tree cut gave a total of 20 co-expressed modules recognized by a hierarchical clustering dendrogram with a range size of 30 (mediumpurple2) to 1313 (brown4) in the A549 dataset and 24 co-expressed modules with a range size of 33 (yellow4) to 841 (darkolivegreen) in the NHBE dataset (Figure S4).
3.3. Module–Trait Association Analysis and Functional Annotation Analysis of Interesting Modules
The evaluation of the relationship between each module eigengene and the trait and module–module association recognized the Turquoise module (r = 1, p-value = 1.00 × 10−5) in the A549 cell type (Table S1 and Figures S5 and S6).
Gene Ontology (GO) and functional and KEGG pathway enrichment analysis were conducted to explore the meaningful biological relevance of the interesting modules using ClueGO (Figure 3). As shown in Figure 3, functional annotations of the Turquoise (A549) and Yellow-green (best module of NHBE ((Table S2), Figure S6)) showed mainly similar enrichments and focused on the immune responses against viral infection. Common pathways were: regulation of viral life cycle, negative regulation of viral genome replication, type 1 interferon signaling pathway, NOD-like receptor signaling pathway, defense response to viruses such as influenza A, measles, Herpes simplex 1 virus, and Epstein–Barr virus.
Figure 3.

Biological processes and pathways detected within the (A) Turquoise module from the A549 dataset and (B) Yellow-green module from the NHBE dataset. Gene Ontology and pathway analysis was performed using significant genes across all datasets. Node size represents the counts (number of genes that are part of each pathway), and node color corresponds to the statistical significance. The darker the pathway node, the more statistically significant it is, with a gradient from red (p-value 0.05–0.005) to black (p-value < 0.0005).
3.4. Hub Gene Identification and Network Analysis of Interesting Modules
The correlation between the features (M.M and G.S) of the selected modules led to the detection of interesting hub genes that were highly associated with the pathogenesis of the disease (Figure 4A and Figure 5A). The 15 genes with maximum M.M and G.S scores in each module were then compared to identify feature genes by DEG analysis, and overlapping genes were considered as final hub genes (Figure 4B and Figure 5B). For the Turquoise module, these genes included OAS1 (2′-5′-Oligoadenylate Synthetase 1), OAS3 (2′-5′-Oligoadenylate Synthetase 3), MX1 (Myxovirus (Influenza) Resistance 1), IFIT1 (Interferon Induced Protein with Tetratricopeptide Repeats 1), IFIT3 (Interferon-Induced Protein With Tetratricopeptide Repeats 3), ISG15 (ISG15 Ubiquitin Like Modifier), IFI6 (Interferon alpha-inducible protein 6), DDX60 (DExD/H-Box Helicase 60), IRF9 (Interferon Regulatory Factor 9), and PARP9 (Poly(ADP-Ribose) Polymerase Family Member 9), and for the Yellow-green module IRF9, PGLYRP4 (Peptidoglycan Recognition Protein 4), IL36G (Interleukin 36 Gamma), SAA2 (Serum Amyloid A2), C15orf48 (Chromosome 15 Open Reading Frame 48), TNFAIP3 (TNF Alpha Induced Protein 3), TNIP1 (TNFAIP3 Interacting Protein 1), HEPHL1 (Hephaestin-Like Protein 1), IRAK2 (Interleukin-1 Receptor-Associated Kinase-Like 2), HBEGF (Heparin Binding EGF Like Growth Factor), LIF (LIF Interleukin 6 Family Cytokine), IL1B (Interleukin 1 Beta), and IL-8 (Interleukin 8) were the hub genes that had a high significance for immune-related COVID-19 response. The critical role of these genes in the host response to various other viruses has been described by many studies [32,33,34,35,36,37,38,39,40,41,42].
Figure 4.
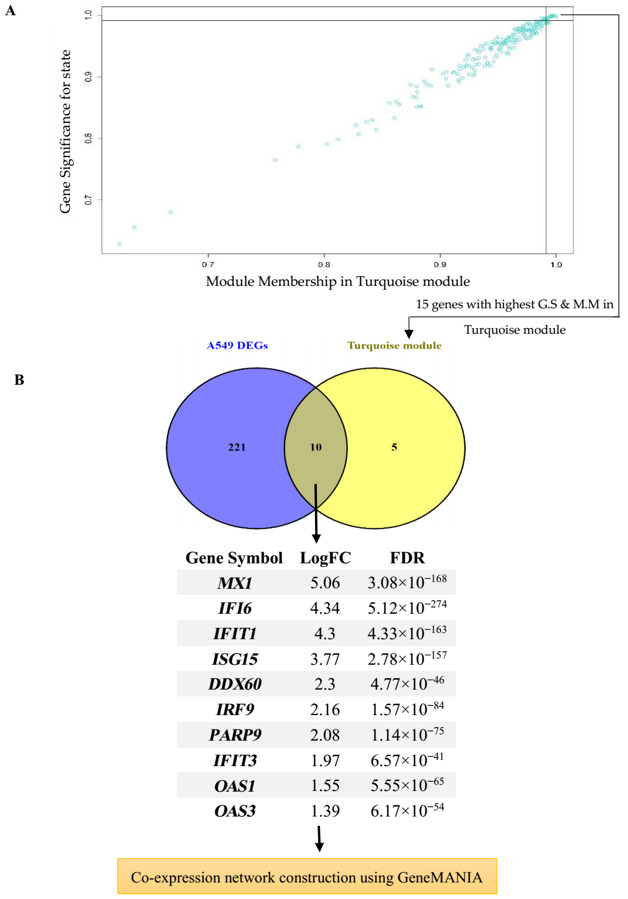
Hub gene detection for the A549 dataset. (A) Turquoise module features of G.S and M.M significantly correlated with the SARS-CoV-2 trait (mock-infected and SARS-CoV-2-infected). Each point represents an individual gene within each module, which is plotted by G.S on the y-axis and M.M on the x-axis; (B) Evaluation of similarities between DEG and hub gene lists using a Venn diagram. Ten genes that were similar in both lists were then imported to GeneMANIA to construct a co-expression network.
Figure 5.

Hub gene detection for NHBE dataset. (A) Yellow-green module features of G.S and M.M significantly correlated with the SARS-CoV-2 trait (mock-infected and SARS-CoV-2-infected). Each point represents an individual gene within each module, which is plotted by G.S on the y-axis and M.M on the x-axis; (B) Evaluation of similarities between DEG and hub gene lists using a Venn diagram. Thirteen genes that were similar in both lists were then imported to GeneMANIA to construct a co-expression network.
3.5. SP-LIME as an Explainable AI Method for Identifying Important Genes
In this paper, we applied the SP-LIME algorithm to detect the importance of genes in the model predictions. We applied multiple classifiers to the collected dataset and ran the SP-LIME algorithm to explain their results. Figure 6 shows the experiments of the SP-LIME algorithm with the random forest classifier, presenting the genes with the highest impact on model prediction. For example, PGLYRP4 and ANXA10 are the most critical genes in the classification process. Interestingly, some feature genes were screened out based on the machine learning algorithm, including IRF9, IFI6, OAS1, PARP9, PGLYRP4, LIF, HEPHL1, and IL8, which were overlapped with identified hub gene candidates, highlighting the potential involvement of these hub genes during COVID-19 pathogenesis. The green and red colors shows the contribution of each gene to have a positive/negative impact for classifying samples towards the COVID-19 state.
Figure 6.

Top genes and their contributions in the model predictions by the SP-LIME algorithm. (A) Turquoise module A549 cell line; (B) Yellow-green module from NHBE cell line.
3.6. Evaluation of Selected Hub Genes’ Behavior in Other Virus-Based Infections
In order to compare differences between SARS-CoV-2, RSV, IAV, and IAV-ΔNS1 molecular pathogenicity, we performed DEG analysis on 22 well-characterized hub genes (10 for A549 cells and 12 for NHBE cells).
As indicated in Table 1 there are no reliable data regarding IAV and IAV-ΔNS1 in the A549 dataset, but expression values in RSV and SARS-CoV-2 showed some critical information. In this regard, eight of ten hub genes in the A549 dataset, including MX1, IFIT1, IFIT3, OAS1, OAS3, ISG15, DDX60, and PARP9, have higher expression values in RSV compared to SARS-CoV-2. On the other hand, two out of ten hub genes, including IFI6 and IRF9, showed higher expression values in SARS-CoV-2 infection.
Table 1.
Comparison of hub gene expression profiles of interesting modules in the response of SARS-CoV-2, RSV, IAV, and IAV-ΔNS1.
| Cell Line | Selected Module | Hub Genes | SARS-CoV-2 | RSV | IAV | IAV-ΔNS1 | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| logFC | p-Value | logFC | p-Value | logFC | p-Value | logFC | p-Value | |||
| A549 | Turquoise | MX1 | 5.06 | 3.08 × 10−168 | 5.54 | 1.76 × 10−110 | −4.46 | ns | - | - |
| IFI6 | 4.29 | 5.12 × 10−274 | 2.92 | 5.97 × 10−43 | −0.57 | ns | - | - | ||
| IFIT1 | 3.76 | 4.33 × 10−163 | 5.09 | 7.98 × 10−105 | 0.03 | ns | - | - | ||
| IFIT3 | 1.97 | 5.28 × 10−44 | 4.99 | 4.88 × 10−101 | 0.34 | ns | - | - | ||
| OAS1 | 1.55 | 2.74 × 10−68 | 3.10 | 8.42 × 10−51 | 0.75 | 0.006989 | - | - | ||
| OAS3 | 1.39 | 3.43 × 10−57 | 2.98 | 1.27 × 10−47 | −0.02 | ns | - | - | ||
| ISG15 | 4.33 | 2.78 × 10−157 | 4.52 | 5.05 × 10−88 | −0.80 | ns | - | - | ||
| DDX60 | 2.30 | 4.77 × 10−46 | 3.10 | 1.61 × 10−38 | −0.06 | ns | - | - | ||
| IRF9 | 2.16 | 1.57 × 10−84 | 1.33 | 1.61 × 10−9 | 0.46 | ns | - | - | ||
| PARP9 | 2.07 | 1.14 × 10−75 | 3.02 | 6.60 × 10−43 | 0.42 | ns | - | - | ||
| NHBE | Yellow-green | IL36G | 2.72 | 1.08 × 10−57 | - | 0.88 | 0.004286 | 4.12 | 7.63 × 10−20 | |
| SAA2 | 2.41 | 1.77 × 10−78 | - | - | 0.74 | 4.02 × 10−7 | 2.69 | 4.02 × 10−07 | ||
| TNIP1 | 1.26 | 1.63 × 10−37 | - | - | 0.01 | ns | 1.46 | 9.28 × 10−8 | ||
| TNFAIP3 | 1.61 | 2.64 × 10−49 | - | - | 1.41 | 4.55 × 10−16 | 4.49 | 1.47 × 10−22 | ||
| HEPHL1 | 1.30 | 2.82 × 10−18 | - | - | 1.45 | 5.47 × 10−8 | 1.56 | 1.86 × 10−5 | ||
| IRAK2 | 1.60 | 2.89 × 10−22 | - | - | 0.97 | 1.25 × 10−5 | 2.86 | 3.05 × 10−23 | ||
| HBEGF | 1.29 | 1.07 × 10−30 | - | - | 1.09 | 0.005477 | 2.07 | 8.60 × 10−24 | ||
| LIF | 1.30 | 4.01 × 10−31 | - | - | 0.17 | ns | 2.89 | 4.10 × 10−9 | ||
| IRF9 | 1.25 | 3.69 × 10−29 | - | - | 1.39 | 9.52 × 10−10 | 2.75 | 3.45 × 10−43 | ||
| IL1B | 1.06 | 7.22 × 10−25 | - | - | −0.04 | ns | 1.24 | 5.98 × 10−9 | ||
| IL8 | 2.32 | 9.20 × 10−96 | - | - | - | - | - | - | ||
| PGLYRP4 | 2.02 | 9.89 × 10−23 | - | - | 1.08 | 0.088695 | 1.98 | 0.004146 | ||
Interestingly, the expression pattern between two lists of SARS-CoV-2 and RSV in A549 cells revealed a diminished anti-viral response to SARS-CoV-2, discriminating this response from common robust interferon induction of anti-viral genes, which had been reported in other studies after viral infections [43]. However, considering obvious suppressed expression levels of IFN-1 and IFN-3 by means of SARS-CoV-2, the underlying mechanism of this unexpected anti-viral response is still to be ascertained. In line with this, IRF9 and IFI6, amongst these hub genes, are very attractive due to their unabated transcriptional response to SARS-CoV-2 when compared to RSV. Considering several recent studies that demonstrated the pivotal role of IRF9 and IFI6 in COVID-19 pathogenesis, these results support a model in which the unique unmuted expression of IRF9 in the response of SARS-CoV-2 infection would be responsible for the observed significant up-regulation of at least a subset of interferon-stimulated genes (ISGs) to escape from robust control over IFN-1 and IFN-3 expression and strengthen the idea that IFI6 up-regulation could be an effective anti-viral strategy to protect healthy respiratory epithelial cells from early apoptosis elicited by surrounding pro-apoptotic cytokines leading to enhanced IFN production [43,44].
The NHBE cell line showed informative data from SARS-CoV-2, IAV, and IAV-ΔNS1 with appropriate p-values. However, no evidence could help to compare the gene expression profile of SARS-CoV-2 and RSV in NHBE cells (Table 1).
In this study, the results showed that the fold change expression of IFI6 in A549 cells was equal to 19.5, with a significant p-value of 5.12 × 10−274. Interestingly, the IFI6 expression value was much lower in cells that were infected with RSV (7.5-fold with a p-value of 5.97 × 10−43) (Table 1).
3.7. Evaluation of Selected Hub Genes’ Behavior in Respiratory Specimens of COVID-19 Patients
The expression patterns of all 22 selected hub genes were investigated in five datasets related to respiratory specimens of COVID-19 patients to understand whether identified transcriptional signatures could be adequately validated by in vitro studies (Figure 7A). Consistent with our in vitro results, most of the selected hub genes displayed significant overexpression in lung and epithelial tissues. Notably, the expression levels of SAA2 and C15orf48 were significantly down-regulated in SARS-CoV-2-infected tracheal aspirate and nasal swab specimens, respectively, which might be candidates for further evaluations. In this regard, recent studies reported an impaired interferon signaling response in tracheal aspirates of patients with severe COVID-19, which may serve as a mechanism underlying reduced SAA2 stimulation in the lower respiratory tract of COVID-19 patients with ARDS [45,46].
Figure 7.

(A) Comparison of the expression level of selected hub genes in respiratory specimens of COVID-19 patients; (B) Evaluation of transcriptional changes in identified hub genes in samples with high versus low SARS-CoV-2 viral load alongside their differential expression over the course of SARS-CoV-2 infection.
3.8. Evaluation of Selected Hub Genes’ Behavior in High and Low Viral Load of Samples from SARS-CoV-2-Infected Patients
As indicated in Figure 7B, these findings further suggest a model of disease evolution in critically ill COVID-19 patients with pneumonia characterized by an early phase of robust IFN host response to the high replication of the virus followed by a later phase of viral clearance with the suppression of IFN signaling, as supported by previous studies [47]. In addition to IFN pathway genes, the higher anti-viral activity of hub genes related to TNF signaling and NAD metabolisms such as TNFAIP3, PARP9, and DTX3L would be able to characterize patients with a high viral load, indicating a clear association of their expression to the presence of the virus and the phase of the disease. Considering the predominant expression of TNFAIP3 in the acute stage as compared to the convalescent stage of SARS-CoV-2 infection, promising repurposable drugs such as methotrexate, ustekinumab, and TNF-α inhibitors targeting TNFAIP3 would likely be more potent in controlling COVID-19 hyper inflammation if administered in the early phase of the disease [48,49,50].
3.9. Drug–Target Network Construction
The two hub gene sets that met the above criteria were then separately imported to the GeneMANIA database to visualize interactions between the genes in each co-expression module. The resulting co-expression networks are visualized as red circles in Figure 8.
Figure 8.

Co-expression network and related drugs of Turquoise (A) and Yellow-green (B) modules.
A drug repositioning analysis was performed on the co-expressed hub genes to identify potential agents suitable for combating COVID-19. As illustrated in Figure 8, hub genes that are a target of detected FDA-approved drugs were ISG15 for the Turquoise module of A549 cells and IL-1β, IL-8, LIF, TNFAIP3, and HBEGF for the Yellow-green module of NHBE cells. However, a large number of these drugs are currently being investigated by several studies to treat SARS-CoV-2 infection [44,46,51,52,53,54,55,56,57,58,59,60]. Thus, we generated a list of 17 novel repurposable drugs that have not yet been explored clinically and need future in vitro and/or in vivo studies to demonstrate their therapeutic properties for the treatment of COVID-19 (Table 2). Overall, our findings provide additional support for drugs that have already reported anti-viral efficacies against COVID-19 and detected promising therapeutic agents that might be potential targets for a closer evaluation.
Table 2.
Novel potent drugs with literature-derived anti-viral evidence for COVID-19 drug repositioning.
| Modules | Genes | Drugs | Drug Class | PMIDs | DGIdb Score | Anti-Viral Properties |
|---|---|---|---|---|---|---|
| Yellow-green module | IL8 | Danazol | Androgen | - | 2 | - |
| IL8 | Cetuximab | Antineoplastic, Anti EGFR | - | 4 | - | |
| IL8 | Aluminum hydroxide | Antacid | - | 2 | - | |
| IL8 | Talc | Sclerosing agents | - | 2 | - | |
| IL8 | Alprazolam | Benzodiazepine | 12218154 | 2 | IAV | |
| IL8 | Pamidronic acid | Bisphosphonate | 21708931 | 2 | IAV | |
| 24154548 | EBV | |||||
| 25220446 | ||||||
| IL1B | Risedronic acid | Bisphosphonate | - | - | ||
| IL1B | Tiludronic acid | Bisphosphonate | - | - | ||
| IL1B | Rilonacept | Interleukin 1 inhibitor | - | - | ||
| IL1B | Pentamidine | Antiprotozoal | - | - | ||
| IL1B | Glucosamine | Non-steroidal anti-inflammatory and antirheumatic | - | - | ||
| IL1B | Ustekinumab | Anti-IL-12 and IL-23 | - | - | ||
| LIF | Capsaicin | Non-narcotic analgesic | 32528827 | - | ||
| 12537981 | - | |||||
| 27574502 | - | |||||
| LIF | Azacitidine | Antineoplastic, cytosine analogue | 32176725 | - | ||
| LIF | Paclitaxel | Antineoplastic, Antimicrotubular | 31797089 | 2 | - | |
| HBEGF | Cetuximab | Antineoplastic, Anti EGFR | - | 4 | - | |
| HBEGF | Panitumumab | Antineoplastic, Anti EGFR | - | - |
PMID: PubMed unique identifier; EBV: Epstein–Barr virus; EGFR: epidermal growth factor receptor; IAV: Influenza A virus.
4. Discussion
The infection of SARS-CoV-2 can cause severe pneumonia as well as other complications with significant morbidity and mortality. Our knowledge about the host’s immune interaction with SARS-CoV-2 is restricted, making it more challenging to manage and develop new therapies. A viral infection typically induces massive changes in the transcriptome of the host, resulting in the aberration of host cells’ metabolism and the modulation of the immune response toward enhancing viral replication [61,62]. Using expression data from human lung epithelial cells, including independent biological triplicates of primary human lung epithelium (NHBE) and transformed lung alveolar (A549) cells, we performed deeper transcriptomic analysis to better understand the molecular basis of COVID-19 and identify putative markers. We found that 20 and 42 genes were differentially expressed (FDR < 0.05, and |log2FC| ≥ 2) when A549 and NHBE samples of COVID-19 cells were compared to mock-treated controls, respectively. GO functional and KEGG pathway enrichment analysis revealed that these genes belong to the type I interferon signaling pathway, IL-17 signaling pathway [63], cytokine-mediated signaling pathway, and defense response to virus categories, all of which play significant roles in restricting viral infection.
It is noteworthy that we observed a significant increase in CCL20 (which displays chemotactic activity for lymphocytes), CXCL8, CXCL2, CXCL3, CXCL5 (as powerful chemoattractants for neutrophils), and CXCL10 (a regulator of leukocyte chemotaxis) in both cells lines studied, suggesting that the presence of these cells is likely to mediate the induction of numerous chemokines and ILs that contribute to the cytokine storm observed in patients with severe COVID-19, requiring intensive care in hospitals [25]. In addition, our data showed a significantly higher expression level of inflammatory mediators consisting of G-CSF, GMCSF, IL-1α, IL-1β, and IL-6 in response to SARS-CoV-2. In parallel, systemic inflammation and cytokine storms with elevated plasma levels of the CXCL family, G-CSF, GMCSF, IL-1β, IL-8, and IL-6 are reported among SARS patients, which are most likely produced by airway epithelial cells as well as a variety of immune cells such as neutrophils, macrophages, lymphocytes, and NK cells [61,64,65,66]. The latest laboratory findings from Wuhan patients also showed that mild COVID-19 patients had elevated levels of IL-1B, IFNγ, CXCL10, and CCL2, whereas in severe cases, G-CSF, CXCL10, CCL2, and CCL3 were elevated [5]. Consistent with our results, a significantly elevated expression of many cytokines was observed in SARS-CoV-2-treated samples compared to mock-treated controls. These findings show that infection with the SARS-CoV-2 virus can result in a cytokine storm that is associated with the severity of the disease. In another study, in patients with SARS disease, IL-6, which is needed to regulate the inflammatory response, B-cell differentiation, and antibody development [67], was increased.
Here, we combined machine learning, WGCNA, as well as DEG analysis to develop a reliable data mining approach to extract or filter valuable targets from the transcriptomic data with high dimension, which leads to the construction of a co-expression network based on the verified hub genes in two independent cell lines. It is noteworthy to highlight that several bioinformatic studies have already analyzed transcriptomic changes in the data of lung cell lines infected with COVID-19 using the GSE147507 dataset to identify key genes involved in COVID-19 that could be used as promising targets for drug repositioning. The dysregulation of a majority of these hub genes was reported in recent bioinformatic studies [36]. However, to our knowledge, only an ordinary DEG analysis has already been applied to this dataset without a cell-specific genome-wide method considering gene–gene connectivity patterns and functional gene significance or employing novel big data analytical techniques, including machine learning, which warrants further studies [58,68,69,70,71]. Moreover, the construction of protein–protein interaction (PPI) networks purely based on online databases like STRING and GeneMania may not accurately reflect the condition-specific interactions of the proteins in a given tissue or under certain disease traits [72].
In this regard, Fagone et al. tried to construct a PPI network based on the most differentially expressed genes using the GeneMania database, which identified MX1, IL8, and IFITM1 as the top hub genes as well as BRDeK23875128, SA-792728, and sirolimus, which are the top three potential predicted drugs targeting DEGs. Inconsistent with our results, the functional enrichment of identified DEGs revealed a specific set of biological pathways, including the IL-17 signaling pathway, cytokine-mediated signaling pathway, and defense response to other organisms [73]. Li et al. performed an integrative DEG analysis of the RNA-seq data from different individual studies of COVID-19, including GSE147507, and identified 233 shared differentially expressed genes as well as LCN2, STAT1, and UBE2L6 genes as novel candidates in COVID-19 pathogenesis [74]. Similar systemic DEG screening analysis has been used to filter significant up- and down-regulated genes as new COVID-19 repurposing opportunities and finally reported more than 50 drug candidates such as Janus kinase and Bruton kinase inhibitors and corticosteroids capable of combating COVID-19 [75]. Another study deployed a bioinformatics workflow detecting a list of DEGs as gene signatures extracted from RNA-seq data of SARS-CoV-2-infected cell lines of tissues to identify potential drug targets as well as disease signatures. The study reported twenty repurposable drug candidates comprising seven drugs currently undergoing trial, such as Cyclosporine, alongside 12 drugs with anti-viral properties with six drugs possessing especial anti-viral properties against the Coronaviruses family [76]. It is worthwhile to note that another sophisticated molecular profiling work provided by a team from the Krogan lab utilized unbiased, global phosphoproteomics approaches to draw a host–viral protein interactions map upon SARS-CoV-2 infection to identify drug targets with therapeutic potential for COVID-19 treatment. They also estimated transcription factor activities from RNA-seq analysis of different SARS-CoV-2-infected human lung cell lines using the GSE147507 dataset and identified that p38/MAPK transcription factors were among the most highly activated transcription factors induced by SARS-CoV-2 infection, as compared with other transcription factors not related to the p38/MAPK pathway. The results showed that the activation of casein kinase II (CK2) and p38 MAPK signaling pathways as a primary host response over the course of SARS-CoV-2 infection causes dramatic phosphorylation changes on host and viral proteins, pro-inflammatory cytokine production, cytoskeleton remodeling and the shutdown of mitotic kinases, resulting in cell cycle arrest to enhance viral replication. Additionally, the study presented 87 drugs and compounds that were reported to target several host kinase activity states in this process, holding promise to possess anti-viral efficacy for COVID-19 therapies [77]. Inconsistent with our results, these findings further highlight the potential anti-viral role of sunitinib for the appropriate balance of the phosphorylation and activation of kinases and pathways, which may target multiple mechanisms hijacked by SARS-CoV-2 infection (Figure 8).
The use of WGCNA for co-expression analysis does not depend on particular contrasts (differential expression). It may establish associations in the study design between the co-expressed genes and essential factors. Based on our analysis of data in the convenient models of bronchial epithelium cells (NHBE and A549 cells), we created the gene co-expression network using WGCNA and recognized two associated modules. The Turquoise module from A549 samples with 196 genes and the Yellow-green module from NHBE samples with 194 genes were the most correlated modules with SARS-CoV-2 infection. KEGG and GO enrichment analysis of these two modules’ genes further confirmed multiple viral infection-related processes, which were both enriched in acute inflammatory response and defense response to virus categories, which are biologically rational and associated with COVID-19 pathogenicity. One of the remarkable pathways enriched in interesting modules is the regulation of the viral life cycle. The genes in this category have a wide range of roles in the SARS-CoV-2 life cycle, such as facilitating survival, attachment, entry, and replicating the virus particles, which are clear candidate targets of anti-viral drugs [78].
Some of the hub genes identified in our study have recently been reported to be particularly related to COVID-19. For example, the functional overexpression of MX1 was a significantly mediated anti-viral response both to SARS-CoV and to SARS-CoV-2 [35,36]. Elevated expression levels of IL-8 and IL-36G have been declared to be closely related to COVID-19 severity [11,79].
Whereas a large number of the identified hub genes had a remarkable enrichment in type I interferon (IFN) pathway genes, PGLYRP4 is known to play a vital IFN-independent role in the regulation of immune responses [80]. PGLYRP4 is a member of the highly conserved human peptidoglycan recognition proteins (PGLYRPs) family, which recognizes not only bacterial peptidoglycan but also shows direct bactericidal activities against a range of Gram-positive and Gram-negative bacteria [81]. In contrast to this well-known canonical function, PGLYRP4 has been conclusively demonstrated to display significant inhibitory effects on the expression of pro-inflammatory mediators and tight junction genes, which attenuates host inflammatory defense, and phagocyte recruitment and activation in the lung result in impaired clearance of bacteria and subsequent bacteremia during pneumococcal pneumonia [82]. Intestinally, it has been shown that PGLYRP4 expression could be stimulated by TLR3 ligand polyI: C as a synthetic viral double-stranded RNA (dsRNA), which suggests a potential immunomodulatory role of PGLYRP4 in response to viral components in addition to its previous anti-bacterial properties [83]. As mentioned earlier, the dysregulation of PGLYRP4 in the NHBC cell line has been reported by previous in silico studies performing DEG analysis on the GSE147507 dataset [84,85,86,87,88]. Intriguingly, a recent in vitro study showed that treatment with an Hsp90 inhibitor, SNX-5422, exerts promising effects in inhibiting SARS-CoV-2 replication as well as regulating the expression of host factors involved in innate immunity such as PGLYRP4. Taken together, these results lead to the hypothesis that PGLYRP4 up-regulation during SARS-CoV-2 infection may be a novel fundamental mechanism underlying the observed early muted immune response that prevents the successful control of viral spread and, hence, an important target for the development of preventive and therapeutic SARS-CoV-2 interventions [89].
However, the anti-inflammatory effects of PGLYRP4 demonstrated significant benefits to reduce early inflammatory processes leading to pulmonary damage and hyper inflammation [81]. Strikingly, PGLYRP4 has been shown to contribute to sphingosine-1-phosphate receptor (S1PR) agonist-mediated disease attenuation during Bordetella pertussis infection, and its beneficial preventive and therapeutic effects on suppressing SARS-CoV-2-related lung damage have been revealed by several studies [90,91,92]. Therefore, these results should be taken with care and warrant further investigations.
HEPHL1 is another interesting hub gene implicated in the regulation of intracellular iron concentrations exerting an oxidoreductase and ferroxidase activity (Fe2+ to Fe3+) at the outer cell membrane [80,93]. The importance of this novel member of the multicopper oxidase family for successful iron homeostasis has been demonstrated with HEPHL1 knockout mouse models as well as the detection of a child with compound heterozygous loss-of-function mutations in HEPHL1 presenting with an abnormal hair phenotype [94]. Considering the fundamental effects of the iron acquisition, transfer, and storage regulating mechanisms in the proper function of diverse cellular proteins and pathways and that preventing the cytotoxicity induced by the donation of electrons to molecular oxygen results in the generation of toxic free radicals, it is not surprising that iron homeostasis is tightly regulated through various control mechanisms. Moreover, imbalanced iron homeostasis with iron overload has been demonstrated to be implicated during infection with various viruses such as SARS-CoV-2, leading to ARDS and pulmonary fibrosis development [95,96]. Notably, the specific role of HEPHL1 in regulating cellular iron efflux is mediated by an enzymatic reaction converting Fe (II) to Fe (III), which requires oxygen consumption as an electron acceptor [94]. Thus, one can speculate that perhaps the persistent up-regulation of HEPHL1 mediated by SARS-CoV-2 replication would participate in gas exchange dysfunction resulting in low oxygen hypoxic conditions and insufficient oxygen supply. Taken together, these findings strengthen the hypothesis that SARS-CoV-2-induced HEPHL1 dysregulation might promote several detrimental effects in SARS-CoV-2-related lung damage, which not only explains the main clinical manifestations of severe COVID-19 patients, including hypoxemia and dyspnea, but also contributes as the possible underlying mechanism of extracellular microenvironment dysregulation and iron accumulation in the lung tissue, which triggers pulmonary fibrosis and lung function decline following SARS-CoV-2 infection.
As shown in Figure 8, further network-based drug repurposing is calculated and novel and potent drugs targeting SARS-CoV-2 are identified. For instance, danazol (antigonadotropic and anti-estrogen) was discovered in our study and other in silico drug screening studies as a repurposable candidate with demonstrated immunomodulatory properties mediating the attenuation of TNFα-induced IL-8 overexpression [97,98]. Additionally, danazol has been shown to be a candidate agent affecting essential pathways involved in SARS-CoV-2 pathogenesis, including mRNA splicing and ubiquitin-mediated proteolysis pathways [98]. As androgens could be suspected as playing a crucial role in driving both specific SARS-CoV-2 entry receptors’ overexpression and anti-viral immune response suppression, therapeutic interventions with danazol must only be administered with great caution and complete consideration [99]. However, the exact mechanisms that may predispose men, especially aged men, to poorer clinical outcomes and higher mortality rates compared with women remain to be delineated. Collectively, these findings suggest a double-sword role of danazol, which dampened the innate immune response against viral infections, resulting in systemic viral spreading with a poor long-term prognosis on one side but preventing hyperinflammatory syndrome characterized by a fulminant and lethal cytokine storm and multiorgan failure on the other side [99].
Although several studies have already been performed to investigate the potential immunoregulatory roles of anti-IL-1 agents such as anakira for treating severe COVID-19, the use of rilonacept as an IL-1 decoy receptor binding to IL-1β and IL-1α has not yet been explored by preclinical and clinical trial studies [100].
Bisphosphonates such as pamidronic acid, risedronic acid, and tiludronic acid are another promising drug group that has been widely utilized to treat osteoporosis, and similar conditions led to increased bone resorption [101]. Treatment with a member of this family, pamidronic acid, can plausibly ameliorate the disease severity and mortality caused by H1N1 and H5N1 viruses as a result of protective γδ T cells’ expansion in a humanized influenza mouse model, which would be repurposed to enhance γδ T cells’ anti-viral function against SARS-CoV-2 [102]. Notably, studies showed that bisphosphonates have unique immunomodulatory properties, which hold promise for their clinical use to address the symptoms resulting from immune response deterioration and systemic hyper-inflammation that occur in severe COVID-19 [103]. Having gained the various anti-SARS-CoV-2 properties of amino-bisphosphonates such as zoledronic acid in γδ T cell immune response stimulation, dendritic cell modulation, and the disruption of SARS-CoV-2-related endosome trafficking, the repositioning of amino-bisphosphonates has been strongly suggested for treating COVID-19 [104]. Taken together, these findings may offer the bisphosphonate family as potential candidates for drug repurposing, and they could prove to be effective additions to the treatment of COVID, warranting further study.
5. Conclusions
To summarize, we used a weighted gene co-expression network analysis to construct a gene co-expression network for COVID-19 and identified a highly correlated hub gene network in A549 and NHBE cell lines. Novel transcriptional signatures of the SARS-CoV-2 virus, including PGLYRP4 and HEPHL1, were detected, and their prospective mechanisms in the pathogenesis of COVID-19 and associated complications were suggested. Additionally, a shortlist of 16 novel candidate repurposable drugs was identified, including drugs that are worthy of further investigation, with the potential to be directly used in clinical trials.
Moving forward, these hub genes may have potential roles in translational medicine and might become promising therapeutic targets.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/jcm10163567/s1, Table S1: Module colors characterization of A549 dataset, Table S2: Module colors characterization of NHBE dataset, Figure S1: Sample clustering to detect outliers, Figure S2: Heatmap of DEGs between mock-treated and SARS-CoV-2-treated cell lines, Figure S3: Selection of the soft-thresholding powers, Figure S4: Cluster dendrogram and module assignment from WGCNA, Figure S5: Module-trait relationship. Each row corresponds to a module eigengene and column corresponds to SARS-CoV-2 trait, Figure S6: Module-module associations. Eigengene dendrogram and Heatmap plot of the adjacencies in the eigengene network including the state represents the relationships among the modules and disease.
Author Contributions
Conceptualization, H.K. and A.D.; formal analysis, H.K., A.D. and M.G.; writing-original draft preparation, M.F., E.M.-M., B.B., N.J.T., S.N., A.G.S., N.S. and A.V.P.; visualization and editing, L.M.M. and S.D.S.; supervision, V.R. and H.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All datasets used in this study have been previously published and are accessible via the Gene Expression Omnibus (GEO) website (https://www.ncbi.nlm.nih.gov/geo/, accessed on 12 February 2021) using the GSE accession numbers listed in the Section 2.
Conflicts of Interest
The authors declare that there is no conflict of interest.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Andersen K.G., Rambaut A., Lipkin W.I., Holmes E.C., Garry R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020;26:450–452. doi: 10.1038/s41591-020-0820-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., Zhang L., Fan G., Xu J., Gu X. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chan J.F.-W., Yuan S., Kok K.-H., To K.K.-W., Chu H., Yang J., Xing F., Liu J., Yip C.C.-Y., Poon R.W.-S., et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: A study of a family cluster. Lancet. 2020;395:514–523. doi: 10.1016/S0140-6736(20)30154-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Young B.E., Ong S.W.X., Kalimuddin S., Low J.G., Tan S.Y., Loh J., Ng O.-T., Marimuthu K., Ang L.W., Mak T.M., et al. Epidemiologic features and clinical course of patients infected with SARS-CoV-2 in Singapore. JAMA. 2020;323:1488–1494. doi: 10.1001/jama.2020.3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arabi Y.M., Fowler R., Hayden F.G. Critical care management of adults with community-acquired severe respiratory viral infection. Intensiv. Care Med. 2020;46:315–328. doi: 10.1007/s00134-020-05943-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cao B., Wang Y., Wen D., Liu W., Wang J., Fan G., Ruan L., Song B., Cai Y., Wei M., et al. A Trial of Lopinavir–Ritonavir in Adults Hospitalized with Severe COVID-19. N. Engl. J. Med. 2020;382:1787–1799. doi: 10.1056/NEJMoa2001282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou P., Yang X.-L., Wang X.-G., Hu B., Zhang L., Zhang W., Si H.-R., Zhu Y., Li B., Huang C.-L., et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fehr A., Channappanavar R., Perlman S. Middle East Respiratory Syndrome: Emergence of a Pathogenic Human Coronavirus. Annu. Rev. Med. 2017;68:387–399. doi: 10.1146/annurev-med-051215-031152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Newton A.H., Cardani A., Braciale T.J. The host immune response in respiratory virus infection: Balancing virus clearance and immunopathology. Semin. Immunopathol. 2016;38:471–482. doi: 10.1007/s00281-016-0558-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xiong Y., Liu Y., Cao L., Wang D., Guo M., Jiang A., Guo D., Hu W., Yang J., Tang Z., et al. Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients. Emerg. Microbes Infect. 2020;9:761–770. doi: 10.1080/22221751.2020.1747363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ren X., Wang S., Chen X., Wei X., Li G., Ren S., Zhang T., Zhang X., Lu Z., You Z. Multiple expression assessments of ACE2 and TMPRSS2 SARS-CoV-2 entry molecules in the urinary tract and their associations with clinical manifestations of COVID-19. Infect. Drug Resist. 2020;13:3977. doi: 10.2147/IDR.S270543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ho J.S.Y., Mok B.W.-Y., Campisi L., Jordan T., Yildiz S., Parameswaran S., Wayman J.A., Gaudreault N.N., Meekins D.A., Indran S.V., et al. Topoisomerase 1 inhibition therapy protects against SARS-CoV-2-induced inflammation and death in animal models. bioRxiv. 2020 doi: 10.1101/2020.12.01.404483. [DOI] [Google Scholar]
- 13.Weingarten-Gabbay S., Klaeger S., Sarkizova S., Pearlman L.R., Chen D.-Y., Bauer M.R., Taylor H.B., Conway H.L., Tomkins-Tinch C.H., Finkel Y., et al. SARS-CoV-2 infected cells present HLA-I peptides from canonical and out-of-frame ORFs. bioRxiv. 2020 doi: 10.1101/2020.10.02.324145. [DOI] [Google Scholar]
- 14.Hoagland D.A., Clarke D.J., Moeller R., Han Y., Yang L., Wojciechowicz M.L., Lachmann A., Oguntuyo K.Y., Stevens C., Lee B., et al. Modulating the transcriptional landscape of SARS-CoV-2 as an effective method for developing antiviral compounds. bioRxiv. 2020 doi: 10.1101/2020.07.12.199687. [DOI] [Google Scholar]
- 15.Daniloski Z., Jordan T.X., Wessels H.-H., Hoagland D.A., Kasela S., Legut M., Maniatis S., Mimitou E.P., Lu L., Geller E., et al. Identification of required host factors for SARS-CoV-2 infection in human cells. Cell. 2021;184:92–105.e16. doi: 10.1016/j.cell.2020.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Y., Liu T., Liu Y., Chen J., Xin B., Wu M., Cui W. Coronary artery disease associated specific modules and feature genes revealed by integrative methods of WGCNA, MetaDE and machine learning. Gene. 2019;710:122–130. doi: 10.1016/j.gene.2019.05.010. [DOI] [PubMed] [Google Scholar]
- 17.Derakhshani A., Hashemzadeh S., Asadzadeh Z., Shadbad M.A., Rasibonab F., Safarpour H., Jafarlou V., Solimando A.G., Racanelli V., Singh P.K., et al. Cytotoxic T-Lymphocyte Antigen-4 in Colorectal Cancer: Another Therapeutic Side of Capecitabine. Cancers. 2021;13:2414. doi: 10.3390/cancers13102414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Malik M., Parikh I., Vasquez J.B., Smith C., Tai L., Bu G., Ladu M.J., Fardo D.W., Rebeck G.W., Estus S. Genetics ignite focus on microglial inflammation in Alzheimer’s disease. Mol. Neurodegener. 2015;10:1–12. doi: 10.1186/s13024-015-0048-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guo S.M., Wang J.X., Li J., Xu F.Y., Wei Q., Wang H.M., Huang H.Q., Zheng S.L., Xie Y.J., Zhang C. Identification of gene expression profiles and key genes in subchondral bone of osteoarthritis using weighted gene coexpression network analysis. J. Cell. Biochem. 2018;119:7687–7695. doi: 10.1002/jcb.27118. [DOI] [PubMed] [Google Scholar]
- 20.Miao L., Yin R.-X., Pan S.-L., Yang S., Yang D.-Z., Lin W.-X. Weighted Gene Co-Expression Network Analysis Identifies Specific Modules and Hub Genes Related to Hyperlipidemia. Cell. Physiol. Biochem. 2018;48:1151–1163. doi: 10.1159/000491982. [DOI] [PubMed] [Google Scholar]
- 21.Langfelder P., Horvath S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Derakhshani A., Mollaei H., Parsamanesh N., Fereidouni M., Miri-Moghaddam E., Nasseri S., Luo Y., Safarpour H., Baradaran B. Gene Co-expression Network Analysis for Identifying Modules and Functionally Enriched Pathways in Vitiligo Disease: A Systems Biology Study. Iran. J. Allergy Asthma Immunol. 2020;19:517–528. doi: 10.18502/ijaai.v19i5.4467. [DOI] [PubMed] [Google Scholar]
- 23.Daamen A.R., Bachali P., Owen K.A., Kingsmore K.M., Hubbard E.L., Labonte A.C., Robl R., Shrotri S., Grammer A.C., Lipsky P.E. Comprehensive transcriptomic analysis of COVID-19 blood, lung, and airway. Sci. Rep. 2021;11:1–19. doi: 10.1038/s41598-021-86002-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blanco-Melo D., Nilsson-Payant B.E., Liu W.-C., Uhl S., Hoagland D., Møller R., Jordan T.X., Oishi K., Panis M., Sachs D., et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell. 2020;181:1036–1045.e9. doi: 10.1016/j.cell.2020.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gentleman R.C., Carey V.J., Bates D.M., Bolstad B., Dettling M., Dudoit S., Ellis B., Gautier L., Ge Y., Gentry J., et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burkart N., Huber M.F. A survey on the explainability of supervised machine learning. J. Artif. Intell. Res. 2021;70:245–317. doi: 10.1613/jair.1.12228. [DOI] [Google Scholar]
- 27.Linardatos P., Papastefanopoulos V., Kotsiantis S. Explainable AI: A Review of Machine Learning Interpretability Methods. Entropy. 2020;23:18. doi: 10.3390/e23010018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ribeiro M.T., Singh S., Guestrin C. “Why should I trust you?” Explaining the predictions of any classifier; Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data mining; San Francisco, CA, USA. 13–17 August 2016; pp. 1135–1144. [Google Scholar]
- 29.Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cao W., Wu W., Yan M., Tian F., Ma C., Zhang Q., Li X., Han P., Liu Z., Gu J., et al. Multiple region whole-exome sequencing reveals dramatically evolving intratumor genomic heterogeneity in esophageal squamous cell carcinoma. Oncogenesis. 2015;4:e175. doi: 10.1038/oncsis.2015.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Asokananthan N., Graham P.T., Fink J., Knight D.A., Bakker A.J., McWilliam A.S., Thompson P.J., Stewart G.A. Activation of Protease-Activated Receptor (PAR)-1, PAR-2, and PAR-4 Stimulates IL-6, IL-8, and Prostaglandin E2Release from Human Respiratory Epithelial Cells. J. Immunol. 2002;168:3577–3585. doi: 10.4049/jimmunol.168.7.3577. [DOI] [PubMed] [Google Scholar]
- 32.Zaas A.K., Chen M., Varkey J., Veldman T., Hero A.O., III, Lucas J., Huang Y., Turner R., Gilbert A., Lambkin-Williams R., et al. Gene expression signatures diagnose influenza and other symptomatic respiratory viral infections in humans. Cell Host Microbe. 2009;6:207–217. doi: 10.1016/j.chom.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fink K., Martin L., Mukawera E., Chartier S., De Deken X., Brochiero E., Miot F., Grandvaux N. IFNβ/TNFα synergism induces a non-canonical STAT2/IRF9-dependent pathway triggering a novel DUOX2 NADPH Oxidase-mediated airway antiviral response. Cell Res. 2013;23:673–690. doi: 10.1038/cr.2013.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wein A.N., Dunbar P.R., McMaster S.R., Li Z.-R.T., Denning T., Kohlmeier J.E. IL-36γ Protects against Severe Influenza Infection by Promoting Lung Alveolar Macrophage Survival and Limiting Viral Replication. J. Immunol. 2018;201:573–582. doi: 10.4049/jimmunol.1701796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leong W., Tan H., Ooi E.E., Koh D., Chow V.T. Microarray and real-time RT-PCR analyses of differential human gene expression patterns induced by severe acute respiratory syndrome (SARS) coronavirus infection of Vero cells. Microbes Infect. 2005;7:248–259. doi: 10.1016/j.micinf.2004.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pillai P.S., Molony R.D., Martinod K., Dong H., Pang I.K., Tal M., Solis A.G., Bielecki P., Mohanty S., Trentalange M., et al. Mx1 reveals innate pathways to antiviral resistance and lethal influenza disease. Science. 2016;352:463–466. doi: 10.1126/science.aaf3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kroeker A. Ph.D. Thesis. University of Manitoba; Winnipeg, MB, Canada: 2012. A Proteomic Approach to Discovering Novel Anti-Influenza Mechanisms in Primary Human Airway Epithelial Cells. [Google Scholar]
- 38.Barik S. Challenges and Opportunities for Respiratory Syncytial Virus Vaccines. Springer; Berlin/Heidelberg, Germany: 2013. Respiratory syncytial virus mechanisms to interfere with type 1 interferons; pp. 173–191. [DOI] [PubMed] [Google Scholar]
- 39.Imajoh M., Hashida Y., Murakami M., Maeda A., Sato T., Fujieda M., Wakiguchi H., Daibata M. Characterization of Epstein–Barr virus (EBV) BZLF1 gene promoter variants and comparison of cellular gene expression profiles in Japanese patients with infectious mononucleosis, chronic active EBV infection, and EBV-associated hemophagocytic lymphohistiocytosis. J. Med. Virol. 2012;84:940–946. doi: 10.1002/jmv.23299. [DOI] [PubMed] [Google Scholar]
- 40.Zhang Y., Mao D., Roswit W.T., Jin X., Patel A.C., Patel D.A., Agapov E., Wang Z., Tidwell R.M., Atkinson J.J., et al. PARP9-DTX3L ubiquitin ligase targets host histone H2BJ and viral 3C protease to enhance interferon signaling and control viral infection. Nat. Immunol. 2015;16:1215–1227. doi: 10.1038/ni.3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tatebe K., Zeytun A., Ribeiro R.M., Hoffmann R., Harrod K.S., Forst C.V. Response network analysis of differential gene expression in human epithelial lung cells during avian influenza infections. BMC Bioinform. 2010;11:170. doi: 10.1186/1471-2105-11-170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blanco-Melo D., Nilsson-Payant B., Liu W.-C., Møller R., Panis M., Sachs D., Albrecht R.A., tenOever B.R. SARS-CoV-2 launches a unique transcriptional signature from in vitro, ex vivo, and in vivo systems. bioRxiv. 2020 doi: 10.1101/2020.03.24.004655. [DOI] [Google Scholar]
- 43.Xu Z., Shi L., Wang Y., Zhang J., Huang L., Zhang C., Liu S., Zhao P., Liu H., Zhu L., et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020;8:420–422. doi: 10.1016/S2213-2600(20)30076-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jumeau C., Awad F., Assrawi E., Cobret L., Duquesnoy P., Giurgea I., Valeyre D., Grateau G., Amselem S., Bernaudin J.-F., et al. Expression of SAA1, SAA2 and SAA4 genes in human primary monocytes and monocyte-derived macrophages. PLoS ONE. 2019;14:e0217005. doi: 10.1371/journal.pone.0217005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sarma A., Christenson S., Mick E., Deiss T., DeVoe C., Pisco A., Ghale R., Jauregui A., Byrne A., Moazed F., et al. COVID-19 ARDS is characterized by a dysregulated host response that differs from cytokine storm and is modified by dexamethasone. Res. Sq. 2021 doi: 10.21203/rs.3.rs-141578/v1. [DOI] [Google Scholar]
- 46.Desai N., Neyaz A., Szabolcs A., Shih A.R., Chen J.H., Thapar V., Nieman L.T., Solovyov A., Mehta A., Lieb D.J., et al. Temporal and spatial heterogeneity of host response to SARS-CoV-2 pulmonary infection. Nat. Commun. 2020;11:1–15. doi: 10.1038/s41467-020-20139-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hemmat N., Asadzadeh Z., Ahangar N.K., Alemohammad H., Najafzadeh B., Derakhshani A., Baghbanzadeh A., Baghi H.B., Javadrashid D., Najafi S., et al. The roles of signaling pathways in SARS-CoV-2 infection; lessons learned from SARS-CoV and MERS-CoV. Arch. Virol. 2021;166:675–696. doi: 10.1007/s00705-021-04958-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Frohman E.M., Villemarette-Pittman N.R., Cruz R.A., Longmuir R., Rowe V., Rowe E.S., Varkey T.C., Steinman L., Zamvil S.Z., Frohman T.C. Part II. High-dose methotrexate with leucovorin rescue for severe COVID-19: An immune stabilization strategy for SARS-CoV-2 induced ‘PANIC’ attack. J. Neurol. Sci. 2020;415:116935. doi: 10.1016/j.jns.2020.116935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stegmann K.M., Dickmanns A., Gerber S., Nikolova V., Klemke L., Manzini V., Schloesser D., Bierwirth C., Freund J., Sitte M., et al. The folate antagonist methotrexate diminishes replication of the coronavirus SARS-CoV-2 and enhances the antiviral efficacy of remdesivir in cell culture models. bioRxiv. 2020 doi: 10.1101/2020.07.18.210013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee V.S., Chong W.L., Sukumaran S.D., Nimmanpipug P., Letchumanan V., Goh B.H., Lee L.-H., Zain S.M., Abd Rahman N. Computational screening and identifying binding interaction of anti-viral and anti-malarial drugs: Toward the potential cure for SARS-CoV-2. Prog. Drug Discov. Biomed. Sci. 2020;3 doi: 10.36877/pddbs.a0000065. [DOI] [Google Scholar]
- 51.Galvez J., Zanni R., Galvez-Llompart M. Drugs Repurposing for Coronavirus Treatment: Computational Study Based On Molecular Topology. Nereis. 2020:15–18. doi: 10.46583/nereis_2020.12.591. [DOI] [Google Scholar]
- 52.Chang Y., Tung Y., Lee K., Chen T., Hsiao Y., Chang H., Hsieh T., Su C., Wang S., Yu J., et al. Potential Therapeutic Agents for COVID-19 Based on the Analysis of Protease and RNA Polymerase Docking. Preprints. 2020 doi: 10.20944/preprints202002.0242.v2. [DOI] [Google Scholar]
- 53.Contini A. Virtual screening of an FDA approved drugs database on two COVID-19 coronavirus proteins. Am. Chem. S. 2020 doi: 10.26434/chemrxiv.11847381.v1. [DOI] [Google Scholar]
- 54.Cava C., Bertoli G., Castiglioni I. In Silico Discovery of Candidate Drugs against COVID-19. Viruses. 2020;12:404. doi: 10.3390/v12040404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jeon S., Ko M., Lee J., Choi I., Byun S.Y., Park S., Shum D., Kim S. Identification of antiviral drug candidates against SARS-CoV-2 from FDA-approved drugs. Antimicrob. Agents Chemother. 2020;64 doi: 10.1128/AAC.00819-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang R., Wang X., Ni L., Di X., Ma B., Niu S., Liu C., Reiter R.J. COVID-19: Melatonin as a potential adjuvant treatment. Life Sci. 2020;250:117583. doi: 10.1016/j.lfs.2020.117583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu X., Li Z., Liu S., Chen Z., Zhao Z., Huang Y.-Y., Zhang Q., Wang J., Shi Y., Xu Y., et al. Therapeutic effects of dipyridamole on COVID-19 patients with coagulation dysfunction. medRxiv. 2020 doi: 10.1101/2020.02.27.20027557. [DOI] [Google Scholar]
- 58.Dong L., Hu S., Gao J. Discovering drugs to treat coronavirus disease 2019 (COVID-19) Drug Discov. Ther. 2020;14:58–60. doi: 10.5582/ddt.2020.01012. [DOI] [PubMed] [Google Scholar]
- 59.Zhang R., Li Y., Pan B., Li Y., Liu A., Li X. Increased expression of hub gene CXCL10 in peripheral blood mononuclear cells of patients with systemic lupus erythematosus. Exp. Ther. Med. 2019;18:4067–4075. doi: 10.3892/etm.2019.8013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Channappanavar R., Fehr A.R., Vijay R., Mack M., Zhao J., Meyerholz D.K., Perlman S. Dysregulated type I interferon and inflammatory monocyte-macrophage responses cause lethal pneumonia in SARS-CoV-infected mice. Cell Host Microbe. 2016;19:181–193. doi: 10.1016/j.chom.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Thaker S.K., Ch’Ng J., Christofk H.R. Viral hijacking of cellular metabolism. BMC Biol. 2019;17:59. doi: 10.1186/s12915-019-0678-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pacha O., Sallman M.A., Evans S.E. COVID-19: A case for inhibiting IL-17? Nat. Rev. Immunol. 2020;20:345–346. doi: 10.1038/s41577-020-0328-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lu L., Zhang H., Zhan M., Jiang J., Yin H., Dauphars D.J., Li S.-Y., Li Y., He Y.-W. Preventing Mortality in COVID-19 Patients: Which Cytokine to Target in a Raging Storm? Front. Cell Dev. Biol. 2020;8:677. doi: 10.3389/fcell.2020.00677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Okabayashi T., Kariwa H., Yokota S.-I., Iki S., Indoh T., Yokosawa N., Takashima I., Tsutsumi H., Fujii N. Cytokine regulation in SARS coronavirus infection compared to other respiratory virus infections. J. Med. Virol. 2006;78:417–424. doi: 10.1002/jmv.20556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hemmat N., Derakhshani A., Baghi H.B., Silvestris N., Baradaran B., De Summa S. Neutrophils, Crucial, or Harmful Immune Cells Involved in Coronavirus Infection: A Bioinformatics Study. Front. Genet. 2020;11:641. doi: 10.3389/fgene.2020.00641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang Y., Li J., Zhan Y., Wu L., Yu X., Zhang W., Ye L., Xu S., Sun R., Wang Y., et al. Analysis of Serum Cytokines in Patients with Severe Acute Respiratory Syndrome. Infect. Immun. 2004;72:4410–4415. doi: 10.1128/IAI.72.8.4410-4415.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xie T., Han M., Su X., Li H., Chen J., Guo X. Identification of Hub genes associated with infection of three lung cell lines by SARS-CoV-2 with integrated bioinformatics analysis. J. Cell. Mol. Med. 2020;24:12225–12230. doi: 10.1111/jcmm.15862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cheng L.-C., Kao T.-J., Phan N.N., Chiao C.-C., Yen M.-C., Chen C.-F., Hung J.-H., Jiang J.-Z., Sun Z., Wang C.-Y. Novel signaling pathways regulate SARS-CoV and SARS-CoV-2 infectious disease. Medicine. 2021;100:e24321. doi: 10.1097/MD.0000000000024321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fang C., Mei J., Tian H., Liou Y.-L., Rong D., Zhang W., Liao Q., Wu N. CSF3 Is a Potential Drug Target for the Treatment of COVID-19. Front. Physiol. 2021;11:605792. doi: 10.3389/fphys.2020.605792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cavalli E., Petralia M.C., Basile M.S., Bramanti A., Bramanti P., Nicoletti F., Spandidos D.A., Shoenfeld Y., Fagone P. Transcriptomic analysis of COVID-19 lungs and bronchoalveolar lavage fluid samples reveals predominant B cell activation responses to infection. Int. J. Mol. Med. 2020;46:1266–1273. doi: 10.3892/ijmm.2020.4702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sevimoglu T., Arga K.Y. The role of protein interaction networks in systems biomedicine. Comput. Struct. Biotechnol. J. 2014;11:22–27. doi: 10.1016/j.csbj.2014.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fagone P., Ciurleo R., Lombardo S.D., Iacobello C., Palermo C.I., Shoenfeld Y., Bendtzen K., Bramanti P., Nicoletti F. Transcriptional landscape of SARS-CoV-2 infection dismantles pathogenic pathways activated by the virus, proposes unique sex-specific differences and predicts tailored therapeutic strategies. Autoimmun. Rev. 2020;19:102571. doi: 10.1016/j.autrev.2020.102571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li G., Wang J., He X., Zhang L., Ran Q., Xiong A., Wu D., Hu L., Song Q., Zhu D. An integrative analysis identifying transcriptional features and key genes involved in COVID-19. Epigenomics. 2020;12:1969–1981. doi: 10.2217/epi-2020-0168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.El-Hachem N., Eid E., Nemer G., Dbaibo G., Abbas O., Rubeiz N., Zeineldine S., Matar G.M., Bikorimana J.-P., Shammaa R., et al. Integrative Transcriptome Analyses Empower the Anti-COVID-19 Drug Arsenal. iScience. 2020;23:101697. doi: 10.1016/j.isci.2020.101697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.O’Donovan S.M., Imami A., Eby H., Henkel N.D., Creeden J.F., Asah S., Zhang X., Wu X., Alnafisah R., Taylor R.T., et al. Identification of candidate repurposable drugs to combat COVID-19 using a signature-based approach. Sci. Rep. 2021;11:1–12. doi: 10.1038/s41598-021-84044-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bouhaddou M., Memon D., Meyer B., White K.M., Rezelj V.V., Marrero M.C., Polacco B.J., Melnyk J.E., Ulferts S., Kaake R.M., et al. The global phosphorylation landscape of SARS-CoV-2 infection. Cell. 2020;182:685–712.e19. doi: 10.1016/j.cell.2020.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pinto B., Oliveira A.E.R., Singh Y., Jimenez L., Gonçalves A.N.A., Ogava R.L.T., Creighton R., Peron J.P.S., I Nakaya H. ACE2 Expression Is Increased in the Lungs of Patients With Comorbidities Associated With Severe COVID-19. J. Infect. Dis. 2020;222:556–563. doi: 10.1093/infdis/jiaa332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gong J., Dong H., Xia S.Q., Huang Y.Z., Wang D., Zhao Y., Liu W., Tu S., Zhang M., Wang Q., et al. Correlation Analysis Between Disease Severity and Inflammation-related Parameters in Patients with COVID-19 Pneumonia. medRxiv. 2020 doi: 10.1101/2020.02.25.20025643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Belinky F., Nativ N., Stelzer G., Zimmerman S., Stein T.I., Safran M., Lancet R. PathCards: Multi-source consolidation of human biological pathways. Database. 2015;2015:bav006. doi: 10.1093/database/bav006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Skerry C., Goldman W.E., Carbonetti N.H. Peptidoglycan Recognition Protein 4 Suppresses Early Inflammatory Responses to Bordetella pertussis and Contributes to Sphingosine-1-Phosphate Receptor Agonist-Mediated Disease Attenuation. Infect. Immun. 2019;87:87. doi: 10.1128/IAI.00601-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dabrowski A.N., Shrivastav A., Conrad C., Komma K., Weigel M., Dietert K., Gruber A.D., Bertrams W., Wilhelm J., Schmeck B., et al. Peptidoglycan Recognition Protein 4 Limits Bacterial Clearance and Inflammation in Lungs by Control of the Gut Microbiota. Front. Immunol. 2019;10:2106. doi: 10.3389/fimmu.2019.02106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ma P., Wang Z., Pflugfelder S.C., Li D.-Q. Toll-like receptors mediate induction of peptidoglycan recognition proteins in human corneal epithelial cells. Exp. Eye Res. 2010;90:130–136. doi: 10.1016/j.exer.2009.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Moni M.A., Quinn J.M., Sinmaz N., Summers M.A. Gene expression profiling of SARS-CoV-2 infections reveal distinct primary lung cell and systemic immune infection responses that identify pathways relevant in COVID-19 disease. Brief. Bioinform. 2021;22:1324–1337. doi: 10.1093/bib/bbaa376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chandrashekar D.S., Manne U., Varambally S. Comparative transcriptome analyses reveal genes associated with SARS-CoV-2 infection of human lung epithelial cells. bioRxiv. 2020 doi: 10.1101/2020.06.24.169268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Al Heialy S., Hachim M.Y., Senok A., Gaudet M., Tayoun A.A., Hamoudi R., Alsheikh-Ali A., Hamid Q. Regulation of Angiotensin- Converting Enzyme 2 in Obesity: Implications for COVID-19. Front. Physiol. 2020;11:11. doi: 10.3389/fphys.2020.555039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Karakurt H.U., Pınar P. Integration of transcriptomic profile of SARS-CoV-2 infected normal human bronchial epi-thelial cells with metabolic and protein-protein interaction networks. Turk. J. Biol. 2020;44:168. doi: 10.3906/biy-2005-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kang K., Kim H.H., Choi Y. Tiotropium Is Predicted to Be a Promising Drug for COVID-19 Through Transcriptome-Based Comprehensive Molecular Pathway Analysis. Viruses. 2020;12:776. doi: 10.3390/v12070776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Goswami R., Russell V.S., Tu J.J., Hughes P.F., Kelly F., Langel S.N., Steppe J., Palmer S.M., Haystead T., Blasi M., et al. Oral Hsp90 inhibitor, SNX-5422, attenuates SARS-CoV-2 replication and dampens inflammation in airway cells. SSRN. 2021 doi: 10.2139/ssrn.3803615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Catanzaro M., Fagiani F., Racchi M., Corsini E., Govoni S., Lanni C. Immune response in COVID-19: Addressing a phar-macological challenge by targeting pathways triggered by SARS-CoV-2. Signal Transduct. Target. Ther. 2020;5:1–10. doi: 10.1038/s41392-020-0191-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Meacci E., Garcia-Gil M., Pierucci F. SARS-CoV-2 infection: A role for S1P/S1P receptor signaling in the nervous system? Int. J. Mol. Sci. 2020;21:6773. doi: 10.3390/ijms21186773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zrzavy T., Wimmer I., Rommer P.S., Berger T. Immunology of COVID-19 and disease-modifying therapies: The good, the bad and the unknown. Eur. J. Neurol. 2020 doi: 10.1111/ene.14578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cronin S.J.F., Woolf C.J., Weiss G., Penninger J.M. The Role of Iron Regulation in Immunometabolism and Immune-Related Disease. Front. Mol. Biosci. 2019;6:116. doi: 10.3389/fmolb.2019.00116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sharma P., Reichert M., Lu Y., Markello T.C., Adams D.R., Steinbach P.J., Fuqua B.K., Parisi X., Kaler S.G., Vulpe C.D., et al. Biallelic HEPHL1 variants impair ferroxidase activity and cause an abnormal hair phenotype. PLoS Genet. 2019;15:e1008143. doi: 10.1371/journal.pgen.1008143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dalamaga M., Karampela I., Mantzoros C.S. Commentary: Could iron chelators prove to be useful as an adjunct to COVID-19 Treatment Regimens? Metabolism. 2020;108:154260. doi: 10.1016/j.metabol.2020.154260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Drakesmith H., Prentice A. Viral infection and iron metabolism. Nat. Rev. Microbiol. 2008;6:541–552. doi: 10.1038/nrmicro1930. [DOI] [PubMed] [Google Scholar]
- 96.Horie S., Harada T., Mitsunari M., Taniguchi F., Iwabe T., Terakawa N. Progesterone and progestational compounds attenuate tumor necrosis factor alpha–induced interleukin-8 production via nuclear factor kappaB inactivation in endometriotic stromal cells. Fertil. Steril. 2005;83:1530–1535. doi: 10.1016/j.fertnstert.2004.11.042. [DOI] [PubMed] [Google Scholar]
- 97.Barh D., Tiwari S., Weener M.E., Azevedo V., Góes-Neto A., Gromiha M.M., Ghosh P. Multi-omics-based identification of SARS-CoV-2 infection biology and candidate drugs against COVID-19. Comput. Biol. Med. 2020;126:104051. doi: 10.1016/j.compbiomed.2020.104051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Xu Y., Liu S., Zhang Y., Zhi Y. Does hereditary angioedema make COVID-19 worse? World Allergy Organ. J. 2020;13:100454. doi: 10.1016/j.waojou.2020.100454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Soy M., Keser G., Atagündüz P., Tabak F., Atagündüz I., Kayhan S. Cytokine storm in COVID-19: Pathogenesis and overview of anti-inflammatory agents used in treatment. Clin. Rheumatol. 2020;39:2085–2094. doi: 10.1007/s10067-020-05190-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cremers S., Papapoulos S. Pharmacology of bisphosphonates. Bone. 2011;49:42–49. doi: 10.1016/j.bone.2011.01.014. [DOI] [PubMed] [Google Scholar]
- 101.Yazdanifar M., Mashkour N., Bertaina A. Making a case for using γδ T cells against SARS-CoV-2. Crit. Rev. Microbiol. 2020;46:689–702. doi: 10.1080/1040841X.2020.1822279. [DOI] [PubMed] [Google Scholar]
- 102.Fujimura T., Kambayashi Y., Furudate S., Kakizaki A., Aiba S. Immunomodulatory Effect of Bisphosphonate Risedronate Sodium on CD163+ Arginase 1+ M2 Macrophages: The Development of a Possible Supportive Therapy for Angiosarcoma. Clin. Dev. Immunol. 2013;2013:1–7. doi: 10.1155/2013/325412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Brufsky A., Marti J.L.G., Nasrazadani A., Lotze M.T. Boning up: Amino-bisphophonates as immunostimulants and endosomal disruptors of dendritic cell in SARS-CoV-2 infection. J. Transl. Med. 2020;18:1–6. doi: 10.1186/s12967-020-02433-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Karami H., Derakhshani A., Fereidouni M., Miri-Moghaddam E., Baradaran B., Silvestris N., Paradiso A.V., Safarpour H., De Summa S. Transcriptional analysis of lung epithelial cells using WGCNA revealed the role of IRF9 and IFI6 genes in SARS-CoV-2 pathogenicity. Res. Sq. 2020 doi: 10.21203/rs.3.rs-31167/v1. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All datasets used in this study have been previously published and are accessible via the Gene Expression Omnibus (GEO) website (https://www.ncbi.nlm.nih.gov/geo/, accessed on 12 February 2021) using the GSE accession numbers listed in the Section 2.