

An efficient amination reaction of allenyl ethers via copper/Lewis acid synergistic catalysis yields functionalized oxalylamines.
Abstract
Allylamines have long been recognized as valuable synthons because of their excellent reactivity in organic synthesis. Here, an efficient amination reaction of allenyl ethers via copper/Lewis acid synergistic catalysis has been established, providing straightforward access to diverse functionalized Z-oxalylamines and E-halogenated oxalylamines in good to excellent yields with high regio- and stereoselectivities. The developed method tolerates more than 100 examples that include late-stage functionalization of bioactive molecules, and features gram-scale synthesis of oxalylamines with high turnover number (TON > 1000) under mild and simple conditions. The applicability of the protocol is further demonstrated with the construction of drug molecules.
INTRODUCTION
Allylamines, especially those with easy-to-transform functional groups, are an indispensable class of building blocks in the field of organic synthesis and the pharmaceutical industry (1). Typically, they are also the versatile synthetic intermediates in the atomic economical construction of alkaloids, amino acids, and complex natural products (2–4). A wide range of drug molecules (5–7) could be obtained by the derivatization of allylamines (Fig. 1A). In recent years, a variety of synthetic methods for accessing allylamine derivatives have been developed (8–14). Among the myriad strategies, the notable way to establish versatile allylamine scaffolds is based on the aminative functionalization of allenes (15–18).
Fig. 1. Examples of biologically active molecules and strategies to obtain allylamine derivatives.
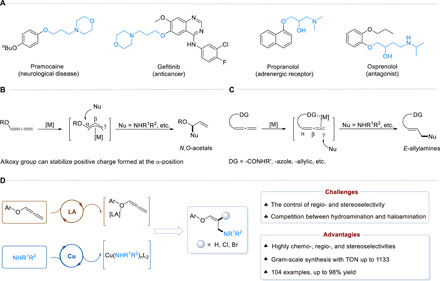
(A) Examples of biologically active molecules. (B) Hydroamination of alkoxyallenes. (C) Hydroamination of allenes with directing group. (D) Our strategy: Copper/Lewis acid synergistic catalysis.
Allenes are a class of unsaturated hydrocarbons with unique structures, which have high reactivity and multiple reaction sites (19–22). The introduction of amines with high regio- and stereoselectivity into the carbon-carbon double bonds of allenes is a fundamental organic transformation (23). To achieve regioselective amination of allenes, some strategies have been proposed. Among them, alkoxyallenes were used as the reaction substrates, taking advantage of their electronic bias to control the regioselectivity, and the aminative functionalization of alkoxyallenes usually generates the N,O-acetal products due to the positive charge formed at α-position stabilized by alkoxy group (Fig. 1B) (24–27). Recently, a substrate-directed strategy has come out as an attractive method to promote the metal-catalyzed regioselective functionalization of allenes. Various directing groups (DGs), such as amide, azole, and allylic, have been introduced to regulate the regioselective amination of allenes. However, E-allylamines are generally obtained, while thermodynamically unstable Z-allylamines are difficult to be constructed in most cases (Fig. 1C) (28, 29). Despite extensive efforts dedicated to developing efficient methods for aminative functionalization of allenes, there are still great challenges owing to regio- and stereocontrol as well as reactivity issues: (i) The unique accumulated diene structural units have greatly limited the selective activation of the two carbon-carbon double bonds of allenes (30). (ii) Efficient selective synthesis of Z-allylamines remains a challenging task for their thermodynamic instability (31–34). (iii) Transition metal catalysts might be poisoned or deactivated because of the high affinity of amines, which are prone to be oxidized under the oxidation reaction conditions (35). Therefore, the aminofunctionalization reactions are usually limited by the protection (and subsequently deprotection) of the amines (36).
To address these challenges, a strategy of copper/Lewis acid synergistic catalysis is proposed, which is able to activate both substrates to drive the bond formation reaction simultaneously and individually (37–39). Lewis acid is known to have an empty orbital, which can coordinate with a lone pair of electrons on the oxygen atom. We envisioned that merging an oxygen atom in alkoxyallene with Lewis acid might effectively tune the selectivity of the accumulated double bonds (40–42). Meanwhile, the synergy effect of copper and Lewis acid would ensure the control of the configuration over the activated intermediate (43). Here, a diverse collection of medicinally relevant oxalylamines via copper/Lewis acid synergistic catalysis is disclosed (Fig. 1D). A wide range of Z-oxalylamines and E-halogenated oxalylamines could be successfully synthesized with high yields and excellent regio- and stereoselectivities. An exciting opportunity offered by the strategy is that drug molecules could be obtained via an operationally simple reduction of the Z-oxalylamines.
RESULTS
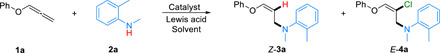
Optimization studies for the aminative functionalization of allenyl ethers
To verify this strategy, phenyl 1,2-propadienyl ether (1a) and N,2-dimethylaniline (2a) were used as the model substrates (Table 1). First, in the presence of Cu(OAc)2 as a catalyst and AgF as Lewis acid in CH2Cl2 at 50°C for 6 hours, the hydroamination product Z-3a was afforded in 24% yield (entry 1). To our delight, different Lewis acids were amenable for this transformation and ZnCl2 served as the most effective additive, with Z-3a obtained in 98% nuclear magnetic resonance (NMR) yield and 93% isolated yield (entries 2 to 5). Studies of the copper salts indicated that Cu(OAc)2 was the most reactive and selective catalyst for direct addition to forming oxalylamine Z-3a. The amounts of Cu(OAc)2 and ZnCl2 could reduce to 5 mole percent (mol %) loading without decreasing the yield (entry 5). Control experiments excluding additives or copper salts gave no corresponding product, demonstrating that Lewis acid and copper catalyst were indispensable for the success of this hydroamination reaction (entries 6 and 7). We believed that CuX2 could work as both the catalyst and Lewis acid, playing the role of halogen source to realize the aminative difunctionalization of allenes. As expected, the desired chloraminated product E-4a was obtained in 24% NMR yield by using 60 mol % of CuCl2 under the oxygen (1 atm) atmosphere at 40°C for 6 hours (entry 8). Further solvent screening revealed that the nonpolar solvents exhibited much higher reactivity than polar solvents, and dioxane was found to be the optimal medium to deliver E-4a in 86% NMR yield with no detected alternative regio- and stereoisomers (entries 9 to 11). Further increasing the amount of CuCl2 has no effect on chloramination reaction (entry 12). However, only a trace amount of E-4a was observed under N2 atmosphere, indicating that O2 played a vital role in the chloramination product formation step (entry 13). More detailed reaction conditions were displayed in the Supplementary Materials.
Table 1. Optimization studies for the aminative functionalization of allenyl ethers.
Exploration of catalyst, Lewis acid, and solvent effects on the aminative functionalization of allenyl ether 1a. DCE, dichloroethane; DMSO, dimethyl sulfoxide; ND, not determined.
| Entry | Catalyst | Lewis acid | Solvent | Yield of Z-3a (%)* | Yield of E-4a (%)† |
| 1‡ | Cu(OAc)2 | AgF | CH2Cl2 | 24 | ND |
| 2‡ | Cu(OAc)2 | Fe(OTf)2 | CH2Cl2 | 42 | ND |
| 3‡ | Cu(OAc)2 | InBr3 | CH2Cl2 | 74 | ND |
| 4‡ | Cu(OAc)2 | CuCl2 | CH2Cl2 | 88 | 12 |
| 5‡,§ | Cu(OAc)2 | ZnCl2 | CH2Cl2 | 98 (93) | ND |
| 6‡ | – | ZnCl2 | CH2Cl2 | ND | ND |
| 7‡ | Cu(OAc)2 | – | CH2Cl2 | Trace | ND |
| 8║ | – | CuCl2 | CH2Cl2 | 18 | 24 |
| 9║ | – | CuCl2 | DCE | 10 | 58 |
| 10║ | – | CuCl2 | DMSO | ND | ND |
| 11 ║ | – | CuCl2 | Dioxane | 10 | 86 (82) |
| 12║,¶ | – | CuCl2 | Dioxane | 10 | 85 |
| 13║,# | – | CuCl2 | Dioxane | Trace | Trace |
*The Z-type to E-type geometric ratio (Z/E) and yield of Z-3a were determined by 1H NMR using CH2BrCl as the internal standard analysis of the crude mixtures. The isolated yield of Z-3a was given in parentheses. Z/E > 20:1 unless otherwise noted.
†The E-type to Z-type geometric ratio (E/Z) was determined by GC-MS analysis of the crude mixtures. Yield of E-4a was determined by 1H NMR using CH2BrCl as the internal standard analysis of the crude mixtures. The isolated yield of E-4a was given in parentheses. E/Z > 20:1 unless otherwise noted.
‡Condition A: All reactions were performed with 1a (0.30 mmol), 2a (0.20 mmol), catalyst (10 mol %), Lewis acid (10 mol %), and solvent (1.0 ml), 50°C, 6 hours.
§Cu(OAc)2 (5 mol %) and ZnCl2 (5 mol %) were used.
║Condition B: All reactions were performed with 1a (0.30 mmol), 2a (0.20 mmol), CuCl2 (60 mol %), solvent (1.0 ml), and O2 (1 atm), 40°C.
¶CuCl2 (1 equiv) was used.
#Under N2 atmosphere.
Substrate scope of hydroamination reactions
With the optimized reaction conditions in hand, the generality of hydroamination reaction was explored and the results were summarized in Fig. 2. Substituents on aromatic secondary amines were initially surveyed. Reactions of substrates bearing electron-donating and electron-withdrawing groups on the benzene ring proceeded well to afford the corresponding products Z-3a to Z-3o in moderate to good yields (32 to 93%). However, electron-deficient N-methyl aromatic amines exhibited lower reactivities (57% for E-3m, 44% for Z-3n, and 32% for Z-3o, respectively). It was noteworthy that the configuration of the major product for the substrate bearing CN group was found to be reversed (Z:E = 1:12). The molecular structure and the relative stereochemistry of Z-3o were unambiguously confirmed by x-ray crystallographic analysis (44). Furthermore, a naphthyl-substituted amine and cyclic aromatic secondary amines, including carbazole and 1,2,3,4-tetrahydroquinoline, could be accommodated well to deliver the hydroamination products Z-3p to Z-3r in 56 to 87% yields. The conversions of diversely substituted amines, such as N-ethylaniline, N-allyl aniline, N-benzylaniline, and N-pentylaniline, proceeded well to produce the desired products Z-3s to Z-3v in satisfactory yields (81 to 90%). However, the hydroamination of aromatic primary amines gave the corresponding products (Z-3w to Z-3z) a slightly lower yield than those of secondary amines.
Fig. 2. Substrate scope of hydroamination reactions.
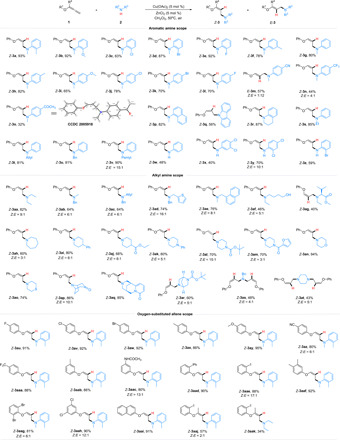
Condition A: 1 (0.3 mmol), 2 (0.2 mmol), Cu(OAc)2 (5 mol %), ZnCl2 (5 mol %), and CH2Cl2 (1.0 ml), 50°C, 6 hours. All yields (given as a percentage) were isolated unless otherwise noted. The Z-type to E-type geometric ratio (Z/E) was determined by 1H NMR using CH2BrCl as the internal standard analysis of the crude mixtures. Z/E > 20:1 unless otherwise noted.
Then, a range of aliphatic secondary amines, including acyclic and cyclic secondary amines, was examined. Pleasingly, the reactivity of the system for acyclic amines enabled the hydroamination of alkyl chain amines with different functional groups (thienyl, naphthylmethyl, hydroxyl, amino acid ester, etc.), which provided the desired hydroamination products (Z-3aa to Z-3ag) in 40 to 82% yields with moderate to excellent stereoselectivities. Moreover, the reactivity of different cyclic amines was investigated. The hydroamination of N-heterocycles, which are commonly used for drug synthesis, afforded the desired products containing cyclohexylamine (2ah), piperidine (2ai and 2aj), piperazine (2ak-2am), morpholine (2an), and thiomorpholine (2ao) in good to high yields with moderate stereoselectivities. Gratifyingly, this process was also compatible with the bridged cyclic amines, which successfully transformed to the desired products (Z-3ap to Z-3ar) in moderate to excellent isolated yields (60 to 86%) with retention of stereochemistry. Notably, evaluation of alkylamines bearing two N-H sites with the same reactivity indicated that they were all suitable for this transformation (Z-3as and Z-3at). However, the tightly binding primary aliphatic amines could not transform to the target products in this system.
The range of oxygen-substituted allenes (1) for hydroamination was subsequently probed. Various functional groups such as -X (1au-1aw, 1aae), which could serve as handles for further functionalization through cross-coupling, -Me (1ax and 1aab), -OMe (1ay), -CN (1az), -CF3 (1aaa), -NHCOCH3 (1aac), and -Ph (1aad) were used and successfully converted into the desired products in satisfying yields (80 to 95%). A notable feature of the reaction was that the electronic effects had little influence on the reaction conversion. The amination was also suitable for the disubstituted allene ethers. Both 3,4- (1aaf) and 2,6- (1aag) as well as 3,5- (1aah) disubstituted allene ethers and even a naphthyloxy-allene (1aai) could transform to the Z-oxalylamine derivatives in high yields with moderate to excellent stereoselectivities. Specifically, when 1-(buta-2,3-dien-2-yloxy)-2-iodobenzene was used to react with N,2-dimethylaniline or diethylamine under the standard conditions, highly substituted allylamines could be obtained. In addition, the configurations of Z-3aaj and Z-3aak were confirmed by nuclear Overhauser effect spectroscopy experiments (see the Supplementary Materials for details).
Substrate scope of chloramination reactions
The scope of the aminochlorination reaction was then evaluated (Fig. 3). A broad range of N-methylanilines with different functional groups such as methyl, methoxyl, chloro, bromo, fluoro, trifluoromethyl, carbonyl at ortho-, meta-, and para-positions of the aromatic ring, as well as naphthyl was well tolerated in this transformation, and the desired products (E-4a to E-4m) were obtained in moderate to high yields. Moreover, reactions of aromatic secondary amines with ethyl (2n), allyl (2o), and benzyl (2p) substituents at the amino position afforded the desired products in synthetically useful yields. However, the aniline partners showed comparatively lower reactivity (40% for E-4q and 45% for E-4r), likely due to the high affinity of primary amines. Overall, oxygen-substituted allene (1) bearing various electron-deficient or electron-rich functionalities on the aryl ring also worked well, giving the desired chloramination products (E-4s to E-4ad) in 52 to 90% yields with high stereoselectivities (E/Z > 20:1). Among them, phenyl allene ether derivatives with disubstituted groups on benzene ring underwent chloramination to afford the corresponding products E-4ae to E-4ag in 80 to 84% yields. In addition, naphthyloxy-allene (1ah) was found to participate readily in this hydroamination reaction with moderate yield. The molecular structure and the relative stereochemistry of E-4ai were unequivocally confirmed by x-ray crystallographic analysis (45).
Fig. 3. Substrate scope of chloramination reactions.
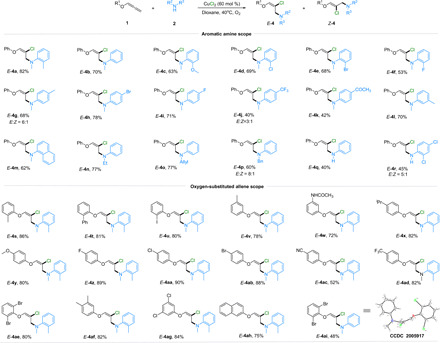
Condition B: 1 (0.3 mmol), 2 (0.2 mmol), CuCl2 (60 mol %), dioxane (1.0 ml), and O2 (1 atm), 40°C, 6 hours. All yields (given as a percentage) were isolated unless otherwise noted. The E-type to Z-type geometric ratio (E/Z) was determined by gas chromatography–mass spectrometry (GC-MS) analysis of the crude mixtures. E/Z > 20:1 unless otherwise noted.
DISCUSSION
Synthetic utilities
To highlight the practicality and synthetic value of this copper/Lewis acid catalyzed aminative functionalization of alkoxyallenes, the reaction was applied for the further transformations to deliver more complex and useful compounds. As delineated in Fig. 4A, the late-stage functionalization of bioactive molecules could manifest the utility of this transformation. Under the hydroamination reaction conditions, a series of bioactive molecules such as aminoglutethimide, atomoxetine (a selective norepinephrine reuptake inhibitor) (46), desipramine, and ibrutinib intermediate could transform to the Z-oxalylamine derivatives (Z-3aal to Z-3aao) in moderate to good yields without a loss of optical purity. In addition, gram-scale (10 mmol) experiments were successfully conducted to confirm the efficiency of the amination functionalization of alkoxyallenes (Fig. 4B). The desired hydroamination products Z-3a and Z-3aj and chloramination product E-4a could all be obtained with high yields and moderate to excellent steoroselectivities. Noteworthily, when reducing the loading of Cu(OAc)2 and ZnCl2 to 0.06 mol %, the desired product Z-3a was formed in 68% yield with high turnover number (TON = 1133). Besides, cross-coupling reaction was achieved with E-4a and phenyl magnesium bromide, as C─C bond formation occurred at the position of alkenyl chloride, leading to the useful phenyl-substituted oxalylamine (6). Moreover, the chloramination reaction could be extended to different halogen sources. For instance, under the conditions of 60 mol % of CuBr2, the bromination reaction proceeded smoothly to deliver the brominated oxalylamine E-4aj in 62% yield with E/Z ratio equaling to 13:1 (Fig. 4B) (47). Given the functionary of this method, the intermediate Z-3aap of drug molecule could be constructed in gram scale. In the presence of Pd/C (5 mol %) as the catalyst, the drug molecule pramoxine (7) could be isolated in 94% yield under H2 atmosphere (Fig. 4C) (48).
Fig. 4. Synthetic utility.
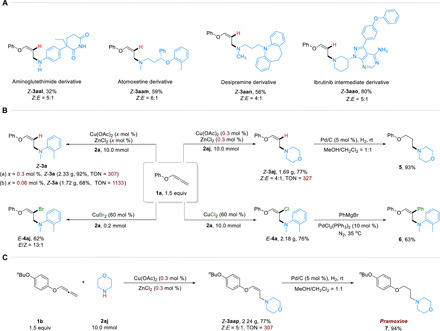
(A) Late-stage hydroamination of bioactive molecules. (B) Gram-scale reactions and further decorations. (C) Synthesis of pramoxine.
Mechanistic studies
To gain more insight into the mechanism of the amination process, a series of control experiments were conducted. First, a deuterium-labeling study was performed under the deuteration reaction condition A′. Applying deuterated diethylamine d-2ab as the amine source, the corresponding deuterated product Z-d-3ab was obtained with a separated yield of 80% and a deuteration rate of 82%, showing that amine served as a hydrogen atom donor (Fig. 5A). Moreover, the D2O labeling experiment proved that amine and D2O might undergo proton exchange in the hydroamination reaction (see the Supplementary Materials for details). Then, several radical trapping agents, such as 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO), 2,6-di-tert-butyl-4-methylphenol (BHT), and 1,1-diphenylethylene, were used to perform the radical trapping experiments. The NMR yield of the products (Z-3a and E-4a) showed that no reactions were inhibited except that TEMPO reduced the yield of the chloramination for its strong oxidation ability. These observations indicated that the free-radical process should not be involved (Fig. 5B). The addition of phenyl allene (1a′), nitrogen-substituted allene (1a″), or sulfur-substituted allene (1a‴) instead of alkoxyallene under the conditions A or B failed to mediate the desired hydroamination or chloramination products (Fig. 5C), which suggested that the coordination between oxygen atom and Lewis acid was important in the aminative functionalization of alkoxyallenes. Furthermore, no chloramination product E-4a was formed in the intermediate experiment when using Z-3a as the substrate, demonstrating that the hydroamination and chloramination reaction might go through different pathways (Fig. 5D). Kinetic analysis experiments for hydroamination were conducted under the most suitable reaction conditions (Fig. 5E). The rate data indicated a first-order dependence only on the concentration of Cu catalyst, which revealed that hydrolysis should be the rate-determining step (49, 50).
Fig. 5. Mechanistic studies.
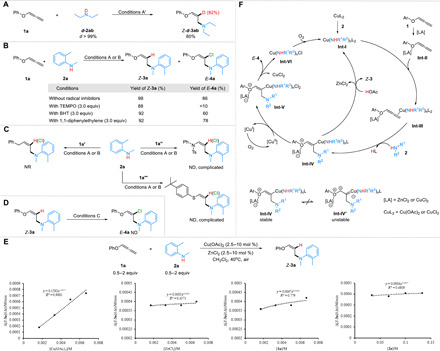
(A) Deuterium-labeling study. (B) Radical trapping experiment. (C) Control experiment. (D) Intermediate experiment. (E) Determination of the order. (F) Proposed mechanism. Condition A′: 1a (0.3 mmol), d-2ab (0.2 mmol), Cu(OAc)2 (5 mol %), ZnCl2 (5 mol %), and dry CH2Cl2 (1.0 ml), 50°C, 6 hours. Condition A: 1 (0.3 mmol), 2a (0.2 mmol), Cu(OAc)2 (5 mol %), ZnCl2 (5 mol %), and CH2Cl2 (1.0 ml), 50°C, 6 hours. Condition B: 1 (0.3 mmol), 2a (0.2 mmol), CuCl2 (60 mol %), dioxane (1.0 ml), and O2 (1 atm), 40°C, 6 hours. Condition C: Z-3a (0.3 mmol), CuCl2 (60 mol %), dioxane (1.0 ml), and O2 (1 atm), 40°C, 6 hours. 1a′: Buta-2,3-dien-1-ylbenzene. 1a″: 4-Methyl-N-phenyl-N-(propa-1,2-dien-1-yl)benzenesulfonamide. 1a‴: 1-tert-Butyl-4-(propa-1,2-dienylthio)benzene.
On the basis of the above investigations and previous literatures (28, 29, 51), a mechanism is proposed in Fig. 5F. Initially, coordination of Cu(II) to the amine substrate 2 generates the intermediate Int-I. At the same time, Int-II is formed by coordination of Lewis acid (ZnCl2 or CuCl2) with alkoxyallene 1. Subsequently, a nucleophilic attack of amine 2 to Cu(II)/allene complex Int-III gives an E-alkenylcopper Int-IV exclusively, which improves the regioselectivity of the reaction. The isomerization of Int-IV to Int-IV′ could be inhibited by the coordination of Lewis acid and oxygen to enhance the stereoselectivity of this process (43). In the case of addition of Cu(OAc)2 and ZnCl2, intermediate Int-IV might undergo hydrolysis to deliver the hydroamination product Z-3. Alternatively, in the case of addition of CuCl2, the resulting alkenyl CuII species Int-IV is oxidized by another equivalent of [CuII] to yield an alkenyl CuIII intermediate Int-V that can undergo facile C─Cl bond formation through reductive elimination. Last, rapid aerobic oxidation of [CuI] regenerates [CuII] to complete the catalytic cycle.
In conclusion, by using a strategy of synergistic catalysis, we have realized a copper/Lewis acid–catalyzed intermolecular hydroamination of alkoxyallenes to produce Z-oxalylamines. The intermolecular aminohalogenation reactions have been developed in succession to obtain a series of E-halogenated oxalylamines by using CuX2 as the transition metal catalyst and Lewis acid as well as halogen sources. This protocol was performed under mild reaction conditions and compatible with a wide scope of alkoxyallenes and amines, simultaneously with high atom economy. The advantages of gram-scale reactions, late-stage functionalization of bioactive molecules, and the synthesis of drug molecules all showcased the potential of this strategy to be widely used.
MATERIALS AND METHODS
General procedure A: Hydroamination of allenyl ethers
The mixture of oxygen-substituted allene 1 (0.3 mmol), amine 2 (0.2 mmol), Cu(OAc)2 (5 mol %), and ZnCl2 (5 mol %) in CH2Cl2 (1.0 ml) was added to a 10-ml dried reaction tube successively. The mixture was stirred at 50°C for 6 hours under air atmosphere. After the reaction was completed, the mixture was cooled to room temperature, diluted with H2O (15 ml), and extracted with CH2Cl2 (10 ml × 3). Combined organic layers were dried over anhydrous MgSO4 and concentrated in vacuum. The resulting crude materials were purified by flash column chromatography on silica gel with petroleum ether/ethyl acetate (1:1 to 500:1) to give the desired products Z-3 in 40 to 95% isolated yields with Z/E ratio from 3:1 to single Z configuration.
General procedure B: Chloramination of allenyl ethers
The mixture of amine 1 (0.2 mmol), oxygen-substituted allene 2 (0.3 mmol), and CuCl2 (60 mol %) in 1,4-dioxane (1.0 ml) was added to a 25-ml dried reaction tube successively. The mixture was stirred at 40°C for 6 hours under O2 atmosphere. After the reaction was completed, the mixture was cooled to room temperature, diluted with H2O (15 ml), and extracted with EtOAc (10 ml × 3). Collected organic layers were dried over anhydrous MgSO4 and concentrated in vacuum. The resulting crude materials were purified by flash column chromatography on silica gel with petroleum ether/ethyl acetate (50:1 to 200:1) to give the desired products E-4 in 40 to 89% isolated yields with E/Z ratio from 3:1 to single E configuration.
General procedure for the preparation of allenyl ethers 1 (52)
Step 1
3-Bromopropyne (16 mmol) was added to a flask containing phenol (14 mmol) and K2CO3 (2.5 equiv) in N,N′-dimethylformamide (20 ml) at room temperature under Ar. After addition, stirring was continued for 12 hours. The mixture was treated with 10 ml of H2O. The resulting mixture was extracted with Et2O (30 ml × 4) and dried over anhydrous Na2SO4. After concentration, the residue was purified by flash column chromatography on silica gel (EtOAc/petroleum ether = 1:100) to give the corresponding alkyne.
Step 2
t-BuOK (0.5 equiv) was added to a solution of the above alkyne (4.0 mmol) in tetrahydrofuran (THF) (20 ml) at 0°C under Ar. The mixture was stirred at room temperature until completion of the reaction [thin-layer chromatography (TLC)]. The solution was filtered through Celite, and the solvent was concentrated under reduced pressure and purified by column chromatography to give the desired products 1 in 70 to 90% yields (petroleum ether). The spectral data of 1 were in accordance with the literature.
General procedure for the preparation of buta-2,3-dien-1-ylbenzene 1a′ (53)
Prop-2-yn-1-ylbenzene (5.0 mmol, 1.0 equiv) and dicyclohexylamine (9.0 mmol, 1.8 equiv) were added to a stirred solution of paraformaldehyde (12.5 mmol, 2.5 equiv) and CuI (2.5 mmol, 0.5 equiv) in dioxane (25 ml) under atmosphere of argon. The resulting mixture was then refluxed for 4 hours. After the reaction was completed as monitored by TLC, the mixture was cooled to room temperature and filtered. The filtrate was concentrated under reduced pressure and then diluted with water and ether, followed by the addition of 1 N HCl to pH 1 to 2. The resulting mixture was extracted three times with ether (30 ml × 3). The organic layer was then washed with brine and dried over Na2SO4, filtrated, and evaporated of the solvent. The residue was purified by flash chromatography with hexane to give the corresponding product 1a′ as a colorless oil in 70% yield (507.5 mg). The spectral data of 1a′ were in accordance with the literature.
General procedure for the preparation of 4-methyl-N-phenyl-N-(propa-1,2-dien-1-yl)benzenesulfonamide 1a″ (54)
t-BuOK (1.5 mmol) was added to a solution of 4-methyl-N-phenyl-N-(prop-2-yn-1-yl)benzenesulfonamide (5 mmol, 1 equiv) in anhydrous THF (15 ml) in three portions under an argon atmosphere at 0°C in an ice water bath. The reaction mixture was allowed to stir at room temperature for 2 hours. The mixture was filtered through Celite, washed with EtOAc, and concentrated. The crude residue was purified by silica gel column chromatography (petroleum ether/ethyl acetate = 30:1 to 20:1) to give 1a″ as a yellow solid in 87% yield (0.124 g). The spectral data of 1a″ were in accordance with the literature.
General procedure for the preparation of 1-tert-butyl-4-(propa-1,2-dienylthio)benzene 1a‴ (55)
Step 1
A solution of sodium thiosulfate pentahydrate (2.7 g, 9.0 mmol) and benzyltriethylammonium chloride (0.2 g, 0.9 mmol) in water (2.0 ml) was added to a solution of propargyl bromide (0.96 ml, 9.0 mmol) in chloroform (8.0 ml), and the reaction mixture was heated at 60°C with vigorous stirring for 4 hours. The reaction mixture was then evaporated to dryness, and the residue was extracted with MeOH (50 ml × 2). The combined methanol extract was evaporated, and the residue was washed with CH2Cl2 to remove the catalyst and the remaining propargyl bromide. The resulting solid was dried in vacuum to give sodium propargylthiosulfate (1.22 g, 78%) as a white-cream solid.
Step 2
The p-tert-butylthiophenol (1.61 mmol) was added under stirring to an aqueous solution (5.0 ml) of KOH (0.10 g, 1.78 mmol), and the reaction mixture was heated to reflux for 1 hour. After cooling to 0°C, an aqueous solution (5.0 ml) of sodium propargylthiosulfate (0.56 g, 3.22 mmol) was added. The resulting mixture was further stirred at room temperature for 1 hour, diluted with diethyl ether (50 ml), washed with 10% KOH (15 ml) and H2O (15 ml), dried over MgSO4, and evaporated to give the crude product 1-tert-butyl-4-(prop-2-ynyldithio)benzene.
Step 3
Triphenylphosphine (0.26 g, 1.0 mmol) was added to a solution of the 1-tert-butyl-4-(prop-2-ynyldithio)benzene (0.5 mmol) in benzene (10.0 ml), and the reaction mixture was heated at 60°C. After completing the reaction (TLC), benzene was evaporated to give the crude reaction mixture as a viscous oil, from which the pure sulfides 1a‴ (yellow oil, 57%) was separated by column chromatography on silica gel with hexane as eluent.
Acknowledgments
Funding: We thank the Ministry of Science and Technology of the People’s Republic of China (2016YFA0602900), the National Nature Science Foundation of China (21871095), and the Fundamental Research Funds for the Central Universities (2020ZYGXZR094) for financial support. Author contributions: Conceptualization: Z.W. Methodology: Z.W. Investigation: Z.W., M.H., Y.J., and J.L. Writing (original draft): Z.W. Writing (review and editing): Z.W., W.W., and H.J. Funding acquisition: H.J. Resources: H.J. Supervision: W.W. and H.J. Competing interests: Z.W., H.J., and W.W. are inventors on pending patents related to this work filed by the China National Intellectual Property Administration (no. CN 112521289 A, filed on 19 March 2021; no. CN 112441934 A., filed on 5 March 2021). The authors declare no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/35/eabh4088/DC1
REFERENCES AND NOTES
- 1.Taghdiri F., Togha M., Jahromi S. R., Refaeian F., Cinnarizine for the prophylaxis of migraine associated vertigo: A retrospective study. Springerplus 3, 231 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu X.-S., Chen Y., Li M.-B., Zhou M.-G., Tian S.-K., Direct substitution of primary allylic amines with sulfinate salts. J. Am. Chem. Soc. 134, 14694–14697 (2012). [DOI] [PubMed] [Google Scholar]
- 3.Yu H., Zhang G., Huang H., Palladium-catalyzed dearomative cyclocarbonylation by C-N bond activation. Angew. Chem. Int. Ed. 54, 10912–10916 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Wang H., Wang Y., Liang D., Liu L., Zhang J., Zhu Q., Copper-catalyzed intramolecular dehydrogenative aminooxygenation: Direct access to formyl-substituted aromatic N-heterocycles. Angew. Chem. Int. Ed. 50, 5678–5681 (2011). [DOI] [PubMed] [Google Scholar]
- 5.Zou H., Chen G., Zhou S., Design, synthesis and biological activity evaluation of benzoate compounds as local anesthetics. RSC Adv. 9, 6627–6635 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen H., Hu B., Lv X., Zhu S., Zhen G., Wan M., Jain A., Gao B., Chai Y., Yang M., Wang X., Deng R., Wang L., Cao Y., Ni S., Liu S., Yuan W., Chen H., Dong X., Guan Y., Yang H., Cao X., Prostaglandin E2 mediates sensory nerve regulation of bone homeostasis. Nat. Commun. 10, 181–193 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gu X., Qiu Y., Lin M., Cui K., Chen G., Chen Y., Fan C., Zhang Y., Xu L., Chen H., Wan J.-B., Lu W., Xiao Z., CuS nanoparticles as a photodynamic nanoswitch for abrogating bypass signaling to overcome gefitinib resistance. Nano Lett. 19, 3344–3352 (2019). [DOI] [PubMed] [Google Scholar]
- 8.Collet F., Lescot C., Dauban P., Catalytic C-H amination: The stereoselectivity issue. Chem. Soc. Rev. 40, 1926–1936 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Ramirez T. A., Zhao B., Shi Y., Recent advances in transition metal-catalyzed sp3 C-H amination adjacent to double bonds and carbonyl groups. Chem. Soc. Rev. 41, 931–942 (2012). [DOI] [PubMed] [Google Scholar]
- 10.Patel S. J., Jamison T. F., Asymmetric catalytic coupling of organoboranes, alkynes, and imines with a removable (trialkylsilyloxy)ethyl group-direct access to enantiomerically pure primary allylic amines. Angew. Chem. Int. Ed. 43, 3941–3944 (2004). [DOI] [PubMed] [Google Scholar]
- 11.Xie Y., Hu J., Wang Y., Xia C., Huang H., Palladium-catalyzed vinylation of aminals with simple alkenes: A new strategy to construct allylamines. J. Am. Chem. Soc. 134, 20613–20616 (2012). [DOI] [PubMed] [Google Scholar]
- 12.Hirata G., Satomura H., Kumagae H., Shimizu A., Onodera G., Kimura M., Direct allylic amination of allylic alcohol catalyzed by palladium complex bearing phosphine-borane ligand. Org. Lett. 19, 6148–6151 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Sweeney J. B., Ball A. K., Lawrence P. A., Sinclair M. C., Smith L. J., A simple, broad-scope Nickel(0) precatalyst system for the direct amination of allyl alcohols. Angew. Chem. Int. Ed. 57, 10202–10206 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Cheng Q., Chen J., Lin S., Ritter T., Allylic amination of alkenes with iminothianthrenes to afford alkyl allylamines. J. Am. Chem. Soc. 142, 17287–17293 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zeng X., Soleilhavoup M., Bertrand G., Gold-catalyzed intermolecular Markovnikov hydroamination of allenes with secondary amines. Org. Lett. 11, 3166–3169 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu T., Mu X., Peng H., Liu G., Silver-catalyzed intramolecular aminofluorination of activated allenes. Angew. Chem. Int. Ed. 50, 8176–8179 (2011). [DOI] [PubMed] [Google Scholar]
- 17.Blieck R., Bahri J., Taillefer M., Monnier F., Copper-catalyzed hydroamination of terminal allenes. Org. Lett. 18, 1482–1485 (2016). [DOI] [PubMed] [Google Scholar]
- 18.Ma S., Xie H., Unexpected facile sequential halolactamization-hydroxylation of 2,3-allenamides with CuX2 for the efficient synthesis of 4-Halo-5-hydroxypyrrol-2(5H)-ones. Org. Lett. 2, 3801–3803 (2000). [DOI] [PubMed] [Google Scholar]
- 19.Alcaide B., Almendros P., Gold-catalyzed cyclization reactions of allenol and alkynol derivatives. Acc. Chem. Res. 47, 939–952 (2014). [DOI] [PubMed] [Google Scholar]
- 20.Huang X., Ma S., Allenation of terminal alkynes with aldehydes and ketones. Acc. Chem. Res. 52, 1301–1312 (2019). [DOI] [PubMed] [Google Scholar]
- 21.Zimmer R., Reissig H.-U., Alkoxyallenes as building blocks for organic synthesis. Chem. Soc. Rev. 43, 2888–2903 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Blieck R., Taillefer M., Monnier F., Metal-catalyzed intermolecular hydrofunctionalization of allenes: Easy access to allylic structures via the selective formation of C-N, C-C, and C-O bonds. Chem. Rev. 120, 13545–13598 (2020). [DOI] [PubMed] [Google Scholar]
- 23.Muńoz M. P., Silver and platinum-catalysed addition of O-H and N-H bonds to allenes. Chem. Soc. Rev. 43, 3164–3183 (2014). [DOI] [PubMed] [Google Scholar]
- 24.Bernar I., Fiser B., Blanco-Ania D., Gomez-Bengoa E., Rutjes F. P. J. T., Pd-catalyzed hydroamination of alkoxyallenes with azole heterocycles: Examples and mechanistic proposal. Org. Lett. 19, 4211–4214 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiong W., Yan D., Qi C., Jiang H., Palladium-catalyzed four-component cascade reaction for the synthesis of highly functionalized acyclic O,O-acetals. Org. Lett. 20, 672–675 (2018). [DOI] [PubMed] [Google Scholar]
- 26.Xiong W., Cheng R., Wu B., Wu W., Qi C., Jiang H., Palladium-catalyzed regioselective cascade reaction of carbon dioxide, amines and allenes for the synthesis of functionalized carbamates. Sci. China Chem. 63, 331–335 (2020). [Google Scholar]
- 27.Wang Y., Jiang M., Liu J.-T., Copper-catalyzed regioselective oxytrifluoromethylation of allenes using a CF3-transfer reagent. Adv. Synth. Catal. 356, 2907–2912 (2014). [Google Scholar]
- 28.Perego L. A., Blieck R., Michel J., Ciofini I., Grimaud L., Taillefer M., Monnier F., Copper-catalyzed hydroamination of N-allenylazoles: Access to amino-substituted N-vinylazoles. Adv. Synth. Catal. 359, 4388–4392 (2017). [Google Scholar]
- 29.Blieck R., Perego L. A., Ciofini I., Grimaud L., Taillefer M., Monnier F., Copper-catalysed hydroamination of N-allenylsulfonamides: The key role of ancillary coordinating groups. Synthesis 51, 1225–1234 (2019). [Google Scholar]
- 30.Xu K., Thieme N., Breit B., Atom-economic, regiodivergent, and stereoselective coupling of imidazole derivatives with terminal allenes. Angew. Chem. Int. Ed. 53, 2162–2165 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Xu Y., Wong J. J., Samkian A. E., Ko J. H., Chen S., Houk K. N., Grubbs R. H., Efficient Z-selective olefin-acrylamide cross-metathesis enabled by sterically demanding cyclometalated ruthenium catalysts. J. Am. Chem. Soc. 142, 20987–20993 (2020). [DOI] [PubMed] [Google Scholar]
- 32.Meek S. J., O’Brien R. V., Llaveria J., Schrock R. R., Hoveyda A. H., Catalytic Z-selective olefin cross-metathesis for natural product synthesis. Nature 471, 461–466 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Singh K., Staig S. J., Weaver J. D., Facile synthesis of Z-alkenes via uphill catalysis. J. Am. Chem. Soc. 136, 5275–5278 (2014). [DOI] [PubMed] [Google Scholar]
- 34.Jiang R., Ding L., Zheng C., You S.-L., Iridium-catalyzed Z-retentive asymmetric allylic substitution reactions. Science 371, 380–386 (2021). [DOI] [PubMed] [Google Scholar]
- 35.Ouyang L., Li J., Zheng J., Huang J., Qi C., Wu W., Jiang H., Access to α-amino acid esters through palladium-catalyzed oxidative amination of vinyl ethers with hydrogen peroxide as the oxidant and oxygen source. Angew. Chem. Int. Ed. 56, 15926–15930 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Butler K. L., Tragni M., Widenhoefer R. A., Gold(I)-catalyzed stereoconvergent, intermolecular enantioselective hydroamination of allenes. Angew. Chem. Int. Ed. 51, 5175–5178 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Allen A. E., MacMillan D. W. C., Synergistic catalysis: A powerful synthetic strategy for new reaction development. Chem. Sci. 3, 633–658 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen D.-F., Han Z.-Y., Zhou X.-L., Gong L.-Z., Asymmetric organocatalysis combined with metal catalysis: Concept, proof of concept, and beyond. Acc. Chem. Res. 47, 2365–2377 (2014). [DOI] [PubMed] [Google Scholar]
- 39.Li M.-L., Yu J.-H., Li Y.-H., Zhu S.-F., Zhou Q.-L., Highly enantioselective carbene insertion into N–H bonds of aliphatic amines. Science 366, 990–994 (2019). [DOI] [PubMed] [Google Scholar]
- 40.Wang Y., Liu L., Zhang L., Combining Zn ion catalysis with homogeneous gold catalysis: An efficient annulation approach to N-protected indoles. Chem. Sci. 4, 739–746 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhu X.-Q., Wang Z.-S., Hou B.-S., Zhang H.-W., Deng C., Ye L.-W., Zinc-catalyzed asymmetric formal [4+3] annulation of isoxazoles with enynol ethers by 6π electrocyclization: Stereoselective access to 2H-azepines. Angew. Chem. Int. Ed. 59, 1666–1673 (2020). [DOI] [PubMed] [Google Scholar]
- 42.Athavale S. V., Simon A., Houk K. N., Denmark S. E., Demystifying the asymmetry-amplifying, autocatalytic behaviour of the soai reaction through structural, mechanistic and computational studies. Nat. Chem. 12, 412–423 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nishina N., Yamamoto Y., Gold-catalyzed intermolecular hydroamination of allenes with arylamines and resulting high chirality transfer. Angew. Chem. Int. Ed. 45, 3314–3317 (2006). [DOI] [PubMed] [Google Scholar]
- 44.CCDC 2005918 (Z-3o) contains the supplementary crystallographic data for this paper.
- 45.CCDC 2005917 (E-4ai) contains the supplementary crystallographic data for this paper.
- 46.Garnock-Jones K. P., Gillian M. K., Atomoxetine: A review of its use in attention-deficit hyperactivity disorder in children and adolescents. Pediatr. Drugs 11, 203–226 (2009). [DOI] [PubMed] [Google Scholar]
- 47.Zeng Y.-F., Ji W.-W., Lv W.-X., Chen Y., Tan D.-H., Li Q., Wang H., Stereoselective direct chlorination of alkenyl MIDA boronates: Divergent synthesis of E and Z α-chloroalkenyl boronates. Angew. Chem. Int. Ed. 56, 14707–14711 (2017). [DOI] [PubMed] [Google Scholar]
- 48.Philippov A. N., Vorobyeva D. V., Monnier F., Osipov S. N., Synthesis of α-CF3-substituted E-dehydroornithine derivatives via copper(i)-catalyzed hydroamination of allenes. Org. Biomol. Chem. 18, 3274–3280 (2020). [DOI] [PubMed] [Google Scholar]
- 49.Li J., Jin L., Liu C., Lei A., Transmetalation of Ar1ZnX with [Ar2-Pd-X] is the rate-limiting step: Kinetic insights from a live Pd-catalyzed Negishi coupling. Org. Chem. Front. 1, 50–53 (2014). [Google Scholar]
- 50.Jin L., Luo X., Lei A., Pd/π-acidic ligand catalyzed ArI and alkyl-in cross-couplingreactions under mild conditions. Acta Chim. Sin. 70, 1538–1542 (2012). [Google Scholar]
- 51.Tang X., Wu W., Zeng W., Jiang H., Copper-catalyzed oxidative carbon-carbon and/or carbon-heteroatom bond formation with O2 or internal oxidants. Acc. Chem. Res. 51, 1092–1105 (2018). [DOI] [PubMed] [Google Scholar]
- 52.Deng G., Li M., Yu K., Liu C., Liu Z., Duan S., Chen W., Yang X., Zhang H., Walsh P. J., Synthesis of benzofuran derivatives through cascade radical cyclization/intermolecular coupling of 2-azaallyls. Angew. Chem. Int. Ed. 58, 2826–2830 (2019). [DOI] [PubMed] [Google Scholar]
- 53.Chanthamath S., Chua H. W., Kimura S., Shibatomi K., Iwasa S., Highly regio- and stereoselective synthesis of alkylidenecyclopropanes via Ru(II)-Pheox catalyzed asymmetric inter- and intramolecular cyclopropanation of allenes. Org. Lett. 16, 3408–3411 (2014). [DOI] [PubMed] [Google Scholar]
- 54.Wang C., Xu G., Shao Y., Tang S., Sun J., Gold-catalyzed intermolecular formal [4 + 2 + 2]-cycloaddition of anthranils with allenamides. Org. Lett. 22, 5990–5994 (2020). [DOI] [PubMed] [Google Scholar]
- 55.Braverman S., Cherkinsky M., Meridor D., Sprecher M., Synthesis and reactivity of dipropargylic disulfides: Tandem rearrangements, cyclization, and oxidative dimerization. Tetrahedron 66, 1925–1930 (2010). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/35/eabh4088/DC1