

Inbreeding depression is linked to persistent epigenetic and gene expression changes, which are reversible by random mating.
Abstract
Inbreeding depression is widespread across plant and animal kingdoms and may arise from the exposure of deleterious alleles and/or loss of overdominant alleles resulting from increased homozygosity, but these genetic models cannot fully explain the phenomenon. Here, we report epigenetic links to inbreeding depression in maize. Teosinte branched1/cycloidea/proliferating cell factor (TCP) transcription factors control plant development. During successive inbreeding among inbred lines, thousands of genomic regions across TCP-binding sites (TBS) are hypermethylated through the H3K9me2-mediated pathway. These hypermethylated regions are accompanied by decreased chromatin accessibility, increased levels of the repressive histone marks H3K27me2 and H3K27me3, and reduced binding affinity of maize TCP-proteins to TBS. Consequently, hundreds of TCP-target genes involved in mitochondrion, chloroplast, and ribosome functions are down-regulated, leading to reduced growth vigor. Conversely, random mating can reverse corresponding hypermethylation sites and TCP-target gene expression, restoring growth vigor. These results support a unique role of reversible epigenetic modifications in inbreeding depression.
INTRODUCTION
Charles R. Darwin documented inbreeding depression as growth disadvantages from self-fertilization compared to outcrossing in many plants (1). Prevailing hypotheses suggest that inbreeding depression results from the exposure of deleterious recessive alleles and/or loss of overdominant alleles due to increased homozygosity (2, 3) or reduced recombination frequency in some regions (4). However, yield in maize inbred lines continues to decline even after 10 generations of self-pollination (5). Moreover, although autotetraploids achieve homozygosity at a slower pace than diploids, inbreeding depression levels appear to be similar between diploid and tetraploid plants (6). Thus, the genetic models cannot completely explain inbreeding depression.
In the absence of genetic diversity, epigenetic variation may contribute to inbreeding depression (7). In pincushion flower (Scabiosa columbaria), inbreeding increases DNA methylation, and treatments with a DNA methylation inhibitor in the inbreds can reverse some depressive growth phenotypes, including reduced photosynthetic efficiency and leaf number (8). Inbreeding depression in Chinook salmon correlates with methylation changes in some specific genes (9). In Arabidopsis thaliana, growth and developmental abnormalities gradually accumulate in the selfing progeny of DNA methylation mutants, including ddm1 (decrease in DNA methylation1) (10), met1 (methyltransferase1) (11), and ros1 (repressor of silencing1) (12). These data suggest association of DNA methylation with inbreeding depression, but the underlying mechanism remains unknown.
In plants, DNA methylation occurs in CG, CHG (H = A, T, or C), and CHH context (13) and can affect binding affinity of transcription factors (14). Teosinte branched1/cycloidea/proliferating cell factor (TCP) proteins control plant growth and development (15, 16), and their consensus binding sites are called TCP-binding sites (TBS), which are predicted to affect chromatin accessibility (17–19). In this study, we found that inbreeding in maize is accompanied by CHH hypermethylation of genomic regions predominately across TBS that correlated with increased H3K9me2, H3K27me2, and H3K27me3 levels and reduced chromatin accessibility of the TCP-target genes involved in photosynthesis (chloroplast), energy metabolism (mitochondrion), and ribosome biosynthesis. Increased methylation levels near the TBS motifs in promoters tended to reduce the binding affinity of ZmTCP proteins and down-regulate the expression of TCP-target genes, leading to reduced growth vigor. Random mating could restore these processes. These results collectively provide molecular evidence for epigenetic regulation of inbreeding depression in maize.
RESULTS
Increased CHH methylation near genes during maize inbreeding depression
Maize F1 hybrids between two inbreds 6958/LX0001 and 1160/3999 were self-pollinated to the sixth generation (S6), from which one single plant was self-pollinated to produce an S7 population (Fig. 1A). Thirty plants in the S7 generation were randomly selected and continuously self-pollinated using the single-seed descent without selection bias to generate an S11 population with 28 lines, excluding two lost during propagation (Fig. 1A). In the S10 generation, 25 lines were randomly mated to generate a random mating (M11) population (n = 75) (Fig. 1A and fig. S1A). The seedlings showed growth reduction from S7, S9, to S11 populations in the third leaf area (Fig. 1B), seedling dry weight, and 100-grain weight (fig. S1, B and C). However, growth phenotypes in the M11 were higher than those in the S11 population and fell between S7 and S9 populations (Fig. 1B and fig. S1, B and C), ranging from low (L) to high (H) variation (fig. S1A). These data were consistent with maize yield reduction during inbreeding (5) and yield regain after mating with the paired inbred lines (20).
Fig. 1. Increased CHH methylation during inbreeding in maize.
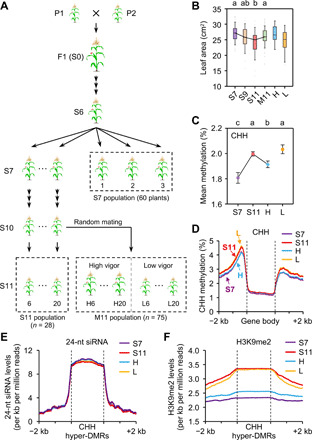
(A) Genetic materials for studying inbreeding depression in maize. Progenies from a single S6 plant were propagated by single-seed descent to generate S7 to S11 populations. Mating between different S10 lines from the common S6 parent generated a random mating (M11) population, with H and L lines indicating high and low/similar levels of growth vigor relative to their S11 lines. Dashed boxes show populations used for phenotypic analyses and lines used for methylome-sequencing analyses. (B) Third-leaf areas of S7 (n = 60), S9 (n = 29), S11 (n = 28), and M11 (n = 75), H (n = 16), and L (n = 11) populations with four to six plants per line. Different letters indicate statistical significance levels at P < 0.05 (Scheffe’s test). (C) CHH methylation levels of S7, S11 (line 6), H (H6), and L (L6) lines. Different letters indicate statistical significance (P < 0.05, Fisher’s LSD test). (D) Average distribution of CHH methylation across genes in S7, S11 (line 6), H (H6), and L (L6) lines. (E and F) Distributions of 24-nt siRNA (E) and H3K9me2 (F) levels across CHH hyper-DMRs and flanking sequences in S7, S11, H and L lines.
Using the MaizeSNP6K array analysis (21), we found that the percentage of genomic homozygosity among different S7 individuals was 99.6 to 99.7%, which was higher than the expected 99.2% (P < 0.05, χ2 test). Furthermore, the genetic uniformity from pairwise comparisons ranged from 99.6 to 99.9% between S7, S11, and M11 H and L lines (table S1). This suggests that phenotypic variation among these lines may result from the changes beyond the primary DNA sequence, which may involve DNA methylation and chromatin modifications (22). Using MethylC sequencing (Methyl-seq), we determined DNA methylomes in the third leaf of S7 (three biological replicates), S11 (line 6), and M11 H (H6) and L (L6) lines, each with two biological replicates (Fig. 1A and table S2). The bisulfite conversion rates were >99.4% with a 5.2 or higher mean coverage of cytosines, while ~70% of cytosines were covered by at least one uniquely mapped read, and the cytosines present in both replicates were used for further analysis. We found that CG and CHG methylation levels were similar among S7, S11, and M11 H and L lines (fig. S1, D and E), whereas CHH methylation levels were higher in S11 than in S7 (similar to M11 L) lines but decreased in the M11 H line [P < 0.05, Fisher’s least significant difference (LSD) test] (Fig. 1C and fig. S1, D and E). Similar trends of CHH methylation changes were also observed in another set of S11 (line 20) and M11 H20 and L20 lines during inbreeding and random mating (fig. S1F). We also confirmed high levels of genetic uniformity (>99.93%) and homozygosity (>99.96%) among all the lines using a model-based caller analysis of bisulfite single-nucleotide polymorphism (SNP) (23), which was in the agreement with SNP array results. Thus, CHH methylation is increased during inbreeding and decreased after random mating.
These CHH hypermethylation (S11 versus S7) and hypomethylation (M11 H versus S11) sites were distributed primarily across gene-rich regions on each chromosome (fig. S2). Moreover, the increased CHH methylation in S11 lines and the reduced CHH methylation in M11 H lines occurred mainly in the 5′ and 3′ regions flanking the coding sequence (Fig. 1D), whereas CG and CHG methylation levels across the genes were similar among S7, S11, and M11 H and L lines (fig. S3, A and B). CHH methylation changes were also observed in transposable elements (TEs) close to (within 2-kb regions) of the genes (fig. S3, C to E), suggesting that CHH methylation changes around these genes and their adjacent TEs during inbreeding.
We identified 8879 CHH hyper-DMRs (differentially methylated regions) between S11 and S7 (table S3) using a threshold value of 5% (0.05) CHH methylation difference. This cutoff value for CHH methylation in plants can vary from 0.05 (24), 0.08 (25), 0.10 to twofold change (26) among different studies because of genomic backgrounds and methylation level differences. In this study, genome-wide CHH methylation levels were ~2%; when the cutoff value was set to 0.05, it included the top 0.4% of all hypermethylated sites (fig. S4A), and most changes were greater than twofold (fig. S4B). As expected, those DMRs at 0.05 included all the DMRs at 0.1 (fig. S4C), while a subset of DMRs could be validated by another method (see below). Thus, we used the cutoff value of 5% for further analysis, unless noted otherwise.
Compared to CG and CHG methylation, the overall CHH methylation levels were relatively low and may exhibit variation between biological replicates and/or inbred lines. To test this, we compared Pearson’s correlation coefficients and found clear separation between S7 and S11 DMRs, despite some variation among replicates within S7 or S11 population (fig. S4D). Specifically, the difference between the inbred lines (S7 and S11) was greater than the difference between biological replicates (P < 1 × 10−200, Wilcoxon rank sum test) (fig. S4E). We also compared S11 hyper-DMRs with variable DMRs between biological replicates in S7 and S11 and found little or no overlap between them (fig. S4F). Using methylation-sensitive quantitative polymerase chain reaction (qPCR) (27), we further tested 15 randomly selected S11 hyper-DMRs and confirmed 14 of the 15 S11 hyper-DMRs examined among three biological replicates, which were higher in S11 than in S7 lines (fig. S4G). During inbreeding, CHH methylation levels were consistently higher in another S11 (S11_20) than in S7 lines (fig. S5A), and moreover, they gradually increased from S7, S9, S10, to S11 lines (P < 1 × 10−200, Wilcoxon rank sum test) (fig. S5B). Collectively, these results suggest that CHH hypermethylation is gradually and persistently established during inbreeding.
DNA hypermethylation during inbreeding via H3K9me2-dependent pathway
CHH hypermethylation may result from RNA-directed DNA methylation (RdDM) (13). Using small RNA sequencing (sRNA-seq) data and the published MethylC-seq data in maize RdDM pathway–defective mutants mop1 (28) and zmet7 (29) (table S2), we found that the amount of 24-nucleotide (nt) small interfering RNAs (siRNAs) was similar in the CHH hyper-DMRs among S7, S11, and M11 H and L lines (Fig. 1E), although most hyper-DMRs showed lower CHH methylation levels with greater reductions in the mop1 and zmet7 mutants than in the wild types (WTs) (fig. S5C). These results indicate that CHH methylation changes during maize inbreeding may be independent of RdDM.
CHH methylation may also depend on the H3K9me2-mediated pathway, which involves CHROMOMETHYLASE 2 (CMT2), CMT3, and DDM1 genes in plants (30, 31). Notably, maize does not have CMT2 orthologs, and CHH methylation may involve two CMT3 homologs, including Zmet2 and Zmet5 (29). Methylation levels in both CHH and CHG contexts were reduced in the CMT-defective mutants, zmet2 and zmet5. By analyzing published data (28, 29) (table S2), we found that CHH methylation levels of hyper-DMRs were reduced in maize zmet2, zmet5, and zmet2, zmet5 mutants (fig. S5D) and the DDM1-like double-mutant chr101 chr106 (fig. S5E). Furthermore, chromatin immunoprecipitation sequencing (ChIP-seq) data (table S2) showed higher H3K9me2 levels of CHH hyper-DMRs in S11 and M11 L lines than in S7 and M11 H lines (P < 5 × 10−67, Wilcoxon rank sum test) (Fig. 1F), consistent with the overall trend of CHH methylation increases during inbreeding (Fig. 1, C and D). Among these CHH hyper-DMRs, CHG methylation levels were also higher in S11 than in S7 lines (P < 1 × 10−100, Wilcoxon rank sum test) (fig. S5F), probably because H3K9me2 is also involved in CHG methylation (31). ZmJMJ8 is an ortholog of Arabidopsis IBM1 encoding histone H3K9 demethylase (32). IBM1 negatively regulates DNA methylation, and ibm1 mutants have morphological defects, such as smaller leaves and slower growth, which become apparent after two selfing generations (32, 33). We found that ZmJMJ8 expression levels were higher in S7 and M11 H lines than in S11 and M11 L lines (fig. S5G), correlating with H3K9me2 level changes from inbreeding to random mating (Fig. 1F). Thus, CHH hypermethylation by inbreeding and hypomethylation by random mating may depend on the H3K9me2 pathway.
CHH hyper-DMRs associated with TBS and reduced chromatin accessibility
As CHH methylation changes mainly occurred around genes, we performed MEME motif analysis (34) and found that these hyper-DMRs had two top-scoring motifs including motif 1 (E = 7.1 × 10−21) and motif 2 (E = 1.0 × 10−40) (Fig. 2A and fig. S6A). Motif 1 resembled TBS (TGGGCY) bound by TCP proteins, including TCP15 (P = 4.86 × 10−3), TCP23 (P = 4.86 × 10−3), and Osl (P = 5.14 × 10−3) (Fig. 2A). Notably, TBS-like motif 1 also matched the binding motif of brain and muscle Arnt-like 1 (BMAL1, also known as Arntl; P = 2.26 × 10−3), which is a pioneer-like circadian transcription regulator in mammals (35), while motif 2 resembled the binding motif of v-rel avian reticuloendotheliosis viral oncogene homolog A (RELA, also known as p65) in humans (P = 4.93 × 10−3) (fig. S6A). BMAL1 and RELA binding promotes chromatin opening (35, 36). In plants, TBS and its adjacent motifs are predicted to modulate chromatin accessibility in Arabidopsis (18), colocalize with deoxyribonuclease (DNase) hypersensitive (HS) in maize (19), and facilitate chromatin accessibility in cucumber (Cucumis sativus L.) (17). Using published data of micrococcal nuclease digestion followed by sequencing (MNase-seq) (37, 38) and DNase-seq (19) (table S2), we found that CHH hyper-DMRs were located adjacent to MNase HS and DNase HS regions approximately 200–base pair (bp) upstream of the transcription start site (TSS) and void of nucleosomes (Fig. 2B). Coincidently, these regions in maize are enriched with cis-regulatory elements and explain ~40% of heritable phenotypic variation in diverse complex traits (19, 37). We predicted that these elements in the hyper-DMRs may affect chromatin accessibility during inbreeding.
Fig. 2. Chromatin accessibility and histone modifications of CHH hyper-DMRs with TBS.
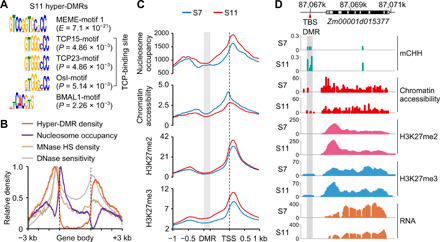
(A) The MEME-motif 1 identified from CHH hyper-DMRs in S11 using MEME suites matched TCP and BMAL1 (Arntl) binding sites in TOMTOM analysis (34). (B) Distributions of CHH hyper-DMRs, nucleosome occupancy, MNase-hypersensitive (MNase HS) regions, and DNase I sensitivity across hyper-DMR–associated genes. (C) Distributions of nucleosome occupancy, chromatin accessibility, and histone modifications across regions from S11 hyper-DMRs to transcriptional start sites (TSS), including 1-kb upstream of the DMR and 1-kb downstream from TSS. The distances between DMRs and TSSs were normalized to the same length. The gray box and black dashed lines indicate DMR and TSS, respectively. (D) An example of the locus Zm00001d015377 shows TBS (red arrowhead), exons (black), introns (white), and 5′ and 3′ untranslated regions (UTRs; gray). In the locus, CHH hypermethylation of the DMR (gray box) correlated with increased levels of H3K27me2 and H3K27me3 and decreased levels of chromatin accessibility and gene expression in S11.
To test the above prediction, we investigated chromatin accessibility using the assay for transposase-accessible chromatin sequencing (ATAC-seq) and ChIP-seq with antibodies against H3K27me2 and H3K27me3 (table S2), two repressive histone marks negatively associated with the chromatin accessibility. ATAC-seq analysis showed an obvious 200-bp periodicity of sequenced fragments, reproducible between biological replicates (fig. S6B). Notably, CHH hyper-DMRs were localized in low–nucleosome density regions, where nucleosome occupancy and H3K27me2 and H3K27me3 levels were increased, but chromatin accessibility levels were decreased from S7 to S11 lines (Fig. 2C). For example, Zm00001d015377 encodes a pentatricopeptide repeat-containing protein and is one of three candidate loci for kernel row number (KRN) (39); TBS within the hyper-DMR had higher levels of CHH methylation and H3K27me2 and H3K27me3 modifications but lower levels of chromatin accessibility, correlating with lower Zm00001d015377 expression levels in S11 than in S7 lines (Fig. 2D).
CHH hypermethylation across TBS affects gene expression
At the genome-wide level, histone modifications and chromatin accessibility from S7 to S11 lines changed more in the genes associated with CHH hyper-DMRs than without DMRs (fig. S6C), suggesting that these changes may affect the expression of DMR-associated genes. Using RNA-seq data (table S2), we found that 763 genes were down-regulated in the S11 relative to S7 lines and enriched in CHH hyper-DMR–associated genes (P < 1 × 10−27, Fisher’s exact test) (Fig. 3A and table S4). These hyper-DMRs were associated with TBS motifs within 0- to 500-bp upstream of the TSS, which were overrepresented among down-regulated genes relative to up-regulated and all genes (P < 5 × 10−29, Wilcoxon rank sum test) (Fig. 3B) and located near the hyper-DMRs (Fig. 3C), from 200 bp (35.8%) to 1 kb (cumulative 80.7%) of TBS, respectively (Fig. 3D).
Fig. 3. Hypermethylation of TBS and down-regulation of TCP-target genes during inbreeding.
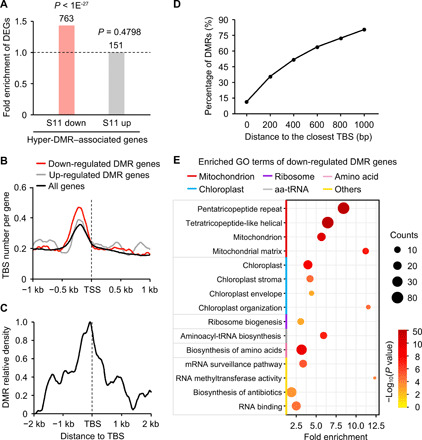
(A) Fold enrichment of differentially expressed genes (DEGs) in CHH hyper-DMR–associated genes. P values were calculated by Fisher’s exact test. (B) Number of TBS per gene within 1 kb of TSS (marked by dashed line). (C) Density of CHH hyper-DMRs within 2 kb of TBS. (D) Percentage of DMRs relative to the distance from the TBS. (E) Down-regulated genes with DMRs were overrepresented by gene ontology (GO) terms involved in mitochondrion, chloroplast, and ribosome biosynthesis (P < 0.01 and counts ≥5) using DAVID analysis (40). aa-tRNA, aminoacyl-tRNA.
These CHH hyper-DMRs across TBS may reduce the expression of TCP-target genes. Using published RNA-seq data of the CMT-defective mutants zmet2 and zmet5 (table S2), we found that most genes with hyper-DMRs were expressed at higher levels with greater fold changes in all methylation-defective mutants tested than in WT (fig. S6D). The data suggest that CHH hypermethylation correlates with down-regulation of TCP-target genes. These 763 down-regulated genes were overrepresented in 15 gene ontology (GO) terms (P < 0.01, a modified Fisher’s exact test) using DAVID analysis (40). These GO terms were classified into six groups based on semantic and functional descriptions, nine of which belonged to three groups related to mitochondrial, chloroplast, and ribosome functions, respectively (Fig. 3E). Similarly, a higher threshold value of 10% (relative to 5%) for identifying DMRs also retained these three groups of genes among DMR-associated and down-regulated genes (fig. S6E). Mitochondria and chloroplasts are critical for energy production and carbohydrate metabolism, while ribosomes are essential for protein biosynthesis, and expression of these genes is coregulated by TBS (41). TBS motifs were enriched in these organelle-related genes (fig. S6F) and adjacent to CHH hyper-DMRs (fig. S6G). Thus, CHH hyper-DMRs accompanied with chromatin accessibility and histone modifications in the TBS may regulate expression of these genes. Notably, CHH hypermethylation and its effect on gene expression were unlikely related to residual heterozygosity. Using Bis-SNP software (23) to identify SNPs from MethylC-seq data, we found only 8 (of 8879) S11 CHH hyper-DMRs and 33 genes in residual heterozygous regions, but they did not overlap with down-regulated and DMR-associated genes (tables S3 and S4). These 33 genes were not likely involved in DNA methylation or leaf development (table S5).
Inbreeding-induced hypermethylation is reversed by random mating
If CHH hyper-DMRs and expression of their associated genes are induced by inbreeding, then they should be reversible by random mating. Compared to S11 lines, DMR methylation levels of randomly mating (M11) lines decreased more in S11 hyper-DMRs of the H (P < 1 × 10−200, Wilcoxon rank sum test) or L (P < 1 × 10−150) lines than in non-DMRs (fig. S7A). More hyper-DMRs were demethylated (reversed) in M11 H (65%) than in M11 L (57%) lines, with a 40% overlap of reversed DMRs between H and L lines (Fig. 4A). Among them, 25% of hyper-DMRs were reversed only in the H line (reversed DMRs) (Fig. 4A) and were associated more with genes than other DMRs (fig. S7B). These reversed DMR-associated genes correlated with increased chromatin accessibility and gene expression levels in M11 H but not in M11 L lines (fig. S7C).
Fig. 4. Inbreeding-induced hypermethylation is reversed by random mating and analysis of ZmTCP binding affinity to methylated TBS.
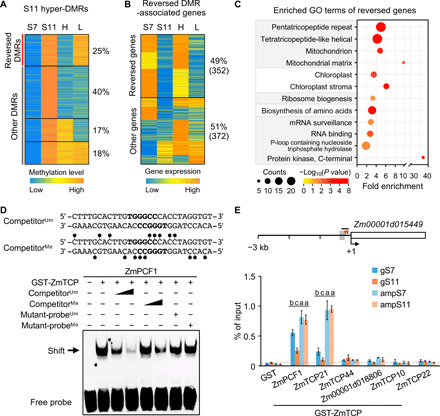
(A) K-means clustering of CHH methylation levels in hyper-DMRs among S7, S11, high (H), and low (L) vigor M11 lines. (B) K-means clustering of expression levels of the genes associated with reversed DMRs. (C) GO-enriched terms of the genes with reversed expression levels (P < 0.05 and counts of ≥5). (D) Reduced binding affinity of recombinant ZmPCF1 protein to methylated probes containing TBS motifs (bold letters) in EMSA. Competition assays used unmethylated probes (competitorUm, 100× and 500× excess), methylated probes (competitorMe, 100× and 500× excess), unmethylated mutant probes (mutant-probeUm, 500× excess), or methylated mutant-probes (mutant-probeMe, 500× excess). CHH methylation sites (lollipop) are shown in the competitorMe sequence. The band shift is indicated by the arrow. (E) DNA affinity purification (DAP)–qPCR analysis of recombinant ZmTCP proteins using genomic DNA of S7 and S11 (gS7 and gS11) and PCR-amplified genomic DNA from S7 and S11 (ampS7 and ampS11). Diagram of the Zm00001d015449 promoter region (3-kb upstream) shows locations of two TBS motifs (red arrowheads), CHH hyper-DMR (gray box), and the fragment (short black line) used in DAP-qPCR assays. Numbers shown are relative to the TSS (+1). Ratio of GST-ZmTCP/GST ≥ 5 was used to show binding of ZmTCP protein to the TBS fragment using the purified GST protein as the negative control. Different letters above each column indicate statistical significance at P < 0.05 (Tukey’s test).
Consistently, expression levels for 352 (~49%) DMR-associated genes were also reversed in M11 H but not in M11 L lines (Fig. 4B). These expression reversal genes were also overrepresented in biological processes of mitochondria, chloroplasts, and ribosome biosynthesis (P < 0.05, a modified Fisher’s exact test) (Fig. 4C), consistent with GO enrichment terms of DMR-associated genes after inbreeding (S11 lines) (Fig. 3E). In contrast, the genes with reversed methylation but not reversed expression in the M11 H line were related to four GO groups with no clear biological inference to growth depression (Fig. 4B and fig. S7D). The data suggest that random mating can reverse DNA methylation and gene expression levels for a subset of CHH hyper-DMR–associated genes in inbred lines. In addition, we identified 1723 DMRs with hypomethylation (hypo-DMRs) in M11 H but not in M11 L lines, and 42% of these DMRs were not hypermethylated during inbreeding (fig. S7E), suggesting that random mating may also induce hypomethylation in some genomic regions.
Biological functions of CHH hypermethylation during inbreeding
DNA methylation can quantitatively alter the binding affinity of transcription factors to their binding sites (14). Using electrophoretic mobility shift assay (EMSA), we determined whether CHH methylation affects the binding affinity of ZmTCP proteins to TBS motifs. Maize has 49 ZmTCP genes that belong to class I (proliferating cell factor, PCF, clade) and class II (42), while the class II genes are further divided into two subclasses, CIN and TB1/CYC (fig. S8). On the basis of RNA-seq data from this study (table S2) and published data of B73 (43), TB1/CYC-related genes were not expressed in leaves. We found that six ZmTCP genes including three PCF-like (ZmPCF1, ZmTCP44, and ZmTCP21) and three CIN-like (Zm00001d018806, ZmTCP10, and ZmTCP22) were highly expressed in leaves (fig. S8). We cloned each of them into a vector to express glutathione S-transferase (GST)–ZmTCP fusion proteins that were subsequently purified (Materials and Methods). EMSA showed that four GST-ZmTCP proteins (ZmPCF1, ZmTCP21, Zm00001d018806, and ZmTCP10) tested preferred to bind unmethylated TBS fragments (Fig. 4D and fig. S9), whereas the other two (ZmTCP44 and ZmTCP22) preferred to bind methylated TBS fragments. These results indicate that ZmTCP proteins bind to TBS motifs and exhibit sensitivity to methylated TBS in vitro.
We further determined whether hypermethylation in S11 lines alters binding affinity of TBS using DNA affinity purification (DAP)–qPCR assays (44). GST-ZmTCP proteins were used to pull down genomic DNA by affinity in S7 and S11 (gS7 and gS11) lines, a fraction of which was amplified by PCR, resulting in amplified DNA (ampS7 and ampS11) with no methylation. We tested promoter fragments containing TBS motifs and hyper-DMRs in five TCP-target genes (Fig. 4E and fig. S10). ZmPCF1 and ZmTCP21 bound to promoters of Zm00001d015449 and Zm00001d044353 (GST-ZmTCP/GST ≥ 5), and the other TCP factor Zm00001d018806 bound to promoters of Zm00001d044353 and Zm00001d036698. In agreement with the EMSA results, these six ZmTCP-TBS interactions showed that methylation on native genomic DNA can reduce the binding of ZmTCP proteins to TBS. For example, the binding affinity of GST-ZmPCF1 proteins to the native genomic DNA of Zm00001d015449 was lower than that to the PCR-amplified DNA (gS7 < ampS7, P < 0.05 and gS11 < ampS11, P < 0.0005, Tukey’s test) (Fig. 4E). Three of six ZmTCP-TBS interactions showed lower binding affinity of the GST-ZmTCP proteins to native genomic targets in S11 than in S7 (gS11 < gS7, P < 0.05, Tukey’s test) (Fig. 4E and fig. S10). Thus, methylation changes in TBS during inbreeding may alter binding affinity of ZmTCP transcription factors, depending on activators or repressors, to alter gene expression.
At the genome-wide level, CHH hyper-DMRs near the TBS motifs correlated negatively with expression levels of their TCP-target genes involved in mitochondrial, chloroplast, and ribosome functions (P < 0.05, Wilcoxon rank sum test) (Fig. 5A). For example, Arabidopsis ADENOSINE DIMETHYL TRANSFERASE 1A (DIM1A) is predicted to mediate nuclear ribosome biogenesis, and in the dim1A mutant, leaf size and root length are reduced (45). Zm00001d015449 (ZmDIM1A) is a maize homolog of DIM1A. During inbreeding from S7 to S11, CHH methylation levels were increased near the two TBS motifs (Fig. 5B), while the binding affinity of ZmPCF1 was decreased (Fig. 4E), and ZmDIM1A expression was down-regulated (Fig. 5B). Moreover, 24-nt siRNAs accumulated near the TBS of ZmDIM1A, which prompted us to use the inverted repeat (IR) sequence (Fig. 5B) for virus-induced gene silencing (VIGS) (46), to modify DNA methylation by RdDM in its promoter region and analyze its biological function (47). Among nine independent VIGS-IR plants tested, we found that DNA methylation levels in TBS were increased (Fig. 5C and fig. S11A), and expression levels of ZmDIM1A were reduced (Fig. 5D). As a result, these VIGS-IR seedlings grew smaller (Fig. 5E and fig. S11B) with reduced height, leaf area, and root length (fig. S11C) compared to VIGS-GFP control plants. As a positive control for VIGS, the seedlings that expressed VIGS of ISPH/Zebra7 (Zb7) displayed yellowing leaf symptoms (Fig. 5E), as observed in the zb7 mutant (48). Furthermore, relative CHH methylation levels in the ZmDIM1A promoter (fig. S11D) were negatively correlated with gene expression levels (fig. S11E) and third-leaf areas (fig. S11F) from S7 (n = 30), S9 (n = 25), to S11 (n = 25) populations. Together, these results suggest that inbreeding-induced CHH hypermethylation of TBS can down-regulate expression of DIM1A and possibly other TCP-target genes that are associated with inbreeding depression in maize.
Fig. 5. DNA hypermethylation across TBS contributes to maize inbreeding depression.
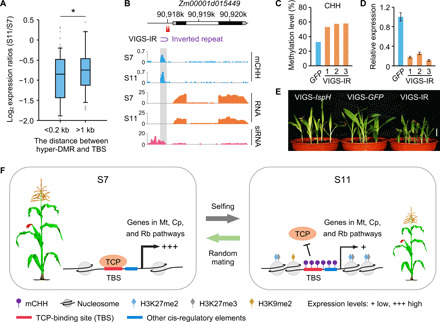
(A) Log2 expression ratios (S11/S7) of the genes involved in mitochondrion, chloroplast, and ribosome biosynthesis, relative to the distances (<0.2 or >1-kb) between CHH hyper-DMRs and TBS motifs. Single asterisk indicates statistical significance (P < 0.05, Wilcoxon rank sum test). (B) Down-regulation of Zm00001d015449 correlated with CHH hypermethylation near TBS motifs. The Zm00001d015449 locus consists of two TBS motifs (red arrowheads), two exons (black), one intron (white), and 5′ and 3′ UTRs (gray). An inverted repeat (IR, purple arc) was used for virus-induced gene silencing (VIGS). Gray box indicates the location of S11 hyper-DMR. (C to E) Levels of increased CHH methylation (C), decreased Zm00001d015449 expression (D), and reduced plant height (E) in VIGS-IR plants (n = 9) compared to the VIGS-GFP control (n = 7). Plants (n = 5) infected with the virus expressing the ZmIspH fragment (VIGS-IspH) were used as the virus infection control. Scale bar, 2 cm. Photo credit: T. Han, Nanjing Agricultural University. (F) A model links DNA methylation to inbreeding depression in maize. From S7 (left) to S11 (right) generations, increased DNA (CHH) methylation levels across TBS and other cis-regulatory elements during selfing (S11 lines) reduces the binding affinity of ZmTCP transcription factors, which is accompanied by reduced chromatin accessibility and increased nucleosome occupancy and H3K27me3 and H3K27me2 levels, and down-regulation of TCP-target genes involved in mitochondrial, chloroplast, and ribosome functions, thereby reducing growth vigor. CHH methylation increase during inbreeding depends on H3K9me2 levels. Arrows (+++ and +) represent high and low expression levels, respectively. Random mating can reverse these effects (green arrow). Mt, mitochondrion; Cp, chloroplast; Rb, ribosome biosynthesis.
DISCUSSION
Results from our study can explain DNA methylation–mediated inbreeding depression in maize (Fig. 5F). Inbreeding increases CHH methylation across TBS and other motifs to reduce the binding affinity of ZmTCP transcription factors, which are accompanied with reduced chromatin accessibility and increased nucleosome occupancy and H3K27me3 and H3K27me2 modifications. As a result, expression of TCP-target genes involved in mitochondrial, chloroplast, and ribosome functions is down-regulated, leading to reduced growth vigor. Conversely, random mating within an inbreeding population may restore the growth vigor by reversing these epigenetic processes in subsets of these TCP-target genes. Notably, other transcription factors may also regulate expression of TCP-target genes involved in mitochondrion, chloroplast, and ribosome biosynthesis functions (41). This is probably because complex TBS motifs consist of binding sites for other transcription regulators such as BMAL1 in mice to remodel chromatin (35). In Arabidopsis, TCP proteins also target circadian clock regulators (22, 49, 50). For example, CIRCADIAN-CLOCK ASSOCIATED1 (CCA1) is a TCP-target gene, and its expression is regulated by CHH methylation across TBS, which correlates with the parent-of-origin effect on biomass heterosis (51). In addition, CCA1 and H3K4me3 modifications comprise a feedback loop for transcriptional regulation (52). In maize, overexpression of ZmCCA1b (a homolog of Arabidopsis CCA1) reduces plant height (53). These results imply that CHH methylation of TBS may also regulate inbreeding depression through modulating circadian clock and its output genes, many of which are involved in photosynthesis and starch metabolism (49). The link between TCP regulators and circadian-mediated growth reduction remains to be investigated. As circadian clock regulation is conserved across plant and animal kingdoms (22, 54, 55), we speculate that DNA methylation and chromatin changes in BMAL/RELA-target genes may also occur during inbreeding in mammals.
In maize, DNA methylation is found to correlate with paramutation of the r1 locus, encoding a helix-by-helix transcription factor in the anthocyanin biosynthesis pathway (56) and silencing of the Mutator (Mu) transposon in a metastable allele of the bronze 2 (bz2) locus, encoding a GST for anthocyanin biosynthesis (57). During inbreeding, metastable r1 expression (58) and Mu activity (59) are decreased, suggesting a possible connection between increased DNA methylation and reduced expression of r1 and Mu loci during inbreeding. We found that inbreeding induces hypermethylation across TEs, which may be silenced to counteract their expansion. However, methylated TEs may, in turn, silence proximal genes and disrupt expression of neighboring genes in Arabidopsis (60) and rice (61). Thus, TEs may affect changes in DNA methylation and expression of TE-associated genes because of inbreeding depression. Last, although sequence variation between high- and low-vigor random mating lines is very limited, inbred lines with high-quality resequencing genome or high-quality reference genome such as B73 should be used to precisely discern relative roles of sequence variation and epigenetic modification in inbreeding depression. In summary, our findings provide molecular evidence that inbreeding is accompanied with reversible epigenetic regulation of TCP-target genes that mediate growth vigor in maize and possibly other plants.
MATERIALS AND METHODS
Maize inbred populations
The F1 hybrids between two maize inbred lines, 6958/LX0001 and 1160/3999, were continuously self-pollinated for six generations (to S6) at Shandong Academy of Agricultural Sciences, and the plants with stable growth traits were selected to produce an elite inbred line. A single individual from the S6 line was self-pollinated to produce an S7 population (Fig. 1A), 30 individuals of which were randomly selected for continuous selfing (single-seed decent) to obtain an S11 population with 28 inbred lines, while two lines were lost during propagation. In each generation, one single individual was randomly chosen from seven individuals planted to ensure that no selection bias was imposed.
In the S10 generation, mating between different lines from the common S6 parent was used to generate a random mating (M11) population (n = 75) (Fig. 1A and fig. S1A). For example, pollens from the line 6 in S10 generation were artificially pollinated to the stigma of itself and other lines, respectively, to generate the S11_6 line (line 6) and its corresponding multiple M11 lines (fig. S1A). A M11 line was classified as the high-vigor (H) line due to significantly increases (P < 0.05, Student’s t test, n = 4 to 6) in leaf area, aerial dry weight, and 100-grain weight compared to its corresponding S11 line. In contrast, a M11 line with low or similar trait performance (lower means or P > 0.95) relative to the S11 line was classified as the low-vigor (L) line.
The populations including S7 (60 plants), S9 (n = 29, 4 to 6 plants per line), S11 (n = 28, 4 to 6 plants per line), and M11 (n = 75, 4 to 6 plants per line) were simultaneously grown in a growth chamber (day, 12 hours, 28°C; night, 12 hours, 22°C, 60% relative humidity) at the Nanjing Agricultural University. Plants were randomly grown in plug trays filled with pindstrup substrate (0 to 10 mm; Pindstrup, Denmark), and positions of plug trays were randomized. The third leaf area and aerial dry weight of each plant were examined at 2 weeks after planting. The third leaves were harvested and immediately frozen in liquid nitrogen for RNA and DNA preparations. Another set of these populations was simultaneously planted in the experimental field of the Nanjing Agricultural University to measure the 100-grain weight.
DNA and RNA extraction
Leaf tissue was flash-frozen in liquid nitrogen and disrupted at least three times using the Tissuelyser-48 at 30 Hz for 30 s (Qiagen, Germantown, MD). DNA and RNA were simultaneously isolated from aliquots of the same sample using a DNeasy plant mini kit (QIAGEN) and a plant DNA/RNA kit (Omega), respectively, following the manufacturer’s recommendations. Unless noted otherwise, each genomic experiment was performed using two to three biological replicates as shown in table S2, and uniquely mapped or normalized data present in two or more replicates were used for further analyses.
MethylC-seq, mRNA-seq, and sRNA-seq libraries
For methylC-seq library construction (62), total genomic DNA (~2 μg) was sonicated to ~300 bp using the Covaris M220 Focused-ultrasonicator (Woburn, MA). DNA after end repair was added an “A” base to the 3′-end and ligated with the methylated sequencing adapters. Ligation products were purified using VAHTS DNA Clean Beads (Vazyme, Nanjing, China), followed by bisulfite treatment using an EZ DNA Methylation-Gold kit (Zymo Research, Irvine, CA) according to the manufacturer’s guidelines. Library enrichment was performed by PCR amplification, and products were purified and subjected to size selection. mRNA-seq library was constructed as follows. mRNA was isolated from total RNA treated by DNase I using magnetic beads with oligo (dT). Then, complementary DNA (cDNA) was synthesized using the mRNA that has been fragmented into short fragments as templates. Double-stranded cDNA was purified for end preparation, single-nucleotide A addition, and connection with adapters. After size selection, purified fragments were amplified for library enrichment. sRNA-seq library construction was made following a published protocol (63). An aliquot of 20 μg of total RNA was subjected to 15% urea-polyacrylamide gel electrophoresis, and sRNAs in the size range of 18 to 30 nt were excised and ligated with adapters and used for library construction.
Nuclei isolation and ATAC-seq library construction
We isolated nuclei as described previously (64). Briefly, 0.5 g of fresh leaves was ground into powder in liquid nitrogen, and the powder was transferred to 10 ml of ice-cold nuclei purification buffer (NPB). This suspension was filtered with a 70-μm cell strainer and then centrifuged at 1200g for 10 min at 4°C. The pellet after discarding the supernatant was resuspended with 1 ml of ice-cold NPB, and this suspension was centrifuged at 1200g for 10 min at 4°C. After removal of supernatant, the pellet was resuspended in 1 ml of ice-cold NEB2. The suspension was centrifuged at 12,000g for 10 min at 4°C. After the supernatant was carefully removed, the pellet was resuspended thoroughly in 300 μl of ice-cold NEB3. The suspension was layered on top of 300 μl of NEB3 in a new 1.5-ml tube, and the tube was centrifuged at 16,000g for 10 min at 4°C. The nuclei pellet after removing the supernatant was resuspended in 1 ml of ice-cold NPB and kept on ice. The hemocytometer (catalog no. 3100, Hausser Scientific, Horsham, PA) was used to count and determine the total yield of the nuclei. Approximately 50,000 purified nuclei were used in each 50 μl of transposase integration reaction to construct the ATAC-seq library using the TransNGS Tn5 DNA Library Prep Kit for Illumina (TransGen Biotech, Beijing, China) according to the manufacturer’s instructions. After amplification for 12 PCR cycles, the products were purified and size-selected using VAHTS DNA Clean Beads (Vazyme, Nanjing, China).
ChIP and ChIP-seq library construction
ChIP experiments were performed as previously described (65), with minor modifications. Briefly, the third leaves from 2-week-old seedlings were cross-linked in 1% formaldehyde under vacuum two times for 10 min, after which the cross-linked leaves were homogenized in liquid nitrogen. Chromatin was extracted and sheared to 200- to 500-bp fragments by sonication (Covaris M220 Focused-ultrasonicator, Woburn, MA). Antibodies against histone H3K9me2 (D85B4, Cell Signaling Technology, Danvers, MA), H3K27me2 (Ab24684, Abcam, Cambridge, UK), and H3K27me3 (Ab6002, Abcam) were added to the sheared chromatin and incubated overnight at 4°C with gentle rotation. Immunoprecipitated DNA was extracted using Protein A/G Magnetic Beads (Thermo Fisher Scientific, Waltham, MA), reversed cross-links at 65°C overnight, and purified using the QIAquick PCR purification Kit (QIAGEN, Germantown, MD). For ChIP-seq, all libraries were constructed using the NEBNext Ultra II DNA Library Prep Kit for Illumina [New England Biolabs (NEB), Ipswich, MA] according to the manufacturer’s instructions. After amplification for 16 PCR cycles, the products were purified and size-selected to obtain the appropriate size for sequencing.
MethylC-seq data analysis
After removing adapter sequences and low-quality reads from raw reads and quality controlling using FastQC (Babraham Institute, Cambridge, UK), clean reads were aligned to the maize B73 reference genome version 4 (B73_RefGen_v4) using Bismark with default parameters (−X 1000). In addition, the unmapped reads were trimmed according to the reports from FastQC using cutadapt (read_1: –u 10 –u -20; read_2: –u 10 –u -50) and then realigned to the genome. Only unique mapping reads were retained for further analysis. The deduplication was carried out using deduplicate_bismark, a script of Bismark package. Methylated cytosines were extracted from Bismark bam files using bismark_methylation_extractor, and some bases were ignored according to the M-bias reports (−comprehensive --ignore 7 --ignore_r2 7 --ignore_3prime 1; and –comprehensive --ignore_3prime 3 --ignore_r2 2). Only the cytosines covered by at least three reads in all compared materials were used to determine mean methylation levels and considered for further analysis.
We used a strategy of fixed C count for creating windows to identify the DMRs for CHH methylation. In brief, we first created sliding windows every 25 CHHs along maize genome. The mean and median of the sliding window length were 200 and 89 bp, respectively. Windows longer than 400 bp (5.38%) were further split though merging CHHs with distances less than 30 bp. Windows with coverage outliers were removed based on a BoxWhisker Outlier Filter (median + the interquartile range × 15). The mean methylation level was calculated for each window containing at least 12 overlapping CHHs in all compared materials. Analysis of variance (ANOVA) tests (P < 0.05) and differences of methylation levels (cutoff value of >0.05) were then performed for windows to identify DMRs. Similarly, Kruskal-Wallis tests were performed to identify the DMRs between the biological replicates (26). DMRs between S11 and S7 lines were validated by methylation-sensitive qPCR (27). Briefly, genomic DNA (~1 μg) was digested with NlaIII (NEB, Ipswich, MA) or glycerol (mock treatment). NlaIII cuts CATG and is inhibited when cytosine is methylated (11). The difference between the mock Ct and digest Ct was calculated to estimate relative CHH methylation levels.
SNP calling from MethylC-seq data
The reads after Bismark deduplication were used to identify SNPs for all plants using Bis-SNP software tool as described previously (23). Briefly, the EMIT_ALL_CONFIDENT_SITES mode of Bis-SNP (−T BisulfiteGenotyper -stand_call_conf 10 -mbq 0 -trim5 7 -trim3 3) was used to obtain all of sites above confident threshold to calculate the genomic homozygosity and the genetic identity between plants compared in pairs. The numbers of visited sites and confident sites were 2,135,082,794 and 1,273,763,252, respectively. In addition, we used the DEFAULT_FOR_TCGA mode to identify all SNP sites above the threshold and then used the VCFpostprocessWalker commend with the default threshold to filter out false positives. SNPs with genotype quality greater than 20 were classified as homozygous and heterozygous, respectively. Residual heterozygous regions were identified by merging continuously heterozygous SNPs within a region of 500 kb (66).
mRNA-seq data analysis
Quality control of sequencing reads was performed using FastQC. Low-quality reads and adapter sequences were trimmed by Trim Galore (Babraham Institute) (--stringency 3 --trim-n --max_n 7) and cutadapt (−u 15) according to the reports from FastQC. Clean reads were mapped to the B73_RefGen_v4 using TopHat (-N 3 --read-edit-dist 3 --no-discordant --no-mixed). Uniquely mapped reads were extracted to determine the gene abundance of gene annotation (Zea_mays.AGPv4.37). After removing 25-bp from the 3′-end of 150-bp sequencing reads, the sequences were analyzed as described above. The differentially expressed genes (DEGs) were identified using both ANOVA tests (P < 0.05) and fold changes (>1.5). The fold enrichment of DEGs in DMR-associated genes was calculated as previously described (67): (#DEGsDMR-associated/#DEGstotal) / (#GenesDMR-associated/#Genestotal).
ATAC-seq data analysis
ATAC-seq reads were removed 3′-end adapters and low-quality reads using Trim Galore (--stringency 3 --trim-n --max_n 7). Clean reads were then aligned to B73_RefGen_v4 using Bowtie2 with the parameters (-k 21 –X 1000 –N 1 --no-mixed --no-discordant). Mapped reads were filtered to obtain uniquely mapped reads and multimapped reads (1 < aligned number ≤ 20 and mapping quality ≥10). Multimapped reads were weighted and assigned to all locations according to the frequencies of uniquely mapped reads as previously described unique-weighting method (68). After assignment of multimapped reads, the reads mapped in either the chloroplast or mitochondrion (<20.62%) were removed and the remaining reads were used for further analysis.
To generate the nucleosome occupancy, reads less than 100 bp were considered nucleosome-free, and reads between 146 and 247 bp, 315 and 473 bp, and 558 and 615 bp were considered to be mono-, di-, and trinucleosomes, respectively (fig. S6B) (69). Di-/trinucleosome reads were split into two/three reads. Reads were analyzed using DANPOS-2.2.2 with the parameters (-m 1 -a 1 -u 0 --mifrsz 0) (70). Nucleosome-free reads were negative weighted and used as backgrounds. In addition, 5′-ends of ATAC-seq reads were used to calculate reads per kilobase per million mapped reads as chromatin accessibility.
ChIP-seq, MNase-seq, and DNase-seq data analysis
Raw sequencing reads were quality controlled and removed adapter sequences. Clean reads were then aligned to B73_RefGen_v4 using Bowtie2. Mapped reads were filtered to obtain reads with mapping quality of ≥20. Histone modifications and nucleosome occupancy were calculated as fragments per kilobase per million mapped fragments. DNase sensitivity indicated reads per kilobase per million mapped reads calculated by 5′-ends of DNase-seq reads. MNase HS regions were identified by subtracting the value of the heavy digest from those of the light digest (cutoff value of ≥6) as previously described (37).
sRNA-seq data analysis
After adapter clipping, sRNA-seq reads (18 to 30 nt) were mapped to the B73_RefGen_v4 using ShortStack (68) (--bowtie_m 70 --nohp). MicroRNAs (miRNAs), pre-miRNAs, ribosomal RNAs, transfer RNAs, small nuclear RNAs, and small nucleolar RNAs were removed, and the abundance of 24-nt siRNAs indicated reads per kilobase per million mapped reads.
Phylogenetic analysis
The MEGA 6 software was used to construct the midpoint-rooted neighbor-joining tree using ZmTCP amino acid sequences aligned by ClustalX. The bootstrap values based on 1000 replicates were shown at the branch points. Using the Poisson correction method, the evolutionary distances were calculated on the basis of the number of amino acid substitutions per site.
Expression and purification of recombinant ZmTCP proteins
The native coding sequences of ZmTCP transcription factors were amplified and cloned into the SalI site of pGEX-4T-3 vector. The recombinant vectors were transformed into Escherichia coli BL21 cells, and the expression of the recombinant GST-tagged proteins was induced by 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG). The GST-ZmTCP fusion proteins were purified by using Pierce Glutathione Magnetic Agarose Beads (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer’s instructions. Last, the GST-ZmTCP proteins were quantified by NanoDrop 2000 (Thermo Fisher Scientific) and SDS–polyacrylamide gel electrophoresis analysis. All proteins were stored at −80°C before assays.
Electrophoretic mobility shift assay
EMSA was performed using the Lightshift Chemiluminescent EMSA Kit (Thermo Fisher Scientific, Waltham, MA). For each reaction, the biotin-labeled unmethylated DNA probes (20 fmol) containing TBS motifs were mixed with the purified GST-tagged ZmTCP proteins (2 μg) in the reaction buffer [1× lightshift binding buffer, 2.5% glycerol, 5 mM MgCl2, poly-dIdC (50 ng/μl), and 0.05% NP-40]. Excessive of unlabeled competitor (unmethylated probes, methylated probes, unmethylated mutant probes, or methylated mutant probes) was added for competition assays. Before adding the biotin-labeled unmethylated probes, the mixture was incubated at room temperature for 10 min and incubated for another 20 min after adding the probes. The products of binding reaction were resolved by electrophoresis on a 6.5% nondenaturing polyacrylamide gel and then transferred onto a nylon membrane presoaked with 0.5× tris-borate ethylenediaminetetraacetic acid (TBE) buffer. The membrane was photographed by the ChemiDoc Touch Imaging System (Bio-Rad Laboratories, Hercules, CA).
DAP-qPCR assay
DAP experiments were performed as described previously (44), with minor modifications. Briefly, we used the NEBNext Ultra II DNA Library Prep Kit for Illumina (NEB, Ispwich, MA) to generate genomic DNA and PCR-amplified genomic DNA samples for affinity purification of GST-ZmTCP proteins. Genomic DNA (~5 μg) extracted from maize leaves was sonicated to 200- to 500-bp fragments, ligated with adapters, and purified by VAHTS DNA Clean Beads (Vazyme, Nanjing, China) to obtain the genomic DNA sample. To generate the PCR-amplified genomic DNA sample, 15 ng of purified DNA was amplified by PCR (11 cycles) to remove methylation. Next, the GST-ZmTCP proteins immobilized on Pierce Glutathione Magnetic Agarose Beads (Thermo Fisher Scientific, Waltham, MA) were incubated with the above DNA samples, respectively. The purified GST protein was used as a negative control. The eluted and recovered DNA was used for quantitative PCR assays using SYBR Green Realtime PCR Master Mix (Toyobo, Osaka, Japan).
Virus-induced gene silencing
We performed VIGS experiments as described previously (46). The IR (table S6) designed according to the promoter sequence of Zm00001d015449 was synthesized by GenScript company and cloned into the cucumber mosaic virus (CMV) VIGS (pCMV201) vector to construct the VIGS-IR plasmid. Plasmids pCMV101, pCMV301, VIGS-IR, VIGS-GFP, and VIGS-ISPH were transformed into the Agrobacterium tumefaciens strain EHA105 by electroporation. Equal amounts of different bacterial infection solutions (pCMV101 and pCMV301 and VIGS-IR or VIGS-GFP or VIGS-ISPH) were infiltrated into leaves of Nicotiana benthamiana. After 4 days, the infiltrated leaves were harvested and ground in 0.1 M phosphate buffer (pH 7.0), and the mixture was centrifuged at 3000g for 3 min at 4°C. Maize seeds were soaked in water at room temperature for 40 min and then inoculated with the supernatant containing virus crude extract using the vascular puncture inoculation method (46). The inoculated maize seeds were kept at 25°C in the dark for 2 days and then transferred into pots, and the seedlings were grown in the growth chamber under the light/dark cycle of 16/8 hours at 20°/18°C (day/night) with 60% humidity. VIGS-GFP plants and VIGS-ISPH plants were used as the control and the positive control for infection, respectively.
Reverse transcriptase qPCR
Reverse transcriptase qPCR (RT-qPCR) assays were carried out using the SYBR Green Realtime PCR Master Mix (Toyobo) in a Bio-Rad CFX96 machine (Bio-Rad Laboratories, Herclues, CA) according to the previous description (71). Gene expression levels were analyzed using the relative quantification method (ΔΔCt), and maize 18S rRNA was used as the internal control. Primers used for RT-qPCR are provided in table S6.
Bisulfite sequencing for individual genes
Genomic DNA was extracted from maize 3-week-old seedlings using the DNeasy Plant Mini Kit (QIAGEN). About 500 ng of genomic DNA was used for bisulfite conversion using the EZ DNA Methylation-Gold kit (Zymo Research, Irvine, CA) according to the manufacturer’s instructions. Bisulfite-treated DNA was then amplified by PCR using Zymo Taq DNA polymerase (Zymo Research) and primers (table S6) targeting the Zm00001d015449 promoter. The purified amplicons were cloned into pGEM-T vectors (Promega, Madison, WI). For VIGS-GFP and VIGS-IR plants, at least 14 clones were randomly chosen for sequencing. Bisulfite DNA sequences and the levels of DNA methylation at the Zm00001d015449 promoter were analyzed using the online Kismeth program (http://katahdin.mssm.edu/kismeth/revpage.pl) (72).
Acknowledgments
We thank T. Zhou at China Agricultural University for providing VIGS vectors and Y. Wu of the Bioinformatics Center at Nanjing Agricultural University for computational support and assistance in data analysis. Funding: Research at Nanjing Agricultural University is supported by grants from the National Natural Science Foundation of China (91631302), the Natural Science Foundation of Jiangsu Province (no. BK20180528), and the Jiangsu Collaborative Innovation Center for Modern Crop Production. Z.J.C. is the D. J. Sibley Centennial Professor in Plant Molecular Genetics. Author contributions: Z.J.C. and T.H. conceived and designed the project. T.H. and F.W. generated the data. T.L., L.W., and W.Y. contributed materials and reagents. Z.J.C., T.H., F.W., and Q.S. analyzed the data. T.H., F.W., Z.J.C. wrote the paper. All authors have read and approved the paper. Competing interests: The authors declare that they have no competing sinterests. Data and materials availability: Sequencing data are accessible under NCBI BioProject number (PRJNA680868) (https://ncbi.nlm.nih.gov/bioproject/PRJNA680868). All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/35/eabg5442/DC1
REFERENCES AND NOTES
- 1.C. R. Darwin, The Effects of Cross- and Self-Fertilization in the Vegetable Kingdom (John Murry, 1876). [Google Scholar]
- 2.Charlesworth D., Willis J. H., The genetics of inbreeding depression. Nat. Rev. Genet. 10, 783–796 (2009). [DOI] [PubMed] [Google Scholar]
- 3.Zhang C., Wang P., Tang D., Yang Z., Lu F., Qi J., Tawari N. R., Shang Y., Li C., Huang S., The genetic basis of inbreeding depression in potato. Nat. Genet. 51, 374–378 (2019). [DOI] [PubMed] [Google Scholar]
- 4.McMullen M. D., Kresovich S., Villeda H. S., Bradbury P., Li H., Sun Q., Flint-Garcia S., Thornsberry J., Acharya C., Bottoms C., Brown P., Browne C., Eller M., Guill K., Harjes C., Kroon D., Lepak N., Mitchell S. E., Peterson B., Pressoir G., Romero S., Oropeza Rosas M., Salvo S., Yates H., Hanson M., Jones E., Smith S., Glaubitz J. C., Goodman M., Ware D., Holland J. B., Buckler E. S., Genetic properties of the maize nested association mapping population. Science 325, 737–740 (2009). [DOI] [PubMed] [Google Scholar]
- 5.Jones D. F., Continued inbreeding in maize. Genetics 24, 462–473 (1939). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yao H., Srivastava S., Swyers N., Han F., Doerge R. W., Birchler J. A., Inbreeding depression in genotypically matched diploid and tetraploid maize. Front. Genet. 11, 564928 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dapp M., Reinders J., Bediee A., Balsera C., Bucher E., Theiler G., Granier C., Paszkowski J., Heterosis and inbreeding depression of epigenetic Arabidopsis hybrids. Nat. Plants 1, 15092 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Vergeer P., Wagemaker N. C., Ouborg N. J., Evidence for an epigenetic role in inbreeding depression. Biol. Lett. 8, 798–801 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Venney C. J., Johansson M. L., Heath D. D., Inbreeding effects on gene-specific DNA methylation among tissues of Chinook salmon. Mol. Ecol. 25, 4521–4533 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Kakutani T., Jeddeloh J. A., Flowers S. K., Munakata K., Richards E. J., Developmental abnormalities and epimutations associated with DNA hypomethylation mutations. Proc. Natl. Acad. Sci. U.S.A. 93, 12406–12411 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mathieu O., Reinders J., Caikovski M., Smathajitt C., Paszkowski J., Transgenerational stability of the Arabidopsis epigenome is coordinated by CG methylation. Cell 130, 851–862 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Gong Z., Morales-Ruiz T., Ariza R. R., Roldan-Arjona T., David L., Zhu J. K., ROS1, a repressor of transcriptional gene silencing in Arabidopsis, encodes a DNA glycosylase/lyase. Cell 111, 803–814 (2002). [DOI] [PubMed] [Google Scholar]
- 13.Stroud H., Do T., Du J., Zhong X., Feng S., Johnson L., Patel D. J., Jacobsen S. E., Non-CG methylation patterns shape the epigenetic landscape in Arabidopsis. Nat. Struct. Mol. Biol. 21, 64–72 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kribelbauer J. F., Laptenko O., Chen S., Martini G. D., Freed-Pastor W. A., Prives C., Mann R. S., Bussemaker H. J., Quantitative analysis of the DNA methylation sensitivity of transcription factor complexes. Cell Rep. 19, 2383–2395 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cubas P., Lauter N., Doebley J., Coen E., The TCP domain: A motif found in proteins regulating plant growth and development. Plant J. 18, 215–222 (1999). [DOI] [PubMed] [Google Scholar]
- 16.Clark R. M., Wagler T. N., Quijada P., Doebley J., A distant upstream enhancer at the maize domestication gene tb1 has pleiotropic effects on plant and inflorescent architecture. Nat. Genet. 38, 594–597 (2006). [DOI] [PubMed] [Google Scholar]
- 17.Yang X., Yan J., Zhang Z., Lin T., Xin T., Wang B., Wang S., Zhao J., Zhang Z., Lucas W. J., Li G., Huang S., Regulation of plant architecture by a new histone acetyltransferase targeting gene bodies. Nat. Plants 6, 809–822 (2020). [DOI] [PubMed] [Google Scholar]
- 18.McIntosh K. B., Degenhardt R. F., Bonham-Smith P. C., Sequence context for transcription and translation of the Arabidopsis RPL23aA and RPL23aB paralogs. Genome 54, 738–751 (2011). [DOI] [PubMed] [Google Scholar]
- 19.Zhao H., Zhang W., Chen L., Wang L., Marand A. P., Wu Y., Jiang J., Proliferation of regulatory DNA elements derived from transposable elements in the maize genome. Plant Physiol. 176, 2789–2803 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jones D. F., The attainment of homozygosity in inbred strains of maize. Genetics 9, 405–418 (1924). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li W., Yang Z., Yao J., Li J., Song W., Yang X., Cellulose synthase-like D1 controls organ size in maize. BMC Plant Biol. 18, 239 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen Z. J., Genomic and epigenetic insights into the molecular bases of heterosis. Nat. Rev. Genet. 14, 471–482 (2013). [DOI] [PubMed] [Google Scholar]
- 23.Liu Y., Siegmund K. D., Laird P. W., Berman B. P., Bis-SNP: Combined DNA methylation and SNP calling for Bisulfite-seq data. Genome Biol. 13, R61 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shen H., He H., Li J., Chen W., Wang X., Guo L., Peng Z., He G., Zhong S., Qi Y., Terzaghi W., Deng X. W., Genome-wide analysis of DNA methylation and gene expression changes in two Arabidopsis ecotypes and their reciprocal hybrids. Plant Cell 24, 875–892 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rigal M., Becker C., Pelissier T., Pogorelcnik R., Devos J., Ikeda Y., Weigel D., Mathieu O., Epigenome confrontation triggers immediate reprogramming of DNA methylation and transposon silencing in Arabidopsis thaliana F1 epihybrids. Proc. Natl. Acad. Sci. U.S.A. 113, E2083–E2092 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhong S., Fei Z., Chen Y. R., Zheng Y., Huang M., Vrebalov J., McQuinn R., Gapper N., Liu B., Xiang J., Shao Y., Giovannoni J. J., Single-base resolution methylomes of tomato fruit development reveal epigenome modifications associated with ripening. Nat. Biotechnol. 31, 154–159 (2013). [DOI] [PubMed] [Google Scholar]
- 27.Eichten S. R., Swanson-Wagner R. A., Schnable J. C., Waters A. J., Hermanson P. J., Liu S., Yeh C. T., Jia Y., Gendler K., Freeling M., Schnable P. S., Vaughn M. W., Springer N. M., Heritable epigenetic variation among maize inbreds. PLOS Genet. 7, e1002372 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fu F. F., Dawe R. K., Gent J. I., Loss of RNA-directed DNA methylation in maize chromomethylase and DDM1-type nucleosome remodeler mutants. Plant Cell 30, 1617–1627 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li Q., Eichten S. R., Hermanson P. J., Zaunbrecher V. M., Song J., Wendt J., Rosenbaum H., Madzima T. F., Sloan A. E., Huang J., Burgess D. L., Richmond T. A., McGinnis K. M., Meeley R. B., Danilevskaya O. N., Vaughn M. W., Kaeppler S. M., Jeddeloh J. A., Springer N. M., Genetic perturbation of the maize methylome. Plant Cell 26, 4602–4616 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang H., Lang Z., Zhu J. K., Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Biol. 19, 489–506 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Du J., Zhong X., Bernatavichute Y. V., Stroud H., Feng S., Caro E., Vashisht A. A., Terragni J., Chin H. G., Tu A., Hetzel J., Wohlschlegel J. A., Pradhan S., Patel D. J., Jacobsen S. E., Dual binding of chromomethylase domains to H3K9me2-containing nucleosomes directs DNA methylation in plants. Cell 151, 167–180 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saze H., Shiraishi A., Miura A., Kakutani T., Control of genic DNA methylation by a jmjC domain-containing protein in Arabidopsis thaliana. Science 319, 462–465 (2008). [DOI] [PubMed] [Google Scholar]
- 33.Stroud H., Greenberg M. V., Feng S., Bernatavichute Y. V., Jacobsen S. E., Comprehensive analysis of silencing mutants reveals complex regulation of the Arabidopsis methylome. Cell 152, 352–364 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bailey T. L., Boden M., Buske F. A., Frith M., Grant C. E., Clementi L., Ren J., Li W. W., Noble W. S., MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 37, W202–W208 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Menet J. S., Pescatore S., Rosbash M., CLOCK:BMAL1 is a pioneer-like transcription factor. Genes Dev. 28, 8–13 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ramos Pittol J. M., Oruba A., Mittler G., Saccani S., van Essen D., Zbtb7a is a transducer for the control of promoter accessibility by NF-kappa B and multiple other transcription factors. PLoS Biol. 16, e2004526 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rodgers-Melnick E., Vera D. L., Bass H. W., Buckler E. S., Open chromatin reveals the functional maize genome. Proc. Natl. Acad. Sci. U.S.A. 113, E3177–E3184 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhao H., Zhu X., Wang K., Gent J. I., Zhang W., Dawe R. K., Jiang J., Gene expression and chromatin modifications associated with maize centromeres. G3 6, 183–192 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.An Y., Chen L., Li Y. X., Li C., Shi Y., Song Y., Zhang D., Li Y., Wang T., Candidate loci for the kernel row number in maize revealed by a combination of transcriptome analysis and regional association mapping. BMC Plant Biol. 19, 201 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang D. W., Sherman B. T., Lempicki R. A., Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009). [DOI] [PubMed] [Google Scholar]
- 41.Welchen E., Garcia L., Mansilla N., Gonzalez D. H., Coordination of plant mitochondrial biogenesis: Keeping pace with cellular requirements. Front. Plant Sci. 4, 551 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ding S., Cai Z., Du H., Wang H., Genome-wide analysis of TCP family genes in Zea mays L. Identified a role for ZmTCP42 in drought tolerance. Int. J. Mol. Sci. 20, 2762 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Walley J. W., Sartor R. C., Shen Z., Schmitz R. J., Wu K. J., Urich M. A., Nery J. R., Smith L. G., Schnable J. C., Ecker J. R., Briggs S. P., Integration of omic networks in a developmental atlas of maize. Science 353, 814–818 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.O'Malley R. C., Huang S. C., Song L., Lewsey M. G., Bartlett A., Nery J. R., Galli M., Gallavotti A., Ecker J. R., Cistrome and epicistrome features shape the regulatory DNA landscape. Cell 165, 1280–1292 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wieckowski Y., Schiefelbein J., Nuclear ribosome biogenesis mediated by the DIM1A rRNA dimethylase is required for organized root growth and epidermal patterning in Arabidopsis. Plant Cell 24, 2839–2856 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang R., Yang X., Wang N., Liu X., Nelson R. S., Li W., Fan Z., Zhou T., An efficient virus-induced gene silencing vector for maize functional genomics research. Plant J. 86, 102–115 (2016). [DOI] [PubMed] [Google Scholar]
- 47.Kanazawa A., Inaba J. I., Shimura H., Otagaki S., Tsukahara S., Matsuzawa A., Kim B. M., Goto K., Masuta C., Virus-mediated efficient induction of epigenetic modifications of endogenous genes with phenotypic changes in plants. Plant J. 65, 156–168 (2011). [DOI] [PubMed] [Google Scholar]
- 48.Lu X. M., Hu X. J., Zhao Y. Z., Song W. B., Zhang M., Chen Z. L., Chen W., Dong Y. B., Wang Z. H., Lai J. S., Map-based cloning of zb7 encoding an IPP and DMAPP synthase in the MEP pathway of maize. Mol. Plant 5, 1100–1112 (2012). [DOI] [PubMed] [Google Scholar]
- 49.Ni Z., Kim E. D., Ha M., Lackey E., Liu J., Zhang Y., Sun Q., Chen Z. J., Altered circadian rhythms regulate growth vigour in hybrids and allopolyploids. Nature 457, 327–331 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Noordally Z. B., Ishii K., Atkins K. A., Wetherill S. J., Kusakina J., Walton E. J., Kato M., Azuma M., Tanaka K., Hanaoka M., Dodd A. N., Circadian control of chloroplast transcription by a nuclear-encoded timing signal. Science 339, 1316–1319 (2013). [DOI] [PubMed] [Google Scholar]
- 51.Ng D. W.-K., Miller M., Yu H. H., Huang T. Y., Kim E. D., Lu J., Xie Q., McClung C. R., Chen Z. J., A role for CHH methylation in the parent-of-origin effect on altered circadian rhythms and biomass heterosis in Arabidopsis intraspecific hybrids. Plant Cell 26, 2430–2440 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Song Q., Huang T. Y., Yu H. H., Ando A., Mas P., Ha M., Chen Z. J., Diurnal regulation of SDG2 and JMJ14 by circadian clock oscillators orchestrates histone modification rhythms in Arabidopsis. Genome Biol. 20, 170 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ko D. K., Rohozinski D., Song Q., Taylor S. H., Juenger T. E., Harmon F. G., Chen Z. J., Temporal shift of circadian-mediated gene expression and carbon fixation contributes to biomass heterosis in maize hybrids. PLOS Genet. 12, e1006197 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bass J., Takahashi J. S., Circadian integration of metabolism and energetics. Science 330, 1349–1354 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wijnen H., Young M. W., Interplay of circadian clocks and metabolic rhythms. Annu. Rev. Genet. 40, 409–448 (2006). [DOI] [PubMed] [Google Scholar]
- 56.Walker E. L., Paramutation of the r1 locus of maize is associated with increased cytosine methylation. Genetics 148, 1973–1981 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chandler V. L., Walbot V., DNA modification of a maize transposable element correlates with loss of activity. Proc. Natl. Acad. Sci. U.S.A. 83, 1767–1771 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Styles E. D., Brink R. A., The metastable nature of paramutable R alleles in maize. I. Heritable enhancement in level of standard R action. Genetics 54, 433–439 (1966). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Robertson D. S., Genetic studies on the loss of mu mutator activity in maize. Genetics 113, 765–773 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hollister J. D., Gaut B. S., Epigenetic silencing of transposable elements: A trade-off between reduced transposition and deleterious effects on neighboring gene expression. Genome Res. 19, 1419–1428 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang L., Cao S., Wang P., Lu K., Song Q., Zhao F. J., Chen Z. J., DNA hypomethylation in tetraploid rice potentiates stress-responsive gene expression for salt tolerance. Proc. Natl. Acad. Sci. U.S.A. 118, e2023981118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Song Q., Zhang T., Stelly D. M., Chen Z. J., Epigenomic and functional analyses reveal roles of epialleles in the loss of photoperiod sensitivity during domestication of allotetraploid cottons. Genome Biol. 18, 99 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lu J., Zhang C., Baulcombe D. C., Chen Z. J., Maternal siRNAs as regulators of parental genome imbalance and gene expression in endosperm of Arabidopsis seeds. Proc. Natl. Acad. Sci. U.S.A. 109, 5529–5534 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bajic M., Maher K. A., Deal R. B., Identification of open chromatin regions in plant genomes using ATAC-Seq. Methods Mol. Biol. 1675, 183–201 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gent J. I., Madzima T. F., Bader R., Kent M. R., Zhang X., Stam M., McGinnis K. M., Dawe R. K., Accessible DNA and relative depletion of H3K9me2 at maize loci undergoing RNA-directed DNA methylation. Plant Cell 26, 4903–4917 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu N., Liu J., Li W., Pan Q., Liu J., Yang X., Yan J., Xiao Y., Intraspecific variation of residual heterozygosity and its utility for quantitative genetic studies in maize. BMC Plant Biol. 18, 66 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Song Q., Guan X., Chen Z. J., Dynamic roles for small RNAs and DNA methylation during ovule and fiber development in allotetraploid cotton. PLOS Genet. 11, e1005724 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Johnson N. R., Yeoh J. M., Coruh C., Axtell M. J., Improved placement of multi-mapping small RNAs. G3 6, 2103–2111 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Buenrostro J. D., Giresi P. G., Zaba L. C., Chang H. Y., Greenleaf W. J., Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen K., Xi Y., Pan X., Li Z., Kaestner K., Tyler J., Dent S., He X., Li W., DANPOS: Dynamic analysis of nucleosome position and occupancy by sequencing. Genome Res. 23, 341–351 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang F., Han T., Song Q., Ye W., Song X., Chu J., Li J., Chen Z. J., The rice circadian clock regulates tiller growth and panicle development through strigolactone signaling and sugar sensing. Plant Cell 32, 3124–3138 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gruntman E., Qi Y., Slotkin R. K., Roeder T., Martienssen R. A., Sachidanandam R., Kismeth: Analyzer of plant methylation states through bisulfite sequencing. BMC Bioinformatics 9, 371 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/35/eabg5442/DC1