

SUMMARY
Cancer relapse begins when malignant cells pass through the extreme metabolic bottleneck of stress from chemotherapy and the byproducts of the massive cell death in the surrounding region. In acute myeloid leukemia, complete remissions are common, but few are cured. We tracked leukemia cells in vivo, defined the moment of maximal response following chemotherapy, captured persisting cells, and conducted unbiased metabolomics, revealing a metabolite profile distinct from the pre-chemo growth or post-chemo relapse phase. Persisting cells used glutamine in a distinctive manner, preferentially fueling pyrimidine and glutathione generation, but not the mitochondrial tricarboxylic acid cycle. Notably, malignant cell pyrimidine synthesis also required aspartate provided by specific bone marrow stromal cells. Blunting glutamine metabolism or pyrimidine synthesis selected against residual leukemia-initiating cells and improved survival in leukemia mouse models and patient-derived xenografts. We propose that timed cell-intrinsic or niche-focused metabolic disruption can exploit a transient vulnerability and induce metabolic collapse in cancer cells to overcome chemoresistance.
Graphical Abstract
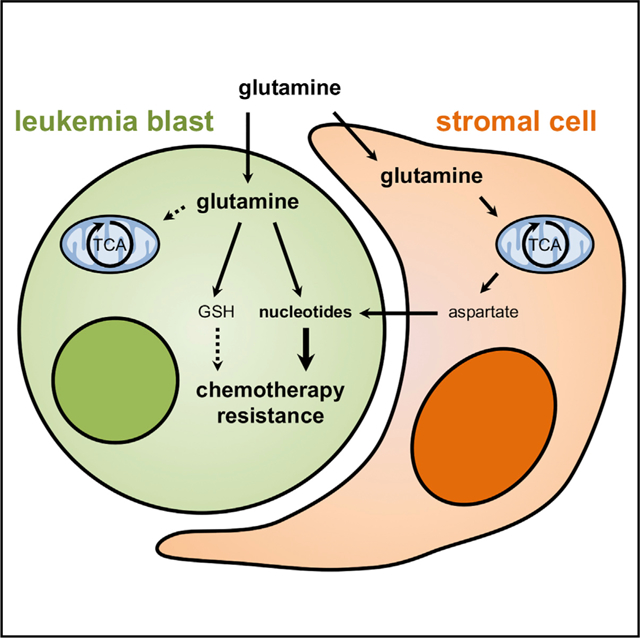
In Brief
Chemotherapy effectively induces remission in acute myeloid leukemia, yet in most patients, a few highly adaptable cells survive and cause relapse. van Gastel et al. show that residual leukemia cells exhibit transient metabolic adaptations enabling their survival. Timed targeting of these programs provokes a metabolic collapse and overcomes chemoresistance.
INTRODUCTION
Per evolutionary biology, when a population is subjected to a stress event or “bottleneck,” only the individuals that have the ability to adapt will survive. Populations of cells behave similarly, as Peter C. Nowell proposed in 1976 to explain clonal evolution in cancer (Nowell, 1976). Clonal evolution has been studied particularly well in acute myeloid leukemia (AML), a highly lethal hematopoietic cancer where residual clones persist through therapy and ultimately cause relapse (Ding et al., 2012a). Yet, genetic analysis has provided only limited insight into chemo-therapy resistance commonly observed in AML, and driver mutations are frequently not amenable to targeted therapies (Magee, 2017). We and others have shown that AML cells have distinctive metabolic dependencies compared with their normal counterparts, often irrespective of the driving mutations (Chen et al., 2016; German et al., 2016; Jacque et al., 2015; Lagadinou et al., 2013; Ni et al., 2019; Pei et al., 2013; Sykes et al., 2016; Wang et al., 2014). Metabolic networks also endow cells with evolutionary-ancient stress-protection systems that are independent of transcriptional changes, providing a first-line defense mechanism to cope with exogenous pressure (Keller et al., 2017; Kültz, 2005; Naviaux, 2014).
We hypothesized that the stress of chemotherapy imposes an extreme metabolic bottleneck through which residual cells must pass to cause relapse. That stress state is transient, represented by the challenges of chemotherapeutic agents and degradation products from massive cell death in the local milieu. It will not be reflected in relapse cells at the time cancer reemergence is clinically evident, since much of the local environment and intracellular imbalances will have re-equilibrated. Rather, it can only be observed in cells extracted from the in vivo context where maximal cell death has occurred and chemotherapy by products are being cleared. This transient moment of stress that drives initial leukemia regeneration has been described in patients as well as patient-derived xenografts and is characterized by the emergence of leukemia-regenerating cells that give rise to relapse (Boyd et al., 2018; Farge et al., 2017). Defining the metabolic dependencies of these persisting cells may reveal vulnerabilities that can be therapeutically exploited. We sought to determine whether timed, targeted perturbations can induce the metabolic collapse of residual malignant cells to overcome chemoresistance.
RESULTS
AML Cells Exhibit Transient Metabolic Changes in Response to Chemotherapy
The point of maximal metabolic stress can be functionally identified as the “point of maximal response” after chemotherapy since thereafter resistant cells would have passed through the bottleneck and begun the regrowth that characterizes relapse. To determine this moment, we used a triple transgenic mouse model in which cells express MLL-AF9, driving AML development, as well as luciferase and GFP (Figure S1A). We transplanted culture-expanded bone marrow cells derived from terminally ill primary mice into secondary wild-type recipients, leading to the establishment of a fast-developing disease that can be monitored through bioluminescence imaging (Figure S1B). We treated mice with an induction chemotherapy (iCT) regimen that closely mimics the one used in clinical care (cytarabine for 5 days + doxorubicin for the first 3 days) or with vehicle, followed disease progression, and discovered that the moment of maximal response occurs approximately 3 days after the last dose of chemotherapy (Figures S1B and S1C).
We then developed a methodology to investigate the metabolic profile of freshly isolated AML cells. We isolated bone marrow of mice subjected to vehicle treatment (day 3 after vehicle), at the time of maximal response after iCT (day 3 after iCT) or after relapse (day 10 after iCT), sorted GFP+ AML cells using fluorescence-activated cell sorting (FACS), and extracted polar metabolites (Figure 1A). Samples were kept at 4°C at all times to minimize metabolic disturbance (Dietmair et al., 2010). The total time for cell processing and sorting was 45 min, kept the same for all samples. We examined the metabolite profile effects of subjecting the cells to FACS (Figure S1D). While many metabolites showed a good correlation between sorted and un-sorted cells, others did change substantially, indicating that some pathways may be more prone to processing artifacts than others. The levels of intracellular metabolites in 25,000 sorted cells were then determined by untargeted metabolomics analysis. We measured 654 metabolites, of which 129 were identified based on their mass spectra using the mzCloud database and another 211 could be putatively identified using their molecular weight by searching the Human Metabolome database and Kyoto Encyclopedia of Genes and Genomes (KEGG) Compound database (Table S1). The 340 putatively identified metabolites were then further analyzed using the MetaboAnalyst software. Principal component analysis showed that while groups clustered closely together, the iCT group separated from the others (Figure S1E). Statistically, 125 metabolites differed significantly between the groups (ANOVA, FDR < 0.01; Table S2), and unsupervised clustering of samples based on the 75 most significantly different metabolites revealed the specific metabolic signature of each group (Figure 1B).
Figure 1. AML Cells Exhibit Transient Metabolic Changes in Response to Chemotherapy.
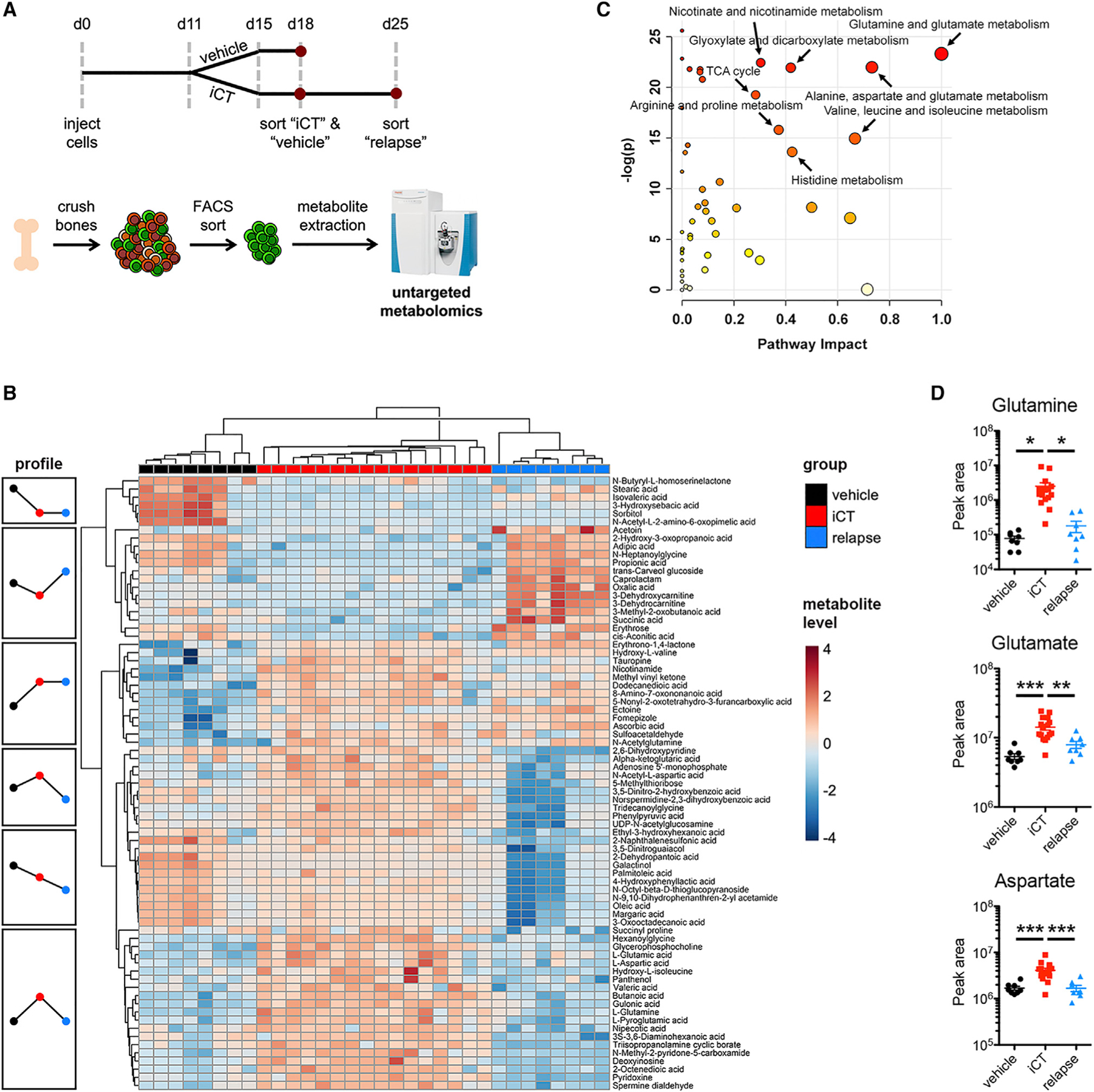
(A) Schematic overview of the experimental design. iCT, induction chemotherapy (5 days of cytarabine 100 mg/kg + 3 days of doxorubicin 3 mg/kg i.p.).
(B) Heatmap of the metabolic profile of MLL-AF9 AML cells obtained from mice with vehicle treatment, at 3 days after iCT (iCT group; representing the moment of maximal response) or at 10 days after iCT (relapse).
(C) Metabolic Pathway Enrichment Analysis using putatively identified metabolites of the iCT versus vehicle and relapse groups. Pathway impact is a measure for the percentage of metabolites that were measured in a given pathway, as well as their relative importance in that pathway.
(D) Levels of glutamine, glutamate, and aspartate in vehicle, iCT, or relapse MLL-AF9 AML cells as measured by untargeted metabolomics. Data are represented as mean ± standard error of the mean (SEM). *p < 0.05, **p < 0.01, ***p < 0.001. See also Figure S1.
Metabolic pathway enrichment analysis revealed the related “glutamine and glutamate metabolism” and “alanine, aspartate, and glutamate metabolism” as the top pathways distinguishing iCT-treated cells from vehicle-treated and relapsed cells (Figure 1C). Glutamine, glutamate, and aspartate, central metabolites in these pathways, are low in vehicle-treated cells, high in the residual cells at the moment of maximal response to iCT, and low again after relapse (Figure 1D), suggesting a role in the immediate stress response to chemotherapy. Tricarboxylic acid (TCA) cycle metabolites, lactate, and other amino acids did not show similar changes (Figure S1F). Targeted analysis corroborated the observed changes in glutamine, glutamate, and aspartate (Figure S1G). Interestingly, when AML cells from vehicle or iCT-treated mice were isolated at the moment of maximal response and cultured in vitro for 24 h, the differences in glutamine, glutamate, and aspartate reversed (Figure S1H). We also did not find a difference in glutamine levels when cells were treated with iCT in culture (Figure S1I), showing that the metabolic profile of AML cells highly depends on the local microenvironment.
Timed Inhibition of Glutamine Metabolism Overcomes Chemoresistance in AML
To determine whether the increase in glutamine metabolism-associated metabolites causes chemoresistance, we assessed whether residual AML cells would be more susceptible to broad inhibition of glutamine metabolism using 6-diazo-5-oxo-L-norleucine (DON), a glutamine analog and antagonist that inhibits all glutamine-dependent enzymes (Lemberg et al., 2018). When DON was given for 5 days at the same time as iCT, no difference in survival was seen between iCT alone and the combination (Figure 2A). However, when DON treatment was started after iCT and covered the moment of maximal response, either daily for a short time (Figure 2B) or continuously every other day (Figure 2C), we observed a clear survival benefit. Short-term treatment with DON after iCT improved elimination of AML cells compared with iCT alone (Figures 2D and 2E), further evidenced by a higher number of apoptotic cells (Figure 2F), and increased double-stranded DNA breaks in residual cells (Figure S2A). Treatment of mice with DON led to the accumulation of glutamine in AML cells while partially reducing the increase in glutamate induced by iCT treatment, showing that DON effectively inhibits glutamine metabolism in AML cells (Figure S2B). Interestingly, aspartate levels did not follow a similar profile as glutamate and remained high in mice treated with iCT followed by DON (Figure S2B). We confirmed the effect of DON on residual cells in a different mouse model of AML, driven by retroviral HoxA9/Meis1 overexpression, which also showed improved survival in mice when treated with DON after completion of the iCT regimen (Figure 2G). These data show that activation of glutamine metabolism protects AML cells at the moment of maximal stress after chemotherapy and reveal the potential of timed inhibition of glutamine metabolism to eliminate persisting AML cells.
Figure 2. Timed Inhibition of Glutamine Metabolism Overcomes Chemoresistance in AML.
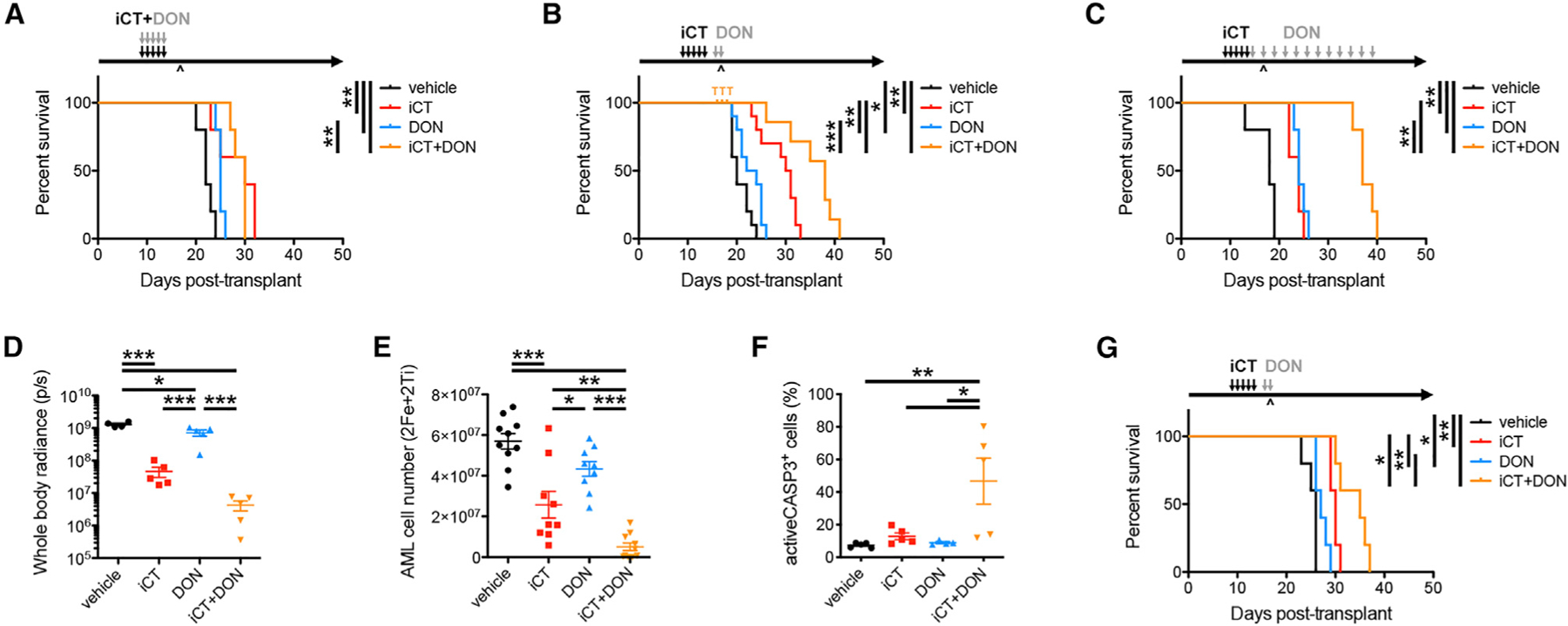
(A–C) Survival curves of MLL-AF9 AML-bearing mice treated with iCT and/or 6-diazo-5-oxo-L-norleucine (DON; 0.3 mg/kg i.p.) concomitantly (A, n = 5 mice per group) or sequentially (B and C). For sequential treatments, mice were either treated with 2 doses of DON at the moment of maximal response (B, n = 10 mice per group) or continuously with DON every other day following iCT (C, n = 5 mice per group). T, death due to drug toxicity; ^, moment of maximal response.
(D and E) Disease burden of MLL-AF9 AML-bearing mice treated sequentially with iCT and/or DON (regimen as in B), determined 1 day after the last dose of DON through bioluminescence imaging (D) or flow cytometry (E).
(F) Percentage of apoptotic cells in MLL-AF9 AML cells obtained from mice treated sequentially with iCT and/or DON (regimen as in B), determined 1 day after the last dose of DON.
(G) Survival curves of HoxA9/Meis1 AML-bearing mice treated sequentially with iCT and/or 2 doses of DON (0.3 mg/kg i.p.) specifically targeting the moment of maximal response (n = 5 mice per group). Data are represented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001. See also Figure S2.
Residual AML Cells Do Not Depend on Glutaminase
While DON has been tested in clinical trials for both solid tumors and leukemia in the 1980s, it was abandoned due to limited efficacy as a single agent and the occurrence of dose-dependent side effects (Lemberg et al., 2018). Due to these toxicity issues (Figure 2B), we sought alternative glutamine pathway inhibitors. The enzyme glutaminase (GLS) has been shown to be a metabolic vulnerability in AML (Gregory et al., 2019; Jacque et al., 2015; Matre et al., 2016), but whether it mitigates the metabolic stress following chemotherapy is unknown. As expected, mouse MLL-AF9 AML cells expressed Gls at much higher levels than Gls2 (Figure S3A) (Jacque et al., 2015). We confirmed that genetic or pharmacological inhibition of GLS reduced AML cell viability in vitro (Figures S3B–S3D), in line with previous studies. Unexpectedly, GLS knockdown had no effect on disease progression or response to iCT in vivo (Figure 3A). In addition, the treatment of mice with a GLS inhibitor following iCT did not enhance AML cell elimination or mouse survival compared with iCT alone (Figure 3B). These results again highlight that cellular metabolism and metabolic dependencies are microenvironment dependent and show that AML cells persisting after chemo-therapy, while critically depending on glutamine metabolism, do not require GLS for their survival.
Figure 3. Residual AML Cells Do Not Depend on Glutaminase.
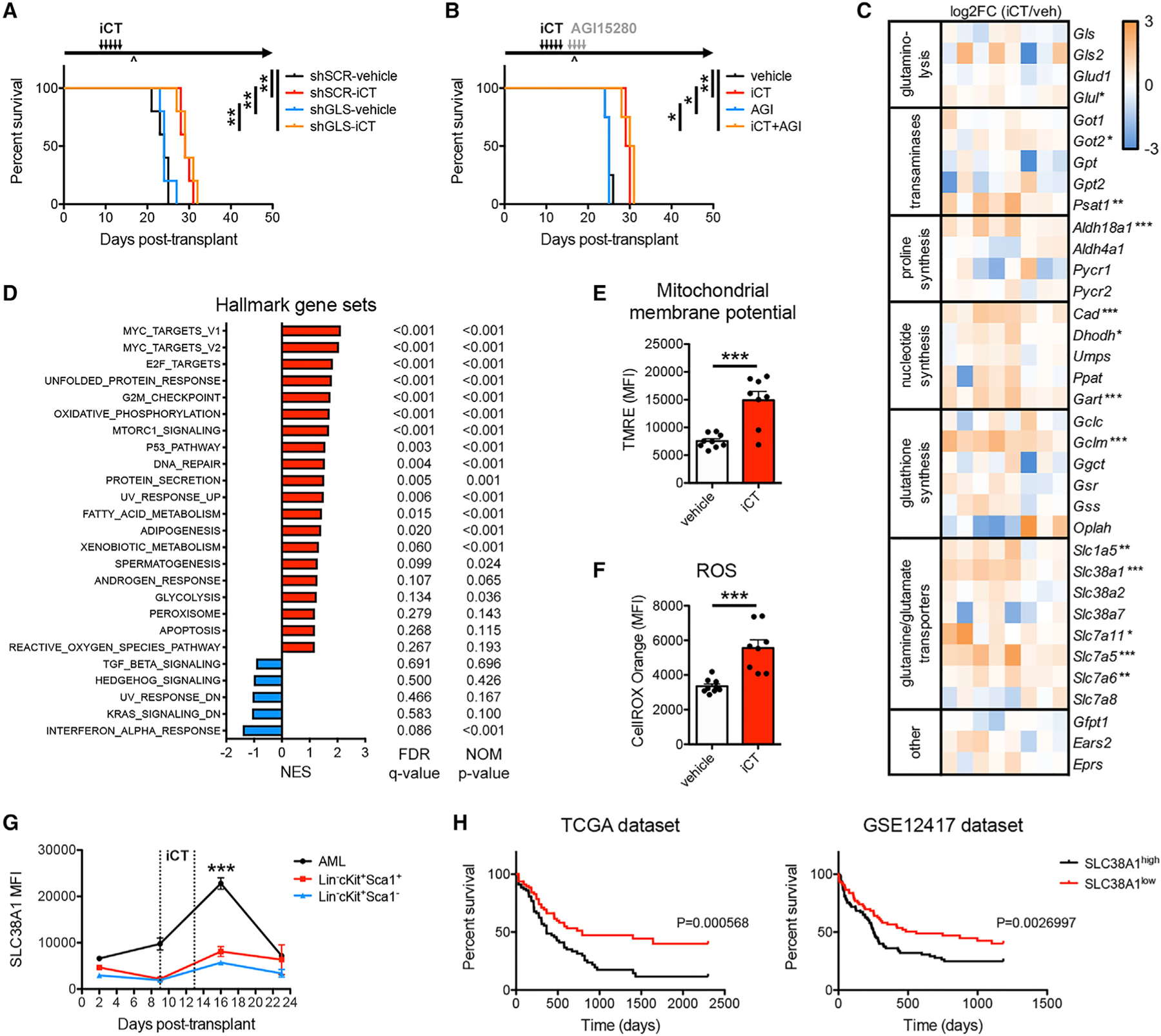
(A) Survival curve of mice engrafted with MLL-AF9 AML cells transduced with scrambled short hairpin RNA (shSCR) or shRNA targeting glutaminase (shGLS), treated with iCT or vehicle (n = 5 mice per group).
(B) Survival curves of MLL-AF9 AML-bearing mice treated sequentially with iCT and/or 4 days of AGI15280 (150 mg/kg twice daily, p.o.) targeting the moment of maximal response (n = 4 mice per group). ^, moment of maximal response.
(C) Heatmap of RNA sequencing data of genes related to glutamine metabolism in MLL-AF9 AML cells obtained from mice treated with vehicle or iCT, at the moment of maximal response.
(D) Gene set enrichment analysis in iCT- versus vehicle-treated MLL-AF9 AML cells obtained at the moment of maximal response. NES, normalized enrichment score; FDR, false discovery rate; NOM, nominal.
(E and F) Mitochondrial membrane potential measured by tetramethylrhodamine ethyl ester (TMRE) staining (E), and levels of ROS measured by CellROX Orange staining (F) in MLL-AF9 AML cells obtained from mice treated with vehicle or iCT, at the moment of maximal response.
(G) Flow cytometric analysis of SLC38A1 protein levels on MLL-AF9 AML cells and on Lin−cKit+Sca1+ and Lin−cKit+Sca1− normal hematopoietic stem and progenitor cells, at different times during the course of iCT treatment.
(H) Survival curves of AML patients grouped according to the expression of SLC38A1 above or below the median. Data were obtained from the TCGA dataset or the GSE12417 dataset. Data are represented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001. See also Figure S3.
To better understand the metabolic fate of glutamine in residual AML cells, the transcriptomic profiles of vehicle- and iCT-treated AML cells at the moment of maximal response were compared. Expression of most enzymes that metabolize glutamine or glutamate was unchanged in persisting cells (Figure 3C). While some individual enzymes showed increased expression post-chemotherapy, no single pathway stood out as particularly altered (Figure 3C). In accordance, gene set enrichment analysis comparing vehicle- and iCT-treated cells did not return any glutamine-related pathways among the top hits (Figures 3D and S3E). In contrast, other metabolic pathways were found to be enriched in residual AML cells at the transcriptomic level, including oxidative phosphorylation, fatty acid metabolism, glycolysis, and the reactive oxygen species (ROS) pathway (Figures 3D and S3F). A recent study has shown that persisting human AML cells require oxidative metabolism fueled by fatty acid oxidation and display high levels of ROS and an increased mitochondrial activity (Farge et al., 2017). We made similar observations in our mouse model of MLL-AF9 AML, with residual cells showing a higher mitochondrial membrane potential and increased ROS levels (Figures 3E and 3F).
Closer examination of glutamine metabolism-related genes revealed increased expression of several glutamine transporters, including Slc1a5, Slc38a1, Slc7a5, and Slc7a6, in residual AML cells (Figure 3C). Flow cytometric analysis confirmed a transient increase in SLC38A1 protein levels after chemotherapy (Figure 3G). The increase in SLC38A1 after iCT was found in all AML cells including immature leukemic granulocyte-monocyte progenitors (L-GMP, also known as leukemia-initiating cells; Krivtsov et al., 2006), but not in normal hematopoietic stem and progenitor cells (Figures 3G and S3G), suggesting that the changes in glutamine metabolism in response to iCT are specific for AML cells. We also analyzed two publicly available human AML survival datasets and found that high expression of SLC38A1 strongly associates with poor overall survival in both datasets (Figure 3H), which was not found for other glutamine metabolism-related genes (Figure S3H). Together, these data show that the transient increase in glutamine metabolism in residual AML cells seen at the metabolite level is not reflected at the mRNA level, with the exception of glutamine transporters.
Glutamine Metabolism Drives AML Chemoresistance by Fueling Pyrimidine Synthesis
To assess the metabolic fate of glutamine in residual AML cells, we performed in vivo stable isotope tracing. Bolus intravenous injection of 13C5-glutamine led to rapid glutamine pool labeling in the peripheral blood and bone marrow plasma, maintained at 45%–55% labeling for 10 min, after which it declined (Figure S4A). Labeling of glutamine in AML cells occurred slower with only 21% of the pool labeled at 10 min, and a rapid decline thereafter. Based on these results, we chose the 10 min time point for analyses. Notably, bolus infusion revealed labeling of glutamine and glutamate pools in AML cells but minimal labeling of the TCA cycle intermediates alpha-ketoglutarate and succinate (Figures S4A and S4B). In contrast, liver tissue showed extensive conversion of glutamine into alpha-ketoglutarate and succinate, in line with the expected pattern for cells using glutamine to fuel the TCA cycle (Figure S4C) (Häussinger and Schliess, 2007). This shows that unlike previous reports describing glutamine contribution to the TCA cycle in vitro in AML cells (Gregory et al., 2019; Matre et al., 2016), in vivo this does not seem to be a predominant fate of glutamine. These results mirror previous findings in lung cancer, where similar discrepancies between in vitro and in vivo glutamine metabolism have been described (Davidson et al., 2016).
To understand how residual AML cells use glutamine, we then injected 13C5-glutamine or 15N2-glutamine intravenously into a vehicle- and iCT-treated mice at the moment of maximal response. Labeling patterns in AML cells showed no changes in the contribution of glutamine carbon or nitrogen to amino acids or TCA cycle metabolites (Figures 4A and 4B). Rather, a substantial increase of glutamine carbon and nitrogen incorporation into glutathione was observed (Figure 4C). In addition, glutamine nitrogen incorporation into nucleotides was markedly elevated (Figure 4D), especially into the pyrimidine uridine 5′-monophosphate (UMP). Investigation of metabolite pool sizes revealed decreased levels of reduced glutathione (GSH) and unchanged levels of oxidized glutathione (GSSG) in residual cells (Figure 4E). Together with the tracing results (Figure 4C), this suggests that residual AML cells ramp up glutathione synthesis to replenish depleted GSH pools. Absolute levels of UMP, in contrast, were increased in residual cells (Figure 4F), in line with increased pyrimidine synthesis fueled by glutamine. AML cells persisting after chemotherapy, thus, require glutamine to fuel glutathione synthesis and generation of pyrimidine nucleotides.
Figure 4. Glutamine Metabolism Drives AML Chemoresistance by Fueling Pyrimidine Synthesis.
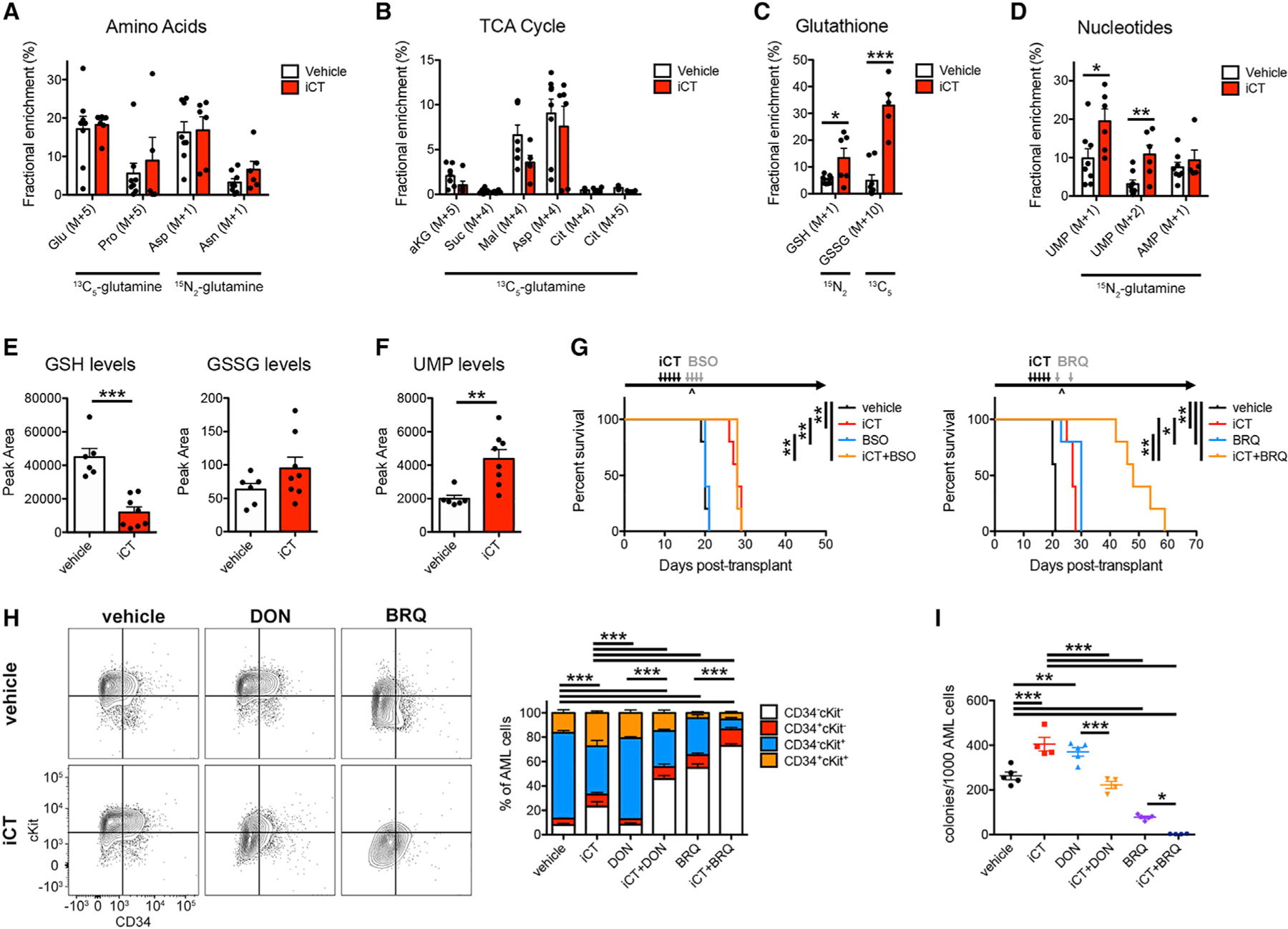
(A–D) Contribution of glutamine carbon and nitrogen to amino acids (A), the TCA cycle (B), glutathione (C), and nucleotides (D) in MLL-AF9 AML cells obtained from vehicle- and iCT-treated mice at the moment of maximal response. Glu, glutamate; Pro, proline; Asp, aspartate; Asn, asparagine; Akg, alpha-ketoglutarate; Suc, succinate; Mal, malate; Cit, citrate; GSH, reduced glutathione; GSSG, oxidized glutathione; UMP, uridine 5′-monophosphate; AMP, adenosine 5′-mono-phosphate.
(E and F) Levels of GSH and GSSG (E) or UMP (F) in MLL-AF9 AML cells obtained from vehicle- and iCT-treated mice at the moment of maximal response.
(G) Survival curves of MLL-AF9 AML-bearing mice treated sequentially with iCT and/or L-buthionine-sulfoximine (BSO, 2.23 mg/kg twice daily, i.p. + 4.45 mg/ml in drinking water; n = 5 mice per group; left) or Brequinar (BRQ, 50 mg/kg, i.p.; n = 5 mice per group; right). ^, moment of maximal response.
(H and I) Flow cytometric analysis of CD34 and cKit levels (H) and colony-forming capacity (I) in AML cells obtained from mice sequentially treated with iCT and/or DON (regimen as in Figure 2B) or BRQ (regimen as in G), determined 4 days after the last dose of iCT. Data are represented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001. See also Figure S4.
Next, we investigated the potential of targeting glutathione or nucleotide synthesis individually. To inhibit glutathione biosyn-thesis, we used L-buthionine-sulfoximine (BSO), which inhibits glutamate-cysteine ligase (GCL) and effectively lowers plasma glutathione levels in mice (Figures S4D and S4E). Treatment of AML-bearing mice with BSO for 4 days following iCT showed no difference in survival compared with iCT alone (Figure 4G). We then blocked pyrimidine synthesis using Brequinar (BRQ), which inhibits the enzyme dihydroorotate dehydrogenase (DHODH) (Figure S4F). Treatment of AML-bearing mice with BRQ induced effective DHODH inhibition shown by the accumulation of upstream metabolites and depletion of downstream products including UMP in AML cells (Figure S4G). Giving just two doses of BRQ following iCT was sufficient to significantly extend the survival of AML-bearing mice compared with iCT treatment alone (Figures 4G and S4H) by eliminating AML cells from the bone marrow (Figure S4I). In line with this, DON prevented the increase of UMP levels in residual AML cells, confirming that they require glutamine to synthesize pyrimidines (Figure S4J). Treatment of mice with DON decreased GSH levels in AML cells but did not further deplete GSH in residual cells when combined with iCT (Figure S4J). These data show that residual AML cells primarily require glutamine-dependent pyrimidine synthesis for their survival during the window of maximal selection pressure.
To test whether our metabolic targeting strategies effectively eliminate leukemia-initiating cells, we next analyzed the expression of the immature cell markers, CD34 and cKit, and tested the colony-forming ability of AML cells obtained from mice treated with vehicle or iCT, alone or in combination with DON or BRQ (Figures 4H, 4I, and S4K). Treatment with iCT mainly depleted CD34−cKit+ cells, leading to an increased percentage of CD34+ cKit+ cells and the number of colony-forming units (CFUs). DON alone had no strong effects on cell numbers or subpopulation percentages but moderately increased the number of CFUs. These strategies thus eliminate bulk AML cells but relatively spare leukemia-initiating cells. The combination of iCT and DON depleted cells across populations, showing that iCT sensitizes both bulk AML cells and leukemia-initiating cells to DON treatment. BRQ, in line with its pro-differentiation effect on AML cells (Sykes et al., 2016), strongly reduced the number and percentage of both CD34+cKit+ and CD34−cKit+ cells as well as the number of CFUs. The combination of iCT and BRQ had similar effects on the percentage of CD34+cKit+ cells compared with BRQ alone but further reduced the percentage of CD34−cKit+ cells, the number of CFUs, and total cell numbers. Leukemia-initiating cells persisting after chemotherapy are thus effectively targeted by inhibitors of glutamine or pyrimidine metabolism.
Bone Marrow Stromal Cell-Derived Aspartate Supports Pyrimidine Synthesis in AML Cells
Our in vivo 13C5-glutamine-tracing experiments showed that AML cells have substantially different labeling patterns compared with those of the liver (Figure S4C). While in liver the TCA cycle is directly fueled by glutamine, AML cells show only labeling of malate and especially aspartate. Since aspartate is essential for pyrimidine synthesis, we further explored this particular labeling pattern. We performed metabolic tracing using 13C5-glutamine in MLL-AF9 and HoxA9/Meis1 AML lines in vivo and in vitro. Interestingly, both AML models showed the unique labeling patterns only in vivo (Figure 5A). Longer in vitro labeling increased tracer enrichment but did not change labeling patterns (Figure S5A). These results show that the 13C5-glutamine labeling pattern seen in AML cells in vivo is not due to technical issues such as labeling time or the cell model used but reflects a unique metabolic program.
Figure 5. Bone Marrow Stromal Cell-Derived Aspartate Supports Pyrimidine Synthesis in AML Cells.
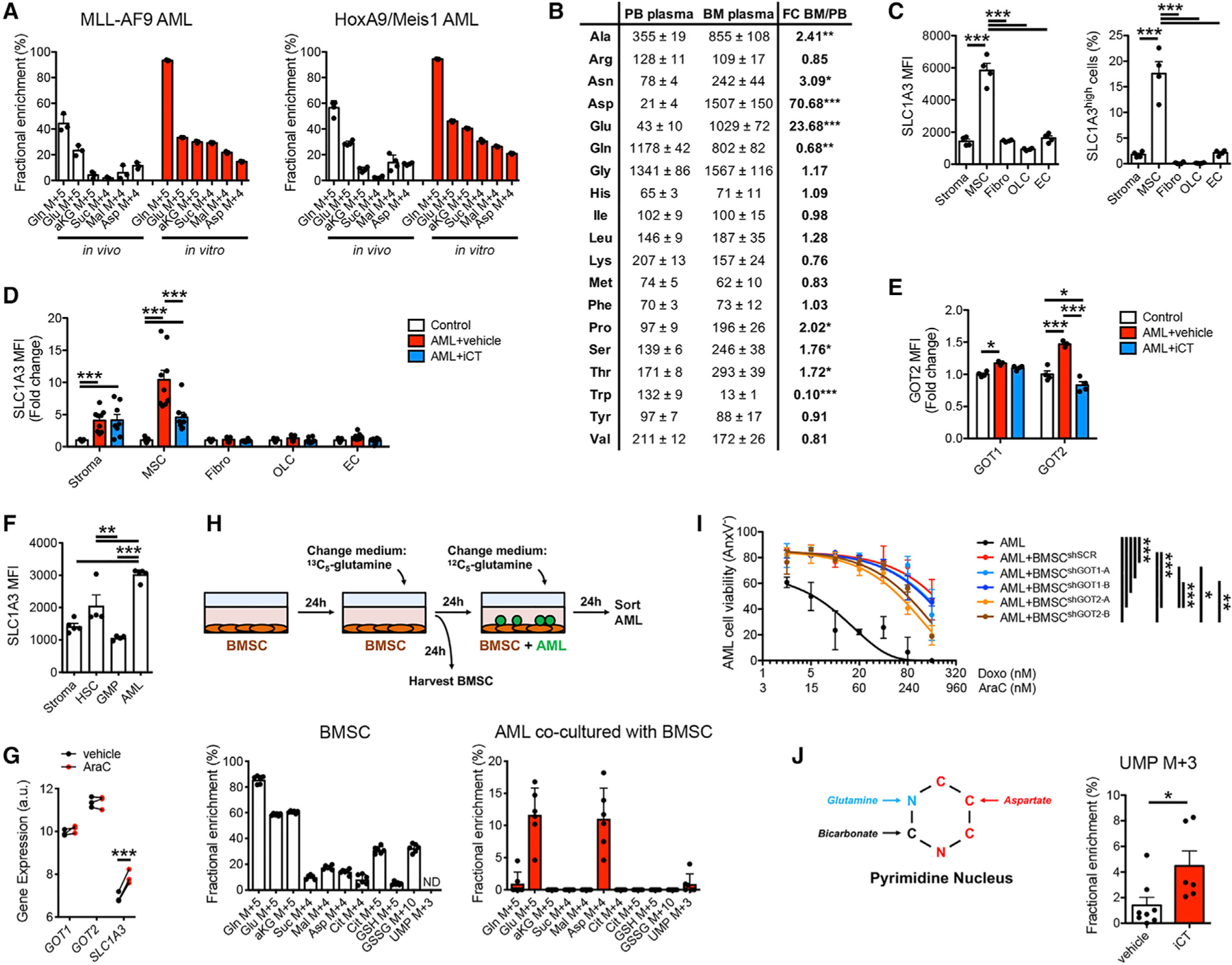
(A) Labeling of glutamine metabolism and TCA cycle metabolites in MLL-AF9 and HoxA9/Meis1 AML cells by 13C5-glutamine in vivo and in vitro (10 min labeling).
(B) LC-MS-based quantification of amino acid levels in PB and BM plasma of control mice. FC, fold change.
(C) Flow cytometric analysis of SLC1A3 protein levels on BMSC subpopulations in control mice. Stroma, total stromal cells (CD45−Ter119−); MSC, mesenchymal stromal cells (CD45−Ter119−CD31−LepR+), Fibro: fibroblasts (CD45−Ter119−CD31−LepR Sca1+CD90+), OLC, osteolineage cells: (CD45−Ter119−CD31−LepR−Sca1−CD90lowCD105+); EC, endothelial cells (CD45−Ter119−CD31+CD105+); MFI, mean fluorescence intensity.
(D) Flow cytometric analysis of SLC1A3 protein levels on BMSC subpopulations obtained from vehicle- or iCT-treated MLL-AF9 AML-bearing mice at the moment of maximal response, compared with control mice.
(E) Flow cytometric analysis of intracellular GOT1 or GOT2 protein levels in MSCs obtained from vehicle- or iCT-treated MLL-AF9 AML-bearing mice at the moment of maximal response, compared with control mice.
(F) Flow cytometric analysis of SLC1A3 protein levels on stromal cells (CD45−Ter119−), hematopoietic stem cells (HSCs; Lin−cKit+Sca1+CD48−CD150+), granulocyte-monocyte progenitor cells (GMP; Lin-cKit+Sca1−CD34+CD16/32+), and MLL-AF9 AML cells.
(G) Expression of GOT1, GOT2, and SLC1A3 by human AML PDX cells isolated from mice treated with cytarabine or vehicle. Data obtained from Farge et al. (2017) (GSE97631).
(H) 13C5-glutamine tracing in co-cultures of BMSC and MLL-AF9 AML cells.
(I) Viability of MLL-AF9 AML cells in monoculture or co-culture with BMSCs transduced with shSCR, shGOT1, or shGOT2, in response to different doses of cytarabine (AraC) and doxorubicin (Doxo).
(J) Labeling of UMP by 13C5-glutamine in MLL-AF9 AML cells obtained from vehicle- or iCT-treated mice at the moment of maximal response. Data are represented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001. See also Figure S5.
Glutamine carbon cannot end up in aspartate without going through alpha-ketoglutarate and either succinate (oxidative glutamine catabolism) or citrate (reductive carboxylation) in the TCA cycle (Figure S4B). Our tracing data, therefore, indicate that while in vitro AML cells direct glutamine carbon into the TCA cycle to synthesize aspartate, they do not do this in vivo, even though aspartate pools are comparable in vitro and in vivo (Figure S5B). AML cells in vivo thus obtain (glutamine-derived) aspartate from another source. Analysis of amino acid levels in peripheral blood revealed that aspartate is the lowest of all amino acids in the circulation (Figure 5B), in accordance with previous reports (Cantor et al., 2017; Tardito et al., 2015). However, we found that bone marrow plasma aspartate levels are 70-fold higher than those in peripheral blood of mice (Figure 5B). Glutamate, also of low abundance in peripheral blood, was 23-fold higher in bone marrow, while glutamine levels were 30% lower. Interestingly, in mice engrafted with AML cells, aspartate levels were further increased in the bone marrow plasma, while after iCT treatment aspartate levels were significantly lower (Figure S5C). Glutamate levels followed a similar trend, while glutamine showed the opposite profile. These data suggest that AML cells may obtain aspartate from a local bone marrow source, which may be of particular importance after iCT, given the increased levels of aspartate in residual AML cells (Figure 1E) and the necessity of this metabolite for pyrimidine synthesis.
To identify the bone marrow cells secreting aspartate, we analyzed the expression of aspartate synthesis enzymes and transporters in a single-cell RNA sequencing dataset of the mouse bone marrow stroma that we recently generated (Figure S5D) (Baryawno et al., 2019). Most enzymes involved in aspartate synthesis from glutamine, including Gls, glutamate-oxaloacetate transaminase 1 (Got1), Got2, and glutamate dehydrogenase (Glud1), were expressed throughout the bone marrow stroma (Figure S5E). In contrast, the aspartate-glutamate transporter Slc1a3 was expressed almost exclusively in a subpopulation of Leptin receptor (LepR)+CXCL12+ mesenchymal stromal cells (cluster 1) (Figure S5E), which we confirmed at the protein level (Figure 5C). The presence of AML cells increased SLC1A3 levels in stromal cells and particularly in LepR+ cells (Figure 5D). In contrast, iCT treatment reduced SLC1A3 levels in LepR+ cells, although they remained significantly higher than mice without AML. The presence of AML cells also increased the levels of the aspartate transaminases, GOT1, and particularly GOT2, in LepR+ cells (Figure 5E). These data are consistent with AML cells inducing bone marrow stromal cells (BMSCs) to increase aspartate production and secretion. The SLC1A3 transporter was also more abundant on MLL-AF9 AML cells than on normal hematopoietic stem cells (HSCs), GMPs, or bulk stromal cells (Figure 5F). Analysis of a publicly available transcriptomics dataset on human AML patient-derived xenografts (PDXs) further showed that expression of SLC1A3, but not of GOT1 or GOT2, increased in AML cells after treatment of mice with cytarabine (Figure 5G).
A possible exchange of aspartate between BMSCs and AML cells was tested by in vitro co-culture. In line with previous studies (Behrmann et al., 2018), we found that BMSCs protect AML cells from iCT treatment (Figure S5F). Of note, we observed an increase in glutamine levels in AML cells exposed to iCT in co-cultures (Figure S5G). This was not seen in AML mono-cultures (Figure S1I) but does mirror the in vivo response of AML cells to iCT (Figures 1D and S1G). The presence of stromal cells thus better mimics the in vivo metabolic microenvironment of AML cells. We then performed 13C5-glutamine-tracing experiments where BMSCs were labeled for 24 h before washing away the tracer and adding AML cells in medium containing unlabeled glutamine. BMSCs metabolized glutamine in the TCA cycle and synthetized aspartate using glutamine carbons with ~15% of the aspartate pool fully labeled (Figure 5H). At least part of this aspartate was then transferred to the AML cells, as evident by 11% of their aspartate pool being labeled. We detected some labeling also in UMP in AML cells, while UMP levels in BMSCs were below the detection limit, suggesting that the transferred aspartate contributes to pyrimidine synthesis in the AML cells. These data confirm the metabolic crosstalk between BMSCs and AML cells. Interestingly, AML cells also showed up-take of labeled glutamate produced by BMSCs, the only other amino acid enriched in the bone marrow plasma (Figures 5B and S5C), highly abundant in AML cells in vivo (Figure S5B) and increased after iCT treatment (Figures 1D and S1G). In contrast, while both GSH and GSSG were clearly labeled by 13C5-glutamine in BMSC, we found no labeling in AML cells, arguing against the transfer of glutathione between these two cell types (Figure 5H). We then analyzed the importance of the aspartate exchange by knocking down GOT1 or GOT2 in BMSCs (Figure S5H). Loss of GOT2 in BMSCs partially sensitized AML cells to iCT treatment, confirming that aspartate exchange contributes to the chemoprotective effect of BMSCs on AML cells (Figure 5I). These data show that BMSCs convert glutamine to aspartate, which is then transferred to AML cells enabling them to survive iCT exposure.
Since aspartate is an essential metabolite for pyrimidine synthesis, we looked for evidence in our in vivo tracing data that glutamine-derived aspartate is used by residual AML cells to fuel UMP synthesis. Our 15N2-glutamine-tracing data showed that both single and double 15N-labeled UMP was increased in residual AML cells (Figure 4D). Since pyrimidines get one nitrogen from glutamine and aspartate each (Figure 5J), this suggests that the glutamine-derived aspartate from BMSCs is indeed used to support increased pyrimidine synthesis. To further confirm this, we looked for UMP containing three 13C atoms in the 13C5-glutamine in vivo tracing data, since aspartate, but not glutamine, contributes three carbons to UMP. We indeed found increased pools of 13C3-UMP in residual AML cells (Figure 5J), further supporting the concept that glutamine fuels pyrimidine synthesis in AML cells persisting after chemotherapy both directly and indirectly via stromal cell-derived aspartate.
Human Chemoresistant AML Cells Are Sensitive to Single-Dose, Timed Inhibition of Pyrimidine Synthesis
As a first step in translating our findings to the clinic, we tested whether timed inhibition of pyrimidine synthesis using BRQ could enhance the elimination of residual AML cells in human PDXs. We used four different PDX models, obtained from patients with distinct mutational backgrounds and age (Figure 6A). Cells were engrafted in immunodeficient mice, animals were treated with a reduced-intensity iCT regimen (Wunderlich et al., 2013) and/or a single dose of BRQ at day 2 post-iCT, and AML burden was measured 2 days later. In all models, the combination of iCT and BRQ improved elimination of AML cells from the peripheral blood and bone marrow and reduced spleen weights compared with either treatment alone (Figures 6B–6D). While differences were noted in the effect size between individual PDX lines and statistical significance was not reached for every parameter, these results indicate that human AML cells have similar metabolic adaptations in response to chemotherapy as their mouse counterparts, and strengthen the clinical potential of our proposed approach.
Figure 6. Human Chemoresistant AML Cells Are Sensitive to Single-Dose, Timed Inhibition of Pyrimidine Synthesis.
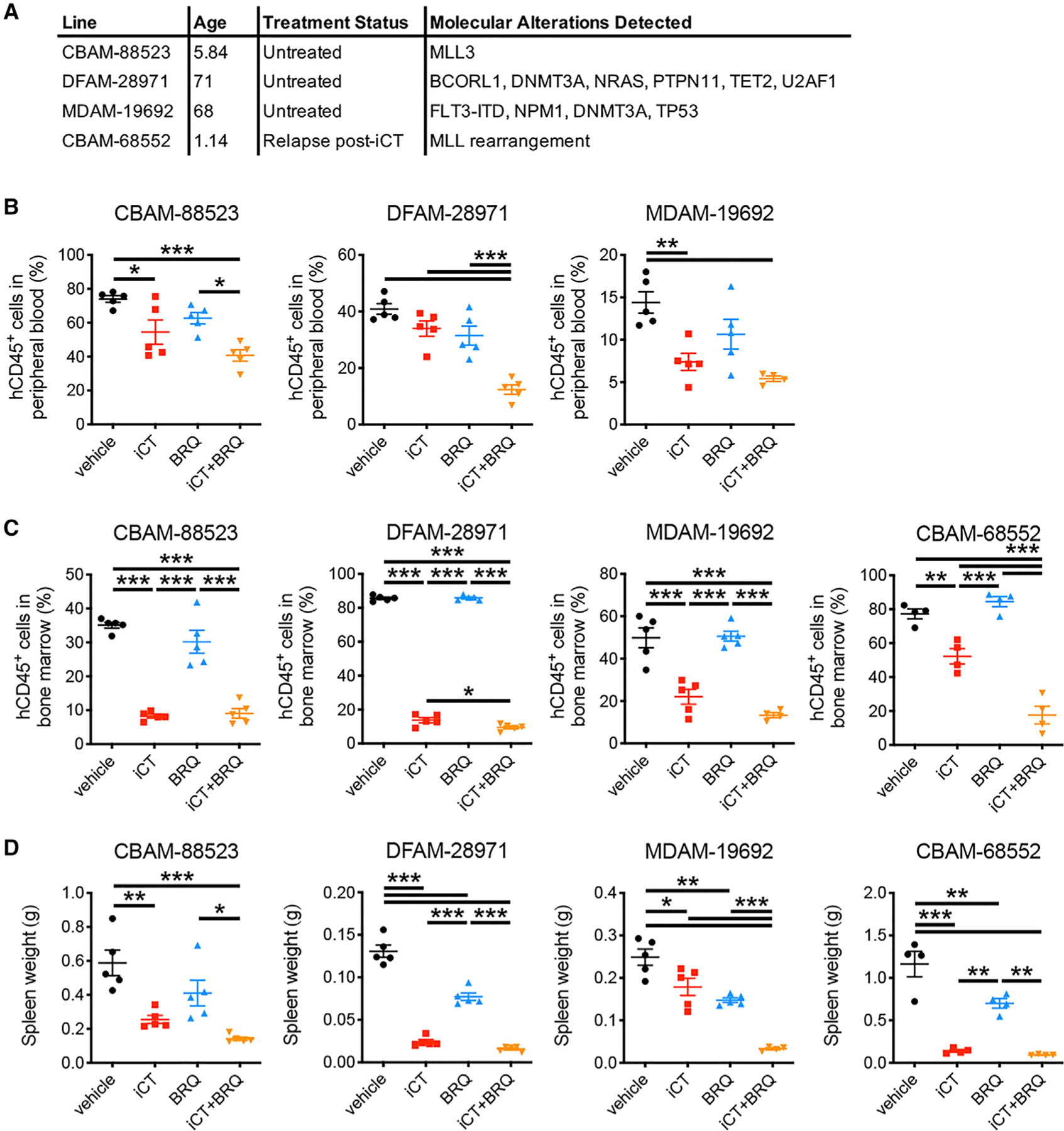
(A) Characteristics of the PDX lines.
(B–D) Percentage of human cells in the peripheral blood (B), bone marrow (C), or spleen weights (D) of PDX-engrafted mice treated sequentially with iCT (5 days of cytarabine 50 mg/kg + 3 days of doxorubicin 1.5 mg/kg i.v.) and/or Brequinar (BRQ, 25 mg/kg i.p.; given at day 2 after iCT), determined at day 4 after iCT. n = 4–5 mice per group. Data are represented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
DISCUSSION
Despite the clear clinical need to prevent or delay cancer relapse, it remains poorly understood how certain tumor cells manage to survive the extreme stress of chemotherapy. In the current study, we defined the metabolic profile of AML cells as they pass through that time of intense selection pressure. We found that residual AML cells exhibit transient metabolic adaptations driving their resistance to chemotherapy and show in mouse models of AML that timed manipulation of specific metabolic pathways such as glutamine metabolism and pyrimidine synthesis holds therapeutic potential. These findings underscore the importance of cell metabolism as a primitive, first-line stress-protection mechanism that can be targeted to eliminate chemo-resistant cancer cells.
To investigate the metabolic profile of AML cells at a time when the milieu of the bone marrow microenvironment is maximally hostile, we developed a pipeline for untargeted metabolomics of freshly isolated AML cells from mice. This approach proved to be highly valuable in revealing the metabolic program of AML cells in their native environment, which would not have been obtained through transcriptomic analysis alone. In addition, a key metabolic feature of residual cells, high glutamine levels, was lost once cells were isolated and cultured and was also not observed when AML cells were treated with iCT in vitro. These results show that the dynamic metabolic adaptations of AML cells persisting after chemotherapy are regulated at the pathway activity level and are highly dependent on the local microenvironment. This notion is further underscored by our metabolic tracer analysis, which revealed striking differences in glutamine metabolism in AML cells in vitro versus in vivo. The low contribution of glutamine to the TCA cycle in AML cells in vivo may explain the poor efficacy of GLS inhibition, even though this appears a promising target based on in vitro assays, and the peculiar nutritional composition of the bone marrow microenvironment with high glutamate levels may even further contribute to this lack of effect. The importance of examining the metabolism of cells in their native environment is supported by other recent studies that have shown how artificial in vitro conditions can create metabolic dependencies that do not exist in vivo (Cantor et al., 2017; Davidson et al., 2016; Muir et al., 2017).
Many studies have investigated the role of the bone marrow niche in AML progression and chemoresistance (Behrmann et al., 2018). However, most have focused on the role of adhesion molecules and cytokines. Very few studies have investigated the nutritional aspects of niche support in leukemia, with one report highlighting a role for BMSC-derived cysteine in chronic lymphocytic leukemia (Zhang et al., 2012) and a second study showing the importance of lipids provided by adipocytes for AML stem cells residing in adipose tissue (Ye et al., 2016). We now show that aspartate provided by LepR+ stromal cells in the bone marrow can fuel nucleotide synthesis in AML cells, which becomes of particular importance in the setting of chemo-therapy treatment. These findings again raise the question of how much of chemoresistance development is determined by genetic alterations versus microenvironmental signals. Are there hotspots of aspartate production in the bone marrow that constitute a protective niche, and is there competition between AML subclones for occupancy of these aspartate-rich locations? Further detailed analysis of our PDX models, which possess more clonal complexity (Potter et al., 2019; Wang et al., 2017), and of patient samples will help answer this question. It will also be of interest to examine whether this metabolic crosstalk with stromal cell subsets plays a role in normal hematopoiesis, given that the bone marrow is unexpectedly rich in aspartate compared to peripheral blood (also true in control mice) and the known role of LepR+ stromal cells in supporting HSCs (Ding et al., 2012b).
The primary reason for increased glutamine uptake in residual AML cells seems to be its role as a nitrogen donor for pyrimidine synthesis. Glutathione synthesis is not a metabolic dependency of AML cells at this time point, but given its pleiotropic metabolic role, we cannot completely rule out that glutamine has other metabolic contributions that support chemoresistance development in AML. While glutamine does not contribute to the TCA cycle in AML cells in vivo, it does support mitochondrial metabolism by fueling pyrimidine synthesis since the rate-limiting DHODH reaction is dependent on electron flux through the mitochondrial respiratory complexes (Boukalova et al., 2020). A dependence on oxidative phosphorylation in chemoresistant AML cells, driven by fatty acid oxidation, has been reported (Farge et al., 2017), and we confirmed this in our model (Figures 3D–3F, S3E, and S3F). Of interest, a recent study showed that reactivation of DHODH-driven pyrimidine biosynthesis was sufficient to restore the growth of respiration-deficient cancer cells (Bajzikova et al., 2019). This suggests that increased glutamine uptake, aspartate uptake, and mitochondrial respiration in chemoresistant AML cells all function in concert to drive pyrimidine synthesis. Translating our findings to the clinical application may thus best be accomplished by exploiting the transient dependency on pyrimidine metabolism. It is currently not clear why chemoresistant AML cells increase pyrimidine but not purine synthesis. Since cytarabine is a pyrimidine analog, cells that can ramp up pyrimidine synthesis may have a larger capacity to repair DNA damage. This is supported by our finding that DON strongly induces double-stranded DNA breaks in persisting AML cells (Figure S2A). Another possibility is the involvement of pyrimidines, but not purines, in O-linked N-acetylglycosylation (Bond and Hanover, 2015). We previously defined that pyrimi-dine synthesis inhibition can induce differentiation of AML cells, indicating a broad range of effects in malignancy (Sykes et al., 2016). Notably, a number of pyrimidine synthesis inhibitors are now in clinical trial, including Brequinar that we show here can remarkably extend animal survival when used for a brief interval post-chemotherapy. We suggest that our findings encourage the testing of such inhibitors in a similar clinical context, given that the transient moment of stress that drives initial leukemia regeneration has also been described in AML patients (Boyd et al., 2018). We also suggest that this work justifies investigating our underlying hypothesis of a unique metabolic bottleneck associated with chemotherapy in the treatment of other tumor types.
Limitations of Study
In the in vivo glutamine-tracing experiments, we chose a bolus injection instead of a slower continuous infusion. The bolus injection allowed investigation of the direct fate of glutamine in AML cells without confounding labeling coming from glutamine-derived metabolites synthetized by other tissues, as will occur with longer glutamine infusion. However, the short labeling time we used may obfuscate some pathways fueled by glutamine due to slower reaction kinetics, and we, therefore, cannot fully exclude that glutamine has additional metabolic fates in chemoresistant AML cells.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, David T. Scadden (david_scadden@harvard.edu).
Materials Availability
This study did not generate new unique materials.
Data and Code Availability
The bulk mRNA sequencing data that support the findings of this study have been deposited in GEO with the accession number GSE139159. The single-cell RNA sequencing data were generated previously (Baryawno et al., 2019) and are deposited in GEO (GSE128423). A portal for exploring the entire single-cell atlas is available (portals.broadinstitute.org/single_cell/study/mouse-bone-marrow-stroma-in-homeostasis). All other data supporting the findings of this study are available within the paper.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
C57BL/6J and NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice were purchased from Jackson Laboratories. Experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of the Faculty of Arts and Sciences of Harvard University.
Syngeneic Leukemia Experiments
The MLL-AF9 and HoxA9/Meis1 leukemia models have been described previously (Sykes et al., 2016). Briefly, the MLL-AF9 model was established by crossing MLL-AF9 knockin mice (Corral et al., 1996) with mice expressing GFP under the control of the ubiquitin promoter as well as mice expressing luciferase under the control of the b-actin promoter. The retroviral transduction model of HoxA9/Meis1 leukemia was generated by the infection of bone marrow mononuclear cells with an MSCV-HoxA9-IRES-Meis1 construct (originally designed by Dr. Guy Sauvageau). For both models, leukemic bone marrow cells from a terminally ill male mouse were harvested and expanded ex vivo in RPMI1640 medium (Lonza) supplemented with 10% fetal bovine serum (FBS; Gibco), 100 I.U./ml penicillin, 100 μg/ml streptomycin, 2 mM L-glutamine (all from Corning), 20 ng/ml recombinant mouse SCF (rmSCF), 10 ng/ml recombinant mouse interleukin 3 (rmIL-3) and 10 ng/ml rmIL-6 (all from R&D Systems).
Recipient male mice (age 8–10 weeks) were injected intravenously (retro-orbital) with 1 million leukemia cells in a volume of 100 μl of saline. Disease progression was tracked intravitally using bioluminescence imaging. Mice were anaesthetized using isoflurane, injected with luciferin (150 μl of a 15 mg/ml solution in PBS), and imaged on an IVIS 200 Spectrum In Vivo Imaging System (PerkinElmer). When indicated, mice were treated with iCT (cytarabine, 100 mg/kg, once daily for 5 days and doxorubicin, 3 mg/kg, once daily for the first 3 days) or vehicle (saline) delivered via I.P. injection. To inhibit glutamine metabolism, mice were treated with DON (0.3 mg/kg, once daily) or vehicle (saline) delivered via I.P. injection. To inhibit GLS, mice were treated with AGI15280 (150 mg/kg, twice daily) or vehicle (20% HP-β-cyclodextrin + 20% Solutol, 50 mM citrate buffer, pH 3) delivered by oral gavage. To inhibit DHODH, mice were treated with BRQ (50 mg/kg, once daily) or vehicle (70% PBS, 30% poly(ethylene glycol)-400, pH 7.2) delivered via I.P. injection. To inhibit GCL, mice were treated with BSO (twice daily by I.P. injection at 2.23 mg/kg + in the drinking water at 4.45 mg/ml) or vehicle (saline by I.P. injection + untreated drinking water). For survival experiments, mice were checked twice daily and euthanized by CO2 asphyxiation when they showed signs of extensive distress or became moribund. All survival experiments were repeated independently at least twice with 4–5 mice per group in each experiment. A representative experiment is shown in the figures.
Patient-Derived Xenograft Experiments
PDX samples were obtained from the Center for Patient Derived Models at the Dana-Farber Cancer Institute and injected intravenously (tail vein) into 6-week-old female NSG mice. Cells were allowed to expand for 3–4 months and AML burden was monitored by measuring the percentage of hCD45+ cells in the peripheral blood. Mice were treated with a reduced intensity iCT regimen (cytarabine, 50 mg/kg, once daily for 5 days and doxorubicin, 1.5 mg/kg, once daily for the first 3 days) or vehicle (saline) delivered via I.P. injection (Wunderlich et al., 2013). To inhibit DHODH, mice were treated with BRQ (25 mg/kg) or vehicle (70% PBS, 30% poly(ethylene glycol)-400, pH 7.2) delivered via I.P. injection on day 2 after the last dose of iCT.
Cell Isolation and Culture
The MLL-AF9 or HoxA9/Meis1 primary leukemia cells were expanded in RPMI1640 medium supplemented with 10% FBS, 100 I.U./ml penicillin, 100 μg/ml streptomycin, 2 mM L-glutamine, 20 ng/ml rmSCF, 10 ng/ml rmIL-3 and 10 ng/ml rmIL-6. Leukemia cells were cultured in a humidified incubator at 37°C in ambient air with 5% CO2.
For BMSC, femurs and tibias of mice (male and female, age 8–10 weeks) were isolated and cleaned to remove muscle. Epiphyses were cut away and bone marrow was flushed out with PBS containing 2% FBS using a 25-gauge needle attached to a 20 ml syringe. Bone marrow fractions were kept on ice. Bones were cut into small pieces using scissors and digested in DMEM medium (Lonza) containing 3 mg/ml collagenase type 2 (Gibco) and 4 mg/ml dispase (Gibco) at 37°C for 45 min. Bone cell suspensions were then pooled with the bone marrow fraction and passed through a 70 μm cell strainer. Cells were used for immediate analysis or cultured in DMEM medium supplemented with 20% FBS, 100 I.U./ml penicillin, 100 μg/ml streptomycin, and 2 mM L-glutamine, in a humid-ified incubator at 37°C in 2% O2 with 7.5% CO2.
For co-cultures, BMSC were seeded in 96-well plates at 15,000 cells/well and AML cells were added at 5,000 cells/well. Cells were cultured in DMEM medium supplemented with 20% dialyzed FBS (Gibco), 100 I.U./ml penicillin, 100 μg/ml streptomycin, 0.4 mM L-glutamine, 20 ng/ml rmSCF, 10 ng/ml rmIL-3 and 10 ng/ml rmIL-6, in a humidified incubator at 37°C in 2% O2 with 7.5% CO2.
METHOD DETAILS
Isolation of Peripheral Blood and Bone Marrow Plasma
Mice were euthanized by CO2 asphyxiation and peripheral blood was obtained by cardiac puncture and collected in K2-EDTA Micro-tainer tubes (BD Biosciences). Peripheral blood plasma was obtained by centrifuging samples for 5 min at 2,500 x g. Femurs and tibias were dissected, cleaned and epiphyses cut off. Bone marrow plasma was obtained by centrifugation for 5 min at 2,500 x g.
Fluorescence-Activated Cell Sorting (FACS)
Mice were euthanized by CO2 asphyxiation and femurs and tibias were isolated and cleaned to remove muscle. Bones were then crushed using a mortar and pestle in ice-cold flow buffer (PBS containing 2% FBS), and the obtained cell suspension was passed through a 70 mm cell strainer. Red blood cells were removed by incubating cells for 5 minutes in ACK lysing buffer (Gibco) on ice, cells were washed and resuspended in ice-cold flow buffer containing the viability dye 7-aminoactinomycin D (7-AAD; BD Biosciences). Viable AML cells (GFP+7-AAD−) were sorted on a BD FACSAria II (BD Biosciences) with the sample collector cooled to 4°C. Sorted cells were immediately processed.
Flow Cytometry
Cells were suspended in flow buffer and stained for 30 min at 4°C in the dark with antibodies against cKit, Sca1, CD34, CD45, Ter-119, CD31, LepR, CD90, CD105, SLC1A3, SLC38A1 or a lineage cocktail (CD3, Gr-1, CD11b, B220, Ter-119). 7-AAD was included as a viability dye to help identify dead cells. When necessary, cells were stained with a secondary antibody (Alexa Fluor 546-conjugated anti-rabbit IgG) or APC-eFluor 780-conjugated streptavidin (eBioscience) for 15 min at 4°C in the dark. For intracellular antibody staining cells were first stained for surface markers and then fixed and permeabilized (BD Cytofix/Cytoperm Kit, BD Biosciences). Samples were then stained with antibodies against GOT1, GOT2 or gamma H2AX in perm/wash buffer washed and, for GOT1 and GOT2, stained with a secondary antibody (Alexa Fluor 546-conjugated anti-rabbit IgG). Cell viability was analyzed using Annexin V-APC (BioLegend) and propidium iodide (PI; ThermoFisher Scientific) with Annexin V−PI− cells considered as viable cells. The number of viable cells was determined by taking into account the total number of cells per well as measured using a Cellometer (Nexcelom). Mitochondrial membrane potential was measured by Tetramethylrhodamine, ethyl ester (TMRE; ThermoFisher) staining, and levels of ROS were measured by CellROX Orange staining. Cells were incubated with 200 nM TMRE or 2.5 μM CellROX Orange in RPMI1640 medium supplemented with 10% FBS, 100 I.U./ml penicillin, 100 μg/ml streptomycin and 2 mM L-glutamine at 37°C for 30 min, washed in flow buffer and analyzed. For all flow cytometry experiments, single color controls were used to set compensations, and fluorescence minus one controls were used to set gates. All flow cytometry data was collected on a BD LSR II flow cytometer and analyzed using FlowJo software.
Colony-Forming Unit Assay
FACS-sorted viable AML cells (GFP+7-AAD−) were resuspended in MethoCult GF M3434 (StemCell Technologies) at 1000 cells/ml and plated in SmartDish 6-well plates (StemCell Technologies) at 1 ml/well in duplicate. Cells were cultured in a humidified incubator at 37°C in ambient air with 5% CO2. After 5-days colonies were counted.
Metabolomics Analysis
FACS-sorted cells or plasma samples were lysed in 100 μl ice-cold methanol and polar metabolites were extracted using methanol-chloroform phase separation (1 ml methanol, 500 μl water, and 1 ml chloroform). Samples were then dried under nitrogen flow, re-suspended in 20–100 μl 70% acetonitrile in water containing 2.5 μM of an internal standard (13C-, 15N-labeled amino acid mix; Cambridge Isotope Laboratories) and run on a ThermoFisher Q-Exactive Orbitrap mass spectrometer equipped with Zic-pHILIC column (150×2.1 mm, 5 μm; Merck). A volume of 5 μl was injected and metabolites were monitored in full-scan, polarity-switching, mode (0 to 45 min, resolution 70,000, AGC target 3×106, m/z range 66 to 990). Mobile phase A for chromatography consisted of 20 mM ammonium carbonate, 0.1% ammonium hydroxide, in water and mobile phase B of 97% acetonitrile in water. For untargeted metabolomics, a pooled sample was created from all samples and used for MS/MS runs. For targeted analysis, a standard mix at 1 μM of each compound of interest was prepared and run after the samples to confirm retention times. Metabolite measurements were normalized to the internal 13C/15N-labelled amino acid standard. For untargeted metabolomics analysis, data were processed with Compound Discoverer 3.0 (ThermoFisher Scientific) using the mzCloud mass spectral library for compound annotation. For compounds for which an MS/MS spectrum was not available, the Kyoto Encyclopedia of Genes and Genomes (KEGG) Compound database (www.genome.jp/kegg; Kanehisa and Goto, 2000) and human metabolome database (HMDB; www.hmdb.ca; Wishart et al., 2007, 2018) were used for putative compound annotation based on the monoisotopic molecular weight. For targeted metabolomics analysis, data were analyzed with Tracefinder4.1 (ThermoFisher Scientific).
For metabolite quantification of total bone marrow cells after brequinar treatment of HoxA9/Meis1 AML-bearing mice, bone marrow was obtained through centrifugation of bones, and samples were extracted in 1 ml ice-cold 80% methanol in water with 500 nM 13C-, 15N-labeled amino acid mix. Cells were vortexed for 10 min at 4°C, and then centrifuged at maximum speed for 10 min at 4°C. Samples were dried under nitrogen flow and resuspended in 50% acetonitrile in water. Metabolites were measured on a Q-Exactive Orbitrap mass spectrometer equipped with Zic-pHILIC column (150×2.1 mm, 5 μm). A volume of 4 μl was injected and metabolites were monitored in full-scan, polarity-switching, mode (resolution 70,000, AGC target 1×106). An additional narrow range full-scan (220–700 m/z) in negative mode only was included to enhance nucleotide detection. Mobile phase A for chromatography consisted of 20 mM ammonium carbonate, 0.1% ammonium hydroxide, in water and mobile phase B of 100% acetonitrile. Relative quantitation of metabolites was performed with XCalibur QuanBrowser 2.2 (Thermo Fisher Scientific) using a 5 ppm mass tolerance and referencing an in-house retention time library of chemical standards. Raw peak areas of metabolites were normalized to the internal 13C/15N-labelled amino acid standard. The protein concentration was measured from the insoluble extraction fraction by a BCA assay, and the metabolite area ratios were further normalized to protein concentration.
In Vitro Metabolic Tracing
Cells in culture were incubated with 2 mM 13C5-glutamine (Cambridge Isotope Laboratories) in glutamine-free media for the indicated times and harvested by centrifugation at 4°C. An aliquot of each well was taken at the time of harvesting to determine cell numbers. After washing with PBS, cells were lysed in ice-cold methanol and polar metabolites were extracted using methanol-chloroform phase separation (1 ml methanol, 500 μl water and 1 ml chloroform). Samples were then dried under nitrogen flow, resuspended in 100 μl 70% acetonitrile in water containing 2.5 mM of an internal standard (13C-, 15N-labeled amino acid mix) and run on a Thermo-Fisher Q-exactive equipped with Zic-pHILIC column (150×2.1 mm, 5 μm; Merck). A volume of 5 μl was injected and the full mass spectrum was obtained in both positive and negative polarity mode (0 to 45 min, resolution 70000, AGC target 3e6, m/z range 66 to 990). Mobile phase A for chromatography consisted of 20 mM ammonium carbonate, 0.1% ammonium hydroxide, in water and mobile phase B of 97% acetonitrile in water. A standard mix at 5 μM of each compound of interest was prepared and run after the samples to confirm retention times and to obtain the experimental value of natural isotopic distribution. Data were analyzed with Tracefinder 4.1. Each compound peak was integrated as the sum of all isotopes. The isotope ratios were then extracted from the mass spectral data.
In Vivo Metabolic Tracing
Mice were injected via the tail vein with 200 μl of a 54.3 mg/ml 13C5-glutamine or 15N2-glutamine (Cambridge Isotope Laboratories) in saline solution. At the indicated times after injection, mice were euthanized by CO2 asphyxiation and femurs and tibias were isolated and processed for FACS. FACS-sorted cells were lysed in 100 μl of ice-cold 80% methanol in water. Samples were dried down using a SpeedVac Vacuum Concentrator (ThermoFisher) and re-suspended in 40 μl mobile phase buffer A (97% H2O, 3% MeOH, 10 mM Tributylamine, 15 mM Glacial Acetic Acid, pH 5.5). Metabolites (15 μl injection) were analyzed on a reverse-phase ion-pairing chromatography (ZORBAX Extend-C18, 2.1 × 150 mm, 1.8 μm; Agilent) coupled to tandem mass spectrometry (Agilent), and analytes were eluted in buffer A and buffer B (10 mM Tributylamine, 15 mM Glacial Acetic Acid in 100% MeOH). Samples were ionized (with negative polarity) using Agilent Jet Spray ionization; nebulizer 45 psi, capillary –2000 V, nozzle voltage: 500 V, sheath gas temperature 325°C, and sheath gas flow 12 L/min. Peaks were integrated in Mass Hunter (Agilent).
RNA Sequencing
Total RNA was isolated using the RNeasy Plus Mini kit (Qiagen), with additional on-column DNase treatment to eliminate traces of genomic DNA. Nucleic acid concentration was quantified using a NanoDrop (Thermo Scientific). Libraries were prepared with an RNA library preparation kit (E7490, NEB) using 100 ng of RNA. RNA-seq libraries were sequenced with a 1 X 50-bp strand-specific protocol on a HiSeq 2500 (Illumina). Data were analyzed using a high-throughput next generation sequencing analysis pipeline: FASTQ files were aligned to the mouse genome (mm9) and gene expression profile for the individual samples was calculated for RPKM values using STAR and Cufflinks.
Single-cell RNA Sequencing
The single-cell RNA sequencing dataset of the mouse long bone and bone marrow stroma was generated previously and detailed information on cell isolation, cell sorting, library preparation, RNA sequencing, and data processing is provided in the original manuscript (Baryawno et al., 2019).
Immunoblotting
Total cell lysates were obtained by lysing equal amounts of cells in RIPA buffer (ThermoFisher Scientific) supplemented with 1x cOmplete protease inhibitor cocktail (Roche). Proteins were separated by SDS-PAGE and transferred to a nitrocellulose membrane (Bio-Rad). Membranes were blocked with 5% dry milk in Tris-buffered saline with 0.1% Tween-20 (TBS-T) for 1 hour at room temperature and incubated overnight at 4°C with primary antibodies (rabbit-anti-GLS, 1/1,000; rabbit-anti-GOT1, 1/1000; rabbit-anti-GOT2, 1/1000; mouse-anti-β-actin, 1/10,000) diluted in 5% BSA (Cell Signaling Technology) in TBS-T. Signals were detected using an Odyssey CLx imaging system (LI-COR) after incubation with IRDye-conjugated secondary antibodies (LI-COR).
shRNAs
To silence GLS, GOT1 and/or GOT2 we transduced cells, in the presence of 7 mg/ml polybrene (Sigma-Aldrich), with a lentivirus carrying a shRNA against mouse GLS (MISSION, Sigma-Aldrich; TRCN0000253167), mouse GOT1 (MISSION, Sigma-Aldrich; TRCN0000119792, TRCN0000119795) and/or mouse GOT2 (MISSION, Sigma-Aldrich; TRCN0000326018, TRCN0000326020). A nonsense scrambled (SCR) shRNA sequence was used as a negative control. Cells were transduced by spinfection (90 minutes at 1,000 x g), incubated for 3 hours, washed and plated in fresh medium.
Histology
Bones were harvested, fixed overnight in 4% paraformaldehyde in PBS, and decalcified using 10% EDTA (pH 8). Bones were then embedded in gelatin-polyvinylpyrrolidone-sucrose and sectioned on a cryostat (50 μm sections) according to a previously published protocol (Kusumbe et al., 2015). Cell nuclei were visualized by Hoechst33342 (ThermoFisher Scientific) and mounted. Images were taken on a Zeiss LSM880 confocal laser scanning microscope.
QUANTIFICATION AND STATISTICAL ANALYSIS
Analysis of the untargeted metabolomics dataset was performed using MetaboAnalyst (www.metaboanalyst.ca; Chong et al., 2018). The “Statistical Analysis” and “Pathway Analysis” nodes were used for analysis. Data was normalized by log transformation (generalized logarithm transformation) and auto-scaling (mean-centered and divided by the standard deviation of each variable). For pathway analysis, the Global Test and Relative-betweenness Centrality algorithms were used, and data were mapped to the mouse KEGG pathway library.
Gene set enrichment analysis was performed using the Gene Set Enrichment Analysis (GSEA) software (Broad Institute; Mootha et al., 2003; Subramanian et al., 2005) using the hallmark and GO biological process gene sets.
Correlation between AML patient survival and gene expression was performed using the PROGgeneV2 prognostics database (Goswami and Nakshatri, 2014). The GSE12417 and TCGA datasets were used for analysis.
All numerical results are reported as mean ± standard error of the mean (SEM). Statistical significance of the difference between experimental groups was analyzed by two-tailed unpaired Student’s t-test, one-way ANOVA with Bonferroni post-hoc test or the log-rank (Mantel-Cox) test for the Kaplan-Meier survival curve analyses using the GraphPad PRISM 5 software. Differences were considered statistically significant for P < 0.05.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Pacific Blue anti-mouse Lineage cocktail | BioLegend | Cat#: 133310 RRID: AB_11150779 |
| APC anti-mouse cKit | BioLegend | Cat#: 135108 RRID: AB_2028407 |
| Brilliant Violet 510 anti-mouse Sca1 | BioLegend | Cat#: 108129 RRID: AB_2561593 |
| eFluor 450 anti-mouse CD34 | eBioscience | Cat#: 48–0341-82 RRID: AB_2043837 |
| PE-Cy7 anti-mouse CD45 | BioLegend | Cat#: 103114 RRID: AB_312979 |
| PE-Cy7 anti-mouse Ter-119 | BioLegend | Cat#: 116222 RRID: AB_2281408 |
| PerCP-Cy5.5 anti-mouse CD31 | BioLegend | Cat#: 102420 RRID: AB_10613644 |
| APC anti-mouse CD90.2 | BioLegend | Cat#: 105312 RRID: AB_313183 |
| Pacific Blue anti-mouse CD105 | BioLegend | Cat#: 120412 RRID: AB_2098890 |
| FITC anti-human CD45 | BioLegend | Cat#: 304006 RRID: AB_314394 |
| Biotin anti-mouse LepR (goat polyclonal) | R&D Systems | Cat#: BAF497 RRID: AB_2296953 |
| Anti-SLC1A3 (rabbit polyclonal) | Novus Biologicals | Cat#: NB100–1869 RRID: AB_531518 |
| Anti-SLC38A1 (rabbit polyclonal) | ThermoFisher Scientific | Cat#: PA5–42420 RRID: AB_2605455 |
| Alexa Fluor 647 anti-H2AX (pS139) (mouse monoclonal) | BD Biosciences | Cat#: 560447 RRID: AB_1645414 |
| Anti-GOT1 (rabbit monoclonal) | Abcam | Cat#: ab170950 |
| Anti-GOT2 (rabbit monoclonal) | Abcam | Cat#: ab171739 |
| Anti-GLS (rabbit monoclonal) | ThermoFisher Scientific | Cat#: 701965 RRID: AB_2633041 |
| Anti-b-actin (mouse monoclonal) | Sigma-Aldrich | Cat#: A2228 RRID: AB_476697 |
| Alexa Fluor 546 anti-rabbit IgG (H+L) | ThermoFisher Scientific | Cat#: A11035 RRID: AB_2633041 |
| IRDye 800CW anti-rabbit IgG (H + L) | LI-COR | Cat#: 925–32211 RRID: AB_2651127 |
| IRDye 680RD anti-mouse IgG (H + L) | LI-COR | Cat#: 925–68070 RRID: AB_2651128 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant Mouse SCF Protein | R&D Systems | 455-MC |
| Recombinant Mouse IL-3 Protein | R&D Systems | 403-ML |
| Recombinant Mouse IL-6 Protein | R&D Systems | 406-ML |
| D-luciferin | GoldBio | LUCK |
| Cytarabine hydrochloride | Sigma-Aldrich | C6645 |
| Doxorubicin hydrochloride | Sigma-Aldrich | D1515 |
| 6-Diazo-5-oxo-L-norleucine (DON) | Sigma-Aldrich | D2141 |
| (2-Hydroxypropyl)-b-cyclodextrin | Sigma-Aldrich | H107 |
| Solutol HS 15 | Sigma-Aldrich | 42966 |
| AGI15280 | Agios Pharmaceuticals | N/A |
| Poly(ethylene glycol)-400 | Sigma-Aldrich | 202398 |
| Brequinar (BRQ) | Broad Institute | N/A |
| 7-aminoactinomycin D (7-AAD) | BD Biosciences | 559925 |
| BPTES | Sigma-Aldrich | SML0601 |
| CB839 | MedKoo Biosciences | 206153 |
| APC Annexin V | BioLegend | 640941 |
| APC-eFluor 780 Streptavidin | eBioscience | 47–4317-82 |
| CellROX Orange | ThermoFisher Scientific | C10443 |
| Tetramethylrhodamine, Ethyl Ester (TMRE) | ThermoFisher Scientific | T669 |
| RPMI 1640 Medium | Lonza | 12–167F |
| DMEM Medium | Lonza | 12–614F |
| Canonical Amino Acid Mix (13C-, 15N- labeled) | Cambridge Isotope Laboratories | MSK-CAA-1 |
| L-Glutamine (13C5, 99%) | Cambridge Isotope Laboratories | CLM-1822-H |
| L-Glutamine (15N2, 98%) | Cambridge Isotope Laboratories | NLM-1328 |
| Critical Commercial Assays | ||
| BD Cytofix/Cytoperm Kit | BD Biosciences | 554714 |
| RNeasy Plus Mini Kit | Qiagen | 74134 |
| MethoCult GF M3434 | StemCell Technologies | 03434 |
| Deposited Data | ||
| RNA sequencing of MLL-AF9 leukemia cells sorted from mice treated with or without iCT, at the moment of maximal response or after relapse | GEO | GSE139159 |
| Single cell RNA sequencing of mouse bone marrow stromal cells | GEO | GSE128423 |
| Experimental Models: Cell Lines | ||
| MLL-AF9 mouse AML | Sykes et al., 2016 | N/A |
| HoxA9/Meis1 mouse AML | Sykes et al., 2016 | N/A |
| PDX lines | Center for Patient-Derived Models, Dana- Farber Cancer Institute | CBAM-88523, DFAM-28971, MDAM- 19692, CBAM-68552 |
| Experimental Models: Organisms/Strains | ||
| C57BL/6J mice | Jackson Laboratories | 000664 |
| NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) | Jackson Laboratories | 005557 |
| Recombinant DNA | ||
| Mouse GLS shRNA (TRCN0000253167) | Sigma-Aldrich | SHCLND-NM_001081081 |
| Mouse GOT1 shRNA (TRCN0000119792, TRCN0000119795) | Sigma-Aldrich | SHCLND-NM_010324 |
| Mouse GOT2 shRNA (TRCN0000326018, TRCN0000326020) | Sigma-Aldrich | SHCLND-NM_010325 |
| Software and Algorithms | ||
| Compound Discoverer (version 3.0) | ThermoFisher Scientific | N/A |
| TraceFinder (version 4.1) | ThermoFisher Scientific | N/A |
| MassHunter (version B.08.00) | Agilent | N/A |
| MetaboAnalyst (version 4.0) | Chong et al., 2018 | N/A |
| GSEA (version 4.0.2) | Mootha et al., 2003; Subramanian et al., 2005 | N/A |
| Prism (version 5.0) | GraphPad Software | N/A |
| FlowJo (version 10) | FlowJo | N/A |
Highlights.
Untargeted metabolomics reveals the in vivo metabolic profile of AML cells
AML cells exhibit transient metabolic changes in response to chemotherapy
Glutamine or pyrimidine metabolism can be targeted to eliminate persisting cells
LepR+ stromal cells provide aspartate to support pyrimidine generation by AML cells
Context and Significance.
Chemoresistance is the difference between remission and cure in cancer patients. This is particularly evident in acute myeloid leukemia where complete remissions are common, but few are cured. In the majority of patients, a few cells persist through the therapeutic selection and cause relapse. Using mouse models, researchers from Harvard University and their colleagues show that leukemia cells pass through extreme metabolic challenges when under selection from chemotherapy. Chemoresistant leukemia cells reprogram their metabolism to survive, a process supported by metabolite exchange with specific bone marrow stromal cells. These metabolic adaptations can be targeted in a timed manner to enhance elimination of residual leukemia cells and overcome chemoresistance.
ACKNOWLEDGMENTS
We thank P. Chea, J. LaVecchio, and N. Kheradmand for technical assistance; S. Biller, G. Kingsbury, S. Sethumadavan, and D. Simons from Agios Pharmaceuticals; the M.D. Anderson Cancer Center and Center for Patient-Derived Models at DFCI for the PDX models; the HSCRB Histology Core; the HSCRB Flow Core; the Harvard Center for Biological Imaging; and the Whitehead Institute Metabolite Profiling Core Facility. N.v.G. was supported by a grant from Alex’s Lemonade Stand Foundation and Tap Cancer Out. D.B.S. was supported by the Koch Institute - DF/HCC Bridge Project. J.M. was supported by the Swedish Research Council. P.P.H. was supported by 2T32CA071345-21A1. M.G.V.H. was supported by 5R35CA242379, the MIT Center for Precision Cancer Medicine, the Ludwig Center at MIT, and a faculty scholars award from HHMI. D.T.S. was supported by the Harvard Stem Cell Institute and the Ludwig Center at Harvard, as well as the Gerald and Darlene Jordan Chair of Medicine at Harvard.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.cmet.2020.07.009.
DECLARATION OF INTERESTS
D.B.S. is co-founder and owns equity in Clear Creek Bio. P.P.H. is a consultant for Auron Therapeutics. M.G.V.H. is a consultant and SAB member for Agios Pharmaceuticals, Aeglea Biotherapeutics, iTeos Therapeutics, and Auron Therapeutics. D.T.S. is a director and equity holder of Agios Pharmaceuticals, Magenta Therapeutics, Editas Medicines, Clear Creek Bio, and LifeVaultBio; he is a founder of Fate Therapeutics and Magenta Therapeutics and a consultant to FOG Pharma, VcanBio, and Flagship Pioneering. N.v.G., A. Schajnovitz, T.O., and D.T.S. are inventors on patents related to this work.
REFERENCES
- Bajzikova M, Kovarova J, Coelho AR, Boukalova S, Oh S, Rohlenova K, Svec D, Hubackova S, Endaya B, Judasova K, et al. (2019). Reactivation of dihydroorotate dehydrogenase-driven pyrimidine biosynthesis restores tumor growth of respiration-deficient cancer cells. Cell Metab 29, 399–416.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baryawno N, Przybylski D, Kowalczyk MS, Kfoury Y, Severe N, Gustafsson K, Kokkaliaris KD, Mercier F, Tabaka M, Hofree M, et al. (2019). A cellular taxonomy of the bone marrow stroma in homeostasis and leukemia. Cell 177, 1915–1932.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrmann L, Wellbrock J, and Fiedler W (2018). Acute myeloid leukemia and the bone marrow niche- take a closer look. Front. Oncol 8, 444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond MR, and Hanover JA (2015). A little sugar goes a long way: the cell biology of O-GlcNAc. J. Cell Biol 208, 869–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boukalova S, Hubackova S, Milosevic M, Ezrova Z, Neuzil J, and Rohlena J (2020). Dihydroorotate dehydrogenase in oxidative phosphorylation and cancer. Biochim. Biophys. Acta Mol. Basis Dis. 1866, 165759. [DOI] [PubMed] [Google Scholar]
- Boyd AL, Aslostovar L, Reid J, Ye W, Tanasijevic B, Porras DP, Shapovalova Z, Almakadi M, Foley R, Leber B, et al. (2018). Identification of chemotherapy-induced leukemic-regenerating cells reveals a transient vulnerability of human AML recurrence. Cancer Cell 34, 483–498.e5. [DOI] [PubMed] [Google Scholar]
- Cantor JR, Abu-Remaileh M, Kanarek N, Freinkman E, Gao X, Louissaint A Jr., Lewis CA, and Sabatini DM (2017). Physiologic medium rewires cellular metabolism and reveals uric acid as an endogenous inhibitor of UMP synthase. Cell 169, 258–272.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WL, Wang YY, Zhao A, Xia L, Xie G, Su M, Zhao L, Liu J, Qu C, Wei R, et al. (2016). Enhanced fructose utilization mediated by SLC2A5 is a unique metabolic feature of acute myeloid leukemia with therapeutic potential. Cancer Cell 30, 779–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong J, Soufan O, Li C, Caraus I, Li S, Bourque G, Wishart DS, and Xia J (2018). MetaboAnalyst 4.0: towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 46, W486–W494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corral J, Lavenir I, Impey H, Warren AJ, Forster A, Larson TA, Bell S, McKenzie AN, King G, and Rabbitts TH (1996). An Mll-AF9 fusion gene made by homologous recombination causes acute leukemia in chimeric mice: a method to create fusion oncogenes. Cell 85, 853–861. [DOI] [PubMed] [Google Scholar]
- Davidson SM, Papagiannakopoulos T, Olenchock BA, Heyman JE, Keibler MA, Luengo A, Bauer MR, Jha AK, O’Brien JP, Pierce KA, et al. (2016). Environment impacts the metabolic dependencies of Ras-driven non-small cell lung cancer. Cell Metab. 23, 517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietmair S, Timmins NE, Gray PP, Nielsen LK, and Krömer JO (2010). Towards quantitative metabolomics of mammalian cells: development of a metabolite extraction protocol. Anal. Biochem 404, 155–164. [DOI] [PubMed] [Google Scholar]
- Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS, Ritchey JK, Young MA, Lamprecht T, McLellan MD, et al. (2012a). Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 481, 506–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L, Saunders TL, Enikolopov G, and Morrison SJ (2012b). Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 481, 457–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farge T, Saland E, de Toni F, Aroua N, Hosseini M, Perry R, Bosc C, Sugita M, Stuani L, Fraisse M, et al. (2017). Chemotherapy-resistant human acute myeloid leukemia cells are not enriched for leukemic stem cells but require oxidative metabolism. Cancer Discov. 7, 716–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- German NJ, Yoon H, Yusuf RZ, Murphy JP, Finley LW, Laurent G, Haas W, Satterstrom FK, Guarnerio J, Zaganjor E, et al. (2016). PHD3 loss in cancer enables metabolic reliance on fatty acid oxidation via deactivation of ACC2. Mol. Cell 63, 1006–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goswami CP, and Nakshatri H (2014). PROGgeneV2: enhancements on the existing database. BMC Cancer 14, 970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory MA, Nemkov T, Park HJ, Zaberezhnyy V, Gehrke S, Adane B, Jordan CT, Hansen KC, D’Alessandro A, and DeGregori J (2019). Targeting glutamine metabolism and redox state for leukemia therapy. Clin. Cancer Res 25, 4079–4090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Häussinger D, and Schliess F (2007). Glutamine metabolism and signaling in the liver. Front. Biosci 12, 371–391. [DOI] [PubMed] [Google Scholar]
- Jacque N, Ronchetti AM, Larrue C, Meunier G, Birsen R, Willems L, Saland E, Decroocq J, Maciel TT, Lambert M, et al. (2015). Targeting glutaminolysis has antileukemic activity in acute myeloid leukemia and synergizes with BCL-2 inhibition. Blood 126, 1346–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, and Goto S (2000). KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller MA, Kampjut D, Harrison SA, and Ralser M (2017). Sulfate radicals enable a non-enzymatic Krebs cycle precursor. Nat. Ecol. Evol 1, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krivtsov AV, Twomey D, Feng Z, Stubbs MC, Wang Y, Faber J, Levine JE, Wang J, Hahn WC, Gilliland DG, et al. (2006). Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature 442, 818–822. [DOI] [PubMed] [Google Scholar]
- Kültz D (2005). Molecular and evolutionary basis of the cellular stress response. Annu. Rev. Physiol 67, 225–257. [DOI] [PubMed] [Google Scholar]
- Kusumbe AP, Ramasamy SK, Starsichova A, and Adams RH (2015). Sample preparation for high-resolution 3D confocal imaging of mouse skeletal tissue. Nat. Protoc 10, 1904–1914. [DOI] [PubMed] [Google Scholar]
- Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M, Ashton JM, Pei S, Grose V, O’Dwyer KM, et al. (2013). BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 12, 329–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemberg KM, Vornov JJ, Rais R, and Slusher BS (2018). We’re not “DON” yet: optimal dosing and prodrug delivery of 6-diazo-5-oxo-L-norleucine. Mol. Cancer Ther 17, 1824–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JA (2017). Comment on: genomics of primary chemoresistance and remission induction failure in paediatric and adult acute myeloid leukaemia. Br. J. Haematol 176, 5–6. [DOI] [PubMed] [Google Scholar]
- Matre P, Velez J, Jacamo R, Qi Y, Su X, Cai T, Chan SM, Lodi A, Sweeney SR, Ma H, et al. (2016). Inhibiting glutaminase in acute myeloid leukemia: metabolic dependency of selected AML subtypes. Oncotarget 7, 79722–79735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E, et al. (2003). PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet 34, 267–273. [DOI] [PubMed] [Google Scholar]
- Muir A, Danai LV, Gui DY, Waingarten CY, Lewis CA, and Vander Heiden MG (2017). Environmental cystine drives glutamine anaplerosis and sensitizes cancer cells to glutaminase inhibition. eLife 6, e27713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naviaux RK (2014). Metabolic features of the cell danger response. Mitochondrion 16, 7–17. [DOI] [PubMed] [Google Scholar]
- Ni F, Yu WM, Li Z, Graham DK, Jin L, Kang S, Rossi MR, Li S, Broxmeyer HE, and Qu CK (2019). Critical role of ASCT2-mediated amino acid metabolism in promoting leukaemia development and progression. Nat. Metab 1, 390–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowell PC (1976). The clonal evolution of tumor cell populations. Science 194, 23–28. [DOI] [PubMed] [Google Scholar]
- Pei S, Minhajuddin M, Callahan KP, Balys M, Ashton JM, Neering SJ, Lagadinou ED, Corbett C, Ye H, Liesveld JL, et al. (2013). Targeting aberrant glutathione metabolism to eradicate human acute myelogenous leukemia cells. J. Biol. Chem 288, 33542–33558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter N, Miraki-Moud F, Ermini L, Titley I, Vijayaraghavan G, Papaemmanuil E, Campbell P, Gribben J, Taussig D, and Greaves M (2019). Single-cell analysis of clonal architecture in acute myeloid leukaemia. Leukemia 33, 1113–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykes DB, Kfoury YS, Mercier FE, Wawer MJ, Law JM, Haynes MK, Lewis TA, Schajnovitz A, Jain E, Lee D, et al. (2016). Inhibition of dihydroorotate dehydrogenase overcomes differentiation blockade in acute myeloid leukemia. Cell 167, 171–186.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardito S, Oudin A, Ahmed SU, Fack F, Keunen O, Zheng L, Miletic H, Sakariassen PØ, Weinstock A, Wagner A, et al. (2015). Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat. Cell Biol 17, 1556–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Sanchez-Martin M, Wang X, Knapp KM, Koche R, Vu L, Nahas MK, He J, Hadler M, Stein EM, et al. (2017). Patient-derived xenotransplants can recapitulate the genetic driver landscape of acute leukemias. Leukemia 31, 151–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YH, Israelsen WJ, Lee D, Yu VWC, Jeanson NT, Clish CB, Cantley LC, Vander Heiden MG, and Scadden DT (2014). Cell-state-specific metabolic dependency in hematopoiesis and leukemogenesis. Cell 158, 1309–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart DS, Feunang YD, Marcu A, Guo AC, Liang K, Vázquez-Fresno R, Sajed T, Johnson D, Li C, Karu N, et al. (2018). HMDB 4.0: the human metabolome database for 2018. Nucleic Acids Res. 46, D608–D617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart DS, Tzur D, Knox C, Eisner R, Guo AC, Young N, Cheng D, Jewell K, Arndt D, Sawhney S, et al. (2007). HMDB: the human metabolome database. Nucleic Acids Res. 35, D521–D526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wunderlich M, Mizukawa B, Chou FS, Sexton C, Shrestha M, Saunthararajah Y, and Mulloy JC (2013). AML cells are differentially sensitive to chemotherapy treatment in a human xenograft model. Blood 121, e90–e97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye H, Adane B, Khan N, Sullivan T, Minhajuddin M, Gasparetto M, Stevens B, Pei S, Balys M, Ashton JM, et al. (2016). Leukemic stem cells evade chemotherapy by metabolic adaptation to an adipose tissue niche. Cell Stem Cell 19, 23–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Trachootham D, Liu J, Chen G, Pelicano H, Garcia-Prieto C, Lu W, Burger JA, Croce CM, Plunkett W, et al. (2012). Stromal control of cystine metabolism promotes cancer cell survival in chronic lymphocytic leukaemia. Nat. Cell Biol 14, 276–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The bulk mRNA sequencing data that support the findings of this study have been deposited in GEO with the accession number GSE139159. The single-cell RNA sequencing data were generated previously (Baryawno et al., 2019) and are deposited in GEO (GSE128423). A portal for exploring the entire single-cell atlas is available (portals.broadinstitute.org/single_cell/study/mouse-bone-marrow-stroma-in-homeostasis). All other data supporting the findings of this study are available within the paper.